
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeted esterase-induced dye (TED) loading supports direct calcium imaging in eukaryotic cell-free systems†
Priyavathi
Dhandapani
a,
Srujan Kumar
Dondapati
a,
Anne
Zemella
a,
Dennis
Bräuer
a,
Doreen Anja
Wüstenhagen
a,
Stefan
Mergler
c and
Stefan
Kubick
 *ab
*ab
aFraunhofer Institute of Cell Therapy and Immunology, Branch of Bioanalytics and Bioprocesses (IZI-BB), Am Muehlenberg 13, Potsdam-Golm, Germany. E-mail: stefan.kubick@izi-bb.fraunhofer.de
bFaculty of Health Sciences, Joint Faculty of Brandenburg University of Technology, Cottbus – Senftenberg, Theodor Fontane Medical School of Brandenburg, University of Potsdam, Germany
cDepartment of Ophthalmology, Charité – Universitätsmedizin Berlin, Campus Virchow-Hospital, Berlin, Germany
First published on 4th May 2021
Abstract
Calcium imaging is an important functional tool for analysing ion channels, transporters and pumps for drug screening in living cells. Depicted eukaryotic cell-free systems utilize microsomes, derived from the endoplasmic reticulum to incorporate the synthesized membrane proteins-like ion channels. Carboxylesterase is required to cleave the acetoxymethyl ester moiety of the chemical calcium indicators in order to ensure its immobility across the endoplasmic reticulum membrane. Absence or an inadequate amount of carboxylesterase in the endoplasmic reticulum of different eukaryotic cells poses a hindrance to perform calcium imaging in microsomes. In this work, we try to overcome this drawback and adapt the cell-based calcium imaging principle to a cell-free protein synthesis platform. Carboxylesterase synthesized in a Spodoptera frugiperda Sf21 lysate translation system is established as a viable calcium imaging tool in microsomes. Cell-free synthesized carboxylesterase inside microsomes is validated with esterase and dye loading assays. Native proteins from the endoplasmic reticulum, such as ryanodine channels and calcium ATPase, are analysed. Cell-free synthesized transient receptor potential channels are used as model proteins to demonstrate the realization of this concept.
1 Introduction
Ion permeable membrane proteins such as ion channels, transporters and pumps contribute to the majority of eukaryotic membrane proteins, serving as viable drug targets for several pathological diseases next to the large family of G-protein coupled receptors.1 Eukaryotic cell-free protein translation overcomes several disadvantages that could be met for overexpression of these ion permeable proteins in cells, such as cell-toxicity, poor expression and deletion due to engineered protein domains, and reduction of expression in permanent cell lines.2 Taken together, the development of functional assays targeting ion channels in eukaryotic cell-free systems is a pre-requisite for rapid pharmacological discoveries.The planar bilayer, radioactive ion-flux assays and fluorescent techniques (ion-sensitive dyes) are common methods to study ion channels. Eukaryotic cell-free systems use microsomes to integrate synthesized membrane proteins. A handful of planar bilayer investigations in eukaryotic cell-free systems have been previously reported.3,4 But the planar bilayer method requires the solubility of ion channels using detergents for subsequent evaluation. Notwithstanding the temporal resolution given by electrophysiological techniques like planar bilayer, fluorescent dye-based methods have been an indispensable tool encompassing a plethora of advantages: ease of applicability for High Throughput Screening (HTS) using a microplate reader, low cost per data point, minimum instrumentation requirements, high spatial resolution and relatively less sophisticated data analysis. Another method to analyse ion permeable proteins in microsomes would be by employing radioactive calcium. Intraluminal calcium is the prime regulator for endoplasmic reticulum (ER) function5,6 and has been analysed concomitantly in microsomes using 45Ca2+ to study native ion channels and pumps.7–11 Apart from the undesirable radioactivity, with a major drawback being an end-point based method, it will also increase the material costs to study the kinetics. Moreover, first the microsomes should be enriched with 45Ca2+ using Ca2+ ATPase (SERCA) for any kind of calcium-efflux studies. Apart from the TED method which we utilize in this work, also other fluorescent methods can be employed, for instance, using membrane impermeable Ca2+ dyes to measure the calcium present in the extra-microsomal solution,12 where luminal calcium levels cannot be monitored directly. As the microsomes are not fixed on the microplate/cuvette in this method, live monitoring of Ca2+ is impracticable while exchanging activator and inhibitor solutions. Considering the disadvantages of the present methods, the necessity to establish direct calcium imaging to monitor the microsomal lumen for ion channels expressed in eukaryotic cell-free platforms is substantiated. The previously reported idea of targeted esterase-induced dye (TED) loading of the endoplasmic reticulum (ER) in mammalian cells,13–15 augmented with the current knowledge of eukaryotic cell-free systems2,16 led us to develop this new method to address the aforementioned shortcomings and also to explore new topics of research.
Eukaryotic cell-free protein synthesis platforms use microsomes in order to incorporate the membrane proteins. The soluble proteins like carboxylesterases can be engineered with signal peptide sequences to be delivered inside the microsomes after synthesis. Carboxylesterase, ubiquitously present in the cytoplasm of most of the eukaryotic cells, is the crucial enzyme for cell-based calcium imaging using acetoxymethyl ester (AM) based chemical dyes. The carboxylesterase enzyme aids in cleaving the AM moiety of the chemical indicator, thereby making the dye immobile across any biological membrane. Immobility of the cleaved dye is caused due to the unmasking of the polar negative charge of the dye. Absence or an inadequate amount of carboxylesterase in the endoplasmic reticulum of different eukaryotic cells, which is necessary to cleave the acetoxymethyl ester moiety of the chemical calcium indicators effectuates the insufficiency of immobile dye formation inside the microsomes. In this work, we try to overcome this drawback and adapt the cell-based calcium imaging principle to a cell-free protein synthesis platform as depicted in Fig. 1. Carboxylesterase synthesized in the Sf21 lysate translation system which is targeted to be delivered inside the microsomes is established as a viable calcium imaging tool to investigate both native proteins present in the ER and also cell-free synthesized ion channels.
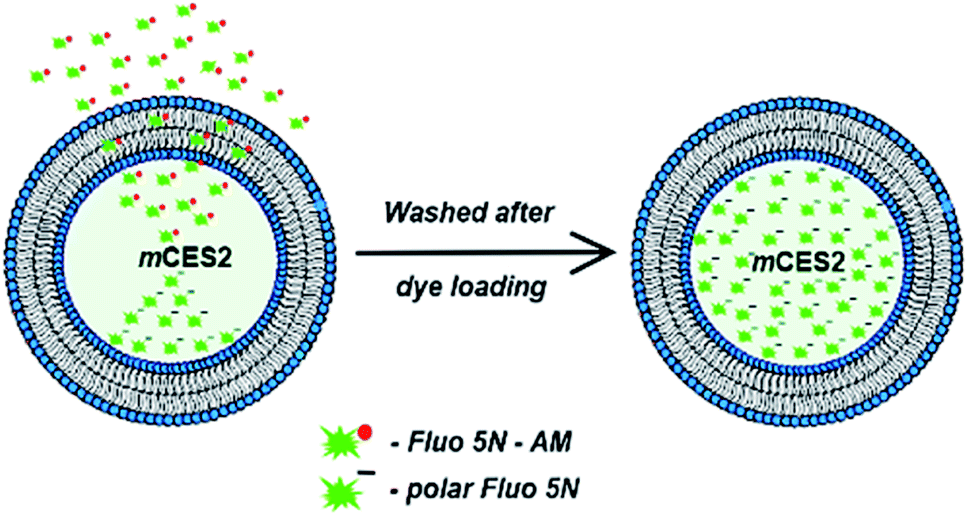 | ||
| Fig. 1 Unmasking polarity of the dye by carboxylesterase mCES2 in microsomes. | ||
2 Materials and methods
2.1. Continuous exchange cell-free (CECF) translation
Eukaryotic CECF translation of proteins was performed using Sf21 lysates in a special dialysis chamber (SCIENOVA) containing two compartments separated by a 10 kDa cut off dialysis membrane in between the reaction mixture and the feeding mixture. A 50 µL standard reaction mixture of a Sf21 cell-free synthesis reaction in the reaction chamber was composed of 40% lysate, 30 mM HEPES-KOH (Merck), 2.5 mM Mg(OAc)2 (Merck), 75 mM KOAc (Merck), 0.25 mM spermidine (Roche), 100 µM each canonical amino acid (Merck), nucleoside triphosphates (1.75 mM ATP, 0.30 mM CTP, 0.30 mM GTP, and 0.30 mM UTP) (Roche), 120 ng µL−1 plasmid DNA (Biocat), 1 U mL−1 T7 RNA-polymerase (Agilent), 20 µM PolyG (Iba Life sciences), 30 µM caspase inhibitor – Z-VAD-FMK (benzyloxycarbonyl-Val-Ala-Asp(OMe)-fluoromethylketone) (Promega), and 0.02% of sodium azide (Merck). 1 mL of the feeding mixture contained all the above components except plasmid, PolyG, T7 RNA polymerase and Sf21 lysate. Caspase inhibitor was used for improving yields and avoiding protein degradation during the incubation step for 24 h. No plasmid was used in NTC (non-template control) samples. For expression of both the proteins, human transient receptor potential channels, vanilloid receptor member 1, hTRPV1 and mouse carboxylesterase 2 (mCES2) were correspondingly sequentially translated for 24 h each. As mCES2 and hTRPV1 contain disulphide bridges, prudently the translation is performed only under non-reducing conditions. As the microsomes obtained from the first translation were used in the second translation, it may occur that the first translated protein is not present in all the microsomes at the end of the second translation. To avoid this issue, we have used only the vesicular fraction obtained after the second translation for all our experiments that require tandem protein translation.Simultaneous translation is not preferred as evaluation of the yields of individual proteins synthesized is not feasible with the same radiolabel. 14C-Labeled leucine (100 dpm per pmol) (Perkin Elmer) was used for the detection of de novo synthesized proteins. For functional analysis like assays and calcium imaging, the proteins were synthesized in the absence of 14C-leucine. Protein translation reactions based on Sf21 lysates were incubated for 24 h at 30 °C, 600 rpm using a thermomixer (Eppendorf). The translation mixture (TM) of cell-free reactions was further fractionated for analysis. The fractionation was realized by centrifugation at 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 10 min at 4 °C in order to separate the ER-derived vesicular fraction (VF) of the cell lysate from the supernatant (SN). The microsomal fraction was suspended in PBS buffer without calcium and magnesium ions for further analysis such as quantification of protein yields. For storage, the total translation mix was snap frozen in liquid nitrogen and stored at −80 °C.
000 × g for 10 min at 4 °C in order to separate the ER-derived vesicular fraction (VF) of the cell lysate from the supernatant (SN). The microsomal fraction was suspended in PBS buffer without calcium and magnesium ions for further analysis such as quantification of protein yields. For storage, the total translation mix was snap frozen in liquid nitrogen and stored at −80 °C.
2.2. Quantification of cell-free synthesized protein yields
Based on the incorporation of 14C-leucine in cell-free synthesized proteins, the respective protein yield can be estimated by scintillation measurement. Therefore, 5 µL aliquots of each translation mixture were mixed with 3 mL of a 10% (v/v) trichloroacetic acid–2% (v/v) casein hydrolysate (Carl Roth) solution in a glass tube and incubated at 80 °C for 15 min. Afterwards, the samples were chilled on ice for 30 min and retained on the surface of glass fiber filter papers using a vacuum filtration system (Hoefer). Filter papers were washed twice with 5% TCA and then vacuum dried with acetone (Carl Roth). Dried filters were placed into a scintillation vial, 3 mL of scintillation cocktail was added and vials were agitated on an orbital shaker for at least 1 h. The scintillation signal was determined using an LS6500 multi-purpose scintillation counter (Beckman Coulter). The protein yields were identified based on the obtained scintillation counts and protein specific parameters including molecular mass and amount of leucine.2.3. SDS-PAGE and autoradiography
The molecular size of radio-labelled, cell-free synthesized protein was analysed using SDS-PAGE followed by autoradiography. First, 5 µL of the respective fraction of a cell-free synthesis reaction including the radio-labelled target protein was subjected to ice-cold acetone. Precipitated protein was separated by centrifugation (16000 × g, 4 °C, 10 min) and then, the protein pellet was dried for at least 30 min at 45 °C. The dried protein pellet was dissolved in LDS sample loading buffer with 50 mM dithiothreitol (DTT, Life Technologies) and loaded on a pre-cast NuPAGE 10% Bis-Tris gel (Life Technologies). If the sample is a soluble protein, then it was also heated at 95 °C for 3 min prior to loading on the gel. The gel was run at 185 V for 35 min according to the manufacturer's protocol. Subsequently, the gel was dried at 70 °C using a gel dryer (Uniequip) and then placed on a phosphor screen for incubation for a minimum of two days. The radioactively labelled proteins were visualized using a Typhoon Trio + variable mode (GE Healthcare).
2.4. Esterase activity using 4-para-nitrophenol
First, mCES2 and NTC proteins were translated using Sf21 CECF reaction as mentioned above. Prior to functional assessment, intensive washing of microsomes was performed to remove the cytosolic carboxylesterase which is carried over from cells to the lysate. This cytosolic carboxylesterase is present outside the microsomes. The esterase activity is preferably analysed for a maximum of 1 h. 50 µL of total translation mix was first centrifuged at 16000 × g for 10 minutes at 4 °C. The microsomal pellet was again washed with Phosphate Buffer Saline (PBS) with no Ca2+ and Mg2+ and centrifuged again to remove the remaining cytosolic carboxylesterases. The pellet was dissolved in esterase assay buffer containing 20 mM Tris–HCl (pH 8.0) (Sigma Aldrich), 150 mM NaCl (Sigma Aldrich), and 0.01% Triton X-100 (Sigma Aldrich). A fresh solution of 4-para-nitrophenylacetate (PNPA) (Sigma Aldrich) was used as the substrate. 250 µL of PNPA substrate solution of varying concentration (mentioned in the figure legend of each experiment) was used to initiate the reaction and the mixture was incubated at 37 °C for 1 hour (or less than 1 hour with varying time for time dependent plot). For the substrate dependence plot, varied concentration of PNPA was used. The para-nitro phenol formed after esterase activity was measured using a Mithras Plate reader (Berthold Technologies) at 410 nm. No protein was added for blank reactions and the esterase activity was evaluated as the percentage of NTC samples.
2.5. Dye loading assays with Fluo-5N AM
First, mCES2 and NTC were translated using Sf21 CECF reaction as mentioned above in the synthesis section. 50 µL of translation mix was first centrifuged at 16000 × g for 10 minutes at 4 °C to obtain the microsomal pellet to remove the cytosolic carboxylesterases present outside the microsomes. The microsomes were resuspended in ATP based calcium imaging buffer to initiate the SERCA activity. The calcium imaging buffer was composed of 75 mM KCl (Sigma Aldrich), 20 mM HEPES-KOH (Merck), 5 mM NaN3 (Merck) and 200 µM CaCl2 (Sigma Aldrich) with pH 7.4. To enhance the SERCA activity, 10 mM adenosine 5′-triphosphate (ATP) (Roche), 1 mM MgCl2 (Sigma Aldrich), 0.5 mM dithiothreitol (DTT) (Life Technologies), 5 mM phosphocreatine (PCr) (Sigma Aldrich), and 20 U mL−1 creatine phosphokinase (CPK) (Sigma Aldrich) were used. SERCA activity induced Ca2+ loading was performed at 37 °C for varying time (mentioned in the figure legend of each experiment) and then stopped by centrifuging and removing the supernatant. The pellet was suspended in 100 µL of Fluo-5N AM dye (Thermo Fischer) of varying concentration (mentioned in the figure legend of each experiment) and then incubated at 37 °C at 500 rpm. The reaction was stopped by centrifugation and removal of the dye. Microsomes were further washed with 100 µL of PBS to purge the un-cleaved Fluo-5N AM at 37 °C for 20 minutes and centrifuged to obtain the microsomal pellet containing only the cleaved dye. Then, the pellet was washed and the fluorescence removed subsequently was measured in a plate reader (Berthold Technologies) with Ex 488 and Em 515 nm with the help of a black plate. The blank sample measurements were subtracted and the data were analysed.
2.6. SERCA activity by fluorescence spectroscopy
50 µL of cell-free translation mix after the reaction was first centrifuged at 16000 × g for 10 minutes at 4 °C to obtain the pellet containing the vesicular fraction. The pellet was suspended in ATP based calcium imaging buffer to initiate the SERCA activity. SERCA activity induced Ca2+ loading was performed at 37 °C for 60 minutes and then stopped by centrifuging and removing the supernatant. To see the effect of thapsigargin (TG) on the SERCA activity, microsomes were also additionally incubated with SERCA buffer in the presence of 100 nM TG, with and without ATP. After the SERCA activity step, the pellet was suspended in 100 µL of Fluo-5N AM dye of 5 µM concentration and then incubated at 37 °C at 500 rpm. The reaction was stopped by centrifugation and removal of the dye. Microsomes were further washed with 100 µL of PBS to remove the uncleaved Fluo-5N AM from the microsomes by incubation at 37 °C for 20 minutes and centrifuged to obtain the microsomal pellet containing only the cleaved dye. Then, the suspended pellet was measured using a plate reader with Ex 488 and Em 515 nm. The blank values were subtracted and data were analysed.
2.7. Calcium imaging using confocal laser microscopy
For all calcium measurements, the microsomes were seeded for attachment on the coverslip coated with poly-D-lysine hydrobromide preferably with a high molecular weight for microsomal membranes. The coverslips were autoclaved and coated overnight with poly-D-lysine hydrobromide (0.1 mg mL−1) (Sigma Aldrich), dried and stored at room temperature. The microsomes were seeded for 1 h at 37 °C and the coverslips were washed with the calcium imaging buffer for the removal of unsettled microsomes. Though 5 µM Fluo-5N AM was used for dye loading experiments performed using a microplate reader, 2 µM Fluo-5N AM was enough for qualitative and quantitative measurements of calcium with confocal microscopy without any loading enhancers like Pluronic F-127, probenecid or saponin. A flow chamber (Warner Instruments) fitted with a coverslip at the bottom with the aid of a vacuum sealing agent was used for all measurements. An argon laser with Alexa 488 with 3% intensity, maximum gain and 5–6 airy units was used for all measurements using the LSM Meta 510 software (Carl Zeiss) time series function with a frequency of one data point per 30 s. A 40× oil immersion objective with a numerical aperture of 1.3 was used. All data are represented as delta F/F, where delta F represents the difference in fluorescence of microsomes and the background. Different regions of interest were selected from each individual experiment from a frame of 512 µM × 512 µM to ensure whether an identical increase or decrease in intensity is observable. The slope of bleaching was drift corrected using the ‘peak and baseline correction protocol’ with the help of OriginPro 2015 software. For comparison of individual experiments with activators and inhibitors, the baseline fluorescence intensity was normalized to 100 a.u. and then an increase or decrease in intensity was analysed. Plotting graphs was performed using OriginPro and Microsoft Excel software.2.8. Statistical analysis
For all statistical analysis, GraphPad Prism Version 5.0 software was used. The data used for statistical analysis were first checked by the normality test (at least one of the tests such as KS normality test, Shapiro–Wilk normality test or D'Agostino and Pearson omnibus test should be passed to consider the data in Gaussian distribution). When the data sets fall under Gaussian distribution, student t tests are performed for paired sample data and unpaired student t tests are performed for unpaired sample data. For datasets that don't fall in Gaussian distribution, non-parametric tests such as the Wilcoxon matched pairs test are performed for paired sample data and the Mann–Whitney test is performed for unpaired sample data. Welch correction is applied when two data sets have non-equal variances. All the tests were performed to evaluate two-tailed p values with a confidence level of 95% and statistical significance is indicated by * or #.3 Results and discussion
3.1. Cell-free synthesis using the Sf21 system
In order to establish a vesicle-based calcium imaging tool, an mCES2 cell-free construct was used to incorporate the carboxylesterase inside the lumen as depicted in Fig. 2a and b. A CrPV IRES translation system with a start codon as GCT, alanine, instead of ATG, methionine in the Sf21 cell-free system was used in the plasmid constructs to improve the translational turnover as shown in Fig. 2a.17 A melittin signal sequence upstream of the gene was utilized in order to deliver the soluble protein inside the microsomal lumen as mentioned previously.18 The mCES2 with a melittin signal sequence is recruited inside the lumen through the SEC translocon and the signal sequence is cleaved by the signal peptidase present in the microsomal membrane as depicted in Fig. 2b. T7 promoter and T7 terminator were used for mRNA transcription. No signal sequence was used for the hTRPV1 construct as hTRPV1 is a poly-transmembrane protein. For the cell-free synthesis of mCES2 and hTRPV1, we used the Continuous Exchange Cell-Free (CECF) mode of our Sf21 translation system.19,20 This mode of synthesis has mainly the amino acids and the energy components in a larger compartment and the eukaryotic translation mix in a smaller compartment separated by a dialysis membrane of 9 kDa cut off. Caspase inhibitor was used for improved yields and avoiding protein degradation during the incubation step for 24 h. PolyG nucleotide (30 nucleotides) was used to enhance the CrPV IRES mediated translation. As hTRPV1 and mCES2 contain disulphide bridges, prudently the translation is performed only under non-reducing conditions. mCES2 and hTRPV1 synthesized in CECF reaction in Fig. 2c and d showed 181 ng µL−1 and 85 ng µL−1 correspondingly. The synthesized mCES2 and hTRPV1 showed a molecular weight of 58 kDa and 98 kDa monomer bands correspondingly in the SDS gel-autoradiogram run under reducing conditions. No protein band was observed in the NTC (Non-Template Control) samples as shown in the autoradiogram in Fig. 2d. | ||
| Fig. 2 Cell-free protein synthesis of hTRPV1 and mCES2: (a) template design of mCES2 and hTRPV1; MEL – melittin signal sequence, T7 P – T7 promoter, T7 T – T7 terminator, and IRES – cricket paralysis virus Internal Ribosome Entry Site; (b) schematic representation of cell-free synthesis of mCES2; (c) protein yields of mCES2 by scintillation counting via CECF reaction for 24 h at 30 °C in the Sf21 system; (d) protein yields of hTRPV1 by scintillation counting via CECF reaction for 24 h at 30 °C in the Sf21 system; (e) autoradiogram of proteins run on SDS MES gel. | ||
3.2. Validation of synthesized mCES2 in the microsomal lumen
Carboxylesterases ubiquitously present in the cytosol also result in lysates but it will not aid in calcium imaging as they are present outside the microsomes. The functionality of the mCES2 synthesized inside the microsomes is assessed using the most commonly used para-nitrophenyl acetate (PNPA) method21,22 (Fig. 3a–c). The PNPA method is an inexpensive and prompt method to assess the esterase activity. mCES2 microsomes showed higher activity relative to the NTC microsomes for 1 h PNPA incubation (p value = 0.0003, *** unpaired t test, two tailed) as shown in Fig. 3a. mCES2 microsomes also showed higher esterase activity relative to NTC microsomes in substrate dependence (Fig. 3b), time dependence and dose dependence (Fig. 3c, ESI Table 1†) measurements. All the above-mentioned plots showed esterase activity, which was linearly dependent on the x-axis parameters.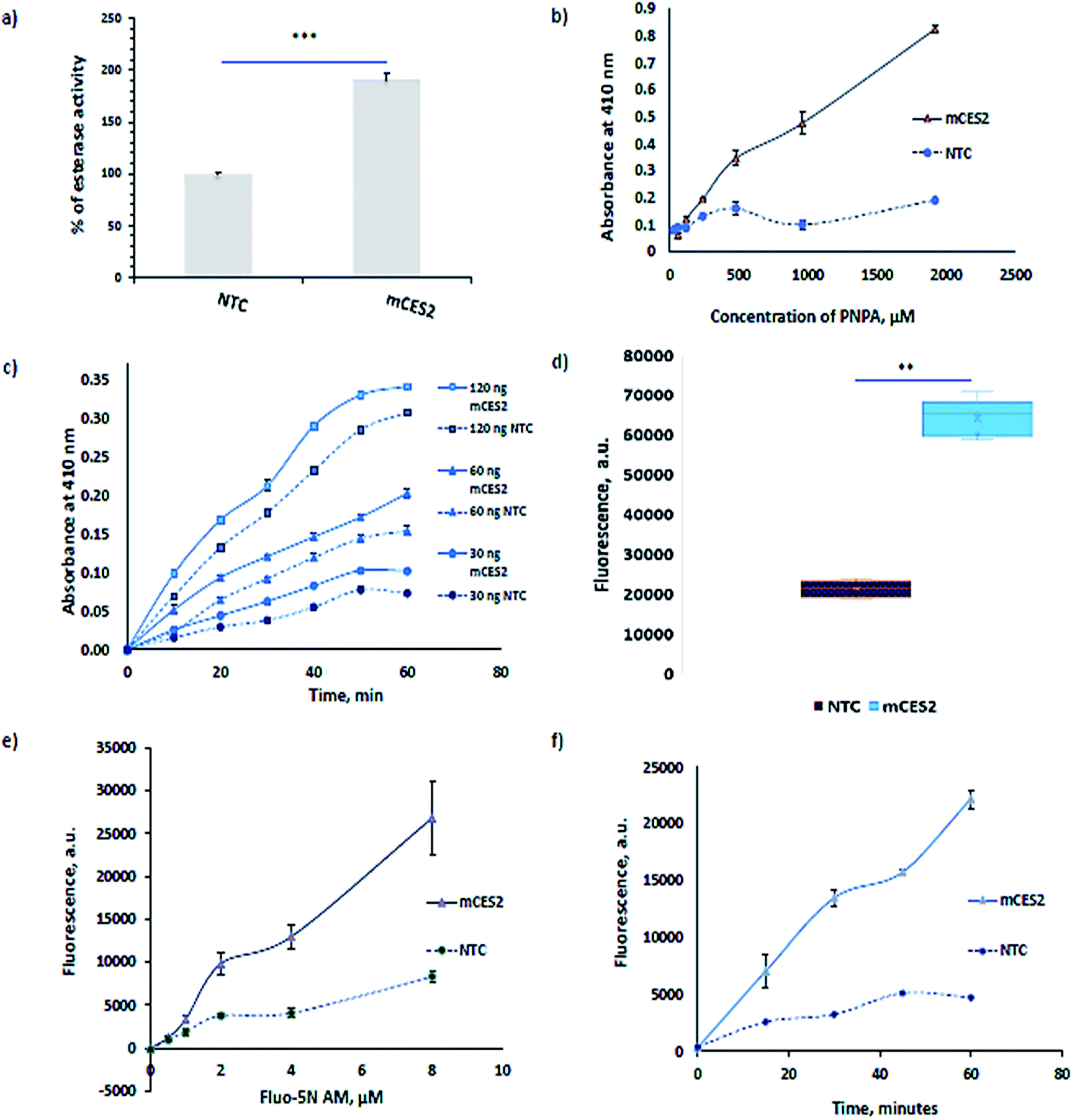 | ||
| Fig. 3 Esterase activity using PNPA: (a) esterase activity of mCES2 using PNPA in the microsomes, 37 °C, 60 min, 100 ng in 0.3 mM PNPA, n = 6; ***, p = 0.0003, unpaired t test, two tailed; (b) substrate concentration dependent esterase activity plot of mCES2, 37 °C, 60 min, 60 ng of mCES2, n = 3; (c) time and enzyme concentration dependence plot of mCES2, 37 °C with 0.3 mM PNPA, n = 3. Calcium dye loading experiments: (d) esterase activity performed with 200 ng of mCES2 each in 100 µL of 5 µM Fluo 5N-AM, 37 °C, 60 min, n = 5; ***, p < 0.0001, unpaired t test, two tailed; (e) Fluo 5N-AM concentration dependent activity with 100 ng of mCES2 protein each in 100 µL reaction, 60 minutes, 37 °C, n = 2; (f) time dependent esterase activity with 100 ng of mCES2 each in 100 µL of 5 µM Fluo 5N-AM, 37 °C, n = 2. | ||
For calcium dye loading experiments, Fluo-5N AM was used due to its low binding affinity (90 µM in buffer) for Ca2+. The masked negative charge of the dye by the acetoxymethyl ester moiety is unveiled with the aid of mCES2 inside the microsomes. Dye loading experiments also reveal a similar pattern shown by PNPA assay. mCES2 microsomes cleaved a significantly higher amount of Fluo-5N AM dye compared to the NTC microsomes when incubated at 5 µM concentration for 1 h as shown in Fig. 3d (p value = <0.0001, *** unpaired t test, two tailed). The mCES2 overexpression inside the microsomes significantly enhances the amount of dye loaded and reduces the dye incubation time relative to NTC microsomes as observed in Fig. 3d–f. Both time dependence plot and substrate dependence plot showed a higher amount of dye loaded and cleaved for the AM moiety by mCES2 microsomes relative to the NTC microsomes, Fig. 3e and f. The dye concentration dependence plot and the time dependence plot both show that the AM-cleaving activity shows a linear profile with the x-axis parameters even at 60 min, with 8 µM of the dye, which is coherent with the expected range of activity for calcium imaging applications.
The activity observed in the NTC microsomes is probably due to the unspecific activity of hydroxylase or a mono-oxygenase class of cytochrome P450 enzymes present in the endoplasmic reticulum.23,24
3.3. Establishment of TED based calcium imaging in microsomes
The Kd of Fluo-5N AM for Sf21 lysate microsomes was calculated to be 265 µM (Fig. 4a). The experiment was performed by first chelating resting calcium levels in the microsomes by 5 mM EGTA (ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetra-acetic acid) and then sequentially increasing the concentration of Ca2+ from 0 to 1 mM every step by 100 µM in the presence of 10 µM ionomycin. The binding affinity, Kd of the dye for calcium in the endoplasmic reticulum is usually higher than phosphate buffer saline buffer 90 µM (ref. 25) and varies congruently with expression levels of calcium binding proteins such as calnexin and calreticulin.26 For example, SR vesicles rabbit ventricular myocytes show a Kd of 400 µM (ref. 27) and mouse skeletal muscle SR has a Kd of 133 µM.14 A change in temperature alters the Kd with a change in the sensitivity range and the fluorescence life time which in turn affects the bleaching rate of the Fluo-5N AM.28 Kd and temperature are inversely related. Hence, all measurements are preferably performed at room temperature.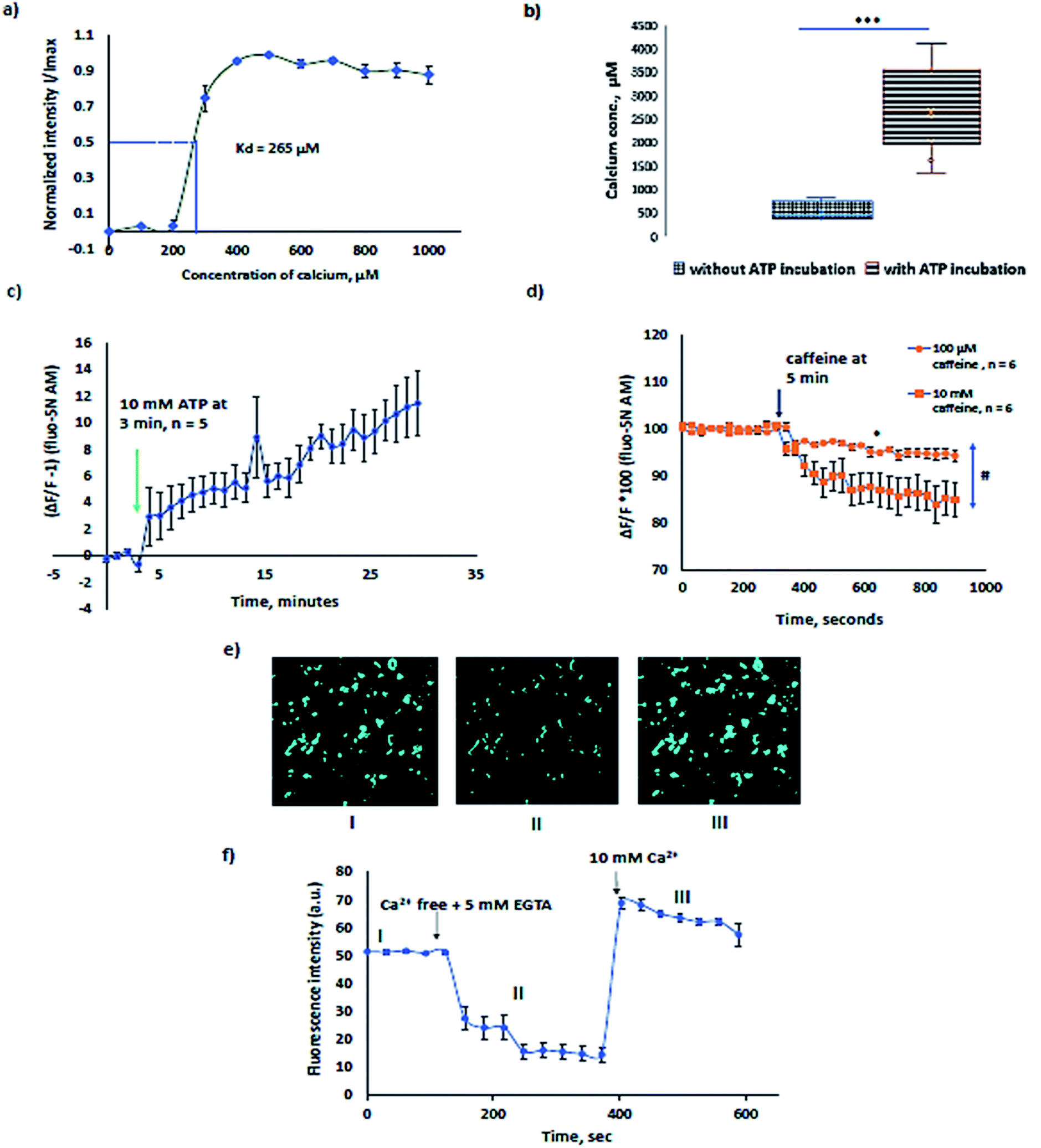 | ||
| Fig. 4 Direct calcium imaging using mCES2 microsomes: (a) Kd evaluation of Fluo 5N-AM in the Sf21 microsomes in the presence of 10 µM ionomycin, n = 4; (b) calcium levels of ATP induced Ca2+ loaded and non-loaded microsomes at 37 °C for 1 h, n = 5 each; the data are represented as a box plot with max and min, and *** indicates significant difference, p < 0.0001, Mann–Whitney test, two tailed; (c) SERCA pump activity in microsomes with 10 mM ATP added at 3 min and measured for 30 min, RT, n = 5. The data before adding ATP are normalized to 0; (d) caffeine induced calcium release in mCES2 microsomes via ryanodine receptor (RyR2) activation, n = 6 each for 10 mM and 100 µM caffeine; * indicates the significant difference of 100 µM caffeine sample data at 150 s and 600 s, p = 0.0313, Wilcoxon matched pair test, two tailed. # indicates the significant difference between 100 µM and 10 mM caffeine samples at 600 s, p value = 0.0247, Mann–Whitney test, two tailed. Calcium was loaded in the mCES2 microsomes with ATP for 60 min at 37 °C prior to caffeine experiments; (e) representative confocal images of I, II and III from different states of graph (f); (f) representative graph of calcium concentration determination using Ca2+ free + 5 mM EGTA and 10 mM Ca2+ in the presence of 10 µM ionomycin. All data in (a), (c), (d), and (f) are presented as mean ± S.E.M. | ||
In order to study the native proteins in microsomes, extensively investigated proteins such as sarcoplasmic reticulum ATPase (SERCA) and ryanodine receptors were choice of interest. In Sf21 microsomes, SERCA activity was previously recorded with radiolabelled 45Ca2+.29,30 The typical [Ca2+] concentration in the ER varies from few µM to mM.31 Calcium binding proteins like calreticulin, calnexin, Glucose Regulated Protein 78 (GRP78), glucose regulated protein 94 (GRP94), Endoplasmic Reticulum Protein (ERp72), protein disulphide isomerase, reticulocalbin, and Endoplasmic Reticulum Calcium binding protein 55 (ERC55) act as buffers, causing multiple orders of variation in the [Ca2+] of the ER due to different individual binding affinities.32,33 The free [Ca2+] in Sf21 microsomes that undergo CECF translation was estimated to be in the range of 100 to 1000 µM (Fig. 4b) which is coherent with previously reported data in other eukaryotic cells. To avoid artifacts caused by temperature, we preferred to evaluate the increase in Ca2+ levels in microsomes at room temperature after treatment of microsomes at 37 °C with ATP. Microsomes treated with 10 mM ATP, 1 mM Mg2+ and 200 µM Ca2+ for 1 h at 37 °C showed a 4–5 times higher range of luminal Ca2+. The sensitivity range of the dye varies from 0.1 to 100 × Kd. Microsomes treated for SERCA activity showed a median value of 2500 µM (Fig. 4b) which is in accordance with the expected sensitivity range (10−7 to 102 M Ca2+ i.e., 0.1 to 100 × Kd) for fluorescent dyes. Experiments with and without ATP shown in Fig. 4b alone can substantiate the pumping of calcium due to SERCA. Further experiments were performed to see the inhibitory function of thapsigargin, a SERCA inhibitor. 100 nM thapsigargin was enough to inhibit SERCA activity. For data, refer to ESI Fig. 1.† SERCA activity at room temperature was observed for about 30 min in the presence of 10 mM ATP with a slow increase in microsomal Ca2+ (Fig. 4c).
Ryanodine receptors RYR1 and RyR2 perform a plethora of functions in mammalian physiology, ranging from skeletal muscle and cardiac muscle contraction to cognitive functions such as learning and memory. Ryanodine channels that deplete the calcium stores in response to ryanodine are ER resident leak channels. RyRs respond to ryanodine at low concentrations and caffeine. Ryanodine receptors have been extensively investigated in eukaryotic cells.34–37 The graph in Fig. 4d presents the calcium response of 100 µM and 10 mM caffeine. 10 mM caffeine induced a higher calcium efflux relative to 100 µM caffeine. Dantrolene inhibited the calcium release induced by caffeine. For inhibitory experiments using dantrolene, refer to ESI Fig. 2.†
In order to estimate the amount of calcium present in the microsomes, we have used 10 µM ionomycin + 5 mM EGTA for Fmin and 10 µM ionomycin + 10 mM Ca2+ for Fmax in the formula,
| [Ca2+] = Kd × (F − Fmin)/(Fmax − F) |
The representative graph for calcium concentration determination is shown in Fig. 4e and f. The sensitivity of the Fluo-5N AM can be visually observed and is shown in Fig. 4e, presenting I, II, and III experimental conditions of Fig. 4f. With the depletion of Ca2+ in microsomes, a decrease in fluorescence is noted in II and when the buffer is completely exchanged to 10 µM ionomycin + 10 mM Ca2+, a increase in intensity is observed in III as recorded in the ESI video file.†
Studying native ER proteins using our method has a couple of advantages: (a) increased signal to noise ratio in microsomes relative to ER in intact cells. As the low affinity AM dyes have to reach the endoplasmic reticulum through the cytosol where the carboxylesterases are abundant, a significant amount of fluorescent dye is cleaved and remains in the cytosol. Despite using low affinity Ca2+ indicators, which diminish the sensitivity to cytosolic Ca2+ levels, it will still contribute to the large noise relative to signal due to the high amount of cleaved dye present in the cytosol. (b) The flexibility to alter the calcium levels outside the microsomes, to reduce the intrinsic calcium leakage due to the potentiation of Ca2+ between the luminal and outer face of the microsomes.38 For all calcium experiments, we used 200 µM calcium in the buffer in order to reduce the spontaneous leakage from microsomes as using 300 nM Ca2+ in the buffer caused extensive leakage relatively (ESI Fig. 3†).
3.4. Analysis of the cell-free synthesized hTRPV1 channel
In pursuance of calcium imaging with cell-free synthesized channels, we have chosen a human Transient Receptor Potential Channel, Vanilloid Receptor member 1 (hTRPV1), as a model protein and also bovine Transient Receptor Potential Channel, Vanilloid receptor member 3, TRPV3 (ESI Fig. 4†). hTRPV1 expressed in cells in the plasma membrane has been studied for calcium entry into the cytoplasm using Fura 2-AM.39,40 hTRPV1 is largely present in the plasma membrane with a large N terminal and the C terminal that stretches towards the cytosol in cells.As expected, in cell-free synthesis, the orientation of the protein incorporated in the microsome is opposite to the native plasma membrane orientation. Hence, the cytoplasmic side is outside the microsomes and the extracellular domain is on the side of the microsomal lumen. Both the N-terminal and the C-terminal cytosolic domains of hTRPV1 are expected to be expressed on the outside of the microsomes during cell-free protein synthesis. Although most of the activators and inhibitors for the TRP channel family are organic membrane permeable compounds, attention should be paid while working with the non-polar activators or inhibitors. It has been reported that TRPV1 expressed both endogenously and heterologously in the ER causes Ca2+ release. TRPV1 is expressed in neurons endogenously in the ER and mediates calcium release from the ER which is not Inositol Triphosphate (IP3) mediated store release.41 Calcium imaging of TRPV1 overexpression in the ER of Sf21 and HEK (Human Embryonic Kidney) 293 cells with the KDEL sequence also showed Ca2+ release into the cytosol without the aid of IP3 mediation from the ER.42 TRPV1 is also expressed endogenously in the ER of non-excitable cells such as myocytes and cancer cells43 and in human lung cells,44 which also show calcium regulation similar to the overexpression systems in the ER.
All experiments depicted in Fig. 5 are performed after calcium loading into the microsomes for 1 h in order to ensure uniform and higher [Ca2+] inside the microsomes. The calcium homeostasis in microsomes along with cell-free synthesized ion channels is depicted in Fig. 5a. Under the above-mentioned experimental conditions, we have observed that the activation of the hTRPV1 caused Ca2+ release from hTRPV1–mCES2 microsomes as shown in Fig. 5b and c. The Ca2+ release by 200 nM CAP was abolished completely by 20 µM capsazepine (CPZ) (Fig. 5b). No Ca2+ release was observed in mCES2 samples even at 10 µM CAP (Fig. 5c). Saturation of capsaicin induced Ca2+ release was observed even with 200 nM CAP in the hTRPV1–mCES2 microsomes. The CAP activation profile of cell-free synthesized hTRPV1 (Fig. 5d) is coherent in the molar range as the cell-based expression.45 Presence of phosphatidylinositol 4,5-bisphosphate, (4,5)PIP2, for the activation of TRP channels has been debated by several researchers.46–48 In our work, we have shown that without external (4,5)PIP2, activation of TRPV1 was feasible. On the other hand, the phenomenon we observe can be supported by the presence of (4,5)PIP2 in the endoplasmic reticulum and in the cytosol.48–50
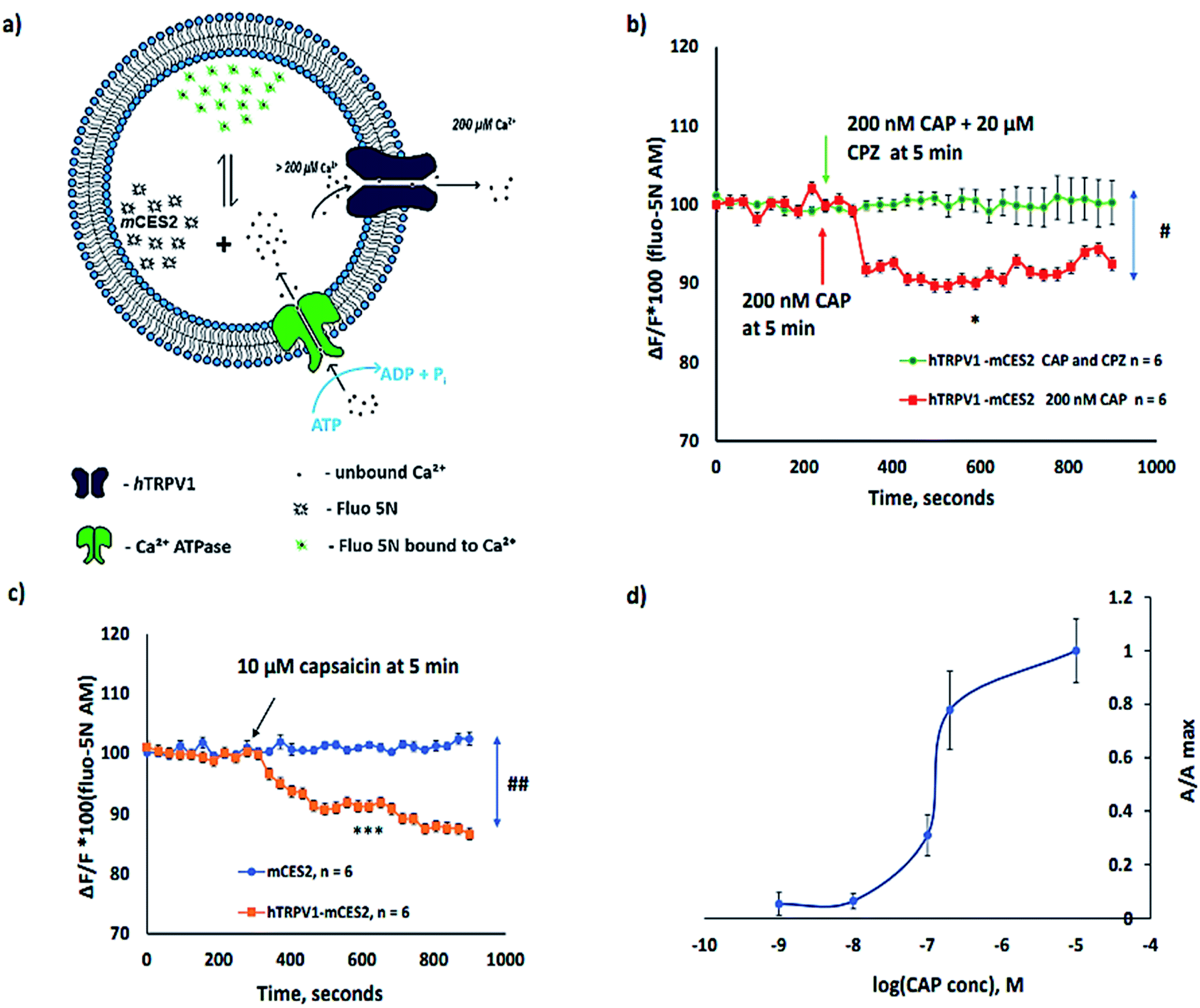 | ||
| Fig. 5 Functional assessment of cell-free synthesized hTRPV1: (a) illustration of calcium dynamics in microsomes expressed with hTRPV1 and mCES2; (b) capsazepine, CPZ (20 µM), mediated inhibition of calcium release caused by capsaicin, CAP, 200 nM, in hTRPV1–mCES2 microsomes, n = 6 each. # indicates the significant difference between two sample groups at 600 s, p = 0.0121, unpaired t test, two tailed. * indicates the significant difference of data sets at 150 s and 600 s of 200 nM CAP stimulation, p = 0.0182, paired t test, two tailed. (c) Comparison plot of hTRPV1–mCES2 and mCES2 microsomes for 10 µM CAP induced Ca2+ release at 5 min from microsomes, n = 6 each. No Ca2+ release was observed in the mCES2 microsomes. ## indicates the significant difference between two sample groups at 600 s, p = 0.0043, Mann–Whitney test, two tailed. *** indicates the significant difference of hTRPV1–mCES2 sample data at 150 s and 600 s, i.e., before and after CAP addition, p < 0.0001, paired t test, two tailed; all samples in (b), (c) and (d) were calcium loaded for 60 min at 37 °C using ATP before CAP and CPZ studies. All data in (b) and (c) are presented as ΔF/F × 100 of the baseline before adding the stimulant; (d) dose dependent response of CAP in hTRPV1–mCES2 microsomes presented as a ratio of area of the curve A/Amax, n = 4 to 7 per each data point. | ||
4 Conclusion and outlook
In conclusion, our work is promising to enhance the following areas of research: (a) to probe other ions such as Cl−, Mg2+ and K+ using AM-based indicators, (b) to study native channels present in the ER membrane by direct ion-sensitive dye imaging of the lumen, (c) drug toxicity studies in microsomes – environmental toxicologists and pharmacologists investigate microsomal Ca2+ levels to estimate the cell toxicity. Ca2+ release by microsomes caused by drug or drug metabolites is the direct indicator of cell toxicity succumbing to higher cytosolic Ca2+ that triggers apoptosis,51–54 (d) potentially it could be used further to develop High Throughput Screening (HTS) for drugs in CFPS platforms for ion permeable proteins that include different families of ion channels, pumps and exchangers. When converted into an HTS assay, the characteristics of the assay will also be changed apparently. For example, it is expected that the spatial resolution of observed calcium or the sensitivity of calcium dye which we observe in confocal microscopy will be reduced in an HTS assay in the microplate. But an HTS platform comes with other advantages such as reduced imaging time and high turnover of measured samples in a short time.55 In summary, in order to cope up with the sustenance needs of the above-mentioned areas, cell-free synthesized carboxylesterases could be a viable calcium imaging platform in microsomes, thereby overcoming the conventional disadvantages of cell-based protein synthesis.Author contributions
PD and SK have framed the basic idea for this work. DAW was responsible for the production and quality control of the cell-free lysates used in this manuscript. AZ, DB, SD, PD and SK have performed all experiments and analysis of the data. PD, SD and SK have contributed to the preparation of the manuscript. SM has provided his decades long expertise in calcium imaging and corrected the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by German Ministry of Education and Research, BMBF (BMBF, No. 031B0078A). We would like to appreciate Dr Rita Sachse for her preliminary attempts in this topic during her Ph.D. thesis in our lab. We would like to acknowledge Ms Dana Wenzel (Fraunhofer IZI, Potsdam-Golm, Germany) for technical support and Dr Lena Thoring and Dr Marlitt Stech (Fraunhofer IZI, Potsdam-Golm, Germany) for providing their insights and discussion in the cell-free synthesis platform in due course of this research work. We would also like to thank Dr Michael Kirschbaum for helping us with the imaging platforms.References
- R. Santos, O. Ursu, A. Gaulton, A. P. Bento, R. S. Donadi and C. G. Bologa, et al., A comprehensive map of molecular drug targets, Nat. Rev. Drug Discovery, 2017, 16(1), 19–34 CrossRef CAS PubMed.
- A. Zemella, L. Thoring, C. Hoffmeister and S. Kubick, Cell-Free Protein Synthesis: Pros and Cons of Prokaryotic and Eukaryotic Systems, ChemBioChem, 2015, 16(17), 2420–2431 CrossRef CAS PubMed.
- R. Sachse, S. K. Dondapati, S. F. Fenz, T. Schmidt and S. Kubick, Membrane protein synthesis in cell-free systems: from bio-mimetic systems to bio-membranes, FEBS Lett., 2014, 588(17), 2774–2781 CrossRef CAS PubMed.
- S. K. Dondapati, M. Stech, A. Zemella and S. Kubick, Cell-Free Protein Synthesis: A Promising Option for Future Drug Development, BioDrugs, 2020, 34(3), 327–348 CrossRef CAS PubMed.
- D. Burdakov, O. H. Petersen and A. Verkhratsky, Intraluminal calcium as a primary regulator of endoplasmic reticulum function, Cell Calcium, 2005, 38(3–4), 303–310 CrossRef CAS PubMed.
- A. Raffaello, C. Mammucari, G. Gherardi and R. Rizzuto, Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes, Trends Biochem. Sci., 2016, 41(12), 1035–1049 CrossRef CAS PubMed.
- C. Betzer, L. B. Lassen, A. Olsen, R. H. Kofoed, L. Reimer, E. Gregersen, J. Zheng, T. Calì, W. P. Gai, T. Chen, A. Moeller, M. Brini, Y. Fu, G. Halliday, T. Brudek, S. Aznar, B. Pakkenberg, J. P. Andersen and P. H. Jensen, et al., Alpha-synuclein aggregates activate calcium pump SERCA leading to calcium dysregulation, EMBO Rep., 2018, 19(5), e44617 CrossRef PubMed.
- D. C. McMullen, W. S. Kean, A. Verma, J. T. Cole and W. D. Watson, A microplate technique to simultaneously assay calcium accumulation in endoplasmic reticulum and SERCA release of inorganic phosphate, Biol. Proced. Online, 2012, 14(1), 4 CrossRef CAS PubMed.
- L. Navazio, M. A. Bewell, A. Siddiqua, G. D. Dickinson, A. Galione and D. Sanders, Calcium release from the endoplasmic reticulum of higher plants elicited by the NADP metabolite nicotinic acid adenine dinucleotide phosphate, Proc. Natl. Acad. Sci. U. S. A., 2000, 97(15), 8693–8698 CrossRef CAS PubMed.
- A. N. K. Yusufi, J. Cheng, M. A. Thompson, J. C. Burnett and J. P. Grande, Differential mechanisms of Ca(2+) release from vascular smooth muscle cell microsomes, Exp. Biol. Med., 2002, 227(1), 36–44 CrossRef CAS PubMed.
- R. Giunti, A. Gamberucci, R. Fulceri, G. Bánhegyi and A. Benedetti, Both translocon and a cation channel are involved in the passive Ca2+ leak from the endoplasmic reticulum: a mechanistic study on rat liver microsomes, Arch. Biochem. Biophys., 2007, 462(1), 115–121 CrossRef CAS PubMed.
- H. M. Dusza, P. H. Cenijn, J. H. Kamstra, R. H. S. Westerink, P. E. G. Leonards and T. Hamers, Effects of environmental pollutants on calcium release and uptake by rat cortical microsomes, Neurotoxicology, 2018, 69, 266–277 CrossRef CAS PubMed.
- S. Samtleben, J. Jaepel, C. Fecher, T. Andreska, M. Rehberg and R. Blum, Direct imaging of ER calcium with targeted-esterase induced dye loading (TED), J. Visualized Exp., 2013,(75), e50317 Search PubMed.
- M. Rehberg, A. Lepier, B. Solchenberger, P. Osten and R. Blum, A new non-disruptive strategy to target calcium indicator dyes to the endoplasmic reticulum, Cell Calcium, 2008, 44(4), 386–399 CrossRef CAS PubMed.
- R. Blum, O. H. Petersen and A. Verkhratsky, Ca2+ Imaging of Intracellular Organelles: Endoplasmic Reticulum, in Calcium Measurement Methods [Internet], ed. A. Verkhratsky and O. H. Petersen, Humana Press, Totowa, NJ, 2010, pp. 147–67, (Neuromethods), DOI:10.1007/978-1-60761-476-0_8.
- M. Stech, A. K. Brödel, R. B. Quast, R. Sachse and S. Kubick, Cell-free systems: functional modules for synthetic and chemical biology, Adv. Biochem. Eng./Biotechnol., 2013, 137, 67–102 CrossRef CAS PubMed.
- A. Zemella, S. Grossmann, R. Sachse, A. Sonnabend, M. Schaefer and S. Kubick, Qualifying a eukaryotic cell-free system for fluorescence based GPCR analyses, Sci. Rep., 2017, 7(1), 3740 CrossRef PubMed.
- A. K. Brödel, A. Sonnabend, L. O. Roberts, M. Stech, D. A. Wüstenhagen and S. Kubick, IRES-mediated translation of membrane proteins and glycoproteins in eukaryotic cell-free systems, PLoS One, 2013, 8(12), e82234 CrossRef PubMed.
- M. Stech, R. B. Quast, R. Sachse, C. Schulze, D. A. Wüstenhagen and S. Kubick, A continuous-exchange cell-free protein synthesis system based on extracts from cultured insect cells, PLoS One, 2014, 9(5), e96635 CrossRef PubMed.
- R. B. Quast, A. Sonnabend, M. Stech, D. A. Wüstenhagen and S. Kubick, High-yield cell-free synthesis of human EGFR by IRES-mediated protein translation in a continuous exchange cell-free reaction format, Sci. Rep., 2016, 6, 30399 CrossRef CAS PubMed.
- D. Gilham and R. Lehner, Techniques to measure lipase and esterase activity in vitro, Methods, 2005, 36(2), 139–147 CrossRef CAS PubMed.
- Y. Zhu, J. Li, H. Cai, H. Ni, A. Xiao and L. Hou, Characterization of a new and thermostable esterase from a metagenomic library, Microbiol. Res., 2013, 168(9), 589–597 CrossRef CAS PubMed.
- A. R. B. do Nascimento, P. Fresia, F. L. Cônsoli and C. Omoto, Comparative transcriptome analysis of lufenuron-resistant and susceptible strains of Spodoptera frugiperda (Lepidoptera: Noctuidae), BMC Genomics, 2015, 16, 985 CrossRef PubMed.
- M. Giraudo, F. Hilliou, T. Fricaux, P. Audant, R. Feyereisen and G. Le Goff, Cytochrome P450s from the fall armyworm (Spodoptera frugiperda): responses to plant allelochemicals and pesticides, Insect Mol. Biol., 2015, 24(1), 115–128 CrossRef CAS PubMed.
- R. M. Paredes, J. C. Etzler, L. T. Watts, W. Zheng and J. D. Lechleiter, Chemical calcium indicators, Methods, 2008, 46(3), 143–151 CrossRef CAS PubMed.
- J. J. Caramelo and A. J. Parodi, Getting in and out from calnexin/calreticulin cycles, J. Biol. Chem., 2008, 283(16), 10221–10225 CrossRef CAS PubMed.
- T. R. Shannon, T. Guo and D. M. Bers, Ca2+ scraps: local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve, Circ. Res., 2003, 93(1), 40–45 CrossRef CAS PubMed.
- A. E. Oliver, G. A. Baker, R. D. Fugate, F. Tablin and J. H. Crowe, Effects of temperature on calcium-sensitive fluorescent probes, Biophys. J., 2000, 78(4), 2116–2126 CrossRef CAS PubMed.
- J. M. Autry and L. R. Jones, Functional Co-expression of the canine cardiac Ca2+ pump and phospholamban in Spodoptera frugiperda (Sf21) cells reveals new insights on ATPase regulation, J. Biol. Chem., 1997, 272(25), 15872–15880 CrossRef CAS PubMed.
- D. L. Winters, J. M. Autry, B. Svensson and D. D. Thomas, Interdomain fluorescence resonance energy transfer in SERCA probed by cyan-fluorescent protein fused to the actuator domain, Biochemistry, 2008, 47(14), 4246–4256 CrossRef CAS PubMed.
- J. Meldolesi and T. Pozzan, The endoplasmic reticulum Ca2+ store: a view from the lumen, Trends Biochem. Sci., 1998, 23(1), 10–14 CrossRef CAS PubMed.
- D. Prins and M. Michalak, Organellar calcium buffers, Cold Spring Harbor Perspect. Biol., 2011, 3(3), a004069 Search PubMed.
- M. Michalak, J. M. Robert Parker and M. Opas, Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum, Cell Calcium, 2002, 32(5–6), 269–278 CrossRef CAS PubMed.
- A. Uehara, T. Murayama, M. Yasukochi, M. Fill, M. Horie and T. Okamoto, et al., Extensive Ca2+ leak through K4750Q cardiac ryanodine receptors caused by cytosolic and luminal Ca2+ hypersensitivity, J. Gen. Physiol., 2017, 149(2), 199–218 CrossRef PubMed.
- E. Camors and H. H. Valdivia, CaMKII regulation of cardiac ryanodine receptors and inositol triphosphate receptors, Front. Pharmacol., 2014, 5, 101 Search PubMed.
- M. Fill and J. A. Copello, Ryanodine receptor calcium release channels, Physiol. Rev., 2002, 82(4), 893–922 CrossRef CAS PubMed.
- O. Vázquez-Martínez, R. Cañedo-Merino, M. Díaz-Muñoz and J. R. Riesgo-Escovar, Biochemical characterization, distribution and phylogenetic analysis of Drosophila melanogaster ryanodine and IP3 receptors, and thapsigargin-sensitive Ca2+ ATPase, J. Cell Sci., 2003, 116(Pt 12), 2483–2494 CrossRef PubMed.
- Y. Kunitomo and D. Terentyev, How to stop the fire? Control of Ca2+-induced Ca2+ release in cardiac muscle, J. Physiol., 2011, 589(Pt 24), 5899–5900 CrossRef CAS PubMed.
- I. Vetter, B. D. Wyse, G. R. Monteith, S. J. Roberts-Thomson and P. J. Cabot, The mu opioid agonist morphine modulates potentiation of capsaicin-evoked TRPV1 responses through a cyclic AMP-dependent protein kinase A pathway, Mol. Pain, 2006, 2, 22 CrossRef PubMed.
- E. R. Grant, A. E. Dubin, S.-P. Zhang, R. A. Zivin and Z. Zhong, Simultaneous intracellular calcium and sodium flux imaging in human vanilloid receptor 1 (VR1)-transfected human embryonic kidney cells: a method to resolve ionic dependence of VR1-mediated cell death, J. Pharmacol. Exp. Ther., 2002, 300(1), 9–17 CrossRef CAS PubMed.
- X.-P. Dong, X. Wang and H. Xu, TRP channels of intracellular membranes, J. Neurochem., 2010, 113(2), 313–328 CrossRef CAS PubMed.
- B. J. Wisnoskey, W. G. Sinkins and W. P. Schilling, Activation of vanilloid receptor type I in the endoplasmic reticulum fails to activate store-operated Ca2+ entry, Biochem. J., 2003, 372(Pt 2), 517–528 CrossRef CAS PubMed.
- A. Haustrate, N. Prevarskaya and V. Lehen’kyi, Role of the TRPV Channels in the Endoplasmic Reticulum Calcium Homeostasis, Cells, 2020, 9(2), 317 CrossRef CAS.
- K. C. Thomas, A. S. Sabnis, M. E. Johansen, D. L. Lanza, P. J. Moos and G. S. Yost, et al., Transient receptor potential vanilloid 1 agonists cause endoplasmic reticulum stress and cell death in human lung cells, J. Pharmacol. Exp. Ther., 2007, 321(3), 830–838 CrossRef CAS PubMed.
- E. N. Senning, M. D. Collins, A. Stratiievska, C. A. Ufret-Vincenty and S. E. Gordon, Regulation of TRPV1 ion channel by phosphoinositide (4,5)-bisphosphate: the role of membrane asymmetry, J. Biol. Chem., 2014, 289(16), 10999–11006 CrossRef CAS PubMed.
- S. Brauchi, G. Orta, C. Mascayano, M. Salazar, N. Raddatz and H. Urbina, et al., Dissection of the components for PIP2 activation and thermosensation in TRP channels, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(24), 10246–10251 CrossRef CAS PubMed.
- T. Rohacs, B. Thyagarajan and V. Lukacs, Phospholipase C mediated modulation of TRPV1 channels, Mol. Neurobiol., 2008, 37(2–3), 153–163 CrossRef CAS.
- S. Kolay, U. Basu and P. Raghu, Control of diverse subcellular processes by a single multi-functional lipid phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2], Biochem. J., 2016, 473(12), 1681–1692 CrossRef CAS PubMed.
- G. R. V. Hammond, G. Schiavo and R. F. Irvine, Immunocytochemical techniques reveal multiple, distinct cellular pools of PtdIns4P and PtdIns(4,5)P(2), Biochem. J., 2009, 422(1), 23–35 CrossRef CAS PubMed.
- M. Schramp, A. Hedman, W. Li, X. Tan and R. Anderson, PIP kinases from the cell membrane to the nucleus, Subcell. Biochem., 2012, 58, 25–59 CAS.
- S. Pentyala, J. Ruggeri, A. Veerraju, Z. Yu, A. Bhatia and D. Desaiah, et al., Microsomal Ca2+ flux modulation as an indicator of heavy metal toxicity, Indian J. Exp. Biol., 2010, 48(7), 737–743 CAS.
- D. A. Stoyanovsky and A. I. Cederbaum, Metabolites of acetaminophen trigger Ca2+ release from liver microsomes, Toxicol. Lett., 1999, 106(1), 23–29 CrossRef CAS PubMed.
- C. G. Coburn, M. C. Currás-Collazo and P. R. S. Kodavanti, In vitro effects of environmentally relevant polybrominated diphenyl ether (PBDE) congeners on calcium buffering mechanisms in rat brain, Neurochem. Res., 2008, 33(2), 355–364 CrossRef CAS PubMed.
- P. R. S. Kodavanti and T. R. Ward, Differential effects of commercial polybrominated diphenyl ether and polychlorinated biphenyl mixtures on intracellular signaling in rat brain in vitro, Toxicol. Sci., 2005, 85(2), 952–962 CrossRef CAS PubMed.
- N. J. Martinez, S. A. Titus, A. K. Wagner and A. Simeonov, High throughput fluorescence imaging approaches for drug discovery using in vitro and in vivo three-dimensional models, Expert Opin. Drug Discovery, 2015, 10, 1347–1361 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary data and video file. See DOI: 10.1039/d0ra08397f |
| This journal is © The Royal Society of Chemistry 2021 |