DOI:
10.1039/D0RA08345C
(Paper)
RSC Adv., 2021,
11, 57-70
Enhancement of air-stability, π-stacking ability, and charge transport properties of fluoroalkyl side chain engineered n-type naphthalene tetracarboxylic diimide compounds†
Received
29th September 2020
, Accepted 26th November 2020
First published on 22nd December 2020
Abstract
In this study, the impact of fluoroalkyl side chain substitution on the air-stability, π-stacking ability, and charge transport properties of the versatile acceptor moiety naphthalene tetracarboxylic diimide (NDI) has been explored. A density functional theory (DFT) study has been carried out for a series of 24 compounds having different side chains (alkyl, fluoroalkyl) through the imide nitrogen position of NDI moiety. The fluoroalkyl side chain engineered NDI compounds have much deeper highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbitals (LUMO) than those of their alkyl substituted compounds due to the electron withdrawing nature of fluoroalkyl groups. The higher electron affinity (EA > 2.8 eV) and low-lying LUMO levels (<−4.00 eV) for fluoroalkyl substituted NDIs reveal that they may exhibit better air-stability with superior n-type character. The computed optical absorption spectra (∼386 nm) for all the investigated NDIs using time-dependent DFT (TD-DFT) lie in the ultra-violet (UV) region of the solar spectrum. In addition, the low value of the LOLIPOP (Localized Orbital Locator Integrated Pi Over Plane) index for fluoroalkyl side chain comprising NDI compounds indicates better π–π stacking ability. This is also in good agreement for the predicted π–π stacking interaction obtained from a molecular electrostatic potential energy surface (ESP) study. The π–π stacking is thought to be of cofacial interaction for the fluoroalkyl substituted compounds and herringbone interaction for the alkyl substituted compounds. The calculated results shed light on why side chain engineering with fluoroalkyl groups can effectively lead to better air-stability, π-stacking ability and improved charge transport properties.
1 Introduction
π-Conjugated organic semiconductors (OSCs) continue to draw appreciable attention from industry as well as academic communities due to their potential optoelectronic device applications such as in organic field-effect transistors (OFETs)1–3 organic light-emitting diodes (OLEDs)4,5 organic photovoltaics (OPVs)6–8 and chemical sensors9–11 etc. In general, depending on the nature of charge carrier, OSCs can be classified as (a) n-type (electron is the majority charge carrier) and (b) p-type (hole is the majority charge carrier). The p-type materials have been studied in detail due to their appreciable air-stability and high mobility. Comparatively lower stability in ambient conditions and high injection barriers are the bottleneck for commercial applications of n-type and ambipolar (having both p-type and n-type character) OSCs. N-type organic materials have significant applications in organic complementary circuits. Hence, currently, the development of high performance and air-stable n-type OSCs is a pivotal challenge for scientific communities.
Several strategies are already reported to design n-type OSCs, such as (i) insertion of electron-withdrawing groups (–F, –Cl, –CN etc.) on the backbone of known p-type materials12 (ii) functionalization of electron-deficient ring systems such as naphthalene tetracarboxylic diimide (NDI),13,14 perylene tetracarboxylic diimide (PDI),15,16 diketopyrrolopyrrole (DPP)17,18 etc. Among the n-type OSCs, naphthalene tetracarboxylic diimides (NDIs) are one of the most promising n-type candidates for their high electron affinity, ambient air-stability and tunable optoelectronic properties. NDI framework is a versatile building block which serves as an acceptor moiety for the synthesis of n-type organic semiconductor.19 Different alkyl chain substitution at the imide nitrogen positions of NDI molecules outstandingly affects the molecular packing and charge carrier mobility.20–27 In NDI, two types of substitutions are reported: (a) substitution at the core of NDI and (b) substitution at imide nitrogen position. The second option of substitution at imide nitrogen position provides several advantages over the first case.28 For example, the substituted alkyl or aryl chains can minimize the steric hindrance by keeping the planarity of the core as they are far from the core. Moreover, the easy synthetic routes for the imide-substituted NDI can be advantageous over the core-substituted NDI. For instance, Zhang et al. reported 0.70 cm2 V−1 s−1 electron mobility for N,N′-bis(4-trifluoromethoxybenzyl) naphthalene-1,4,5,8-tetracarboxylic acid diimide.29 See et al. also showed good air-stability and an electron-accepting moiety of N,N′-substituted 1,4,5,8-naphthalene tetracarboxylic diimide.30 The presence of short side groups and highly crystalline film of this moiety leads to better electron carrier than hole carrier.31 Oh et al. have reported electron mobility of 0.26 ± 0.02 cm2 V−1 s−1 in the air for N,N′-bis-(heptafluorobutyl)-1,4,5,8-naphthalene tetracarboxylic diimide.21 They also reported the field effect mobility of NDI-based compounds having di- and tetrachloro substitution at the bay position of NDI with perfluoro group attached to the N-imide position. Dichloro-substituted compound shows better mobility than the tetrachloro-substituted one, which was supported by the intramolecular packing of fluoroalkyl groups at the N-imide position leading to higher air stability in the former case.
In our study, we have investigated the electronic properties and π-stacking ability of a series of compounds where the N atom of imide group in NDI compound is attached with alkyl, and fluoroalkyl groups. The position of electron-withdrawing fluoroalkyl groups is varied by the insertion of methylene group, to further understand its impact on the NDI core. Our study shows that a simple modification of the side chain in NDI compounds may significantly vary their electronic properties.
2 Computational details
In the present work, the geometries of the designed compounds were optimized using the popular hybrid functional B3LYP (Becke's three-parameter functional and the Lee–Yang–Parr functional) at 6-31+G(d,p) basis set level within the framework of density functional theory (DFT). B3LYP/6-31+G(d,p) theoretical level is a good choice for geometry optimization and electronic property calculations for NDI derivatives.21,32 The DFT calculations are performed using Gaussian 09 program package.33 The vibrational frequency analysis was performed at the same level of theory and the resulted positive frequencies confirmed that the optimized geometries were found at the real minima on the potential energy surfaces. Further, electron affinity in both vertical (EAv) and adiabatic (EAa) excitations, ionization potential in both vertical (IPv) and adiabatic (IPa) excitations and hole and electron reorganization energy (λh and λe, respectively) were calculated according to the following formula:| | |
λh = ENGC − ENGN + ECGN − ECGC
| (5) |
| | |
λe = ENGA − ENGN + EAGN − EAGA
| (6) |
where ENGN, ECGC and EAGA are the energies of neutral molecules, cations, and anions, respectively, in their optimized geometries and ECGN and EAGN are the energy of cations and anions, respectively with the geometry of neutral molecules; ENGC and ENGA are the energy of neutral compounds with the geometry of cations and anions, respectively.
The total density of states (DOS) and partial density of states (PDOS) of the investigated compounds were evaluated using GaussSum.34 The energy interval was taken in the range of −15 eV to 6 eV, and the value of FWHM in the Gaussian curve was kept fixed at 0.3 eV, to convolute the DOS spectra. The PDOS of the compounds were calculated by considering each atom/orbitals present in the molecule as a “group”, such as naphthalene tetracarboxylic diimide (NDI) fragment, alkyl chains, and fluoroalkyl chain. For electronic absorption spectra and vertical singlet electronic transitions, time-dependent density functional theory (TD-DFT) was employed at B3LYP/6-31+G(d,p) level in dichloromethane (ε = 8.93) solvent. To figure out the variation of electron density over the whole molecular backbone, molecular electrostatic potential surface (ESP) analysis was performed. The LOLIPOP (Localized Orbital Locator Integrated Pi Over Plane) index was computed to estimate the π-stacking ability of the designed NDI-based compounds using Multiwfn.35,36
3 Results and discussion
3.1 Geometry
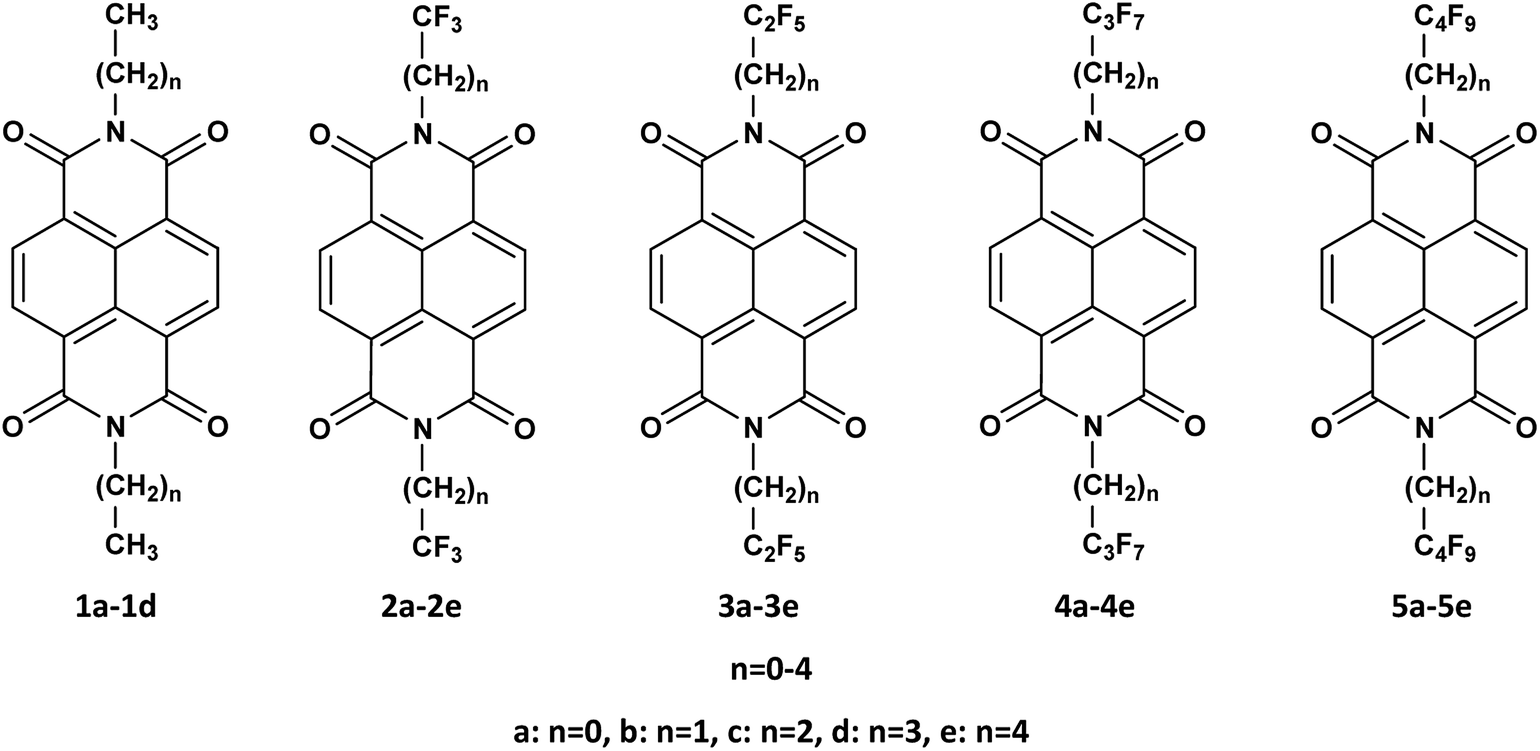
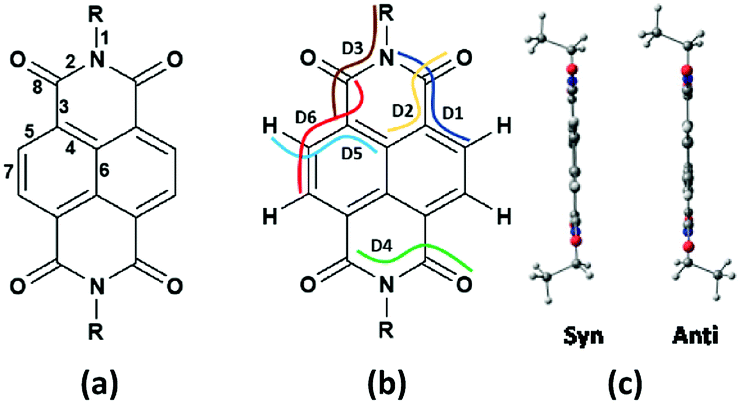
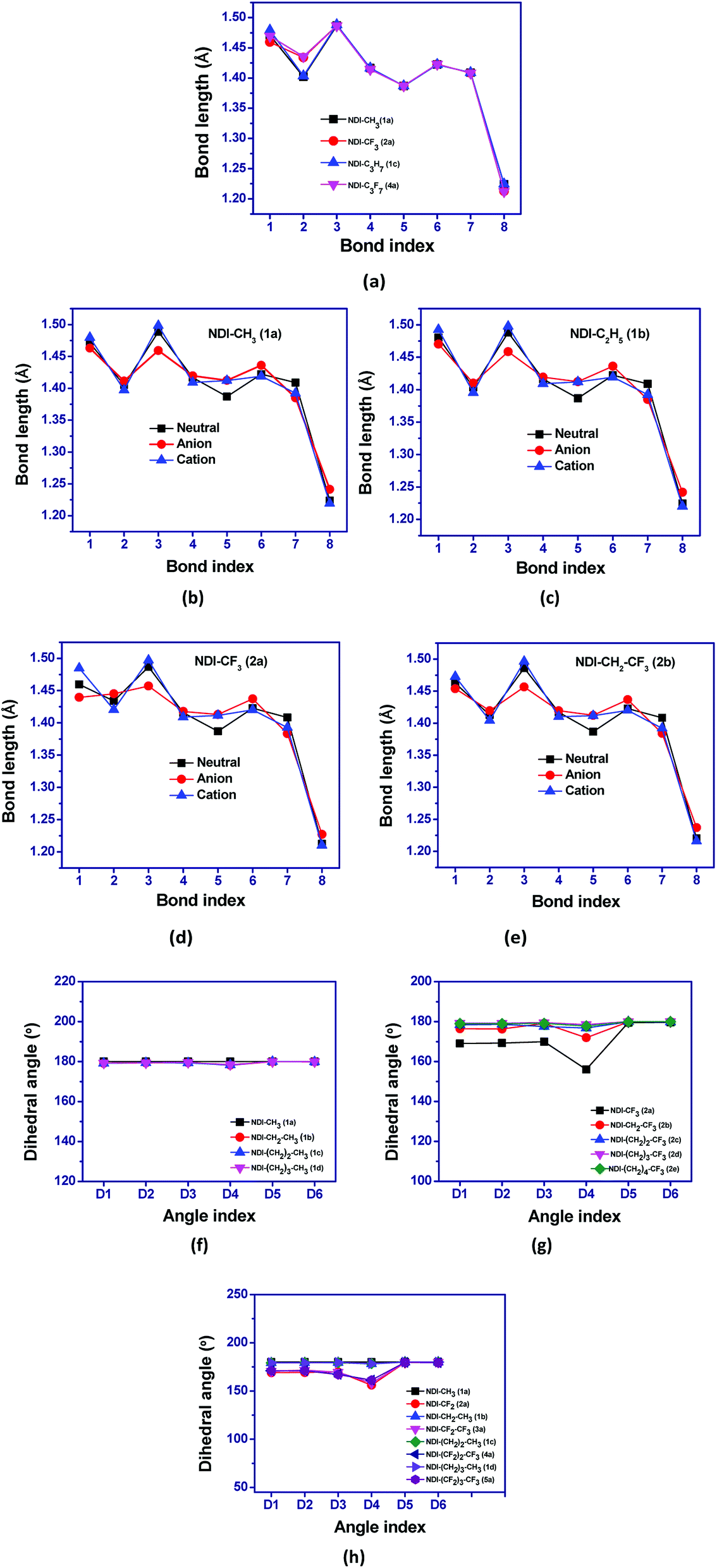
The structures of naphthalene tetracarboxylic diimide (NDI) derivatives studied in the present work are shown in Fig. 1, and the optimized structures are provided in Fig. S1 in the ESI.† The various alkyl and fluoroalkyl groups substituted at the NDI core can be of either in syn position or anti position. The anti and syn conformation for the compound 1b (NDI–C2H5) is shown in Fig. 2(c). As the difference between the syn and anti-conformation is very less (0.01–0.05 kcal mol−1), therefore, all the calculations are done in anti-conformation of substituted groups to the NDI core. The NDI core is found to be completely planar for all the alkyl substituted compounds. However, the compounds having perfluoro groups directly attached to the NDI-core have non-planar structure on account of the N atom of the imide group being shifted away from the molecular plane of NDI-core. Thus, the direct incorporation of electron-withdrawing perfluoro groups (such as –CF3, –C2F5) to the NDI-core through imide N leads to distortion in the planar structure of the compounds. The bending of the imide N from the plane of the NDI core decreases as the fluoroalkyl or perfluoroalkyl group moves farther away from the NDI core by the insertion of methylene groups. For instance, the value of dihedral angle, D1 for the compounds 1a (NDI–CH3), 2a (NDI–CF3), 2b (NDI–CH2–CF3), 2c (NDI–(CH2)2–CF3), 2d (NDI–(CH2)3–CF3) and 2e (NDI–(CH2)4–CF3) are found to be −179.9°, −169.1°, −176.5°, 178.5°, −179.5° and −179.0° respectively. This is probably due to the more electrostatic hindrance induced for the perfluoroalkyl substituted compounds as compared to the alkyl substituted one. In addition, substitution of alkyl groups with perfluoroalkyl groups in the imide-N position leads to change in the bond lengths. The molecular framework showing the bond index and dihedral angle is shown in Fig. 2(a and b) along with the graphical representations of the variation in bond lengths and dihedral angles of some studied compounds which are displayed in Fig. 3(a–h). On comparing the bond parameters of fluoroalkyl substituted compounds with the alkyl substituted one, it is seen that the bond index 2 (C–N imide bond) is significantly elongated in the former as shown in the Fig. 3(a). The bond parameters of the studied compounds have been compared in their neutral and charged states. From the Fig. 3(b and c), it is seen that the C–C bonds adjacent to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of imide moiety (bond index 3 and 5) have highest deviation in their bond lengths in charged states as compared to the neutral state. The bond index 3, i.e. the C–C bond adjacent to the CO group, is significantly shortened in anionic state as compared to the neutral and cationic state. In addition, the bond index 5 is elongated in the charged states when compared to the neutral compound. Interestingly, the C–N bond (bond index 1) connecting fluoroalkyl group to imide nitrogen is also found to show differ in lengths in the charged and neutral states for the perfluoroalkyl substituted compounds (Fig. 3(d)). This deviation in bond length decreases if the perfluoroalkyl group is attached to the imide N-atom through methylene bridges (Fig. 3(e)). The maximum deviation is found to be in the order of ∼0.009–0.033 Å for carbon–heteroatom bonds: C–N, CO bonds (bond index 1, 2 and 8) in the compounds by substituting fluoroalkyl with alkyl in the studied compounds. For instance, bond index 2 for compound 1a (NDI–CH3) is 1.402 Å, and for compound 2a (NDI–CF3), it is 1.434 Å (a difference of 0.03 Å). Also, for compound 1c (NDI–C3H7), this bond index 2 is 1.404 Å, and for compound 4a (NDI–C3F7), it is 1.436 Å (a difference of 0.03 Å). Further, as shown in Fig. 3(b–e), it is also observed that, the geometry relaxation in reduction state is slightly more than that of oxidation state. However, this difference is minimal, as it is also confirmed by the hole and electron reorganization energy. The plot for remaining compounds are shown in the Fig. S3 in the ESI.† This suggests that the most adjacent bond to the N-imide linkage is the reason for the differences in their planarity also. The trend in the variation of the bond index is also found similar for the previously reported crystal structure of NDI-based compounds.37–43 A table is given for the same to visualize the trend in the Table S1 of the ESI.† This is in line with the earlier reported crystal structure of some of the studied compounds.
O group of imide moiety (bond index 3 and 5) have highest deviation in their bond lengths in charged states as compared to the neutral state. The bond index 3, i.e. the C–C bond adjacent to the CO group, is significantly shortened in anionic state as compared to the neutral and cationic state. In addition, the bond index 5 is elongated in the charged states when compared to the neutral compound. Interestingly, the C–N bond (bond index 1) connecting fluoroalkyl group to imide nitrogen is also found to show differ in lengths in the charged and neutral states for the perfluoroalkyl substituted compounds (Fig. 3(d)). This deviation in bond length decreases if the perfluoroalkyl group is attached to the imide N-atom through methylene bridges (Fig. 3(e)). The maximum deviation is found to be in the order of ∼0.009–0.033 Å for carbon–heteroatom bonds: C–N, CO bonds (bond index 1, 2 and 8) in the compounds by substituting fluoroalkyl with alkyl in the studied compounds. For instance, bond index 2 for compound 1a (NDI–CH3) is 1.402 Å, and for compound 2a (NDI–CF3), it is 1.434 Å (a difference of 0.03 Å). Also, for compound 1c (NDI–C3H7), this bond index 2 is 1.404 Å, and for compound 4a (NDI–C3F7), it is 1.436 Å (a difference of 0.03 Å). Further, as shown in Fig. 3(b–e), it is also observed that, the geometry relaxation in reduction state is slightly more than that of oxidation state. However, this difference is minimal, as it is also confirmed by the hole and electron reorganization energy. The plot for remaining compounds are shown in the Fig. S3 in the ESI.† This suggests that the most adjacent bond to the N-imide linkage is the reason for the differences in their planarity also. The trend in the variation of the bond index is also found similar for the previously reported crystal structure of NDI-based compounds.37–43 A table is given for the same to visualize the trend in the Table S1 of the ESI.† This is in line with the earlier reported crystal structure of some of the studied compounds.
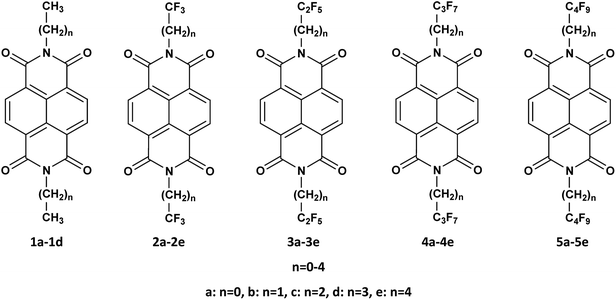 |
| | Fig. 1 Chemical structure of the studied NDI compounds. | |
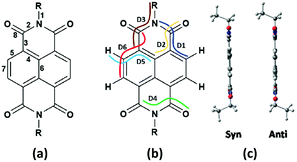 |
| | Fig. 2 (a) NDI-core showing bond index, (b) dihedral angle index and (c) syn- and anti conformation in NDI. | |
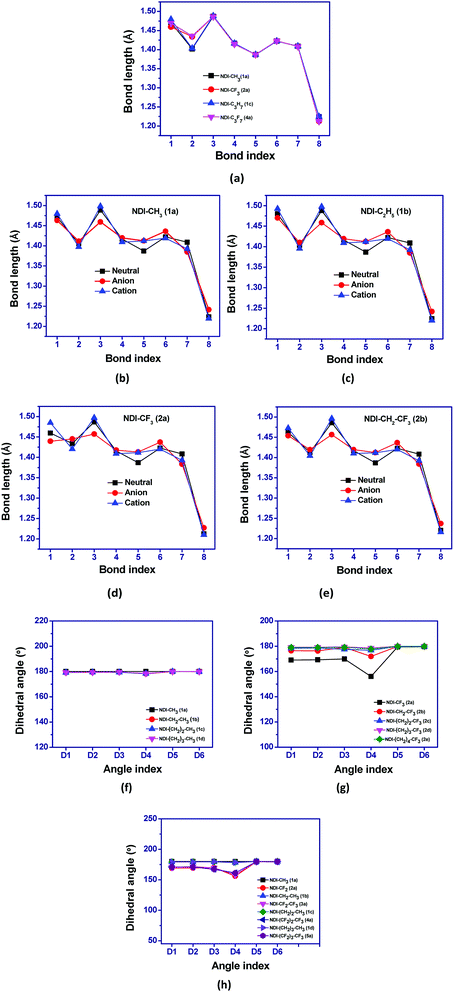 |
| | Fig. 3 Graphical representation of (a–e) bond lengths in neutral and ionic states of NDI compounds and (f–h) comparision of dihedral angles for some of the studied NDI compounds. | |
3.2 Frontier molecular orbitals (FMOs) and density of states (DOS)
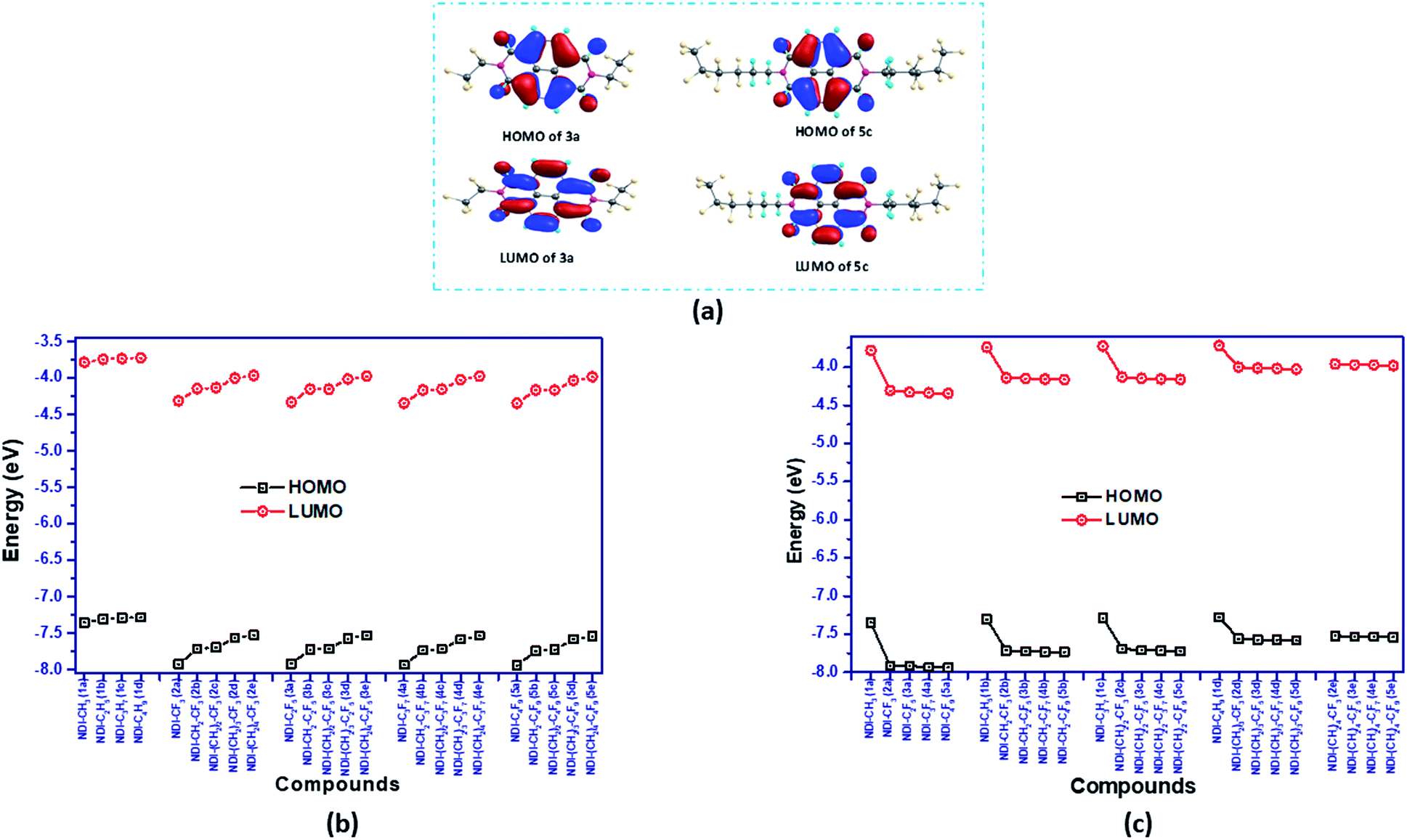
The energy of the frontier molecular orbitals (FMOs) of organic semiconductor materials is one of the key factors influencing the charge transport properties and the intrinsic air-stability of the materials. The computed HOMO/LUMO energies of all the NDI derivatives are shown in Fig. 4(b and c) and the isosurfaces for representative compounds 3a (NDI–C2F5) and 5c (NDI–(CH2)2–C4F9) are displayed in Fig. 4(a). The remaining isosurfaces are provided in Fig. S4 in the ESI.† The plot of isosurfaces showed that the HOMO and LUMO of all the NDI derivatives are predominantly π in character and are localized mostly on the NDI core of the compounds. Comparing the frontier molecular orbital energy levels of fluoroalkyl substituted compounds with their alkyl counterparts, it is seen that fluoroalkyl substituted compounds have much deeper HOMO and LUMO energy levels. The deepening of HOMO and LUMO energy levels in perfluoroalkyl compounds is due to the electron withdrawing nature of the fluoroalkyl groups. For instance, the HOMO and LUMO energy levels of compound 2a (NDI–CF3) are −7.92 eV and −4.31 eV, respectively which are significantly lower in energy than HOMO and LUMO levels of −7.35 eV and −3.78 eV for 1a (NDI–CH3). Similarly, the HOMO/LUMO energy levels of compounds 1b (NDI–C2H5), 1c (NDI–C3H7), 1d (NDI–C4H9) and 3a (NDI–C2F5), 4a (NDI–C3F7), 5a (NDI–C4F9) are −7.30/−3.74 eV, −7.29/3.73 eV, −7.28/−3.72 eV and −7.92/−4.33 eV, −7.93/−4.34 eV, −7.94/−4.34 eV, respectively. As the distance between the perfluoroalkyl group and NDI core is increased by the intervening methylene groups, the HOMO and LUMO energy also increases. For example, the HOMO/LUMO levels of 2a (NDI–CF3), 2b (NDI–CH2–CF3), 2c (NDI–(CH2)2–CF3), 2d (NDI–(CH2)3–CF3), and 2e (NDI–(CH2)4–CF3), increases as −7.92/−4.31 eV, −7.71/−4.14 eV, −7.69/−4.13, −7.56/−4 eV, and −7.52/−3.96 eV, respectively, on moving from 2a to 2e. This observation ensures that the strength of electron withdrawing perfluoroalkyl groups get reduced with increasing distance (or intervening methylene groups) from the NDI core. So, the energy levels get more stabilized when the fluoroalkyl group is directly attached to the N-atom of the NDI-core. In addition, increasing the length of perfluoroalkyl group beyond CF3, does not lead to significant changes in the energy levels. For instance, the HOMO energy of 2a (NDI–CF3) which has CF3 group attached to the imide N of NDI is −7.92 eV while the energy of HOMO of 5a (NDI–C4F9) is −7.94 eV. Similarly, the corresponding LUMO energies of 2a and 5a are −4.31 eV and −4.34 eV, respectively. Interestingly, the HOMO–LUMO gaps of the studied compounds are nearly the same; the range of the band gap is 3.56–3.61 eV for all the studied compounds.
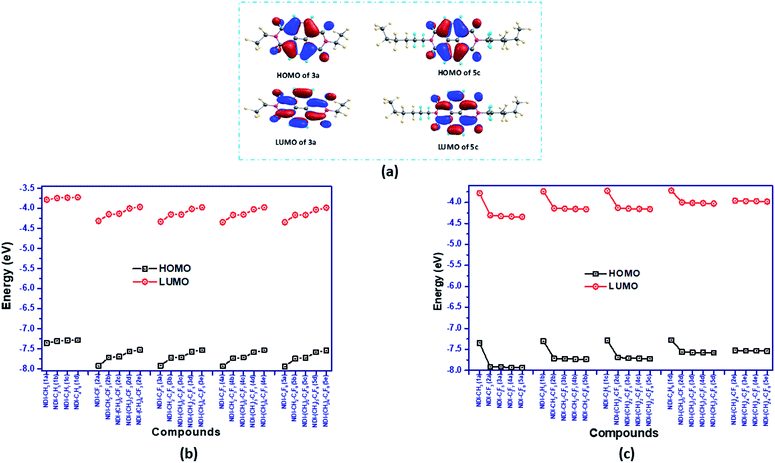 |
| | Fig. 4 Frontier molecular orbital picture of compounds (a) 3a (NDI–C2F5) and 5c (NDI–(CH2)2–C4F9) and the graphical representation of HOMO/LUMO energy levels of the investigated compounds (b) with increasing alkyl groups (by keeping fluoroalkyl group constant), (c) with increasing fluoroalkyl groups (by keeping alkyl group constant). | |
Besides, it is found that the LUMO energy levels of all the investigated fluoroalkyl substituted NDI derivatives lie in the range of −4.0 eV to −4.4 eV, which suggests their ambient air-stability and hence, they can be used as an air-stable organic semiconductor.19,44,45 This is in accordance with the earlier reported results, that the incorporation of the electron-withdrawing groups such as; –F, –Cl, –CF3, and –CN in naphthalene diimide, perylene diimide, acene derivatives lower the LUMO energy level and hence improve the ambient stability.20,46–49 In addition, the HOMO levels of the compounds are well within the air oxidation threshold value (ca. −5.27 eV or 0.57 V vs. SCE)50,51 value, thus signifying their aerial stability.
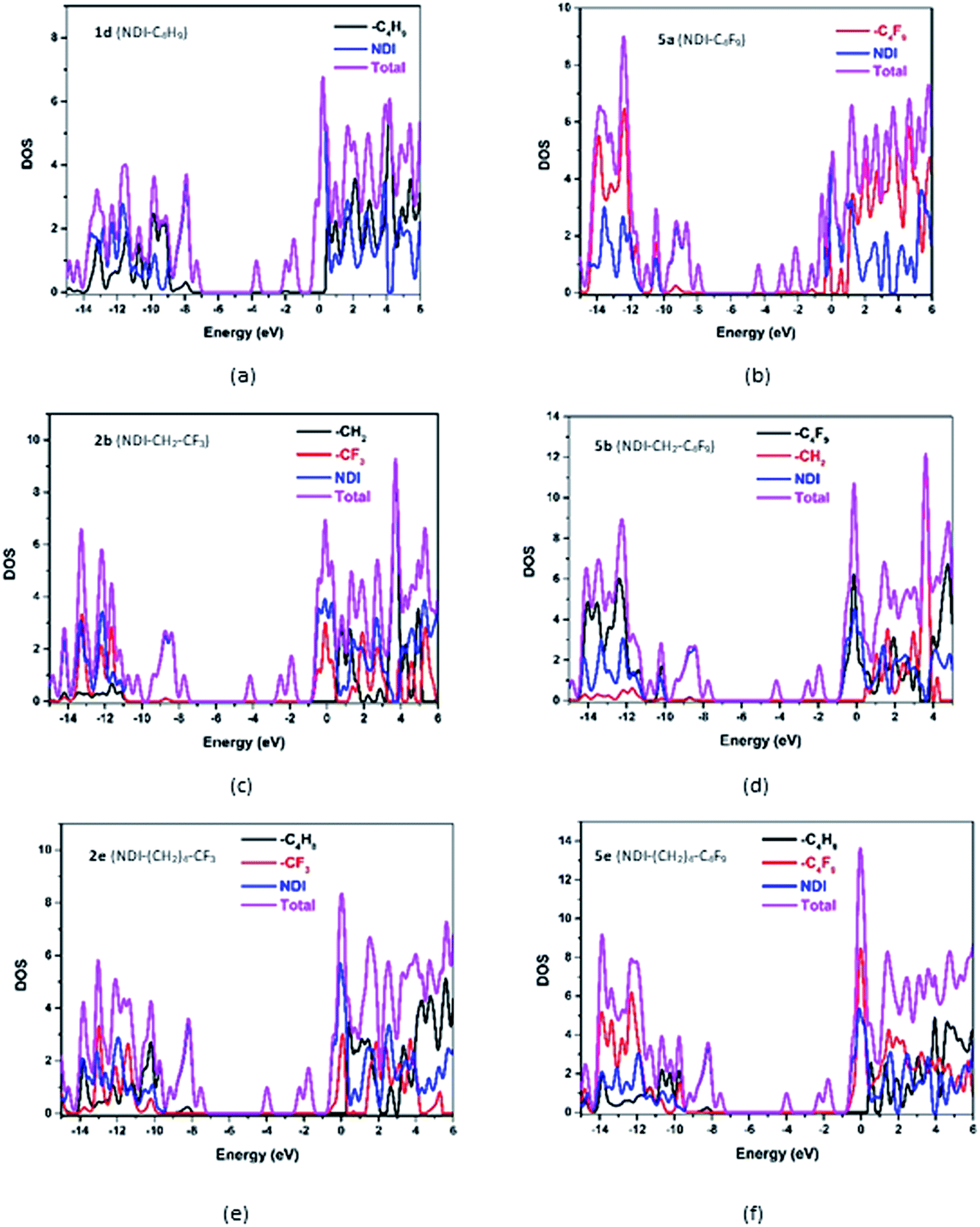
For a detailed comparative study of electronic structures, along with FMOs, the total density of states (DOS) and partial density of states (PDOS) of the investigated NDI derivatives are calculated. The computed DOS and PDOS spectra of some of the investigated NDI derivatives are shown in Fig. 5, and the spectra of the remaining compounds are provided in Fig. S5 in the ESI.† From this study, it is seen that among all the investigated NDI derivatives, the NDI core offers the maximum contribution (99%) to both HOMOs and LUMOs of the compound. The side chains such as alkyl, and fluoroalkyl groups which are being substituted at the N-positions of the NDI core have negligible (∼0.01%) contribution to HOMO/LUMO densities. The above observation is also supported by the frontier molecular orbital analysis, where the isosurfaces of molecular orbitals are found to be localized mostly over the NDI core of all the compounds.
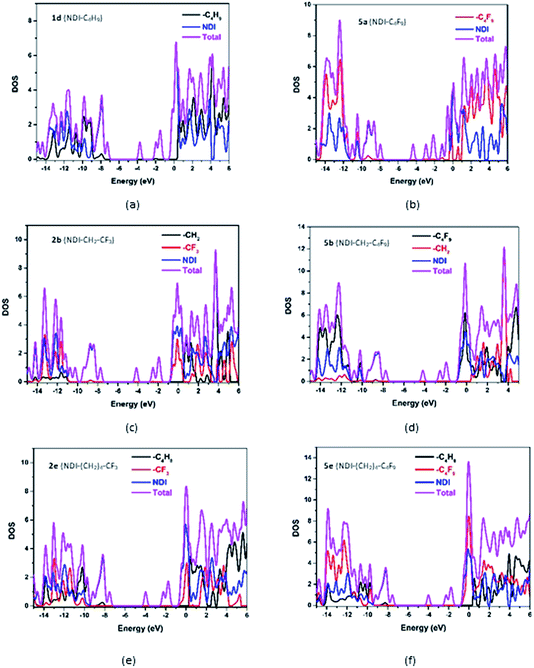 |
| | Fig. 5 Density of states of representative NDI-based compounds. | |
3.3 Electron affinity (EA) and ionization potential (IP)
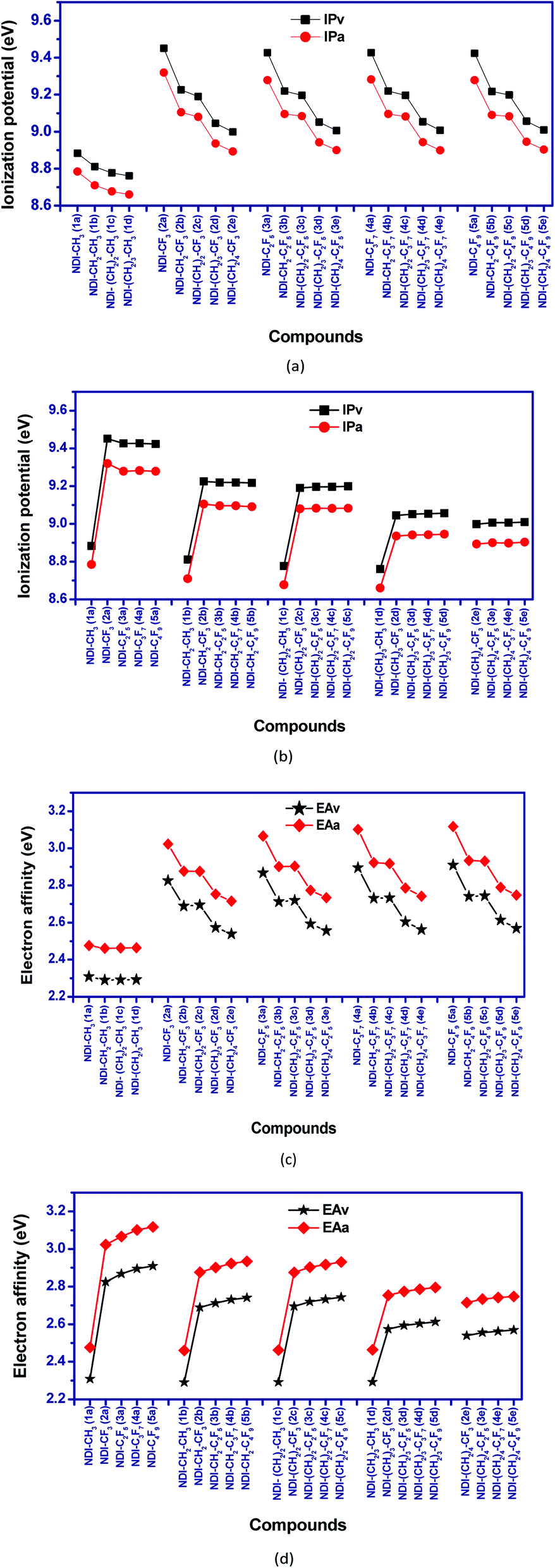
Ionization potential (IP) and electron affinity (EA) are the key factors which determine the ease of charge injection of an organic semiconductor. Both these factors are calculated from the potential energy surfaces in the two forms; adiabatic (IPa and EAa) and vertical (IPv and EAv) way, respectively. The calculated IP and EA values are presented as graphical representation in Fig. 6. The adiabatic ionization potential (IPa) and vertical ionization potential (IPv) of the studied compounds show similar trend. The ionization potential of purely alkyl substituted NDI's is significantly lower than the perfluoroalkyl substituted compounds. This effect is prominent when the carbon atom of perfluoroalkyl group is directly attached to the NDI core (as in NDI–CF3, NDI–CF2–CF3 etc.). For example, IPa of 1a (NDI–CH3) and 2a (NDI–CF3) are 8.78 eV and 9.32 eV, respectively. Similarly, IPa of 1c (NDI–C3H7) and 4a (NDI–C3F7) are 8.68 eV and 9.28 eV, respectively. In all the investigated compounds, a reduction in the calculated vertical and adiabatic ionization potential is observed as the fluoroalkyl group is moved away from the NDI core by inserting methylene (CH2)n group in between them. For example, IPv/IPa of compounds 2a (NDI–CF3), 2b (NDI–CH2–CF3) and 2c (NDI–(CH2)2–CF3) are 9.45/9.32 eV, 9.23/9.11 eV and 9.19/9.08 eV respectively in which n in (CH2)n increases from 0 to 2. Interestingly, increasing the length of perfluoroalkyl groups in a (CH2)n (n = 1–4) containing NDI series does not significantly change the IP's. For instance, IPa for the compounds 2b (NDI–CH2–CF3), 3b (NDI–CH2–C2F5) and 4b (NDI–CH2–C3F7) are 9.11 eV, 9.10 eV and 9.10 eV respectively, and hence is nearly constant.
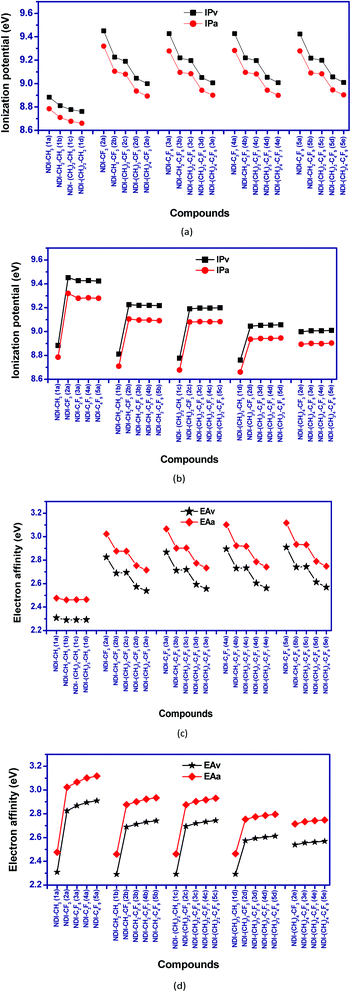 |
| | Fig. 6 Calculated vertical and adiabatic (a and b) ionization potential (c and d) electron affinity for the studied NDI compounds. | |
The incorporation of EWGs (–F atoms) in the NDI core, enhances the electron affinity (EA) values of the compounds. The EA values are found to be highest when the electron withdrawing perfluoroalkyl group is directly attached to the NDI core. For example, EAa for compound 2a (NDI–CF3) is 3.02 eV as compared to 2.48 eV for 1a (NDI–CH3). Similarly, 3a (NDI–C2F5) has EAa of 3.07 eV as compared to 2.46 eV for 1b (NDI–C2H5). So, introduction of perfluoroalkyl groups directly into the NDI core leads to significant increment in the electron affinity values as compared to their alkyl counterparts.
For purely alkyl substituted NDIs, EA doesn't vary significantly with chain length and remains nearly constant. In case of fluoroalkyl substituted derivatives, there is slight increase in EA with increase in chain length of perfluoroalkyl groups. For example, the EAa of the compounds 2a (NDI–CF3) and 5a (NDI–C4F9) are 3.02 eV and 3.12 eV, respectively; which is enhanced by the order ∼0.10 eV with the increase of chain length of perfluoroalkyl groups from –CF3 to –C4F9. Similarly, on moving from the compound 2b (NDI–CH2–CF3) to 5b (NDI–CH2–C4F9), an enhancement of the electron affinity values by order of 0.06 eV is observed. The increment of the electron affinity values with the increase of EWGs in the compounds indicates the enhancement electron transfer nature.52
High EA (adiabatic) (>2.80 eV) and low LUMO energy level (<−4.00 eV) are the important parameters for ambient air-stability and n-type behaviour of organic semiconductors.53,54 In our study, it is found that purely alkyl substituted NDIs (1a, 1b, 1c, 1d) have EA value less than 2.8 eV and LUMO levels higher than −4.00 eV. Higher EA (>2.8 eV) and low LUMO levels (<−4.00 eV) of perfluoroalkyl substituted NDIs (2a, 3a, 4a, 5a) indicates their better air-stable character with superior n-type behaviour than the alkyl substituted ones. In case of fluoroalkyl substituted NDIs, EA values are found to be decreased and LUMO energy level increases, with increasing the distance between the fluoroalkyl group and the NDI core. For example, 2b (NDI–CH2–CF3) has EA of 2.88 eV and LUMO of −4.14 eV whereas, value of EA and LUMO for 2e (NDI–(CH2)4–CF3) are 2.72 eV and −3.96 eV, respectively. Thus, introduction of (poly)methylene groups between the NDI and perfluoroalkyl group reduces the air-stability and n-type character to some extent.
3.4 Reorganization energy (λ)
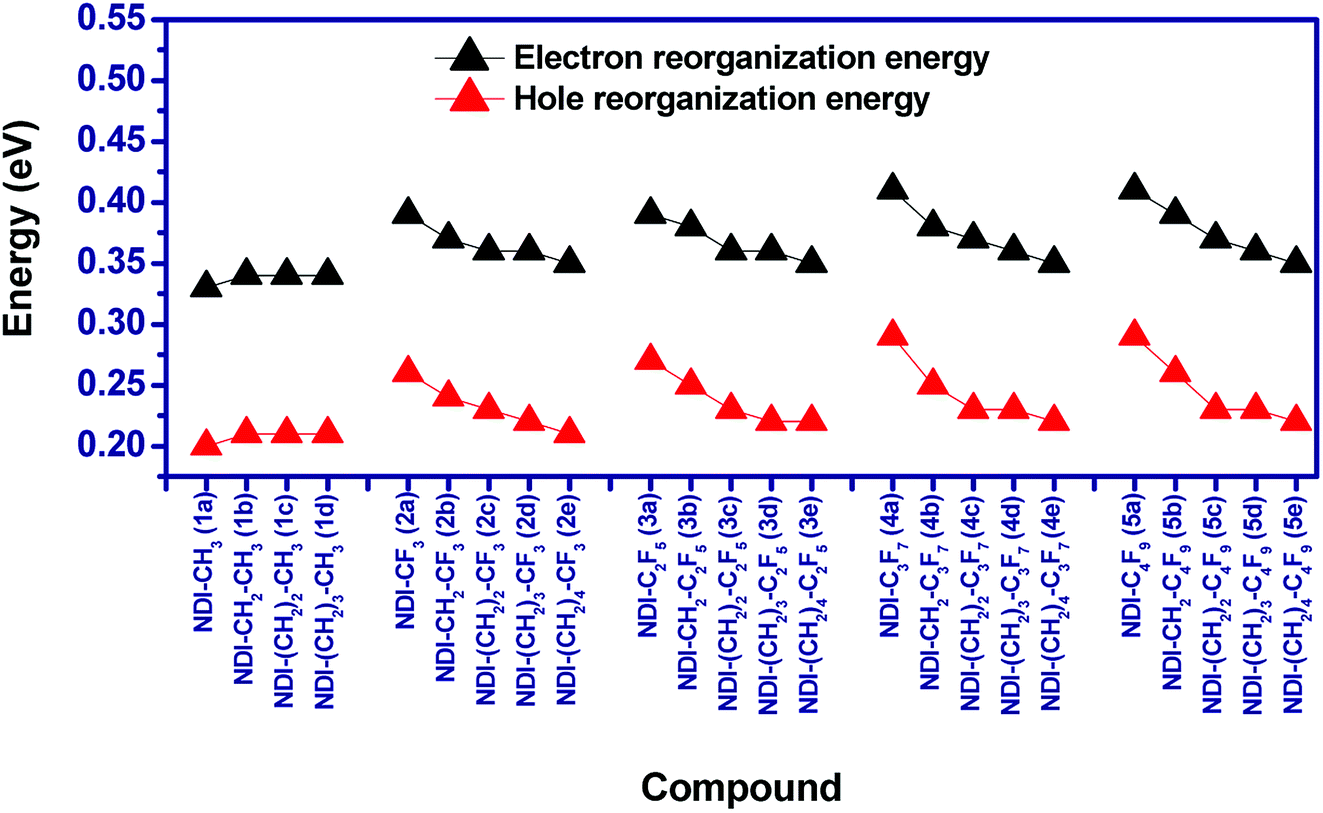
Reorganization energy is considered as an important parameter that affects the charge mobility calculation of organic semiconductors. In this study, both the hole (λh) and electron reorganization energies (λe) are calculated from the adiabatic potential energy surface method. The calculated reorganization energies of the compounds have been shown in Fig. 7. It is found that for the alkyl substituted NDIs i.e. 1a–1d, the increase in length of alkyl chains do not lead to any significant change in λh and λe. For example, the calculated λh/λe for the compounds 1a–1d varies from 0.20–0.21/0.33–0.34. In case of perfluoroalkyl compounds as well, increasing the chain length of perfluoro group does not lead to significant changes in reorganization energies. In addition, for a given perfluoroalkyl group, λh/λe decreases as the distance between the perfluoroalkyl group and NDI core increases. For example, λh/λe for compounds 2a (NDI–CF3), 2b (NDI–CH2–CF3) and 2c (NDI–(CH2)2–CF3) are 0.26/0.39 eV, 0.24/0.37 eV and 0.23/0.36 eV respectively. Due to the low difference between the electron and hole reorganization energy, charge injection barrier is also evaluated to have an idea about the charge transport properties of the studied compounds.
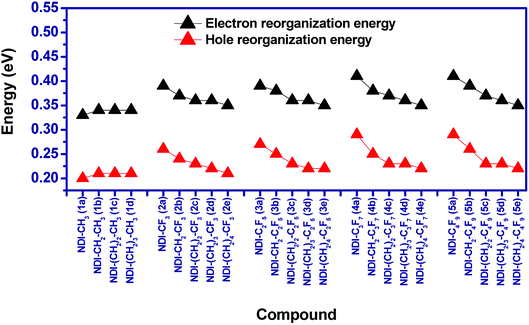 |
| | Fig. 7 Graphical presentation of the calculated reorganization energies of the compounds. | |
The charge injection barrier can be used to understand the ease with which charge can be injected through source-drain electrode. As, in majority of the OFETs, Au electrodes having work function (φ) of 5.1 eV are usually employed as source and drain electrodes, we have considered it for calculating injection barrier. Considering the LUMO energy levels of the materials, the electron injection for the Au electrode is calculated as φe = 5.1 − |LUMO|, where |LUMO| is the absolute value of the energy of LUMO of the materials. In the case of the investigated compounds, electron injection barrier is observed in the range of 0.76–1.32 eV. The hole injection barrier with the same electrode is found to be very much higher in the range of 2.18–2.83 eV, which is tabulated in ESI (Table S6).†
3.5 Optical absorption spectra
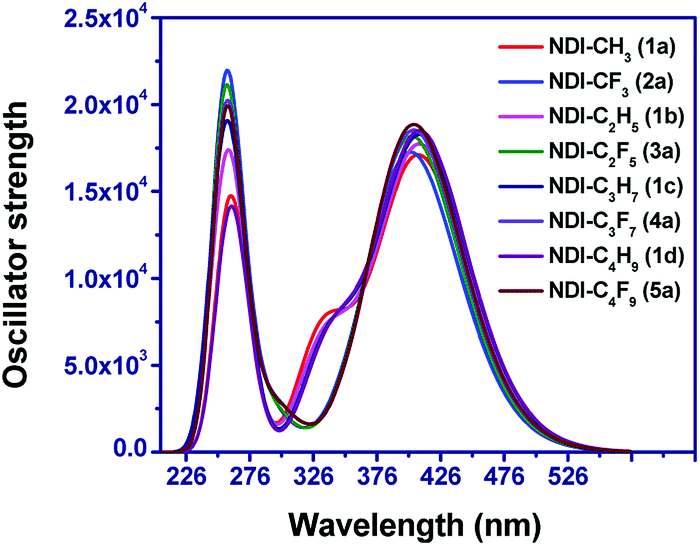
The optical absorption spectra were computed at B3LYP and CAM-B3LYP functional. The computed spectra of the compounds at B3LYP/6-31+G(d,p) level showed absorption maxima in the range of 382–386 nm and which are in good agreement with the reported experimental spectra.20,33,55,56 The computed absorption spectra for eight compounds at B3LYP/6-31+G(d,p) level are displayed in Fig. 8. The computed absorption spectra of remaining compounds are shown in Fig. S6† and the values obtained at CAM-B3LYP level are shown in Table S5 of the ESI† respectively. The corresponding transition energy (E), absorption wavelength (λabs), oscillator strength (f) and major transitions for the eight compounds and others are listed in Tables 1 and S4 in ESI,† respectively. For all the compounds, the computed absorption spectra ranges from 180 nm to 600 nm and mostly lies in the ultra-violet (UV) region in the solar spectra. It is observed that the main contribution for electronic transitions is from the ground state (S0) and the first excited state (S1) and mostly attributed to HOMO to LUMO transition. This trend also affirms a linear correlation between the transition energy and HOMO–LUMO energy gap.
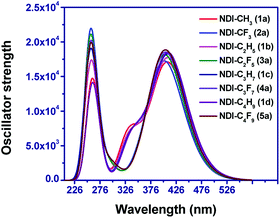 |
| | Fig. 8 Simulated absorption spectra of the eight NDI-based compounds in dichloromethane solvent. | |
Table 1 Computed electronic transition energy (E), absorption wavelength (λ), oscillator strength (f) and transition and major composition for dominant excitations in dichloromethane solvent at B3LYP/6-31+G(d,p) level
| Compound |
State |
E (eV) |
λ (nm) |
f |
Major configuration |
| 1a (NDI–CH3) |
S1 |
3.2196 |
385 |
0.3999 |
HOMO → LUMO (99%) |
| S14 |
5.2686 |
235 |
0.3645 |
HOMO → L+2 (81%) |
| 1b (NDI–C2H5) |
S1 |
3.2134 |
386 |
0.4161 |
HOMO → LUMO (99%) |
| S14 |
5.2574 |
236 |
0.3488 |
HOMO → L+2 (80%) |
| 1c (NDI–C3H7) |
S1 |
3.2117 |
386 |
0.4301 |
HOMO → LUMO (99%) |
| S16 |
5.2516 |
236 |
0.3292 |
HOMO → L+2 (80%) |
| 1d (NDI–C4H9) |
S1 |
3.2116 |
386 |
0.4362 |
HOMO → LUMO (99%) |
| S18 |
5.2500 |
236 |
0.3113 |
HOMO → L+2 (78%) |
| 2a (NDI–CF3) |
S1 |
3.2443 |
382 |
0.3714 |
HOMO → LUMO (98%) |
| S16 |
5.3414 |
232 |
0.5174 |
HOMO → L+3 (80%) |
| 3a (NDI–C2F5) |
S1 |
3.2398 |
383 |
0.3939 |
HOMO → LUMO (98%) |
| S16 |
5.3486 |
232 |
0.498 |
HOMO → L+3 (80%) |
| 4a (NDI–C3F7) |
S1 |
3.2296 |
384 |
0.4057 |
HOMO → LUMO (98%) |
| S16 |
5.3340 |
232 |
0.4827 |
HOMO → L+3 (81%) |
| 5a (NDI–C4F9) |
S1 |
3.2292 |
384 |
0.4143 |
HOMO → LUMO (98%) |
| S16 |
5.3325 |
233 |
0.4754 |
HOMO → L+3 (81%) |
All the computed predominant electronic transitions are noted to be π–π* in nature and it is also supported by molecular orbital analysis (Fig. 4(a) and S4†). As the observed absorption maximas are found at approximately similar range (382–386 nm) for all the studied compounds, the compounds can be suitable candidates as an organic photo-detector in the UV region.
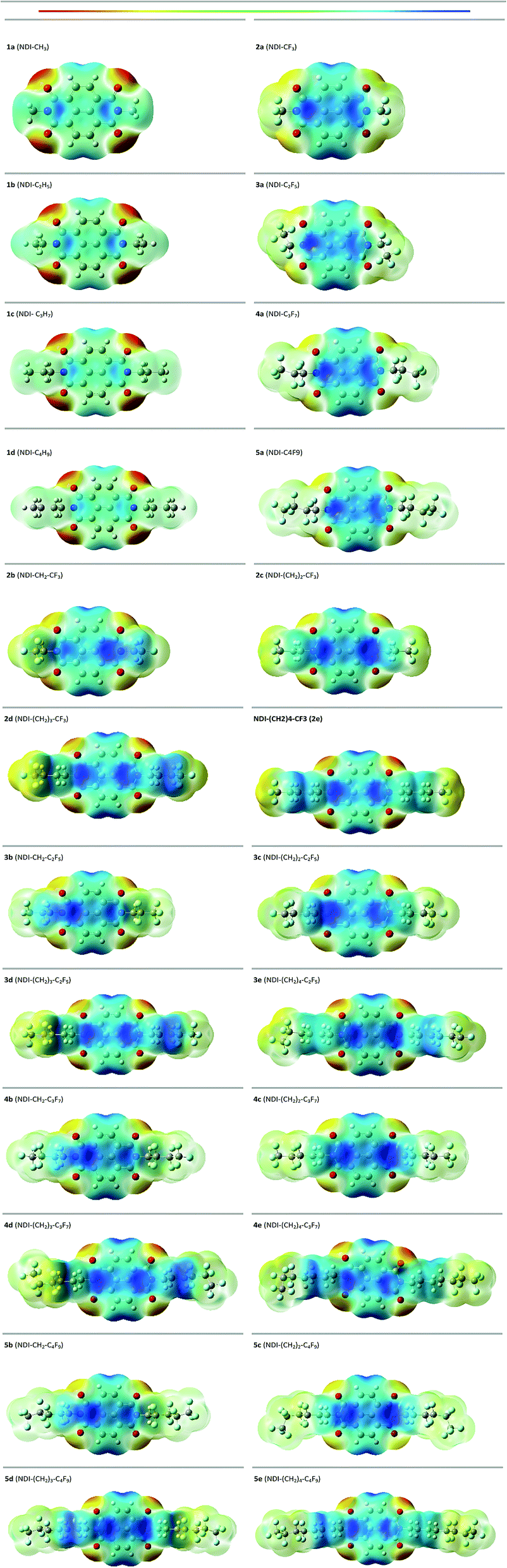
3.6 Molecular electrostatic potential (ESP) surface
The molecular electrostatic potential (ESP) surface is evaluated to understand the electron density regions in the molecular framework.57,58 The evaluated ESP surface of the studied compounds are shown in Fig. 9. Here, the blue colour signifies electron-poor region and the red colour signifies an electron rich region. The positive potential increases on moving from red to blue as red < orange < yellow < green < blue shown in the colour bar of Fig. 9. From the ESP surface, it is seen that the compounds with alkyl substitution have more negative potential located mainly on the carbonyl group of the compound than its corresponding fluoroalkyl substituted compounds. For instance, 1a (NDI–CH3) has more negative potential as compared with 2a (NDI–CF3) on the carbonyl group of the compound. For the alkyl-substituted compounds, the terminal side chain is more electropositive showing the blue color in the ESP surface. But this appears greenish yellow in the fluoroalkyl substituted compounds indicating towards electronegative nature. This implies that, the negative potential increases for the fluoroalkyl substituted compounds towards the side chain as compared with its alkyl substituted counterparts. This is in good agreement with the higher EA value observed for the former case. It was reported that, the presence of more electronegative region on the NDI-core restrict the cofacial interaction, rather it favours the herringbone interaction pattern with more electrostatic repulsion.59–61 The distribution of the more electronegative region towards the terminal chain and less electronegative region in the NDI-core might lead to cofacial interaction for fluoroalkyl substituted compounds. This, in turn, may affect the charge transport properties of the fluoroalkylated compounds vis-a-vis the alkyl substituted one.
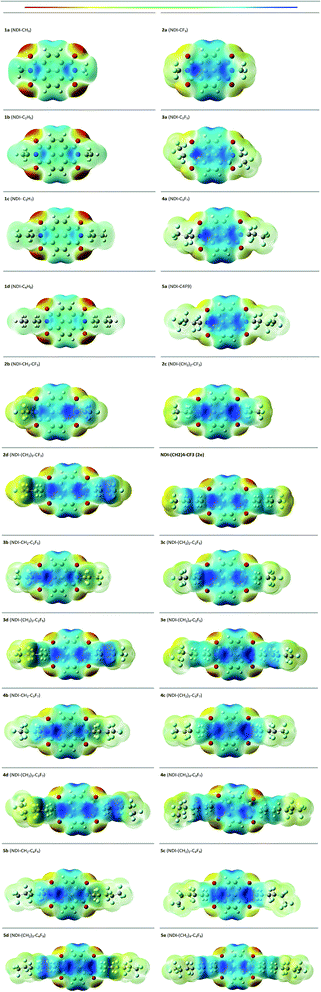 |
| | Fig. 9 ESP surface of the studied compounds. | |
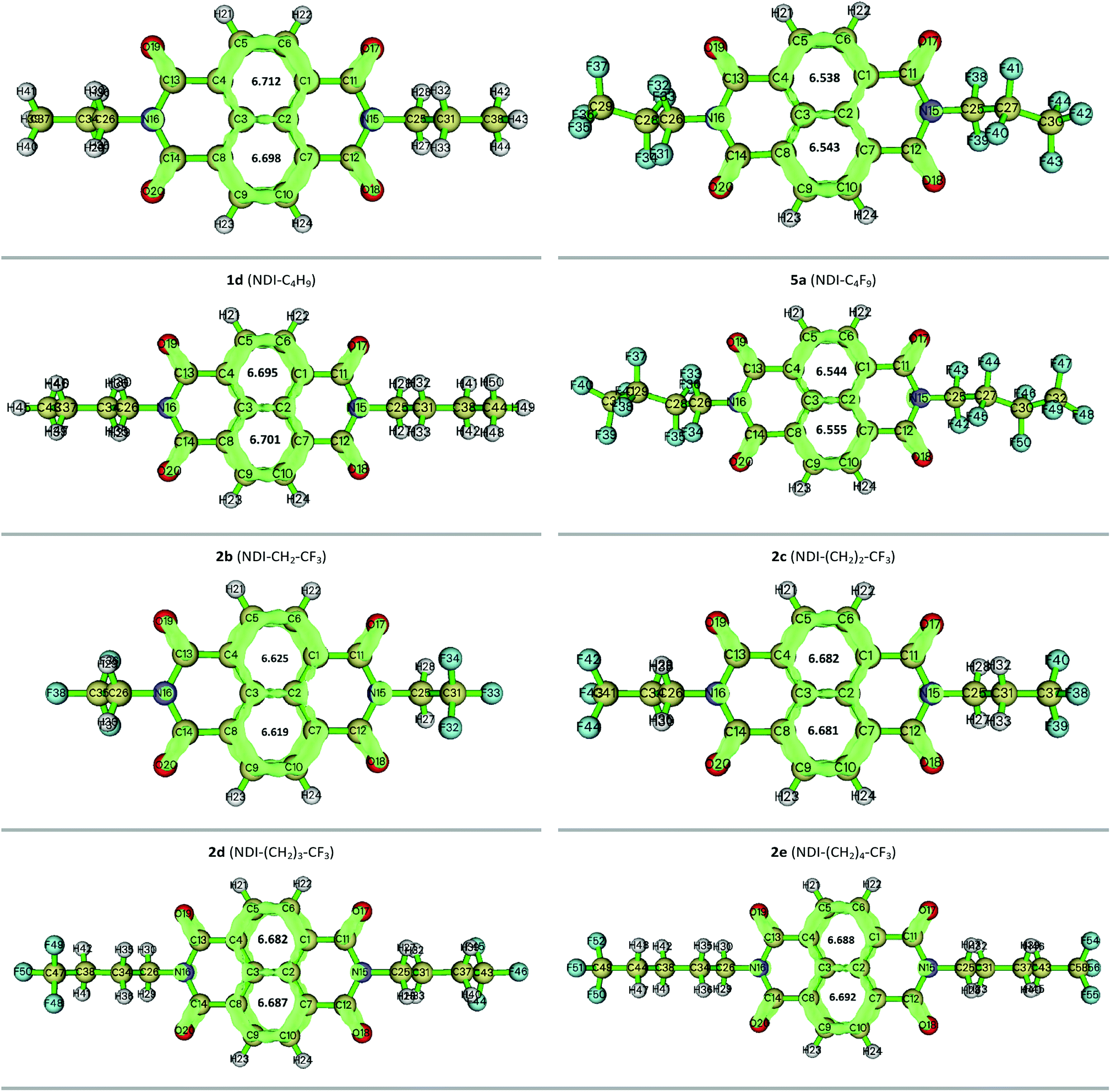
3.7 LOLIPOP calculation
The LOLIPOP (Localized Orbital Locator Integrated Pi Over Plane) index was first introduced by Gonthier et al.62 to characterize the π-electron nature and π-stacking ability of aromatic systems. The low values of LOLIPOP imply that a molecule might exhibit relatively stronger π–π interaction and better π-stacking ability.30,31 In the present study, the LOLIPOP calculation was carried out based on Localized Orbital Locator-π (LOL-π) approach.63,64 The isosurface map of the Localized Orbital Locator-π (LOL-π) based on the automatically detected π-LMOs are generated to get an idea about the π-delocalization pathway with the isosurface value of 0.55. The obtained figures along with the computed LOLIPOP values of the corresponding aromatic rings for twelve compounds are given in Fig. 10 and the rest are given in the Fig. S7 of the ESI.†
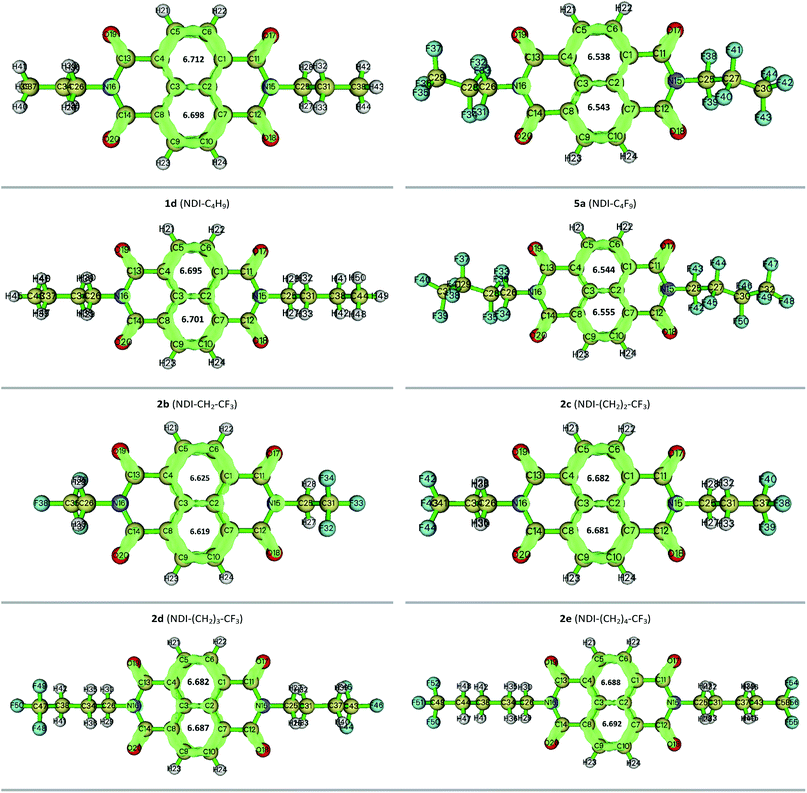 |
| | Fig. 10 Localized orbital locator surface along with the LOLIPOP value for twelve NDI-based compounds. | |
In our study, the LOLIPOP calculation was performed at B3LYP/6-31G(d,p) theoretical level using Pipek–Mezey orbital localization method.30 From the calculated LOLIPOP values, it is found that, on substitution of fluoroalkyl groups with the alkyl groups in the compounds, the central rings or core rings of the designed compounds have relatively smaller LOLIPOP values and thus they are more favourable for π–π stacking. This may lead to better π-stacking ability of the fluoroalkyl substituted compounds as compared with the alkyl substituted compounds. For instance, the LOLIPOP values for the central rings of 1a (NDI–CH3) is found to be 7.050 and 7.052 whereas it comes at 6.516 and 6.515 for the central rings of 2a (NDI–CF3). From the LOL-π isosurface maps, π-depletion is observed in the central rings for fluoroalkyl substituted compounds and it is supported by their smaller LOLIPOP values. In addition, as the distance between the fluoroalkyl group and NDI increases by the incorporation of methylene groups to the N-atom of NDI core, LOLIPOP value increases. For example, for compound 2a (NDI–CF3), the LOLIPOP values for the central rings are 6.516 and 6.515. For compounds 2b (NDI–CH2–CF3) and 2e (NDI–(CH2)4–CF3) the corresponding values are 6.625, 6.619 and 6.688, 6.692, respectively. The observation indicates that the incorporation of methylene groups to the N-atom of NDI reduces the π–π interaction ability. Therefore, side chain engineering might have a strong impact on the π–π stacking ability and fluoroalkyl substituted NDI compounds can be a better candidate than the alkyl substituted ones in organic semiconductor applications for having higher π-stacking ability which can lead to better charge transport properties.65,66 A table is given in the ESI (Table S7)† to summarize the mobility obtained for some NDI-based compounds experimentally reported earlier.
4 Conclusion
Herein, we explore the effect of electron-withdrawing side chains viz., fluoroalkyl group attached to the N atom of imide functionality in NDI on their structures and electronic properties. The NDI core for the alkyl substituted compounds is found to be completely planar. Similar observation is observed in case of the fluoroalkyl substituted compounds having the higher number of the intervening methylene groups between the NDI core and the fluoroalkyl group (such as 2d (NDI–(CH2)3–CF3), 2e (NDI–(CH2)4–CF3)). In contrast, a small deviation (∼12°) from planarity is seen for all the perfluoroalkyl substituted compounds. Compounds having a smaller number of the intervening methylene groups between the NDI core and the fluoroalkyl group (such as 2b (NDI–CH2–CF3), 3b (NDI–CH2–C2F5)) also exhibited small deviation from planarity. Incorporation of fluoroalkyl group to the NDI-core can significantly lower the HOMO, LUMO levels as compared to the alkyl substituted one. As the distance between the fluoroalkyl group and NDI increases, HOMO and LUMO energy increases due to reduced electron withdrawing nature of fluorine atom/group and thus reduce the air-stability to some extent. The computed electron affinity is found to be enhanced after incorporation of EWG fluoroalkyl groups in the NDI core. In case of pure alkyl substituted NDIs, side chain length has no significant impact on IP and EA. However, for fluoroalkyl substituted derivates, IP and EA increase with the increment of fluoroalkyl groups attached to the core. Reorganization energy, another key parameter for charge transport properties, is influenced by simultaneous increment of alkyl, fluoroalkyl groups in NDI derivatives. High EA (adiabatic) (>2.80 eV) and low LUMO energy (<–4.00 eV) ensures fluoroalkyl substituted NDIs are more stable and have more n-type nature than their alkyl analogues. This trend is also confirmed by the observed negative potential on the side chains from the ESP surface analysis. All the investigated compounds show optical absorption spectra in the range of 180–600 nm, with intense absorption in UV-region. Hence, the compounds may be considered as the suitable candidate for organic photodetector applications in the UV region of solar spectrum. Further, the LOLIPOP values point towards the π-stacking ability of the studied compounds, as comparatively low LOLIPOP value of the fluoroalkyl substituted compounds leads to better π-stacking capability in fluoroalkyl substituted compounds than the alkyl substituted ones. The results obtained in this work show the importance of side chains attached to the NDI-core. We believe that our present work will contribute towards a better understanding of the structure–property relationships and will provide an insight for further development of side chain substituted NDI derivatives for optoelectronic applications.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
SS thank the Department of Science and Technology (DST), Govt. of India for the INSPIRE Faculty Grant (DST/INSPIRE/04/2014/000185). The authors also thank Institute of Advanced Study in Science and Technology (IASST) for the research facility and fellowship. SS wishes to thank Rev. Fr. Dr Stephen Mavely, Vice Chancellor, Assam Don Bosco University for the encouragement and providing necessary research infrastructure.
References
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Semiconducting π-Conjugated Systems in Field-Effect Transistors: A Material Odyssey of Organic Electronics, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS.
- S. Allard, M. Forster, B. Souharce, H. Thiem and U. Scherf, Organic Semiconductors for Solution-Processable Field-Effect Transistors (OFETs), Angew. Chem., Int. Ed., 2008, 47, 4070–4098 CrossRef CAS.
- H. Sirringhaus, 25th Anniversary Article: Organic Field-Effect Transistors: The Path Beyond Amorphous Silicon, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS.
- B. Geffroy, P. l. Roy and C. Prat, Organic light-emitting diode (OLED) technology: materials, devices and display Technologies, Polym. Int., 2006, 55, 572–582 CrossRef CAS.
- R. K. Gupta, H. Ulla, M. N. Satyanarayan and A. A. Sudhakar, A Perylene-Triazine-Based Star-Shaped Green Light Emitter for Organic Light Emitting Diodes, Eur. J. Org. Chem., 2018, 1608–1613 CrossRef CAS.
- A. Facchetti, Polymer donor–polymer acceptor (all-polymer) solar cells, Mater. Today, 2013, 16, 123–132 CrossRef CAS.
- G. Li, W. H. Chang and Y. Yang, Low-bandgap conjugated polymers enabling solution-processable tandem solar cells, Nat. Rev. Mater., 2017, 2, 1–13 Search PubMed.
- T. M. Clarke and J. R. Durrant, Charge Photogeneration in Organic Solar Cells, Chem. Rev., 2010, 110, 6736–6767 CrossRef CAS.
- M. Kielar, T. Hamid, L. Wu, F. Windels, P. Sah and A. K. Pandey, Organic Optoelectronic Diodes as Tactile Sensors for Soft-Touch Applications, ACS Appl. Mater. Interfaces, 2019, 11, 21775–21783 CrossRef CAS.
- Y. Yan, X. Wu, Q. Chen, Y. Liu, H. Chen and T. Guo, High-Performance Low-Voltage Flexible Photodetector Arrays Based on All-Solid-State Organic Electrochemical Transistors for Photosensing and Imaging, ACS Appl. Mater. Interfaces, 2019, 11, 20214–20224 CrossRef CAS.
- L. Kergoat, B. Piro, M. Berggren, G. Horowitz and M. C. Pham, Advances in Organic Transistor-Based Biosensors: From Organic Electrochemical Transistors to Electrolyte-Gated Organic Field-Effect Transistors, Anal. Bioanal. Chem., 2012, 402, 1813–1826 CrossRef CAS.
- C. R. Newman, C. D. Frisbie, D. A. da Silva Filho, J. L. Bredas, P. C. Ewbank and K. R. Mann, Introduction to Organic Thin Film Transistors and Design of n-Channel Organic Semiconductors, Chem. Mater., 2004, 16, 4436–4451 CrossRef CAS.
- Z. Chen, Y. Zheng, H. Yan and A. Facchetti, Naphthalenedicarboximide- vs. Perylenedicarboximide-Based Copolymers. Synthesis and Semiconducting Properties in Bottom-Gate N-Channel Organic Transistors, J. Am. Chem. Soc., 2009, 131, 8–9 CrossRef CAS.
- C. Thalacker, C. Roger and F. Wurthner, Synthesis and Optical and Redox Properties of Core-Substituted Naphthalene Diimide Dyes, J. Org. Chem., 2006, 71, 8098–8105 CrossRef CAS.
- H. Z. Chen, M. M. Ling, X. Mo, M. M. Shi, M. Wang and Z. Bao, Air Stable n-channel Organic Semiconductors for Thin Film Transistors Based on Fluorinated Derivatives of Perylene Diimides, Chem. Mater., 2007, 19, 816–824 CrossRef CAS.
- C. Huang, S. Barlow and S. R. Marder, Perylene-3,4,9,10-tetracarboxylic Acid Diimides: Synthesis, Physical Properties, and Use in Organic Electronics, J. Org. Chem., 2011, 76, 2386–2407 CrossRef CAS.
- S. Y. g. Liu, W. Q. Liu, J. Q. Xu, C. C. Fan, W. F. Fu, J. Ling, J. Y. Wu, M. M. Shi, A. K. Y. Jen and H. Z. Chen, Pyrene and Diketopyrrolopyrrole-Based Oligomers Synthesized via Direct Arylation for OSC Applications, ACS Appl. Mater. Interfaces, 2014, 6, 6765–6775 CrossRef CAS.
- J. Y. Back, H. Yu, I. Song, I. Kang, A. Hyungju, T. J. Shin, S. Kwon, J. H. Oh and Y. H. Kim, Investigation of Structure–Property Relationships in Diketopyrrolopyrrole-Based Polymer Semiconductors via Side-Chain Engineering, Chem. Mater., 2015, 27, 1732–1739 CrossRef CAS.
- J. G. Laquindanum, H. E. Katz, A. Dodabalapur and A. J. Lovinger, n-Channel Organic Transistor Materials Based on Naphthalene Frameworks, J. Am. Chem. Soc., 1996, 118, 11331–11332 CrossRef CAS.
- Y. Jung, K. J. Baeg, D. Y. Kim, T. Someya and S. Y. Park, A thermally resistant and air-stable n-type organic semiconductor: naphthalene diimide of 3,5-bis-trifluoromethyl aniline, Synth. Met., 2009, 159, 2117–2121 CrossRef CAS.
- J. H. Oh, S. L. Suraru, W. Y. Lee, M. Konemann, H. W. Hoffken, C. Roger, R. Schmidt, Y. Chung, W. C. Chen, F. Wurthner and Z. Bao, High-Performance Air-Stable n-Type Organic Transistors Based on Core-Chlorinated Naphthalene Tetracarboxylic Diimides, Adv. Funct. Mater., 2010, 20, 2148–2156 CrossRef CAS.
- Z. Ma, H. Geng, D. Wang and Z. Shuai, Influences of alkyl side-chain length on the carrier mobility in organic semiconductors: herringbone vs. pi-pi stacking, J. Mater. Chem. C, 2016, 4, 4546–4555 RSC.
- H. E. Katz, T. Siegrist, J. Hendrik Schon, C. Kloc, B. Batlogg, A. J. Lovinger and J. Johnson, Solid-State Structural and Electrical Characterization of N-Benzyl and N-Alkyl Naphthalene 1,4,5,8-Tetracarboxylic Diimides, ChemPhysChem, 2001, 2, 167–172 CrossRef CAS.
- H. E. Katz, J. Johnson, A. J. Lovinger and W. Li, Naphthalenetetracarboxylic Diimide-Based n-Channel Transistor Semiconductors: Structural Variation and Thiol-Enhanced Gold Contacts, J. Am. Chem. Soc., 2000, 122, 7787–7792 CrossRef CAS.
- H. E. Katz, A. J. Lovinger, J. Johnson, C. Kloc, T. Siegrist, W. Li, Y.-Y. Lin and A. Dodabalapur, A soluble and air-stable organic semiconductor with high electron mobility, Nature, 2000, 404, 478–481 CrossRef CAS.
- Y. L. Lee, H.-L. Hsu, S.-Y. Chen and T.-R. Yew, Solution-Processed Naphthalene Diimide Derivatives as n-Type Semiconductor Materials, J. Phys. Chem. C, 2008, 112, 1694–1699 CrossRef CAS.
- B. J. Jung, K. Lee, J. Sun, A. G. Andreou and H. E. Katz, Air-Operable, High-Mobility Organic Transistors with Semifluorinated Side Chains and Unsubstituted Naphthalenetetracarboxylic Diimide Cores: High Mobility and Environmental and Bias Stress Stability from the Perfluorooctylpropyl Side Chain, Adv. Funct. Mater., 2010, 20, 2930–2944 CrossRef CAS.
- X. Guo, A. Facchetti and T. J. Marks, Imide- and Amide-Functionalized Polymer Semiconductors, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS.
- D. Zhang, L. Zhao, Y. Zhu, A. Li, C. He, H. Yu, Y. He, C. Yan, O. Goto and H. Meng, Effects of p-(Trifluoromethoxy)benzyl and p-(Trifluoromethoxy)phenyl Molecular Architecture on the Performance of Naphthalene Tetracarboxylic Diimide-Based Air Stable n-Type Semiconductors, ACS Appl. Mater. Interfaces, 2016, 8, 18277–18283 CrossRef CAS.
- K. C. See, C. Landis, A. Sarjeant and H. E. Katz, Easily Synthesized Naphthalene Tetracarboxylic Diimide Semiconductors with High Electron Mobility in Air, Chem. Mater., 2008, 20, 3609–3616 CrossRef CAS.
- T. B. Singh, S. Erten, S. Gunes, C. Zafer, G. Turkmen, B. Kuban, Y. Teoman, N. S. Sariciftci and S. Icli, Soluble derivatives of perylene and naphthalene diimide for n-channel organic field-effect transistors, Org. Electron., 2006, 7, 480–489 CrossRef CAS.
- S. Canola and F. Negri, Anisotropy of the n-type charge transport and thermal effects in crystals of a fluoro-alkylated naphthalene diimide: a computational investigation, Phys. Chem. Chem. Phys., 2014, 16, 21550–21558 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 in Revision E.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, cclib: A Library for Package-Independent Computational Chemistry Algorithms, J. Comput. Chem., 2008, 29, 839–845 CrossRef.
- T. Lu and F. Chen, Multiwfn: A Multifunctional Wavefunction Analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- T. Lu and Q. Chen, A simple method of identifying π orbitals for non-planar systems and a protocol of studying π electronic structure, Theor. Chem. Acc., 2020, 139, 1–12 Search PubMed.
- G. R. Krishna, R. Devarapalli, G. Lal and C. M. Reddy, Mechanically Flexible Organic Crystals Achieved by Introducing Weak Interactions in Structure: Supramolecular Shape Synthons, J. Am. Chem. Soc., 2016, 138, 13561–13567 CrossRef CAS.
- X. Zhao, Y. Xiong, J. Ma and Z. Yuan, Rylene and Rylene Diimides: Comparison of Theoretical and Experimental Results and Prediction for High-Rylene Derivatives, J. Phys. Chem. A, 2016, 120, 7554–7560 CrossRef CAS.
- M. Pandeeswar, H. Khare, S. Ramakumar and T. Govindaraju, Biomimetic molecular organization of naphthalene diimide in the solid state: tunable (chiro-) optical, viscoelastic and nanoscale properties, RSC Adv., 2014, 4, 20154–20163 RSC.
- R. Devarapalli, S. B. Kadambi, C. T. Chen, G. R. Krishna, B. R. Kammari, M. J. Buehler, U. Ramamurty and C. Malla Reddy, Remarkably Distinct Mechanical Flexibility in Three Structurally Similar Semiconducting Organic Crystals Studied by Nanoindentation and Molecular Dynamics, Chem. Mater., 2019, 31, 1391–1402 CrossRef CAS.
- P. M. Alvey, J. J. Reczek, V. Lynch and B. L. Iverson, A Systematic Study of Thermochromic Aromatic Donor-Acceptor Materials, J. Org. Chem., 2010, 75, 7682–7690 CrossRef CAS.
- A. Lv, Y. Li, W. Yue, L. Jiang, H. Dong, G. Zhao, Q. Meng, W. Jiang, Y. He, Z. Li, Z. Wang and W. Hu, High performance n-type single crystalline transistors of naphthalene bis(dicarboximide) and their anisotropic transport in crystals, Chem. Commun., 2012, 48, 5154–5156 RSC.
- D. Shukla, M. Rajeswaran, W. G. Ahearn and D. M. Meyer, N,N-Bis(2,2,3,3,4,4,4-heptafluorobutyl)-naphthalene-1,4:5,8-tetracarboximide, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o2327 CrossRef CAS.
- H. Usta, C. Risko, Z. Wang, H. Huang, M. K. Deliomeroglu, A. Zhukhovitskiy, A. Facchetti and T. J. Marks, Design, synthesis, and characterization of ladder-type molecules and polymers. Air-stable, solution-processable n-channel and ambipolar semiconductors for thin-film transistors via experiment and theory, J. Am. Chem. Soc., 2009, 131, 5586–5608 CrossRef CAS.
- Y. C. Chang, M. Y. Kuo, C. P. Chen, H. F. Lu and I. Chao, On the air stability of n-channel organic field-effect transistors: a theoretical study of adiabatic electron affinities of organic semiconductors, J. Mater. Chem. C, 2010, 114, 11595–11601 CAS.
- B. A. Jones, A. Facchetti, M. R. Wasielewski and T. J. Marks, Effects of Arylene Diimide Thin Film Growth Conditions on n-Channel OFET Performance, Adv. Funct. Mater., 2008, 18, 1329–1339 CrossRef CAS.
- J. H. Oh, S. Liu and Z. Bao, Air-stable n-channel organic thin-film transistors with high field-effect mobility based on N,N′-bis(heptafluorobutyl)-3,4:9,10-perylene diimide, Appl. Phys. Lett., 2007, 91, 212107 CrossRef.
- C. C. Liu, S. W. Mao and M. Y. Kuo, Cyanated Pentaceno[2,3-c]chalcogenophenes for Potential Application in Air-Stable Ambipolar Organic Thin-Film Transistors, J. Phys. Chem. C, 2010, 114, 22316–22321 CrossRef CAS.
- C. H. Li, C. H. Huang and M. Y. Kuo, Halogenated 6,13-bis(triisopropylsilylethynyl)-5,7,12,14-tetraazapentacene: applications for ambipolar air-stable organic field-effect transistors, Phys. Chem. Chem. Phys., 2011, 13, 11148–11155 RSC.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletete, G. Durocher, Y. Tao and M. Leclerc, Toward a Rational Design of Poly(2,7-Carbazole) Derivatives for Solar Cells, J. Am. Chem. Soc., 2008, 130, 732–742 CrossRef CAS.
- D. M de Leeuw, M. M. J. Simenon, A. R. Brown and R. E. F. Einhard, Stability of n-type doped conducting polymers and consequences for polymeric microelectronic devices, Synth. Met., 1997, 87, 53–59 CrossRef.
- T. Sutradhar and A. Misra, Role of Electron-Donating and Electron-Withdrawing Groups in Tuning the Optoelectronic Properties of Difluoroboron–Napthyridine Analogues, J. Phys. Chem. A, 2018, 122, 4111–4120 CrossRef CAS.
- Y. C. Chang, M. Y. Kuo, C. P. Chen, H. F. Lu and I. Chao, On the Air Stability of n-Channel Organic Field-Effect Transistors: A Theoretical Study of Adiabatic Electron Affinities of Organic Semiconductors, J. Phys. Chem. C, 2010, 114, 11595–11601 CrossRef CAS.
- U. Purushotham and G. N. Sastry, Conjugate acene fused buckybowls: evaluating their suitability for p-type, ambipolar and n-type air stable organic semiconductors, Phys. Chem. Chem. Phys., 2013, 15, 5039–5048 RSC.
- G. Andric, J. F. Boas, A. M. Bond, G. D. Fallon, K. P. Ghiggino, C. F. Hogan, J. A. Hutchison, M. A. P. Lee, S. J. Langford, J. R. Pilbrow, G. J. Troup and C. P. Woodward, Spectroscopy of Naphthalene Diimides and Their Anion Radicals, Aust. J. Chem., 2004, 57, 1011–1019 CrossRef CAS.
- L. Liu, Z. Ren, C. Xiao, B. He, H. Dong, S. Yan, W. Hu and Z. Wang, Epitaxially-crystallized oriented naphthalene bis(dicarboximide) morphology for significant performance improvement of electron transporting thin-film transistors, Chem. Commun., 2016, 52, 4902–4905 RSC.
- J. S. Murray and K. Sen, Molecular electrostatic potentials: concepts and applications, Amsterdam, The Netherlands, 1996 Search PubMed.
- G. Gogoi, S. R. Sahoo, B. K. Rajbongshi, S. Sahu, N. S. Sarma and S. Sharma, New types of organic semiconductors based on diketopyrrolopyrroles and 2,1,3-benzochalcogenadiazoles: a computational study, J. Mol. Model., 2019, 25, 1–12 CrossRef CAS.
- R. K. Raju, J. W. G. Bloom and S. E. Wheeler, Broad transferability of substituent effects in π-stacking interactions provides new insights into their origin, J. Chem. Theory Comput., 2013, 9, 3479–3490 CrossRef CAS.
- Z. F. Yao, J. Y. Wang and J. Pei, Control of π–π Stacking via Crystal Engineering in Organic Conjugated Small Molecule Crystals, Cryst. Growth Des., 2018, 18, 7–15 CrossRef CAS.
- Y. Y. Lai, V. H. Huang, H. T. Lee and H. R. Yang, Stacking principles on π- and lamellar stacking for organic semiconductors evaluated by energy decomposition analysis, ACS Omega, 2018, 3, 18656–18662 CrossRef CAS.
- J. F. Gonthier, S. N. Steinmann, L. Roch, A. Ruggi, N. Luisier, K. Severin and C. Corminboeuf, pi-Depletion as a criterion to predict π-stacking ability, Chem. Commun., 2012, 48, 9239–9241 RSC.
- Y. C. Chang, M. Y. Kuo, C. P. Chen, H. F. Lu and I. Chao, On the Air Stability of n-Channel Organic Field-Effect Transistors: A Theoretical Study of Adiabatic Electron Affinities of Organic Semiconductors, J. Phys. Chem. C, 2010, 114, 11595–11601 CrossRef CAS.
- U. Purushotham and G. N. Sastry, Conjugate acene fused buckybowls: evaluating their suitability for p-type, ambipolar and n-type air stable organic semiconductors, Phys. Chem. Chem. Phys., 2013, 15, 5039–5048 RSC.
- F. Gamier, G. Horowitz, D. Fichou and A. Yassar, Molecular order in organic-based field-effect transistors, Synth. Met., 1996, 81, 163–171 CrossRef.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Semiconducting π-Conjugated Systems in Field-Effect Transistors: A Material Odyssey of Organic Electronics, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08345c |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *d
*d