
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidating the influence of molten salt chemistries on the synthesis and stability of perovskites oxides†
Benjamin Levitas a,
Spencer Piligianb,
Thomas Irelandc and
Srikanth Gopalan*ad
a,
Spencer Piligianb,
Thomas Irelandc and
Srikanth Gopalan*ad
aDivision of Materials Science and Engineering, Boston University, Brookline, Massachusetts 02445, USA. E-mail: sgopalan@bu.edu
bDepartment of Electrical and Computer Engineering, Boston University, Boston, Massachusetts 02215, USA
cDepartment of Earth and Environment, Boston University, Boston, Massachusetts 02215, USA
dDepartment of Mechanical Engineering, Boston University, Boston, Massachusetts 02215, USA
First published on 1st September 2021
Abstract
In this work, we investigate the synthesis of (La0.8Sr0.2)MnO3 (LSM) in various molten salts to gain insight on the influence of molten salt ions for synthesizing materials critical for energy applications. LSM nanoparticles with a size range of ∼10–200 nm and with target stoichiometries were formed from oxide precursors via feeding into KNO3. Furthermore, feeding precursors into the melt compared to mixing and heating from room temperature results in complete formation of LSM that was otherwise unattainable using conventional molten salt synthesis methods. In LiCl–KCl eutectic, the high Lux acidity of Li+ and Cl− establishes a thermodynamic barrier that impedes Sr from reacting with other precursors in solution and increases Sr stability in the melt compared to the perovskite phase. As a result, LSM will not form in a LiCl–KCl eutectic under ambient conditions. Thus, this study further explicates the molten salt synthesis for perovskites and can serve as a guide for future syntheses.
Introduction
Perovskite type oxides (PTOs) are prevalent in numerous applications such as superconductors, catalysts, electronics, magnets, and fuel cells.1–4 In particular, La1−xSrxMnO3−δ (LSM) is a widely used oxygen electrode material for solid oxide fuel cells (SOFCs) and solid oxide electrolysis cells (SOECs). Synthesizing LSM, however, requires sintering temperatures above 1000 °C for as long as 10 hours.5–8 The molten salt synthesis (MSS) method is a viable option to form PTOs at reduced temperatures. The reaction occurs in the liquid state and proceeds rapidly since diffusion in a liquid is orders of magnitude faster than in a solid and thus induces intimate and rapid contact of the precursor species. For example, the diffusion coefficient for solid state sintering of gadolinium doped ceria ranges from 10−12 to 10−8 cm2 s−1 from 800–1100 °C.9 On the other hand, in a molten LiCl–KCl eutectic at 500 °C, the diffusion coefficients for lanthanides and actinides range from 10−3 to 10−5 cm2 s−1.10 By contrast, at 500 °C, diffusion in the solid state in inorganic oxides is immeasurably slow. The rapid diffusion and reaction in the liquid solvent results in reduced temperatures and times required to form PTOs.11,12 One limitation of the MSS is that the salt system be chemically inert and have a low (but non-zero) solubility for the precursors and the product.Molten salt systems have been widely studied and are known to contain rich and diverse chemical environments. For example, species such as superoxides and dissolved metals, which are usually nonexistent in a solid-state PTO synthesis, can be found in the MSS.13 The overarching chemical basis of molten salt chemistry and synthesis is understood through the Lux-Flood acid base theory:14
| Base → acid + O2− | (1) |
The base is the oxygen ion donor and the acid the oxygen ion acceptor. The fundamental thermodynamic principles driving the formation of the product phase in the molten salt are thoroughly described by Kimura.15 Essentially, reactions occur between the dissolved species in the molten salt and products precipitate out of solution once the concentration of the product species in solution in the molten salt exceeds its solubility limit. Solubilities of species within the solvent are dependent on their acidity and the oxygen ion concentration (basicity) within the melt.16 Many studies have been done to synthesize perovskites using the MSS,17–19 including LSM,20–22 but how a particular molten salt influences the thermodynamics and kinetics of nucleation and growth has not been explicitly studied to the best of our knowledge. Moreover, studies that have presented the MSS of LSM using salts such as LiCl–KCl23 do not provide elemental analysis of the formed powders to determine if LSM of the target composition truly forms.
This study investigates the thermodynamic influence of molten salt cations and anions to synthesize the PTO, LSM, in five different salt systems. These salts include halide and oxosalt eutectics such as LiCl–KCl, LiNO3–KNO3, and Li2CO3–K2CO3, as well as single oxosalts such as LiNO3 and KNO3. Oxosalts directly provide oxygen ions to the precursors in solution and thus increase the basicity, whereas halides do not provide oxygen ions and increase the acidity. The cations are thus varied between Li+ and K+ to directly compare the high acidity of Li+ to the more basic K+.24 This study focuses on using oxide precursors for LSM. Past studies detailing LSM MSS use nitrate precursors,20–22 which are hygroscopic, require specific storing conditions, and lack easy bulk preparation for scalable production of LSM. Furthermore, we apply a feeding procedure that has been previously employed to synthesize LaCoO3 nanoparticles with dramatically reduced particle size compared to when LaCoO3 precursors were heated up to the dwell temperature25(see ESI† for all methodologies). However, the present study also highlights how effectively this feeding procedure facilitates the formation of LSM. Moreover, the feeding procedure provides a way to bypass certain pitfalls of conventional MSS methods for LSM production. A combination of X-ray diffraction (XRD), Energy Dispersive X-ray Spectroscopy (EDX), and Inductively Coupled Plasma Mass Spectrometry (ICP-MS) reveals that PTO structures can form in all salts tested, but the increased Lux acidity of a molten salt impedes pure phase formation of LSM. In doing so, we present a deeper understanding of molten salt chemistries in the context of LSM formation, which can be applied to synthesize other relevant highly ordered, complex PTOs for energy applications.
Results and discussion
Influence of molten salt anion on LSM formation
The XRD patterns for high temperature calcined (HTC) LSM, molten salt attempted synthesis in Li2CO3–K2CO3 eutectic, LiNO3–KNO3 eutectic, and LiCl–KCl eutectic are shown together in Fig. 1. Each of the three attempted MSS LSM patterns contain peaks that correspond to LSM when compared to the HTC pattern, yet there are a significant number of peaks matched to secondary phases besides LSM. In particular, when the synthesis was performed in Li2CO3–K2CO3 and LiNO3–KNO3, peaks for leftover SrCO3 are present. Li2CO3 is identified in the Li2CO3–K2CO3 pattern since Li2CO3 has an extremely low solubility in water26 and remained in the powder mixture even after washing for more than 20 times. While the LiCl–KCl synthesis pattern does not contain any leftover reactants, there are peaks matched to Li2MnO3. In fact, Li2MnO3 is indexed for all three of the patterns of molten salt synthesized powders. Since the LiCl–KCl synthesized powder did not contain any leftover SrCO3 based on XRD, this powder was analyzed using SEM EDX to compare the elemental composition to HTC LSM. As shown in Fig. 1b and c, the Sr L-line around 1.8 keV in the LiCl–KCl synthesized powder is substantially lower than the corresponding peak for HTC LSM. Moreover, the major Mn K-line around 5.9 keV for LiCl–KCl synthesized powder is diminished to that in the HTC LSM spectrum. These two results indicate that (1) LSM does not appreciably form in any of the eutectic molten salts analyzed under these conditions and (2) the La1−xMnO3 perovskite that forms in LiCl–KCl is Mn-deficient compared to HTC LSM perovskite as a result of the Li+ reacting with the leftover Mn2O3 precursor.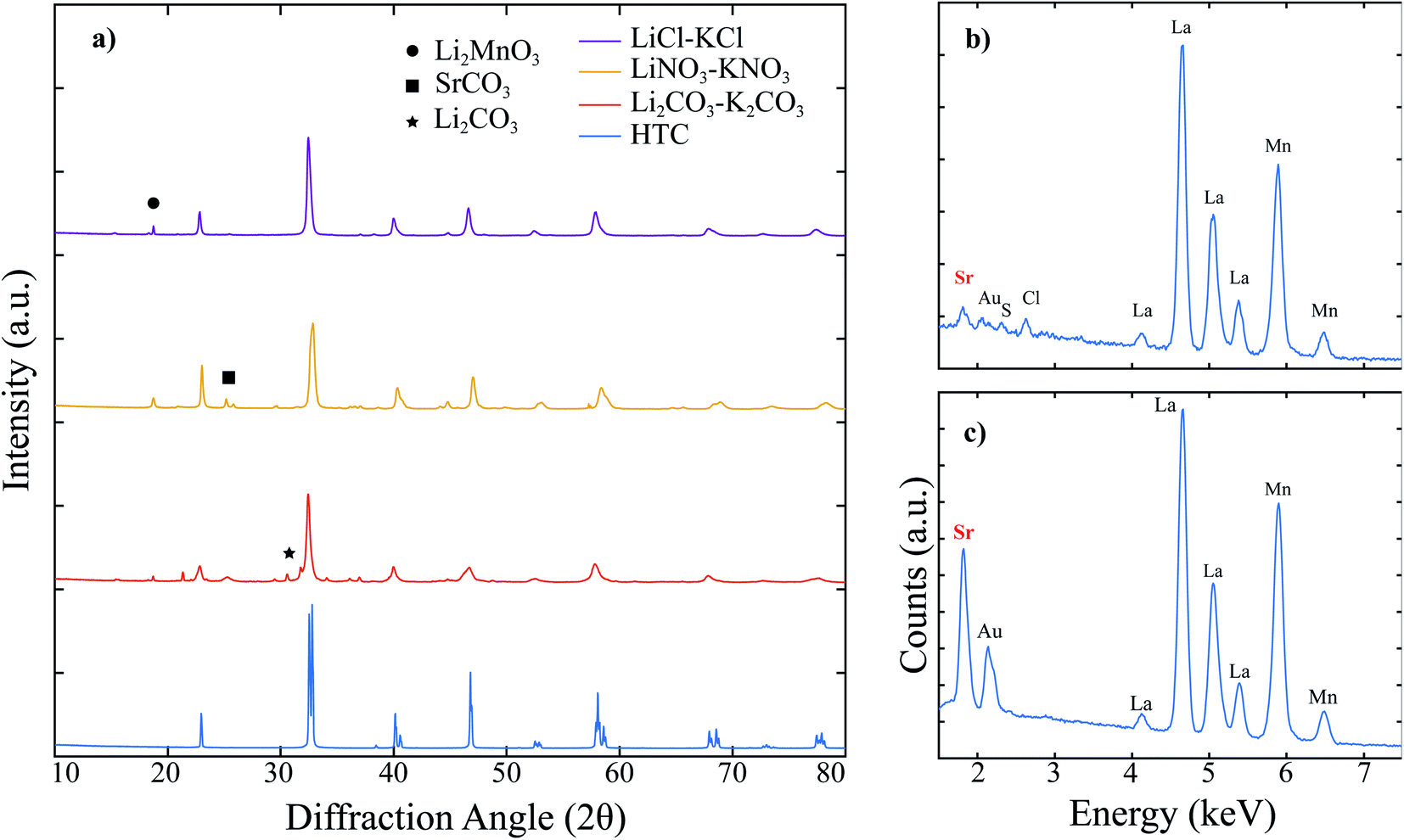 | ||
Fig. 1 (a) XRD patterns of attempted molten salt synthesized (MSS) LSM powders in different molten salts at 600 °C for 2 hours compared to LSM synthesized via high temperature calcination (HTC) at 1200 °C for 10 hours. The precursor powders were mixed oxides in stoichiometric amounts, and the powder to salt mass ratio was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8. (b) EDX spectra for the LiCl–KCl attempted MSS and (c) EDX spectra for the HTC LSM, which compare the intensities of the Sr L-line highlighted in red to show LSM does not appreciably form when LiCl–KCl is used. :8. (b) EDX spectra for the LiCl–KCl attempted MSS and (c) EDX spectra for the HTC LSM, which compare the intensities of the Sr L-line highlighted in red to show LSM does not appreciably form when LiCl–KCl is used. | ||
The molten salt syntheses were repeated, but instead the amount of molten salt eutectics was adjusted relative to the LSM oxide precursors. The XRD patterns are presented Fig. S2–S4.† Adjusting the amount of Li2CO3–K2CO3 has no apparent effect on the formation of LSM, since amorphous SrCO3 is present in all three patterns and pure phase LSM is not present. The peaks for Li2CO3 are present in all three patterns for the same reasoning as described above. Again, Li2CO3 requires a significant amount of water to completely separate the product powders from salt and thus is not a suitable candidate for large scale productions using the MSS. As a result, this salt system was not investigated further. Adjusting the amount of LiNO3–KNO3 relative to LSM oxide precursors yields many secondary phases coinciding with the perovskite peaks. As seen in Fig. S3,† pure phase LSM is not achieved for any salt amount tested. The patterns for attempted LSM in LiNO3–KNO3 contain numerous peaks besides those corresponding to LSM as shown in the HTC pattern. When the amount of LiCl–KCl was varied relative to LSM precursors, the XRD patterns indicate small peaks corresponding to Li2MnO3. However, no peaks for SrCO3 are present, which suggests that LSM may form to some extent as opposed to LiNO3–KNO3 or Li2CO3–K2CO3, which had SrCO3 peaks present in the respective XRD patterns. As a result, additional syntheses were conducted for 8 hours to further investigate the possibility of LSM formation in LiCl–KCl. Nonetheless, the EDX spectra for each attempted synthesis shown in Fig. S4b–e† shows that the Sr L-lines in each are substantially lower in intensity than HTC LSM. Therefore, despite the XRD patterns suggesting LSM formation in LiCl–KCl, EDX confirms that the perovskite that forms is Sr-deficient. Since SrCO3 is not present in the XRD pattern of the collected powder, it is possible that this precursor was dissolved and washed away with the salt, post synthesis.
Pure LSM was determined to be unable to form in any of the eutectic molten salt systems tested thus far. To gain more insight on the thermodynamics and interactions of LSM perovskite within the molten salts, HTC LSM was dwelt in LiNO3–KNO3 and LiCl–KCl respectively, washed, and collected for XRD measurements. The XRD patterns shown in Fig. 2a indicate that LSM is not stable when exposed to either molten salt. Specifically, SrCO3 is present after dwelling in LiNO3–KNO3, and Li2MnO3 is present after dwelling in both LiNO3–KNO3 and LiCl–KCl. The SEM images and EDX spectra for the HTC powder, post dwell LiNO3–KNO3, and post dwell LiCl–KCl in Fig. 2b–g. After dwelling in LiNO3–KNO3, the particle surfaces are significantly more faceted than the as is and dwelt-LSM in LiCl–KCl. Nonetheless, the Sr L-lines in both the dwelt powders have diminished Sr L-lines compared to the HTC LSM as indicated by the red dashed lines. The difference in surface morphology in the LiNO3–KNO3-dwelt powder could be a result of the NO3− ions in solution stabilizing different crystal facets27 after Sr is leached out of the perovskite. The LiCl–KCl-dwelt spectrum has an even lower Sr L-line intensity compared to that of the LiNO3–KNO3-dwelt spectrum, which indicates that the chloride containing salt (the more Lux acidic molten salt) leaches out more Sr from the perovskite lattice. The more acidic solution would dissolve the segregated SrCO3 to then be removed from the powder during the washing step as outlined in the experimental procedures (see ESI†), and thus not appear in the XRD pattern in Fig. 2. As a result, LSM of a target La0.8Sr0.2MnO3 stoichiometry is not stable in any of the molten salt solvents used to determine the influence of the anionic character of the salts. Pure phase LSM was also found to not form in any of the above investigated molten salts. Thus, while the anion does play a role in the MSS of LSM, it is not the deciding factor.
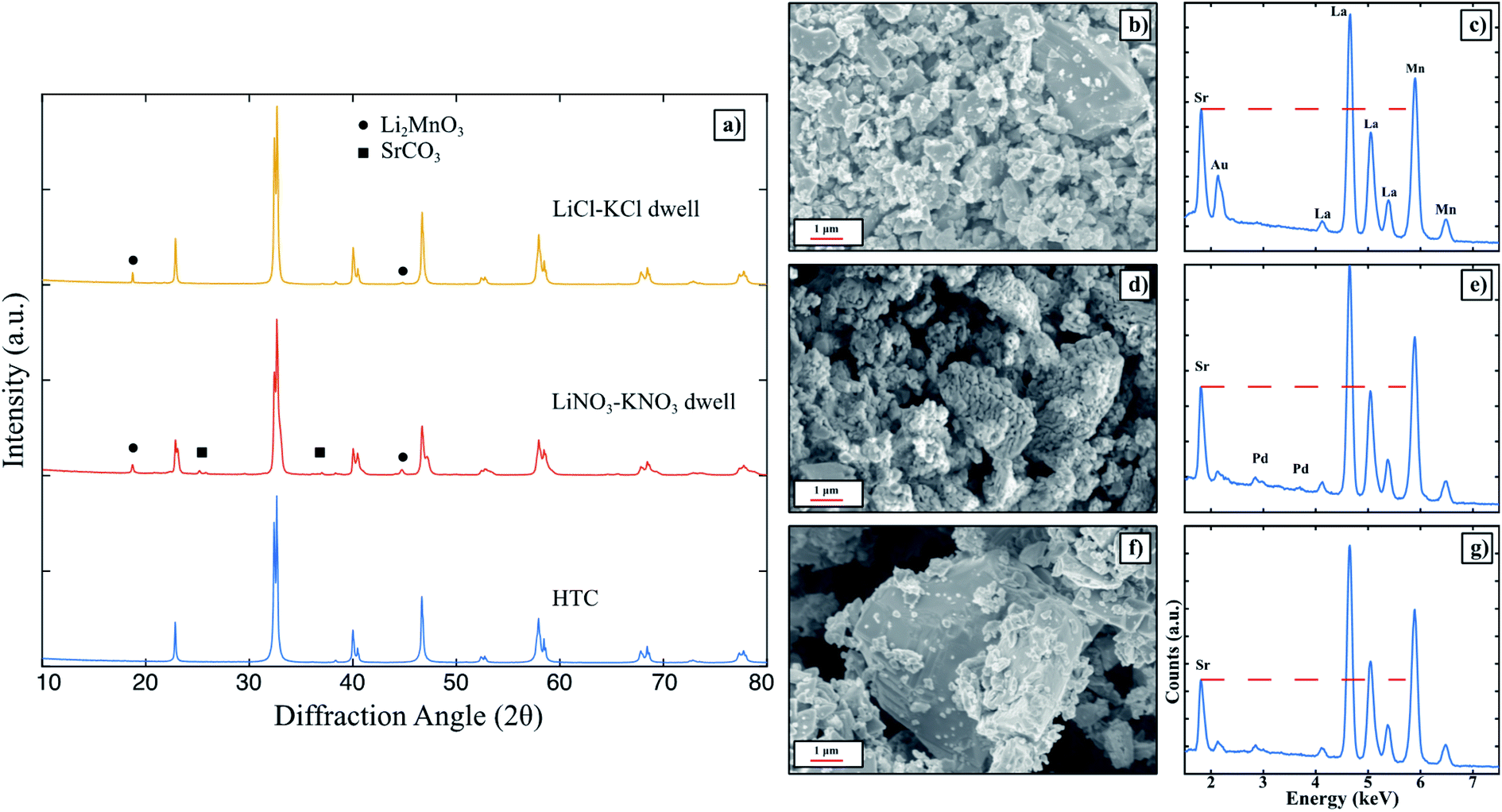 | ||
| Fig. 2 (a) XRD patterns of HTC LSM compared to HTC LSM dwelt in LiNO3–KNO3 and LiCl–KCl eutectics at 600 °C for 2 hours. The mass ratio of powder to salt was 1:8. SEM images and EDX patterns of (b and c) HTC LSM, (d and e) LiNO3–KNO3 dwelt LSM, and (f and g) LiCl–KCl dwelt LSM. The dashed red lines indicate diminishing Sr L-line intensities compared to HTC LSM. | ||
Influence of molten salt cation on LSM formation
In this section, the molten salts investigated were single salt nitrate systems (LiNO3 and KNO3) since each of these salts had desirably low melting points and would give direct insight on the influence of the cation on LSM formation. HTC LSM was dwelt in LiNO3 and KNO3 to determine any influence on the acidic character of the cation on LSM perovskite. The XRD patterns in Fig. 3 clearly indicate identical patterns for the HTC LSM as is, and the HTC LSM dwelt in KNO3. When dwelt in LiNO3, the corresponding pattern contains peaks for Li2MnO3 and SrCO3 as identified before for the LSM dwelt in Li+-containing molten salts. The EDX spectrum for LiNO3-dwelt LSM clearly identifies a diminished Sr L-line compared to that in the HTC LSM spectrum (Fig. 3e and g). However, while the KNO3-exposed LSM has a significantly more intense Sr L-line, it is still slightly lower than the Sr-L line for HTC LSM (Fig. 3b and g). These results indicate that the molten salt cation has a significant effect on the retention of Sr in the LSM perovskite. Even though HTC LSM experienced slight Sr depletion when exposed to the KNO3 melt, it is possible that prolonged exposure during the heat up and cool down furnace steps exacerbated the salt leaching Sr from the LSM lattice. Furthermore, a small peak for the potassium K-line around 3.3 keV is present, which suggests that Sr near the surface of the perovskite has been replaced by a K+. We do not expect this to be detrimental for LSM from the standpoint of application in solid state devices such as fuel cells and electrolyzers, since the incorporation of a small amount of K+ into the melt-synthesized perovskites would be incorporated in either the A (La) or B (Mn) sublattice as acceptor dopants, resulting in the formation of oxygen vacancies or holes, which are already part of the native point defect structure of LSM.28,29 Nonetheless, KNO3 is the most promising molten salt to preserve LSM perovskite phase with a desired stoichiometry and was then investigated further for the MSS of LSM.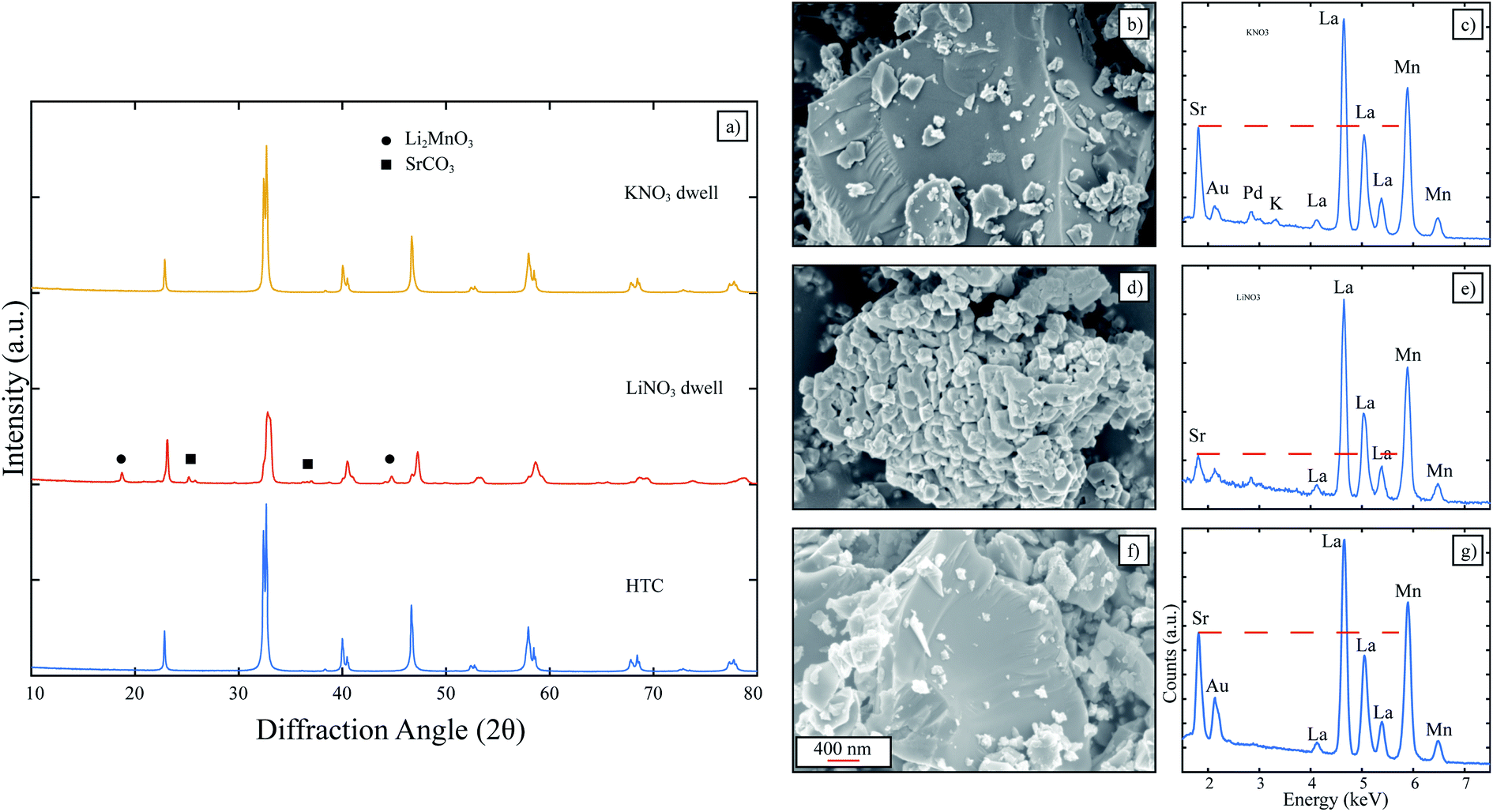 | ||
| Fig. 3 (a) XRD patterns of HTC LSM compared to HTC LSM dwelt in LiNO3 and KNO3. The dwell conditions were 600 °C for 2 hours, and the mass ratio of powder to salt was 1:8. SEM images and EDX spectra are presented for (b and c) KNO3-dwelt HTC LSM, (d and e) LiNO3-dwelt HTC LSM, and (f and g) HTC LSM. The dotted red line shows a direct comparison between the Sr L-line intensities for each spectrum. | ||
The XRD patterns for the attempted LSM MSS in KNO3 are shown in Fig. S5.† The mass ratio of LSM oxide precursors to KNO3 was varied from 3:1 (mass powder to mass KNO3) up to 1:12, yet pure phase LSM was not obtained in any instance. At greater amounts of salt to LSM precursor ratios (the 1:8 and 1:12 samples), the SrMnO3 phase is observed. In all patterns, a K0.5Mn2O4·2H2O phase is present with variable intensity. Since the HTC LSM was shown to remain stable in KNO3 at the same temperature and time in a 1:8 powder to salt mass ratio, the most thermodynamically stable phase at such conditions is LSM perovskite. The experimental conditions of mixing stoichiometric amounts of La2O3, Mn2O3, and SrCO3 precursors with KNO3, heating to 600 °C, dwelling, then cooling back down to room temperature, may instead facilitate the reaction between K+ and Mn2O3 at a lower temperature to form a complex. This is observed in Fig. S6† with varying the amount of Mn2O3 precursor dwelt in KNO3 at 600 °C for 2 hours, and collecting XRD patterns of the resulting powder. Counterintuitively, when the amount of KNO3 relative to Mn2O3 decreases a far greater amount of K0.5Mn2O4·2H2O forms, whereas Mn2O3 is the major phase when the amount of KNO3 relative to Mn2O3 increases. The SrMnO3 phase may form at greater amounts of KNO3 to LSM precursors as a result of this change in Mn2O3 solubility and consistently high La2O3 solubility in KNO3.20
Given that mixing and heating from room temperature may provide avenues for undesired secondary phases to form, there is a need to instead introduce the mixed LSM precursors at the same time directly into equilibrated KNO3 at 600 °C. This feeding procedure25 was followed as described in the ESI.† The XRD pattern (see Fig. S7†) for the fed LSM into equilibrated KNO3 shows completely pure phase LSM formation after dwelling for 1 hour before removing the crucible from the hot plate. SEM/EDX also show a particle size distribution ranging from ∼10 to 200 nm, with the majority of particles spanning 15–45 nm in diameter (see Fig. S8†), and a comparable Sr L-line intensity to that of HTC LSM with a slight peak for K that may be substituted for Sr near the surface of the perovskite. Thus, the feeding procedure is the more successful method to expeditiously form pure phase LSM nanoparticles of desired stoichiometry, when the appropriate molten salt KNO3 is identified.
Thermodynamic basis for molten salt chemistry: LSM formation in KNO3 and not LiCl–KCl
Of the molten salt systems tested for the MSS of LSM, LiCl–KCl and KNO3 were the closest to successful considering the extent of secondary phases that formed and EDX analysis. The failure to form LSM in the LiCl–KCl eutectic melt can be understood from the context of supersaturation in the molten salt. The driving thermodynamic factor regarding nucleation and perovskite formation within a molten salt is heavily influenced by solubilities within the melt.15,30 Specifically, the central equations are as follows:
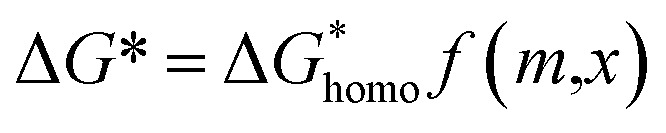 | (2) |
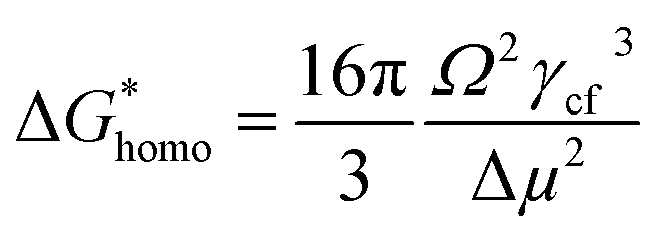 | (3) |
|
Δμ = kTln(1 + σ)
| (4) |
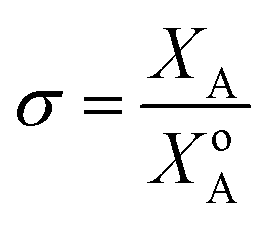 | (5) |
Explanations of the above equations along with key thermodynamic parameters for nucleation can be found elsewhere.30 Briefly, ΔG* is the nucleation energy barrier, 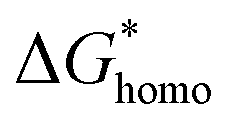 is the homogeneous nucleation energy barrier, f(m,x) is the heterogeneous nucleation shape factor which is a function of nucleus radius and wettability, Ω is the volume of a nucleus, γcf is the interfacial free energy between the crystal nucleus and the molten salt phase, and σ is the supersaturation. The actual solubility of component A in a mixture comprising the molten salt and A is denoted as XA. When a product, P, is formed by the reaction between reactants A and B, XoA is the solubility of A in the molten salt when equilibrium is achieved between P, B, A, and the molten salt. Here, we focus on the change in chemical potential between the actual and equilibrium state, Δμ, and the supersaturation, σ. Regarding the formation of LSM, it is more useful and elucidating to apply this formulation to each reactant and calculate respective supersaturation values. Fig. 4 shows the measured concentrations of ions in the melt (samples drawn from the melt) as a function of time. These concentrations are the respective equilibrium concentrations, XoA, for each reactant and were collected at the 8 h mark since the measured concentrations were relatively stable. The actual solubilities, equilibrium solubilities in the molten salt in the presence of the desired product phase, and calculated supersaturations are presented in Table 1. The σLa and σMn values are around 2 orders of magnitude larger than σSr, which indicates that there is much less of a thermodynamic driving force to incorporate Sr into the perovskite lattice. This is observed in Fig. 4a and b where La and Mn concentrations in the salt melt over the course of perovskite formation are significantly lower than Sr, which was expected as less La and Mn would be solubilized as more product perovskite precipitates out of solution. Sr concentrations in the salt melt remained relatively constant throughout the duration of the synthesis, which further demonstrates Sr is not reacting with the other precursors and therefore not being accommodated in the product perovskite phase. However, since Sr L-lines were observed in the EDX spectra in Fig. 1b and S4b–e† for the LiCl–KCl MSS samples, it is possible that the non-zero supersaturation for Sr allows for trace amounts of Sr to react with the other precursors and incorporate into the perovskite lattice.
is the homogeneous nucleation energy barrier, f(m,x) is the heterogeneous nucleation shape factor which is a function of nucleus radius and wettability, Ω is the volume of a nucleus, γcf is the interfacial free energy between the crystal nucleus and the molten salt phase, and σ is the supersaturation. The actual solubility of component A in a mixture comprising the molten salt and A is denoted as XA. When a product, P, is formed by the reaction between reactants A and B, XoA is the solubility of A in the molten salt when equilibrium is achieved between P, B, A, and the molten salt. Here, we focus on the change in chemical potential between the actual and equilibrium state, Δμ, and the supersaturation, σ. Regarding the formation of LSM, it is more useful and elucidating to apply this formulation to each reactant and calculate respective supersaturation values. Fig. 4 shows the measured concentrations of ions in the melt (samples drawn from the melt) as a function of time. These concentrations are the respective equilibrium concentrations, XoA, for each reactant and were collected at the 8 h mark since the measured concentrations were relatively stable. The actual solubilities, equilibrium solubilities in the molten salt in the presence of the desired product phase, and calculated supersaturations are presented in Table 1. The σLa and σMn values are around 2 orders of magnitude larger than σSr, which indicates that there is much less of a thermodynamic driving force to incorporate Sr into the perovskite lattice. This is observed in Fig. 4a and b where La and Mn concentrations in the salt melt over the course of perovskite formation are significantly lower than Sr, which was expected as less La and Mn would be solubilized as more product perovskite precipitates out of solution. Sr concentrations in the salt melt remained relatively constant throughout the duration of the synthesis, which further demonstrates Sr is not reacting with the other precursors and therefore not being accommodated in the product perovskite phase. However, since Sr L-lines were observed in the EDX spectra in Fig. 1b and S4b–e† for the LiCl–KCl MSS samples, it is possible that the non-zero supersaturation for Sr allows for trace amounts of Sr to react with the other precursors and incorporate into the perovskite lattice.
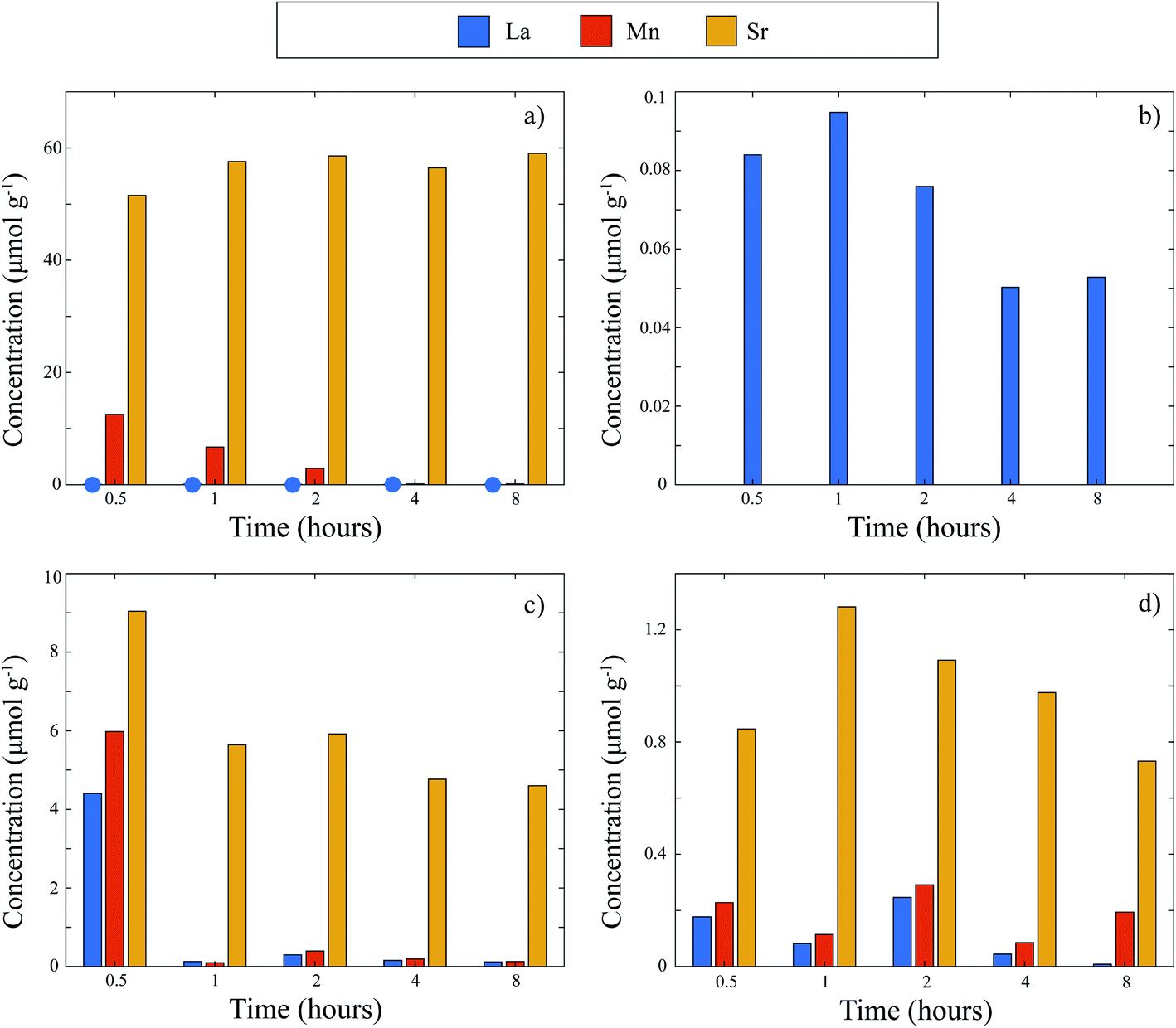 | ||
| Fig. 4 Concentrations of metal ions present during MSS of LSM. (a) LiCl–KCl eutectic at 550 °C. Blue circles indicate La concentrations. (b) Concentrations of La in LiCl–KCl at 550 °C plotted on a magnified scale. (c) KNO3 at 600 °C. (d) Feed into KNO3 at 600 °C. | ||
| LSM | La mole ratio | Sr mole ratio | Sr/La mole ratio | Mn mole ratio |
|---|---|---|---|---|
| HTC LSM | 0.81 ± 0.09 | 0.206 ± 0.002 | 0.25 ± 0.01 | 1.1 ± 0.1 |
| LiCl–KCl | 0.76 ± 0.09 | 0.019 ± 0.002 | 0.025 ± 0.001 | 1.0 ± 0.1 |
| KNO3 feed | 0.74 ± 0.04 | 0.183 ± 0.009 | 0.247 ± 0.001 | 1.00 ± 0.05 |
| LiCl–KCl LSM | La | Sr | Mn |
|---|---|---|---|
| XA (μmol g−1 salt) | 8.28 | 174.62 | 41.54 |
| XoA (μmol g−1 salt) | 0.053 | 59.044 | 0.155 |
| σ | 160 | 2.9575 | 268 |
While a high solubility of a component in the molten salt, XA, may appear favorable for a larger supersaturation, it is important that the XoA value be lower than XA to drive the reaction forward. In other words, the chemical potential of the component in the desired product phase must be lower than the chemical potential of the precursor phase A alone in equilibrium with the molten salt. If this is satisfied, the change in chemical potential of A will be negative and indicate a favorable reaction (Fig. S9†). The greater this difference, the more spontaneous the reaction and hence, a smaller nucleation activation energy barrier prevails as shown in eqn (2) and (3). In LiCl–KCl eutectic, the high concentration of Sr in the melt can be attributed to the Lux acidity of both the Li+ and Cl−, which have been shown to significantly solubilize alkaline earth metals and oxides.31,32 Furthermore, the Lux-acidic environment induces a less favorable environment for Sr within the perovskite lattice. Fig. S10† shows XRD patterns for HTC La0.6Sr0.4Co0.2Fe0.8O3 (LSCF) as synthesized, dwelt in LiCl–KCl eutectic, and in KNO3 at 600 °C for 4 hours. The patterns for as-synthesized and KNO3-dwelt LSCF are nearly identical, yet noticeable down-shifted peaks appear on the shoulders of the main LSCF peaks in the LiCl–KCl pattern. These down-shifted peaks indicate lattice relaxation33 and suggest Sr is leached out of the lattice when exposed to an acidic molten salt. Elemental analysis via ICP-MS indicates that Sr concentrations are indeed diminished for the LiCl–KCl eutectic-dwelt LSCF, whereas the KNO3-dwelt and as-synthesized LSCF are similar (ESI Table 1†). Since Sr is less stable in the perovskite lattice, these results suggest that μSrProduct in the LiCl–KCl eutectic is increased compared to μSrProduct in the KNO3 melt. As a result, the magnitude of ΔμSr in LiCl–KCl eutectic is smaller than that in KNO3 so there is less of a driving force for Sr to nucleate into the perovskite phase in LiCl–KCl. Stated another way, the partition coefficient of Sr between LiCl–KCl and the perovskite phase is very high and thus, LSM does not form.
KNO3 is a desirable molten salt since K+ has the lowest acidity among alkali metals according to Lux-Flood theory, and nitrates are less acidic than chlorides. Thus, KNO3 is overall less acidic than LiCl–KCl eutectic.32 Therefore, it was expected that more of the Sr would be retained in the perovskite phase when equilibrated in the less acidic KNO3 melt. From Fig. 4c, it is readily apparent that Sr melt solubility is indeed drastically reduced in the KNO3 melt compared to LiCl–KCl eutectic over the course of LSM formation. Moreover, the decreasing concentration of each metal as time progressed reflects that each precursor is able to react and form LSM. The XRD patterns of KNO3-dwelt HTC LSM and LSCF demonstrate that the less acidic properties of KNO3 favor Sr remaining in the perovskite phase instead of Sr in solution, as opposed to the more acidic properties of LiCl–KCl eutectic, and are essential for the successful synthesis of phase pure perovskite (Fig. 3, S10 and Table S1†). Thus, the formation of LSM in a KNO3 melt gives credence to the greater stability of Sr in the LSM phase in the molten salt compared to Sr in the molten salt. It is postulated that the supersaturation of Sr, σSr, is now higher than that in the LiCl–KCl eutectic. As a result, it is clear from eqn (3) and (4) that the nucleation activation energy barrier for LSM in a KNO3 melt will be lower in magnitude than that in a LiCl–KCl eutectic. The chemistry of the molten salt and how it affects the reactants and products is thus an important aspect in the MSS of LSM.
When the oxide precursors were fed to the KNO3 melt equilibrated at 600 °C, the concentrations of each metal in the melt were drastically lower than those in the experiment where the precursors and salt mixtures were mixed and heated together from room temperature. Specifically, Fig. 4d indicates each of the precursors have relatively stabilized in solution even at the first sample collection at 30 minutes. Thus, the precursors are rapidly reacting and forming the target perovskite when introduced to the melt at 600 °C, which drastically accelerates LSM formation compared to heating from room temperature. The higher concentrations of Sr that prevail in the mix and heat method may be a result of the precursors being exposed to and reacting at lower temperatures in the molten salt during the ramp schedule as explained earlier. The marked decrease in precursor concentrations displayed in Fig. 4c at the 1 h mark, and then consistent concentrations from there on, suggest a lag where the melt was still heating to 600 °C despite the heating program reaching 600 °C earlier. By contrast, since all precursors in the feed method are directly introduced to the melt at 600 °C, all three can rapidly react to form the desired perovskite phase. In this case, Fig. 4d shows no drastic decrease in precursor concentrations, indicating firstly that melt is indeed at 600 °C upon drawing the first sample, and secondly that the reaction has likely already occurred and equilibrated in the molten salt. This rapid kinetics of formation of PTO has important implications to not only further elucidate critical factors for PTO formation in molten salts, but also facilitate mass production of PTOs for industrial applications.
Conclusions
In conclusion, the synthesis of La0.8Sr0.2MnO3 from oxide precursors in various molten salts was investigated to determine the influence of molten salt chemistry on the efficacy of LSM formation. It was determined that varying the molten salt anion alone did not facilitate LSM formation and thus indicates that the molten salt anion is not the critical factor. The less acidic K+ cation compared to Li+ is preferred for the MSS of LSM, but pure phase LSM was only obtained by feeding oxide precursors into an equilibrated KNO3 melt. The size distribution of the LSM formed via feeding and dwelling for 1 hour spanned from ∼10–200 nm, with the majority of particles ranging from 15–45 nm in diameter. LSM did not form in LiCl–KCl as a result of the large thermodynamic barrier impeding LSM formation due to high acidity of the salt ions in solution creating a more favorable environment for Sr in the molten salt instead of the product perovskite phase. The reaction mixture instead resulted in the formation of A-site deficient lanthanum manganite perovskite phase. Conversely, the relatively low acidic KNO3 has a lower nucleation barrier and thus results in a lower partition coefficient of Sr between the melt and the perovskite phase. Phase pure LSM with the desired composition can therefore rapidly form when fed into in this molten salt medium. Essentially, an appropriate molten salt for LSM and other perovskite synthesis requires the molten salt ions, with an emphasis on the cation, to be less acidic than the precursors. In a broader context for the MSS of other PTOs, researchers should consider the inherent chemistry of each salt system and, if necessary, select those with low Lux acidity.Disclaimer
This work was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, nor any of their contractors, subcontractors or their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or any third party's use or the results of such use of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof or its contractors or subcontractors. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof, its contractors or subcontractors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Department of Energy, National Energy Technology Laboratory for funding this project through cooperative agreement numbers DE-FE0031205 and DE-FE0031652. The authors also acknowledge the Boston University Materials Science and Engineering Core Research Facility for access to XRD instrumentation, which was funded by NSF Award Number 1337471. The authors thank Professor Katsuyoshi Kakinuma at the University of Yamanashi for his insight and assistance in TEM analysis of the formed nanoparticles (not included in this paper).References
- E. Grabowska, Appl. Catal., B, 2016, 186, 97 CrossRef CAS.
- C. Moure and O. Peña, Prog. Solid State Chem., 2015, 43, 123 CrossRef CAS.
- A. Chroneos, R. V. Vovk, I. L. Goulatis and L. I. Goulatis, J. Alloys Compd., 2010, 494, 190 CrossRef CAS.
- J. Shi and L. Guo, Prog. Nat. Sci.: Mater. Int., 2012, 22, 592 CrossRef.
- S. C. Singhal, Solid State Ionics, 2012, 152, 405 Search PubMed.
- Y. Jiang, S. Z. Wang, Y. H. Zhang, J. W. Yan and W. Z. Li, Solid State Ionics, 1998, 110, 111 CrossRef CAS.
- S. P. Simner, M. D. Anderson, M. H. Engelhard and J. W. Stevenson, Electrochem. Solid-State Lett., 2006, 9, A479 CrossRef.
- A. Mai, M. Becker, W. Assenmacher, F. Tietz, D. Hathiramani, E. Ivers-Tiffee, D. Stover and W. Mader, Solid State Ionics, 2006, 177, 1965 CrossRef CAS.
- E. Jud, C. B. Huwiler and L. J. Gauckler, J. Am. Ceram. Soc., 2005, 88, 3013 CrossRef CAS.
- D. Yamada, T. Murai, K. Moritani, T. Sasaki, I. Takagi, H. Moriyama, K. Kinoshita and H. Yamana, J. Alloys Compd., 2007, 444, 557 CrossRef.
- X. Liu, N. Fechler and M. Antonietti, Chem. Soc. Rev., 2013, 42, 8237 RSC.
- R. H. Arendt, J. H. Rosolowski and J. W. Szymaszek, Mater. Res. Bull., 1979, 14, 703 CrossRef CAS.
- J. T. S. Irvine, Faraday Discuss., 2016, 190, 551 RSC.
- H. Flood and T. Forland, Acta Chem. Scand., 1947, 1, 592 CrossRef CAS PubMed.
- T. Kimura, in Molten Salt Synthesis of Ceramic Powders, ed. C. Sikalidis, IntechOpen, London, United Kingdom, 2011, ch. 4, pp. 75–100 Search PubMed.
- P. Afanasiev and C. Geantet, Coord. Chem. Rev., 1998, 178, 1725 CrossRef.
- H. Zhou, Y. Mao and S. S. Wong, Chem. Mater., 2007, 19, 5238 CrossRef CAS.
- R. E. Rojas-Hernandez, F. Rubio-Marcos, R. H. Gonçalves, M. A. Rodriguez, E. Véron, M. Allix, C. Bessade and J. F. Fernandez, Inorg. Chem., 2015, 54, 9896 CrossRef CAS PubMed.
- H. L. T. N'Goc, L. D. N. Mouafo, C. Etrillard, A. Torres-Pardo, J. F. Dayen, S. Rano, G. Rousse, C. Laberty-Robert, J. G. Calbet, M. Drillon, C. Sanchez, B. Doudin and D. Portehault, Adv. Mater., 2017, 29, 1604745 CrossRef PubMed.
- C. Matei, D. Berger, P. Marote, S. Stoleriu and J. P. Deloume, Prog. Solid State Chem., 2007, 35, 203 CrossRef CAS.
- M. Kačenka, O. Kama, Z. Jirák, M. Maryško, P. Žvátora, S. Vratislav and I. Lukeš, J. Appl. Phys., 2014, 115, 17B525 CrossRef.
- F. Luo, Y. H. Huang, C. H. Yan, S. Jiang, X. H. Li, Z. M. Wang and C. S. Liao, J. Magn. Magn. Mater., 2003, 260, 173 CrossRef CAS.
- S. Gu, H. Shin, Y. Hong, D. Yeo, J. Kim, S. Nahm and S. Yoon, Met. Mater. Int., 2012, 18, 723–726 CrossRef CAS.
- A. M. Shams el Din and A. A. El Hosary, J. Electroanal. Chem. Interfacial Electrochem., 1968, 16, 551–562 CrossRef.
- L. Vradman, E. Friedland, J. Zana, R. Vidruk-Nehemya and M. Herskowitz, J. Mater. Sci., 2017, 52, 11383 CrossRef CAS.
- K. Othmer, Encyclopedia of Chemical Technology, John Wiley and Sons, New York, NY, 1991 Search PubMed.
- X. Wang, K. Huang, L. Yuan, S. Li, W. Ma, Z. Liu and S. Feng, ACS Appl. Mater. Interfaces, 2018, 10, 28219–28231 CrossRef CAS PubMed.
- N. Miniajluk, J. Trawczyński, M. Zawadzki and W. Tylus, Adv. Mater. Phys. Chem., 2018, 8, 193 CrossRef CAS.
- S. Royer, H. Alamdari, D. Duprez and S. Kaliaguine, Appl. Catal., B, 2005, 58, 273 CrossRef CAS.
- X. Y. Liu, J. Cryst. Growth, 2002, 237, 1806 CrossRef.
- V. N. Gaprindashvili and N. V. Kanashvili, Soobshch. Akad. Nauk Gruz. SSR, 1970, 58, 357 CAS.
- V. L. Cherginets and T. P. Rebrova, Zh. Fiz. Khim., 1999, 70, 687 Search PubMed.
- M. Pajot, V. Diffort, E. Capoen, A. S. Mamede and R. N. Vannier, J. Power Sources, 2020, 450, 227649 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04324a |
| This journal is © The Royal Society of Chemistry 2021 |