
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn aza-Diels–Alder approach to chlorinated quinolines, benzoquinolines, and polybenzoquinolines†
Juhwan Kima,
Mehran J. Umeranib,
Reina Kurakakeb,
Huiting Qina,
Joseph W. Zillerc,
Alon A. Gorodetsky *abc and
Young S. Park*ad
*abc and
Young S. Park*ad
aDepartment of Chemical and Biomolecular Engineering, University of California, Irvine, Irvine, CA 92697, USA. E-mail: alon.gorodetsky@uci.edu
bDepartment of Materials Science and Engineering, University of California, Irvine, Irvine, CA 92697, USA
cDepartment of Chemistry, University of California, Irvine, Irvine, CA 92697, USA
dDepartment of Chemistry, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulsan, 44919, Republic of Korea. E-mail: youngspark@unist.ac.kr
First published on 14th April 2021
Abstract
Quinolines and quinoline-containing macromolecules are renowned for their valuable biological activities and excellent materials properties. Herein, we validate a general strategy for the synthesis of chloro-containing quinoline, benzoquinoline and polybenzoquinoline variants via the aza-Diels–Alder reaction. The described findings could be ultimately implemented in other synthetic pathways and may open new opportunities for analogous quinoline-derived materials.
1 Introduction
Quinolines and quinoline-containing macromolecules have been studied for over a century because of their desirable biological activities and favourable combinations of materials properties.1–12 In particular, quinoline-containing molecules are famous for their strong antimalarial activity and for their critical roles in anticancer drug development.1–6 Moreover, quinoline-derived macromolecules have found applications as active materials in organic electronic devices, stable heat-resistant coatings in packaging, and fluorescent probes in various sensing schemes.7–18 As such, substantial effort has focused on the preparation of quinolines and quinoline-based polymers, with the former often synthesized via the Combes, Conrad–Limpach, Skraup, Doebner–Miller, Friedländer, Pfitzinger, or Povarov reactions19–25 and the latter primarily synthesized via Suzuki, Sonogashira, oxidative coupling, Friedländer, or Povarov polymerization methodologies.7,8,10,26–35 Given the importance and value of quinolines and their derivatives, there exists powerful motivation for the continued improvement and refinement of the aforementioned synthetic strategies.Within the context of quinoline-based materials, our laboratory has focused on developing and implementing various synthetic strategies34–40 that leverage the Povarov reaction.21,41–43 This classic reaction employs an electron-deficient diene (e.g., an aryl imine formed from an aniline and a benzaldehyde) and an electron-rich conjugated dienophile (e.g., an alkyne).42,43 The diene coordinates with a Lewis acid and reacts with the dienophile in a formal aza-Diels–Alder [4+2]-cycloaddition, with the resulting dihydro intermediate oxidized to generate a quinoline moiety.42,43 To date, we have used the Povarov reaction to synthesize quinolines and benzoquinolines (in typical yields of >60%) as well as polyquinolines and polybenzoquinolines (in more modest yields of ∼8% to ∼34%).34–36 These materials have demonstrated interesting photophysical properties and have been envisioned as both model compounds and polymeric precursors for nitrogen-doped graphene nanoribbons.34–36 Moreover, we have used the Povarov reaction to synthesize biquinolines and diquinolineanthracenes (in typical yields of >60%) as well as polybiquinolines and polydiquinolineanthracenes (in variable yields of ∼9% to ∼87%).37–40 These materials have demonstrated promising electronic and electrochemical characteristics and have been converted into nitrogen-containing tetrabenzopentacenes and nitrogen-doped graphene nanoribbons.37–40 In general, our Povarov-based methodology has required few steps and mild conditions, featured excellent regioselectivity, enabled the incorporation of a variety of substituents, readily yielded crowded and congested molecular architectures, and afforded polymers with otherwise challenging-to-access connectivities.34–40 However, in our previous studies, we have not explored the installation of synthetically-valuable chloro functional groups for a wide range of quinoline derivatives and quinoline-based macromolecules.
Herein, we conceived building upon the previous efforts with regard to the synthesis of quinolines, benzoquinolines, and polybenzoquinolines and establishing generalizable straightforward routes to these molecules' and polymers' chlorinated variants. As such, we pictured synthesizing a series of quinolines with variable moieties at their 2 positions and chloro groups at their 5 positions, as illustrated in Fig. 1a. Furthermore, we envisaged synthesizing multiple crowded benzoquinoline derivatives with chloro groups at their 5 positions, as illustrated in Fig. 1b. Last, we envisioned synthesizing congested polybenzoquinolines featuring substituted outer peripheries and chloro group-functionalized interiors, as illustrated in Fig. 1c. We theorized that the successful preparation of these targets would have the potential to open new opportunities for quinoline-derived systems.
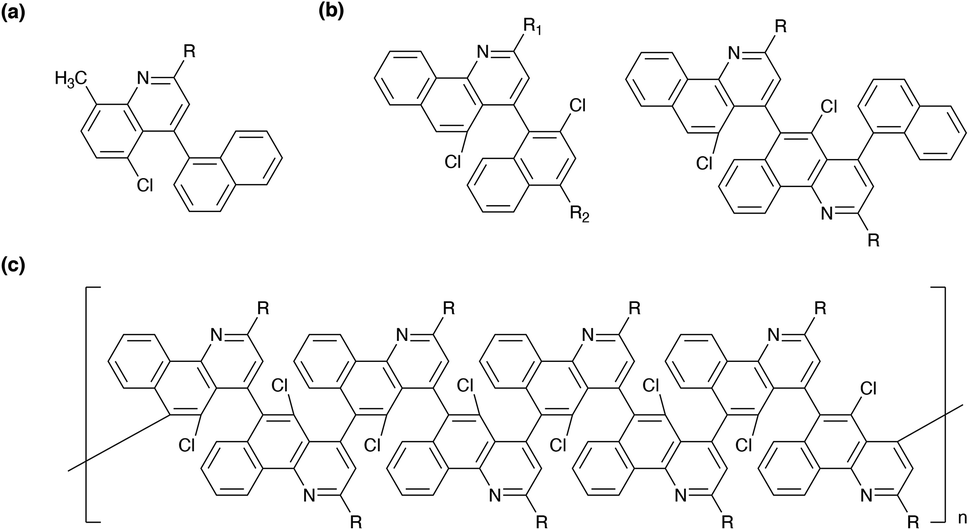 | ||
| Fig. 1 (a) The general chemical structure of the proposed chlorinated quinolines. (b) The general chemical structures of the proposed chlorinated benzoquinolines. (c) The general chemical structure of the proposed chlorinated polybenzoquinolines. Note that the pendant R groups are variable in each instance. | ||
2 Results
We began our efforts by leveraging the aza-Diels–Alder (Povarov) reaction for the preparation of a series of chlorinated quinolines under standard conditions, as shown in Fig. 2.34–40 First, we reacted 5-chloro-2-methylaniline with various commercially-available benzaldehyde derivatives (with glacial acetic acid as the catalyst and toluene as the solvent), obtaining aldimines 1a–1f in reasonable yields of ≥60% (Fig. 2 and S1–S12†). Next, we reacted 1-ethynylnaphthalene with each of the isolated aldimines 1a–1f (in the presence of molecular sieves and with BF3·OEt2 as the Lewis acid mediator, chloranil as the oxidant, and chloroform as the solvent), obtaining functionalized quinolines 2a–2f in moderate to good yields of between ∼26% and ∼89% (Fig. 2 and S13, S24†). Notably, our reaction conditions furnished only the desired regioisomers, in analogy to previous findings for quinolines and biquinolines.34,37 Moreover, the protocol enabled the installation of electron withdrawing group-modified, electron donating group-modified, or sterically-hindered aryl substituents. However, we found some variability in the reaction yields for the aldimines and quinolines, which could not be easily rationalized by considering electronic/steric effects and likely arose from differences in solubility and/or ease of purification. Nonetheless, the described strategy readily afforded chlorinated quinolines in two synthetic steps from commercial reagents under mild reaction conditions.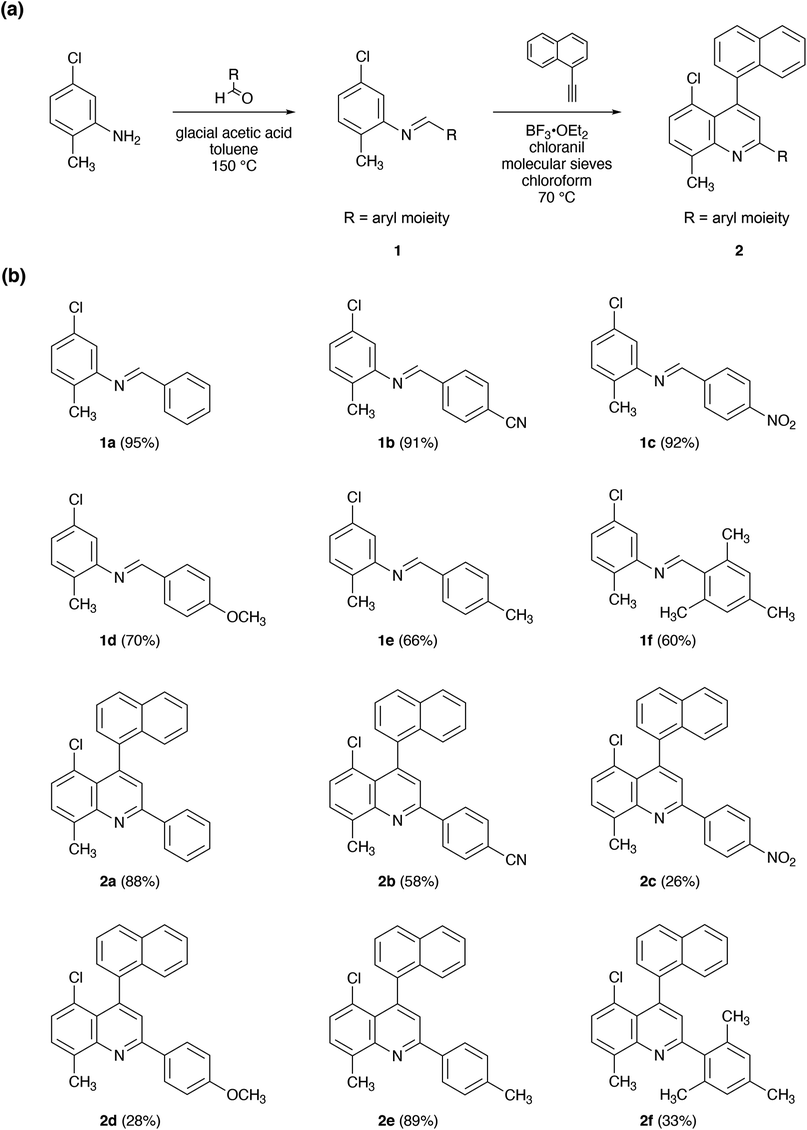 | ||
| Fig. 2 (a) General synthesis of chlorinated quinoline derivatives substituted with different aryl moieties. (b) The chemical structures of aldimines 1a–1f and chlorinated quinolines 2a–2f. | ||
To broaden the utility of our approach, we evaluated the preparation of chlorinated quinolines via an alternative procedure under modified Povarov reaction conditions (see Experimental).34–40 Towards this end, we focused our efforts on 5-chloro-8-methyl-4-(naphthalen-1-yl)-2-(p-tolyl)quinoline 2e, which initially had been obtained in the highest yield of ∼89%. First, we reacted 5-chloro-2-methylaniline with 4-methylbenzaldehyde (with p-toluenesulfonic acid (PTSA) as the catalyst and benzene as the solvent), obtaining aldimine 1e in a good yield of ∼74%. Next, we reacted 1-ethynylnaphthalene with the isolated aldimine 1e (in the absence of molecular sieves and with BF3·OEt2 as the Lewis acid mediator, chloranil as the oxidant, and toluene as the solvent), obtaining functionalized quinoline 2e in a good yield of ∼75%. Notably, these comparatively simplified reaction conditions, which did not require molecular sieves and used only aromatic solvents, i.e., toluene and benzene, again readily furnished the desired regioisomer in high overall yield. Consequently, we anticipated that the described alternative approach could facilitate the synthesis of larger and thus likely less soluble pi-conjugated systems.
To enable the preparation of chlorinated benzoquinolines and polybenzoquinolines, we next synthesized multiple functionalized naphthalene derivatives, as shown in Scheme 1. Initially, we directly chlorinated commercially-available 4-nitronaphthalen-1-amine according to reported protocols,44 furnishing 2-chloro-4-nitronaphthalen-1-amine 3 in a high yield of ∼94 % (Scheme 1 and Fig. S25†). We then selectively converted 3's amine group to an iodo group by following well-known procedures with slight modifications,45 furnishing 2-chloro-1-iodo-4-nitronaphthalene 4 in a good yield of ∼79% (Scheme 1 and Fig. S26, S27†) (note that the identity of 4 was further confirmed via X-ray crystallography, as described in the ESI†). In one subsequent procedure, we simultaneously removed 4's iodo group and converted 4's nitro group to an amine by means of a Zn-mediated reduction reaction,46 obtaining 3-chloronaphthalen-1-amine 5 in a moderate yield of ∼30% (Scheme 1 and Fig. S28, S29†). In a parallel subsequent procedure, we coupled 4's iodo group and trimethylsilylacetylene (TMS acetylene) in a Sonogashira reaction,47 obtaining ((2-chloro-4-nitronaphthalen-1-yl)ethynyl)trimethylsilane 6 in quantitative yield (Scheme 1 and Fig. S30, S31†), and we then further deprotected 6, i.e., removed the TMS group, with tetrabutylammonium fluoride (TBAF) under routine conditions, obtaining ethynylnaphthalene 7 in a high yield of ∼83% (Scheme 1 and Fig. S32, S33†). Altogether, our minimalist strategy produced the key starting materials required for the synthesis of chlorinated benzoquinolines and polybenzoquinolines.
 | ||
| Scheme 1 The synthesis of functionalized naphthalene derivatives 5, 6, and 7, as starting materials for chlorinated benzoquinolines and polybenzoquinolines. PTSA (p-toluenesulfonic acid). TPGS (DL-α-tocopherol methoxypolyethylene glycol succinate). | ||
With our naphthalene starting materials in hand, we synthesized chlorinated benzoquinolines by leveraging the aza-Diels–Alder (Povarov) reaction, as shown in Scheme 2. For this purpose, we used variants of the alternative conditions implemented for the preparation of 1e and 2e (see Experimental) and the approach validated for the conversion of 4 to 5 (see Scheme 1), thus facilitating protocol efficiency and ensuring appropriate intermediate/product solubility. First, we reacted 3-chloronaphthalen-1-amine 5 with 4-octylbenzaldehyde, presumably forming the corresponding imine in situ, and we further reacted this imine with ethynylnaphthalene 7 under Povarov conditions, furnishing chlorinated benzoquinoline 8 in a moderate overall yield of ∼24% (Scheme 2 and Fig. S34, S35†). Next, we converted 8's nitro group to an amine via a Zn-mediated reduction reaction, obtaining chlorinated benzoquinoline 9 in a high yield of ∼93% (Scheme 2 and Fig. S36, S37†). Last, we reacted benzoquinoline 9 with 4-octylbenzaldehyde, once again presumably forming the corresponding imine in situ, and we further reacted this imine with 1-ethynylnaphthalene under Povarov conditions, furnishing chlorinated benzoquinoline dimer 10 in a moderate overall yield of ∼33% (Scheme 2 and Fig. S38, S39†). Notably, the demonstrated one-pot approach was material-efficient and obviated the need for time-consuming and occasionally-challenging imine isolation. Compared to our previously-reported routes to benzoquinolines,35,36 the strategy validated here afforded chlorinated benzoquinolines in analogous yields but required fewer synthetic steps and employed more convenient protocols.
 | ||
| Scheme 2 The synthesis of chlorinated benzoquinoline derivatives 8, 9, and 10. | ||
We proceeded to demonstrate the preparation of chlorinated polybenzoquinolines via an aza-Diels–Alder (Povarov) reaction-based methodology, as shown in Scheme 3. Towards this end, we slightly modified our reported reaction conditions and also used starting materials with branched alkyl chains to further enhance product solubility.35,36 We initially converted 6's nitro group to an amine, obtaining 3-chloro-4-((trimethylsilyl)ethynyl)naphthalen-1-amine 11 in a high yield of ∼94% (Scheme 3 and Fig. S40, S41†). We then further reacted 11 with 4-(2-hexyldecyl)benzaldehyde and removed the TMS group with TBAF in situ, furnishing (E)-N-(3-chloro-4-ethynylnaphthalen-1-yl)-1-(4-(2-hexyldecyl)phenyl)methanimine 12, which constituted an AB-type bifunctional monomer, in a reasonable yield of ∼47% (Scheme 3 and Fig. S42, S43†). We subsequently polymerized monomer 12 under standard Povarov conditions and terminated the reaction through addition of phenylacetylene (Scheme 3).35 This procedure furnished polymer P1 as a crude mixture of macromolecules in a high yield of ∼81% (Scheme 3 and Fig. S44†). Our overall strategy provided access to the targeted chlorinated polybenzoquinoline in straightforward fashion.
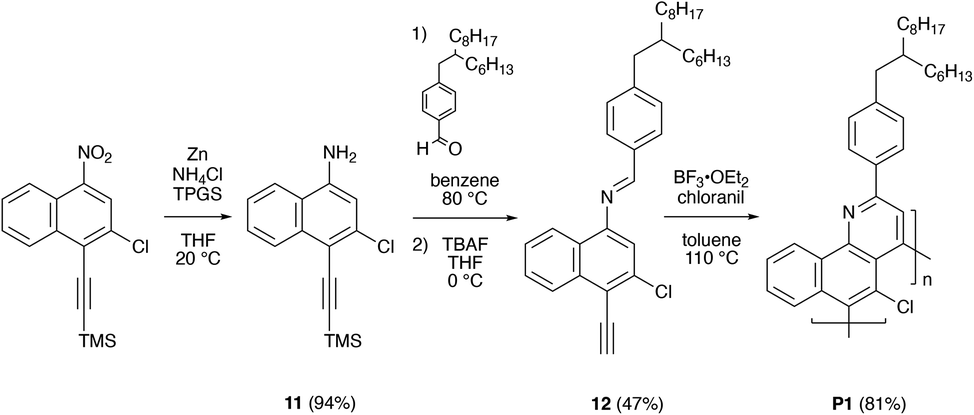 | ||
| Scheme 3 The synthesis of chlorinated polybenzoquinoline P1. | ||
We subsequently characterized both crude and purified polymers via size exclusion chromatography with a refractive index detector (SEC-RI). First, we analyzed the polymers in the initially obtained crude mixture. For this material, we found a number average molecular weight (Mn) of 5347 g mol−1, a weight average molecular weight (Mw) of 5986 g mol−1, and a polydispersity index (PDI) of 1.12 (Fig. S45†). In addition, we analyzed the purified polymers isolated via additional steps. For this material, we found an Mn of 7315 g mol−1, an Mw of 7670 g mol−1, and a PDI of 1.05 (Fig. S45†). Here, we note that the purified chlorinated polybenzoquinoline consisted of ≥14 benzoquinoline subunits, which was fewer than the ≥21 subunits previously reported for analogous polybenzoquinolines, suggesting that further optimization of our reaction and isolation conditions would be possible.35 These measurements provided added insight into the identities and sizes of the obtained benzoquinoline-based macromolecules.
3 Conclusion
In summary, by building upon our prior efforts,34–40 we have established a general Povarov reaction-based approach to chlorinated quinoline-derived molecular and macromolecular frameworks. Our strategy relies upon easily-accessible starting materials, generally mild reaction conditions, a minimal number of synthetic steps, and robust well-known chemical transformations. The strategy also accommodates the installation of varying functionalities on the targeted frameworks, including aryl rings modified with electron-donating, electron-withdrawing, and sterically-hindered groups. The developed protocols furthermore provide access to chlorinated molecules (i.e., quinolines and benzoquinolines) and polymers (i.e., polybenzoquinolines) in generally reasonable overall yields. However, the described approach appears limited by the variability among the isolated yields and/or solubility in some instances. Consequently, there exists an opportunity for further improving and more broadly implementing our methodology in other synthetic pathways.Last, we note that the ability to controllably incorporate chloro groups in our quinolines, benzoquinolines, and polybenzoquinolines may prove particularly valuable from a materials development perspective. For example, varying the degree of chlorination may enable tuning of our materials' electronic and photophysical properties for organic solar cell and transistor applications.48,49 In addition, the installed chloro handles should facilitate post-synthetic functionalization of our molecules via a variety of modern palladium-catalyzed cross-coupling reactions.50,51 Moreover, the chloro functionalities could allow for cyclodehydrodehalogenation of our polymers into well-defined nitrogen-doped N = 7 graphene nanoribbons.35,36 Consequently, the described findings appear poised to broadly enhance the utility of our quinoline-derived molecular and macromolecular frameworks as functional materials.
4 Experimental
4.1. Synthetic protocols for chlorinated quinolines
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 to 1:9, ethyl acetate:hexanes).:100 to 1:2, dichloromethane:hexanes).
:100 to 1:9, ethyl acetate:hexanes).:100 to 1:2, dichloromethane:hexanes).4.2. Synthetic protocols for chlorinated benzoquinolines
:1, hexanes:ethyl acetate). When the starting materials had been consumed, the reaction mixture was cooled to room temperature and then poured into a sodium bicarbonate solution. The resulting precipitate was collected via filtration and thoroughly washed with deionized water. The crude product was isolated and used without further purification (6.27 g, 94%): 1H NMR (500 MHz, CDCl3) δ 8.94 (d, J = 8.8 Hz, 1H), 8.50 (s, 1H), 7.83 (d, J = 8.5 Hz, 1H), 7.76–7.71 (m, J = 8 Hz, 1H), 7.64–7.59 (m, 1H), 5.36 (br s, 2H); HRMS (ESI) m/z calcd for C10H6ClN2O2 [M − H]− 221.0123, found 221.0111.:100 to 1:1, chloroform:hexanes) and isolated as a yellow powder (3.56 g, 79%): 1H NMR (500 MHz, CDCl3) δ 8.45–8.38 (m, 2H), 8.22 (s, 1H), 7.77–7.70 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 137.0, 136.8, 134.5, 130.4, 129.9, 123.7, 123.4, 123.1, 111.8 ppm; HRMS (ESI) m/z calcd for C10H4ClINO2 [M − H]− 331.8980, found 331.8986.:1, hexanes:ethyl acetate). When the starting materials had been consumed, the reaction mixture was filtered through a pad of Celite. The products were extracted by using dichloromethane and a sodium bicarbonate solution, and the organic layer was further rinsed with brine. The organics were dried with sodium sulfate, and the solvent was removed in vacuo to afford the crude product. The crude product was purified via gradient flash chromatography (0:100 to 3:7, ethyl acetate:hexanes) and isolated as a red oil (0.15 g, 30%): 1H NMR (500 MHz, CD2Cl2) δ 7.78 (d, J = 8.4 Hz, 1H), 7.70 (d, J = 7.8 Hz, 1H), 7.52–7.42 (m, 2H), 7.25 (d, J = 1.6 Hz, 1H), 6.73 (d, J = 1.9 Hz, 1H), 4.34 (br s, 2H); 13C NMR (126 MHz, CD2Cl2) δ 144.5, 135.4, 132.3, 128.2, 127.6, 125.5, 122.4, 121.4, 117.4, 110.0 ppm.:1, hexanes:ethyl acetate). When the starting materials had been consumed, the solvent was removed in vacuo. The crude product was isolated and used without further purification (0.91 g, 100%): 1H NMR (500 MHz, CDCl3) δ 8.51–8.47 (m, 1H), 8.46–8.42 (m, 1H), 8.22 (s, 1H), 7.75–7.69 (m, 2H), 0.38 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 145.9, 135.2, 133.4, 129.7, 129.2, 127.2, 126.3, 124.7, 123.6, 123.4, 111.8, 98.2, 0.2 ppm; HRMS (ESI) m/z calcd for C15H14ClNO2SiNa [M + Na]+ 326.0375, found 326.0367.:1, hexanes:ethyl acetate). When the starting materials had been consumed, the reaction was quenched by addition of methanol (10 mL). The products were extracted by using chloroform and a sodium bicarbonate solution. The organics were dried with sodium sulfate, and the solvent was removed in vacuo to afford the crude product. The crude product was purified via gradient flash chromatography (0:100 to 3:7, ethyl acetate:hexanes) and isolated as a yellow powder (1.00 g, 83%): 1H NMR (500 MHz, CDCl3) δ 8.52–8.45 (m, 2H), 8.22 (s, 1H), 7.78–7.71 (m, 2H), 4.04 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 146.5, 135.4, 134.0, 129.8, 129.5, 127.0, 125.2, 124.4, 123.7, 123.3, 92.1, 77.6 ppm; HRMS (ESI) m/z calcd for C12H5ClNO2 [M − H]− 230.0014, found 230.0002.:1, hexanes:ethyl acetate). When the starting materials had been consumed, the mixture was cooled to room temperature, and the solvent was removed in vacuo. The obtained materials were dissolved in toluene (50 mL); 2-chloro-1-ethynyl-4-nitronaphthalene (7) (1.56 g, 6.78 mmol), BF3·OEt2 (0.962 g, 6.78 mmol), and chloranil (2.08 g, 8.48 mmol) were added to this solution; and the resulting mixture was stirred at 100 °C for 24 hours. The products were extracted by using chloroform and a sodium bicarbonate solution. The organics were dried with sodium sulfate, and the solvent was removed in vacuo to afford the crude product. The crude product was purified via gradient flash chromatography (0:100 to 1:3, chloroform:hexanes) and isolated as a yellow powder (0.82 g, 24%): 1H NMR (500 MHz, CD2Cl2) δ 9.60 (d, J = 7.6 Hz, 1H), 8.59 (d, J = 8.8 Hz, 1H), 8.37 (s, 1H), 8.31–8.26 (m, 2H), 7.88–7.71 (m, 6H), 7.49 (td, J = 6.5, 1 Hz, 1H) 7.41–7.36 (m, 3H), 2.71 (t, J = 7.8 Hz, 2H), 1.67 (quint, J = 7.7 Hz, 2H), 1.42–1.21 (m, 10H), 0.88 (t, J = 7.0 Hz, 3H); 13C NMR (126 MHz, CD2Cl2) δ 155.9, 148.7, 147.5, 146.2, 144.2, 143.1, 136.1, 135.3, 133.7, 131.9, 130.1, 130.0, 129.79, 129.77, 129.6, 129.2, 128.1, 127.9, 127.71, 127.69, 127.4, 126.2, 125.1, 124.0, 123.7, 122.4, 122.1, 36.3, 32.4, 32.0, 30.0, 29.9, 29.8, 23.2, 14.4 ppm; HRMS (ESI) m/z calcd for C37H33Cl2N2O2 [M + H]+ 607.1914, found 607.1924.:1, hexanes:ethyl acetate). When the starting materials had been consumed, the reaction mixture was filtered through a pad of Celite. The products were extracted by using chloroform and a sodium bicarbonate solution, and the organic layer was further rinsed with brine. The organics were dried with sodium sulfate, and the solvent was removed in vacuo to afford the crude product. The resulting product was purified via gradient flash chromatography (0:100 to 3:7, ethyl acetate:hexanes) and isolated as a red oil (0.65 g, 93%): 1H NMR (500 MHz, CD2Cl2) δ 9.58 (d, J = 8.7 Hz, 1H), 8.29 (d, J = 8.3 Hz, 2H), 7.93 (s, 1H), 7.90 (d, J = 8.5 Hz, 1H), 7.84–7.72 (m, 4H), 7.47 (td, J = 6.5, 1.0 Hz, 1H), 7.37 (d, J = 8.3 Hz, 2H), 7.32 (td, J = 7.0, 1.5 Hz, 1H), 7.17 (d, J = 8.5 Hz, 1H), 6.94 (s, 1H), 4.48 (s, 2H), 2.70 (t, J = 7.8 Hz, 2H), 1.67 (quint, J = 7.7 Hz, 2H), 1.45–1.19 (m, 10H), 0.88 (t, J = 7.0 Hz, 3H); 13C NMR (126 MHz, CD2Cl2) δ 155.7, 148.6, 145.8, 145.3, 144.5, 136.5, 135.0, 133.6, 132.1, 131.7, 129.54, 129.52, 129.4, 128.5, 127.8, 127.74, 127.72, 127.3, 127.2, 126.1, 125.4, 124.1, 123.3, 122.5, 121.6, 110.1, 36.3, 32.5, 32.0, 30.0, 29.9, 29.8, 23.2, 14.4 ppm; HRMS (ESI) m/z calcd for C37H34Cl2N2Na [M + Na]+ 599.2002, found 599.2004.:1, hexanes:ethyl acetate). When the starting materials had been consumed, the mixture was cooled to room temperature, and the solvent was removed in vacuo. The obtained materials were dissolved in toluene (50 mL); 1-ethynylnaphthalene (0.361 g, 2.37 mmol), BF3·OEt2 (0.336 g, 2.37 mmol), and chloranil (0.729 g, 2.97 mmol) were added to this solution; and the resulting mixture was stirred at 85 °C for 24 hours. The products were extracted by using chloroform and a sodium bicarbonate solution. The organics were dried with sodium sulfate, and the solvent was removed in vacuo to afford the crude product. The crude product was purified via gradient flash chromatography (0:100 to 1:4, dichloromethane:hexanes) and isolated as a yellow powder containing a mixture of atropisomers (0.60 g, 33%): 1H NMR (500 MHz, CD2Cl2) δ 9.75 (dd, J = 8.0, 2.5 Hz, 1H), 9.52 (dd, J = 7.5, 3.5 Hz, 1H) 8.35 (d, J = 7.7 Hz, 2H), 8.23 (d, J = 8.3 Hz, 1H), 8.19 (d, J = 8.3 Hz, 1H), 8.06 (d, J = 10.4 Hz, 1H), 7.92–7.66 (m, 8H), 7.59–7.30 (m, 10H), 7.20 (dd, J = 8.2, 2.1 Hz, 1H), 2.73 (t, J = 7.7 Hz 2H), 2.68 (t, J = 7.7 Hz, 2H), 1.77–1.60 (m, 4H), 1.46–1.19 (m, 20H), 0.91–0.86 (m, 6H); 13C NMR (126 MHz, CD2Cl2) δ 155.84, 155.81, 155.44, 155.40, 148.5, 148.3, 148.14, 148.11, 145.86, 145.83, 145.81, 145.6, 145.5, 140.4, 140.2, 138.83, 138.79, 136.54, 136.52, 136.31, 136.30, 134.5, 133.7, 133.6, 133.5, 133.3, 133.1, 131.91, 131.88, 131.6, 129.6, 129.50, 129.48, 129.36, 128.7, 128.6, 128.52, 128.50, 128.3, 127.91, 127.89, 127.80, 127.78, 127.6, 127.23, 127.22, 127.20, 126.91, 126.89, 126.85, 126.8, 126.7, 126.66, 126.40, 126.37, 126.32, 126.27, 126.08, 126.05, 125.7, 125.6, 124.1, 124.0, 123.1, 123.04, 122.82, 122.80, 122.6, 122.5, 36.34, 36.28, 32.5, 32.4, 32.00, 31.95, 30.1, 30.0, 29.92, 29.86, 29.8, 23.3, 23.2, 14.5, 14.4 ppm; HRMS (ESI) m/z calcd for C64H61Cl2N2 [M + H]+ 927.4207, found 927.4236.4.3. Synthetic protocols for chlorinated polybenzoquinolines
:1, hexanes:ethyl acetate). When the starting materials had been consumed, the reaction mixture was filtered through a pad of Celite. The products were extracted by using chloroform and a sodium bicarbonate solution. The organics were dried with sodium sulfate, and the solvent was removed in vacuo to afford the crude product. The product was purified via gradient flash chromatography (0:100 to 1:2, ethyl acetate:hexanes) and isolated as a red oil (0.77 g, 94%): 1H NMR (500 MHz, CDCl3) δ 8.29 (d, J = 8.4 Hz, 1H), 7.73 (d, J = 8.4 Hz, 1H), 7.58 (t, J = 7.6 Hz, 1H), 7.47 (t, J = 7.6 Hz, 1H), 6.76 (s, 1H), 4.39 (br s, 2H), 0.33 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 143.8, 136.2, 135.3, 128.0, 127.2, 125.5, 121.6, 121.0, 110.1, 110.0, 102.9, 100.5, 0.4 ppm; HRMS (ESI) m/z calcd for C15H16ClNSiNa [M + Na]+ 296.0643, found 296.0648.:1, hexanes:ethyl acetate). When the starting materials had been consumed, the reaction mixture was cooled to room temperature, and the solvent was removed in vacuo. The obtained materials were dissolved in THF (30 mL), TBAF (1 M in THF, 6.055 mL, 6.055 mmol) was added to this solution, and the resulting reaction mixture was stirred at 0 °C for 40 minutes. The consumption of the reagents and reaction progress were monitored via TLC analysis (9:1, hexanes:ethyl acetate). When the starting materials had been consumed, the reaction was quenched by addition of methanol (10 mL). The products were extracted by using dichloromethane and a sodium bicarbonate solution. The organics were dried with sodium sulfate, and the solvent was removed in vacuo to afford the crude product. The product was purified via gradient flash chromatography using silica gel deactivated by 10% trimethylamine in hexanes (0:100 to 3:7, ethyl acetate:hexanes) and isolated as a yellow powder (1.5 g, 47%): 1H NMR (500 MHz, CDCl3) δ 8.49 (s, 1H), 8.33 (dd, J = 12.8, 8.3 Hz, 2H), 7.92 (d, J = 8.1 Hz, 2H), 7.68–7.60 (m, 1H), 7.57–7.49 (m, 1H), 7.30 (d, J = 8.1 Hz, 2H), 7.06 (s, 1H), 3.82 (s, 1H), 2.62 (d, J = 7.1 Hz, 2H), 1.74–1.64 (m, 1H), 1.42–1.18 (m, 24H), 0.89 (t, J = 7.0 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 161.9, 151.1, 147.1, 135.9, 134.9, 133.6, 130.0, 129.3, 128.5, 127.3, 126.4, 126.0, 124.7, 115.9, 113.9, 87.5, 79.0, 40.9, 39.9, 33.4, 33.3, 32.07, 32.05, 30.1, 29.82, 29.77, 29.5, 26.70, 26.69, 22.8, 14.3 ppm; HRMS (ESI) m/z calcd for C35H45ClN [M + H]+ 514.3235, found 514.3254.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Office of Naval Research (N00014-12-1-0491, N00014-13-1-0650, and N00014-15-1-2680). Authors thank Dr Anthony Burke, Ethan Ryan Peng, and Henry Hoa B. Dihn for assistance with writing, technical editing, and proofreading.Notes and references
- S. Kumar, S. Bawa and H. Gupta, Mini-Rev. Med. Chem., 2009, 9, 1648–1654 CrossRef CAS PubMed.
- V. R. Solomon and H. Lee, Curr. Med. Chem., 2011, 18, 1488–1508 CrossRef CAS PubMed.
- X.-F. Shang, S. L. Morris-Natschke, Y.-Q. Liu, X. Guo, X.-S. Xu, M. Goto, J.-C. Li, G.-Z. Yang and K.-H. Lee, Med. Res. Rev., 2018, 38, 775–828 CrossRef CAS PubMed.
- X. Nqoro, N. Tobeka and B. A. Aderibigbe, Molecules, 2017, 22, 2268 CrossRef PubMed.
- S. Jain, V. Chandra, P. K. Jain, K. Pathak, D. Pathak and A. Vaidya, Arabian J. Chem., 2019, 12, 4920–4946 CrossRef CAS.
- J. K. Baird, Clin. Microbiol. Rev., 2019, 32, e00011–e00019 CrossRef PubMed.
- C. J. Lee, J. Macromol. Sci., Polym. Rev., 1989, 29, 431–560 CrossRef.
- A. L. Rusanov, L. G. Komarova, M. P. Prigozhina and D. Y. Likhatchev, Russ. Chem. Rev., 2005, 74, 671–683 CrossRef CAS.
- A. P. Kulkarni, A. P. Gifford, C. J. Tonzola and S. A. Jenekhe, Chem. Mater., 2004, 16, 4556–4573 CrossRef CAS.
- A. Kimyonok, X. Wang and M. Weck, J. Macromol. Sci., Part C: Polym. Rev., 2006, 46, 47–77 CrossRef CAS.
- X. Zhao and X. Zhan, Chem. Soc. Rev., 2011, 40, 3728–3743 RSC.
- K. Ilina and M. Henary, Chem.–Eur. J., 2020, 26, 1–20 CrossRef.
- X. Zhang, A. S. Shetty and S. A. Jenekhe, Macromolecules, 1999, 32, 7422–7429 CrossRef CAS.
- C. J. Tonzola, A. P. Kulkarni, A. P. Gifford, W. Kaminsky and S. A. Jenekhe, Adv. Funct. Mater., 2007, 17, 863–874 CrossRef CAS.
- N. V. Matyushina, V. M. Svetlichniyi, L. A. Myagkova, E. L. Aleksandrova, E. N. Popova, I. V. Gofman, M. E. Vylegzhanina, A. Y. Volkov, T. E. Sukhanonva and V. V. Kudryavtsev, Polym. Sci., Ser. B, 2017, 59, 718–729 CrossRef CAS.
- N. H. Hendricks, M. L. Marrorco, D. M. Stoakley and A. K. S. Clair, Int. SAMPE Electron. Conf., 1990, 4, 544–555 CAS.
- X. Zhou, P. Li, Z. Shi, X. Tang, C. Chen and W. Liu, Inorg. Chem., 2012, 51, 9226–9231 CrossRef CAS PubMed.
- J. V. Jun, E. J. Petersson and D. M. Chenoweth, J. Am. Chem. Soc., 2018, 140, 9486–9493 CrossRef CAS PubMed.
- M. G.-A. Shvekhgeimer, Chem. Heterocycl. Compd., 2004, 40, 257–294 CrossRef CAS.
- S. A. Yamashkin and E. A. Oreshkina, Chem. Heterocycl. Compd., 2006, 42, 701–718 CrossRef CAS.
- V. V. Kouznetsov, Tetrahedron, 2009, 65, 2721–2750 CrossRef CAS.
- J. Marco-Contelles, E. Pérez-Mayoral, A. Samadi, M. do C. Carreiras and E. Soriano, Chem. Rev., 2009, 109, 2652–2671 CrossRef CAS PubMed.
- S. M. Prajapati, K. D. Patel, R. H. Vekariya, S. N. Panchal and H. D. Patel, RSC Adv., 2014, 4, 24463–24476 RSC.
- G. A. Ramann and B. J. Cowen, Molecules, 2016, 21, 986 CrossRef PubMed.
- A. Weyesa and E. Mulugeta, RSC Adv., 2020, 10, 20784–20793 RSC.
- B. Siemssen, K. W. Kim, M. S. Kim, B. S. Kim, S. J. Cho, D. K. Park, H. S. Woo and T. W. Kwon, Mol. Cryst. Liq. Cryst., 2006, 462, 159–167 CrossRef.
- M. Tomar, N. T. Lucas, H. Kim, F. Laquai, K. Müllen and J. Jacob, Polym. Int., 2012, 61, 1318–1325 CrossRef CAS.
- C. G. Bangcuyo, M. E. Rampey-Vaughn, L. T. Quan, S. M. Angel, M. D. Smith and U. H. F. Bunz, Macromolecules, 2002, 35, 1563–1568 CrossRef CAS.
- G. Jégou and S. A. Jenekhe, Macromolecules, 2001, 34, 7926–7928 CrossRef.
- A. Bilici, F. Doğan, M. Yıldırım and İ. Kaya, React. Funct. Polym., 2011, 71, 675–683 CrossRef CAS.
- İ. Kaya, F. Kolcu, G. Demiral, H. Ergül and E. Kiliç, Des. Monomers Polym., 2015, 18, 89–104 CrossRef.
- J. K. Stille, Macromolecules, 1981, 14, 870–880 CrossRef CAS.
- A. K. Agrawal and S. A. Jenekhe, Macromolecules, 1993, 26, 895–905 CrossRef CAS.
- D. J. Dibble, M. J. Umerani, A. Mazaheripour, Y. S. Park, J. W. Ziller and A. A. Gorodetsky, Macromolecules, 2015, 48, 557–561 CrossRef CAS.
- D. J. Dibble, Y. S. Park, A. Mazaheripour, M. J. Umerani, J. W. Ziller and A. A. Gorodetsky, Angew. Chem., Int. Ed., 2015, 54, 5883–5887 CrossRef CAS PubMed.
- A. Mazaheripour, D. J. Dibble, M. J. Umerani, Y. S. Park, R. Lopez, D. Laidlaw, E. Vargas, J. W. Ziller and A. A. Gorodetsky, Org. Lett., 2016, 18, 156–159 CrossRef CAS PubMed.
- M. J. Umerani, D. J. Dibble, A. G. Wardrip, A. Mazaheripour, E. Vargas, J. W. Ziller and A. A. Gorodetsky, J. Mater. Chem. C, 2016, 4, 4060–4066 RSC.
- Y. S. Park, D. J. Dibble, J. Kim, R. C. Lopez, E. Vargas and A. A. Gorodetsky, Angew. Chem., Int. Ed., 2016, 55, 3352–3355 CrossRef CAS PubMed.
- D. J. Dibble, R. Kurakake, A. G. Wardrip, A. Bartlett, R. Lopez, J. A. Linares, M. Firstman, A. M. Schmidt, M. J. Umerani and A. A. Gorodetsky, Org. Lett., 2018, 20, 502–505 CrossRef CAS PubMed.
- Z. Feng, A. Mazaheripour, D. J. Dibble, P. Wagner, G. Czap, G. Kladnik, A. Cossaro, A. Verdini, L. Floreano, G. Bavdek, W. Ho, G. Comelli, D. Cvetko, A. Morgante and A. A. Gorodetsky, Carbon, 2020, 170, 677–684 CrossRef CAS.
- L. S. Povarov, Russ. Chem. Rev., 1967, 36, 656–670 CrossRef.
- M. Fochi, L. Caruana and L. Bernardi, Synthesis, 2014, 46, 135–157 CrossRef.
- L. R. Domingo, M. J. Aurell, J. A. Sáez and S. M. Mekelleche, RSC Adv., 2014, 4, 25268 RSC.
- J. Li, D. Smith, S. Krishnananthan, R. A. Hartz, B. Dasgupta, V. Ahuja, W. D. Schmitz, J. J. Bronson, A. Mathur, J. C. Barrish and B.-C. Chen, Org. Process Res. Dev., 2012, 16, 156–159 CrossRef CAS.
- D. Guan, L. Han, L. Wang, H. Song, W. Chu and Z. Sun, Chem. Lett., 2015, 44, 743–745 CrossRef CAS.
- S. M. Kelly and B. H. Lipshutz, Org. Lett., 2014, 16, 98–101 CrossRef CAS PubMed.
- M. J. Mphahlele and L. G. Lesenyeho, J. Heterocycl. Chem., 2013, 50, 1 CrossRef CAS.
- H. Chen, Z. Hu, H. Wang, L. Liu, P. Chao, J. Qu, W. Chen, A. Liu and F. He, Joule, 2018, 2, 1623–1634 CrossRef CAS.
- M. L. Tang, J. H. Oh, A. D. Reichardt and Z. Bao, J. Am. Chem. Soc., 2009, 131, 3733–3740 CrossRef CAS PubMed.
- R. F. Heck, Org. React., 1982, 27, 345–390 CrossRef CAS.
- M. Oestreich, The Mizoroki–Heck Reaction, John Wiley & Sons, Ltd, Chichester, UK, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2021146. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra06744j |
| This journal is © The Royal Society of Chemistry 2021 |