
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpectroscopic sensing and quantification of AP-endonucleases using fluorescence-enhancement by cis–trans isomerization of cyanine dyes†
JunHo Choa,
Sanghoon Ohb,
DongHun Leea,
Jae Won Hana,
Jungmin Yooa,
Daeho Parkac and
Gwangrog Lee *abc
*abc
aSchool of Life Sciences, Gwangju Institute of Science and Technology, Gwangju, 500-712, Korea. E-mail: glee@gist.ac.kr; Fax: +82-62-715-2484
bDepartment of Biomedical Science and Engineering, Gwangju Institute of Science and Technology, Gwangju, 500-712, Korea
cCell Mechanobiology Research Center, Gwangju Institute of Science and Technology, Gwangju, 61005, Republic of Korea
First published on 18th March 2021
Abstract
Apurinic/apyrimidinic (AP) endonucleases are vital DNA repair enzymes, and proposed to be a prognostic biomarker for various types of cancer in humans. Numerous DNA sensors have been developed to evaluate the extent of nuclease activity but their DNA termini are not protected against other nucleases, hampering accurate quantification. Here we developed a new fluorescence enhancement (FE)-based method as an enzyme-specific DNA biosensor with nuclease-protection by three functional units (an AP-site, Cy3 and termini that are protected from exonucleolytic cleavage). A robust FE signal arises from the fluorescent cis–trans isomerization of a cyanine dye (e.g., Cy3) upon the enzyme-triggered structural change from double-stranded (ds)DNA to single-stranded (ss)DNA that carries Cy3. The FE-based assay reveals a linear dependency on sub-nanomolar concentrations as low as 10−11 M for the target enzyme and can be also utilized as a sensitive readout of other nuclease activities.
1. Introduction
AP endonucleases (apurinic/apyrimidinic) are key enzymes involved in DNA base excision repair (BER), oxidative stress regulation, and demethylation pathways. Moreover, many studies have recently reported that AP endonuclease in humans (APE1) is overexpressed in most cancer cells such as lung,1 ovarian,2–6 osteosarcoma,7 colorectal8,9 and breast10–12 cancers. APE1 thus has been proposed to be a therapeutic target in various types of human cancer, and its overexpression has been suggested to be a biomarker of cancer progression. To date, numerous techniques have been developed to evaluate the expression level of AP endonucleases, but each one has its own advantages and disadvantages. For example, conventional western blotting (WB)11,13–15 involves multiple steps and is time-consuming; and fluorescence-based sensors16–23 and electrochemical biosensors24 recently developed have high sensitivity to AP endonucleases but do not have absolute selectivity and specificity because terminal ends of DNA sensors are not protected against other exonucleases. Thus, there is an urgent need for a new methodology capable of meeting sensitivity, selectivity, and specificity for early cancer diagnosis and cancer prognosis.Fluorescence enhancement (FE) is a photo-physical effect in which the emission of a fluorophore becomes brighter due to a local environmental change that raises the quantum yield of the fluorophore. The photo-physical properties of cyanine dyes are well characterized and their intensities vary depending on cis–trans isomerization between fluorescent trans-state and non-fluorescent cis-state.25,26 In particular, when a protein binds to the vicinity of Cy3, protein-induced fluorescence enhancement (PIFE) occurs due to an increase of the lifetime and quantum yield through isomerization events.25 Recent single-molecule studies have quantified the extraordinary sensitivity of PIFE in a range between 1 to 4 nm at the single molecule level and also found that PIFE appears to be as a function of a distance between the protein and the fluorophore.27,28 This proximity-dependent effect has been utilized in various biophysical studies, e.g., dynamics of polymerases29,30 and mechanisms of helicases31,32 at the single molecule level as well as a motor protein in bulk ensemble.33 Although PIFE has been used to study activities of numerous enzymes at the single molecule level, its transient fluorescent signal has limited the application under steady-state experimental conditions.
In an effort to solve this unmet need, it is thus very important to develop a rapid, reliable, and sensitive assay with analytical capabilities that can make a large impact on a wide range of applications from the rapid detection of biomarkers to the unravelling of expression level of enzymes. Here, we report a simple, rapid and sensitive technique that not only detects the activity of type II AP endonuclease as a general cancer biomarker but also accurately quantifies the amount of the target enzyme. Our method can be achieved with the specific dsDNA probe that confers not only its target specificity via protection of the termini from exonucleolytic cleavage but also the FE-based readout via a change in the isomerization of a cyanine fluorophore, Cy3, upon the structural conversion from dsDNA to ssDNA. The purpose of biotin–NeutrAvidin end-capping shown in Scheme 1 is to physically prevent all exonucleolytic degradation. Therefore, the AP site of our probe is the only site in which DNA can be endo-nucleolytically degraded by the enzyme-specific activities of AP-endonucleases, endowing specificity and selectivity toward APE1. In short, our DNA sensor provides a new readout-platform of the FE that results from the fluorescent cis–trans isomerization of a cyanine dye, Cy3. The three functional groups of our probe (an AP-site, Cy3 and termini protected from exonucleolytic cleavage) provide high specificity and selectivity to the target AP endonuclease in the presence of other nucleases.
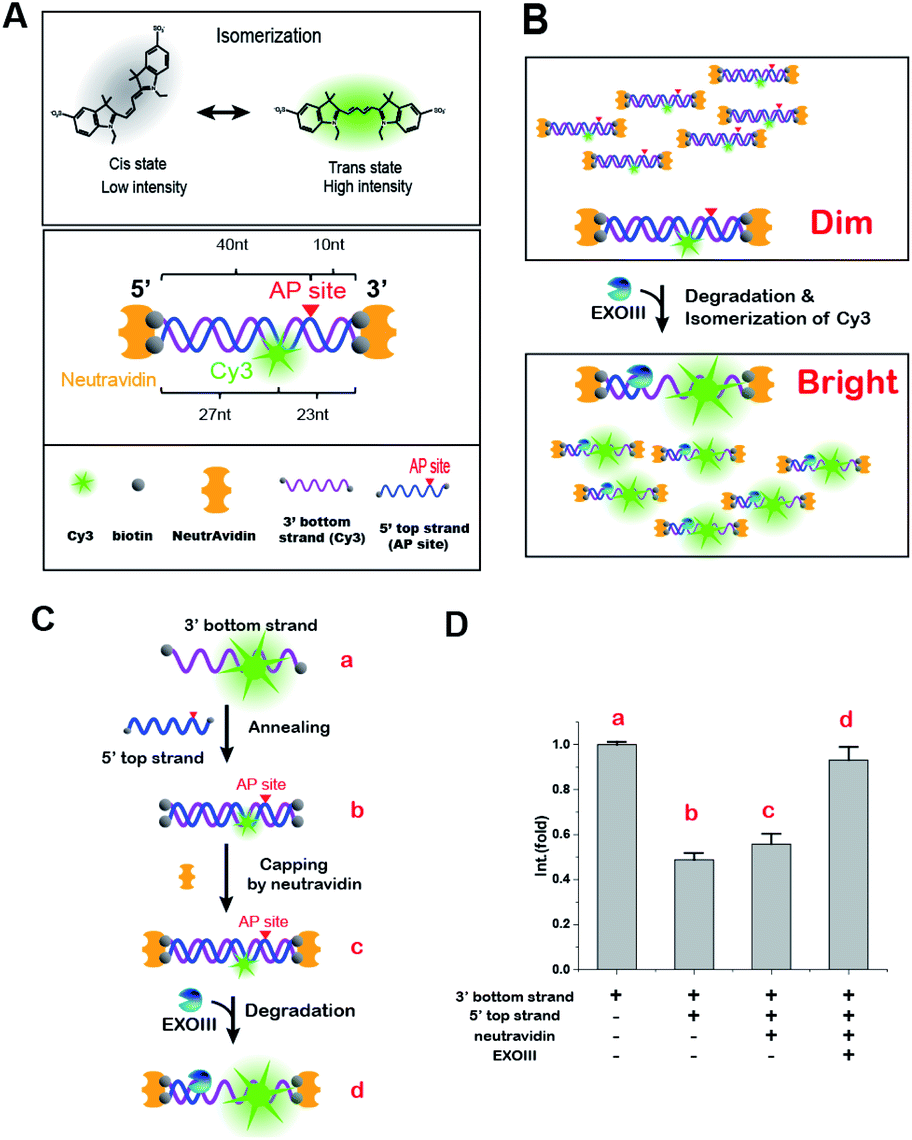 | ||
| Scheme 1 Schematic illustration of fluorescence enhancement (FE). (A) Cis–trans isomerization of the cyanine fluorophore, Cy3, and a biosensor capable of producing FE on Cy3 (dsDNA-FE) with an AP site and exonucleolytic protection-antibody. (B) Enzymatic FE reaction for type II AP endonucleases containing the activities of AP endo- and exo-nucleases. (C and D) Step-by-step procedures for reverting FE and their corresponding results, illustrating that the molecular origin of FE is cis–trans isomerization induced by a DNA structural change from dsDNA to ssDNA. Error bars are means ± SEM, and are produced by three independent replications. | ||
2. Materials and methods
2.1 Materials and apparatus
DNA oligonucleotides were purchased from IDT (Integrated DNA Technologies: Coralville, Iowa, USA), and their sequences can be found in Table S1 in the ESI.† NeutrAvidin and NHS-Cy3 were received from Pierce (USA) and GE Healthcare (USA), respectively. T4 DNA polymerase and T7 exonuclease were purchased from New England Biolabs (NEB: Ipswich, MA, USA). Fluorescent measurements were recorded on a fluorescence spectrometer (Model: FS-2, SCINCO, Inc., Seoul, Korea) using a quartz cuvette with a path length of 10 mm (Starna Scientific: 16.100-F-Q-10/Z15) that contained reaction buffers in a range between 50 to 100 μL.2.2 Protein purification
Exonuclease lll,34 Lambda exonuclease,35 and Phi29 polymerase36 were purified as previously described. Briefly, they were expressed in E. coli BL21(De3) star cells in 1 L of LB (Conda) media with 100 μg mL−1 of ampicillin (Duchefa) and 1 mM IPTG (LPS solution) at 18 °C for overnight, and APE1 (human) was expressed in E. coli BL21(De3) star cells in 1 L of LB medium with 100 μg mL−1 of ampicillin and 1 mM IPTG at 37 °C for 1 h. These proteins were FPLC-purified using Akta Prime (GE Healthcare). For ExoIII, standard A-buffer (20 mM Tris–HCl, 200 mM NaCl, pH 7.5) and B-buffer (20 mM Tris–HCl, 200 mM NaCl, Imidazole 500 mM, pH 7.5) were applied to Ni–NTA (HisTrap HP) for affinity purification. For APE1, standard A-buffer (50 mM Tris–HCl, 200 mM NaCl, pH 7.5) and B-buffer (50 mM Tris–HCl, 200 mM NaCl, imidazole 500 mM, pH 7.5) were used for affinity purification. The purified proteins were dialysed each A-buffer combined with glycerol (30%), aliquoted and frozen in liquid nitrogen.2.3 Cy3 labelling and annealing
For the fluorescent Cy3 conjugation, the non-hydrolysed strand containing an internal amine-modification at various positions was fluorescently labelled with Cy3 NHS ester (Ex/Em = 550/570 nm), as previously reported.37,38 The fluorescently labelled oligonucleotide was annealed with its complementary strand (molar ratio, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in Tris buffer (pH 8.0) with 100 mM NaCl by heating at 95 °C for 2 min and cooling slowly to room temperature for ∼3 hours in a tube covered with aluminium foil. After fluorescence labelling, the fluorescence efficiencies were determined using Nanodrop™ and were more than 90%.
:1) in Tris buffer (pH 8.0) with 100 mM NaCl by heating at 95 °C for 2 min and cooling slowly to room temperature for ∼3 hours in a tube covered with aluminium foil. After fluorescence labelling, the fluorescence efficiencies were determined using Nanodrop™ and were more than 90%.
2.4 Gel-based degradation assay by electrophoresis
The activities of the enzymes (e.g., ExoIII, hAPE1, and T4 DNA polymerase) were confirmed by gel electrophoresis using the Cy3-labelled AP DNA substrate (Fig. S1†). To prevent digestion of the biotinylated ends, we further blocked these ends with NeutrAvidin, taking advantage of the power of the NeutrAvidin–biotin interaction. With NeutrAvidin in place, the exonuclease reaction by ExoIII was successfully prevented (Fig. S3†). Next, the ExoIII reaction was performed in buffer C (10 mM Tris, 10 mM MgCl2, and 1 mM DTT, pH 7) for 3 min, and the hAPE1 reaction was performed with the AP-DNA substrate in buffer D (50 mM potassium acetate, 20 mM tris-acetate, 10 mM magnesium acetate, and 1 mM DTT, pH 7.9) for 3 min. For the reaction by hAPE1, hAPE1 was combined with the AP-DNA substrate in a tube for 10 min, and T4 DNA polymerase (100 mM) was subsequently added to the same tube for 3 min. All reactions were conducted at room temperature (25 °C), and formamide was used to stop the enzymatic reactions. The gel assays were run using denaturing gels (20% acrylamide, urea) at room temperature (25 °C) for 1 h at 250 V.2.5 NeutrAvidin attachment
For the capping-reaction to the biotinylated DNA termini using NeutrAvidin, the NeutrAvidin (5 mg mL−1) was diluted 10×. Biotinylated dsDNA and NeutrAvidin (molar ratio, 1:10) were mixed in a tube with 10 μL of DNA (10 μM), and 10 μL of NeutrAvidin (500 μg mL−1) in the total volume of 30 μL of 1× binding buffer (12% glycerol, 12 mM HEPES–NaOH [pH 7.9], 4 mM Tris–HCl [pH 7.9], 60 mM KCL, 1 mM EDTA, 1 mM DTT). After mixing, the sample was placed in a 37 °C incubator with shaking for 30 min in a dark tube. With NeutrAvidin in place, the degradation reaction by various exonucleases was successfully prevented.39
2.6 Fluorescence spectrometer measurement
Fluorescence measurements were carried out using a fluorescence spectrometer (Model: FS-2, SCINCO, Inc., Nonhyeon-dong, Gangnam-gu, Seoul, Korea) at room temperature. To collect the Cy3 fluorescence signal, the excitation wavelength was 532 nm and the emission-scanning range was 550 to 570 nm. All intensity measurements were conducted for 100 μL of the solution in a quartz cuvette containing 1× reaction enzyme buffer. To determine how PIFE increased the Cy3 intensity from the basal level, the fluorescently labelled DNA probe was incubated in the quartz cuvette with either ExoIII or the sequential addition of APE1 and T4 DNA polymerase for 10 min at room temperature. After the completion of the reaction, the Cy3 intensities were measured to evaluate the enhancement of the Cy3 fluorescence intensity by the reactions.3. Results and discussion
3.1 Design of a FE-based flaring biosensor
For a proof of concept, we chose E. coli ExoIII, which belongs to the type II family, as a representative AP endonuclease. ExoIII is a multifunctional enzyme that is capable of performing as either an AP endonuclease or a 3′ → 5′ exonuclease, or both, depending on the form of the DNA substrate.40–42 For instance, in the presence of a dsDNA lacking an AP-site, it serves as a 3′ → 5′ exonuclease whereas in the presence of a dsDNA with an AP-site in the middle of the dsDNA, the enzyme first recognizes and severs 5′ to the AP-site as an AP endonuclease and then subsequently degrades one strand of dsDNA in the 3′ to 5′ direction as a 3′ → 5′ exonuclease, generating a 5′ ss-overhang. For ssDNA containing an AP-site, ExoIII cuts the AP-site and acts as a ss-endonuclease.34Biochemical affinity studies43–45 have indicated that an AP site can be employed as a bait for sensing AP endonucleases, and recent technical developments have demonstrated a distance-dependent PIFE.27,28 However, the PIFE method was limitedly applied only to real-time single-molecule measurements but the limitation came from the fact that PIFE transiently appears and spatiotemporally disappears when the distance between a protein and Cy3 exceeds the proper proximity. In addition, single molecule measurements require special instruments and expertise to use the PIFE as a readout.
To overcome these limitations, we conceived a new FE methodology by which the FE remains stable even after the completion of the reaction. This methodology would make experiments much easier and improve their fidelity and reproducibility because the FE would become final product-dependent but not transient interaction-dependent. To develop such a fluorescence sensor, we paid attention to two characteristics: (1) ExoIII converts dsDNA to ssDNA as a reaction product and (2) Cy3 intensity is strongly affected by the local environment around Cy3 so that it becomes brighter as the following order: free Cy3 <Cy3 on dsDNA <Cy3 on ssDNA.25 The molecular basis of the photo-physical effect was found to be redistribution of Cy3 cis–trans isomerization between fluorescent trans-state and non-fluorescent cis-state (top in Scheme 1A). If the isomerization of Cy3 takes place concomitant with a structural change of the DNA probe, a new product-based stable FE will be achieved.
For that purpose, an AP-site and Cy3 were incorporated into dsDNA as the binding-hot-spot for AP endonucleases and a fluorescent signal-generator, respectively. We constructed five DNA probes in which the positions of Cy3 and the AP-site varied and measured their fluorescent spectra in the presence of ExoIII and 10 mM Mg2+ using a FS-2 SCINCO fluorimetre™ (Fig. S2†). Consistently, the Cy3 emission peaks appeared at 570 nm.
We found that one of the DNA probes had a persistent FE (termed as dsDNA-FE). The dsDNA-FE (bottom in Scheme 1A) was composed of a non-hydrolysed fluorescence strand (3′ bottom strand) with Cy3 at 23 nt from the 3′ end; a hydrolysed AP strand (5′ top strand) with an AP-site at 5 nt from the 3′ end; and NeutrAvidin antibodies that blocked the dsDNA at all ends. We thought that the stable FE resulted from the Cy3-bearing ssDNA produced in the course of the hydrolysed strand degradation starting from the AP-site, upon mixing ExoIII with the dsDNA-FE (Scheme 1B).
In contrast, the A2 probe, which has the NeutrAvidin but not the AP site, did not produce any FE (Fig. S2†). We found that it was not digested by ExoIII (Fig. S3†). These experiments confirmed that the terminal protection by NeutrAvidin prevented the activity of exonucleases from the ends of dsDNA and that both AP endo- and exo-activities were required for the degradation of the dsDNA-FE. Overall, the data indicated that the structural conversion from dsDNA to ssDNA was the source of a stable FE, owing to a change in the isomerization frequency between fluorescent trans-state and non-fluorescent cis-state by transforming local flexibility and mobility in the vicinity of Cy3.
3.2 Molecular origin of the persistent FE
To confirm that the FE arises from the Cy3 isomerization, we reversed the degradation reaction step-by-step from ssDNA to dsDNA to ascertain whether the reconstitution into the dsDNA-FE suppressed the fluorescence signal (Scheme 1C). Upon annealing of the 3′ bottom strand (Cy3-labelled ssDNA) and the 5′ top strand (complementary strand with AP-site), the fluorescence signal was reduced by half (Scheme 1D). The addition of NeutrAvidin for terminal capping of DNA did not significantly change the intensity of Cy3, but the subsequent addition of ExoIII increased the FE two-fold afterward. Therefore, we concluded that the origin of the stable FE is indeed the isomerization of Cy3 caused by the structural conversion from dsDNA to ssDNA.The occurrence of the FE here reports whether an enzyme to be tested contains both activities of AP endo- and exo-nucleases (Scheme 1B). Importantly, the current FE methodology is photo-physically different from the canonical PIFE, in that it is not directly caused by the protein-Cy3 interaction. Rather, our FE stems from the fact that the quantum yield of Cy3 in the final product, ssDNA, is higher than that in dsDNA. Thus, the FE is stable. A simple fluorescent dsDNA-FE with the AP-site functions as a biosensor that is sensitive enough to identify type II AP endonucleases.
3.3 Quantitative sensing for bacterial AP endonuclease (ExoIII)
We further investigated the effect of the presence or absence of the AP-site and NeutrAvidin on the FE. Unlike the dsDNA-FE (green in Fig. 1A), other dsDNA probes in which either the NeutrAvidin (blue) or the AP-site (magenta), or both (orange) were omitted did not exhibit a marked FE (Fig. 1A and B). The results suggested again that both the AP-site and NeutrAvidin were essential for the stable FE by ExoIII.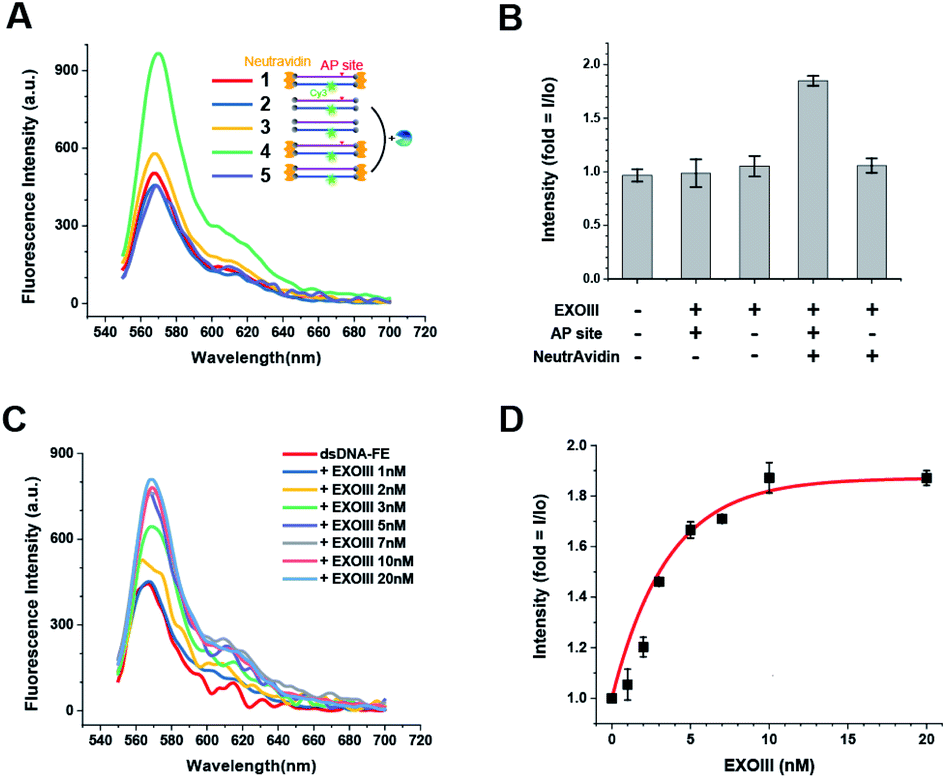 | ||
| Fig. 1 Quantification of the amount of AP endonuclease by FE. (A) Fluorescence spectra obtained from various DNA probes with different functional units, highlighting that both the AP site and NeutrAvidin play essential roles in generating FE. (B) The effect of the presence or absence of either the AP site or NeutrAvidin (or both) on FE. (C) Fluorescence spectra obtained as a function of [ExoIII]. (D) Fluorescence intensity vs. [ExoIII] curve, showing the fold change in the FE. Error bars are means ± SEM, and are produced by three independent replications. | ||
Next, we examined the capability to quantify the amount of ExoIII based on the FE. The spectra were obtained as a function of the concentration of ExoIII (hereafter [ExoIII]) after ExoIII was incubated with 80 nM dsDNA-FE probe (50 μL) for ∼10 min. We found that the FE occurred in a [ExoIII]-dependent manner as [ExoIII] was increased from 1 to 100 nM (Fig. 1C and D). A quantitation for ExoIII could be spectroscopically determined by means of the characteristic [ExoIII]-dependent curve (Fig. 1C and D).
3.4 Quantitative sensing for human AP endonuclease (hAPE1)
To develop a biosensor as a new tool for human applications, we applied the same protocol developed for ExoIII to hAPE1. Although hAPE1 is structurally homologous to bacterial ExoIII, hAPE1 possesses strong activity of AP endonuclease but has very weak 3′ to 5′ exonuclease activity compared to ExoIII due to the alteration in W280 residue46 (left of Fig. 2A). For this reason, the conversion from dsDNA to ssDNA did not take place following the addition of hAPE1 alone (Fig. 2B and C), and thus did not exhibit any FE. We discovered that hAPE1 supplemented with a 3′ to 5′ exonuclease activity by T4 DNA polymerase (DNAP), recapitulated the multi-functional ExoIII activities of both the AP endo- and exo-nucleases (right of Fig. 2A). Further tests confirmed that neither hAPE1 or T4 DNAP resulted in the FE but the combination of both the proteins resulted in a marked FE, comparable to that by ExoIII (Fig. 2B and C).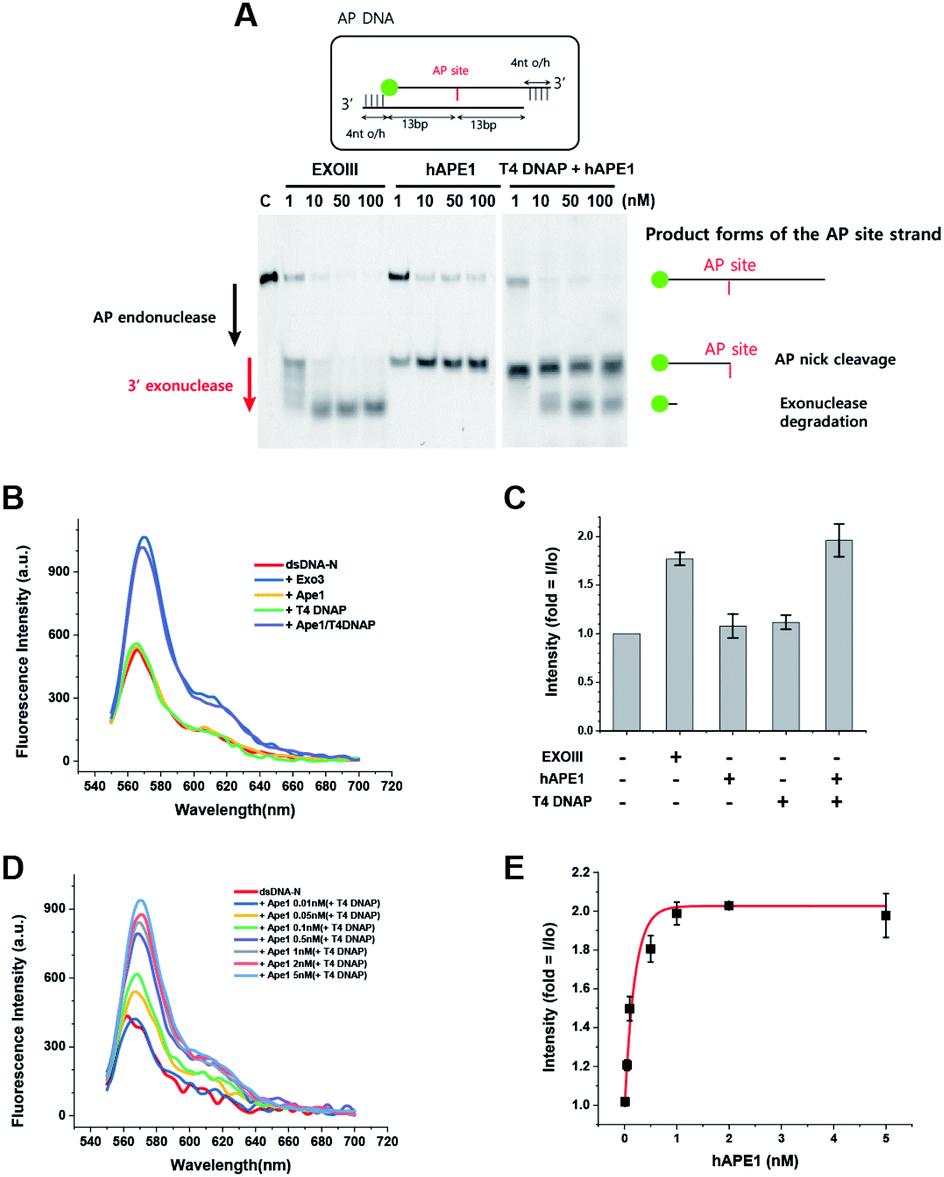 | ||
| Fig. 2 Quantification of the amount of human AP endonuclease 1 (hAPE1). (A) A gel-electrophoresis assay showing the different activities of ExoIII and APE1. ExoIII first cleaves the AP site and continues to degrade dsDNA, whereas hAPE1 only possesses the activity of AP endonuclease but not that of 3′ → 5′ exonuclease. However, the addition of T4 DNAP, carrying the domain of 3′ → 5′ exonuclease, recovers the combinatorial activity of both endo- and exo-nucleases. (B and C) Neither hAPE1 nor T4 DNAP showed combinatorial activity, but the addition of both showed this function, the same activity as ExoIII. (D) Fluorescence spectra obtained as a function of [hAPE1] supplemented with 100 mM T4 DNAP. (E) Intensity vs. [hAPE1] curve showing the fold change in FE. Error bars are means ± SEM, and are produced by three independent replications. | ||
To quantitatively detect APE1, we increased [APE1] from 0.01 to 5 nM in the presence of 100 nM T4 DNAP. The [APE1] dependence on FE appeared in a wide range from 0.01 to 1 nM (Fig. 2D and E), and saturated beyond 1 nM APE1. The high sensitivity with the 10-fold range indicated that the quantitative determination of APE1 can be spectroscopically assessed by the characteristic [APE1]-dependent curve (Fig. 2E).
3.5 Specificity of the DNA sensor for type II AP endonucleases
The dsDNA-FE should be specific only for type II AP endonucleases and not detect other nucleases abundant in cells (e.g., 3′ → 5′ and 5′ → 3′ exonucleases; and site-specific and site-nonspecific endonucleases). However, it has been reported that no other endonuclease exists in human cells except for hAPE1 as a sequence-specific or nonspecific endonuclease, even though many endo-nucleases are present in prokaryotic systems. Therefore, we only considered exonucleases as the other types of nucleases that cleave the DNA probe. We tested hAPE1 alone; T4 DNAP and Phi29 DNAP as examples of 3′ → 5′ exonucleases; and T7 exonuclease and lambda exonuclease as examples of 5′ → 3′ exonucleases. We found that the FE occurred only when ExoIII or hAPE/T4 DNAP were added to the reaction buffer that contained dsDNA-FE and 10 mM Mg2+, but not when other exonucleases were added (Fig. 3A and B). This result suggested that the dsDNA-FE provides high specificity for sensing type II AP endonucleases even in the presence of other exonucleases.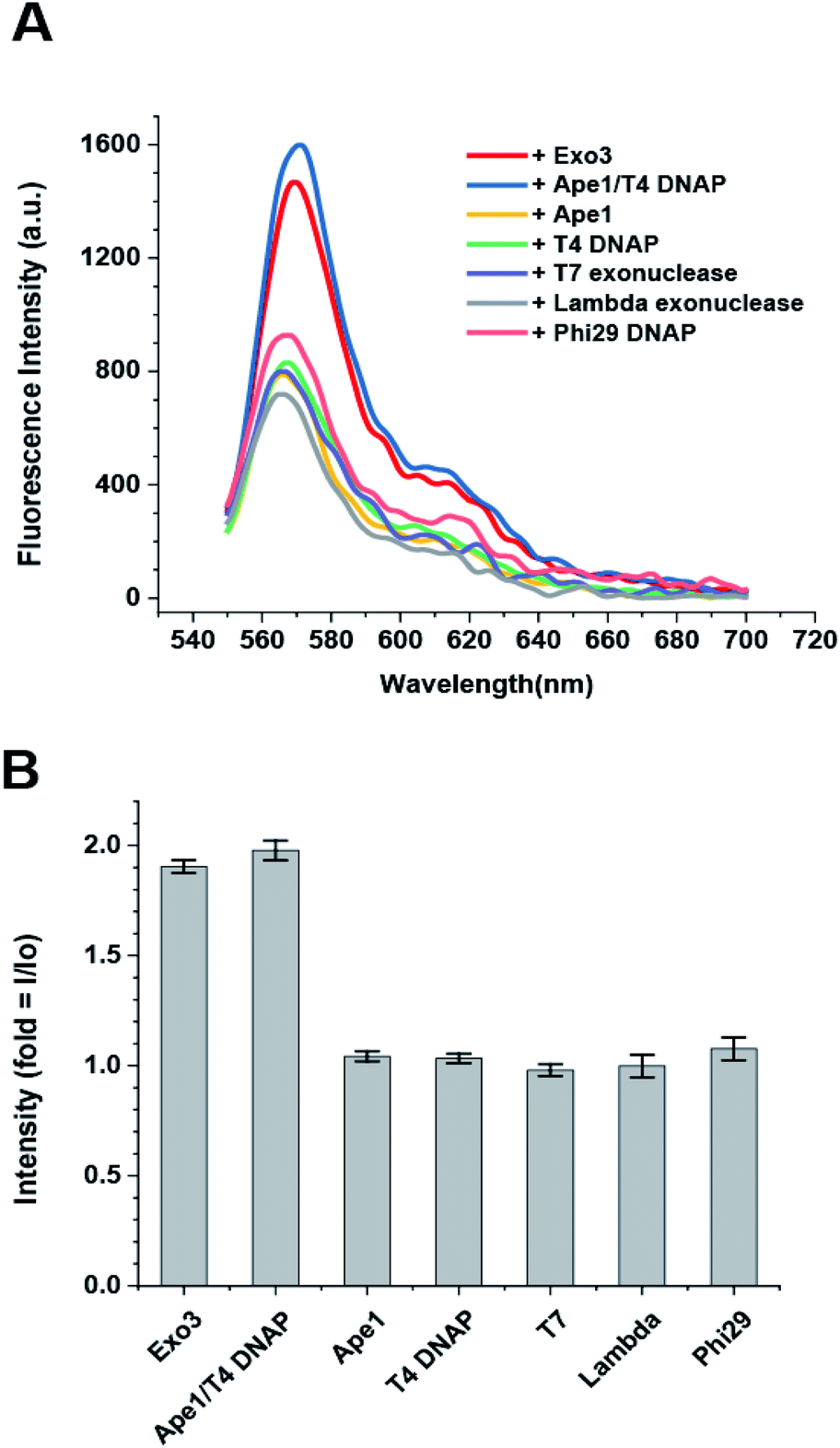 | ||
| Fig. 3 Enzymatic specificity of the biosensor (dsDNA-FE). (A) Fluorescence spectra were obtained from various exonucleases, demonstrating that the dsDNA-FE selectively detects type II AP endonucleases over the activity of other 3′ → 5′ or 5′ → 3′ exonucleases. Error bars are means ± SEM, and are produced by three independent replications. (B) Quantification of fluorescence intensity of (A) at the wavelength of 568 nm. | ||
4. Conclusion
We developed a sensitive fluorometric assay not only for detecting AP endonucleases (e.g., bacterial ExoIII and human APE1) but also for assaying the amount of AP endonucleases based on [enzyme]-dependent characteristic FE-curves (Fig. 1D and 2E). The persistent FE-based approach provides an accurate platform for characterizing the type II family of AP endonucleases due to resistance to the nonspecific digestion by other nucleases (Fig. 1B and 2C). Fig. 2E shows the linear response of our sensor in the range of nanomolar (10−9 M) to tens of picomolar (10−11 M). The detection limit is 5 × 10−11 M hAPE1, determined by the target enzyme concentration, corresponding to the second lowest concentration. The fold-change intensity is 20% higher than that of no enzyme control (dsDNA-N only), confidently detecting hAPE1 within the statistical error bars (1.2 ± 0.05, mean ± SD with three independent replications). The detection limit of our method is much better than other popular methods, such as 10−7 M for surface plasmon resonance47 and 10−6 M for an electrophoretic gel mobility assay.48 The sensitivity of FE is as low as 5 × 10−11 M to detect hAPE1 due to its sub-nanomolar sensitivity (see, blue and orange curves in Fig. 2D). This sensitivity is ∼100 fold more sensitive compared to other fluorescence-based methods with current sensitivity16,17,19 of 10−8 M. This extremely high sensitivity may result from the fact that the AP endonuclease recognizes and tightly binds to AP-sites via topological coupling between the AP-substrate and enzymes49,50 and the subsequent reaction converts the DNA structural change to the change in Cy3 isomerization state of a single fluorophore.25,26 Another advantage of our method is that it is relatively simper than fluorescent energy-transfer based methods between two fluorophores (e.g., FRET or quencher-based sensors) in that it utilizes the isomerization property of a single cyanine dye.In short, our method is a new analytical tool that uses a robust readout of the fluorescent cis–trans isomerization of a cyanine dye (Cy3) to detect AP endonucleases at very low concentrations. More importantly, the three functional groups incorporated in our probe (an AP-site, Cy3 and exonucleolytic protected termini) provide high specificity and selectivity to target AP endonucleases relative to other nucleases due to the terminal protection that prevents degradation if any other exonucleases are present in the sample (Fig. 3A and B). This fluorometric platform can be extended for rapid and high-throughput applications with cell lysate samples in the presence of other nucleases, for example, in detecting disease-causing enzymes, quantifying enzyme activity, and screening therapeutic inhibitors, by incorporating a multi-well plate reader.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the GIST Research Institute (GRI) in 2021 and the National Research Foundation of Korea (NRF) grant funded by the Korea government (NRF-2020R1A2C2006712 and NRF-2019R1A4A1028802) and by the grant from the Korean Health Technology R&D project, Ministry of Health and Welfare, Republic of Korea (HA17C0031: #1720050).Notes and references
- F. Puglisi, G. Aprile, A. M. Minisini, F. Barbone, P. Cataldi, G. Tell, M. R. Kelley, G. Damante, C. A. Beltrami and C. Di Loreto, Anticancer Res., 2001, 21, 4041–4049 CAS.
- Q. Sheng, Y. Zhang, R. Wang, J. Zhang, B. Chen, J. Wang, W. Zhang and X. Xin, Med. Oncol, 2012, 29, 1265–1271 CrossRef CAS PubMed.
- M. L. Fishel, Y. He, A. M. Reed, H. Chin-Sinex, G. D. Hutchins, M. S. Mendonca and M. R. Kelley, DNA Repair, 2008, 7, 177–186 CrossRef CAS PubMed.
- X. Fan, L. Wen, Y. Li, L. Lou, W. Liu and J. Zhang, APMIS, 2017, 125, 857–862 CrossRef CAS PubMed.
- A. P. Londero, M. Orsaria, G. Tell, S. Marzinotto, V. Capodicasa, M. Poletto, C. Vascotto, C. Sacco and L. Mariuzzi, Am. J. Clin. Pathol., 2014, 141, 404–414 CrossRef PubMed.
- X. Wen, R. Lu, S. Xie, H. Zheng, H. Wang, Y. Wang, J. Sun, X. Gao and L. Guo, Cancer Biomarkers, 2016, 17, 313–322 CAS.
- D. Wang, M. Luo and M. R. Kelley, Mol. Cancer Ther., 2004, 3, 679–686 CAS.
- E. Canbay, B. Cakmakoglu, U. Zeybek, S. Sozen, C. Cacina, M. Gulluoglu, E. Balik, T. Bulut, S. Yamaner and D. Bugra, Curr. Med. Res. Opin., 2011, 27, 1295–1302 CrossRef CAS PubMed.
- S. H. Zhang, L. A. Wang, Z. Li, Y. Peng, Y. P. Cun, N. Dai, Y. Cheng, H. Xiao, Y. L. Xiong and D. Wang, World J. Gastroenterol., 2014, 8700–8708 CrossRef CAS PubMed.
- T. M. A. Abdel-Fatah, C. Perry, P. Moseley, K. Johnson, A. Arora, S. Chan, I. O. Ellis and S. Madhusudan, Breast Cancer Res. Treat., 2014, 143, 411–421 CrossRef CAS PubMed.
- E. Coskun, P. Jaruga, P. T. Reddy and M. Dizdaroglu, Biochemistry, 2015, 54, 5787–5790 CrossRef CAS PubMed.
- J. Woo, H. Park, S. H. Sung, B. I. Moon, H. Suh and W. Lim, PLoS One, 2014, 9(6), e99528 CrossRef PubMed.
- S. A. Jin, H. J. Seo, S. K. Kim, Y. R. Lee, S. Choi, K. T. Ahn, J. H. Kim, J. H. Park, J. H. Lee, S. W. Choi, I. W. Seong, B. H. Jeon and J. O. Jeong, Korean Circ. J., 2015, 45, 364–371 CrossRef PubMed.
- J. H. Shin, S. Choi, Y. R. Lee, M. S. Park, Y. G. Na, K. Irani, S. D. Lee, J. B. Park, J. M. Kim, J. S. Lim and B. H. Jeon, Cancer Res. Treat., 2015, 47, 823–833 CrossRef CAS PubMed.
- S. Choi, J. H. Shin, Y. R. Lee, H. K. Joo, K. H. Song, Y. G. Na, S. J. Chang, J. S. Lim and B. H. Jeon, Dis. Markers, 2016, 2016, 7276502 CrossRef PubMed.
- X. Li, M. Xiong, Y. Huang, L. Zhang and S. Zhao, Anal. Methods, 2019, 11, 739–743 RSC.
- Y. Huang, Y. Ma, Y. Li, M. Xiong, X. Li, L. Zhang and S. Zhao, New J. Chem., 2017, 41, 1893–1896 RSC.
- J. Zhai, Y. Liu, S. Huang, S. Fang and M. Zhao, Nucleic Acids Res., 2017, 45(6), e45 CrossRef PubMed.
- B. Liu and L. Peng, Anal. Methods, 2016, 8, 862–868 RSC.
- J. Li, D. Svilar, S. McClellan, J. H. Kim, E. E. Ahn, C. Vens, D. M. Wilson III and R. W. Sobol, Oncotarget, 2018, 9, 31719–31743 CrossRef PubMed.
- D. Dorjsuren, D. Kim, V. N. Vyjayanti, D. J. Maloney, A. Jadhav, D. M. Wilson Ii and A. Simeonov, PLoS One, 2012, 7(10), e47974 CrossRef CAS PubMed.
- A. A. Kuznetsova, O. S. Fedorova and N. A. Kuznetsov, Molecules, 2018, 23, 2101 CrossRef PubMed.
- A. A. Kuznetsova, A. G. Matveeva, A. D. Milov, Y. N. Vorobjev, S. A. Dzuba, O. S. Fedorova and N. A. Kuznetsov, Nucleic Acids Res., 2018, 46, 11454–11465 CrossRef CAS PubMed.
- M. Zhou, C. Feng, D. Mao, S. Yang, L. Ren, G. Chen and X. Zhu, Biosens. Bioelectron., 2019, 142, 111558 CrossRef CAS PubMed.
- M. E. Sanborn, B. K. Connolly, K. Gurunathan and M. Levitus, J. Phys. Chem. B, 2007, 111, 11064–11074 CrossRef CAS PubMed.
- E. Lerner, E. Ploetz, J. Hohlbein, T. Cordes and S. Weiss, J. Phys. Chem. B, 2016, 120, 6401–6410 CrossRef CAS PubMed.
- H. Hwang and S. Myong, Chem. Soc. Rev., 2014, 43, 1221–1229 RSC.
- H. Hwang, H. Kim and S. Myong, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7414–7418 CrossRef CAS PubMed.
- G. Luo, M. Wang, W. H. Konigsberg and X. S. Xie, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12610–12615 CrossRef CAS PubMed.
- M. Sorokina, H. R. Koh, S. S. Patel and T. Ha, J. Am. Chem. Soc., 2009, 131, 9630–9631 CrossRef CAS PubMed.
- S. Myong, M. M. Bruno, A. M. Pyle and T. Ha, Science, 2007, 317, 513–516 CrossRef CAS PubMed.
- S. Myong, S. Cui, P. V. Cornish, A. Kirchhofer, M. U. Gack, J. U. Jung, K. P. Hopfner and T. Ha, Science, 2009, 323, 1070–1074 CrossRef CAS PubMed.
- C. J. Fischer and T. M. Lohman, J. Mol. Biol., 2004, 344, 1265–1286 CrossRef CAS PubMed.
- T. Shida, M. Noda and J. Sekiguchi, Nucleic Acids Res., 1996, 24, 4572–4576 CrossRef CAS PubMed.
- J. Yoo and G. Lee, Nucleic Acids Res., 2015, 43, 10861–10869 CrossRef CAS PubMed.
- H. Takahashi, H. Yamazaki, S. Akanuma, H. Kanahara, T. Saito, T. Chimuro, T. Kobayashi, T. Ohtani, K. Yamamoto, S. Sugiyama and T. Kobori, PLoS One, 2014, 9(2), e82624 CrossRef PubMed.
- R. Roy, S. Hohng and T. Ha, Nat. Methods, 2008, 5, 507–516 CrossRef CAS PubMed.
- G. Lee, M. A. Bratkowski, F. Ding, A. Ke and T. Ha, Science, 2012, 336, 1726–1729 CrossRef CAS PubMed.
- J. Song, V. M. Hoa, J. Yoo, S. Oh, H. Im, D. Park and G. Lee, RSC Adv., 2017, 7, 14917–14922 RSC.
- D. Brutlag and A. Kornberg, J. Biol. Chem., 1972, 247, 241–248 CrossRef CAS.
- B. Luckow, R. Renkawitz and G. Schutz, Nucleic Acids Res., 1987, 15, 417–429 CrossRef CAS PubMed.
- Y. Liu, R. Prasad, W. A. Beard, P. S. Kedar, E. W. Hou, D. D. Shock and S. H. Wilson, J. Biol. Chem., 2007, 282, 13532–13541 CrossRef CAS PubMed.
- Q. Xu, A. Cao, L. F. Zhang and C. Y. Zhang, Anal. Chem., 2012, 84, 10845–10851 CrossRef CAS PubMed.
- J. W. Hill, T. K. Hazra, T. Izumi and S. Mitra, Nucleic Acids Res., 2001, 29, 430–438 CrossRef CAS PubMed.
- R. L. Maher and L. B. Bloom, J. Biol. Chem., 2007, 282, 30577–30585 CrossRef CAS PubMed.
- K. Kaneda, J. Sekiguchi and T. Shida, Nucleic Acids Res., 2006, 34, 1552–1563 CrossRef CAS PubMed.
- T. Jiang, M. Minunni, P. Wilson, J. Zhang, A. P. F. Turner and M. Mascini, Biosens. Bioelectron., 2005, 20, 1939–1945 CrossRef CAS PubMed.
- L. M. Hellman and M. G. Fried, Nat. Protoc., 2007, 2, 1849–1861 CrossRef CAS PubMed.
- C. D. Mol, C. F. Kuo, M. M. Thayer, R. P. Cunningham and J. A. Tainer, Nature, 1995, 374, 381–386 CrossRef CAS PubMed.
- C. D. Mol, T. Izumi, S. Mitra and J. A. Talner, Nature, 2000, 403, 451–456 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08051a |
| This journal is © The Royal Society of Chemistry 2021 |