
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxygen doped graphitic carbon nitride nanosheets for the degradation of organic pollutants by activating hydrogen peroxide in the presence of bicarbonate in the dark†
Tian-Jiao Jiang‡
a,
Chao Xie‡a,
Huai-De Penga,
Bo Lei a,
Qing-Qing Chena,
Gang Lia and
Cai-Wu Luo*abcd
a,
Qing-Qing Chena,
Gang Lia and
Cai-Wu Luo*abcd
aSchool of Resource Environmental and Safety Engineering, University of South China, 421000, China. E-mail: luocaiwu00@126.com; Tel: +86-734-8282345
bState Key Laboratory of Safety and Health for Metal Mines, Sinosteel Maanshan General Institute of Mining Research Co., Ltd, 243000, China
cKey Laboratory of Clean Energy Material, LongYan University, 364012, China
dResearch Center for Eco-Environmental Sciences, Chinese Academy of Sciences, 100085, China
First published on 24th December 2020
Abstract
The development of novel wastewater treatment processes that use heterogeneous catalysts to activate hydrogen peroxide (H2O2) with bicarbonate (HCO3−) has been a subject of great interest in recent years; however, significant challenges remain, despite research into numerous metal-based catalysts. The work presented herein employed oxygen-doped graphitic carbon nitride (O/g-C3N4) as a non-metal catalyst for activating H2O2 in the presence of HCO3−, and this method represented the first system capable of removing organic pollutants in the dark, to our knowledge. The catalysts were characterized using several microscopic imaging, spectroscopic, electrochemical, and crystallographic techniques, as well as N2-physorption procedures. Analysis of the results revealed that the O/g-C3N4 catalyst possessed a high specific surface area and many defect sites. Various operational parameters, including the relative amounts of HCO3−, H2O2, and O/g-C3N4, were systemically investigated. A clear performance enhancement was observed in the degradation of organic contaminants when subjected to the HCO3−–H2O2–O/g-C3N4 system, and this result was ascribed to the synchronous adsorption and chemical oxidation processes. The novel system presented herein represented a new water treatment technology that was effective for removing organic contaminants.
1. Introduction
The bicarbonate anion, HCO3−, is relatively nontoxic and is common in natural water ecosystems.1 For example, its concentration is known to reach 50–200 ppm in biological systems, and it may be presented at 14.7–25 mM in humans. It is worthwhile to note that HCO3− is always generated from CO2 as a main product of advanced oxidation processes, which are typically impractical for certain applications. Directly introducing HCO3− into a solution containing H2O2 often inhibits the degradation reaction; however, a few cases demonstrate that this process can promote the removal of organic pollutants.2–11 Despite this promising result, the huge required dosage is one of the main challenges associated with this approach because it significantly increases the cost of the treatment. To reduce this input cost, various transition metal ions, such as cobalt(II)12–18 and copper(II),19,20 have been tested as homogeneous catalysts. The results of those studies demonstrate that such metal catalysts accelerate the degradation of organic pollutants and distinctly reduce the required concentrations of HCO3− and H2O2. However, it is extremely difficult to recover the catalyst from the product solution, which is a significant drawback of this approach. As an alternative, various heterogeneous catalysts have been carefully developed to avoid such problems. To date, these systems have involved metal-based materials, such as Co-based21–25 and Cu-based catalysts.26–28 For example, Guo et al.23 reported an S-modified CoFe2O4 catalyst, which degraded acid orange II in a reactive system containing HCO3− and H2O2. Similarly, Pi et al.25 showed that their CoxMn3−xO4 material was an effective catalyst for removing chlorophenols in the presence of a naturally-occurring concentration of HCO3−. Despite these advances, the leaching of toxic metal ions must be confronted during these reactions, because this phenomenon easily caused a second contamination and may reduce the stability of the catalyst. Therefore, other useful strategies have been explored.29,30 For example, Pétrier et al.29 adopted sonochemical technology to enhance the degradation of bisphenol in the presence of HCO3−. However, the required complex equipment input increased the cost. Therefore, in order to replace metal-based heterogeneous methods, there is a pressing need to develop simple non-metal-based heterogeneous catalysts that are inexpensive and green, and have excellent activity in combination with H2O2 and HCO3−.Among non-metal heterogeneous catalysts, graphitic carbon nitride is an advantageous choice because it is safety, low cost, and has high stability. Importantly, it exhibits excellent catalytic performance under visible light illumination. Therefore, this type of catalyst has been applied for various purposes, including hydrogen production,31–34 but there are limited reports describing its activity for dark Fenton-like reaction. Cui et al.32 reported the degradation of organic dyes using g-C3N4 in the presence of H2O2; however, this system showed hardly any catalytic activity in the dark. To attain the aforementioned target activity, it is necessary to first explore the catalytic sites in g-C3N4 to evaluate H2O2 decomposition into reactive oxygen species (ROS). In general, this type of catalyst is characterized by extremely low specific surface area, which plays a vital role in the adsorption of organic pollutants. This factor significantly hinders the broad application of such catalysts in industry. To overcome this challenge, our group34 developed a novel g-C3N4 catalyst using oxygen modification. This O/g-C3N4 material possessed a huge specific surface area and exhibited some catalytic activity for the dark Fenton-like reaction. To further improve its catalytic performance, further system modification is necessary. As mentioned above, it can be expected that introducing HCO3− to the system should strengthen its capability to degrade organic pollutants in wastewater. This modification creates a weakly alkaline environment, which is favorable for decomposing H2O2 into ROS.21 However, it may generate new radicals, such as CO3˙−. The surface of g-C3N4 has a negative charge under near-alkaline conditions; therefore, it generates a counteracting force whereby HCO3− leaves the surface of the catalyst, thus inevitably capturing ROS, such as ˙OH radicals. Once the ˙OH radicals enter the solution, they are immediately captured by HCO3− to produce CO3˙− radicals, which have a longer lifetime. A literature survey35,36 confirmed that CO3˙− radicals could directly remove organic pollutants. Still, the relationships between the specific surface area, nitrogen defective sites, and catalytic activity must be further elucidated, especially in terms of the dark Fenton-like reaction.
Herein, we describe a novel treatment technique for removing organic pollutants during dark Fenton-like reaction. To our knowledge, this is the first time that O/g-C3N4 has been used as a heterogeneous catalyst in a system containing HCO3− and H2O2. Various factors, including the specific surface area and nitrogen defective sites, are systemically studied and discussed in terms of how they influence the overall catalytic activity. A clear enhancement in organic pollutant degradation is observed using this novel system.
2. Experimental
2.1. Materials and reagents
All chemical reagents were purchased with analytical purity and used without any further purification. Deionized water (resistivity 18.2 MΩ) was filtered using a Millipore Milli-Q water purification system.2.2. Preparation of O/g-C3N4 catalyst
The synthesis of the O/g-C3N4 catalyst has been reported previously by our group.34 Specifically, melamine powder was placed in a crucible with a cover, and then calcinated in a static air atmosphere at 550 °C for 4 h. The obtained yellow sample was denoted as g-C3N4. This g-C3N4 was added to deionized water, then transferred into a Teflon-sealed autoclave and maintained at 180 °C for 4 h. After cooling to room temperature, the obtained sample was washed with more deionized water and dried. Finally, the dried sample was placed in a crucible with a cover and calcinated for 4 h at different temperatures (350, 450, and 550 °C) in a static air atmosphere. The resulting samples were denoted as O/g-C3N4-T (where T = 350, 450, or 550 °C). Unless otherwise stated, the sample at 550 °C was denoted as simply, O/g-C3N4. Additionally, the Na–O/g-C3N4 variant was synthesized using a wet impregnation method. Essentially, the O/g-C3N4 catalyst was added to a solution containing NaHCO3, which was stirred vigorously for 24 h at room temperature. Next, they were dried. Finally, the sample was placed in a crucible with a cover, and calcinated for 4 h at 300 °C in a static air atmosphere (the theory value of Na+ is 10 wt%).2.3. Characterization
X-ray diffraction (XRD) spectroscopy was carried out with a Bruker D8-Advance X-ray diffraction instrument; N2-physisorption was conducted on a Quantachrome Autosorb-1 instrument at liquid-N2 temperature; scanning electron microscopy (SEM) was carried out on a JEOL JSM 6700 F operating at an accelerating voltage of 10 kV; transmission electron microscope (TEM) was conducted on TALOS F200 X instrument operating at an accelerating voltage of 200 kV; electron paramagnetic resonance (EPR) was conducted on Bruker A300; X-ray photoelectron spectra (XPS) was examined on Thermo Fisher Scientific using Al Kα; zeta potential was carried out on Zetasizer Nano ZSP instrument.2.4. Degradation of organic pollutants in the dark and under illumination
The degradation reactions were performed in a flask at 25 °C, at atmospheric pressure. The reaction solution contained a certain amount of catalyst, NaHCO3, H2O2, and organic pollutants. The reaction mixture was stirred vigorously in the dark unless otherwise stated. For the photocatalytic reactions, the reaction mixture was placed under LED illumination, using all other reaction conditions identical to the experiments conducted in the dark (i.e., same catalyst dosages and concentrations of H2O2 and HCO3−). In all cases, a certain aliquot of each reaction solution was extracted at fixed intervals and then separated. The liquid was collected, and they were analyzed using a UV-vis spectrophotometer to quantify the concentration of residual organic pollutants.3. Results and discussion
3.1. Characterization
Fig. 1 displayed the XRD patterns of the g-C3N4 and O/g-C3N4 catalysts. The diffraction peaks at 2θ = 13.0° and 27.5° were observed for the g-C3N4 species, and these were assigned to the repeating tri-s-triazine units within the g-C3N4 unit layer (100), and the inter-planar stacking of unit layers (002),32 respectively. Besides the characteristic peaks associated with g-C3N4, the O/g-C3N4 catalyst didn't exhibit any diffraction peaks corresponding to other phases, indicating that the original structure of g-C3N4 was largely retained following the oxygen modification. However, the strongest diffraction peaks for the O/g-C3N4 catalyst shifted toward higher angles (i.e., toward 2θ = 27.8°), relative to those observed for g-C3N4, representing the decreased distances within the layered structure.11 This change was attributed to the doping effect of oxygen atoms and the resulting distortion of the graphite structure.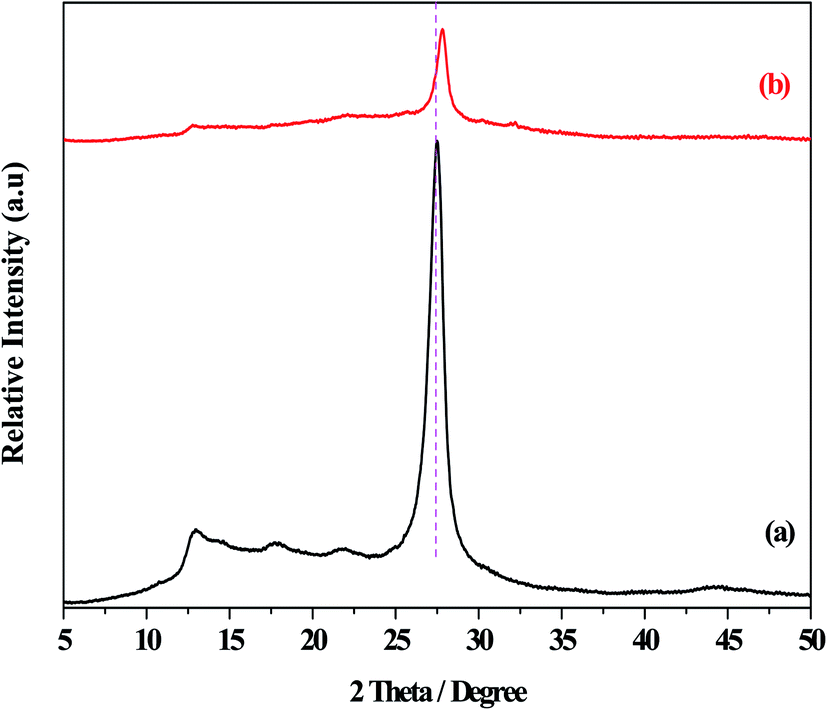 | ||
| Fig. 1 The XRD patterns of different catalysts. (a) g-C3N4, (b) O/g-C3N4. | ||
The images of the g-C3N4 and O/g-C3N4 catalysts captured using SEM characterization were shown in Fig. S1.† It is clear from these images that the g-C3N4 catalyst contained agglomerated particles, and its surface appeared relatively rough. Compared to the bulk g-C3N4, a cotton-like morphology was visible in the O/g-C3N4 image. This is likely because the nanosheets of g-C3N4 were partially decomposed following the hydrothermal-calcination treatment. This difference was further confirmed through characterization using TEM characterization (Fig. S2†). Table S1† listed the contents of carbon, nitrogen, and oxygen atoms, which were determined by elemental analysis of the g-C3N4 and O/g-C3N4 samples from SEM characterization. It is observed that O/g-C3N4 contained lower nitrogen atom content but a higher atomic ratio of carbon to nitrogen, relative to g-C3N4. These results indicated that delamination and depolymerization processes were happened, causing loss of nitrogen atoms and creation of nitrogen defect sites. To confirm the existence of such defect sites, EPR measurements were carried out, and the results were presented in Fig. S3.† There were six large positive peaks observed in the spectrum of the g-C3N4 catalyst, but only four positive peaks in the O/g-C3N4 catalyst's spectrum. These results suggested that the latter had a greater quantity of unpaired electrons than the former. This can be justified based on the fact that some nitrogen atoms have been removed and other nitrogen atoms were replaced by oxygen atoms, in agreement with the XRD and elemental analysis. The analysis of all of these results led to the conclusion that the nitrogen content, and especially the morphology, of O/g-C3N4 was altered relative to those of g-C3N4.
The textural properties of g-C3N4 and the series of O/g-C3N4-T catalysts, which were determined from N2 adsorption–desorption experiments, were compiled in Table 1. Relative to the g-C3N4, and within the series of O/g-C3N4-T catalysts, the specific surface area (SBET), first slightly increased and then increased remarkably, up to >23 times. Simultaneously, the total pore volume (Vtotal) first increased, and then significantly increased, up to approximately 10 times. It is well-known that g-C3N4 produced from various precursors can be easily obtained via high-temperature calcination, but the resulting SBET was typically below 10 m2 g−1. This is because the interactions between the layers of g-C3N4 were too strong, owing to the van der Waals forces and/or hydrogen bonds in the material, which led to serious aggregation of the particles. After the hydrothermal treatment, these interactions were weakened due to the attacks from water molecules at high temperature and pressure. The resulting exposed nanosheets were further attacked by oxygen during the high-temperature calcination, thus creating a new morphology and increasing the SBET. Based on the SEM, TEM, and pore size distribution results (see Fig. S4†), we determined that the increased SBET was mainly attributed to the change in the morphology of the catalyst. In general, the large SBET is an important factor for strengthening catalytic performance, because this represents a greater proportion of exposed adsorption and catalytic active sites.
| Catalysts | SBET (m2 g−1) | Vtotal (cc g−1) |
|---|---|---|
| a SBET was denoted as the specific surface area; Vtotal was denoted as the total pore volume. | ||
| g-C3N4 | 11.5 | 0.0693 |
| O/g-C3N4-350 °C | 42.2 | 0.3056 |
| O/g-C3N4-450 °C | 50.7 | 0.3107 |
| O/g-C3N4-550 °C | 236.4 | 0.6344 |
Fig. 2 displayed the binding energies of the N 1s in g-C3N4 and the series of O/g-C3N4-T samples, which were determined based on XPS. In the bulk g-C3N4, four different peaks at 398.9, 399.8, 401.5, and 404.6 eV were observed, corresponding to the sp2-hybridized nitrogen (N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N; denoted as sp2(N)), the sp3-hybridized nitrogen (N–[C]3; denoted as sp3(N)), the N–H bonds, and the π band localized in heterocycles, respectively.10,11,14–18 The N 1s spectra associated with the series of O/g-C3N4-T samples contained similar peaks to the bulk g-C3N4. However, the ratio of sp2(N) to sp3(N) first increased (see Table S2†) and then decreased with increasing temperature in the O/g-C3N4-T series (i.e., 350 °C vs. 450 °C vs. 550 °C), and as compared to g-C3N4. This result illustrated that loss of N occurred preferentially, thus forming some nitrogen defect sites.
N; denoted as sp2(N)), the sp3-hybridized nitrogen (N–[C]3; denoted as sp3(N)), the N–H bonds, and the π band localized in heterocycles, respectively.10,11,14–18 The N 1s spectra associated with the series of O/g-C3N4-T samples contained similar peaks to the bulk g-C3N4. However, the ratio of sp2(N) to sp3(N) first increased (see Table S2†) and then decreased with increasing temperature in the O/g-C3N4-T series (i.e., 350 °C vs. 450 °C vs. 550 °C), and as compared to g-C3N4. This result illustrated that loss of N occurred preferentially, thus forming some nitrogen defect sites.
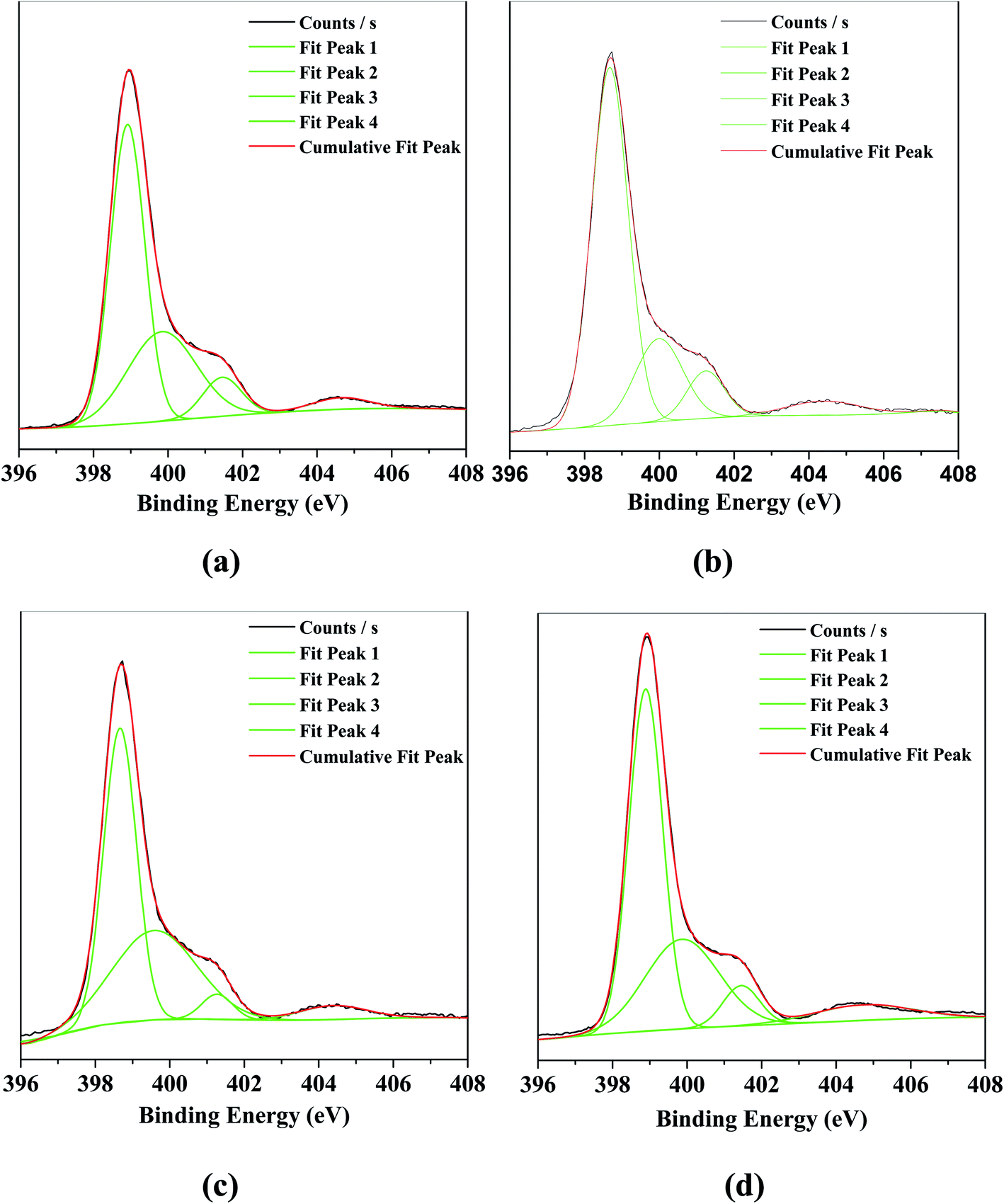 | ||
| Fig. 2 The XPS characterization for N 1s of various catalysts. (a) g-C3N4, (b) O/g-C3N4-350 °C, (c) O/g-C3N4-450 °C, (d) O/g-C3N4-550 °C. | ||
3.2. Degradation of organic pollutants under different reaction conditions
(i) Effect of the HCO3− concentration. Fig. 3 illustrated the influence of the concentration of HCO3− on the outcome of the dark Fenton-like reaction. It is determined that the rate of RhB degradation in the reaction solution accelerated significantly following the addition of HCO3−. Moreover, the RhB degradation rate increased with increasing concentrations of HCO3−, achieving the fastest value with 10.0 mM HCO3−. Upon increasing the concentration of HCO3− even further, the degradation rate was reduced, although it remained prominently higher than that measured in the absence of HCO3− (i.e., when [HCO3−] > 10 mM, the reaction rate was still enhanced). These results showed that the concentration range for HCO3− was very broad under these experimental conditions. Literature reports16,19 demonstrated that the impact of HCO3− concentration on the organic pollutant degradation varied depending on the system. For example, Cheng et al.19 found that the degradation of acid orange II using a Cu(II)–H2O2 system was restrained when the concentration of HCO3− was greater than 5 mM. This indicated that another reaction may have taken place in solution, besides that involving the catalyst. Surprisingly, the solution pH was found to increase slightly (∼1.0 pH unit) after the reaction, as shown in Fig. S6.† Lei et al.28 observed the same phenomenon in their catalytic system containing a mixture of CuO–FeO and persulfate. According to eqn (1)–(6), singlet oxygen species can be obtained on the basis of pH changes after the reaction.
| HCO3− + ˙OH → CO3˙− + H2O | (1) |
| CO3˙− + H2O2 → HCO3− + ˙OOH | (2) |
| ˙OOH → H+ + O2˙− | (3) |
| O2˙− + ˙OH → 1O2 + OH− | (4) |
| O2˙− + ˙OOH → 1O2 + OOH− | (5) |
| ˙OOH + ˙OOH → 1O2 + H2O2 | (6) |
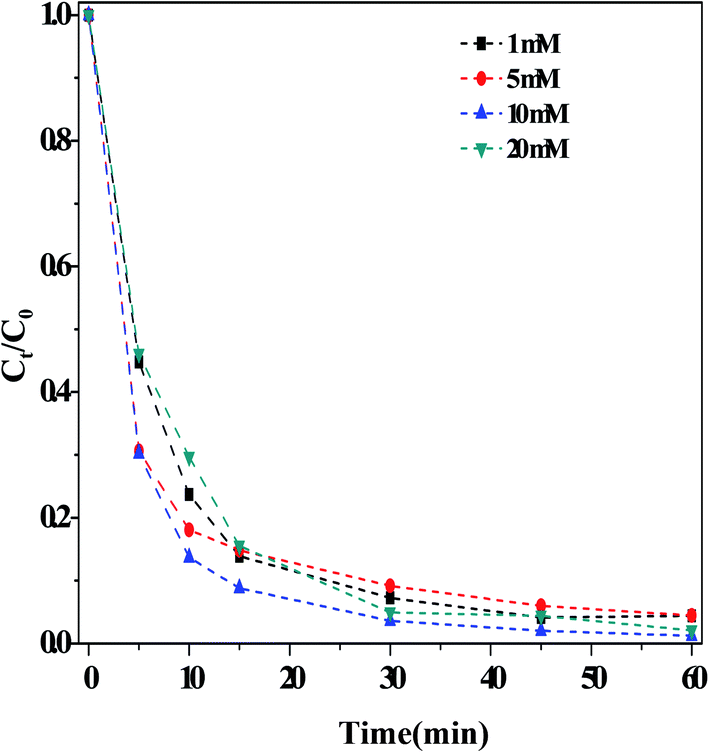 | ||
| Fig. 3 Effect of the concentrations of HCO3− on the RhB degradation. Reaction conditions: [H2O2] = 36 mM, [O/g-C3N4] = 1.0 g L−1, [RhB] = 10 mg L−1, 25 °C and 60 min. | ||
(ii) Effect of the H2O2 concentration. Fig. 4 depicted the influence of the concentration of H2O2 on the dark Fenton-like reaction. It is clear that the RhB degradation efficiency increased with increasing concentrations of H2O2, arriving at a maximum rate when [H2O2] = 15 mM. Increasing the H2O2 concentration further did not lead to any change in the efficiency of RhB degradation. As mentioned above, the ROS were generated from the decomposition of H2O2, so it followed that, when the concentration of H2O2 was relatively lower, fewer ROS could be formed. Therefore, by increasing the concentration of H2O2, the efficiency of RhB degradation was enhanced. However, the H2O2 concentration was increased further, the negligible influence on the RhB degradation indicated that the overall reaction predominately relied on the action of the catalyst.
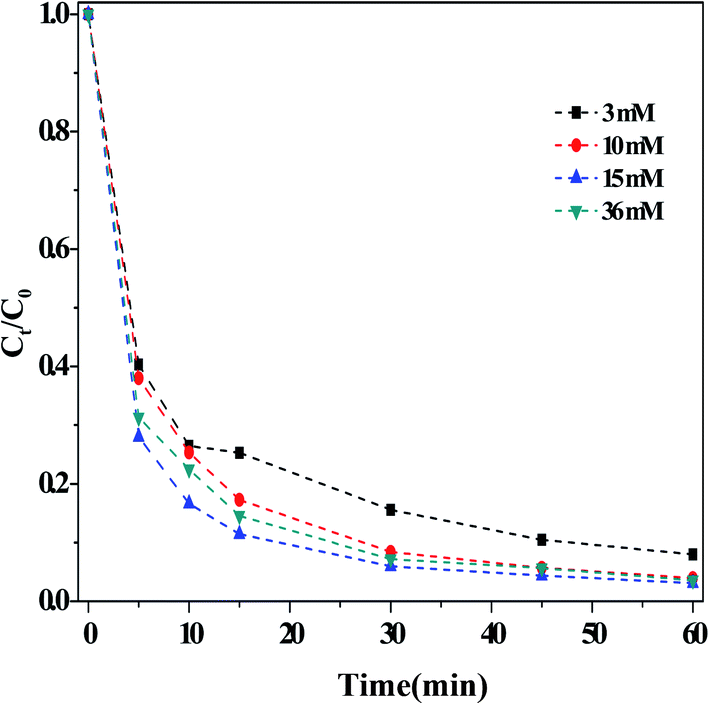 | ||
| Fig. 4 Effect of the concentration of H2O2 on the RhB degradation. Reaction conditions: [HCO3] = 10 mM, [O/g-C3N4] = 1.0 g L−1, [RhB] = 10 mg L−1, 25 °C, pH ≈ 8.4 and 60 min. | ||
(iii) Effect of the O/g-C3N4 catalyst. Fig. 5 illustrated the impact of the O/g-C3N4 catalyst on RhB degradation in the dark Fenton-like reaction. The degradation of RhB clearly increased with increasing catalyst loading, reaching a maximum value at a catalyst concentration of 0.8 g L−1. The rate remained stable when additional catalyst was added. It is evident that the removal of RhB was closely related to the catalyst dosage, as shown in Fig. S7.† This is because a larger quantity of catalyst offered more adsorption sites, thus enabling more RhB removal. However, addition of too much catalyst led to agglomeration and increased counter forces between the particles, ultimately reducing RhB adsorption. In contrast, an insufficient amount of catalyst was introduced, there would not have enough available sites to activate H2O2 for producing ROS, so the RhB degradation rate stabilized or may decrease slightly.
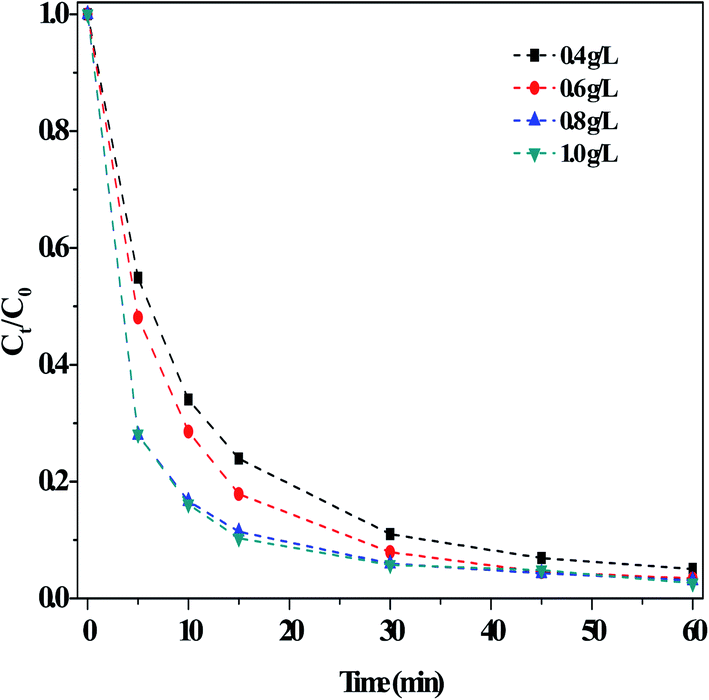 | ||
| Fig. 5 Effect of the usages of O/g-C3N4 on the RhB degradation. Reaction conditions: [NaHCO3] = 15 mM, [H2O2] = 10 mM, [RhB] = 10 mg L−1, 25 °C, pH ≈ 8.4 and 60 min. | ||
Based on the described systematic screening experiments, the optimal reaction conditions were determined: [NaHCO3] = 10 mM, [H2O2] = 15 mM, and [O/g-C3N4] = 0.8 g L−1. Applying these conditions, other important parameters were investigated.
Fig. S8† showed the influence of metal ions (with Cl− as the counter anion) on RhB degradation in the dark Fenton-like reaction. Based on this figure, there appeared to be no clear difference in RhB degradation among the tested metal ions, indicating that these ions had no appreciable effect on the RhB degradation reaction. This is because they were not involved the catalytic reaction, but rather, only impact solubility. For example, Ca2+ decreased the solubility of reaction solution, resulting in a decreased concentration of HCO3−. Many previous studies12–19 demonstrated that certain transition metal ions, such as Co(II) and Cu(II), effectively degraded organic pollutants due to the formation of M[HCO3−-H2O2]2RhB complexes. These complexes were formed when, for example, Co2+ interacted with HCO3− to form a polymeric Mn+[HCO3−] species, which then adsorbed H2O2. As a result, the ˙OH radicals were immediately generated, and attacked the RhB molecules on the surface of the catalyst. These processes led to a remarkable enhancement in RhB degradation. In contrast to transition metal ions, the metal ions studied herein were widely distributed in natural water environments, and they did not form M[HCO3−–H2O2]2RhB complexes. We additionally studied the influence of several anions (using Na+ as the metal ion), and the results were displayed in Fig. S9.† It is determined that most anions, except for CO3− and CH3COO−, positively impacted the RhB degradation. These results demonstrated that the studied catalytic system was resilient in terms of metal ions and anions, so it represented great potential for applications in industry. The removal of other organic pollutants was also investigated, and the results were shown in Fig. S10.† All of the tested species were removed effectively using this system, further verifying that it was favorable for degrading various organic contaminants.
Fig. S11† displayed the impact of darkness versus LED illumination on the degradation of RhB. It is evident that the system containing O/g-C3N4, HCO3−, and H2O2 exhibited better catalytic activity under LED illumination, relative to in the dark. This is because additional light-induced reactions occurred simultaneously with the dark Fenton-like reaction, as described by eqn (7)–(9); these represented a typical photo-Fenton-like reaction process. Specifically, under LED light, the photo-generated hole (h+) directly reacted with RhB, and the photo-generated electron (e−) interacted with the dissolved oxygen in aqueous solution to form the O2˙− species. These species could remove RhB, so the degradation of RhB was enhanced under LED illumination. The results of these experiments clearly showed that this catalytic degradation process could proceed under both light and dark conditions. This is a particularly useful finding for this new non-metal catalytic system, which is capable of eliminating organic pollutants, thus reducing environmental contamination as much as possible. On the contrary, traditional carbon-based materials that promote Fenton-like reactions doesn't happen under light illumination, so the system developed herein clearly has an advantage in its ability to function in the dark.
| Catalyst + hv → h+ + e− | (7) |
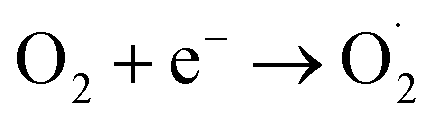 | (8) |
| h+ (or O2˙−) + organics → CO2 + H2O | (9) |
The stability of the O/g-C3N4 catalyst was investigated, and the results were presented in Fig. 6. Cyclic RhB degradation experiments were conducted under dark conditions. Specifically, after the reaction, the catalyst was carefully washed with deionized water and ethanol, respectively, and then dried. Afterward, it was employed for the next cycle following the addition of a fresh reaction solution containing HCO3− and H2O2, wherein the concentration of these components remained the same as in the first reaction. As shown in Fig. 6, the RhB degradation rate hardly decreased, even after several reaction cycles, thereby verifying the high stability of the system and repeatability of the process.
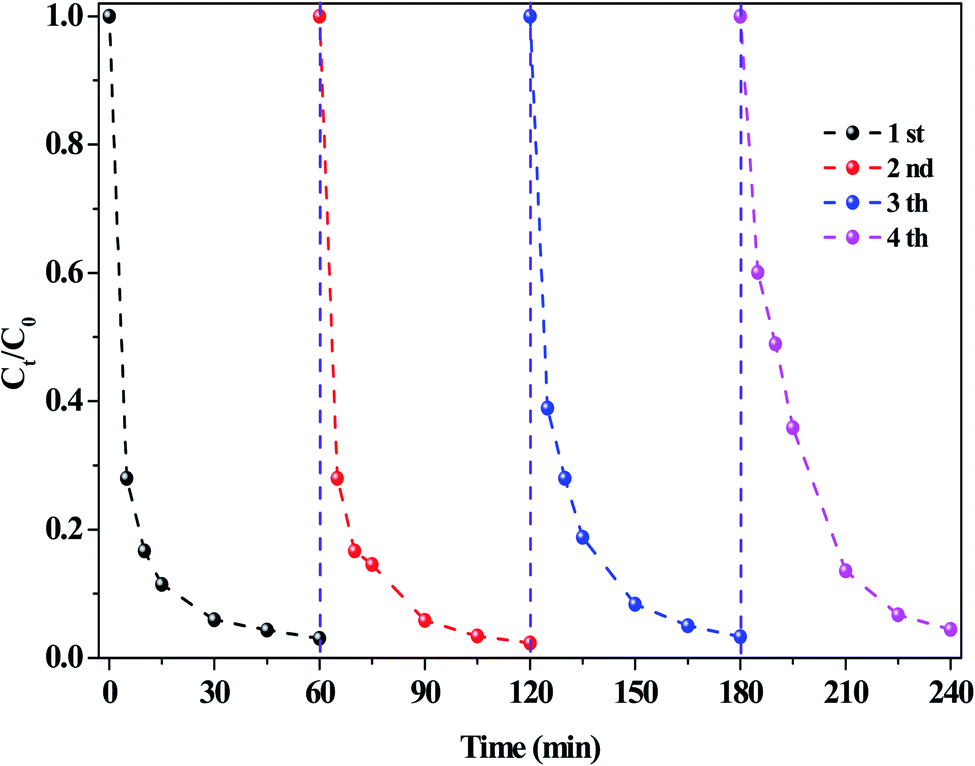 | ||
| Fig. 6 The stability of catalysts on the RhB degradation. Reaction conditions: [NaHCO3] = 10 mM, [H2O2] = 15 mM, [O/g-C3N4] = 0.8 g L−1, [RhB] = 10 mg L−1, 25 °C, pH ≈ 8.4 and 60 min. | ||
The degradation of organic dyes in wastewater was completed using the system containing HCO3− and H2O2, and the results were compiled in Table 2. Based on these reactions, the HCO3− and H2O2 consumptions were significant in the absence of catalyst. When Co(II) or Cu(II) was added into the reaction solution, significant degradation of the organic dyes was always observed. The main drawbacks associated with these catalysts included their cost, high toxicity, and difficult recovery. In order to find a suitable substitute for homogeneous catalysts, one current method involved using heterogeneous catalysts. For example, diatomite-supported cobalt,21 CoMgAl–Na–Y,22 and S/CoFe2O4 (ref. 23) have been applied for removing organic pollutants; however, these options also faced problems, such as their cost and complex preparation. Importantly, leaching of metal ions from these heterogeneous catalysts was inevitable and caused secondary contamination. These factors constituted the main challenges that must be overcome to allow their industrial application. Relative to such heterogeneous variants, the HCO3−–H2O2–O/g-C3N4 catalytic system was green, inexpensive, and easy to operate. These advantages confirmed that the novel technology presented herein represented high potential for practical applications in the removal of organic pollutants from wastewater.
| Catalysts | Organics | Reaction conditions | Degradation | Ref. |
|---|---|---|---|---|
| a A fixed-bed reactor.b LED illumination. | ||||
| No | MB, MO, RhB | C(H2O2) = 0.1 M, C(NaHCO3) = 0.5 M | ∼100%, 300 min | 5 |
| Co(II) | MB | C(Co(II)) = 20 μM, C(H2O2) = 20 mM, C(NaHCO3) = 25 mM | ∼100%, ∼50 min | 13 |
| AOII | C(Co(II)) = 5 μM, C(H2O2) = 4 mM, C(NaHCO3) = 10 mM | >90%, 10 min | 15 | |
| X-3B | >90%, 40 min | 16 | ||
| Cu(II) | AOII | C(Cu(II)) = 0.03 mM, C(H2O2) = 4 mM, C(NaHCO3) = 10 mM | ∼100%, 10 min | 19 |
| Diatomite-supported cobalt | MB | C(Catal.) = 0.4 g L−1, C(H2O2) = 60 mM, C(NaHCO3) = 25 mM | ∼99%, 80 min | 21 |
| RhB | 70%, 5 h | |||
| CoMgAl–Na–Y | MO | C(Catal.) = 0.6 g L−1, C(H2O2) = 50 mM, C(NaHCO3) = 25 mM | 100%, 10–60 min | 22 |
| MBa | F(Catal.) = 3 mL g−1 h−1, C(H2O2) = 25 mM, C(NaHCO3) = 48 mM | 100%, 0–132 h; 96–98%, 132–312 h | ||
| S/CoFe2O4 | AOII | C(Catal.) = 0.1 g L−1, C(H2O2) = 3 mM, C(NaHCO3) = 0.1 g L−1 | ∼99%, 15 min | 23 |
| O/g-C3N4 | RhB | C(Catal.) = 0.8 g L−1, C(H2O2) = 15 mM, C(NaHCO3) = 10 mM | ∼90%, 15 min | In this work |
| MB | >90%, 5 min | |||
| RhBb | C(Catal.) = 0.1 g L−1, C(H2O2) = 15 mM, C(NaHCO3) = 10 mM | >90%, 30 min | ||
In order to probe the production of O2˙− along the reaction pathway, the influence of different concentrations of the scavenger, Trion, on the RhB degradation was investigated. It is clear from Fig. S14† that the RhB degradation in the presence of 5 mM Trion decreased remarkably at first and then stabilized with increasing reaction time. This trend was also observed at higher concentrations (50 and 200 mM) of Trion. These results suggested that the O2˙− radical was an important ROS. Another radical scavenger, benzoquinone, was also studied in this reaction, and the results were shown in Fig. S15.† It is determined that the degradation of RhB decreased with the addition of 3 and 6 mM of benzoquinone, respectively. This confirmed that the O2˙− radical had an important role in the removal of organic pollutants in this system.
Fig. S16†shows the effect of different concentrations of NaN3 (used for singlet oxygen detection) on the RhB degradation. In this case, the change toward the RhB degradation was not significance at two different NaN3 concentrations (4 and 10 mM). Therefore, we determined that 1O2 was not a major ROS in this degradation mechanism. In order to support this conclusion, another experiment was carried out by adding furfuryl alcohol (FFA), rather than NaN3, into the reaction, and the results were shown in Fig. S17.† Under these conditions, the RhB degradation decreased only slightly relative to that of the control experiment. This fact confirmed that 1O2 was not a primary participant in this reaction, and it was consistent with the analysis of Fig. S6.† Overall, the mechanistic analysis indicated that ˙OH and O2˙− radicals were important ROS for this reactivity, but the 1O2 species was little involved.
| HCO3− + H2O2 ↔ HCO4− + H2O | (10) |
 | ||
| Fig. 7 Comparison results of different catalysts on the RhB degradation. Reaction conditions: [NaHCO3] = 10 mM, [H2O2] = 15 mM, [O/g-C3N4] = 0.8 g L−1, [RhB] = 10 mg L−1, 25 °C, pH ≈ 8.4 and 60 min. | ||
The activated pathway, comprised of HCO3− + H2O2 + O/g-C3N4, demonstrated an increased degradation efficiency (>90% after 60 min). As reported previously,37 the electrons near nitrogen defect sites could elongate the O–O bonds in H2O2 molecules, thereby reducing the activation energy. We have already demonstrated that the O/g-C3N4 catalyst contained many such defect sites, which could directly activate H2O2 in the dark (Fig. S5†). As a result, some ROS, including ˙OH radicals, were generated and served to enhance RhB degradation.34 Similarly, the addition of NaOH rather than NaHCO3 into the reaction solution generated a similar pH environment (∼8.4) and enhanced the degradation of RhB, although not as much as HCO3−. In addition, the ∼50% enhancement in degradation efficiency over O/g-C3N4 was obtained due to HCO3−, and this increase was mainly attributed to the adsorption aspect.
Overall, there are two main reasons for the enhancement exhibited by this system. The first involved the extremely high specific surface area of the O/g-C3N4 catalyst, which was beneficial for improving the removal of RhB via an adsorption process. In theory, the pKa of g-C3N4 obtained from melamine was about 3,38 meaning that it could easily combine with RhB through electrostatic interactions (RhB was a positively charged molecule, and the surface of g-C3N4 presented a negative charge). The zeta potential of the catalyst was determined at pH ≈ 8.4, and this value was about −19 mV. In general, the specific surface area of the catalyst was high, so its adsorption capacity for RhB was large; consequently, the enhanced degradation was attributed partially to the high specific surface area of the catalyst. The second reason for the enhanced activity of the developed catalytic system involved ROS production. In advanced oxidation technologies, ˙OH radicals are well-known catalytic active species for the degradation of various organic pollutants, with redox potentials in acidic and alkaline conditions of +2.8 V and +1.5 V, respectively.21 The NaOH or NaHCO3 environment provided a weak alkaline medium and also served to activate H2O2, thus inducing the formation of more ROS. However, some ROS were produced as a result of the synergistic behavior among NaHCO3, H2O2, and O/g-C3N4. As previously stated,21 the CO3˙− radicals were formed by the reaction shown as eqn (1), and these radicals may be even more effective for the degradation of organic dye molecules than ˙OH radicals. The HCO3− anion was often employed as scavenger to detect whether ˙OH radicals were presented in the reaction solution. However, it has been reported that HCO3− could accelerate the degradation of organic pollutants under certain reaction conditions, i.e., with specific catalysts or at specified concentrations.8,11–27 This is because the ˙OH radicals (i) had shorter lifetimes due to the potential self-combination process,32 and (ii) could react with H2O2 to produce O2.32 In contrast, CO3˙− radicals had longer lifetimes due to coulombic repulsion.21 In fact, it has been demonstrated that CO3˙− radicals could accelerate the degradation of organic pollutants,21 or generate other effective radicals (e.g., ˙OOH, O2˙− and 1O2) that were active for organic pollutant degradation under different conditions (eqn (2)–(6)).21
In this work, the ˙OH radicals were first generated by activating H2O2 at the defective sites on the O/g-C3N4 catalyst. Some ˙OH radicals left the surface of the catalyst and entered the reaction solution. As mentioned above, some of these ˙OH radicals immediately reacted with each other, thus contributing to their extremely short measured lifetimes. However, if the reaction solution contained an adequate concentration of HCO3− ions, the ˙OH radicals may react with HCO3− ions to form CO3˙− radicals. In this case, the RhB degradation continued, and the pollutant removal efficiency increased further. This phenomenon was evidenced by the relationship between HCO3− concentration and RhB degradation, as illustrated in Fig. 3. Based on eqn (1), the formation of CO3˙− radicals was mainly due to the reaction between HCO3− and ˙OH. Therefore, the increased concentrations of HCO3− theoretically led to increased quantities of CO3˙− radicals, thus improving RhB degradation. However, if the concentration of HCO3− is too high, more CO3˙− and related radicals were generated (eqn (2)–(6)), resulting in a remarkable reduction in the concentration of ˙OH radicals. These conditions led to a lesser degree of RhB degradation, and clearly, this phenomenon was not observed in the NaOH-containing system.
Overall, the optimal RhB degradation efficiency was obtained using the HCO3−–H2O2–O/g-C3N4 system. In this case, ROS, including ˙OH, O2˙−, 1O2, and CO3˙−, attacked the organic pollutants through a chemical process, leading to the destruction of the RhB structure. The structural change was further confirmed based on UV-vis results, as shown in Fig. S18.† It is worth noting that there was a large difference in the Na+ content in the NaOH and NaHCO3 systems (negligible (μM) vs. 10 mM, respectively), and its role was not thoroughly taken into consideration. To certify this assumption, preliminary experiments were conducted using Na–O/g-C3N4 as the catalyst, and the results were displayed in Fig. S19,† clearly showing that there was little influence on the removal of RhB.
In summary, the high degradation capability of the HCO3−–H2O2–O/g-C3N4 system was mainly ascribed to the synergistic effect between adsorption and chemical oxidation. The HCO3− and O/g-C3N4 components were responsible for activating H2O2 and removing RhB. Specifically, the O/g-C3N4 catalyst acted as a bifunctional material for dispelling RhB, thus contributing to the simultaneous adsorption and chemical oxidation processes. The huge specific surface area of the O/g-C3N4 catalyst allowed significant adsorption via a non-radical pathway, but the chemical oxidation was carried out through a radical route, which relied on the defective sites of O/g-C3N4 for activating H2O2 to ROS in the presence of HCO3−. Chemical oxidation also occurred in the reaction solution itself, and this mechanistic pathway was outlined in Scheme 1.
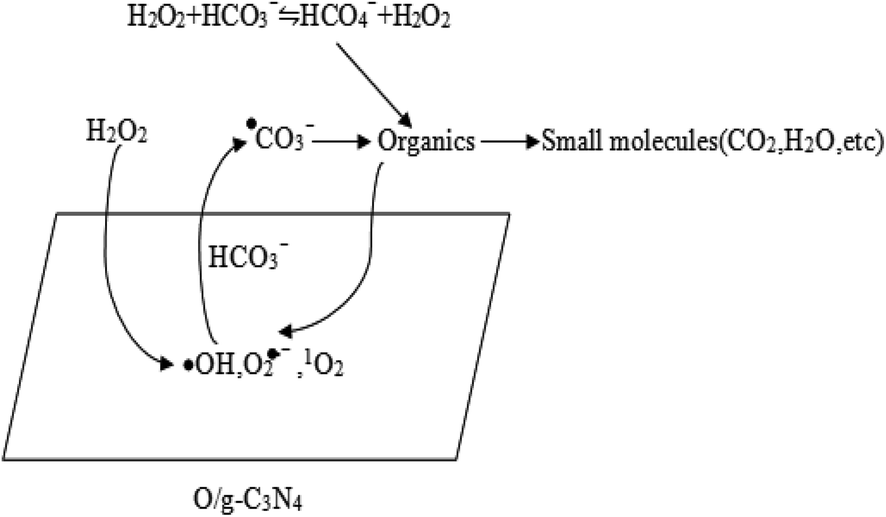 | ||
| Scheme 1 The degradation pathway of organic pollutants. | ||
4. Conclusions
This report discussed a newly-developed strategy for treating wastewater polluted with organic contaminants. The employed system consisted of HCO3−, H2O2, and an O/g-C3N4 catalyst, which together demonstrated efficient degradation of organic compounds. Various influencing factors were investigated, and the reaction conditions were optimized in terms of the catalyst loading and preparation, concentrations of HCO3− and H2O2, and light vs. dark operation. The catalytic system retained high stability and exhibited sufficient reproducibility after running several reaction cycles. Mechanistic probing revealed that the degradation pathway was dominated by the synergistic effect between adsorption and chemical oxidation processes. The adsorption was closely related to the tunable specific surface area of the catalyst, and the chemical oxidation was achieved by various ROS. Overall, this work presented a novel, widely-applicable method for removing organic pollutants from wastewater.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 51978648), Open Project of State Key Laboratory of Safety and Health for Metal Mines (No. 2019-JSKSSYS-04), Hunan Environmental Protection Scientific Research (No. Hunan Finance and construction (2019)0011), Open Project of Program of Provincial Key Laboratory of Clean Energy Material, LongYan University (No. QJNY-201802), China Postdoctoral Science Foundation (No. 2019M660824) and Hunan Provincial Natural Science Foundation of China (No. 2018JJ3427).References
- J. Ali, Z. Chen and G. Yin, Bicarbonate activation of hydrogen peroxide: A new emerging technology for wastewater treatment, Chin. J. Catal., 2016, 37, 810–825 CrossRef.
- H. Yao and D. E. Richardson, Epoxidation of alkenes with bicarbonate-activated hydrogen peroxide, J. Am. Chem. Soc., 2000, 122, 3220–3221 CrossRef CAS.
- D. E. Richardson, H. Yao, K. M. Frank and A. Deon, Bennett Equilibria, kinetics, and mechanism in the bicarbonate activation of hydrogen peroxide:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) oxidation of sulfides by peroxymonocarbonate, J. Am. Chem. Soc., 2000, 122, 1729–1739 CrossRef CAS.
oxidation of sulfides by peroxymonocarbonate, J. Am. Chem. Soc., 2000, 122, 1729–1739 CrossRef CAS. - H. Yao and D. E. Richardson, Bicarbonate surfoxidants: micellar oxidations of aryl sulfides with bicarbonate-activated hydrogen peroxide, J. Am. Chem. Soc., 2003, 125, 6211–6221 CrossRef CAS.
- A. Xu, X. Li, H. Xiong and G. Yin, Efficient degradation of organic pollutants in aqueous solution with bicarbonate-activated hydrogen peroxide, Chemosphere, 2011, 82, 1190–1195 CrossRef CAS.
- J. Wu, H. Xiao, T. Wang, T. Hong, B. Fu, D. Bai, Z. He, S. Peng, X. Xing, J. Hu, Pu Guo and X. Zhou, N6-Hydroperoxymethyladenosine: a new intermediate of chemical oxidation of N6-methyladenosine mediated by bicarbonate-activated hydrogen peroxide, Chem. Sci., 2015, 6, 3013–3017 RSC.
- M. Puiu, T. Galaon, L. Bondilǎ, A. Rǎducan and O. Dumitru, Feed-back action of nitrite in the oxidation of nitrophenols by bicarbonate-activated peroxide system, Appl. Catal., A, 2016, 516, 90–99 CrossRef CAS.
- D. Alok, Bokare, Wonyong Choi. Bicarbonate-induced activation of H2O2 for metal-free oxidative desulfurization, J. Hazard. Mater., 2016, 304, 313–319 CrossRef.
- H. Fakhraian and F. Valizadeh, Activation of hydrogen peroxide via bicarbonate, sulfate, phosphate and urea in the oxidation of methyl phenyl sulfide, J. Mol. Catal. A: Chem., 2010, 333, 69–72 CrossRef CAS.
- S. Zhao, H. Xi, Y. Zuo, Qi Wang, Z. Wang and Z. Yan, Bicarbonate-activated hydrogen peroxide and efficient decontamination of toxic sulfur mustard and nerve gas simulants, J. Hazard. Mater., 2018, 344, 136–145 CrossRef CAS.
- H. Kan, S. Heng, Z. Yang, R. Wu, J. Shen, G. Qu and T. Wang, Purification of dye wastewater using bicarbonate activated hydrogen peroxide: reaction process and mechanisms, Sep. Purif. Technol., 2020, 232, 115974 CrossRef CAS.
- S.-X. Liang, Li-X. Zhao, Bo-T. Zhang and J.-M. Lin, Experimental studies on the Chemiluminescence reaction mechanism of carbonate/bicarbonate and hydrogen peroxide in the presence of cobalt (II), J. Phys. Chem. A, 2008, 112, 618–623 CrossRef CAS.
- A. Xu, X. Li, S. Ye, G. Yin and Q. Zeng, Catalyzed oxidative degradation of methylene blue by in situ generated cobalt (II)-bicarbonate complexes with hydrogen peroxide, Appl. Catal., B, 2011, 102, 37–43 CrossRef CAS.
- X. Li, Z. Xiong, X. Ruan, D. Xia, Q. Zeng and A. Xu, Kinetics and mechanism of organic pollutants degradation with cobalt-bicarbonate-hydrogen peroxide system: Investigation of the role of substrates, Appl. Catal., A, 2012, 411–412, 24–30 CAS.
- X. Long, Z. Yang, H. Wang, M. Chen, K. Peng, Q. Zeng and A. Xu, Selective degradation of orange II with the cobalt(II)-bicarbonate-hydrogen peroxide system, Ind. Eng. Chem. Res., 2012, 51, 11998–12003 CrossRef CAS.
- Z. Yang, H. Wang, M. Chen, M. Luo, D. Xia, A. Xu and Q. Zeng, Fast Degradation and biodegradability improvement of reactive brilliant red X-3B by the cobalt(II)/bicarbonate/hydrogen peroxide system, Ind. Eng. Chem. Res., 2012, 51, 11104–11111 CrossRef CAS.
- M. Luo, L. Lv, G. Deng, Y. Wen, R. Yang, X. Li and A. Xu, The mechanism of bound hydroxyl radical formation and degradation pathway of acid orange II in Fenton-like Co2+-HCO3− system, Appl. Catal., A, 2014, 469, 198–205 CrossRef CAS.
- I. F. Macías-Quiroga, E. F. Rojas-Méndez, G. I. Giraldo- Gómez and N. R. Sanabria-González, Experimental data of a catalytic decolorization of Ponceau 4R dye using the cobalt(II)/NaHCO3/H2O2 system in aqueous solution, Data Brief, 2020, 30, 105463 CrossRef.
- L. Cheng, M. Wei, L. Huang, F. Pan, D. Xia, X. Li and A. Xu, Efficient H2O2 oxidation of organic dyes catalyzed by simple copper(II) ions in bicarbonate aqueous solution, Ind. Eng. Chem. Res., 2014, 53, 3478–3485 CrossRef CAS.
- J. Peng, H. Shi, J. Li, L. Wang, Z. Wang and S. Gao, Bicarbonate enhanced removal of triclosan by copper(II) catalyzed Fenton-like reaction in aqueous solution, Chem. Eng. J., 2016, 306, 484–491 CrossRef CAS.
- Li Zhou, W. Song, Z. Chen and G. Yin, Degradation of organic pollutants in wastewater by bicarbonate-activated hydrogen peroxide with a supported cobalt catalyst, Environ. Sci. Technol., 2013, 47, 3833–3839 CrossRef CAS.
- J. Ali, Y. Li, X. Lu, Z. Chen, W. Liu and G. Yin, Controlled leaching with prolonged activity for Co-LDH supported catalyst during treatment of organic dyes using bicarbonate activation of hydrogen peroxide, J. Hazard. Mater., 2015, 289, 165–173 CrossRef.
- X. Guo, H. Li and S. Zhao, Fast degradation of acid orange II by bicarbonate-activated hydrogen peroxide with a magnetic S-modified CoFe2O4 catalyst, J. Taiwan Inst. Chem. Eng., 2015, 55, 90–100 CrossRef CAS.
- L. Duan, Y. Chen, K. Zhang, H. Luo, J. Huang and A. Xu, Catalytic degradation of acid orange 7 with hydrogen peroxide using CoxOy-N/GAC catalysts in a bicarbonate aqueous solution, RSC Adv., 2015, 5, 84303–84310 RSC.
- P. Liu, N. Yang, W. Han, W. Xiao, D. Wang, Y. Xiong, M. Zhou, H. Hou and X. Mao, Heterogeneous activation of peroxymonocarbonate by Co-Mn oxides for the efficient degradation of chlorophenols in the presence of a naturally occurring level of bicarbonate, Chem. Eng. J., 2018, 334, 1297–1308 CrossRef.
- Y. Li, J. Ali, A. Khan, X. Lu, Z. Chen, W. Liu and G. Yin, Synergistic degradation of phenols by bimetallic CuO-Co3O4@γ-Al2O3 catalyst in H2O2/HCO3− system, Chin. J. Catal., 2016, 37, 963–970 CrossRef CAS.
- J. Ali, Y. Li, L. Guo, A. Khan, Z. Chen, J. Wang, J. Yang, W. Liu and G. Yin, Bimetallic synergistic degradation of chlorophenols by CuCoOx-LDH catalyst in bicarbonate-activated hydrogen peroxide system, RSC Adv., 2016, 6, 72643–72653 RSC.
- L. Yang, C. Chuh-Shun, Y.-J. Tu, Y.-H. Huang and H. Zhang, Heterogeneous degradation of organic pollutants by persulfate activated by CuO-Fe3O4: mechanism, stability, and effects of pH and bicarbonate ions, Environ. Sci. Technol., 2015, 49, 6838–6845 CrossRef.
- C. Pétrier, R. Torres-Palma, E. Combet, G. Sarantakos, S. Baup and C. Pulgarin, Enhanced sonochemical degradation of bisphenol-A by bicarbonate ions, Ultrason. Sonochem., 2010, 17, 111–115 CrossRef.
- W. Hu, S. Chen, D. Wu, J. Zheng and X. Ye, Ultrasonic-assisted citrus pectin modification in the bicarbonate- activated hydrogen peroxide system: Chemical and microstructural analysis, Ultrason. Sonochem., 2019, 58, 104576 CrossRef CAS.
- D. Wu, S. Hu, H. Xue, X. Hou, H. Du, G. Xu and Y. Yuan, Protonation and microwave-assisted heating induced excitation of lone-pair electrons in graphitic carbon nitride for increased photocatalytic hydrogen generation, J. Mater. Chem. A, 2019, 7, 20223–20228 RSC.
- Y. Cui, Z. Ding, P. Liu, M. Antonietti, X. Fu and X. Wang, Metal-free activation of H2O2 by g-C3N4 under visible light irradiation for the degradation of organic pollutants, Phys. Chem. Chem. Phys., 2012, 14, 1455–1462 RSC.
- Hu, D. Jiang, L. Gu, G. Xu, Z. Li and Y. Yuan, Awakening n → π* electronic transition by breaking hydrogen bonds in graphitic carbon nitride for increased photocatalytic hydrogen generation, Chem. Eng. J., 2020, 399, 125847 CrossRef CAS.
- T.-J. Jiang, C.-W. Luo, C. Xie, Y.-H. Wei and Li An, Synthesis of oxygen-doped graphitic carbon nitride and its application for the degradation of organic pollutants via dark Fenton-like reactions, RSC Adv., 2020, 10, 32906–32918 RSC.
- Y. Liu, X. He, X. Duan, Y. Fu, D. Fatta-Kassinos and D. D. Dionysios, Significant role of UV and carbonate radical on the degradation of oxytetracycline in UV-AOPs: Kinetics and mechanism, Water Res., 2016, 95, 195–204 CrossRef CAS.
- Y. Huang, M. Kong, D. Westerman, E. G. Xu, C. Scott, K. H. Cochran, Y. Liu, S. D. Richardson, D. Schlenk and D. D. Dionysios, Effects of HCO3− on degradation of toxic contaminants of emerging concern by UV/NO3−, Environ. Sci. Technol., 2018, 52, 12697–12707 CrossRef CAS.
- S. Zhang, Y. Liu, P. Gu, R. Ma, T. Wen, G. Zhao, L. Li, Y. Ai, C. Hu and X. Wang, Enhanced photodegradation of toxic organic pollutants using dual-oxygen doped porous g-C3N4: Mechanism exploration from both experimental and DFT studies, Appl. Catal., B, 2019, 248, 1–10 CrossRef CAS.
- W. Fang, J. Liu, Yu Lei, Z. Jiang and W. Shangguan, Novel (Na, O) co-doped g-C3N4 with simultaneously enhanced absorption and narrowed bandgap for highly efficient hydrogen evolution, Appl. Catal., B, 2017, 209, 631–636 CrossRef CAS.
- Ai-Y. Zhang, L. Tan, Y.-Y. He and Y.-X. Mou, Heterogeneous activation of H2O2 by defect-engineered TiO2−x single crystals for refractory pollutants degradation: A Fenton-like mechanism, J. Hazard. Mater., 2016, 3115, 81–90 CrossRef.
- N. Zhang, E. P. Tsang, J. Chen, Z. Fang and D. Zhao, Critical role of oxygen vacancies in heterogeneous Fenton oxidation over ceria-based catalysts, J. Colloid Interface Sci., 2020, 558, 163–172 CrossRef.
- P. Gao, X. Chen, H. Mengjie, F. Xiao and S. Yang, Oxygen vacancy enhancing the Fe2O3-CeO2 catalysts in Fenton-like reaction for the sulfamerazine degradation under O2 atmosphere, Chemosphere, 2019, 228, 521–527 CrossRef CAS.
- Q. Xia, Z. Yao, D. Zhang, D. Li, Z. Zhang and Z. Jiang, Rational synthesis of micronano dendritic ZVI@Fe3O4 modified with carbon quantum dots and oxygen vacancies for accelerating Fenton-like oxidation, Sci. Total Environ., 2019, 671, 1056–1065 CrossRef CAS.
- Xi Xiao, T. T. Hu, Bo Meng, Y. Sun and Yi-F. Han, Catalytic degradation of anthraquinones-containing H2O2 production effluent over layered Co-Cu hydroxides: defects facilitating hydroxyl radicals generation, Appl. Catal., B, 2020, 260, 118157 CrossRef.
- S. Song, H. Yang, R. Rao, H. Liu and A. Zhang, Defects of multi-walled carbon nanotubes as active sites for benzene hydroxylation to phenol in the presence of H2O2, Catal. Commun., 2010, 11, 783–787 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07893j |
| ‡ They contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |