
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntrinsically flame-retardant polyamide 66 with high flame retardancy and mechanical properties
Jingnan Zhang†
 a,
Siming Lian†b,
Yifan Hea,
Xinyu Caoa,
Jiaming Shanga,
Qingyun Liuc,
Gang Yea,
Kun Zheng*a and
Yongmei Ma*ad
a,
Siming Lian†b,
Yifan Hea,
Xinyu Caoa,
Jiaming Shanga,
Qingyun Liuc,
Gang Yea,
Kun Zheng*a and
Yongmei Ma*ad
aInstitute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: maym@iccas.ac.cn; zhengkunwd@iccas.ac.cn
bZhuhai Green Electric Co., Ltd, Guangdong 519070, China
cShandong University of Science and Technology, Shandong 266590, China
dBeijing National Laboratory for Molecular Sciences (BNLMS), Beijing, 100190, China
First published on 23rd December 2020
Abstract
The key factor in the synthesis of intrinsic flame retardant polymers is the thermal stability and reactivity of phosphorus-based flame retardants. However, it is difficult to realize both thermal stability and high reactivity by using one phosphorus-based flame retardant. Herein, we proposed a strategy to improve the thermal stability of highly reactive flame-retardant, 4-(2-(((2-carboxyethyl)(phenyl)phosphoryl)oxy)ethoxy)-4-oxohexanoic acid (CPPOA), by reacting it with 1,6-diaminohexane to obtain CPPOA salt, which then was copolymerized with PA66 salt to obtain intrinsic flame-retardant polyamide 66 (FRPA66). The thermal stability of CPPOA was significantly improved. The LOI and vertical combustion grade of FRPA66 with 6 wt% CPPOA reached 27.2% and V-0 rating, respectively. Furthermore, the tensile strength and impact strength of the FRPA66 reached 70 MPa and 5.6 kJ m−2, respectively. Our work presents an efficient approach to synthesize polymers having high flame retardancy and good mechanical properties, showing high potential for real applications.
Introduction
Polyamide 66 (PA66) is an important engineering-plastic that possesses excellent thermal and mechanical properties, strong chemical and electrical resistance, and durability against fatigue and abrasion. Hence, it is widely used in engineering applications, such as transportation, aerospace, the army, and so on.1–4 However, flammability of PA66 severely limits its application in fields where flame-retardancy is required. Thus, the improvement of flame retardancy of PA66 is extremely important.The most effective method to improve the flame retardancy of PA66 is to incorporate a flame retardant material into the PA66 matrix. Halogen-based compounds and the phosphorus-based compounds are two main types of flame retardants generally used. Although halogen-based flame retardants have high efficiency and low cost, they release toxins, carcinogenic dioxins, and furans and obscure the smoke emitted during combustion. This causes environmental pollution and is also a health risk to humans. Hence, environmentally friendly compound, viz. phosphonate ester, has been extensively studied.5,6
Generally, a flame retardant PA66 (FRPA66), containing phosphonate group, can be prepared by blending or copolymerization.7–13 Compared with blending, copolymerization can impart PA66 with intrinsic flame-retardancy and enhanced durability. However, flame-retardant monomers with high thermal stability are required to prevent decomposition under the harsh polymerization conditions of high temperature,14 since the phosphonate structure is susceptible to decomposition at high temperature.15 Presently, the thermal stability of phosphonate structure is generally enhanced by linking it to large sterically hindered groups.16,17 For instance, the thermal stability of phosphonate ester can be significantly improved by its linking with multiple benzene rings, such as 9,10-dihydro-10-[2,3-di(hydroxycarbonyl)propyl]-10-phosphaphenanthrene-10-oxide (DDP), bis(4-carboxyphenyl)phenyl phosphine oxide (BCPPO), and bis(4-carboxyanilino)phenyl phosphine oxide (BCNPO).18–24 However, the bulky structure makes it difficult to copolymerize with PA66 salt, which results in decreased content of retardant in the copolymers. Recently, 2-carboxyethyl phenyl phosphinic acid (CEPPA), having only one benzene ring, was introduced into PA66 to enhance its flame-retardancy.7 However, the terminal groups in these compounds are carboxylic acids and phosphoric acid, respectively, and have different reactivities, which makes it difficult to control the extent of reaction during the copolymerization process. Therefore, there is great demand for a flame retardant with high thermal stability and high reactivity.
The compound, 4-(2-(((2-carboxyethyl)(phenyl)phosphoryl)oxy)ethoxy)-4-oxohexanoic acid (CPPOA) (Fig. 1), belongs to a new class of phosphonate flame-retardants, which has high reactivity and good flame retardancy.25 However, CPPOA has a relatively low thermal stability and decomposes during high temperature polymerization.25 If thermal stability of CPPOA can be improved, it would become a highly efficient flame retardant.
 | ||
| Fig. 1 Chemical formula of CPPOA. | ||
In this study, the thermal stability of CPPOA was improved by reacting it with 1,6-diaminohexane to obtain CPPOA salt, making it suitable for melt polymerization with PA66. Further, the flame retardant PA66 (FRPA66) was prepared by copolymerization of PA66 salt with CPPOA salt. The thermal stability, flame retardancy, combustion behavior, mechanical properties, material structure, and crystallization behavior of FRPA66 were investigated.
Experimental section
Materials
1,6-Diaminohexane and m-cresol were purchased from Sinopharm Chemical Reagent Co. Ltd. Hypophosphorous acid was obtained from Beijing Hengye Zhongyuan Chemical Co. Ltd. 4-(2-(((2-Carboxyethyl)(phenyl)phosphoryl)oxy)ethoxy)-4-oxohexanoic acid (CPPOA) was supplied by Chengdu Tuoli Chemical Industry Co. Ltd. Polyamide 66 salt solution (50 wt%) was purchased from Pingdingshan Shenma Engineering Plastics Co. Ltd. All of them were of commercial grade and were used as received without further purification.Sample preparation
The synthesis of FRPA66 was conducted in two steps: synthesis of CPPOA salt and synthesis of FRPA66, as shown in Fig. 2.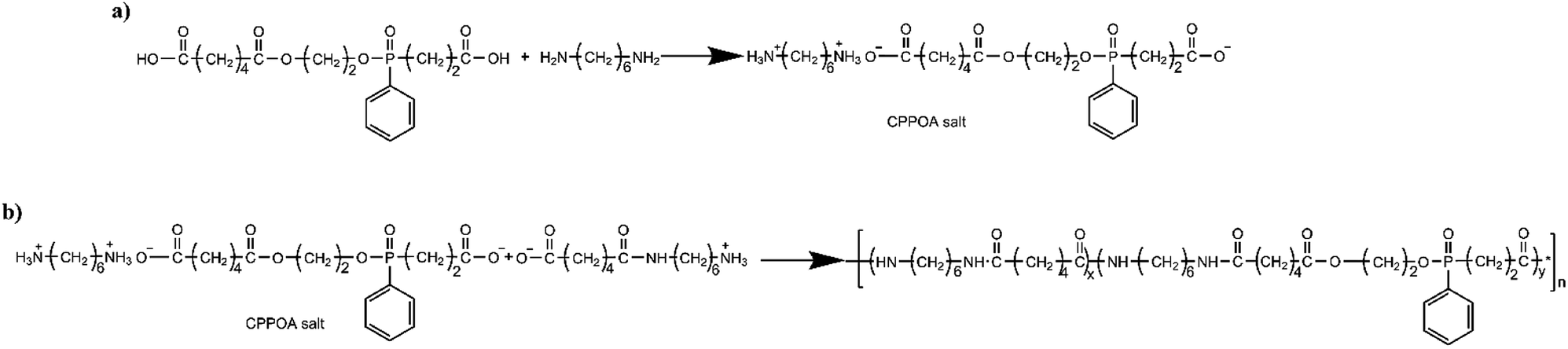 | ||
| Fig. 2 Route for synthesis of (a) CPPOA salt and (b) FRPA66. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio were added with appropriate amount of deionized water in a three-necked bottle and stirred mechanically at 60 °C for one hour under nitrogen atmosphere. The pH of the CPPOA salt solution was adjusted to 7–8 with 1,6-diaminohexane.
:1 molar ratio were added with appropriate amount of deionized water in a three-necked bottle and stirred mechanically at 60 °C for one hour under nitrogen atmosphere. The pH of the CPPOA salt solution was adjusted to 7–8 with 1,6-diaminohexane.The obtained product was first dried at 60 °C by air blowing for 3 hours and then at 90 °C under vacuum for 4 hours. The sample for testing was obtained by injection molding. The injection molding was carried out at 260 °C and 70 MPa pressure.
Characterization
Results and discussion
Chemical structure and thermal stability of CPPOA salt
The chemical structure of CPPOA salt was investigated by FTIR and 31P-NMR spectral analyses. The FTIR spectra of CPPOA, HMD, and CPPOA salt are shown in Fig. 3(a). In the spectrum of pure CPPOA, peak at 1734 cm−1 was assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations. Moreover, peaks at 1235 cm−1 and 1138 cm−1 corresponded to PO stretching vibrations, whereas peaks at 1059 cm−1 and 969 cm−1 corresponded to characteristic P–O vibrations.26 After salt formation, the CO peak shifted to lower wavenumbers (red-shift), from 1734 cm−1 in CPPOA to 1726 cm−1 in CPPOA salt, due to the electron-withdrawing inductive effect of amino group. Similarly, peaks of PO and P–O also shifted from 1235 cm−1 to 1128 cm−1 and 1059 cm−1 to 1034 cm−1. The peak for vibrations of N–H bond also shifted from 3339 cm−1 in HMD to 3410 cm−1 in CPPOA salt. The FTIR results confirmed the reaction between CPPOA and HMD to form CPPOA salt. The interactions of groups in CPPOA salt were further verified from the 31P-NMR spectra of CPPOA and CPPOA. In Fig. 3(b), spectrum of CPPOA showed peaks at 44.1 ppm and 44.4 ppm. However, the peaks in CPPOA salt shifted high field to 35.7 ppm and 34.9 ppm, respectively. This indicated a chemical reaction between CPPOA and HMD, since the chemical environmental changes of P caused peak shifting. This was based on the electron cloud density around the core and the degree of spherical asymmetry that affected the peak positions in the phosphorus spectrum. To sum up, combination of FTIR and 31P-NMR results confirmed the reaction of CPPOA with HMD to form CPPOA salt, having the formula shown in Fig. 2(a).
O stretching vibrations. Moreover, peaks at 1235 cm−1 and 1138 cm−1 corresponded to PO stretching vibrations, whereas peaks at 1059 cm−1 and 969 cm−1 corresponded to characteristic P–O vibrations.26 After salt formation, the CO peak shifted to lower wavenumbers (red-shift), from 1734 cm−1 in CPPOA to 1726 cm−1 in CPPOA salt, due to the electron-withdrawing inductive effect of amino group. Similarly, peaks of PO and P–O also shifted from 1235 cm−1 to 1128 cm−1 and 1059 cm−1 to 1034 cm−1. The peak for vibrations of N–H bond also shifted from 3339 cm−1 in HMD to 3410 cm−1 in CPPOA salt. The FTIR results confirmed the reaction between CPPOA and HMD to form CPPOA salt. The interactions of groups in CPPOA salt were further verified from the 31P-NMR spectra of CPPOA and CPPOA. In Fig. 3(b), spectrum of CPPOA showed peaks at 44.1 ppm and 44.4 ppm. However, the peaks in CPPOA salt shifted high field to 35.7 ppm and 34.9 ppm, respectively. This indicated a chemical reaction between CPPOA and HMD, since the chemical environmental changes of P caused peak shifting. This was based on the electron cloud density around the core and the degree of spherical asymmetry that affected the peak positions in the phosphorus spectrum. To sum up, combination of FTIR and 31P-NMR results confirmed the reaction of CPPOA with HMD to form CPPOA salt, having the formula shown in Fig. 2(a).
 | ||
| Fig. 3 Structural characterization and thermal analysis of flame-retardants: (a) FTIR spectra of CPPOA, HMD, and CPPOA salt; (b) 31P-NMR spectra of CPPOA and CPPOA salt; (c) TGA and (d) DTG curves of CPPOA and CPPOA salt. | ||
Thermal stability of CPPOA salt was very crucial for conducting melt polymerization at high temperature. The TGA and DTG curves of CPPOA and CPPOA salts are shown in Fig. 3(c and d). At about 200 °C, the CPPOA salt underwent significant decomposition, due to the sublimation of excess of 1,6-diaminohexane. After 282.6 °C, the decomposition rate of CPPOA salt was slower than that of CPPOA, and a decomposition platform appeared. The residual char of the CPPOA salt (at 600 °C) increased from 2.0 wt% to 8.3 wt%. The DTG curves of both CPPOA and CPPOA salt showed two thermal degradation processes. Compared with pure CPPOA, the maximum rates of thermal decomposition of CPPOA salt were delayed by over 40 °C, from 334.8 °C and 394.1 °C to 376.9 °C and 427.7 °C, respectively. The onset decomposition temperature of pure CPPOA was about 185 °C, while that of CPPOA salt was about 300 °C, which was satisfactory for the polymerization reaction (270–280 °C) of FRPA66. In conclusion, the thermal stability of CPPOA was significantly improved by formation of salt.
Chemical structure of FRPA66
In Fig. 4(a), the FTIR spectra of PA66 and FRPA66 showed a peak at 1630 cm−1 due to CO stretching vibrations of the amide bond (–CONH–).27 Meanwhile, the peak at 3295 cm−1 was assigned to N–H stretching and bending vibrations.23 The other peaks included 2927–2931 cm−1 (stretching vibrations of methyl group, CH3–), 2857–2860 cm−1 (stretching vibrations of methylene, CH2–), and 1533–1535 cm−1 (deformation vibration peak of N–H).27 In addition to the characteristic peaks of pure PA66, FRPA66 showed several new peaks between 1000–1500 cm−1. As shown in Fig. 4(b), the absorption peaks at 1015 cm−1 and 1033 cm−1 corresponded to the P–O bond in the phosphonate unit. Besides, peaks at 1178 cm−1 and 1189 cm−1 were assigned to PO stretching vibrations, whereas peaks at 1436 cm−1 and 1418 cm−1 were attributed to the benzene ring. The FTIR results suggested that CPPOA was successfully introduced into PA66 chain.
 | ||
| Fig. 4 (a) FTIR spectra of PA66 and FRPA66. (b) Enlarged view of 900–1500 cm−1 spectral range. (c) 31P-NMR spectra of CPPOA salt, blend-PA66, and FRPA66. (d) Relative viscosities of PA66 and FRPA66. (e) Number average molecular weight (Mn) of PA66 and FRPA66. | ||
31P-NMR spectroscopy was employed to gain a better insight into the reaction between CPPOA and PA66. In Fig. 4(c), the 31P-NMR spectrum of FRPA66 was totally different from those of blend-PA66 and CPPOA salt. This was possibly due to the change in chemical environment of the phosphorus element in the phosphonate unit, as a result of copolymerization reaction. Thus, 31P-NMR once again proved the reaction between CPPOA and PA66 salt.
The relative viscosities of PA66 and FRPA66 are shown in Fig. 4(d). As the content of flame-retardant increased, its relative viscosity decreased, which could be attributed to the incorporation of CPPOA. The phenyl groups of CPPOA were bulky, which partially hindered polymerization,24,28 and hence the viscosity decreased.
The number average molecular weights (Mn) of PA66 and FRPA66 are shown in Fig. 4(e). As the content of flame-retardant increased, the Mn decreased. The addition of CPPOA decreased the degree of polymerization of FRPA66. This result is consistent with the viscosity data.
All the results of structural characterization confirmed the successful copolymerization of CPPOA into PA66 chains.
Crystallization and melting behaviors of FRPA66
The DSC curves of pure PA66 and FRPA66 are shown in Fig. 5 and the results are presented in Table 1. With increase in CPPOA content, both the melting peak and the crystallization peak of FRPA66 shifted to lower temperature, and gradually broadened. Compared with pure PA66, the melting temperature (Tm) and crystallization temperature (Tc) of FRPA66 with 8 wt% CPPOA decreased by 9 °C and 11.5 °C, respectively. This occurred due to the introduction of CPPOA monomer, which reduced the structural symmetry and regularity of the PA66 molecular chains. This further hindered the growth of the macromolecular folded chains into large crystals, leading to a lower melting point.29 The decrease of the melting temperature and crystallization temperature also indirectly indicated that the phosphonate unit was well copolymerized with the PA66 chain.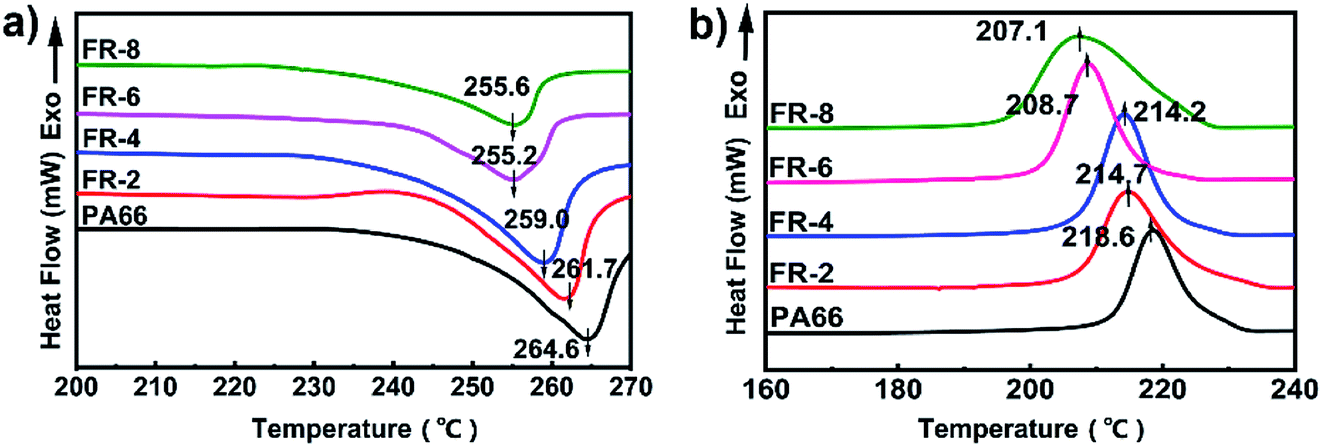 | ||
| Fig. 5 DSC curves of PA66 and FRPA66 (a) heating (b) cooling. | ||
| Sample | Tm/°C | Tc/°C | ΔHm/J g−1 | Xc/% |
|---|---|---|---|---|
| PA66 | 264.6 | 218.6 | 65.7 | 34.6 |
| FRPA66-2 | 261.7 | 214.7 | 58.3 | 30.5 |
| FRPA66-4 | 259.0 | 214.2 | 57.3 | 30.2 |
| FRPA66-6 | 255.2 | 208.7 | 56.3 | 29.6 |
| FRPA66-8 | 255.6 | 207.1 | 54.6 | 28.7 |
In Fig. 6, pure PA66 showed only α crystals. With increase in loading of CPPOA, the diffraction peaks of the two crystal planes that were characteristic of FRPA66 shifted towards a lower angle. The relative intensity of the α1 peak gradually decreased and eventually reached the same height as that of α2 peak. This was probably due to introduction of the phosphonate ester monomer into the PA66 chain and changes in the hydrogen bond structure between the molecular chains, which hindered the growth of α1 surface. However, the crystal growth at the α2 plane was not affected, since there was no change in the van der Waals forces.30
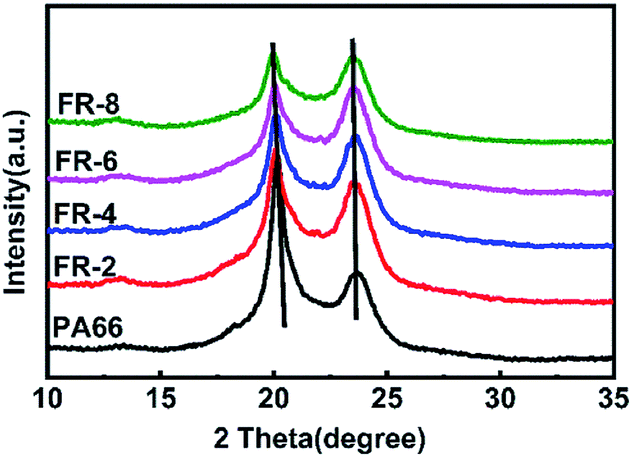 | ||
| Fig. 6 XRD patterns of PA66 and FRPA66. | ||
The above results suggested that CPPOA had negative effect on crystallization, which is consistent with other previous reports.11
Thermal stability of FRPA66
The thermal stabilities of FRPA66 copolymers were evaluated by TGA. T5% and T80% were the temperatures corresponding to 5 wt% loss and 80 wt% loss respectively. T5% was also the initial decomposition temperature, which indicated the thermal stability. From Fig. 7(a and b), it was evident that FRPA66 had lower T5% than that of pure PA66. This could be mainly attributed to the early decomposition of the phosphorus-containing moieties that produced compounds such as phosphoric acid and polyphosphoric acid. These substances formed a viscous molten layer and protected the polymeric matrix from burning or oxidation.31 The initial decomposition temperature of FRPA66 was still above 370 °C, suggesting that FRPA66 had relatively good thermal stability. | ||
| Fig. 7 (a) TGA-DTG curves of PA66 and FRPA66; (b) plots for T5% and Tend values of FRPA66 and FRPA66; (c) the relationship between residual char content and phosphorus content; IR spectra of (d) pure PA66 at different pyrolysis temperatures; (e) 6 wt% FRPA66 at different pyrolysis temperatures. | ||
The T80% values were in order of PA66 (469.25 °C) < FR-2 (476.02 °C) < FR-6 (478.98 °C) < FR-8 (480.23 °C) < FR-4 (482.02 °C). This suggested that the viscous molten layer that remained in the condensed phase could improve the thermal stability of the residue at high temperatures. From Fig. 7(c), with increase in CPPOA content, the residual carbon ratio of the polymer increased. The char residue of FRPA66 copolymer with 0.73 wt% of phosphorus was 3.2% at 600 °C, while that of pure PA66 was 1.9%. These results suggested that due to the carbonization of copolymer and increase in phosphorus content, the durability of the copolymer against thermal oxidation at high temperatures was improved, which was favorable for flame retardancy.
TG-IR analysis was used to investigate the changes in chemical structure with temperature, as shown in Fig. 7(d and e), for pure PA66 and FRPA66, respectively. The characteristic absorption peaks at 2936 cm−1 and 2870 cm−1 were ascribed to the C–H bond. For pure PA66, the characteristic absorption peaks for CO and N–H appeared at 1762 cm−1 and 3332 cm−1, respectively.23 Since these peaks disappeared, the maximum decomposition temperature was between 440 °C and 470 °C. This was consistent with results with the thermal degradation. For FRPA66, the characteristic absorption peaks of P–H, CO, and N–H appeared at 2314 cm−1, 1762 cm−1, and 3332 cm−1, respectively, between 380 °C and 410 °C.23 Above this temperature range, the aforementioned peaks disappeared. This was ascribed to the thermal decomposition of phosphorus-based flame-retardant, which produced polyphosphoric acid and PH3 gas and promoted carbon formation. The decomposition temperature of FRPA66 was about 410 °C, which was also consistent with the TGA results and it suggested a relatively high thermal stability.
Flame retardancy of FRPA66
The LOI and UL-94 tests are two widely used methods for evaluating the flame-retardancy of a material. As shown in Fig. 8, the LOI value and UL-94 grades of FRPA66 were higher than those of pure PA66. In Fig. 8(a), the UL-94 test for pure PA66 showed that the melt dripping was severe after it was ignited and it reached only the UL-94 V-2 rating. After the introduction of a flame-retardant material, the melt dripping of PA66 showed great improvement and simultaneously the number of droplets also decreased significantly. As the carbon content in residual char increased, the melt dripping material became substantially flameless when it dropped onto the absorbent cotton and it could not ignite the absorbent cotton. When the CPPOA content reached 8 wt%, the FRPA66 with 28.5% of LOI value, could achieve the UL-94 V-0 rating. The appearance of the FRPA66 spline after the completion of UL-94 test is shown in Fig. 8(b). Pure PA66 was completely burnt during the evaluation of grade of flame-retardant. Moreover, the density of the carbon layer of FRPA66 was effectively improved with increase in CPPOA content.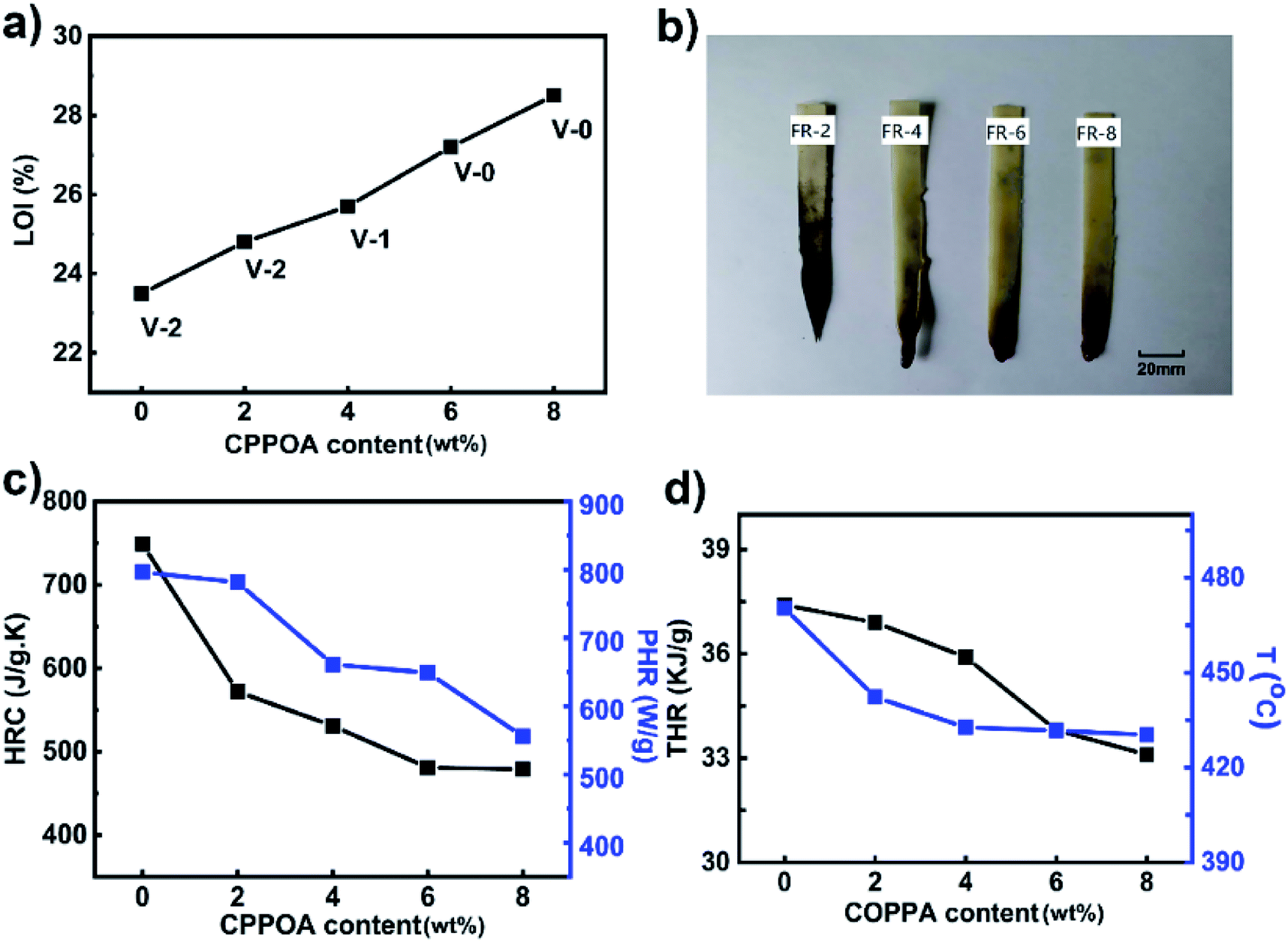 | ||
| Fig. 8 Flame retardancy and combustion behavior of FRPA66: (a) results of LOI and UL-94 tests of PA66 and FRPA66; (b) photographs of PA66 and FRPA66 samples after UL-94 tests; (c) and (d) are MCC combustion data of samples. | ||
Micro-scale combustion calorimetry (MCC) was used to quantitatively evaluate the combustion properties of a material under fire conditions. Parameters such as heat release capacity (HRC), peak heat release rate (pHRR), and total heat release (THR) could be evaluated from MCC. Compared with pure PA66, the HRC, pHRR, THR, and Tp (the temperature corresponding to the peak heat release rate) of FRPA66 with 8 wt% CPPOA decreased by 36.0%, 30.2%, 11.5%, and 40 °C, respectively, as shown in Fig. 8(c and d). The results suggested that addition of CPPOA significantly reduced the flammability of PA66 and was beneficial for controlling the fire. This could be attributed to the phosphonate esters that could increase the amount of the carbon formed from the materials at high temperature.
To explain the effect of coke formation on the flame retardancy of FR-PA66, the coke residue of FR-PA66 after the LOI test was analyzed by SEM and FTIR. In Fig. 9, the carbon layer of pure PA66 showed a honeycomb structure with loosened texture, whereas the carbon surface of FRPA66 was relatively flat and dense. The formation of dense carbon layer on the polymer surface could effectively prevent the burning of the underlying matrix.17 The elemental analysis of the residual carbon surface (Fig. 9) clearly indicated the presence of phosphorus. Compounds such as phosphoric acid (as the degradation product of carbon residue) could promote the formation of carbon and thus improve the flame retardancy of FRPA66.32
 | ||
| Fig. 9 Micromorphologies of the char layer: (a) PA66; (b) FRPA66; (c) elemental analysis of the residual carbon surface. | ||
The FTIR spectra of the carbon residues of PA66 and FRPA66 (remaining in the LOI test) are shown in Fig. 10. Peaks at 2930 cm−1 and 2860 cm−1 were assigned to CH2– stretching vibrations. The peaks of 1640 cm−1 and 1550 cm−1 corresponded to the CO and N–H stretching vibrations, respectively. In addition to the peaks of pure PA66, the spectrum of FRPA66 showed several new peaks. Peaks at 1129 cm−1 and 915 cm−1 were assigned to P–O stretching vibrations and P–Ph stretching vibrations, respectively.33,34 The results indicated the presence of degradation products of CPPOA in residual carbon, which were probably phosphoric acid, polyphosphoric acid, etc. They could promote the dehydration into carbon and make the carbon layer denser, thus improving the flame-retardancy of FRPA66.24
 | ||
| Fig. 10 Infrared spectra of carbon residues of PA66 and FRPA66. | ||
In summary, introduction of CPPOA to PA66 could effectively improve the flame retardancy.
Mechanical properties FRPA66
Mechanical performance of a material is important for its practical applications. Normally, the mechanical performance of flame retardant PA66 would decrease significantly with the introduction of a flame retardant, due to its relatively lower molecular weight. As shown in Fig. 11, the tensile strength and impact strength decreased slightly from 88 MPa and 8 kJ m−2 for pure PA66 to 70 MPa and 5.6 kJ m−2 for FR-6, which also achieved V-0 rating. It should be noted that the mechanical properties of FRPA66 in this study with 6% CCOPA outperformed others from most of the reported works which contained 6% CCOPA or even lower content.18–22 The excellent mechanical performance of our composites was mainly because of the highly reactive CPPOA, which could easily react with PA chains. | ||
| Fig. 11 Mechanical properties of PA66 and FRPA66. | ||
Conclusion
In this study, a flame-retardant with high thermal stability and high reactivity was synthesized. The reaction of 4-(2-(((2-carboxyethyl)(phenyl)phosphoryl)oxy)ethoxy)-4-oxohexanoic acid (CPPOA) with 1,6-diaminohexane produced CPPOA salt, which was then copolymerized with PA66 salt to obtain FRPA66. The thermal stability of CPPOA was significantly improved from 185 to 300 °C after salt formation. FTIR and 31P-NMR results showed that CPPOA was successfully incorporated into PA66 chains. The as-prepared FRPA66 showed excellent flame retardancy with LOI of 27.2% and the vertical combustion grade of V-0, when the CPPOA content was 6 wt%. Furthermore, the FRPA66 had high tensile strength and impact strength of 70 MPa and 5.6 kJ m−2, respectively. This work demonstrated that CPPOA can simultaneously improve the flame retardancy and mechanical properties of PA66, which has high potential for real applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Key R&D Program of China (2016YFB0303000 and 2016YFB1100800) and the NSFC (Grants 51803218, 51373184, and 51373179).References
- Y. P. Bai, L. Huang, T. Huang, J. Long and Y. F. Zhou, Polymer, 2013, 54(16), 4171–4176 CrossRef CAS.
- J. Peng, P. J. Walsh, R. C. Sabo, L.-S. Turng and C. M. Clemons, Polymer, 2016, 84, 158–166 CrossRef CAS.
- J. Xiao, Y. Tan, Y. Song and Q. Zheng, RSC Adv., 2016, 6(47), 41392–41403 RSC.
- R. Yuan, S. Fan, D. Wu, X. Wang, J. Yu, L. Chen and F. Li, Polym. Chem., 2018, 9(11), 1327–1336 RSC.
- F. Sheng, X. Tang, S. Zhang, X. Ding, Z. Yu and Z. Qiu, Polym. Adv. Technol., 2012, 23(2), 137–142 CrossRef CAS.
- A. D. Naik, G. Fontaine, F. Samyn, X. Delva, J. Louisy, S. Bellayer, Y. Bourgeois and S. Bourbigot, RSC Adv., 2014, 4(35), 18406–18418 RSC.
- S. Tan, L. Ma, X. Sun and X. Tang, Adv. Polym. Sci., 2014, 27(1), 65–73 Search PubMed.
- X. M. Fu, Y. Liu, Q. Wang, Z. J. Zhang, Z. Y. Wang and J. Z. Zhang, Polym.-Plast. Technol. Eng., 2011, 50(15), 1527–1532 CrossRef CAS.
- Z. S. Zhan, B. Li, M. J. Xu and Z. H. Guo, Adv. Polym. Sci., 2015, 28(2), 140–146 Search PubMed.
- C. Zhang, J. Huang, S. Liu and J. Zhao, Polym. Adv. Technol., 2011, 22(12), 1768–1777 CrossRef CAS.
- H. Ge, W. Wang, Y. Pan, X. Yu, W. Hu and Y. Hu, RSC Adv., 2016, 6(85), 81802–81808 RSC.
- K. Liu, Y. Li, L. Tao and R. Xiao, RSC Adv., 2018, 8(17), 9261–9271 RSC.
- J. C. Markwart, A. Battig, T. Kuckhoff, B. Schartel and F. R. Wurm, Polym. Chem., 2019, 10(43), 5920–5930 RSC.
- K. Dai, Z. Deng, G. Liu, Y. Wu, W. Xu and Y. Hu, Polymers, 2020, 12(7), 1441–1456 CrossRef CAS.
- X. Zhang, Y. Yuan, W. W. Ye, Y. Yang and Y. H. Qian, Eng. Plast. Appl., 2015, 43(11), 112–117 Search PubMed.
- Y. C. Shi and G. J. Wang, Polym. Mater.: Sci. Eng., 2016, 32, 167–175 CAS.
- T. Ranganathan, B.-C. Ku, J. Zilberman, M. Beaulieu, R. J. Farris, E. B. Coughlin and T. Emrick, J. Polym. Sci., Part A: Polym. Chem., 2007, 45(20), 4573–4580 CrossRef CAS.
- W. Lv, Y. Cui, X. Zhang, Y. Cheng, X. Zhang, J. Yuan and W. Zhang, Eng. Plast. Appl., 2015, 43(7), 20–24 Search PubMed.
- W. Lyu, Y. Cui, X. Zhang, J. Yuan and W. Zhang, Des. Monomers Polym., 2016, 19(5), 420–428 CrossRef CAS.
- W. Lyu, Y. Cui, X. Zhang, J. Yuan and W. Zhang, J. Appl. Polym. Sci., 2016, 133(24), 43538–43547 CrossRef.
- X. Zhang, Y. Cui, W. Lv and H. Yin, Eng. Plast. Appl., 2015, 43(11), 6–10 CAS.
- W. Lv, Y. Cu, X. Zhang, Y. Cheng, J. Yuan and W. Zhang, Plastic, 2016, 45(1), 60–63 Search PubMed.
- X. Yang, Q. Li, Z. Chen and H. Han, Bull. Mater. Sci., 2009, 32(4), 375–380 CrossRef CAS.
- Y. Y. Li, K. Liu, J. D. Zhang and R. Xiao, Polym. Adv. Technol., 2018, 29(2), 951–960 CrossRef CAS.
- W. Lv, J. Zhang, F. Yang, Y. Ma, D. Yi and H. Liu, Plastic, 2017, 46(4), 1–5 Search PubMed.
- H. Ge, G. Tang, W. Hu, B. Wang, Y. Pan, L. Song and Y. Hu, J. Hazard. Mater., 2015, 294, 186–194 CrossRef CAS.
- X. Lu, X. Qiao, T. Yang, K. Sun and X. Chen, J. Appl. Polym. Sci., 2011, 122(3), 1688–1697 CrossRef CAS.
- Z. Wei, C. Zhou, Y. Yu and Y. Li, Polymer, 2015, 71, 31–42 CrossRef CAS.
- P. Flory, Trans. Faraday Soc., 1955, 51(6), 848–857 RSC.
- X. Y. Zhao and M. Z. Wang, J. Appl. Polym. Sci., 2006, 100(4), 3116–3122 CrossRef CAS.
- M. Jimenez, N. Lesaffre, S. Bellayer, R. Dupretz, M. Vandenbossche, S. Duquesne and S. Bourbigot, RSC Adv., 2015, 5(78), 63853–63865 RSC.
- S. Jiang, Y. Zhu, Y. Hu, G. Chen, X. Shi and X. Qian, Polym. Adv. Technol., 2016, 27(2), 266–272 CrossRef CAS.
- A. Šehić, B. Tomšič, I. Jerman, J. Vasiljević, J. Medved and B. Simončič, Polym. Degrad. Stab., 2016, 128, 245–252 CrossRef.
- B. Schartel, A. I. Balabanovich, U. Braun, U. Knoll, J. Artner, M. Ciesielski, M. Döring, R. Perez, J. K. W. Sandler, V. Altstädt, T. Hoffmann and D. Pospiech, J. Appl. Polym. Sci., 2007, 104(4), 2260–2269 CrossRef CAS.
Footnote |
| † Jingnan Zhang and Siming Lian contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |