
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn aza-Diels–Alder route to quinoline-based unnatural amino acids and polypeptide surrogates†
M. J. Umerani a,
H. Yangb,
P. Pratakshyab,
J. S. Nowickb and
A. A. Gorodetsky*abc
a,
H. Yangb,
P. Pratakshyab,
J. S. Nowickb and
A. A. Gorodetsky*abc
aDepartment of Materials Science and Engineering, University of California, Irvine, Irvine, CA 92697, USA. E-mail: alon.gorodetsky@uci.edu
bDepartment of Chemistry, University of California, Irvine, Irvine, CA 92697, USA
cDepartment of Chemical and Biomolecular Engineering, University of California, Irvine, Irvine, CA 92697, USA
First published on 19th April 2021
Abstract
Macromolecules composed of quinoline building blocks are viewed as valuable synthetic targets due to their potential as functional materials in optoelectronic devices and promise as therapeutic compounds for the treatment of disease. As such, a number of routes to polyquinolines, quinoline-decorated oligopeptides, and quinoline-containing oligoamides have been developed to date. Herein, by drawing inspiration from prior efforts, we synthesize quinoline-based unnatural amino acid building blocks via the aza-Diels–Alder (Povarov) reaction and then prepare polypeptide surrogates through iterative coupling of these building blocks on solid support. The described strategy uses economical procedures, requires straightforward conditions, and affords the targeted constructs in reasonable yields. Overall, our findings may expand the scope of possibilities for quinoline-based bioinspired polymers and facilitate the further development of quinoline-based functional materials.
Introduction
The synthesis of quinoline-containing macromolecules has been studied extensively because of their potential as functional materials in optoelectronic devices and promise as therapeutic compounds for the treatment of disease.1–10 For instance, polyquinolines have been typically obtained via approaches that leverage the Suzuki reaction, Sonogashira coupling, oxidative polycondensation, or the Friedländer synthesis.1,2,11–21 These polymers have demonstrated a number of highly desirable features, including tunable electronic properties, chemical inertness, mechanical robustness, thermal stability, and facile processability.1–4,21–23 Additionally, quinoline-decorated oligopeptides and quinoline-containing oligoamides have been frequently prepared via the conjugation of quinoline derivatives to amino acids or via the iterative coupling of small molecular building blocks on solid support.24–28 These oligopeptides and oligoamides have exhibited various favorable characteristics, including sequence modularity, resistance to proteolytic degradation, ready cellular uptake, controllable conformational stability, and compatibility with a wide range of solvents.28–34 Consequently, there has emerged an opportunity for the design and synthesis of alternative quinoline-based polypeptide surrogates that draw inspiration from the reported quinoline-containing macromolecules.Recently, our laboratory has focused on the preparation of quinoline-based materials via the aza-Diels–Alder (Povarov) reaction.35–43 For instance, we have demonstrated the synthesis of polymers with difficult-to-access architectures and/or connectivities, such as 4,6-linked polyquinolines, zigzag polyquinolines, polydiquinolineanthracenes, and crowded polybenzoquinolines.35–39 We have moreover studied the likely conformations and spectroscopic characteristics of these polymers (while also making judicious comparisons to corresponding model compounds).35–39 In addition, we have described the synthesis of both nitrogen-containing expanded acenes and nitrogen-doped graphene nanoribbons from quinoline-substituted precursors.40,41 We have furthermore interrogated the electronic structures of the acenes and nanoribbons via computational, electrochemical, scanning probe microscopy, and/or spectroscopic techniques.40,41 Together, these efforts have established a utilitarian synthetic toolkit that can provide access to a broad range of quinoline-based small molecular and macromolecular frameworks.
Herein, we report the preparation and characterization of unnatural quinoline-based amino acids and polypeptide surrogates. First, we outline a synthetic route to judicious polypeptide targets by drawing inspiration from prior studies of quinoline-containing oligoamide foldamers. Second, we synthesize the requisite quinoline-based amino acid building blocks via the aza-Diels–Alder (Povarov) reaction. Third, we confirm the diastereomeric purity of our unnatural amino acids via standard peptide chemistry analytical methods. Fourth, we synthesize polypeptide surrogates by means of iterative coupling of such small molecular building blocks on solid support. Fifth, we characterize the polypeptide surrogates by using size exclusion chromatography and mass spectrometry. Sixth, we study our surrogates' electronic and photophysical properties with ultraviolet-visible absorption and fluorescence spectroscopy. Last, we assess our surrogates' likely solution-phase conformations with circular dichroism spectroscopy. Overall, our findings may expand the scope of possibilities for quinoline-based bioinspired polymers and facilitate the further development of quinoline-based functional materials.
Results
We began our experiments by designing a straightforward route to quinoline-based polypeptide surrogates. Towards this end, we drew inspiration from the classic strategy reported for the preparation of quinoline-containing oligoamide foldamers,26–34 which is illustrated in Fig. 1A. This approach entails (1) the synthesis of a 2,4,8-substituted quinoline monomer in 6 synthetic steps from 2-nitroaniline as the starting material and (2) the iterative deprotection/coupling of this building block on solid support to generate oligoamide foldamers. Thus, we envisioned an alternative strategy based on our prior efforts,35–41 which is illustrated in Fig. 1B. This approach entails (1) the synthesis of a 2,4,6-substituted quinoline-based unnatural amino acid in 2 synthetic steps from Fmoc-4-amino-(L)-phenylalanine as the starting material and (2) the iterative deprotection/coupling of this building block on solid support to generate polypeptide surrogates. We postulated that our methodology would readily afford macromolecules wherein pendant quinolines are arranged on a standard peptide backbone.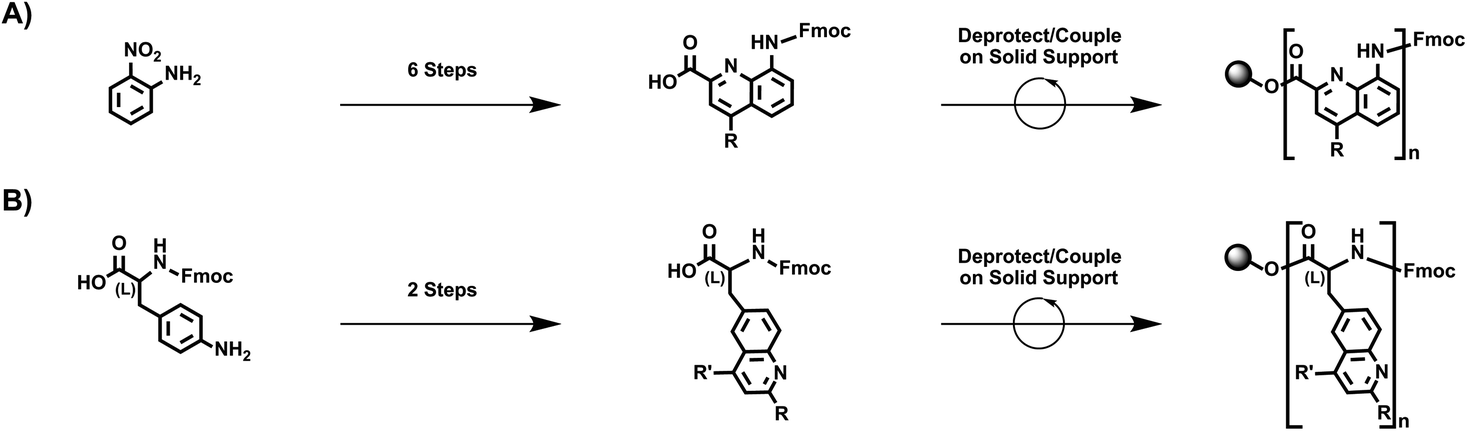 | ||
| Fig. 1 (A) The approach reported for the preparation of quinoline-containing oligoamide foldamers. (B) The alternative approach envisioned for the preparation of quinoline-based polypeptide surrogates. | ||
We proceeded to synthesize the requisite model quinoline-based amino acid building blocks via the aza-Diels–Alder (Povarov) reaction.35–43 We reacted Fmoc-protected 4-amino-(L)-phenylalanine with 4-octylbenzaldehyde according to literature procedures, obtaining aldimine 1 in good yields of 79% that were comparable to those reported for analogous imines (Scheme 1).35–39 We next reacted 1 with phenylacetylene under our standard Povarov conditions, forming Fmoc-protected unnatural amino acid 2a in reasonable yields of 46% that were slightly lower than those reported for analogous heterocycles (Scheme 1).35–39 We then used a routine peptide chemistry protocol to remove the Fmoc group, obtaining deprotected unnatural amino acid 2b in good yields of 83% (Scheme 1).44,45 We confirmed the identities of 1, 2a, and 2b through a combination of 1H nuclear magnetic resonance (NMR) spectroscopy, 13C NMR spectroscopy, and matrix assisted laser desorption/ionization (MALDI) mass spectrometry (Fig. S1–S9†). We also characterized 2a and 2b with ultraviolet-visible (UV-Vis) absorption spectroscopy, obtaining spectra consistent with a 2,4,6-substituted quinoline core35,36 (Fig. S10 and S11†). We moreover determined the overall purity of 2a and 2b to be >99% by means of high-performance liquid chromatography (HPLC) (Fig. S12 and S13†). We last validated the assignment of 2b's aromatic protons' resonances via two-dimensional 1H–1H correlation spectroscopy (COSY), which provided further confirmation for our deprotected amino acid's structure (Fig. S14†). Altogether, our approach furnished the desired quinoline-based unnatural building blocks under mild and straightforward reaction conditions, from commercially-available reagents via a minimal number of steps, and in overall yields that were reasonable for our crowded molecules.
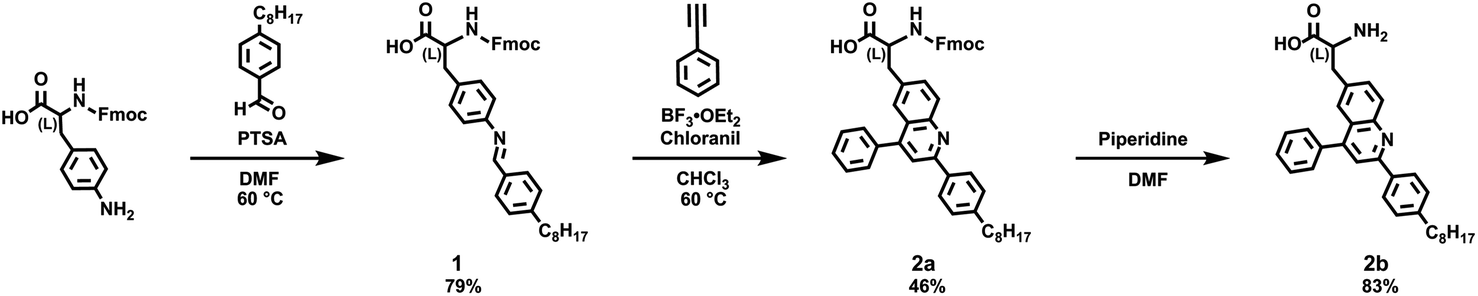 | ||
| Scheme 1 Synthesis of unnatural quinoline amino acids 2a and 2b. | ||
We next sought to further evaluate the overall configuration and identity of our unnatural quinoline-based amino acids. We initially loaded 2-chlorotrityl chloride resin with Fmoc-protected L-alanine, coupled protected unnatural amino acid 2a to the deprotected resin-bound L-alanine, and cleaved the deprotected dyad 3 from the resin (Scheme 2). We initially analyzed the crude dyad 3 via HPLC, with the observed well-resolved peak indicating the presence of a single diastereomer (Fig. S15†). We next confirmed the identity of pure dyad 3 through a combination of 1H NMR spectroscopy, 13C NMR spectroscopy, and MALDI mass spectrometry (Fig. S16–S18†). We in turn characterized pure dyad 3 with UV-Vis absorption spectroscopy, observing changes in the spectrum consistent with the presence of a quinoline core and a natural amino acid (Fig. S19†). These experiments not only confirmed the diastereomeric purity of our quinoline-based amino acid but also validated a procedure for conjugating the building block to other amino acids on solid support.
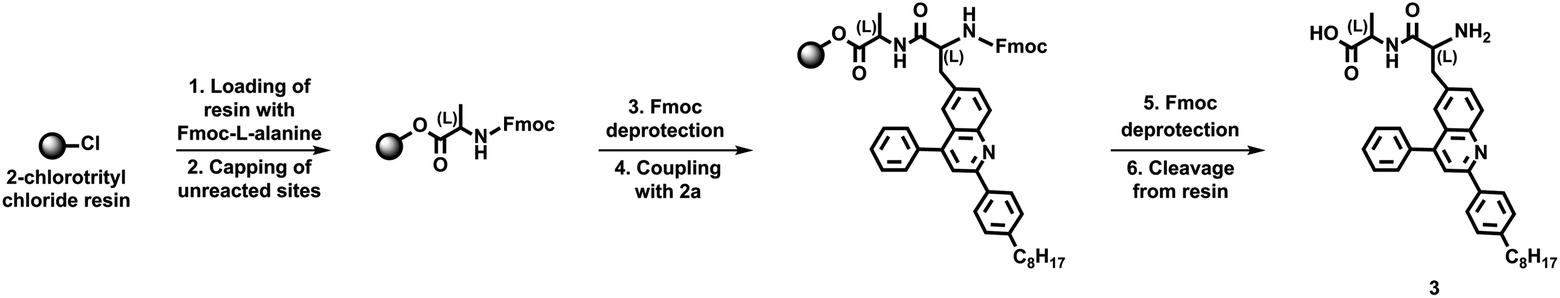 | ||
| Scheme 2 Synthesis of dyad 3. | ||
We continued our efforts by synthesizing polypeptide surrogates consisting of variable numbers of quinoline building blocks via our validated solid support-based protocols. First, we loaded 2-chlorotrityl chloride resin with Fmoc-protected unnatural amino acid 2a and afterwards capped all of the unreacted sites (Scheme 3). Next, we removed the Fmoc protecting groups from the resin-bound amino acids and coupled protected unnatural amino acid 2a once to the loaded resin (Scheme 3). In turn, we directly cleaved the Fmoc-protected dimer 4a from the resin, or we removed the terminal Fmoc group and then cleaved the deprotected dimer 4b from the resin (Scheme 3). Furthermore, we used the same general protocol to couple our protected unnatural amino acid 2a four, nine, or nineteen times to the loaded resin (Scheme 3). Subsequently, we directly cleaved the Fmoc-protected pentamer 5a, decamer 6a, and eicosamer 7a from the resin, or we removed the terminal Fmoc group and then cleaved the deprotected pentamer 5b, decamer 6b, and eicosamer 7b from the resin (Scheme 3). Last, we purified all our polypeptide surrogates via flash chromatography, isolating 4a, 5a, 6a, and 7a in estimated yields of 54%, 49%, 42%, and 34%, respectively, and 4b, 5b, 6b, and 7b in estimated yields of 47%, 39%, 37%, and 55%, respectively. This general iterative strategy furnished the targeted quinoline-based polypeptide surrogates in straightforward fashion and in reasonable total yields.
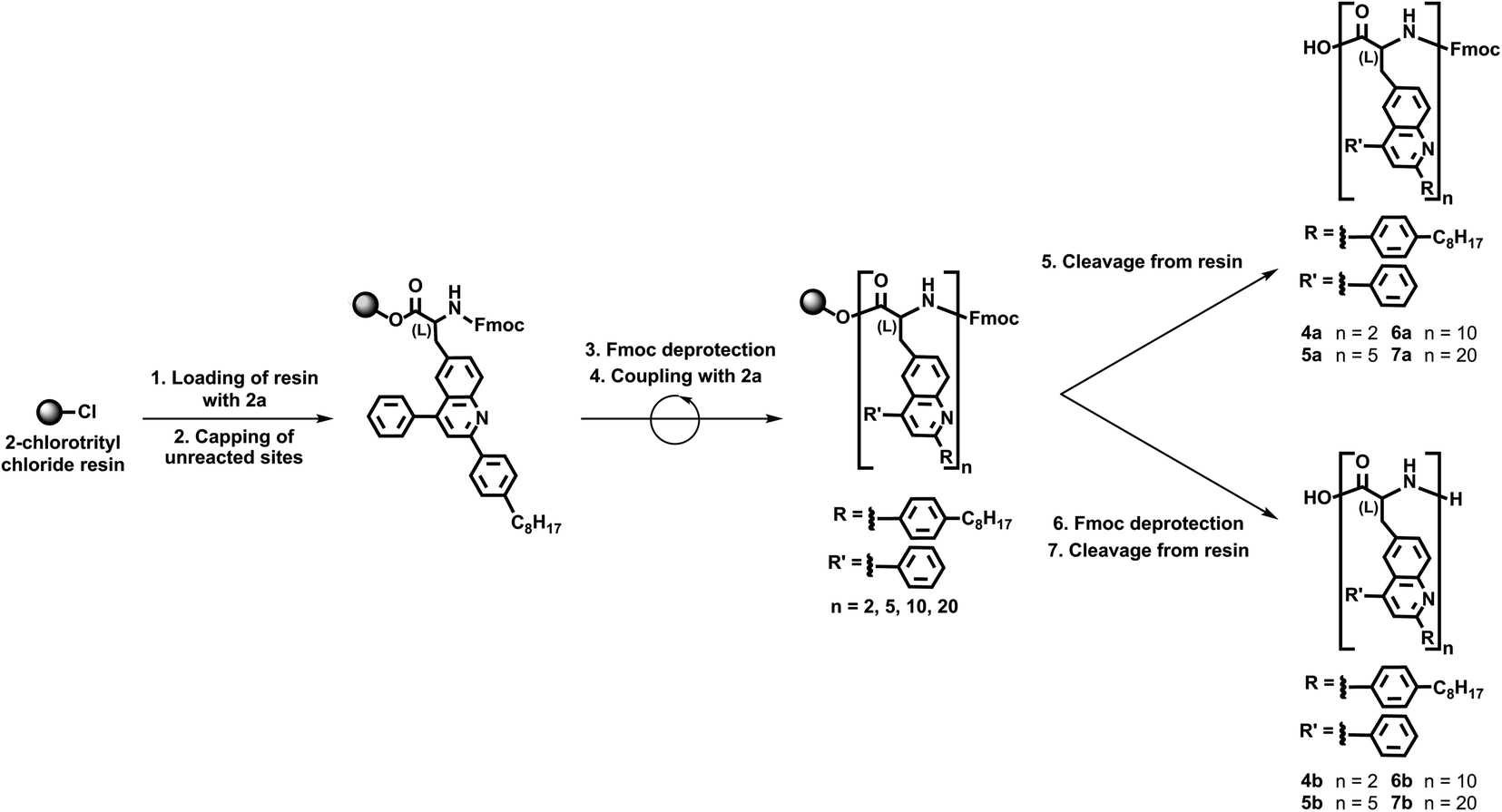 | ||
| Scheme 3 Synthesis of quinoline-based polypeptide surrogates 4a–7a and 4b–7b. | ||
We systematically characterized our polypeptide surrogates by using size exclusion chromatography (SEC). The representative SEC chromatograms obtained for deprotected surrogates 4b, 5b, 6b, and 7b are shown in Fig. 2. For dimer 4b, the chromatogram featured a sharp peak at a retention volume (Vr) of ∼10.47 mL and indicated a number average molecular weight (Mn) of ∼1.52 kg mol−1, a weight average molecular weight (Mw) of ∼1.6 kg mol−1, and a polydispersity index (PDI) of ∼1.06 (Fig. 2, green trace). For pentamer 5b, the chromatogram again featured a sharp peak at a Vr of ∼9.8 mL and indicated a Mn of ∼3.68 kg mol−1, a Mw of ∼3.88 kg mol−1, and a PDI of ∼1.05 (Fig. 2, blue trace). For decamer 6b, the chromatogram featured a broadened peak at a Vr of ∼9.29 mL and indicated a Mn of ∼5.88 kg mol−1, a Mw of ∼6.98 kg mol−1, and a PDI of ∼1.19 (Fig. 2, orange trace). For eicosamer 7b, the chromatogram featured a broadened, skewed peak at a Vr of ∼8.98 mL and indicated a Mn of ∼10.96 kg mol−1, a Mw of ∼12.33 kg mol−1, and a PDI of ∼1.13 (Fig. 2, red trace). In general, the number average molecular weights measured for our deprotected polypeptide surrogates were consistent with expectations for macromolecules from increasing numbers of quinoline-based building blocks. Notably, the representative chromatograms obtained for Fmoc-protected surrogates 4a, 5a, 6a, and 7a revealed number average molecular weights, weight average molecular weights, and polydispersities comparable to those of their deprotected analogues (Fig. S20†). Additionally, the MALDI mass spectra obtained for the relatively shorter polypeptides, i.e., 4a, 5a, 6a, 4b, 5b, and 6b, further confirmed their identities (Fig. S21–S26†). When considered together, these experiments confirmed the synthesis of the targeted variable-length, quinoline-based polypeptide surrogates.
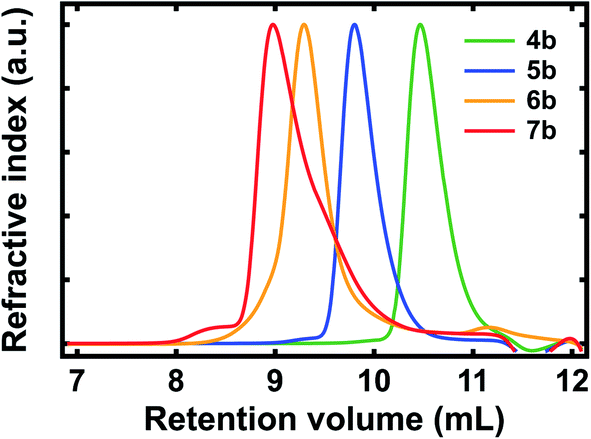 | ||
| Fig. 2 Representative SEC chromatograms obtained for quinoline-based polypeptide surrogates 4b (green trace), 5b (blue trace), 6b (orange trace), and 7b (red trace). | ||
With our polypeptide surrogates in hand, we studied their electronic and photophysical properties with UV-Vis absorption spectroscopy and fluorescence spectroscopy. The representative UV-Vis absorption and fluorescence emission spectra obtained for deprotected surrogates 4b, 5b, 6b, and 7b are shown in Fig. 3. First, for dimer 4b, the absorption spectrum revealed a maximum at ∼270 nm, a cluster of multiple sharp peaks between ∼300 nm and ∼350 nm, and an onset at ∼360 nm, with these features attributed to the pendant substituted quinolines (Fig. 3, green solid trace).35,36 For pentamer 5b, decamer 6b, and eicosamer 7b, the analogous spectra revealed multi-peak clusters that somewhat broadened and onsets that slightly red-shifted as the surrogates' lengths increased, in agreement with prior observations for polyquinolines and polybenzoquinolines (Fig. 3, blue, orange, and red solid traces).35,36,38 Moreover, for dimer 4b, the fluorescence spectrum revealed an emission maximum at ∼376 nm, in agreement with prior observations for biquinolines (Fig. 3, green dashed trace).36 For pentamer 5b, decamer 6b, and eicosamer 7b, the analogous spectra revealed emission maxima that slightly red-shifted as the polypeptide surrogates' lengths increased, in agreement with prior observations for zigzag polyquinolines (Fig. 3, blue, orange, and red dashed traces).36 Here, the representative UV-Vis absorption and fluorescence emission spectra obtained for terminal Fmoc-protected surrogates 4a, 5a, 6a, and 7a exhibited characteristics generally comparable to those of their deprotected analogues (Fig. S27†). These measurements elucidated and benchmarked our quinoline-based polypeptide surrogates' electronic and photophysical properties.
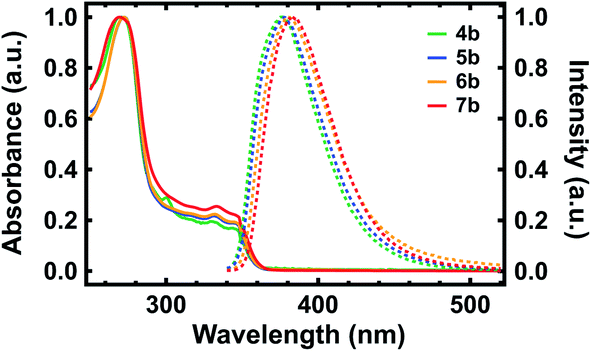 | ||
| Fig. 3 Representative UV-Vis absorption spectra obtained for quinoline-based polypeptide surrogates 4b (green solid trace), 5b (blue solid trace), 6b (orange solid trace), and 7b (red solid trace), and representative fluorescence emission spectra obtained for quinoline-based polypeptide surrogates 4b (green dashed trace), 5b (blue dashed trace), 6b (orange dashed trace), and 7b (red dashed trace). | ||
Last, we evaluated the optical activity of our polypeptide surrogates with circular dichroism (CD) spectroscopy. The representative CD spectra obtained for deprotected surrogates 4b, 5b, 6b, and 7b are shown in Fig. 4. First, for dimer 4b, the spectrum revealed positive peaks with maxima at ∼222 nm and ∼252 nm as well as a negative peak with a maximum at ∼276 nm (Fig. 4, green trace). Similarly, for pentamer 5b, the spectrum revealed positive peaks with maxima at ∼228 nm and ∼248 nm as well as a negative peak with a maximum at ∼286 nm (Fig. 4, blue trace). However, for decamer 6, the spectrum revealed a negative peak with a maximum at ∼220 nm, a positive peak with a maximum at ∼258 nm, and a negative peak with a maximum at ∼282 nm (Fig. 4, orange trace). Analogously, for eicosamer 7, the spectrum revealed a negative peak with a maximum at ∼234 nm, a positive peak with a maximum at ∼260 nm, and a negative peak with a maximum at ∼280 nm (Fig. 4, red trace). Notably, the representative CD spectra obtained for Fmoc-protected surrogates 4a, 5a, 6a, and 7a revealed peaks quite similar to those of the deprotected analogues (Fig. S28†). In general, the spectra suggested that the longer quinoline-based polypeptide surrogates, i.e. 6a, 7a, 6b, and 7b, exhibited more helical character when compared to the shorter surrogates, i.e. 4a, 5a, 4b, and 5b, in agreement with previous reports for quinoline-based oligoamide foldamers.46–48 These measurements afforded insight into the evolution of our polypeptide surrogates' length-dependent conformations in solution.
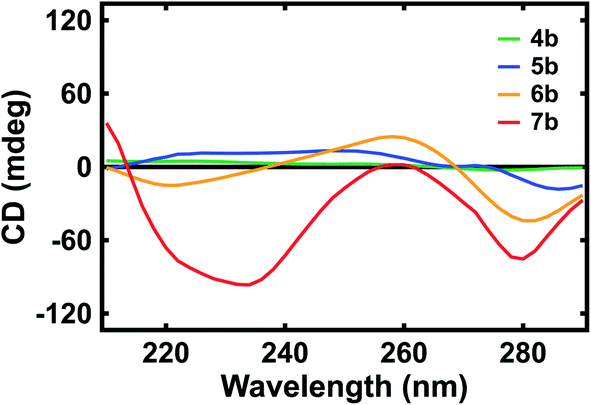 | ||
| Fig. 4 Representative CD spectra obtained for quinoline-based polypeptide surrogates 4b (green trace), 5b (blue trace), 6b (orange trace), and 7b (red trace). | ||
Discussion
In summary, we have designed, synthesized, and characterized quinoline-based polypeptide surrogates inspired by quinoline-containing oligoamide foldamers, and as such, it appears instructive to comparatively assess the current findings with respect to the relevant precedent. First, the described Povarov reaction-based approach to 2,4,6-substituted quinoline-based unnatural amino acid building blocks, which requires only 2 straightforward steps, mild reaction conditions, and commercially-available starting materials, may prove valuable from a methodology perspective with respect to those reported for the inspiring 2,4,8-substituted quinoline monomers.26–28,31,32 Second, the validated solid support-based strategy affords polypeptide surrogates for which the repeating units resemble those of oligoamide foldamers26–28,31,32 but the standard protein backbone resembles that of typical quinoline-decorated oligopeptides,24,25 thus complementing the reported classes of macromolecules. Third, although the absorption spectra obtained for our quinoline-based polypeptide surrogates generally matched the ones reported for comparable quinoline-containing oligoamide foldamers, the fluorescence spectra obtained for the surrogates revealed emission maxima that were blue-shifted with respect to those found for the oligoamide foldamers, suggesting relatively weaker pi–pi stacking interactions among our surrogates' quinoline-based building blocks.48,49 Last, the CD spectra obtained for our polypeptide surrogates and for oligoamide foldamers46–48 intimated that both types of macromolecules' longer variants adopted helical conformations in solution. Overall, the above analysis suggests that our findings are complementary to the ones in the prior studies and may expand the scope of possibilities for quinoline-based bioinspired polymers.In general, our ability to prepare quinoline-based unnatural amino acids and polypeptide surrogates could ultimately prove significant for several reasons. For instance, the described Povarov reaction-based approach is not only scalable but also amenable to further optimization for improved yields, portending favorably for the production of large quantities of building blocks with varying substituents on their quinoline moieties. This degree of synthetic tractability and modularity could open opportunities for the high-throughput construction of libraries of polypeptide surrogates with programmable lengths and sequences on solid support. In addition, our constructs possess a standard protein backbone and effectively consist of L-phenylalanine or L-tryptophan analogues, making them in principle compatible with integration of long tracts of canonical amino acids. Such inherent compatibility could facilitate the preparation of quinoline-based biomolecular materials that resemble block copolymers via traditional peptide chemistry protocols. Furthermore, our macromolecules exhibit electronic and spectroscopic characteristics analogous to those reported not only for quinoline-containing foldamers but also for various polyquinolines. These observations thus raise the intriguing possibility of designing and synthesizing extended polypeptide surrogates with valuable materials properties, such as thermal stability or mechanical robustness. Given the above considerations, our findings may ultimately enable the rational design and molecular engineering of new types of hybrid quinoline-based functional materials.
Experimental
Detailed synthetic protocols
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, chloroform:methanol), suspended in hexanes, and isolated by filtration as a yellow solid (2.4 g, 79%): 1H NMR (600 MHz, DMF-d7) δ 8.58 (s, 1H), 7.93 (d, J = 7.6 Hz, 2H), 7.89 (d, J = 7.8 Hz, 2H), 7.76 (d, J = 8.6 Hz, 1H), 7.72 (dd, J = 7.1, 3.8 Hz, 2H), 7.43 (m, 4H), 7.39 (d, J = 7.8 Hz, 2H), 7.34 (m, 2H), 7.25 (d, J = 7.9 Hz, 2H), 4.48 (m, 1H), 4.24 (m, 3H), 3.29 (dd, J = 13.8 Hz, 4.2 Hz, 1H), 3.08 (dd, J = 13.5 Hz, 10.4 Hz, 1H), 2.69 (t, J = 7.7 Hz, 2H), 2.12 (s, 1H), 1.65 (m, 2H), 1.39–1.19 (m, 10H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (151 MHz, DMF) δ 174.5, 160.9, 157.4, 151.6, 147.8, 145.29, 145.28, 142.22, 142.20, 137.0, 135.5, 131.2, 129.9, 129.8, 128.8, 128.2, 126.6, 126.5, 122.0, 121.2, 67.4, 57.1, 48.1, 37.9, 36.6, 32.8, 32.3, 31.13, 30.4, 30.21, 30.15, 23.5, 14.7. MALDI m/z calcd for C39H43N2O4 [M + H]+ 603.314, found 603.249.:5, chloroform: methanol) and isolated by precipitation from ethanol as a white solid (2.47 g, 46%): 1H NMR (600 MHz, DMF-d7) δ 8.53 (d, J = 7.9 Hz, 2H), 8.17 (d, J = 8.7 Hz, 1H), 8.05 (s, 1H), 7.98 (s, 1H), 7.90 (d, J = 7.8 Hz, 2H), 7.86 (m, 2H), 7.73 (d, J = 7.2 Hz, 2H), 7.66 (t, J = 7.6 Hz, 2H), 7.58 (m, 3H), 7.43 (d, J = 8.1 Hz, 1H), 7.40 (q, J = 7.5 Hz, 2H), 7.27 (t, J = 7.5 Hz, 1H), 7.23 (t, J = 7.5 Hz, 1H), 4.57 (m, 1ppH), 4.21 (m, 3H), 3.44 (dd, J = 13.9, 4.5 Hz, 1H), 3.26 (dd, J = 13.8, 10.1 Hz, 1H), 2.71 (t, J = 7.7 Hz, 2H), 1.68 (p, J = 7.5 Hz, 2H), 1.20–1.42 (m, 10H), 0.87 (t, J = 6.9 Hz, 3H). 13C NMR (151 MHz, DMF) δ 174.3, 157.4, 156.9, 149.7, 149.0, 145.6, 145.3, 145.2, 142.20, 142.18, 139.3, 137.9, 137.7, 132.5, 130.9, 129.9, 129.8, 129.6, 128.8, 128.7, 128.5, 128.2, 128.1, 126.7, 126.50, 126.46, 126.3, 121.1, 120.1, 67.4, 56.9, 48.1, 38.4, 36.5, 32.8, 32.4, 23.6, 14.8. MALDI m/z calcd for C47H47N2O4 [M + H]+ 703.346, found 703.206.:10, chloroform:methanol) and isolated as a white solid (0.57 g, 83%): 1H NMR (600 MHz, chloroform-d) δ 8.15 (d, J = 8.2 Hz, 1H), 7.94 (d, J = 7.8 Hz, 2H), 7.77 (s, 1H), 7.69 (s, 1H), 7.59 (d, J = 8.5 Hz, 1H), 7.52 (d, J = 7.2 Hz, 2H), 7.46 (t, J = 7.2 Hz, 2H), 7.41 (t, J = 7.2 Hz, 1H), 7.27 (d, J = 7.6 Hz, 2H), 3.99 (s, 1H), 3.27 (d, J = 13.5 Hz, 1H), 3.06 (dd, J = 14.4, 8.8 Hz, 1H), 2.64 (t, J = 7.2 Hz, 2H), 1.64–1.59 (m, 2H), 1.33–1.23 (m, 10H), 0.86 (t, J = 6.2 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 173.7, 156.7, 150.3, 146.9, 145.2, 137.8, 135.8, 134.2, 131.8, 129.7, 129.3, 129.1, 128.8, 128.7, 127.9, 126.6, 125.7, 120.2, 56.1, 36.6, 35.9, 31.9, 31.4, 29.6, 29.4, 29.4, 22.7, 14.2. MALDI m/z calcd for C32H37N2O2 [M + H]+ 481.278, found 481.161.:2:1, 8 mL) was added to the resin in the column. The resulting suspension was agitated for 1 hour to cap the unreacted resin sites. The solution was drained from the column, and the loaded and capped resin was washed with dry CH2Cl2 (×3) and with MeOH (×3) to remove excess reagents. The resin was dried by purging nitrogen gas through the chromatography column. The resin loading was typically 0.08 mmol [0.27 mmol g−1], as determined by cleavage and UV-Vis analysis of the Fmoc protecting groups. Second, the resin loaded with Fmoc-L-alanine was suspended in dry DMF and transferred to a 25 mL solid-phase peptide synthesis reaction vessel. Dyad 3 was synthesized on the solid support by (1) deprotection of the terminal amine, i.e., removal of the Fmoc, by treatment of the resin with 20% (v/v) piperidine in DMF (3 mL) for 30 min; (2) thorough washing of the resin with dry DMF (×3) to remove residual reagents; (3) coupling to the free terminal amines on the resin by treatment with 2a (0.54 mmol, 168 mg) and HCTU (0.54 mmol, 105 mg) in 20% (v/v) 2,4,6-collidine/dry DMF (3 mL) for 1 hour; and (4) thorough washing of the resin with dry DMF (×3) to remove residual reagents; (5) deprotection of the terminal amine, i.e., removal of the Fmoc, by treatment of the resin with 20% (v/v) piperidine in DMF (3 mL) for 30 min; and (6) thorough washing of the resin with dry DMF (×3) to remove residual reagents. The success of the coupling of 2a to the free terminal amine on the resin was routinely assessed by cleavage of the product and subsequent MALDI-TOF and chromatographic analyses. Third, the resin modified with dyad 3 was transferred to a 10 mL Bio-Rad Poly-Prep chromatography column. The resin was washed with dry DMF (×3) and dry CH2Cl2 (×3). Dyad 3 was cleaved from the solid support by addition of 20% hexafluoroisopropanol in dry CH2Cl2 (6 mL) and agitation of this suspension for 1 hour. The resulting filtrate from the column was collected in a round-bottom flask. The resin was washed with a second aliquot of 20% hexafluoroisopropanol in dry CH2Cl2 (6 mL) and with additional dry CH2Cl2 (×3). The resulting filtrate from the column was again collected in a round-bottom flask. The combined filtrates were concentrated under reduced pressure to afford a white residue, which was stored under low vacuum (<100 mTorr) to remove residual solvent. The dry filtrate was purified via flash chromatography (99:1, chloroform:methanol), and the product was isolated as a white solid (31 mg, 54%): 1H NMR (600 MHz, DMSO-d6) δ 8.9 (d, J = 7.2 Hz, 1H), 8.25 (d, J = 8.4 Hz, 2H), 8.15 (m, 3H), 8.02 (s, 1H), 7.85 (s, 1H), 7.82 (m, 1H), 7.73–7.69 (m, 2H), 7.65–7.60 (m, 2H), 7.62–7.55 (m, 1H), 7.38 (d, J = 8.0 Hz, 2H), 4.30 (p, J = 7.2 Hz, 1H), 4.14–4.05 (m, 1H), 3.32 (dd, J = 14.2, 5.0 Hz, 1H), 3.06 (dd, J = 14.2, 8.6 Hz, 1H), 2.67 (t, J = 7.6 Hz, 2H), 1.62 (m, 2H), 1.35–1.21 (m, 14H), 0.86 (t, J = 6.9 Hz, 3H). 13C NMR (151 MHz, DMSO) δ 173.7, 167.7, 158.2, 157.9, 155.4, 148.7, 144.4, 137.5, 135.6, 133.4, 131.7, 129.8, 128.8, 127.4, 126.2, 124.9, 119.0, 116.7, 114.8, 53.3, 47.7, 40.0, 39.9, 39.8, 39.7, 39.5, 39.4, 39.2, 39.1, 34.9, 31.3, 30.8, 28.8, 28.7, 22.1, 17.3, 13.9. MALDI m/z calcd for C35H42N3O3 [M + H]+ 552.315, found 552.231.:2:1, 8 mL) was added to the resin in the column. The resulting suspension was agitated for 1 hour to cap the unreacted resin sites. The solution was drained from the column, and the loaded and capped resin was washed with dry CH2Cl2 (×3) and with MeOH (×3) to remove excess reagents. The resin was dried by purging nitrogen gas through the chromatography column. The resin loading was typically 0.08 mmol [0.27 mmol g−1], as determined by cleavage and UV-Vis analysis of the Fmoc protecting groups. Second, the resin loaded with 2a was suspended in dry DMF and transferred to a 25 mL solid-phase peptide synthesis reaction vessel. Quinoline-based polypeptide surrogates 4a–7a were synthesized on the solid support by (1) deprotection of the terminal amine, i.e., removal of the Fmoc, by treatment of the resin with 20% (v/v) piperidine in DMF (3 mL) for 30 minutes; (2) thorough washing of the resin with dry DMF (×3) to remove residual reagents; (3) coupling to the free terminal amines on the resin by treatment with 2a (126 mg, 0.18 mmol) and HCTU (0.54 mmol, 105 mg) in 20% (v/v) 2,4,6-collidine/dry DMF (3 mL) for 1 hour; (4) thorough washing of the resin with dry DMF (×3) to remove residual reagents; and (5) repetition of steps (1) through (4) a total of 1, 4, 9, or 19 times to generate Fmoc-protected dimer 4a, Fmoc-protected pentamer 5a, Fmoc-protected decamer 6a, or Fmoc-protected eicosamer 7a, respectively. The success of the coupling to the free terminal amines on the resin was routinely assessed by cleavage of the product and subsequent MALDI-TOF and chromatographic analyses. Third, the resin modified with 4a, 5a, 6a, or 7a was transferred to a 10 mL Bio-Rad Poly-Prep chromatography column. The resin was washed with dry DMF (×3) and dry CH2Cl2 (×3). Polypeptide surrogates 4a, 5a, 6a, or 7a were cleaved from the solid support by addition of 20% hexafluoroisopropanol in dry CH2Cl2 (6 mL) and agitation of this suspension for 1 hour. The resulting filtrates from 4a, 5a, 6a, or 7a were collected in a round-bottom flask. The different resins were washed with a second aliquot of 20% hexafluoroisopropanol in dry CH2Cl2 (6 mL) and with additional dry CH2Cl2 (×3). The resulting filtrates from 4a, 5a, 6a, or 7a were again collected in a round-bottom flask. The filtrates from 4a, 5a, 6a, or 7a were individually concentrated under reduced pressure and stored independently under vacuum (<100 mTorr) to remove any residual solvents. The dry filtrates were purified via flash chromatography (99:1, chloroform:methanol), and the products were isolated as white solids (4a, 0.063 g, 54%), (5a, 0.126 g, 49%), (6a, 0.202 g, 42%), and (7a, 0.258 g, 34%).:1, chloroform:methanol), and the products were isolated as white solids (4b, 0.044 g, 47%), (5b, 0.175 g, 39%), (6b, 0.173 g, 37%), and (7b, 0.406 g, 55%).
:5, chloroform:methanol), suspended in hexanes, and isolated by filtration as a yellow solid (2.4 g, 79%): 1H NMR (600 MHz, DMF-d7) δ 8.58 (s, 1H), 7.93 (d, J = 7.6 Hz, 2H), 7.89 (d, J = 7.8 Hz, 2H), 7.76 (d, J = 8.6 Hz, 1H), 7.72 (dd, J = 7.1, 3.8 Hz, 2H), 7.43 (m, 4H), 7.39 (d, J = 7.8 Hz, 2H), 7.34 (m, 2H), 7.25 (d, J = 7.9 Hz, 2H), 4.48 (m, 1H), 4.24 (m, 3H), 3.29 (dd, J = 13.8 Hz, 4.2 Hz, 1H), 3.08 (dd, J = 13.5 Hz, 10.4 Hz, 1H), 2.69 (t, J = 7.7 Hz, 2H), 2.12 (s, 1H), 1.65 (m, 2H), 1.39–1.19 (m, 10H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (151 MHz, DMF) δ 174.5, 160.9, 157.4, 151.6, 147.8, 145.29, 145.28, 142.22, 142.20, 137.0, 135.5, 131.2, 129.9, 129.8, 128.8, 128.2, 126.6, 126.5, 122.0, 121.2, 67.4, 57.1, 48.1, 37.9, 36.6, 32.8, 32.3, 31.13, 30.4, 30.21, 30.15, 23.5, 14.7. MALDI m/z calcd for C39H43N2O4 [M + H]+ 603.314, found 603.249.:5, chloroform: methanol) and isolated by precipitation from ethanol as a white solid (2.47 g, 46%): 1H NMR (600 MHz, DMF-d7) δ 8.53 (d, J = 7.9 Hz, 2H), 8.17 (d, J = 8.7 Hz, 1H), 8.05 (s, 1H), 7.98 (s, 1H), 7.90 (d, J = 7.8 Hz, 2H), 7.86 (m, 2H), 7.73 (d, J = 7.2 Hz, 2H), 7.66 (t, J = 7.6 Hz, 2H), 7.58 (m, 3H), 7.43 (d, J = 8.1 Hz, 1H), 7.40 (q, J = 7.5 Hz, 2H), 7.27 (t, J = 7.5 Hz, 1H), 7.23 (t, J = 7.5 Hz, 1H), 4.57 (m, 1ppH), 4.21 (m, 3H), 3.44 (dd, J = 13.9, 4.5 Hz, 1H), 3.26 (dd, J = 13.8, 10.1 Hz, 1H), 2.71 (t, J = 7.7 Hz, 2H), 1.68 (p, J = 7.5 Hz, 2H), 1.20–1.42 (m, 10H), 0.87 (t, J = 6.9 Hz, 3H). 13C NMR (151 MHz, DMF) δ 174.3, 157.4, 156.9, 149.7, 149.0, 145.6, 145.3, 145.2, 142.20, 142.18, 139.3, 137.9, 137.7, 132.5, 130.9, 129.9, 129.8, 129.6, 128.8, 128.7, 128.5, 128.2, 128.1, 126.7, 126.50, 126.46, 126.3, 121.1, 120.1, 67.4, 56.9, 48.1, 38.4, 36.5, 32.8, 32.4, 23.6, 14.8. MALDI m/z calcd for C47H47N2O4 [M + H]+ 703.346, found 703.206.:10, chloroform:methanol) and isolated as a white solid (0.57 g, 83%): 1H NMR (600 MHz, chloroform-d) δ 8.15 (d, J = 8.2 Hz, 1H), 7.94 (d, J = 7.8 Hz, 2H), 7.77 (s, 1H), 7.69 (s, 1H), 7.59 (d, J = 8.5 Hz, 1H), 7.52 (d, J = 7.2 Hz, 2H), 7.46 (t, J = 7.2 Hz, 2H), 7.41 (t, J = 7.2 Hz, 1H), 7.27 (d, J = 7.6 Hz, 2H), 3.99 (s, 1H), 3.27 (d, J = 13.5 Hz, 1H), 3.06 (dd, J = 14.4, 8.8 Hz, 1H), 2.64 (t, J = 7.2 Hz, 2H), 1.64–1.59 (m, 2H), 1.33–1.23 (m, 10H), 0.86 (t, J = 6.2 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 173.7, 156.7, 150.3, 146.9, 145.2, 137.8, 135.8, 134.2, 131.8, 129.7, 129.3, 129.1, 128.8, 128.7, 127.9, 126.6, 125.7, 120.2, 56.1, 36.6, 35.9, 31.9, 31.4, 29.6, 29.4, 29.4, 22.7, 14.2. MALDI m/z calcd for C32H37N2O2 [M + H]+ 481.278, found 481.161.:2:1, 8 mL) was added to the resin in the column. The resulting suspension was agitated for 1 hour to cap the unreacted resin sites. The solution was drained from the column, and the loaded and capped resin was washed with dry CH2Cl2 (×3) and with MeOH (×3) to remove excess reagents. The resin was dried by purging nitrogen gas through the chromatography column. The resin loading was typically 0.08 mmol [0.27 mmol g−1], as determined by cleavage and UV-Vis analysis of the Fmoc protecting groups. Second, the resin loaded with Fmoc-L-alanine was suspended in dry DMF and transferred to a 25 mL solid-phase peptide synthesis reaction vessel. Dyad 3 was synthesized on the solid support by (1) deprotection of the terminal amine, i.e., removal of the Fmoc, by treatment of the resin with 20% (v/v) piperidine in DMF (3 mL) for 30 min; (2) thorough washing of the resin with dry DMF (×3) to remove residual reagents; (3) coupling to the free terminal amines on the resin by treatment with 2a (0.54 mmol, 168 mg) and HCTU (0.54 mmol, 105 mg) in 20% (v/v) 2,4,6-collidine/dry DMF (3 mL) for 1 hour; and (4) thorough washing of the resin with dry DMF (×3) to remove residual reagents; (5) deprotection of the terminal amine, i.e., removal of the Fmoc, by treatment of the resin with 20% (v/v) piperidine in DMF (3 mL) for 30 min; and (6) thorough washing of the resin with dry DMF (×3) to remove residual reagents. The success of the coupling of 2a to the free terminal amine on the resin was routinely assessed by cleavage of the product and subsequent MALDI-TOF and chromatographic analyses. Third, the resin modified with dyad 3 was transferred to a 10 mL Bio-Rad Poly-Prep chromatography column. The resin was washed with dry DMF (×3) and dry CH2Cl2 (×3). Dyad 3 was cleaved from the solid support by addition of 20% hexafluoroisopropanol in dry CH2Cl2 (6 mL) and agitation of this suspension for 1 hour. The resulting filtrate from the column was collected in a round-bottom flask. The resin was washed with a second aliquot of 20% hexafluoroisopropanol in dry CH2Cl2 (6 mL) and with additional dry CH2Cl2 (×3). The resulting filtrate from the column was again collected in a round-bottom flask. The combined filtrates were concentrated under reduced pressure to afford a white residue, which was stored under low vacuum (<100 mTorr) to remove residual solvent. The dry filtrate was purified via flash chromatography (99:1, chloroform:methanol), and the product was isolated as a white solid (31 mg, 54%): 1H NMR (600 MHz, DMSO-d6) δ 8.9 (d, J = 7.2 Hz, 1H), 8.25 (d, J = 8.4 Hz, 2H), 8.15 (m, 3H), 8.02 (s, 1H), 7.85 (s, 1H), 7.82 (m, 1H), 7.73–7.69 (m, 2H), 7.65–7.60 (m, 2H), 7.62–7.55 (m, 1H), 7.38 (d, J = 8.0 Hz, 2H), 4.30 (p, J = 7.2 Hz, 1H), 4.14–4.05 (m, 1H), 3.32 (dd, J = 14.2, 5.0 Hz, 1H), 3.06 (dd, J = 14.2, 8.6 Hz, 1H), 2.67 (t, J = 7.6 Hz, 2H), 1.62 (m, 2H), 1.35–1.21 (m, 14H), 0.86 (t, J = 6.9 Hz, 3H). 13C NMR (151 MHz, DMSO) δ 173.7, 167.7, 158.2, 157.9, 155.4, 148.7, 144.4, 137.5, 135.6, 133.4, 131.7, 129.8, 128.8, 127.4, 126.2, 124.9, 119.0, 116.7, 114.8, 53.3, 47.7, 40.0, 39.9, 39.8, 39.7, 39.5, 39.4, 39.2, 39.1, 34.9, 31.3, 30.8, 28.8, 28.7, 22.1, 17.3, 13.9. MALDI m/z calcd for C35H42N3O3 [M + H]+ 552.315, found 552.231.:2:1, 8 mL) was added to the resin in the column. The resulting suspension was agitated for 1 hour to cap the unreacted resin sites. The solution was drained from the column, and the loaded and capped resin was washed with dry CH2Cl2 (×3) and with MeOH (×3) to remove excess reagents. The resin was dried by purging nitrogen gas through the chromatography column. The resin loading was typically 0.08 mmol [0.27 mmol g−1], as determined by cleavage and UV-Vis analysis of the Fmoc protecting groups. Second, the resin loaded with 2a was suspended in dry DMF and transferred to a 25 mL solid-phase peptide synthesis reaction vessel. Quinoline-based polypeptide surrogates 4a–7a were synthesized on the solid support by (1) deprotection of the terminal amine, i.e., removal of the Fmoc, by treatment of the resin with 20% (v/v) piperidine in DMF (3 mL) for 30 minutes; (2) thorough washing of the resin with dry DMF (×3) to remove residual reagents; (3) coupling to the free terminal amines on the resin by treatment with 2a (126 mg, 0.18 mmol) and HCTU (0.54 mmol, 105 mg) in 20% (v/v) 2,4,6-collidine/dry DMF (3 mL) for 1 hour; (4) thorough washing of the resin with dry DMF (×3) to remove residual reagents; and (5) repetition of steps (1) through (4) a total of 1, 4, 9, or 19 times to generate Fmoc-protected dimer 4a, Fmoc-protected pentamer 5a, Fmoc-protected decamer 6a, or Fmoc-protected eicosamer 7a, respectively. The success of the coupling to the free terminal amines on the resin was routinely assessed by cleavage of the product and subsequent MALDI-TOF and chromatographic analyses. Third, the resin modified with 4a, 5a, 6a, or 7a was transferred to a 10 mL Bio-Rad Poly-Prep chromatography column. The resin was washed with dry DMF (×3) and dry CH2Cl2 (×3). Polypeptide surrogates 4a, 5a, 6a, or 7a were cleaved from the solid support by addition of 20% hexafluoroisopropanol in dry CH2Cl2 (6 mL) and agitation of this suspension for 1 hour. The resulting filtrates from 4a, 5a, 6a, or 7a were collected in a round-bottom flask. The different resins were washed with a second aliquot of 20% hexafluoroisopropanol in dry CH2Cl2 (6 mL) and with additional dry CH2Cl2 (×3). The resulting filtrates from 4a, 5a, 6a, or 7a were again collected in a round-bottom flask. The filtrates from 4a, 5a, 6a, or 7a were individually concentrated under reduced pressure and stored independently under vacuum (<100 mTorr) to remove any residual solvents. The dry filtrates were purified via flash chromatography (99:1, chloroform:methanol), and the products were isolated as white solids (4a, 0.063 g, 54%), (5a, 0.126 g, 49%), (6a, 0.202 g, 42%), and (7a, 0.258 g, 34%).:1, chloroform:methanol), and the products were isolated as white solids (4b, 0.044 g, 47%), (5b, 0.175 g, 39%), (6b, 0.173 g, 37%), and (7b, 0.406 g, 55%).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Mass Spectrometry Facility and the Laser Spectroscopy Labs at University of California, Irvine for access to experimental facilities. This work was supported by the Office of Naval Research N00014-16-1-2741 and the Air Force Office of Scientific Research FA9550-16-1-0296.References
- A. L. Rusanov, L. G. Komarova, M. P. Prigozhina and D. Y. Likhatchev, Russ. Chem. Rev., 2005, 74, 671–683 CrossRef CAS.
- A. Kimyonok, X. Wang and M. Weck, J. Macromol. Sci., Part C: Polym. Rev., 2006, 46, 47–77 CrossRef CAS.
- A. P. Kulkarni, A. P. Gifford, C. J. Tonzola and S. A. Jenekhe, Chem. Mater., 2004, 16, 4556–4573 CrossRef CAS.
- X. Zhao and X. Zhan, Chem. Soc. Rev., 2011, 40, 3728–3743 RSC.
- S. Kumar, S. Bawa and H. Gupta, Mini-Rev. Med. Chem., 2009, 9, 1648–1654 CrossRef CAS PubMed.
- V. R. Solomon and H. Lee, Curr. Med. Chem., 2011, 18, 1488–1508 CrossRef CAS PubMed.
- M. Kukowska, Eur. J. Pharm. Sci., 2017, 109, 587–615 CrossRef CAS PubMed.
- R. Gopalakrishnan, A. I. Frolov, L. Knerr, W. J. Drury III and E. Valeur, J. Med. Chem., 2016, 59, 9599–9621 CrossRef CAS PubMed.
- M. Pasco, C. Dolain and G. Guichard, in Comprehensive Supramolecular Chemistry II, ed. J. L. Atwood, Elsevier, 2nd edn, 2017, ch. 5, pp. 89–125 Search PubMed.
- S. Kumar, M. Birol, D. E. Schlamadinger, S. P. Wojcik, E. Rhoades and A. D. Miranker, Nat. Commun., 2016, 7, 11412 CrossRef CAS PubMed.
- B. Siemssen, K. W. Kim, M. S. Kim, B. S. Kim, S. J. Cho, D. K. Park, H. S. Woo and T. W. Kwon, Mol. Cryst. Liq. Cryst., 2006, 462, 159–167 CrossRef.
- M. Tomar, N. T. Lucas, H. Kim, F. Laquai, K. Müllen and J. Jacob, Polym. Int., 2012, 61, 1318–1325 CrossRef CAS.
- C. G. Bangcuyo, M. E. Rampey-Vaughn, L. T. Quan, S. M. Angel, M. D. Smith and U. H. F. Bunz, Macromolecules, 2002, 35, 1563–1568 CrossRef CAS.
- G. Jégou and S. A. Jenekhe, Macromolecules, 2001, 34, 7926–7928 CrossRef.
- A. Bilici, F. Doğan, M. Yıldırım and İ. Kaya, React. Funct. Polym., 2011, 71, 675–683 CrossRef CAS.
- W. H. Beever and J. K. Stille, Macromolecules, 1979, 12, 1033–1038 CrossRef CAS.
- J. Marco-Contelles, E. Pérez-Mayoral, A. Samadi, M. D. C. Carreiras and E. Soriano, Chem. Rev., 2009, 109, 2652–2671 CrossRef CAS PubMed.
- D. M. Sutherlin, J. K. Stille and W. B. Alston, Macromolecules, 1986, 19, 257–266 CrossRef CAS.
- A. K.-Y. Jen, X. Wu and H. Ma, Chem. Mater., 1998, 10, 471–473 CrossRef CAS.
- H. Ma, X. Wang, X. Wu, S. Liu and A. K.-Y. Jen, Macromolecules, 1998, 31, 4049–4052 CrossRef CAS.
- J. K. Stille, Macromolecules, 1981, 14, 870–880 CrossRef CAS.
- W. Wrasidlo, S. O. Norris, J. F. Wolfe, T. Katto and J. K. Stille, Macromolecules, 1976, 9, 512–516 CrossRef CAS.
- A. K. Agrawal and S. A. Jenekhe, Chem. Mater., 1992, 4, 95–104 CrossRef CAS.
- P. Liu, B. Li, M. Xi, Z. Chen, H. Sun, X. Huan, X. Xu, Y. Zhang, K. Zou, X. Jiang, Z. Miao, J. Liu, J. Shen, K. Chen and W. Zhu, Green Chem., 2019, 21, 4231–4237 RSC.
- M. Krishnamurthy, B. D. Gooch and P. A. Beal, Org. Biomol. Chem., 2006, 4, 639–645 RSC.
- S. J. Dawson, X. Hu, S. Claerhout and I. Huc, Methods Enzymol., 2016, 580, 279–300 CAS.
- X. Hu, S. J. Dawson, Y. Nagaoka, A. Tanatani and I. Huc, J. Org. Chem., 2016, 81, 1137–1150 CrossRef CAS PubMed.
- B. Baptiste, C. Douat-Casassus, K. Laxmi-Reddy and I. Huc, J. Org. Chem., 2010, 75, 7175–7185 CrossRef CAS PubMed.
- E. R. Gillies, F. Deiss, J.-M. Schmitter and I. Huc, Angew. Chem., Int. Ed., 2007, 46, 4081–4084 CrossRef CAS PubMed.
- J. Iriondo-Alberdi, K. Laxmi-Reddy, B. Bouguerne, C. Staedel and I. Huc, ChemBioChem, 2010, 11, 1679–1685 CrossRef CAS PubMed.
- H. Jiang, J. M. Léger, C. Dolain, P. Guionneau and I. Huc, Tetrahedron, 2003, 59, 8365–8374 CrossRef CAS.
- H. Jiang, J.-M. Leger and I. Huc, J. Am. Chem. Soc., 2003, 125, 3448–3449 CrossRef CAS PubMed.
- N. Delsuc, T. Kawanami, J. Lefeuvre, A. Shundo, H. Ihara, M. Takafuji and I. Huc, ChemPhysChem, 2008, 9, 1882–1890 CrossRef CAS PubMed.
- T. Qi, V. Maurizot, H. Noguchi, T. Charoenraks, B. Kauffmann, M. Takafuji, H. Ihara and I. Huc, Chem. Commun., 2012, 48, 6337–6339 RSC.
- D. J. Dibble, M. J. Umerani, A. Mazaheripour, Y. S. Park, J. W. Ziller and A. A. Gorodetsky, Macromolecules, 2015, 48, 557–561 CrossRef CAS.
- M. J. Umerani, D. J. Dibble, A. G. Wardrip, A. Mazaheripour, E. Vargas, J. W. Ziller and A. A. Gorodetsky, J. Mater. Chem. C, 2016, 4, 4060–4066 RSC.
- D. J. Dibble, R. Kurakake, A. G. Wardrip, A. Bartlett, R. Lopez, J. A. Linares, M. Firstman, A. M. Schmidt, M. J. Umerani and A. A. Gorodetsky, Org. Lett., 2018, 20, 502–505 CrossRef CAS PubMed.
- D. J. Dibble, Y. S. Park, A. Mazaheripour, M. J. Umerani, J. W. Ziller and A. A. Gorodetsky, Angew. Chem., Int. Ed., 2015, 54, 5883–5887 CrossRef CAS PubMed.
- A. Mazaheripour, D. J. Dibble, M. J. Umerani, Y. S. Park, R. Lopez, D. Laidlaw, E. Vargas, J. W. Ziller and A. A. Gorodetsky, Org. Lett., 2016, 18, 156–159 CrossRef CAS PubMed.
- Y. S. Park, D. J. Dibble, J. Kim, R. C. Lopez, E. Vargas and A. A. Gorodetsky, Angew. Chem., Int. Ed., 2016, 55, 3352–3355 CrossRef CAS PubMed.
- Z. Feng, A. Mazaheripour, D. J. Dibble, P. Wagner, G. Czap, G. Kladnik, A. Cossaro, A. Verdini, L. Floreano, G. Bavdek, W. Ho, G. Comelli, D. Cvetko, A. Morgante and A. A. Gorodetsky, Carbon, 2020, 170, 677–684 CrossRef CAS.
- L. S. Povarov, Russ. Chem. Rev., 1967, 36, 656–670 CrossRef.
- V. V. Kouznetsov, Tetrahedron, 2009, 65, 2721–2750 CrossRef CAS.
- I. Coin, M. Beyermann and M. Bienert, Nat. Protoc., 2007, 2, 3247–3256 CrossRef CAS PubMed.
- H. Yang, K. H. Chen and J. S. Nowick, ACS Chem. Biol., 2016, 11, 1823–1826 CrossRef CAS PubMed.
- M. Kudo, V. Maurizot, B. Kauffmann, A. Tanatani and I. Huc, J. Am. Chem. Soc., 2013, 135, 9628–9631 CrossRef CAS PubMed.
- M. Kudo, V. Maurizot, H. Masu, A. Tanatani and I. Huc, Chem. Commun., 2014, 50, 10090–10093 RSC.
- D. Zheng, L. Zheng, C. Yu, Y. Zhan, Y. Wang and H. Jiang, Org. Lett., 2019, 21, 2555–2559 CrossRef CAS PubMed.
- X. Li, N. Markandeya, G. Jonusauskas, N. D. McClenaghan, V. Maurizot, S. A. Denisov and I. Huc, J. Am. Chem. Soc., 2016, 138, 13568–13578 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04783j |
| This journal is © The Royal Society of Chemistry 2021 |