
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDrug dual-release matrix proprieties and the correlations with nanostructure aggregation kinetics for siloxane-polyether/hydrogel nanocomposites†
Mac-Kedson Medeiros Salviano Santos *abc,
Marcelo Henrique Sousaab and
Juliano Alexandre Chakerab
*abc,
Marcelo Henrique Sousaab and
Juliano Alexandre Chakerab
aInstitute of Biological Sciences, University of Brasilia, Brasilia, Brazil. E-mail: mac-kedson.santos@unieuro.edu.br
bFaculty of Ceilandia, University of Brasilia, Brasilia, Brazil
cCentro Universitário Unieuro, Brasília, DF, Brazil
First published on 19th January 2021
Abstract
The influence of hydrogels on the nanostructural formation of siloxane-polyether nanocomposites was examined. The nanostructure was studied with small-angle X-ray scattering (SAXS) to determine the siloxane nanostructure aggregation mechanisms. The interactions between matrix and drug were examined by infrared spectroscopy to verify the compatibility of the drug with the matrix. For in vitro release tests Piroxicam was used as a model molecule. The variation of the different types of hydrogels, bis-acrylamide (BIS), poly(acrylamide-co-acrylic acid) (PAM) and polyvinylpyrrolidone (PVP) can modify the drug release profiles. The release behaviour was determined to be composed of two concomitant release mechanisms. The first is in the early stages of drug release, governed by erosion, diffusion and swelling and the second, in advanced stages of release, typical of diffusion through pores. These dependencies were found to be correlated to the physical and chemical properties of the nanocomposites, including the interactions disturbing polycondensation formation. The release rate depends on intramolecular matrix–matrix and intermolecular drug–matrix interactions, as well as a crystalline state of the matrix.
Introduction
Sustained drug delivery systems have become a fundamental part of drug development. Drug excipient matrices and tablet coatings are fundamental for delivery dosage control and they drive the absorption of the drug. In particular, matrices for dermal treatments have improved properties, associating the sustained release of the active molecule with flexibility, non-occlusivity, and efficacy as a microbiological barrier and wound healing capacity.1Innovations in sustained drug release matrices have grown enormously, and these advances take into account the development of specific polymers to control release over long periods of time, cyclical releases and concomitant delivery of hydrophilic and hydrophobic drugs. Other aspects equally governed by the molecular and structural characteristics of the polymers, such as the potential for bioadhesion, water-insolubility and swelling should ideally be adjusted to the final properties.2–5
Hybrid nanocomposite polymers offer the ability to manipulate both organic and inorganic groups to produce a final compound with adjustable physical and chemical properties.6 The versatility of the colloidal method in the matrix release preparations allows, for example, for modification of the polymer side chain structure in order to tune the solubility, and delivery kinetics of host from the matrix.7 It has been found in the literature that drug release mechanisms are mainly governed by interactions between the host molecule and the matrix, e.g., inter-chain crosslinking, intermolecular interactions and matrix porosity. Besides, polymers with swelling and relaxation characteristics are very suitable choices for hosting hydrophilic drugs.8–11 In other cases, responsive polymers can be used as stimuli release devices with a different pH, temperature or light trigger.12–15
A special class of hybrid organic–inorganic nanocomposites that has been studied as a hosting matrix for sustainable release drugs, despite its unique properties, is derived from a nanoscale bi-phased organic and inorganic nanocomposite. The organic phase material prepared from polymer blend polypropylene (PPO)–polyethylene (PEO) oxides induces flexibility, wettability, solubility and diffusion of hydrophilic and hydrophobic drugs through the matrix. The inorganic phase, obtained from a modified silane alkoxide, allows skin film formation, improving chemical and mechanical resistance, transparency and bioadhesion.16 The crosslinked siloxane nanoaggregates are interconnected to each other through polymeric PPO–PEO–PPO, resulting in a porous hybrid matrix in which the porosity and the interactions of the host with the organic phase act together in the kinetics of diffusion and release of the drug. Some studies17,18 demonstrate the release of drugs from a hybrid matrix of siloxane-polyether with PPO. However, the matrix containing PPO has a more hydrophobic character compared to polyethylene oxide PEO, and the combination with compounds with more hydrophilic properties, such as hydrogels, has not been well explored in the production of these release devices.19
Structurally, nanocomposite formation has influenced their capacity polycondensation reaction. Hydrogels can perturb the three-dimensional arrangement of their molecules due to interactions with drugs or other chemical groups. The extremities of these gels have hydroxyl groupings (–OH), carboxylic acids (–COOH), amines (–NH) and other hydrophilic groups.20 The interaction between the matrix and the incorporated drug determines the release profile and, depending on the material, it may be governed by various release mechanisms.7 In addition, hydrogels can interfere in the formation process of siloxane-polyether matrices, resulting in hydrophilic and hydrophobic chains and thus allowing the incorporation of different drugs.
Therefore, the objective here was to produce a hybrid nanocomposite prepared with different hydrogels as an additive in a hybrid siloxane-polyether matrix, in order to obtain new materials for the drug release topical system and correlate the influence of polycondensation properties with a release mechanism. The nanocomposites were prepared with a sol–gel process and used Piroxicam (4-hydroxy-2-methyl-1,1-dioxo-N-pyridine-2-yl-1λ6,2-benzothiazine-3-carboxamide) as the model drug for release experiments. Piroxicam is an anti-inflammatory used in the treatment of osteoarthritis and rheumatoid arthritis, but it is well known for its gastrotoxic and duodenotoxic effects when used orally.21
Experimental
Synthetic procedures
The synthesis procedure of siloxane-polyether nanocomposites was adapted by protocols found in the literature.18,19,22 Firstly, the hybrid polymer (SP) was obtained from the mixing of a polyether (polypropylene oxide (PPO)) of molecular weight (4000 g mol−1) with a silicon alkoxide (3-isocyanate propyl)triethoxysilane in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. The reagents were stirred together in anhydrous ethanol (ETOH) under reflux for 24 h. The ETOH was removed by evaporation at 60 °C. The hybrid polymer underwent hydrolysis and polycondensation through the sol–gel process with different hydrogels for nanocomposite formation. Mixtures of 200 mg of hybrid polymer were prepared for each sample, with different volumes of hydrogel, bis-acrylamide (BIS), poly(acrylamide-co-acrylic acid) (PAM) and polyvinylpyrrolidone (PVP) solutions in volume ratio of 70:30 ethanol:water (v/v), with the anti-inflammatory Piroxicam (PRX) 1% (w/w) used with the following mass proportions: 25%, 15%, 5%, 2.5%, 1.5%, 0.5% and 0% (w/w). For each sample, Triton X-100 (100 μL) was added, mixed, and stirred to form a homogeneous solution; HCl was added to the 0.05 M ethanol solution (50 μL) to catalyze the hydrolysis and condensation reaction. After 60 minutes, the formation of a monolithic film was observed by crosslinking the siloxane groups, resulting in the samples of SP-BIS, SP-PAM, SP-PVP and SP. The process of hydrolysis and polycondensation formation shows in Fig. 1(a). The multi-scale scheme of the nanocomposites is shown in Fig. 1(b).
:2. The reagents were stirred together in anhydrous ethanol (ETOH) under reflux for 24 h. The ETOH was removed by evaporation at 60 °C. The hybrid polymer underwent hydrolysis and polycondensation through the sol–gel process with different hydrogels for nanocomposite formation. Mixtures of 200 mg of hybrid polymer were prepared for each sample, with different volumes of hydrogel, bis-acrylamide (BIS), poly(acrylamide-co-acrylic acid) (PAM) and polyvinylpyrrolidone (PVP) solutions in volume ratio of 70:30 ethanol:water (v/v), with the anti-inflammatory Piroxicam (PRX) 1% (w/w) used with the following mass proportions: 25%, 15%, 5%, 2.5%, 1.5%, 0.5% and 0% (w/w). For each sample, Triton X-100 (100 μL) was added, mixed, and stirred to form a homogeneous solution; HCl was added to the 0.05 M ethanol solution (50 μL) to catalyze the hydrolysis and condensation reaction. After 60 minutes, the formation of a monolithic film was observed by crosslinking the siloxane groups, resulting in the samples of SP-BIS, SP-PAM, SP-PVP and SP. The process of hydrolysis and polycondensation formation shows in Fig. 1(a). The multi-scale scheme of the nanocomposites is shown in Fig. 1(b).
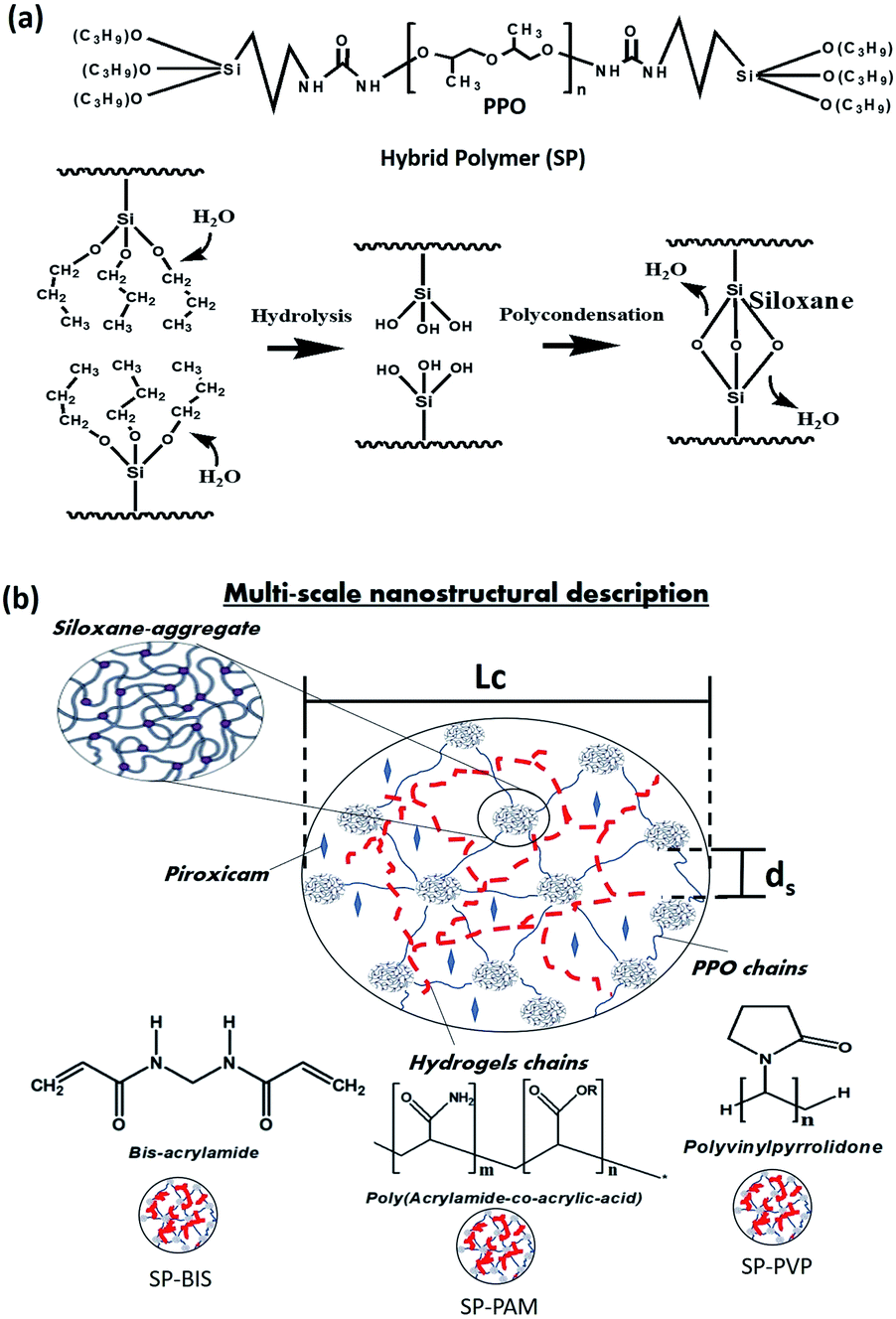 | ||
| Fig. 1 (a) Structure molecular of hybrid polymer and process of hydrolysis and polycondensation. (b) Scheme represents multiscale nanostructural description of polymeric nanocomposites. ds represent the spatial correlation distance between the siloxane aggregates and Lc represents the spatial correlation limit of these aggregates. | ||
Characterization
Molecular characterization
The chemical bonds formed by the interaction of hydrogels and chemical groups present in the nanocomposite were evaluated by vibrational spectroscopy in the infrared. For spectroscopic analysis in the infrared region of the nanocomposites, the measurements were performed in two steps: first, for samples of Piroxicam powder compressed into tablet form with KBr, a Shimadzu spectrometer Model 21 IR Prestige was used. A resolution of 4 cm−1 was used, and 32 scans were measured in a range of 4000 to 400 cm−1. Second, for nanocomposites, the spectra were obtained directly by diffuse reflectance, in a tag device Varian 640-IR model, measured in the range of 4000 to 600 cm−1 with 16 scans and 4 cm−1 resolution. For these measurements, the sample was divided transversely and arranged to measure the interior of the monolith obtained.Nanostructural characterization
The Small Angle X-ray Scattering (SAXS) measurements were performed in the D11A beamline of the National Synchrotron Light Laboratory (LNLS, Campinas, Brazil). The beamline is equipped with a side-bounce W/B4C multilayer (500 double layers) monochromator on a silicon substrate that yields a monochromatic (λ = 1.54 Å) and horizontally focused beam. A Pilatus 2D detector operating in a single-photon-counting mode, located at 540 mm from the sample, was used to record the SAXS intensity, I(q), in a 172 × 172 μm2 pixels image area as a function of the modulus of the scattering vector, q = (4π/λ)sin(ξ/2), with ξ the scattering angle. Because of the small size of the incident beam cross-section on the detection plane, no mathematical de-smearing of the experimental SAXS intensity function was needed. The film samples were filed in a 1 mm sample holder with Kapton windows. The scattering curve was corrected for an empty sample holder contribution.Crystallinity characterization
The X-ray diffraction (XRD) analysis was performed to verify the crystallinity of the nanocomposites. The film samples were placed in an aluminium sample holder, and the measurements were carried out from 5° to 35°, with step 0.02° and speed of 10° per minute in Rigaku equipment, model Miniflex 600.Statistical analysis
A proper linear fit to Porod power law was conducted, using Origin Pro 7.0 software, and an adjustment was made to determine parameters such as radius gyration (Rg) and the fractal dimension (df). The aggregates can be described as fractal mass, and this phenomenon is observed in the measured samples. According to the mass fractal model, the fractal dimension variant is ≥1 and ≤3 and is related to the size of the dispersed object. Thus, for example, a cylinder dimension is given as df = 1, because it has only one dimension, length. Likewise, for a disk and a ball the values are df = 2 and 3, respectively. The radius gyration (Rg) can be defined as the mean square distance of the centre of gravity where the mass is seen by the electron density.23 These parameters were obtained by fitting the experimental curve in accordance with the model, using the SASfit DLCAggregation program. The information shown on the appearance of a peak correlation of samples was taken using the PseudoVoigt Model 2, available from Origin Pro 7.0 software, where one can extract parameters such as the maximum q (qmax) of the correlation peak and the half-width (W½). The ds values represent the distances between the siloxane aggregate, which are correlated with the size of the polyether chain of the hybrid precursor (SP); by being covalently bonded, siloxane requires the distance between the aggregates.In vitro drug release studies (UV-vis absorption)
The influence of the composite matrix composition of Piroxicam (PRX) on the drug release profile was performed by studies of samples BIS-SP, SP-SP-PAM and PVP containing 25, 15 and 5% by weight of each of hydrogels. For measurement, spectroscopy in ultraviolet-visible (UV-vis) was used. The samples were prepared inside a quartz cuvette of 10 mm, which adhered to the time of 24 hours of gel processing. After gel processing, 3 mL of ultrapure water was added. Using a HITACHI U-3900H spectrophotometer model the sample was measured in the range of 200 to 410 nm, in 2 intervals of 2 minute cycles for 3 hours, and 30 minutes cycles for 45 hours at a controlled temperature of 25 °C, thus determining the amount of Piroxicam released in a period of 48 hours. The release curves were studied by the mathematical model postulated by Korsmeyer–Peppas,24,25 since the curves obtained by the assay are similar to those defined by the cited models.Results and discussion
Fig. 2 shows the infrared absorption curves from 1200 to 800 cm−1 for siloxane-polyether hybrid polymer (SP) with different hydrogels, bis-acrylamide (BIS), poly(acrylamide-co-acrylic acid) (PAM) and polyvinylpyrrolidone (PVP), SP-BIS, SP-PAM, and SP-PVP samples, and the bands' deconvolution. Table 1 displays the deconvolution results for Si–O–Si asymmetric stretching envelopes at 1086–1049 cm−1 and β Si–OH β plane-symmetric stretching envelopes at 930–910 cm−1 range, respectively.26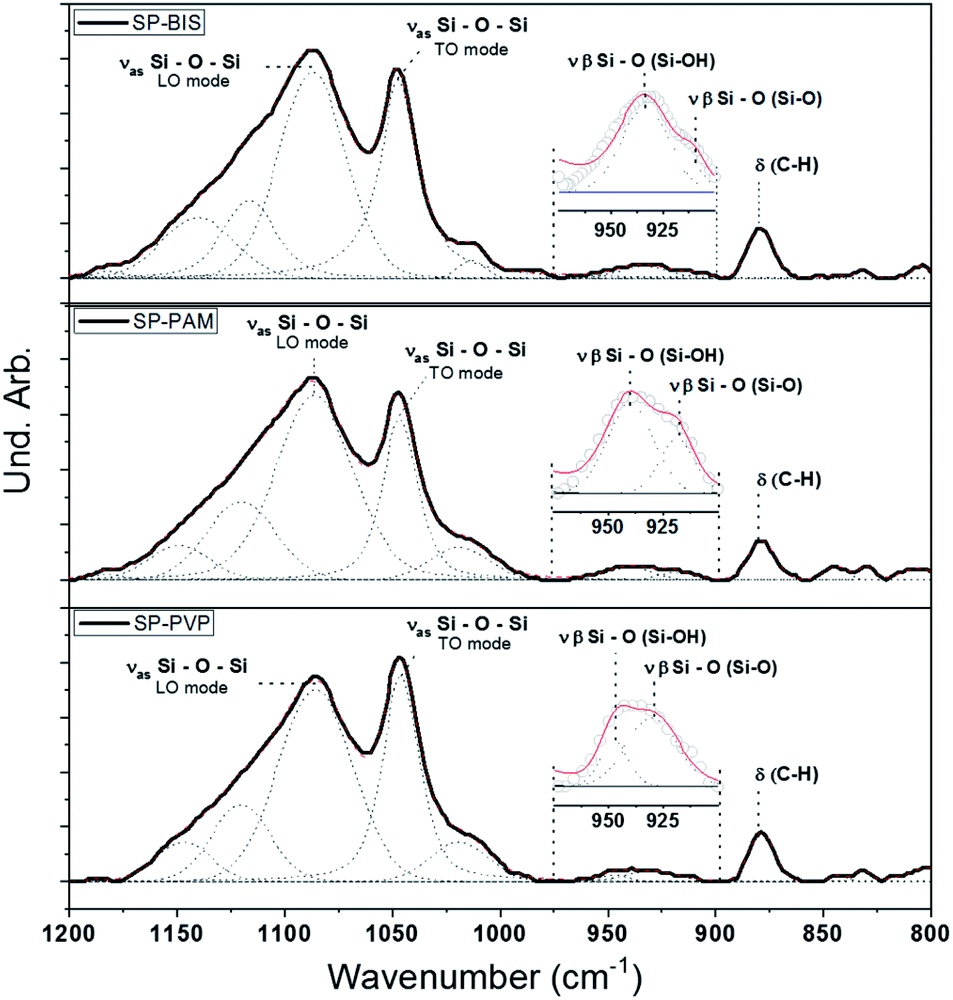 | ||
| Fig. 2 Infrared spectrum and deconvolution adjustments for nanocomposite samples SP-BIS 25% (w/w), SP-PAM 25% (w/w), and SP-PVP 25% (w/w). | ||
| Attribution | Samples (area%) | |||
|---|---|---|---|---|
| SP | SP-BIS | SP-PAM | SP-PVP | |
| νas Si–O–SiLO mode | 46.36 | 55.47 | 48.87 | 47.17 |
| νas Si–O–SiTO mode | 17.51 | 56.51 | 26.37 | 30.54 |
| ν β Si–O(Si–OH) | 3.88 | 3.22 | 1.90 | 0.57 |
| ν β Si–O(Si–O–) | ∞ | 0.37 | 1.01 | 1.63 |
| Ratio νas Si–O–Si(LO mode/TO mode) | 2.65 | 0.98 | 1.85 | 1.54 |
| Ratio ν β Si–O(Si–OH/Si–O–) | ∞ | 8.66 | 1.88 | 0.35 |
The relative contribution between the area of silanol Si–OH and Si–O– in-plane stretching vibration modes is shown in Table 1. The ratio, the area between Si–OH modes and Si–O– modes reveals a higher Si–O– mode contribution for the SP-PVP sample compared to the other samples, suggesting non-polycondensed silanol groups as the major species. Furthermore, the relative contribution to the TO mode (transversal optical) in the asymmetric stretching Si–O–Si absorption band is higher compared to the LO mode (longitudinal optical) for the same sample.26,27 These results indicate that the polycondensed reaction kinetics for the SP-PVP sample is less pronounced.
In all spectra the presence of a band is observed at 879 cm−1, referring to the aromatic stretch ring C–H for Piroxicam, showing the presence of the drug in the matrices produced. According to the Fig. S1 in ESI,† is it possible to observe the aromatic C–H absorption peak at 879 cm−1 (dashed line). However, it was not possible to observe other Piroxicam interactions through infrared spectra. On the other hand, evidence of interactions between the silanols and carbonyl groups of PVP polymer has already been observed in other works,28 which could explain the low polycondensation of silanol groups.
The nanostructural characterization by SAXS analysis was performed in order to verify the nature of the nanocomposites SP; SP-PVP; SP-PAM and SP-BIS regarding their nanostructural organization. The experimental small angle X-ray scattering for the SP sample (open circles) in the range 0.05 ≤ q ≤ 2.70 nm−1 can be observed in Fig. 3. The SAXS curves for SP-BIS, SP-PAM, and SP-PVP are not shown in the figure; nevertheless, in all of them, three main regimes are observed: (I) for q ≤ 0.18 nm−1 there is a linear regime due to larger particle domains; (II) in the range 0.18 ≤ q ≤ 0.8 mass their is fractal behaviour and (III) for q ≥ 0.8 a correlation peak is seen, due to the scattering of ordered siloxane aggregates.29 The solid lines represent the theoretical adjustment for each SAXS model regime, and the inset shows the results obtained from the experimental adjustment of the regime (II). In general, the system is described as biphasic, composed of particles of high electron density, dispersed phase in a lower electron density. In such a system, larger particles coexist with smaller particles in an interconnected or non-aggregated form. The behaviour of the nanostructure sample without hydrogel has already been observed in similar composites containing organic pigments,30 or others like silica and titanium particles.23 In region (I), a q−4 slope can be adjusted with a linear function, which is described as a smooth and defined interface between the larger and smaller particles, as described by Porod's law.31
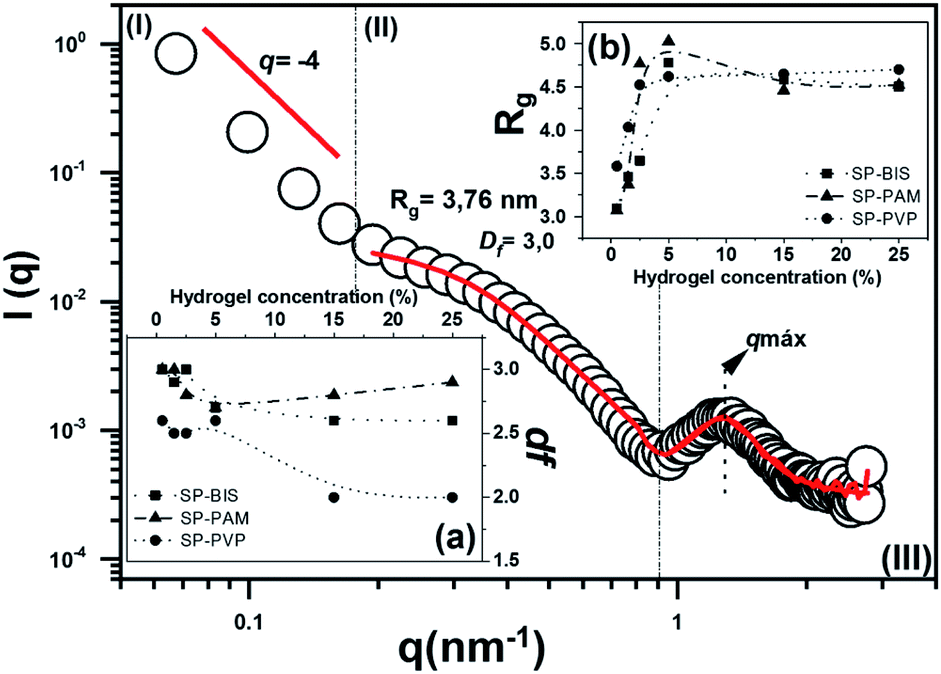 | ||
| Fig. 3 The experimental curve of SAXS for sample SP with data fit. Observed (a) fractal dimension (df) results; (b) radius gyration (Rg) results for samples SP-BIS, SP-PAM and SP-PVP. | ||
According to the model for region (II), the growth kinetics can be established from the fractal dimension.32 The inset (a) in Fig. 3 shows fractal dimension (df) values as a function of hydrogel concentration. A df close to 3.0 is related to the typical monomer-cluster limited by the reaction growth, and df close to 2.0 is related to growth limited by diffusion. In the first case, the growth kinetics of the aggregates generate dense and poorly branched structures, while for limited diffusion growth results in the formation of more open, branched and less dense structures. The curve shows df values as almost invariant and close to 3.0 for SP-PAM and SP-BIS. On the other hand, for SP-PVP samples the df values decrease from 3.0 to 2.0 with PVP hydrogel concentration. The results reveal different siloxane aggregate growth mechanisms depending on the hydrogel type utilized in the nanocomposites. The growth process can also be verified with the radius gyration (Rg) variation, which increases from 3.0 to 5.0 nm range.
The adjustment results from region (III), in Fig. 3, are displayed in Table 2. For all studied samples, the ds (correlation distance) value remains constant, close to 5.0, and it represents the chain length related to the organic polymer used in nanocomposite preparation.33 A decrease in Lc (correlation limit) values is observed with hydrogel concentration. This effect is more pronounced in the SP-PVP sample and may be related to the decrease of the nanostructure spatial correlation with hydrogel concentration.
| Concentration (%) | SP-BIS | SP-PAM | SP-PVP | |||
|---|---|---|---|---|---|---|
| Ds | Lc | Ds | Lc | Ds | Lc | |
| 0.5 | 4.89 | 23.27 | 4.89 | 24.32 | 4.87 | 24.62 |
| 1.5 | 4.91 | 22.65 | 5.00 | 24.31 | 4.90 | 24.51 |
| 2.5 | 4.93 | 23.26 | 5.05 | 19.08 | 4.87 | 23.15 |
| 5 | 4.90 | 20.98 | 5.10 | 19.38 | 4.93 | 22.92 |
| 15 | 4.84 | 22.36 | 5.00 | 22.89 | 4.74 | 18.03 |
| 25 | 4.94 | 18.90 | 5.00 | 18.86 | 4.93 | 11.43 |
In vitro drug release studies with UV-vis absorption
The release profile (scatter) of the Piroxicam model molecule for all the samples and the adjustment (solid lines) according to the Korsmeyer–Peppas release model is shown in Fig. 4. The curves for SP-BIS, SP-PAM, and SP-PVP samples show kinetics release behaviour that is dependent on hydrogel concentration (5%, 15%, and 25%), and the maximum concentration released is between 1.88% for PVP samples and 6.88% for BIS samples. However, for all samples, a sustained release of 48 hours is observed. This result indicates the drug's affinity for the hydrophobic matrix. The hydrophobic characteristic of Piroxicam limits its diffusion, due to possible interactions of hydrophobic association with the polypropylene matrix.7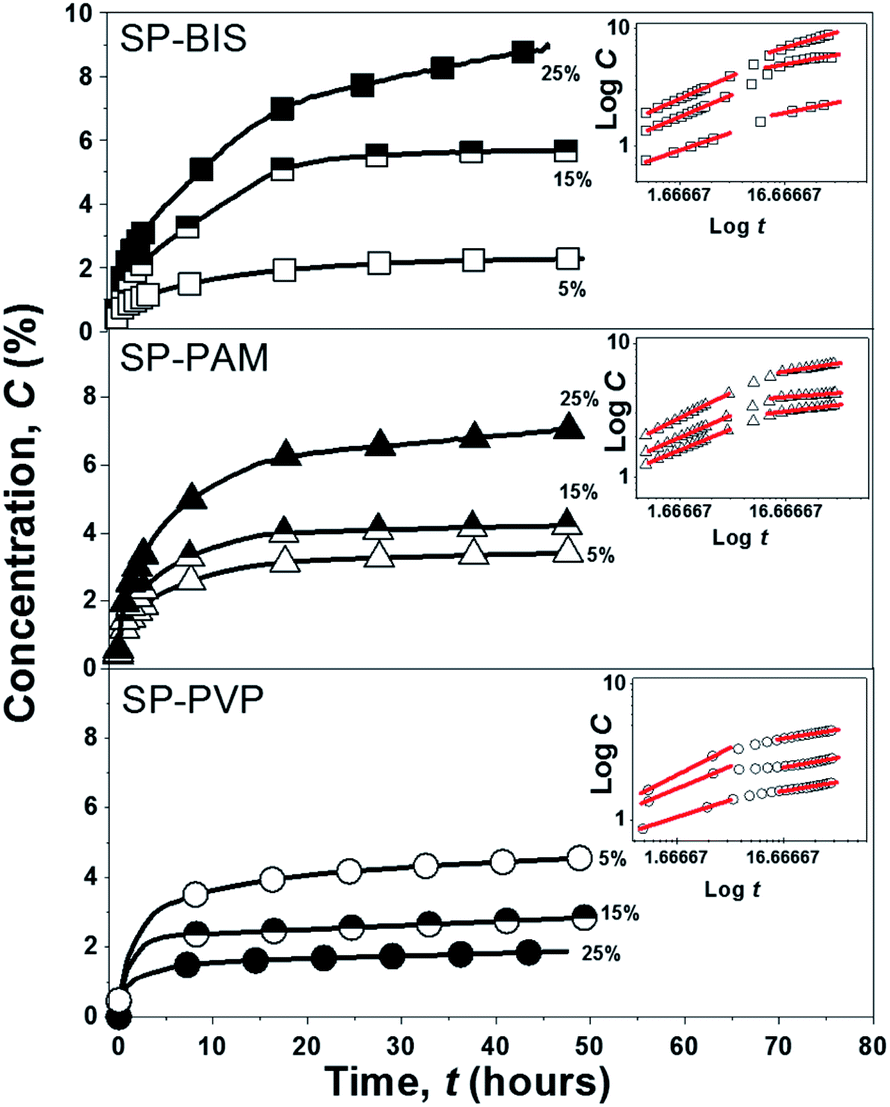 | ||
| Fig. 4 Experimental curves release kinetics of SP-BIS, SP-PAM and SP-PVP and the inset curves of the bi-logarithmic scale samples under the linear adjustments according to the Korsmeyer–Peppas model. | ||
Additionally, for the SP-BIS and SP-PAM samples, an increase in Piroxicam release with hydrogels concentration can be observed. However, an opposite effect can be observed for SP-PVP, i.e., the increase of hydrogel concentration results in a lower release of Piroxicam. Some authors affirm that the difference in the release pattern derives from drug-matrix intermolecular interaction and also matrix viscosity.34 The increase in intermolecular hydrogen bonding between amide or carbonyl groups from PVP hydrogels with protonated pyridine groups from Piroxicam could explain the decrease in the release process with hydrogel concentration (5% > 15% ≫ 25%).34 We cannot elucidate clearly but observing the interactions of Piroxicam with the binding site of the proteins 3DY9, 4NZ2 and 2AYR reveal that the chemical groups SO2 and pyridine ring of the drug interacts via hydrogen and pi-alkyl bonding respectively.35 So, we can suggest that Piroxicam can interacts with: (1) hydrogel: the –OH groups from hydrogel structure interacts by hydrogen bonding with SO2 group from Piroxicam. (2) nanocomposite matrix: the non-hydrolyzed silanol –Si–OH as well ether type oxygen of the PPO groups produces hydrogen bond interactions with SO2 group of Piroxicam. Additionally, the same interaction between the –OH and –SO2 groups in the crystalline form of Piroxicam has been reported in the literature.36 It is also important to indicate that these interactions are pH dependent, which indicates that the release process can be modulated as a function of pH.
The BIS hydrogels possess an amide group in their composition; however, we suggest that in the bisacrylamide hydrogel the carbonyl groups are not available to carry out interactions with Piroxicam. This hypothesis is supported by XRD data.
As observed with the XRD results in Fig. 5 for the crystalline pattern, in all samples, the presence of the extended peak centred around 2θ = 22°, referring to the contribution of amorphous siloxane aggregates.6 There are no crystalline peaks referring to Piroxicam in the samples, indicating good solubility of the active principle in the nanocomposites. However, it is noted that for higher concentrations of bisacrylamide, the presence of two peaks centred on 2θ = 8.29° and 2θ = 16.1° may be related to the packaging of crystals of needle-shaped molecules, due to the strong interactions of hydrogen formed between the parts of the N–H amide or atoms of the carbonyl groups in adjacent molecules, as proposed by other authors.37 Nevertheless, for the PAM and PVP samples, we observed that crystallization did not occur.
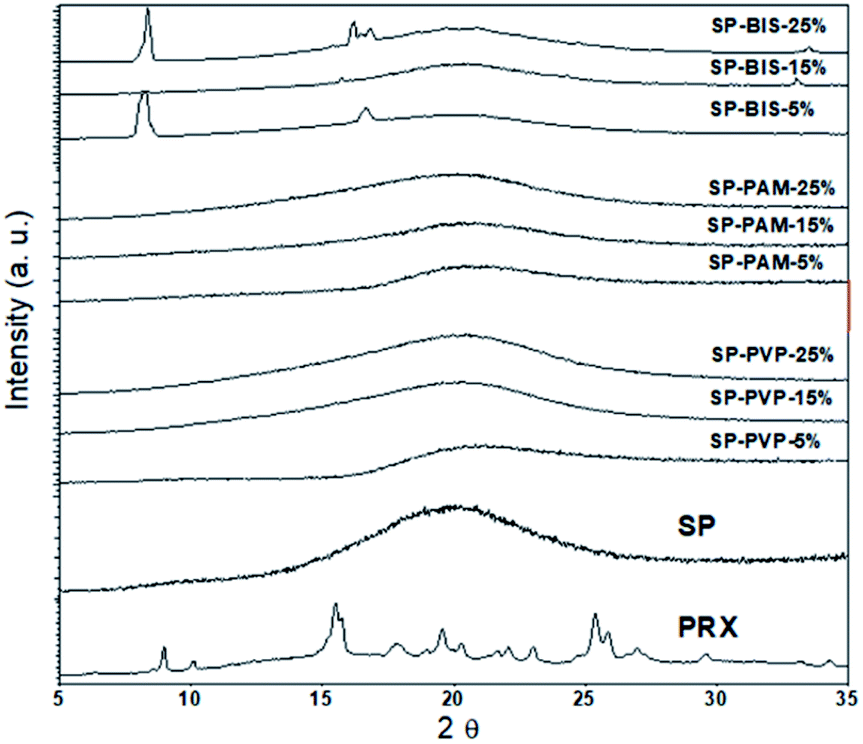 | ||
| Fig. 5 XRD pattern of nanocomposites SP-BIS, SP-PAM, SP-PVP with variation of 25%, 15% and 5% hydrogels concentration, SP and Piroxicam (PRX). | ||
The release process shown in the inset of Fig. 4 by the bi-logarithmic release curves describes two distinct release behaviour profiles independently of the hydrogels used in a preparation: (i) the first period for logt < 6 h reveals fast release kinetics and (ii) the second period for logt > 6 h shows slower release kinetics.38,39 According to Korsmeyer–Peppas models, the values of n obtained from a linear regression of bi-logarithmic curves can be related to the release mechanisms. The n values obtained for all samples are shown in Table 3.
| Samples | n values (first period) | n values (second period) | |
|---|---|---|---|
| SP-BIS | 25% | 0.39 | 0.25 |
| 15% | 0.38 | 0.20 | |
| 5% | 0.31 | 0.12 | |
| SP-PAM | 25% | 0.32 | 0.05 |
| 15% | 0.34 | 0.05 | |
| 5% | 0.39 | 0.13 | |
| SP-PVP | 25% | 0.25 | 0.1 |
| 15% | 0.32 | 0.1 | |
| 5% | 0.40 | 0.15 | |
The values presented show that all the samples respect the same mechanisms of dissolution of porous materials with the influence of the dissolution and erosion of the hydrogels.25 In the case of the values of the second period, it is observed that the values of n are lower for all the samples, showing that after erosion and dissolution of the hydrogel in the receiving medium, the preponderant release mechanism is the dissolution by pores. The swelling process of the nanocomposites can be seen in the photos in Fig. S2(a) and (b) presented in the ESI.† This result indicates that the nanocomposite is governed by a dual-release mechanism, where the first period is accompanied by erosion of the hydrogel chains and the second period through a dissolution by the polyether matrix of a more hydrophobic character, limiting the release of Piroxicam.
Conclusions
We can conclude that nanocomposites with different hydrogels have properties that can modulate molecular interaction, nanostructural arrangement and release rate, under the same dual-release mechanism. Likewise, it can be concluded that the hydrogels are eroded and released in a subsequent step with the drug. As expected, the nanostructure is directly affected by the composition of the nanocomposite, as well as the mechanisms of formation of the nanometric arrangements of the siloxane-polyether matrix. The release rate is strongly dependent on the intramolecular matrix–matrix and intermolecular drug–matrix interactions, as well on the crystalline state of the matrix but although these results are related to the erosion of the matrix, further studies are needed regarding the influence of temperature, irradiation, and pH.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge financial support from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação de Apoio à Pesquisa do Distrito Federal (FAPDF), Decanato de Pesquisa e Inovação (DPI-UnB) and Fundação de Empreendimentos Científicos e Tecnológicos (FINATEC). The authors are also greatly indebted to Laboratório Nacional de Luz Síncotron (LNLS) for the SAXS experiments.Notes and references
- S. Saghazadeh, C. Rinoldi, M. Schot, S. S. Kashaf, H. Derakhshandeh, K. Yue and W. Swieszkowski, Adv. Drug Delivery Rev., 2019, 138–166 Search PubMed.
- T. M. M. Ways, W. M. Lau and V. V. Khutoryanskiy, Polymers, 2018, 10, 138–166 CrossRef.
- N. Mondal, Int. J. Appl. Pharm., 2018, 10, 1–6 CrossRef CAS.
- W. L. Guerra-Ponce, S. L. Gracia-Vásquez, P. González-Barranco, I. A. Camacho-Mora, Y. A. Gracia-Vásquez, E. Orozco-Beltrán and L. A. Felton, Braz. J. Pharm. Sci., 2016, 52, 751–760 CrossRef CAS.
- L. N. M. Ribeiro, A. C. S. Alcântara, G. H. Rodrigues Da Silva, M. Franz-Montan, S. V. G. Nista, S. R. Castro, V. M. Couto, V. A. Guilherme and E. De Paula, Int. J. Polym. Sci., 2017, 2017, 1–16 CrossRef.
- J. A. Chaker, C. V. Santilli, S. H. Pulcinelli, K. Dahmouche, V. Briois and P. Judeinstein, J. Mater. Chem., 2007, 17, 744–757 RSC.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 1–18 Search PubMed.
- K. W. Seo, D. J. Kim and K. N. Park, J. Ind. Eng. Chem., 2004, 10, 794–800 CAS.
- C. Özeroglu and A. Birdal, eXPRESS Polym. Lett., 2009, 3, 168–176 CrossRef.
- P. A. Steward, J. Hearn and M. C. Wilkinson, Adv. Colloid Interface Sci., 2000, 86, 195–267 CrossRef CAS.
- I. Zurdo Schroeder, P. Franke, U. F. Schaefer and C. M. Lehr, Eur. J. Pharm. Biopharm., 2007, 65, 111–121 CrossRef CAS.
- K. S. Chen, Y. A. Ku, H. R. Lin, T. R. Yan, D. C. Sheu, T. M. Chen and F. H. Lin, Mater. Chem. Phys., 2005, 91, 484–489 CrossRef CAS.
- B. Ahmad, S. Abbas, Z. Iqbal, S. Bashir and J. Ali, Middle East J. Sci. Res., 2013, 14, 273–283 CAS.
- S. Mandal, R. Ray, S. K. Basu and B. Sa, J. Biomater. Sci., Polym. Ed., 2010, 21, 1799–1814 CrossRef CAS.
- H. Priya James, R. John, A. Alex and K. R. Anoop, Acta Pharm. Sin. B, 2014, 4, 120–127 CrossRef.
- J. A. Oshiro, M. P. Abuçafy, E. B. Manaia, B. L. Da Silva, B. G. Chiari-Andréo and L. A. Chiavacci, Polymers, 2016, 8(4), 91 CrossRef.
- C. V. Santilli, L. A. Chiavacci, L. Lopes, S. H. Pulcinelli and A. G. Oliveira, Chem. Mater., 2009, 21, 463–467 CrossRef CAS.
- L. K. Souza, C. H. Bruno, L. Lopes, S. H. Pulcinelli, C. V. Santilli and L. A. Chiavacci, Colloids Surf., B, 2013, 101, 156–161 CrossRef CAS.
- E. F. Molina, S. H. Pulcinelli, C. V. Santilli, S. Blanchandin and V. Briois, J. Phys. Chem. B, 2010, 114, 3461–3466 CrossRef CAS.
- Q. V. Nguyen, D. P. Huynh, J. H. Park and D. S. Lee, Eur. Polym. J., 2015, 72, 602–619 CrossRef CAS.
- N. J. Joseph, S. Lakshmi and A. Jayakrishnan, J. Controlled Release, 2002, 79, 71–79 CrossRef CAS.
- P. Judeinstein, M. E. Brik, J. P. Bayle, J. Courtieu and J. Rault, Mater. Res. Soc. Symp. Proc., 1994, 346, 937–942 CrossRef CAS.
- J. Hyeon-Lee, G. Beaucage, S. E. Pratsinis and S. Vemury, Langmuir, 1998, 14, 5751–5756 CrossRef CAS.
- N. A. Peppas and B. Narasimhan, J. Controlled Release, 2014, 190, 75–81 CrossRef CAS.
- M. L. Bruschi, Strateg. to Modify Drug Release from Pharm. Syst., 2015, pp. 63–86 Search PubMed.
- R. Al-Oweini and H. El-Rassy, J. Mol. Struct., 2009, 919, 140–145 CrossRef CAS.
- A. Duran, J. M. Fernandez Navarro, P. Casariego and A. Joglar, J. Non-Cryst. Solids, 1986, 82, 391–399 CrossRef CAS.
- O. E. Voronina, M. L. Malysheva and L. V Nosach, Ann. Univ. Mariae Curie-Skłodowska Lublin – Pol., 2017, LXXII, 51–66 Search PubMed.
- K. Dahmouche, C. V. Santilli, S. H. Pulcinelli and A. F. Craievich, J. Phys. Chem. B, 1999, 103, 4937–4942 CrossRef CAS.
- G. Beaucage, L. Clapp, J. Ilavsky, P. Jemian, D. J. Kohls, G. Long, R. J. Schwartz and G. Skillas, APS Sci., 1999, 4–5 Search PubMed.
- J. Oberdisse, W. Pyckhout-hintzen and E. Straube, Recent Adv. Polym. Nanocompos., 2009, 1, 397 Search PubMed.
- D. W. Schaefer, MRS Bull., 1988, 13, 22–27 CrossRef CAS.
- J. A. Oshiro, C. R. Scardueli, G. J. P. L. de Oliveira, R. A. C. Marcantonio and L. A. Chiavacci, Biomed. Phys. Eng. Express, 2017, 3, 015019 CrossRef.
- V. Tantishaiyakul, N. Kaewnopparat and S. Ingkatawornwong, Int. J. Pharm., 1999, 181, 143–151 CrossRef CAS.
- Y. S. Mary, Y. S. Mary, K. S. Resmi and R. Thomas, Heliyon, 2019, 5, e02175 CrossRef.
- J. Jayaselli, J. M. S. Cheemala, D. P. Geetha Rani and S. Pal, J. Braz. Chem. Soc., 2008, 19, 509–515 CrossRef CAS.
- C. Graiff, D. Pontiroli, L. Bergamonti, C. Cavallari, P. P. Lottici and G. Predieri, J. Appl. Crystallogr., 2015, 48, 550–557 CrossRef CAS.
- A. Bose, T. W. Wong and N. Singh, Saudi Pharm. J., 2013, 21, 201–213 CrossRef.
- H. G. Lee, Y. S. Park, J. H. Jeong, Y. Bin Kwon, D. H. Shin, J. Y. Kim, Y. S. Rhee, E. S. Park, D. W. Kim and C. W. Park, Drug Des., Dev. Ther., 2019, 13, 2459–2474 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08270h |
| This journal is © The Royal Society of Chemistry 2021 |