
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Graphene and its electrochemistry – an update
Adriano
Ambrosi†
,
Chun Kiang
Chua†
,
Naziah Mohamad
Latiff
,
Adeline Huiling
Loo
,
Colin Hong An
Wong
,
Alex Yong Sheng
Eng
,
Alessandra
Bonanni
and
Martin
Pumera
*
Division of Chemistry & Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore. E-mail: pumera.research@gmail.com; Fax: +65-6791-1961
First published on 7th April 2016
Abstract
The electrochemistry of graphene and its derivatives has been extensively researched in recent years. In the aspect of graphene preparation methods, the efficiencies of the top-down electrochemical exfoliation of graphite, the electrochemical reduction of graphene oxide and the electrochemical delamination of CVD grown graphene, are currently on par with conventional procedures. Electrochemical analysis of graphene oxide has revealed an unexpected inherent redox activity with, in some cases, an astonishing chemical reversibility. Furthermore, graphene modified with p-block elements has shown impressive electrocatalytic performances in processes which have been historically dominated by metal-based catalysts. Further progress has also been achieved in the practical usage of graphene in sensing and biosensing applications. This review is an update of our previous article in Chem. Soc. Rev. 2010, 39, 4146–4157, with special focus on the developments over the past two years.
 Adriano AmbrosiAuthors contributed equally. Adriano AmbrosiAuthors contributed equally. | Pumera's group originated in the wonderful National Institute for Materials Science, Tsukuba, Japan, in 2006. In 2010, this group, having two members, moved to Nanyang Technological University, Singapore (M. Pumera renounced ERC-StG offer at EPFL at that time) where quickly grew to current 20+ members. It consists of Martin, four postdoctoral researchers, a lecturer, thirteen PhD students, three master students and countless undergraduates. This group takes pride in the fact that it has produced three young faculty members, who are strategically located from the Iberian to Malay Peninsula, thus spanning the whole Eurasian continent; we have trained 17 PhD students and over forty undergraduate students. Pumera's group has broad interests in nanomaterials and microsystems, in the specific areas of electrochemistry and synthetic chemistry of carbon nanomaterials, layered anisotropic materials, nanotoxicity, micro- and nanomachines and 3D printing. This group produced over 400 papers (some of them are reviewed here) which received over 14 |
1. Introduction
Graphene and its derivatives, such as graphene oxide, doped and functionalized graphene materials, is the major focus in the area of materials science research.1,2 The high conductivity, large surface area and high electron transfer rate of graphene are the underlying reasons for its extensive usage in modern electrochemistry related technologies. This is further enhanced by the fact that the carbon backbone of graphene can be covalently or non-covalently functionalized with heteroatoms (non-carbon atoms), as it unlocks the potential of tailoring its electrochemical and electrical properties to cater to various applications.In 2010,3 when research on the electrochemistry of graphene was just taking off, we have contributed a tutorial review on the topic. Now six years later, we wish to provide a more comprehensive overview on the electrochemistry of graphene, focusing mostly, but not exclusively, on its developments over the past two years. In the first section of this review, an overview on the preparation of graphene based on electrochemical techniques, mainly by electrochemical exfoliation of graphite and electrochemical delamination of chemical vapour deposited graphene, would be provided. Subsequently, the modification of graphene via electrochemically-induced reactions as well as the major factors that influence the capacitance of graphene would be reviewed. Next, the variations of electrocatalytic properties of graphene materials, as conferred by the introduction of heteroatoms (also known as doping) such as p-block (i.e., B, N, S, P, halogens) and d-block elements, would be addressed. Lastly, the usage of graphene for sensing and biosensing applications would be critically reviewed to also include important practical considerations.
We believe that this review is a timely insight into the quintessence of graphene electrochemistry.
2. Electrochemically assisted fabrication of graphene
The use of chemical methods currently available for the preparation of graphene and graphene materials guarantees certain positive outcomes but suffers from major drawbacks as well. For example, liquid-phase exfoliation of graphite in organic solvents assisted by sonication and/or mechanical shearing can provide extremely high quality graphene with low density of defects and negligible presence of oxygen functionalities, but the exfoliation efficiency is extremely low with yields generally of <1%.4 Higher efficiencies can be achieved if an intercalation step is introduced prior to exfoliation. The formation of such graphite intercalation compounds (GIC) normally requires strong oxidants and acids which assist in splitting the graphene layers apart to facilitate exfoliation.5 However, this is achieved at the cost of significant functionalization with various oxygen groups and/or structural damage to the resulting graphene.5 If we take into consideration the bottom-up chemical vapor deposition (CVD) of graphene onto catalytic metallic substrates, this allows greater control over the quality and quantity of graphene sheets produced. However, it is limited by the need for post-production chemical etching of the metal substrate in order to separate graphene for transfer to more useful surfaces. Such chemical etching is time-consuming, and more importantly, cannot guarantee the complete removal of the catalytic metal substrate which always remains strongly adsorbed onto the graphene in the form of atomic species and/or nanometer-sized metal impurities. These impurities have profound influences on the electronic and electrochemical properties of graphene.6–8The electrochemical technique has recently regained great interest due to very promising results not only in assisting the exfoliation of graphite but also as a powerful method to control the delamination of CVD graphene. Much higher yields have been achieved with the electrochemical exfoliation of graphite apart from the possibility to control the chemical and structural features of the produced graphene. As for the electrochemical assisted delamination of CVD graphene, this method has demonstrated good control, minimal structural damage and functionalization, no residual contamination and more importantly, it is an extremely fast process compared to the aforementioned chemical etching technique. A detailed review of the most recent methods based on electrochemistry to either exfoliate graphite or to delaminate CVD graphene from the metal substrate is presented in the following sub-sections.
2.1 Electrochemical exfoliation of graphite
The electrochemically assisted exfoliation of graphite consists of the application of a cathodic or anodic potential to a graphite working electrode in aqueous or organic electrolytes. A second Pt electrode is generally used in a two-electrode electrochemical system while Pt and reference (e.g. Ag/AgCl, SCE, etc.) electrodes are used in a standard three-electrode electrochemical system. The usage of positive (anodic) potential facilitates the intercalation of negative anions into the graphite layers. This results in the gradual expansion of graphite leading to successive exfoliation to form graphene flakes. One of the drawbacks of this method is the introduction of a significant amount of oxygen functionalities into the produced graphene material with consequences for its electrical properties. On the other hand, a negative (cathodic) potential drives the intercalation of positive ions from the electrolyte into graphite layers followed by expansion and exfoliation. In general, this approach is less efficient and slower as compared to anodic exfoliation. However, cathodic exfoliation produces graphene flakes of higher quality. Some methods combine both the anodic and cathodic potentials, in which the initial potential is applied to drive intercalation while the reverse potential is used to facilitate exfoliation.The use of electrochemical methods to intercalate ions and compounds inside graphite layers was first demonstrated more than thirty years ago (1980s) when graphite intercalation compounds (GICs) were prepared with Li+ ions,9 F− ions,10,11 Ni2+ ions12 or sulfuric acid,13 in aqueous or organic solvents. Such intercalation methods are aimed exclusively at the preparation of “graphitic salts” as promising materials for batteries, mainly to serve as catalysts and also as materials with tunable electrical and electronic properties. However, it is only after the discovery of the extraordinary properties of graphene that the electrochemical approach experienced renewed scientific interest as a promising scalable method for the preparation of graphene. The first attempt was made by Wang and collaborators in 2009 where they employed poly(styrenesulfonate) (PSS) as an electrolyte and two graphite rods as cathode and anode electrodes.14 A 5 V bias to the electrodes was applied for over 4 h during which a black product was released from the anode. After characterization, graphene flakes showed quite a significant density of defects and presence of oxygen functionalities as well as residual PSS molecules strongly adsorbed on them.14 Since treatment with anodic potentials produces more defective graphene materials containing significant amounts of oxygen functional groups, cathodic exfoliation procedures have been attempted. It is worth mentioning the work by Morales et al. which carefully studied the effects of different reducing potentials applied to graphite foils in aqueous perchloric acid.15 The produced graphene indeed showed a low D-band in the Raman spectra but due to the scarce efficiency of the exfoliation process, post-processing microwave thermal treatment and prolonged sonication in NMP were required to obtain a final dispersion. Based on similar cathodic processes, Loh et al. obtained remarkable results by employing a highly negative potential of −15 V in the presence of Li+ ions in an organic propylene carbonate (PC) electrolyte. After prolonged sonication, an impressive value of over 70% of the graphene flakes had thickness of less than five layers although with reduced lateral size due to prior sonication treatment.16 An improved cathodic procedure was later proposed by Zhong and Swager who employed a milder cathodic potential of −5 V, but in two successive steps. The first expansion was done in the presence of Li+ ions in PC, followed by a second treatment with the same potential but in the presence of a larger cation, tetra-n-butylammonium (TBA) (Fig. 1a). After extensive washing to remove the residual Li ions, the material showed extremely low oxygen content (Fig. 1b and c). However the presence of defects was still detected by Raman spectroscopy (Fig. 1e).17 Cooper et al. used tetramethylammonium perchlorate (TMA ClO4), tetraethylammonium tetrafluoroborate (TEA BF4), and tetrabutylammonium tetrafluoroborate (TBA BF4) as electrolytes to study the expansion and the exfoliation of different graphite sources (HOPG and graphite rods) using cyclic voltammetry in a three-electrode electrochemical system.18
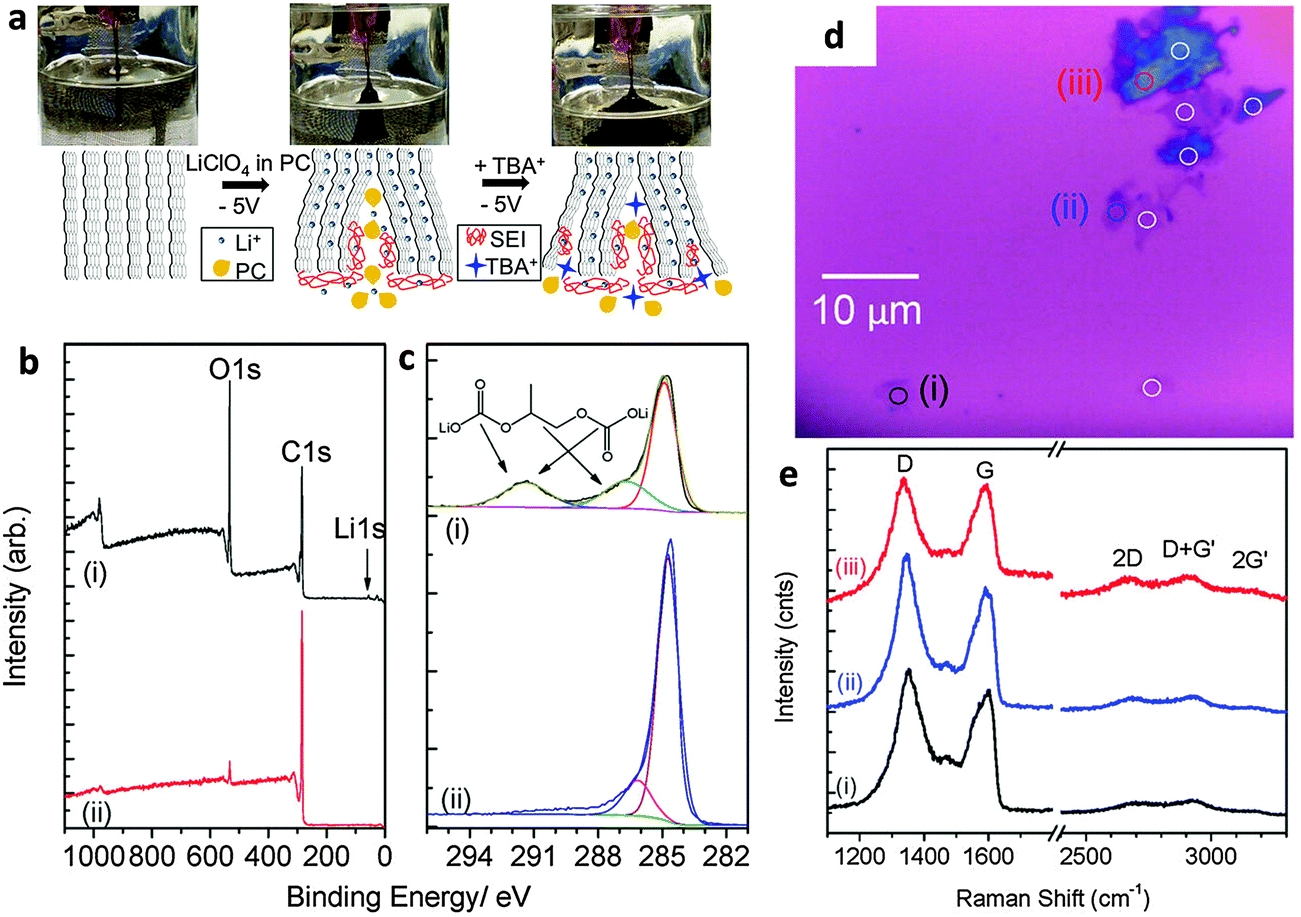 | ||
| Fig. 1 (a) Schematic and images of electrochemical expansion of graphite. (b) XPS survey scans and (c) C1s XPS spectrum of electrochemically expanded graphene (i) after rinsing once with DMF and (ii) after extensive washing. (d) Optical micrograph of EFG spin-coated on silicon before laser ablation. (e) Raman spectra of selected spots (i) to (iii) as marked in panel d. Adapted with permission from ref. 17. Copyright (2012) American Chemical Society. | ||
In other procedures, a combination of both anodic and cathodic potentials has been used to facilitate the intercalation of electrolyte ions in the first step followed by the application of a reverse potential to drive the exfoliation process. For example, using an aqueous sodium dodecyl sulfate (SDS) solution, graphene sheets have been obtained from graphite rods by first applying a preliminary anodic potential of +1.6 V in order to obtain SDS-intercalated graphite followed by the application of −1 V to exfoliate the intercalated graphite.19 Graphene flakes of 1–2 layers thick and around 0.5 μm in size were obtained in a stable graphene/SDS suspension. Apart from that, an ionic liquid [triethyl sulfonium bis (trifluromethyl sulfonyl) imide] (IL) electrolyte was used for the first time to provide greener alternatives to conventional methods that employ organic electrolytes. In such a study, a pencil graphite electrode was subjected to a potential of +8 V for 600 s followed by a step of 600 s with the reverse (−8 V) potential. The produced graphene flakes were 1–2 layers thick and have unexpectedly lower density of defects compared to the initial pencil graphite electrode.20 In addition, cycles of anodic/cathodic potentials of ±10 V have been used in the presence of NaClO4 in PC to exfoliate microcrystalline natural graphite minerals. In this work, the authors showed that high quality impurity-free graphene can be obtained despite the fact that the starting graphite material is rich in silicate and other impurities from the ore, which thus avoids complex preliminary purification steps.21
It is undoubtedly recognized that the electrochemical exfoliation of graphite electrodes using anodic potentials is far more efficient than using cathodic potentials, particularly in aqueous electrolytes. This is because the intercalation of anions is facilitated by the concomitant formation of radical species (e.g. ˙OH and ˙O) by the oxidation of water. These radical species oxidize/break the graphite structure starting from the edges and result in the surge of available openings for anions to penetrate. The successive formation of oxygen gas splits the graphene layers apart to provide a suspension of graphene. Therefore, such an exfoliation process is significantly accelerated but at the cost of a more defective and oxygen-functionalized graphene. A fast anodic exfoliation was demonstrated in sulfuric acid by Su and collaborators who applied a preliminary potential of +1 V for 5–10 min to wet a graphite sample and initiate the intercalation of SO42− ions into the graphite. This was then followed by the application of a +10 V bias to the graphite anode which resulted in a massive exfoliation over 1 min. Between 5–8% of the collected product was single layer graphene and over 60% was bilayer graphene; oxygen content was quite low but according to Raman analysis, the graphene sheets presented an ID/IG ratio of around 0.7.22 A similar procedure was proposed by Parvez et al. who by using a positive potential of +10 V in the H2SO4 electrolyte obtained graphene sheets with a lower ID/IG ratio (0.4) and low oxygen content. The graphene sheets were subsequently applied for organic electronics.23 The same group later demonstrated a more efficient exfoliation method and production of higher quality graphene by using an aqueous solution of inorganic salts such as ammonium sulfate ((NH4)2SO4), sodium sulfate (Na2SO4), and potassium sulfate (K2SO4) at neutral pH.24 It is however important to notice that while the presence of edge plane defects on graphene materials can be detrimental for their mechanical properties and for some electronic applications, they are beneficial for other applications, such as electrochemistry in particular. It is well-known, in fact, that electron transfer processes on carbon materials occur at much higher rates at edge plane defects than on basal planes25 and therefore anodic exfoliation of graphite could be the preferred method for electrochemical applications. In our group, we investigated the use of different aqueous electrolytes (i.e., H2SO4, Na2SO4 and LiClO4) for anodic exfoliation of graphite foils (Fig. 2a–g). Subsequent structural characterization showed the presence of defects regardless of the electrolyte used (Fig. 2i), but interestingly the oxygen content could be tuned ranging from a very low C/O ratio of 8.8 using Na2SO4 to a value of 4.0 using LiClO4 which is close to the values usually observed for the chemically produced graphene oxide (GO) materials (Fig. 2h). By varying the chemical features using different exfoliating electrolytes, the electrochemical properties of the produced graphene are also altered, which might suit different electrochemical applications (Fig. 2j–m).26
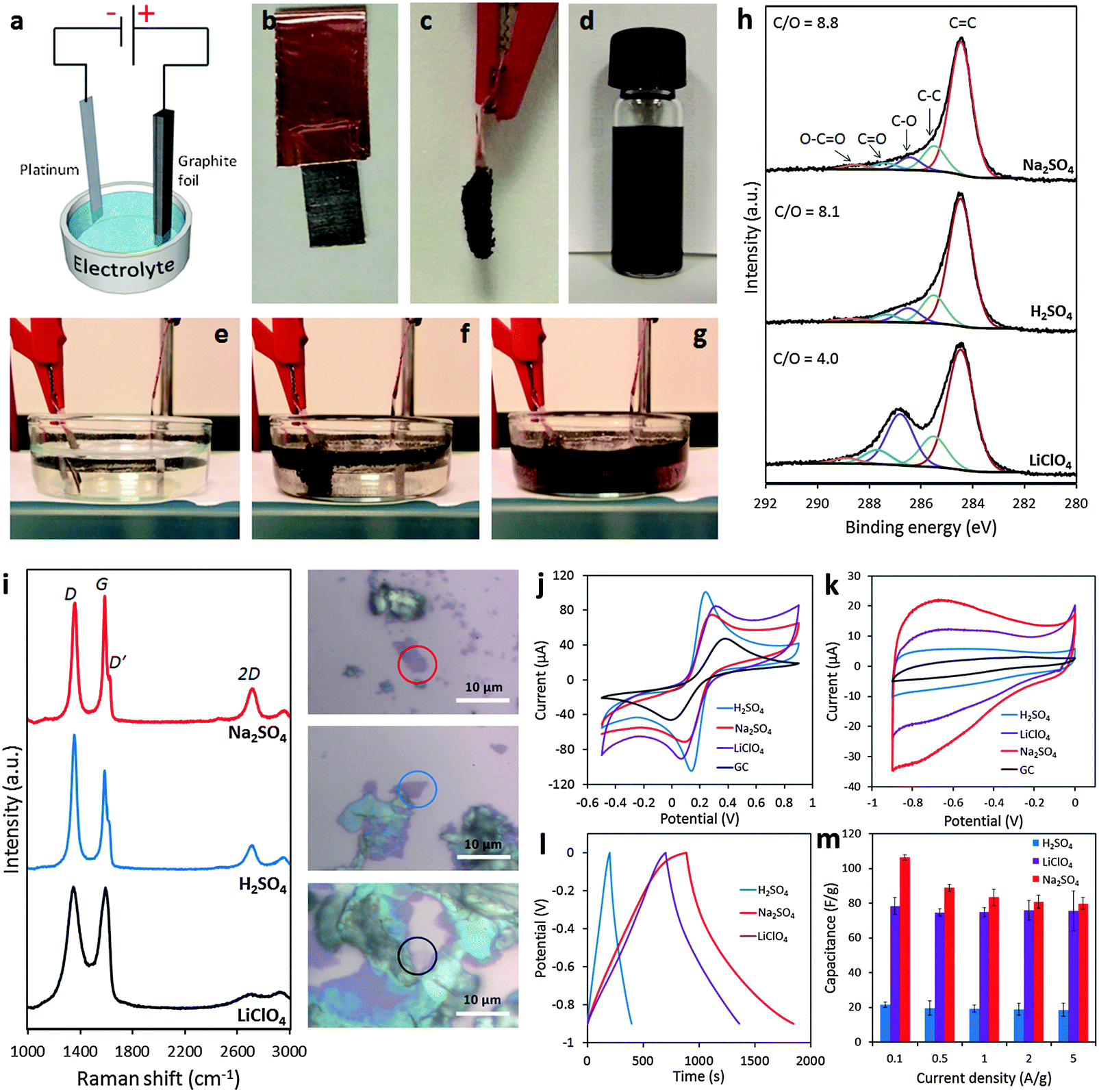 | ||
| Fig. 2 (a) Schematic illustration of the experimental setup. Photos of graphite foil (b) before and (c) after the exfoliation process. (d) Graphene dispersion in DMF solution (1 mg mL−1). Photos illustrating the exfoliation process at (e) time zero, (f) after 5 min, and (g) after 20 min. (h) High-resolution C1s XPS spectra of the electrochemically exfoliated graphene materials. Fitting peaks corresponding to different functional groups along with the C/O ratios for each material are also indicated. (i) Representative Raman spectra recorded for the graphene material obtained in Na2SO4 (red), H2SO4 (blue), and LiClO4 (black) corresponding to the material portion as indicated in the optical images on the right. (j) Representative cyclic voltammograms recorded by using a GC electrode modified with graphene obtained in Na2SO4 (red), H2SO4 (blue), and LiClO4 (purple) in the presence of a 5 mM Fe(CN)63−/4− redox probe in 0.1 M KCl electrolyte. (k) Cyclic voltammograms at a 100 mV s−1 scan rate in 6 M KOH solution recorded for the graphene material obtained in Na2SO4 (red), H2SO4 (blue), and LiClO4 (purple). The voltammogram of the bare GC electrode (black) is also shown for comparison. (l) Galvanostatic charge/discharge curves recorded for all graphene materials at a current density of 0.1 A g−1 in 6 M KOH solution. (m) Summary of the gravimetric capacitance measured for all graphene materials at different current densities between 0.1 and 5 A g−1. Adapted with permission from ref. 26. Copyright (2016) John Wiley and Sons. | ||
An improved exfoliation process proposed by Liu et al. applied anodic potentials in solutions of protonic acids (i.e., H2SO4, H3PO4 or H2C2O4) as electrolytes in a vertical cell configuration with the graphite rod placed at the bottom of the cell. This system allowed for multiple exfoliation steps to improve the yield and quality of graphene.27
Rao et al. employed milder anodic conditions (+1–3 V) in the presence of an electrolyte solution containing NaOH, H2O2 and H2O to obtain an impressive 95% yield of graphene with thickness between 3–6 layers. The authors highlighted the crucial role of H2O2 in the process as it reacts with hydroxyl ions to form the peroxide ion O22−, which is a strong nucleophile that can effectively penetrate into the graphene layers.28 The same group later proposed a more efficient exfoliation method that provides graphene of similar quality by using glycine–H2SO4 ionic complex electrolyte solution.29 A recent interesting work by the Müllen group showed that the introduction of reducing agents during the anodic exfoliation process in aqueous electrolytes can eliminate highly reactive radicals that are generated from H2O. Different reducing agents such as (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO), ascorbic acid and sodium borohydride were tested and the most effective additive which gave the highest quality graphene was TEMPO. The produced graphene sheets have large dimensions (5–10 μm), outstanding hole mobilities (∼ 405 cm2 V−1 s−1), impressively low Raman ID/IG ratios (<0.1), and extremely high carbon to oxygen (C/O) ratios (∼25.3).30
In addition, a recently published study aimed to investigate the effects of changing different parameters of the anodic exfoliation process on the quality of graphene. In this study, by varying the exfoliation time, the potential applied and the type of electrolyte, the authors carefully evaluated the efficiency of exfoliation, the density of defects and the electrical properties of graphene. As this study provides further insights into the crucial steps occurring during the electrochemical exfoliation process, it can certainly be a valuable reference to control the properties of electrochemically exfoliated graphene.31
2.2 Electrochemical delamination of CVD grown graphene
Electrochemical methods can also be applied to the transfer of CVD graphene, which is grown on catalytic metal substrates. The most commonly used transfer process typically starts with the spin-coating of a polymer support layer onto the graphene surface, followed by the removal of the metal foil by wet-etching in solutions such as iron chloride and ammonium persulfate. This chemical etching step takes several hours for the metal foil to be completely dissolved and can contaminate the graphene film with dissolved metals from either the foil or the etchant.8 Furthermore, the metal waste incurs additional costs from both economical as well as environmental standpoints. In a bid to overcome these problems, Wang et al. pioneered an electrochemical method to delaminate poly(methyl methacrylate) (PMMA)/graphene films from Cu foil.32 A PMMA/graphene/Cu stack was immersed in aqueous K2S2O8 and held at a voltage bias of −5 V, causing the reduction of water and producing hydrogen bubbles as shown in the equation: 2H2O + 2e− → H2 + 2OH−. The H2 bubbles generated at the graphene/Cu interface serve to delaminate the PMMA/graphene film from the Cu foil, starting from the edges exposed to the electrolyte and progressing to the rest of the film due to electrolyte permeation into the interlayers (Fig. 3A). Increasing the potential applied to the cathode as well as the concentration of electrolyte used resulted in quicker delamination. Complete delamination followed by transfer to desired substrates could be achieved in 60 min, a mere fraction of the duration required for traditional wet-etching techniques. The authors noted that only a small amount of Cu was electrochemically etched during each CVD growth–delamination–transfer cycle (<40 nm), allowing the Cu foil to be reused for hundreds of cycles for a 25 μm thick foil. Additionally, repeated etching/heating cycles caused the Cu foil to become increasingly smoother, due to the preferential etching of step edges and grain boundaries, coupled with the electrodeposition of Cu nanoparticles in trenches and concavities on the foil. This resulted in the improvement of the quality and charge carrier mobility of the grown graphene films with each successive cycle (Fig. 3B). Gao et al. later improved on this method by growing CVD graphene on Pt foil, which exhibits faster growth rates as well as lower temperature requirements than Cu foil.33 The PMMA/graphene/Pt stack then served as a cathode in a cell with aqueous NaOH, in which bubbling delamination took place and was completed in 30 s for a 1 × 3 cm2 film under a constant current of 1 A. Owing to the chemical inertness of Pt, this delamination could be carried out without etching of the foil. Atomic terraces on the Pt substrate were maintained without any degradation even after several hundred growth and delamination cycles, implying that the Pt foils could be re-used indefinitely. Graphene films grown on the re-used Pt foils under the same growth conditions showed no obvious structural differences even after 100 transfers. Despite the great potential of the high-speed delamination of CVD graphene from metal foils via electrochemical hydrogen bubbling, there is a possibility of mechanical damage to the graphene film due to the surface tension of bubbles at the polymer/graphene interface.34 To avoid the formation of hydrogen bubbles, Cherian et al. developed an electrochemical delamination method at a less negative potential compared to bubbling delamination.35 This method exploits the selective etching of adventitious cuprous oxide on the Cu foil surface (Cu2O + H2O + 2e− → 2Cu + 2OH−) by holding the PMMA/graphene/Cu stack at −2.6 V in an unreferenced two-electrode electrochemical system, with 0.5 M NaCl as the electrolyte. To speed up the delamination in the absence of mechanical forces originally provided by bubbles, a gradual immersion technique was used, where the initially delaminated PMMA/graphene stack floating on the solution provided the driving force instead. Under these conditions, delamination of an ∼1.5 cm2 sample could be completed after 1–2 min without any visible formation of bubbles (Fig. 3C). Vigorous hydrogen evolution typically occurred at potentials more negative than −2.8 V. The graphene film obtained using “bubble-free” delamination adhered more uniformly to the final substrate, had a negligible amount of mechanical damage, and had lower sheet resistance than that obtained from hydrogen bubbling delamination (Fig. 3D). The authors also commented that their “bubble-free” method did not exclude the evolution of hydrogen, but noted that the very slow rate of hydrogen production enabled hydrogen to diffuse into the solution instead of forming a gas phase at the graphene/Cu interface. A similar method was demonstrated by Pizzocchero et al. for CVD graphene grown on copper catalyst surfaces, in particular copper thin films deposited on support substrates.36 Such thin films tend to delaminate at the Cu/support interface rather than the graphene/Cu interface in the presence of small amounts of hydrogen bubbling; it is thus important to ensure that hydrogen production from the thin film is kept to an absolute minimum, if not completely eliminated. In this procedure, CVD graphene was grown on Cu thin films sputtered on a SiO2/Si support, and then spin-coated with cellulose acetate butyrate (CAB). Once immersed in a supporting electrolyte (1 M KCl), the Cu thin film oxidised to Cu2O by dissolved oxygen present in the electrolyte, which started to weaken the adhesion of graphene to the catalyst surface. The CAB/graphene/Cu stack was then held at a potential of −0.4 V (vs. Ag/AgCl), which is sufficient to reduce Cu2O back to Cu but at the same time preventing the evolution of hydrogen. A localised excess of hydroxide ions was produced and capillary forces aided in drawing more electrolyte further between the graphene film and Cu, which ultimately resulted in the delamination of the graphene film without removing Cu from the support substrate. No change in the mass or thickness of the Cu catalyst layer could be detected even after being exposed to the delamination conditions for 100 h. The Cu/support could then be reused for further CVD growth and delamination cycles, limited only by the loss of small amounts of catalyst through evaporation of Cu during CVD growth. However, time taken for complete delamination of graphene from Cu was significantly slower due to the milder conditions used, with the Cu oxidation front proceeding at an average rate of 24 mm per hour (Fig. 3E).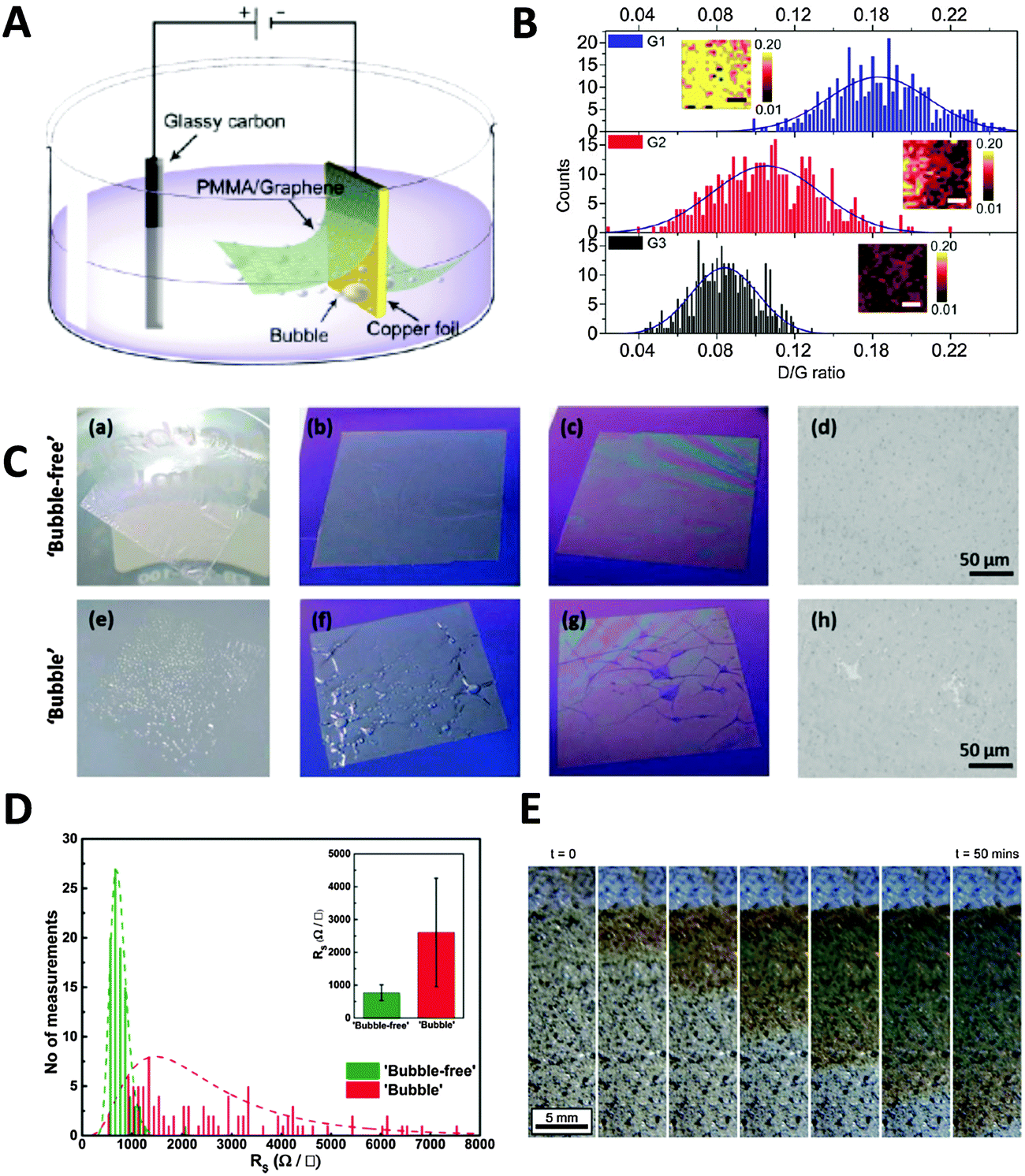 | ||
| Fig. 3 Electrochemical delamination of CVD graphene from Cu foil. (A) Schematic of the electrochemical setup used in bubbling delamination. (B) Raman ID/IG ratios of graphene films G1, G2, and G3 during bubbling delamination (G1, G2, and G3 refer to the first, second, and third cycles of electrochemical delamination); inset: Raman ID/IG maps obtained from an area of 10 × 10 μm2, scale bar = 3 μm. Reproduced with permission from ref. 32. Copyright (2011) American Chemical Society. (C) Comparison of films obtained from bubble-free (top) and bubbling (bottom) delamination at various stages of the transfer process: PMMA/graphene film floating on DI water after rinsing (a and e); immediately after transfer to Si wafer (b and f); after drying at 80 °C (c and g), as well as representative optical microscope images of the films (d and h; scale bars = 50 μm). (D) Histograms of sheet resistance measurements of bubble-free and bubbling delaminated graphene films. Inset: Average sheet resistance values of delaminated graphene films as well as their standard deviations. Reproduced with permission from ref. 35. Copyright (2015) John Wiley and Sons. (E) Optical images showing progression of the Cu oxidation front underneath CAB/graphene. Reproduced with permission from ref. 36. Copyright (2015) Elsevier. | ||
Alternatively, the Cu foil itself can also be electrochemically etched away to leave just the PMMA/graphene film, using a fraction of the time needed for conventional wet-etching techniques (although the Cu foil is wasted as well). Yang et al. showed that etching could be accelerated by holding a PMMA/graphene/Cu stack at a potential of +0.5 V (vs. SCE) in 0.5 M sulfuric acid.37 In this acidic environment, Cu was oxidised to Cu2+ ions, which readily diffused into the bulk solution (Fig. 4A). As the electrochemical reaction proceeded and Cu was etched away, the current initially decreased slowly (Fig. 4B). A rapid drop in current values signified the point where the foil was etched into increasingly discontinuous Cu islands; however, the continuous and highly conductive graphene film then served as an electrode to facilitate the further etching of these Cu islands. In the final stage, current values dropped to almost zero, indicating that the electrochemical etching process was complete (within 10 min). Visual observation of the electrochemical etching process could be carried out using optical microscopy at different stages of etching (Fig. 4C–E). Electrochemical oxidation afforded the graphene film showing less p-doping than traditional wet-etching, was significantly faster, and avoided the additional contamination of iron impurities from Fe-based chemical etchants. Since the oxidative etching of Cu occurs at the anode, this process can be coupled with the abovementioned bubbling delamination (at the cathode) to improve transfer efficiency. Shi et al. employed such a bi-electrode electrochemical transfer system by using identical PMMA/graphene/Cu stacks as both electrodes, immersing them in 0.1 M (NH4)2S2O8, and then applying a voltage of +2 V to the anode (Fig. 4F).38 This electrolyte and concentration were chosen because while an acidic environment was necessary for the dissolution of Cu2O to Cu2+ at the anode, the generation of H2 bubbles at the cathode would be too vigorous if the H+ ion concentration is high, potentially damaging the graphene film. Under these experimental conditions, two graphene films could be obtained simultaneously from a single setup within 20 min. The cathode (electrochemical oxidative etching) graphene showed higher p-doping than traditional metal-etched graphene, while the anode (bubbling delamination) graphene had less p-doping, which the authors attributed to differing electron loss/gain of graphene at their respective electrodes. The measured sheet resistance of both electrochemically-derived graphene samples was somewhat higher than that of metal-etched graphene, although their resistance distributions were narrower, hinting at more uniform graphene films being obtained via the electrochemical methods.
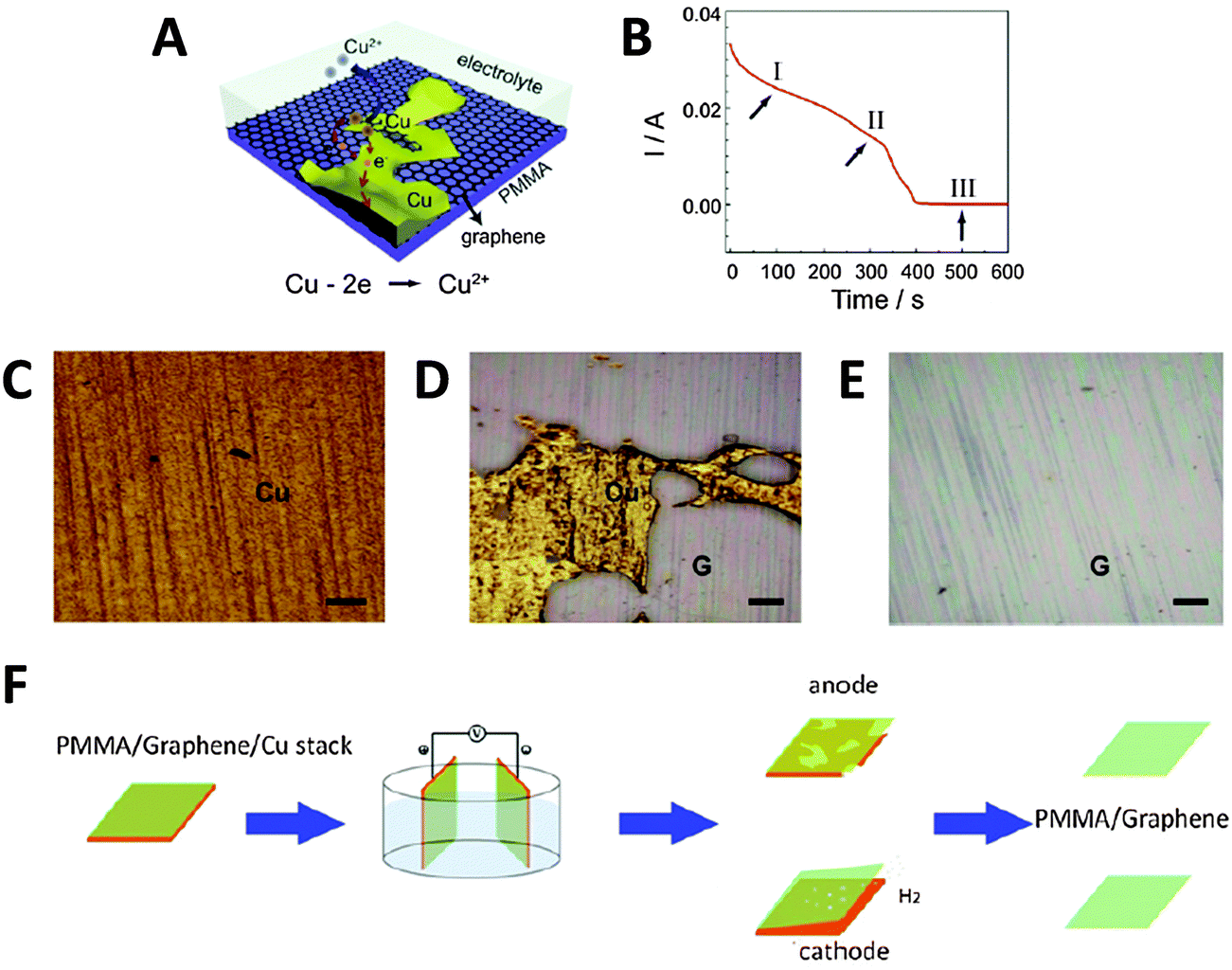 | ||
| Fig. 4 Transfer of graphene thin films by electrochemical etching of a metal substrate. (A) Schematic of PMMA/graphene/Cu stack and the electrochemical reactions that occur during the etching process. (B) Evolution of current values measured during the etching process at +0.5 V (vs. SCE) in 0.5 M H2SO4. (C–E) Optical micrographs of the sample taken during stages I, II, and III of the etching process as shown in (B). G denotes the graphene film, scale bars = 200 μm. Reproduced with permission from ref. 37. Copyright (2013) Elsevier. (F) Schematic of the bi-electrode setup used for coupling electrochemical etching (anode) with bubbling delamination (cathode) for the simultaneous production of two PMMA/graphene films. Reproduced with permission from ref. 38. Copyright (2014) IOP Publishing. | ||
3. Graphene oxide as an electrochemically active material
3.1 Inherent redox activity of oxygen functional groups
A popular “top-down” approach to graphene production is the exfoliation and reduction of graphite oxide. This first involves graphite oxidation by chemical means using a combination of strong acids and oxidants, followed by subsequent reduction/exfoliation via thermal, chemical or electrochemical means. Ultrasonication may also be employed to facilitate exfoliation, which produces graphene oxide (GO). In contrast to pristine graphene, the chemical (and electrochemical) characteristics of GO are exuberant due to its many reactive oxygen functionalities that include hydroxyls, epoxides, carbonyls, quinones, and carboxylic acids. Electrochemical reduction of GO was first explored in depth by Dong and co-workers, who used linear sweep voltammetry for GO electroreduction in aqueous solution at varying pH (Fig. 5A).39 With application of a reductive potential, optical images show the gradual colour change from the yellow-brown of GO (Fig. 5B-i) to black for reduced-graphene near the electrode tip (Fig. 5B-ii) due to regeneration of π-conjugation. Although electrode morphology remains relatively unchanged (Fig. 5B, right panel), the deoxygenation of oxygen groups is confirmed by XPS and elemental analysis.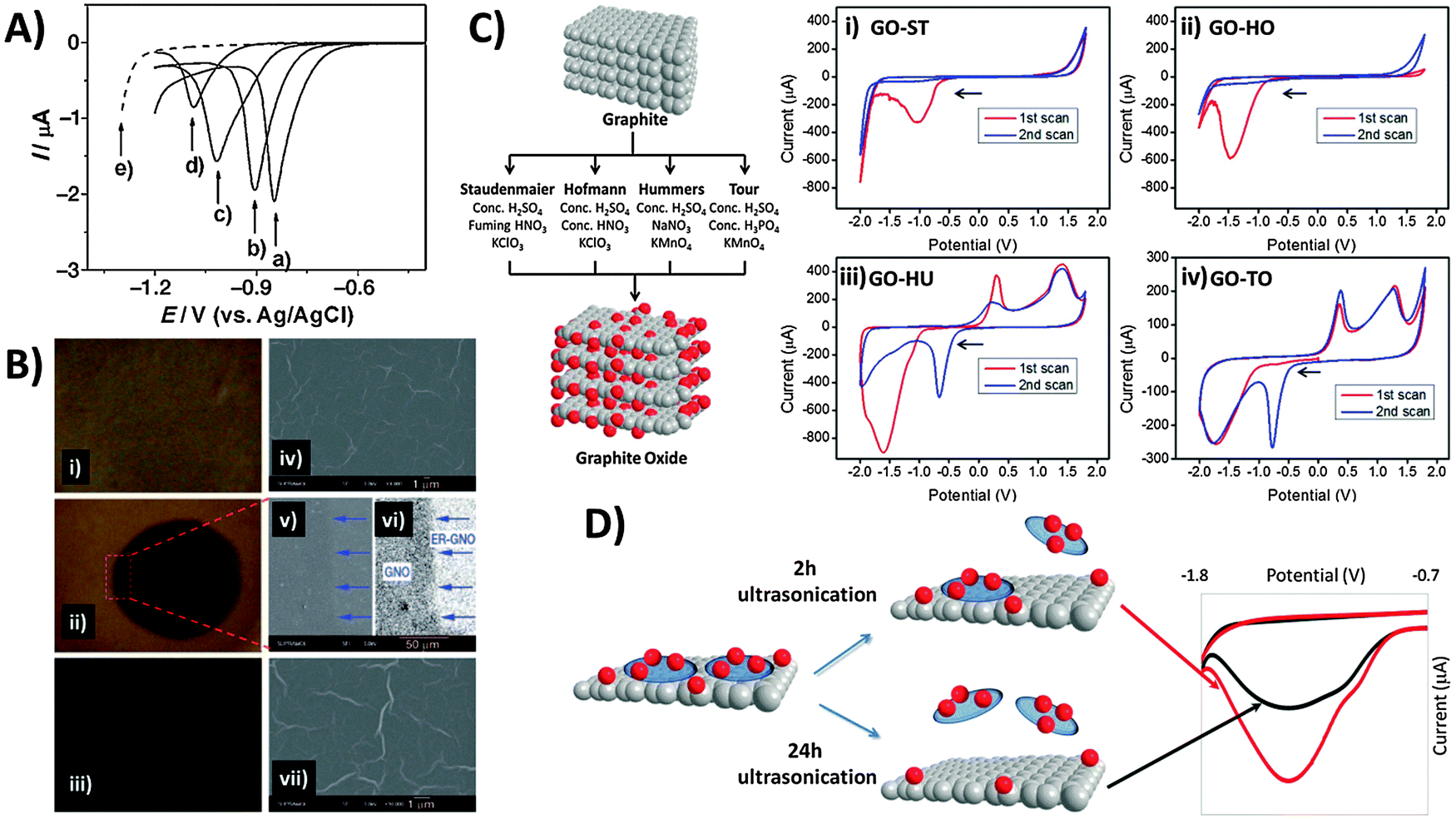 | ||
| Fig. 5 Electrochemical behavior of inherent oxygen functionalities present in graphene oxide. (A) Linear sweep voltammograms of the GC electrode in contact with graphene oxide films on quartz, measured at pH values of (a) 4.12, (b) 7.22, (c) 10.26, and (d) 12.11. (e) Linear sweep voltammogram of the GC electrode in PBS (1 M, pH 4.12). (B) Optical (i–iii) and SEM images (iv–vii) of GO films before (top panel), during (1000 s; middle panel) and after electrolysis (5000 s; bottom panel). Image (vi) was obtained from image (v) using contrast enhancement. Arrows indicate the boundary between the reduced circular area and the unreduced surrounding GO. Reprinted with permission from ref. 39. Copyright (2009) John Wiley and Sons. (C) Scheme illustrating oxidation methods used for graphite oxide production. Cyclic voltammograms of GOs prepared from the methods of (i) Staudenmaier, (ii) Hofmann, (iii) Hummers, and (iv) Tour, with starting cathodic scan. Conditions: 50 mM PBS, pH 7.2, scan rate: 100 mV s−1. All potentials are relative to the Ag/AgCl reference electrode. Reprinted with permission from ref. 40. Copyright (2013) John Wiley and Sons. (D) Effect of 2 h (red line) versus 24 h (black line) ultrasonication times on the cyclic voltammograms of GO as performed in 0.1 M PBS at pH 7. Reprinted with permission from ref. 41. Copyright (2014) American Chemical Society. | ||
It is also crucial to note that differences in the composition of oxygen functionalities exist between GOs produced by various preparation methods (Fig. 5C).40 All GOs exhibit characteristic reduction peaks and the peak potential typically correlates with the oxidation extent of GO in the order: Staudenmaier (ST) < Hofmann (HO) < Hummers (HU) < Tour (TO). This is because groups like carbonyls require stronger reductive overpotentials than epoxyls, peroxyls or aldehydes. More importantly, reductions occur irreversibly for the Staudenmaier and Hofmann GOs appearing only within the first cathodic sweep (Fig. 5C-i and ii). Interestingly however, chemically reversible behavior is observed for the Hummers and Tour GOs, with new oxidation and reduction waves that persist after activation from the initial cathodic sweep (Fig. 5Ciii and iv). It was noticed that this disparity primarily arises from the choice of oxidising agent: either chlorate from the Staudenmaier and Hofmann methods or permanganate from the Hummers and Tour methods. The low Mn content measured from ICP-MS as compared to the large reduction charge passed additionally invalidates the possibility that manganese-based esters or impurities may be responsible. High resolution carbon-1s XPS and data from pH studies further suggest quinone–hydroquinone couples as likely sources of the reversible electrochemistry in GO-HU and GO-TO. As GO production is essentially a bulk oxidation and exfoliation process, it is also easy to envisage some heterogeneity in the size and stoichiometry of GO sheets. Particularly, small and highly oxidised sheets, known to be oxidative debris (OD), have been previously shown to exhibit fluorescence but are also likely the major contributors to the observed electroactivity.41 OD fragments are typically strongly adsorbed on larger GO sheets due to π–π stacking, therefore requiring extended sonication times for desorption. Increasing sonication times in principle dislodge larger amounts of OD from the GO sheets, analogous to a cleaning procedure. Thus as seen in Fig. 5D, intensity of the reduction peak for precipitated GO sheets decreases with longer sonication due to OD removal. In this regard, one should always consider possible variations in GO electrochemistry as a result of either the oxidation method or experimental procedures (e.g. sonication times) employed during the material preparation process.
3.2 Applications of graphene oxide activity and its practical limitations
In light of the chemical reactivity of GO, several applications have been developed that exploit it for specific uses. The majority of graphene-modified electrodes to date are prepared through the technique of drop-casting, but also through spin-coating to a smaller extent. Subsequently, Luo and co-workers showed in a 2011 report and in subsequent studies that a reduced-graphene film may be prepared through electrochemical deposition for sensing applications.42 The technique capitalizes on the solubility of GO in aqueous media due to its polar oxygen groups, compared to the poor dispersibility of graphene. Potential cycling of a 1.0 mg mL−1 GO dispersion in phosphate buffer at pH 9.18 (Fig. 6A) demonstrates a reduction at approximately −1.0 V vs. SCE, and a redox pair at 0.0 V similar to the inherent electroactivity of permanganate-oxidized GOs earlier discussed. The increase in peak currents with each cycle of deposition and film formation can be easily tracked using SEM. Consequently, sensing of a hydroquinone and catechol mixture was found to be significantly enhanced with the deposited graphene film in contrast to the lower sensitivity of bare glassy carbon electrodes (Fig. 6B) whether CV or DPV techniques were employed. In addition to detection of redox analytes, biological sensing is also feasible through the use of GO as an electrochemically-active label.43 Selective thrombin aptasensing is achieved through a protocol first involving modification of a commercial carbon electrode with the thrombin aptamer (Fig. 6C). Upon exposure to thrombin, specific binding with the physically immobilised thrombin aptamer results in its partial release from the electrode surface, subsequently resulting in large uncovered surfaces for charge transfer between GO and the electrode. Detection is then based on voltammetric reduction of the inherent oxygen groups from the nanoplatelets, with a larger signal occurring in the case with thrombin.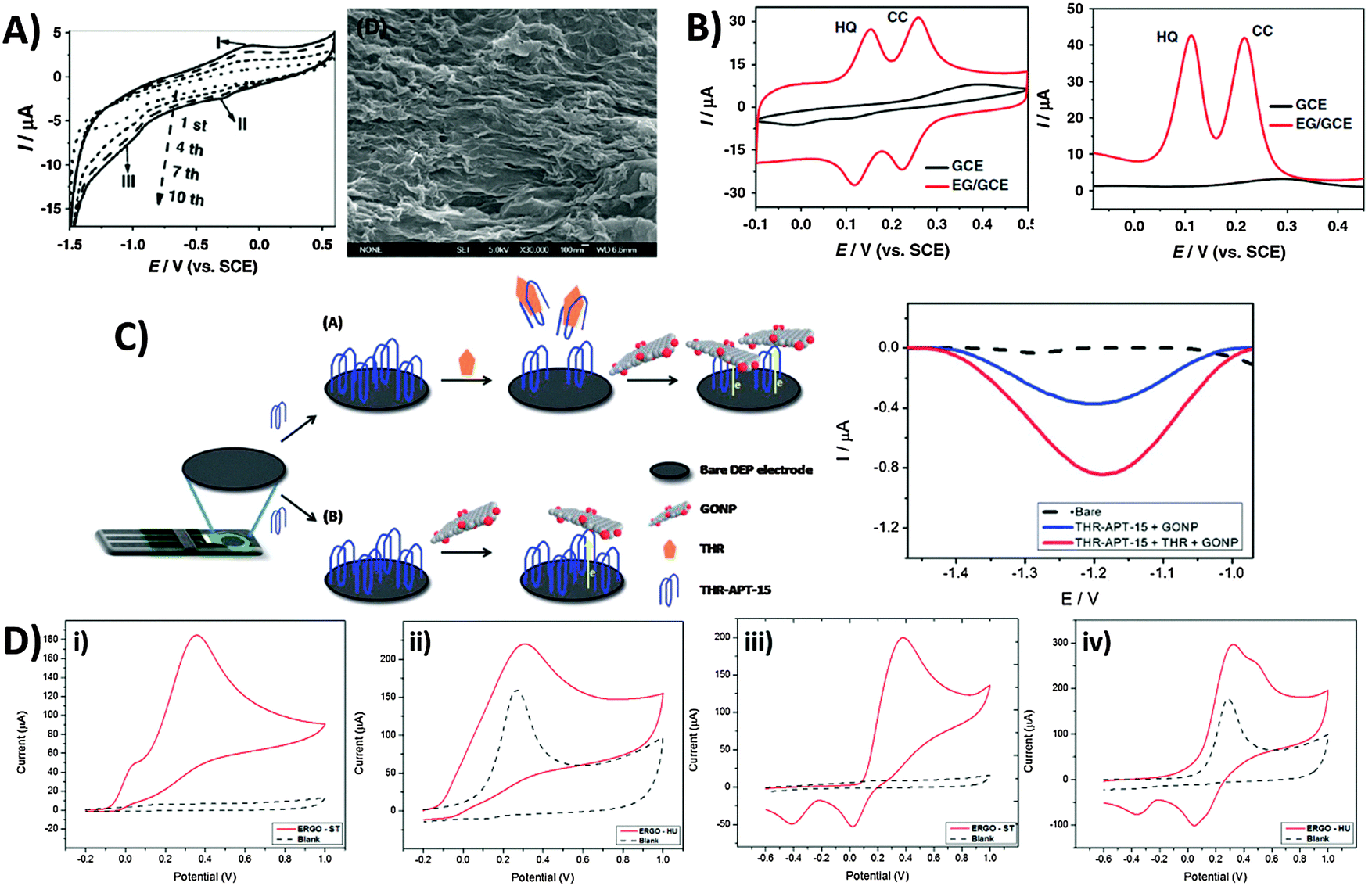 | ||
| Fig. 6 Employment of inherent GO activity towards film deposition, sensing and its associated limitations. (A) Cyclic voltammogram illustrating electrochemical reduction of GO at 1.0 mg mL−1 concentration onto GC electrodes (0.067 M PBS, pH 9.18, scan rate: 10 mV s−1), and the SEM image of the electro-deposited graphene film. (B) Cyclic voltammetry and differential pulse voltammetry of a mixed sample of hydroquinone (0.2 mM) and catechol (0.2 mM) in 0.2 M acetate buffer (pH 5.8) on bare GC and electro-deposited graphene film. CV parameters: a scan rate of 50 mV s−1; DPV parameters: a scan rate of 4 mV s−1, 50 mV pulse amplitude, and 20 ms pulse width. Reprinted with permission from ref. 42. Copyright (2011) Elsevier. (C) Use of graphene oxide nanoplatelets (GONPs) as electroactive labels for the detection of thrombin (THR). In the presence of THR, the aptamer (THR-APT-15) binds specifically to THR and results in the partial release of immobilized THR-APT-15 from the electrode surface, uncovering larger areas for electron transfer between conjugate GONPs and the electrode. Differential pulse voltammograms of GO reduction in the presence (red) and absence (blue) of THR. Reprinted with permission from ref. 43. Copyright (2013) Royal Society of Chemistry. (D) Inherent activity of permanganate-oxidised GO limits potential window for analyte sensing. Cyclic voltammograms of (i and ii) 10 mM ascorbic acid and (iii and iv) 10 mM dopamine on graphene oxides prepared by the (i and iii) Staudenmaier and (ii and iv) Hummer's methods. Voltammograms in a blank supporting electrolyte (dotted) are shown for comparison. Conditions: 50 mM PBS; pH 7.2; scan rate 100 mV s−1. Reprinted with permission from ref. 44. Copyright (2014) American Chemical Society. | ||
Despite the usefulness of inherent GO activity towards sensing, it nonetheless places certain restrictions on its own applicability. The primary concern is that reliable detection cannot be achieved for any analyte with electrochemical activity occurring in the same region as the activity of inherent GO functionalities. This difficulty in distinguishing the voltammetric waves of the analyte and the material limits the usable potential window of the electrode. Such a situation is demonstrated for chlorate-based GO-ST against the permanganate-based GO-HU.44 Anodic detection of molecules like ascorbic acid and dopamine are possible for GO-ST with no influence from the underlying electrode (Fig. 6Di and iii). For GO-HU however, its inherent oxidation overlaps with those from both analytes (Fig. 6Dii and iv), preventing any quantification data from being extracted. Hence, special care should be exercised when working with redox active graphene materials such as GO especially in sensing applications, and a simple measurement of the electrode material in a blank electrolyte is highly recommended for experimentalists before they proceed with any sensing protocols.
3.3 Electrochemical modification/activation of graphene oxide for tuning its electron transfer properties towards sensing and capacitance
Whilst we have seen that the inherent activity of GO can either be a boon or bane, it is reassuring to note that the oxygen content can in fact be controlled or tuned through prior electrochemical treatment. Electrochemical activation can be simply performed by application of a cathodic or anodic potential which tunes the surface oxygen composition and consequently the electron transfer properties of the material. In most cases with GO, electro-reduction of the oxygen groups greatly improves the heterogeneous electron transfer (HET) rate. Using XPS to identify changes in surface oxygen functionalities for a GO film on a screen-printed electrode (Fig. 7A), we observed the gradual loss of C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups with increasingly reductive potentials applied.45 Peak separations for the ferro/ferricyanide redox couple decreased correspondingly, with HET rates increasing 1000-fold from 1.6 × 10−6 cm s−1 for the untreated GO film to 4.0 × 10−3 cm s−1 with an applied potential of −1.50 V. It should be noted however that strongly reducing potentials past −1.50 V vs. Ag/AgCl cause irreversible film damage for the screen-printed electrode due to hydrogen evolution. Additionally, changes in electron transfer properties due to electrochemical activation can also differ between various redox probes. For example, while both Fe(CN)63−/4− and Fe2+/3+ are typically described as inner-sphere probes due to their sensitivity to surface oxygen groups,46 a strong dependence of HET rates on the activation potential exists only for the former.47 In its hydrated form, Fe2+/3+ experiences strong interactions specifically with carbonyl groups similar to a chelate, and thus shows faster HET with anodic activation on glassy carbon.47 However, a similar trend was not observed for GO since additional carbonyl groups cannot be introduced as these can only exist at sheet edges which are likely saturated from the beginning (Fig. 7B). Reduction of GO further removes such edge carbonyls which thus lower the observed HET rate. In comparison, ruthenium hexamine Ru(NH3)62+/3+ is an outer-sphere redox probe due to its surface insensitivity and is only affected by factors such as surface area and the electronic density of states (DOS). It is therefore largely uninfluenced by electrochemical activation. Finally in the case of ascorbic acid (AA), cathodic activation treatment of GO reduced the overpotential for AA oxidation as it removes oxygen moieties that would otherwise experience electrostatic repulsion with the negatively charged AA under neutral pH conditions.
O groups with increasingly reductive potentials applied.45 Peak separations for the ferro/ferricyanide redox couple decreased correspondingly, with HET rates increasing 1000-fold from 1.6 × 10−6 cm s−1 for the untreated GO film to 4.0 × 10−3 cm s−1 with an applied potential of −1.50 V. It should be noted however that strongly reducing potentials past −1.50 V vs. Ag/AgCl cause irreversible film damage for the screen-printed electrode due to hydrogen evolution. Additionally, changes in electron transfer properties due to electrochemical activation can also differ between various redox probes. For example, while both Fe(CN)63−/4− and Fe2+/3+ are typically described as inner-sphere probes due to their sensitivity to surface oxygen groups,46 a strong dependence of HET rates on the activation potential exists only for the former.47 In its hydrated form, Fe2+/3+ experiences strong interactions specifically with carbonyl groups similar to a chelate, and thus shows faster HET with anodic activation on glassy carbon.47 However, a similar trend was not observed for GO since additional carbonyl groups cannot be introduced as these can only exist at sheet edges which are likely saturated from the beginning (Fig. 7B). Reduction of GO further removes such edge carbonyls which thus lower the observed HET rate. In comparison, ruthenium hexamine Ru(NH3)62+/3+ is an outer-sphere redox probe due to its surface insensitivity and is only affected by factors such as surface area and the electronic density of states (DOS). It is therefore largely uninfluenced by electrochemical activation. Finally in the case of ascorbic acid (AA), cathodic activation treatment of GO reduced the overpotential for AA oxidation as it removes oxygen moieties that would otherwise experience electrostatic repulsion with the negatively charged AA under neutral pH conditions.
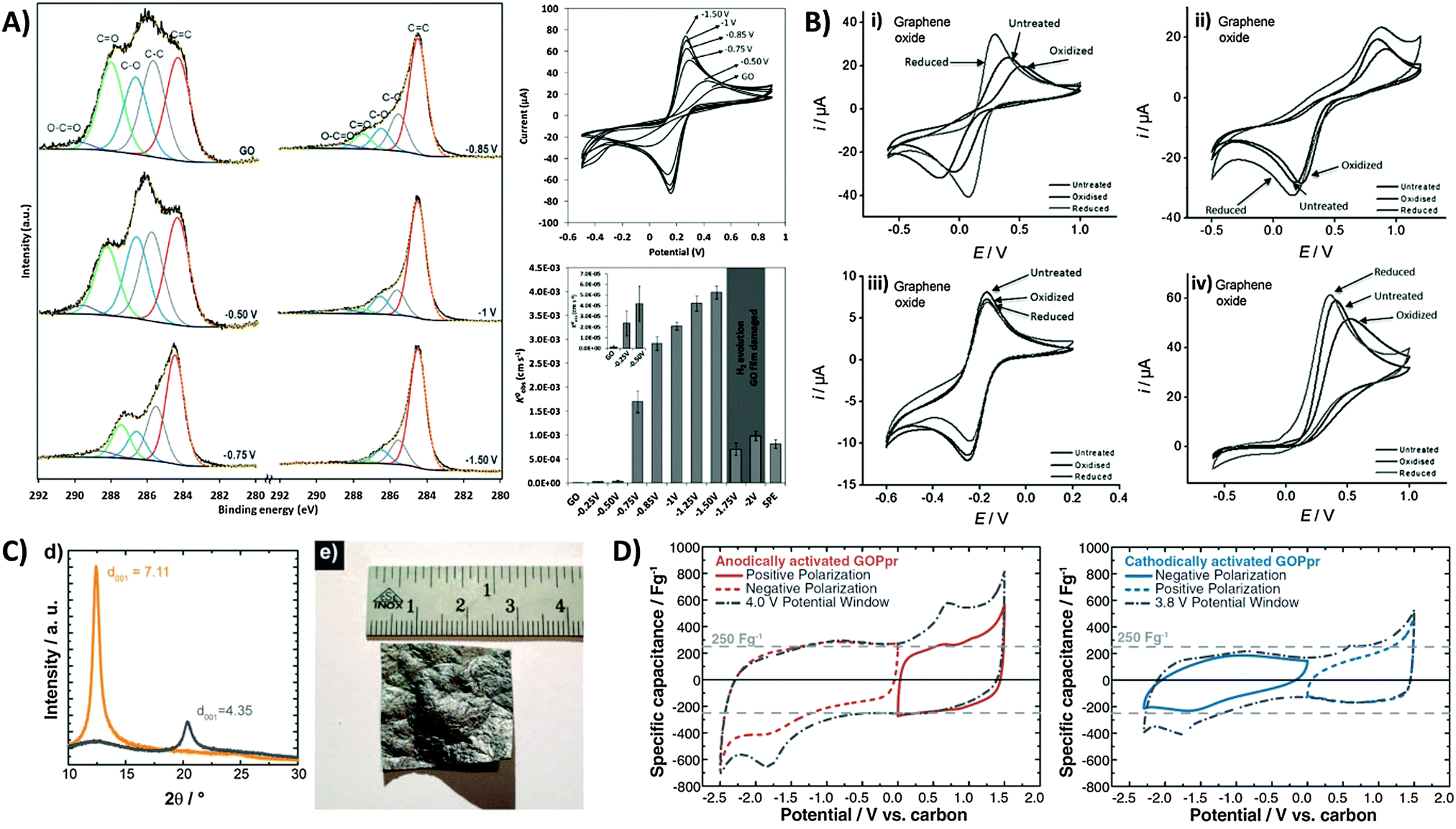 | ||
| Fig. 7 Electrochemical modification and activation of graphene oxide towards applications. (A) High-resolution carbon-1s XPS spectra of GO-modified screen printed electrodes after application of different potentials for 5 min. Cyclic voltammograms of 5 mM ferro/ferricyanide obtained on GO-modified electrodes after electrochemical treatments at different potentials. Conditions: 0.1 M KCl; scan rate, 0.1 V s−1; potentials are with reference to Ag/AgCl. HET rate constants (k0obs) calculated from the peak-to-peak separation of ferro/ferricyanide. Inset: enlarged graph of untreated GO film and after application of −0.25 and −0.50 V. Reprinted with permission from ref. 45. Copyright (2013) John Wiley and Sons. (B) Cyclic voltammograms of (i) Fe(CN)63−/4−, (ii) Fe2+/3+, (iii) Ru(NH3)62+/3+, and (iv) ascorbic acid redox probes on GO after electrochemical oxidation or reduction activation treatments. Scan rate: 100 mV s−1; supporting electrolyte: 50 mM PBS at pH 7.2. Reprinted with permission from ref. 47. Copyright (2012) John Wiley and Sons. (C) XRD spectra of GO paper before (interlayer distance: 7.11 Å) and after (4.35 Å) thermal reduction treatment. Image of a large 7 cm2 partially-reduced GO membrane. (D) Cyclic voltammograms demonstrating capacitances from positive and negative polarizations, and full sweep of anodically-activated GOPpr (grey dot-dashed line). Negative and positive polarizations, and full sweep of cathodically-activated GOPpr (grey dot-dashed line). Conditions: 1 mV s−1 scan rate, 1 M TEABF4 in acetonitrile. Reprinted with permission from ref. 48. Copyright (2013) The Electrochemical Society. | ||
Electrochemical activation has also been used to improve the performance of graphene-based capacitors. There are several reports by Kötz and co-workers who employed GO as a capacitor material.48 As shown in Fig. 7C, large centimetre-sized GO paper (GOP) can be produced by flow-directed filtration, with subsequent thermal treatment to give partially reduced GO paper (GOPpr). This effectively reduces the interlayer distance measured by X-ray diffraction. With an electrolyte of TEABF4 in acetonitrile, the authors reported potentials of 1.31 V and −1.13 V vs. carbon for anodic and cathodic activations, respectively. It was particularly noted that anodic activation (Fig. 7D) resulted in an enhanced capacitive discharge of up to 270 F g−1 during the discharge sweep at 0.0 V regardless of polarization. Redox peaks were also seen at approximately −1.8 V during initial cathodic sweeps and at 0.7 V with anodic sweeps, but do not contribute to the overall capacitance. Cathodic activation in contrast produced slightly distorted voltammograms with less obvious redox peaks, and a lower specific capacitance of ca. 150 F g−1. Although both activations were said to improve capacitor behavior, anodic activation resulted in a superior enhancement not only due to the different types of intercalated ions but was also proposed to be due to the remaining oxygen functional groups.48
4. Capacitance of graphene
The high conductivity, the exceptional mechanical properties and more importantly its high theoretical specific surface area (SSA) calculated as 2630 m2 g−1 have certainly suggested the use of graphene as an electrode material for the construction of supercapacitors. However, several limitations still impede the full exploitation of all the extraordinary properties of graphene, as a result, leading to a much lower experimentally measured capacitance than expected. One of such limitations is the tendency of graphene sheets to restack after the production reducing significantly the active surface area. A gravimetric capacitance in the order of only tens of F g−1 has been measured for a graphene-based electrode which is similar to that obtained using graphite as the electrode material.49Also, it was discovered that the interfacial capacitance measured for both sides of a single graphene sheet is much lower than the one measured on only one side indicating a quantum component in the charge storage mechanism.50 Both these phenomena make the achievement of the theoretical capacitance value of 550 F g−1 for pure graphene51 extremely hard. In order to overcome such limitations different strategies have been adopted in the last few years, which can be summarized as follows: production of 3D graphene structures with high conductivity and high surface area, introduction of spacers to avoid graphene re-stacking, introduction of heteroatoms (doping), functionalization of graphene sheets with redox molecules and preparation of graphene composites in combination with other materials.
An activation method using KOH at high concentrations is known to increase significantly the active surface area of carbon materials.52 Such a procedure was employed in 2011 by Zhu et al. to activate a microwave-exfoliated graphene oxide (MEGO). An extraordinary SSA value of 3100 m2 g−1 and a high conductivity of 500 S m−1 were measured for the activated MEGO material which resulted in a capacitance of 166 F g−1 in 1-butyl-3-methyl-imidazolium tetrafluoroborate/acetonitrile (BMIM BF4)/AN electrolyte at a current density of up to 5.7 A g−1.53 A similar activation procedure using KOH was utilized by Ma and coworkers to prepare a graphene-activated carbon composite (GAC) with a high SSA of 798 m2 g−1 giving a capacitance of 122 F g−1 in an aqueous electrolyte (6 M KOH).54 The same group later measured a SSA of 3523 m2 g−1 for a porous graphene material obtained by hydrothermal synthesis and carbonization of a mixture of GO and carbon sources such as biomass, phenol-formaldehyde (PF), and polyvinyl alcohol (PVA), followed by activation in KOH. Such porous graphene exhibited a high specific capacitance of 202 F g−1 in 1 M tetraethylammonium tetrafluoroborate in AN (TEA BF4/AN) and 231 F g−1 in 1-ethyl-3-methylimidazolium (EMIM) BF4, electrolyte.55 Very recently a graphene aerogel has been activated using phosphoric acid and thermal annealing at 800 °C obtaining a porous material of about 1145 m2 g−1 SSA exhibiting a gravimetric capacitance of 204 F g−1.56 Physical activation was proposed by Yun et al. who firstly prepared a trimodal porous graphene structure by self-assembly of graphene sheets, followed by CO2 activation at 900 °C which produced micropores. A SSA of 829 m2 g−1 was measured for this material which gave a capacitance of 278.5 F g−1 in an aqueous H2SO4 electrolyte (Fig. 8).57
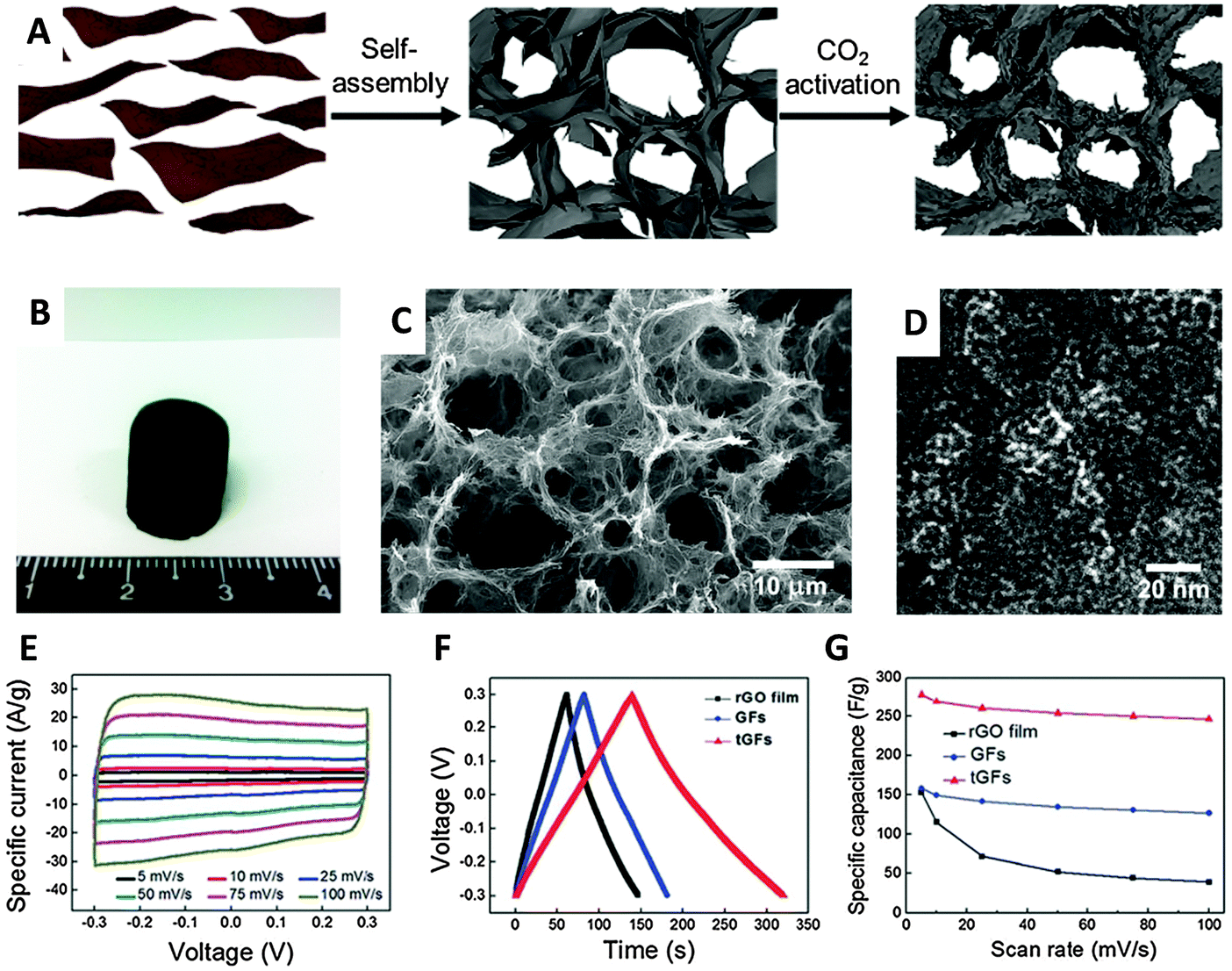 | ||
| Fig. 8 (A) Schematic illustration of preparation of the tGFs through self-assembly and CO2 activation. (B) Optical image of the resultant tGFs. (C) Low magnification SEM and (D) HR-TEM image of tGFs. (E) CV curves of tGF at the scan rates of 1 to 100 mV s−1. (F) Galvanostatic charge/discharge curve of tGF at 1 A g−1. (G) Rate capability of tGF at a scan rate of 5–100 mV s−1. Reproduced with permission from ref. 57. Copyright (2014) Royal Society of Chemistry. | ||
A solution processable holey graphene oxide was easily obtained by heating a GO solution to 100 °C in the presence of H2O2 which, according to authors, etches the oxygenated carbons present on the basal plane. These holey graphene oxide nanosheets have then been processed into reduced porous 3D hydrogels and 2D layered papers with a SSA of 1330 m2 g−1 and 217 m2 g−1, respectively. Both materials have been tested for capacitors obtaining a gravimetric capacitance of 283 F g−1 for the hydrogel and 209 F g−1 for the paper at a current density of 1 A g−1 in an aqueous electrolyte.58
To deal with the tendency of graphene to restack after exfoliation, different spacers have been introduced such as Au nanoparticles59 and carbon nanotubes (CNTs).60–66 Interestingly some reports showed a significant increase of capacitance after the preparation of CNT/graphene hybrid materials compared to that of the individual components60–65 while others demonstrated that the final capacitance corresponded to the average between them.66 Functionalization of graphene could also be used as methodology to prevent restacking of graphene sheets. Recent studies compared different graphene preparation methods in terms of the capacitance of the resulting materials.67–69 Chemical, electrochemical or thermal reduction of graphene oxide significantly influences the capacitance of the final graphene material due to the different amount and types of oxygen functionalities. Conductivity is certainly another factor influencing the capacitance and from this study it resulted that the graphene material with the lowest amount of oxygen groups exhibited the largest capacitance due to the superior conductivity of the material.67–69 Water has also been used as an effective spacer to prevent graphene restacking and fabricate highly porous graphene aerogels. Following a crystallization route which forms ice spacers a free-standing aerogel structure exhibited a capacitance of 172 F g−1 at a current density of 1 A g−1.70,71
A different approach to improve the capacitive properties of graphene has been the introduction of heteroatoms into the graphene structure. These include nitrogen and boron as the most widely investigated, but also sulfur and phosphorous. It is interesting to notice that both the introduction of nitrogen (n-type electron donating atom)72–75 and boron (p-type electron withdrawing atom)76 has resulted in an increased capacitance. The mechanism is still not completely clear although with regard to the nitrogen it is believed that the increased capacitance is due to the changes in the electronic structure of graphene which is directly linked to the quantum capacitance.77 Comparing different boron- and nitrogen-doped graphene materials and after careful chemical and structural characterization, we recently demonstrated that the density of structural defects (edge planes) in the graphene structure represents the dominating factor for the resulting capacitive behavior regardless of the type and amount of doping.78 Sulfur-doping of graphene resulted in an increased capacitance due to the fact that the sulfur species decreased the ability of graphene to adsorb water, enhancing the electrosorption of the electrolyte ions.79 Doping carbon materials with phosphorus has been proposed in the past using phosphoric acid as an activation agent during thermal treatment at 800 °C.80 In this work the authors demonstrated the significant influence of the phosphorous groups on the capacitive behavior of the carbon substrate, which was greatly improved. However, only very recently phosphorus doping has been applied to graphene. In one work, Thirumal et al. proposed a simple electrochemical procedure to exfoliate a graphite electrode under anodic conditions in the presence of phosphoric acid. The resulting electrochemically exfoliated graphene presented about 0.7% content of phosphorus in the form of phosphate groups and exhibited a specific capacitance of 290 F g−1 at a current density of 0.5 A g−1.81 Wen and collaborators proposed an annealing procedure in the presence of phosphoric acid to prepare P-doped graphene. The authors obtained a graphene with about 1.3% P content which allowed graphene to be used with a larger potential window of 1.7 V and with great stability. The measured capacitance was found to be 115 F g−1 at a current density of 0.05 A g−1 in an aqueous sulfuric acid electrolyte.82
The graphene materials described so far are able to store electrical charges at the double layer electrode/electrolyte interface, without any faradaic process to occur and therefore are mainly influenced by conductivity and surface area. Another method of electrical storage is the one which exploits redox reactions with also electrons involved. This type of capacitor is based on the so-called pseudocapacitance and since a faradaic process occurs it can store a larger amount of electrical charges. Pseudocapacitors can be obtained by the introduction of redox active molecules and/or materials within a graphene structure. A common combination involves the use of metal oxides or hydroxides with a highly porous graphene. A graphene/Ni(OH)2 nanocomposite has been recently obtained hydrothermally giving a capacitance value of 1212 F g−1 and 813 F g−1 at a current density of 2 and 16 A g−1, respectively.83 Graphene oxide decorated with Ni(OH)2 nanoparticles was obtained by the decomposition of Ni(CH3COO)2 at 80 °C followed by hydrothermal treatment resulting in an impressive capacitance of 1335 F g−1.84 Cobalt oxide/graphene composites have been considered as one of the most promising materials for next-generation supercapacitors. Zhou and collaborators prepared a Co3O4/graphene composite which showed a specific capacitance of 159 F g−1 at a scan rate of 5 mV s−1.85 A 3D Co3O4/graphene aerogel material has been recently fabricated using hydrothermal synthesis at 180 °C followed by a freeze-drying process. The material showed a capacitance of 660 F g−1 at a current density of 0.5 A g−1 in an aqueous electrolyte.86 RuO2 nanoparticles have been grown on the defects of a reduced graphene oxide using the atomic layer deposition (ALD) method. The rGO–RuO2 material showed a specific capacitance of 1132 F g−1 at a scan rate of 50 mV s−1.87 Other studies proposed the combination of graphene with Fe2O3,88 MnO2,89,90 V2O5,91 Fe3O492 and SnO293,94 among others. Another strategy to enhance the capacitive properties of graphene is to combine it with conducting polymers. Zhang et al. fabricated a polyaniline nanofiber/graphene composite which demonstrated a specific capacitance of 480 F g−1.95In situ anodic electropolymerization (AEP) was used by Wang et al. to prepare polyaniline/graphene composite paper (GPCP) material which showed a specific capacitance of 233 F g−1 at a scan rate of 2 mV s−1 (Fig. 9).96 Lately similar polyaniline/graphene composites achieved a capacitance of 286 F g−1 at 5 mV s−1.97 In recent years excellent capacitive behavior was obtained by combining graphene with other 2D materials, particularly transition metal dichalcogenides. Firmiano and collaborators deposited by microwave heating layered MoS2 onto reduced graphene oxide (rGO) obtaining a composite material with capacitance as high as 265 F g−1 at 10 mV s−1 in acidic media.98 The good capacitive behavior is due to a combined faradaic and non-faradaic capacitive process coupled with the high conductivity of graphene.98 In another work, MoS2 was firstly obtained by liquid phase exfoliation of bulk MoS2 and then mixed with a dispersion of exfoliated graphene. The MoS2/graphene composite material was assembled as a thin film and produced a capacitance of 11 mF cm−2 at a scan rate of 5 mV s−1.99
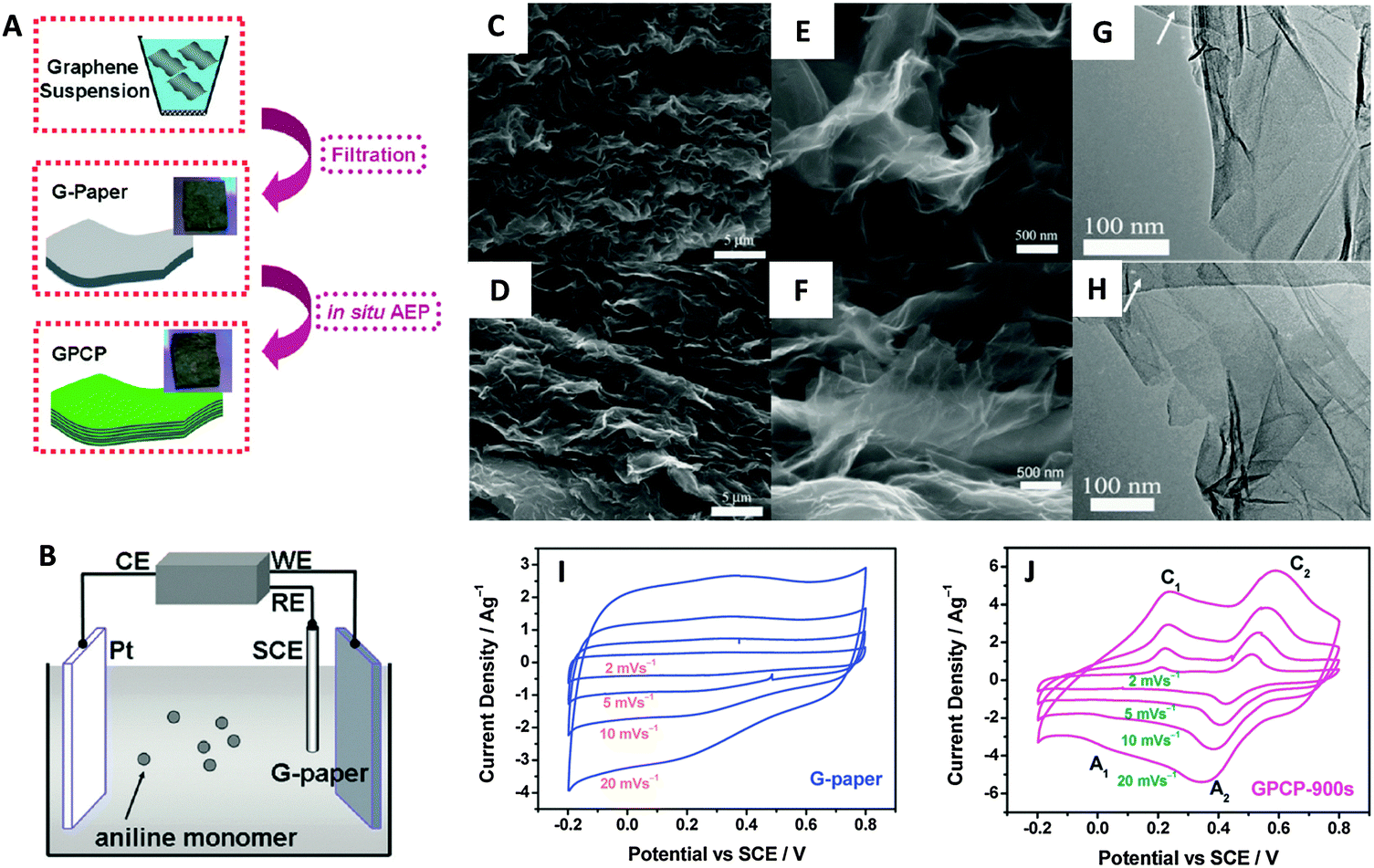 | ||
| Fig. 9 (A) Illustrative fabrication process toward graphene/polyaniline composite paper (GPCP). (B) Cartoon illustrating the anodic electropolymerization (AEP) of an aniline monomer on G-paper. CE: counter electrode (Pt plate). WE: working electrode (G-paper). RE: reference electrode (SCE). SEM and TEM images of the G-paper (C, E and G) and GPCP-900s (D, F and H). (C and D) Low-magnification SEM images showing the stacked layer-by-layer structure. (E and F) High-magnification SEM images and (G and H) low-magnification TEM images showing the morphology of graphene and graphene/PANi sheets. Arrows in G and H denote the amorphous carbon film deposited on the copper grid. Cyclic voltammograms recorded from 2 to 20 mV s−1 in 1 M H2SO4 for (I) G-paper and (J) GPCP. Reproduced with permission from ref. 96. Copyright (2009) American Chemical Society. | ||
5. Electrochemistry of graphene modified with p-block elements
5.1 Electronic and electrochemical properties
Graphene is a zero-gap semiconductor. Due to the intersecting valence and conduction bands at two inequivalent points, K and K′, in the reciprocal space, graphene exhibits excellent conductivity and a distinct electric field effect with high charge concentrations and mobilities.100,101 Pristine graphene shows a p-type behavior due to the presence of adsorbed oxygen or water on its surface.102 Doping graphene with p-block elements (i.e., nitrogen, boron, sulfur, hydrogen, oxygen and fluorine) disrupts the planar structure and introduces foreign elements which would alter its electronic properties. Despite that, such modifications have subsequently widened its applications in various technological devices.103,104In the first part of this section, we will discuss the electronic and electrochemical properties of N-doped, B-doped, S-doped, hydrogenated graphene (graphane), hydroxylated graphene (graphol) and fluorinated graphene (fluorographene) (Fig. 10). Following that, the applications of these graphene materials for oxygen reduction and hydrogen evolution reactions would be evaluated. Readers interested in the synthesis methods of these graphene materials are directed to several review articles available in the literature.105–108
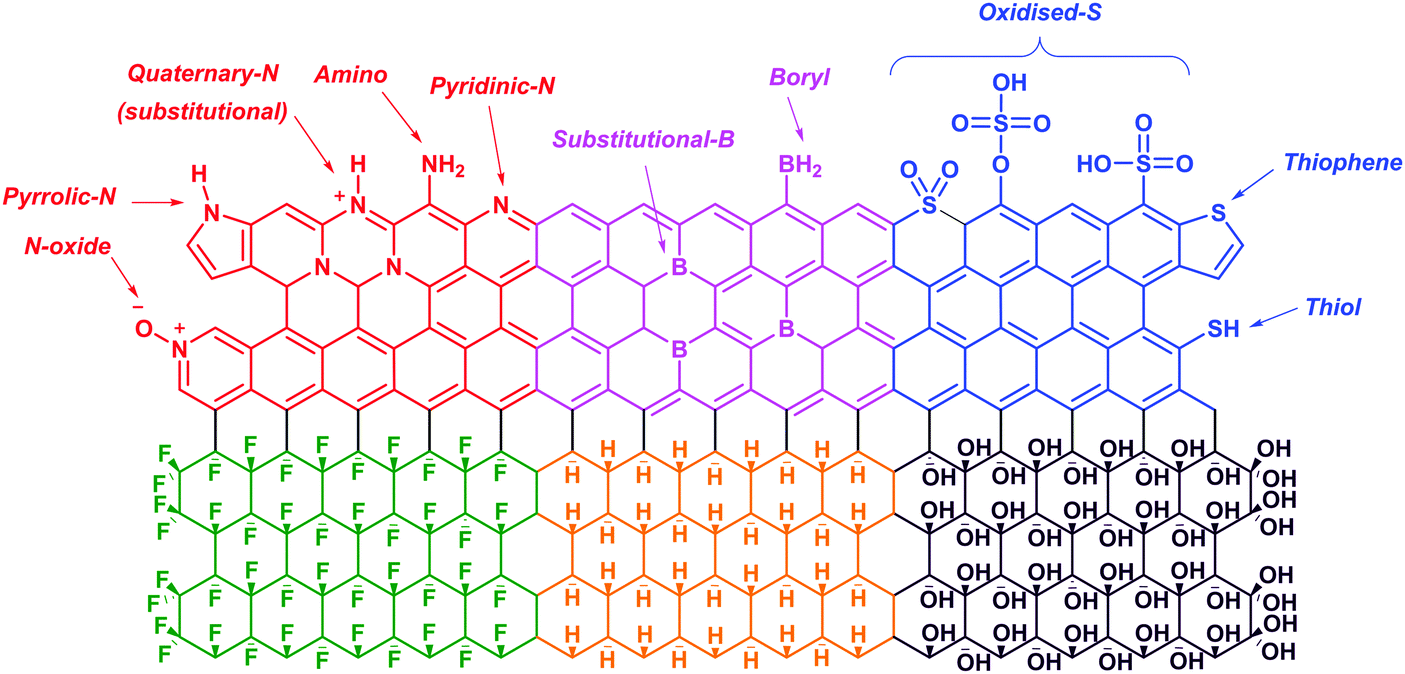 | ||
| Fig. 10 Illustrations of the structures of N-doped (red), B-doped (pink), S-doped (blue), fluorinated (green), hydrogenated (yellow) and hydroxylated (purple) graphene. | ||
Based on the nature of the substitutionally-doped p-block elements (i.e., carbon atoms in the graphene lattice is replaced by dopants), which could be electron-withdrawing or donating, the electronic properties of the resulting graphene materials would vary. Additional effects from additive dopants (i.e., the graphene lattice is functionalized and resulted in the conversion of sp2- to sp3-hybridized carbon atoms) or topological defects could introduce more variables into the electronic properties. More often than not, such modifications vary the density of state (DOS) near the Fermi energy (Fm) level and thus also the conductivities of the doped graphene materials. In electrochemistry, as electron transfer does not generally occur between an electrode and redox systems with E° values lying in the band gap region, the electrochemical properties of the doped graphene materials would differ greatly among themselves.109
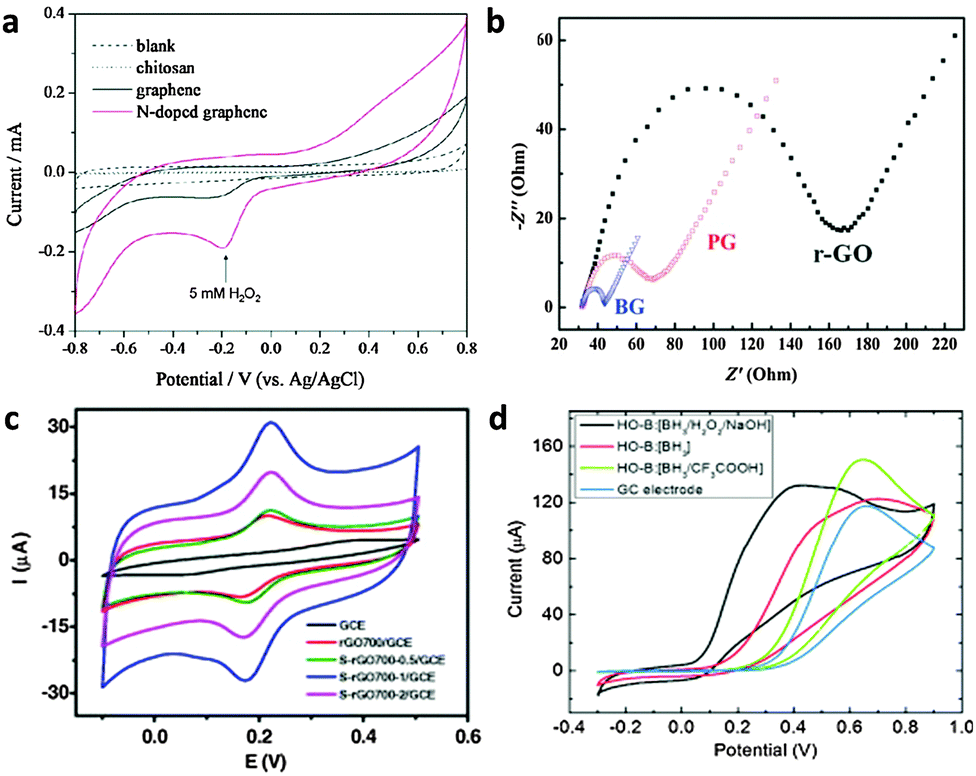 | ||
| Fig. 11 (a) Cyclic voltammograms of 5 mM H2O2 in N2-saturated 100 mM PBS (pH 7.0) on the chitosan electrode (dotted line), the graphene electrode (black line), and the N-doped graphene electrode (red line). Adapted with permission from ref. 112. Copyright (2010) American Chemical Society. (b) The typical electrochemical Nyquist plots of the reduced-GO (r-GO), pristine graphene (PG) and B-doped graphene electrodes. Adapted with permission from ref. 115. Copyright (2011) Royal Society of Chemistry. (c) CVs of GCE, rGO700/GCE, S-rGO700-0.5/GCE, S-rGO700-1/GCE and S-rGO700-2/GCE in 0.5 mM DA (200 mM PBD, pH 6.0) at a scan rate of 50 mV s−1. Adapted with permission from ref. 124. Copyright (2015) Elsevier. (d) Cyclic voltammetry of ascorbic acid (10 mM) in 50 mM PBS buffer (pH 7.0) at the HO-B:[BH3/H2O2/NaOH] (C1O0.78H0.75)n electrode. Adapted with permission from ref. 133. Copyright (2015) John Wiley and Sons. | ||
5.2 Oxygen reduction reaction
The oxygen reduction reaction (ORR) is a critical reaction in fuel cells and air–zinc batteries. Unfortunately, the slow kinetics of ORR at the cathode sides of fuel cells, which are typically composed of platinum-based catalysts, has hindered the practical application of this technology. Given the high cost and sensitivity of platinum to CO poisoning and methanol crossover, carbon-based catalysts especially doped graphene materials are vigorously explored as inexpensive and robust alternative electrocatalysts. While doping can modify the electronic and chemical properties of graphene to improve O2 reduction, the ORR performances of the doped graphene materials, in most cases, exceed that of platinum electrocatalysts only in alkaline solutions.In alkaline (and acidic) solutions, oxygen reduction can proceed by either a two- or four-electron pathway (shown below). Pure carbon-based materials typically catalyse ORR reaction through the two-electron pathway, which involves the formation of a metastable intermediate H2O2. However, doped-graphene materials are expected to proceed via the four-electron pathway, in both alkaline and acidic solutions.
(A) Alkaline solutions
(a) Direct four-electron pathway:
| O2 + 2H2O + 4e− → 4OH− |
(b) Two-step two-electron pathway through peroxide formation:
| O2 + 2H2O + 2e− → HO2− + OH− |
| HO2− + H2O + 2e− → 3OH− |
(B) Acidic solutions
(a) Direct four-electron pathway:
| O2 + 4H+ + 4e− → 2H2O |
(b) Two-step two-electron pathway through peroxide formation:
| O2 + 2H+ + 2e− → H2O2 |
| H2O2 + 2H+ + 2e− → H2O |
Despite understanding the basic mechanism of ORR, further enhancement of the catalytic activity of doped graphene materials can only be achieved by contemplating on the structure of the electrocatalysts and catalytic active sites. Recent advances have ventured into three-dimensional (3D) graphene materials as such electrocatalysts can provide high specific surface areas, strong mechanical strength as well as fast mass and electron transport kinetics due to the combination of 3D porous structures and the excellent intrinsic properties of graphene materials.134 Further dual- and tri-doped graphene materials have been introduced to exploit the benefits of each type of dopant for oxygen reduction.108 These would, however, not be covered in this review.
On top of all these, the presence of metallic impurities in doped graphene materials should not be overlooked. Most graphene materials derived from graphite oxide are contaminated with a considerable amount of metallic impurities.135–137 It has previously been shown that manganese-based impurities, which are usually introduced in excess during oxidation of graphite using Hummer's oxidation method, are able to catalyse oxygen reduction.138 As a result, the electrocatalytic effects observed for oxygen reduction could be mistakenly assigned as the inherent performance of graphene materials. Readers are thus cautioned on the possibilities of manganese-based impurities masking the actual electrocatalytic effect of doped-graphene materials.
The electronegative nitrogen atoms are capable of inducing charge polarisation on the surrounding carbon atoms resulting in positively charged carbon centres. Such high spin density and hybridization freedom on carbon atoms are deduced to improve the adsorption of oxygen molecules on N-doped graphene resulting from charge transfer, which is a critical step for ORR activity. In fact, density functional theory (DFT) studies highlighted that the bonding interactions between oxygen and N-doped graphene grew stronger with increasing concentration of nitrogen, whereby the endothermicity of oxygen adsorption became exothermic.140
Although energy separation between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) was expected to function as a simple indicator of the kinetic stability and chemical reactivity for ORR activity, it fell-short as a conclusive indicator. In fact, the availability of catalytic active sites on N-doped graphene is determined by the spin density distribution and atomic charge distribution.141
On the other hand, pyridinic-N and quaternary-N moieties were determined as the catalytic active sites of N-doped graphene for ORR.142 Ruoff and co-workers have concluded that the electrocatalytic activity of N-doped graphene is dependent on the presence of these two nitrogen moieties, whereby the quaternary-N determines the limiting current density, while the pyridinic-N improves the onset potential for ORR.143 A further study by Murakoshi highlighted that the quaternary-N reduces oxygen via a two-electron pathway while pyridinic-N reduces oxygen via a four-electron pathway, in alkaline solutions (Fig. 12). The determination of active ORR catalytic sites in N-doped graphene has, as such, spurred further research in the synthesis of pyridinic-N-rich graphene materials.144,145
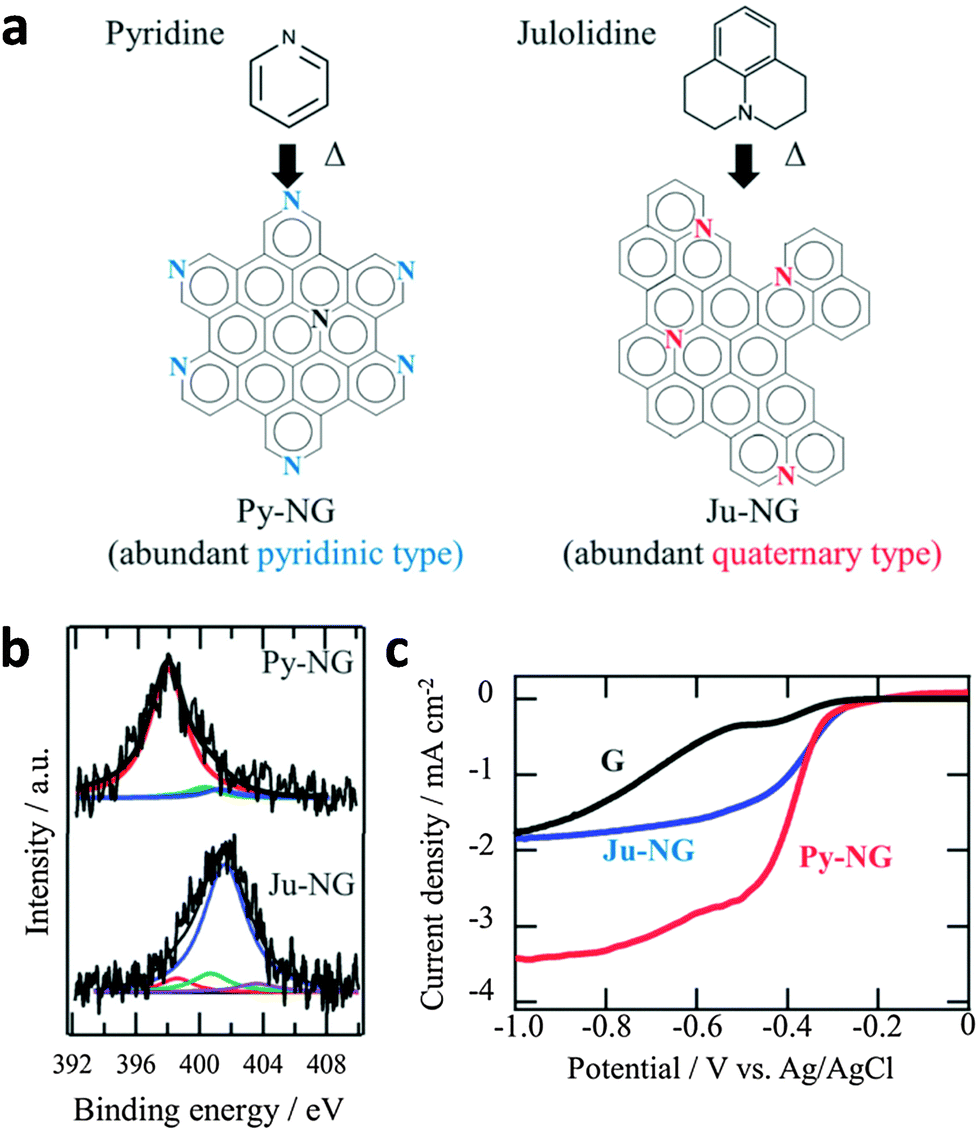 | ||
| Fig. 12 (a) Selective nitrogen doping in graphene. (b) N1s spectra of Py-NG and Ju-NG. (c) RDE voltammetry curves for oxygen reduction on pristine graphene, Ju-NG, and Py-NG in an O2-saturated 0.1 M KOH solution. The electrode rotating rate and scan rate was 625 rpm and 0.01 V s−1, respectively. Reproduced with permission from ref. 146. Copyright (2013) Royal Society of Chemistry. | ||
5.3 Hydrogen evolution reaction
The hydrogen evolution reaction (HER) is another important energy reaction of interest. It occurs in the cathodic side of electrolyser systems which provides a clean supply of hydrogen gas for fuel cells, without any carbon emissions. Similar to ORR, HER has thermodynamic and kinetic constraints; the kinetic constraints are typically resolved by using efficient platinum-based electrocatalysts. However, their high cost and limited abundance hinder their widespread practical implementation. To improve cost-competitiveness, it is critical to develop alternative efficient HER electrocatalysts based on earth-abundant elements. Previously, the search had mainly revolved around non-noble metal-based materials (i.e., Mo, W, Ni, Fe). However, recently, the research community has witnessed numerous carbon-based electrocatalysts reported with good HER activities. Compared to non-noble metals, carbon-based materials are generally cheaper, more abundant, and less susceptible to corrosion and oxidation. For these reasons, they are attractive as possible alternative HER electrocatalysts. In particular, doped graphene and related materials have been developed with promising HER performances close to that reported for platinum in acidic media.HER occurs via a two-electron pathway (2H+ + 2e− → H2). It is generally accepted to involve two steps; namely electrochemical hydrogen adsorption (Volmer step) and desorption which can occur either electrochemically (Heyrovský step) or chemically (Tafel step).152–155 In the Volmer step, a proton and an electron combine to produce adsorbed hydrogen (H*) on the active site of the electrocatalyst surface (M). Subsequently, the reaction can proceed by either the Heyrovský or Tafel step.152 In the Heyrovský step, adsorbed hydrogen merges with a proton in the presence of an electron to produce hydrogen gas, whereas in the Tafel step, two adsorbed hydrogen atoms combine for hydrogen gas formation. Since H* is present regardless which route is followed, the Gibbs free energy of hydrogen adsorption (ΔGH*) greatly influences the overall rate of reaction and is often described as the main criterion for assessment of the process. Optimal HER electrocatalysts, such as platinum, have ΔGH* close to zero which indicates that H* binds neither too strongly (when ΔGH* is largely negative) nor too weakly (when ΔGH* is largely positive) onto the electrocatalyst surface.156
In order to determine which pathway the HER process proceeds after the first step of electrochemical hydrogen adsorption, a Tafel plot obtained from the HER polarization curve can be used.157 The Tafel plot relates the overpotential (η) as a function of logarithm of current density (j). By fitting the linear portion of the graph to the Tafel equation (η = b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logj + a), the Tafel slope value can be obtained from the value ‘b’. Tafel slope values close to 40 mV per decade (mV dec−1) indicate that the Heyrovský step is the rate-determining step (RDS), while values close to 30 mV dec−1 suggest that the Tafel step is the RDS. From here, we can propose whether the reaction pathway proceeded through the electrochemical desorption or the chemical desorption process. In fact, Tafel slope is an inherent property of a material. For Pt, its Tafel slope value is measured to be 30 mV dec−1155 which shows that it catalyses HER via the Volmer–Tafel pathway, with the Tafel step being the RDS.
logj + a), the Tafel slope value can be obtained from the value ‘b’. Tafel slope values close to 40 mV per decade (mV dec−1) indicate that the Heyrovský step is the rate-determining step (RDS), while values close to 30 mV dec−1 suggest that the Tafel step is the RDS. From here, we can propose whether the reaction pathway proceeded through the electrochemical desorption or the chemical desorption process. In fact, Tafel slope is an inherent property of a material. For Pt, its Tafel slope value is measured to be 30 mV dec−1155 which shows that it catalyses HER via the Volmer–Tafel pathway, with the Tafel step being the RDS.
Besides acidic media, HER can also be performed under alkaline conditions (2H2O + 2e− ⇌ H2 + 2OH−) in electrolyser systems. However, comparing acidic and alkaline electrolysers, the former is generally more superior to the latter in terms of industrial operation.153 This could be due to an additional water dissociation step required for hydrogen generation under basic conditions, which increases the energy requirement of the process.155 A summary and comparison of the HER mechanism in acidic and basic media is presented in Table 1.
| Reaction pathway | Acidic media | Basic media | Tafel slope (mV dec−1) |
|---|---|---|---|
| Volmer step | M + H+ + e− ⇌ M − H* | M + H2O + e− ⇌ M − H* + OH− | 120 |
| Heyrovský step | M − H* + H+ + e− ⇌ M + H2 | M − H* + H2O + e− ⇌ M + H2 + OH− | 40 |
| Tafel step | 2(M − H*) ⇌ H2 + 2M | 2(M − H*) ⇌ H2 + 2M | 30 |
| Overall: | 2H+ + 2e− ⇌ H2 | 2H2O + 2e− ⇌ H2 + 2OH− | — |
In the pursuit of finding alternative HER electrocatalysts to replace Pt, numerous theoretical studies based on the established mechanisms behind HER have been subsequently conducted. With the possible tailoring of the electronic and electrochemical properties of graphene through doping, computational studies predicted that doping graphene materials with p-block elements can successfully tailor the ΔGH* to enhance their HER activities.158 Qiao's group presented one of the earliest explorations on density functional theory (DFT) calculations of doped graphene materials for HER applications.158 They showed that the presence of a p-block element dopant can produce noticeable differences in the charge population of adjacent carbon atoms to tune their electron donor–acceptor properties for different H* adsorption behaviors. Using molecular orbital theory, they proposed that the lowest valence orbital of the activated carbon atom hybridizes with the bonding orbital of the adsorbed H* to form bonding and antibonding states. With lower and more stable valence-band energy levels, a stronger bonding between H* and the active carbon can be achieved for a reduced ΔGH*. Indeed, experimentally, there have been several reports where p-block element doped graphene materials show enhanced HER electrocatalysis compared to their undoped counterparts.158–160
However, the sole presence of a dopant in graphene materials is often insufficient to compete with the performances of state-of-the-art metal HER electrocatalysts. As such, various strategies have been explored to further improve the catalytic activity of doped graphene materials for hydrogen generation. Here, we review such strategies investigated. Even though some of these novel HER electrocatalysts may be presented as metal-free, readers are cautioned that there may be traces of metallic impurities in the graphene materials which can arise from the starting material or preparation method that could play a role in altering their electrochemical properties. As a parameter of comparison among reported electrocatalysts, overpotential at a current density of −10 mA cm−2 is commonly used.153 To provide a general overview, Pt-based electrocatalysts exhibit excellent activity with almost zero overpotential, while many state-of-the-art metal-based HER electrocatalysts require between 100 to 200 mV to reach this current density.153 Unless otherwise stated, the term ‘overpotential’ used subsequently here refers to the overpotential required to achieve a current density of −10 mA cm−2. Besides overpotential and Tafel slope, other parameters frequently reported to characterize HER catalyst materials include onset potential, exchange current density and turnover frequency. However, for simplicity, overpotential and Tafel slope are chosen as the more relevant aspects of HER activity for comparison in our discussion here. Readers interested to know more about the other parameters of HER performance can refer to ref. 153 and 154. Since most of the HER experiments reported are conducted under acidic conditions (0.5 M H2SO4), the performances depicted that follows would refer to acidic conditions unless otherwise stated. For ease of comparison, the main details of various reports to be presented below on doped graphene and their related materials for HER applications are summarized in Table 2.
| Electrocatalyst | Remarks (e.g. dopant amounts, surface area, particle size, etc.) | Overpotential at −10 mA cm−2 current density (mV) | Tafel slope (mV dec−1) | Ref. |
|---|---|---|---|---|
| a HER experiment was conducted in 0.1 M H2SO4. b The overpotential at a current density of −10 mA cm−2 was not mentioned, but approximated from the polarization curve. c The overpotential at a current density of −10 mA cm−2 was not mentioned, but the overpotential at a current density of −5 mA cm−2 was approximated from the polarization curve. | ||||
| Under acidic conditions (0.5 M H2SO4) | ||||
| p-block element doped graphene | ||||
| B-doped graphene | 1.85 at% B | ∼440b | ∼99 | 159 |
| N-doped mesoporous graphene | 3.93 at% N, 927 m2 g−1 | ∼245b | ∼109 | 162 |
| N,P-codoped graphene | 4.60 at% N, 1.63 at% P | ∼420 | ∼91 | 158 |
| N,S-codoped nanoporous graphene | 5.06 at% S, 3.79 at% N, 0.07 at% Ni, 1320 m2 g−1 | ∼280 | ∼81 | 160 |
| Highly N,P-dual doped multilayer nanoporous graphene | 3 at% P, 11 at% N, 1102.1 m2 g−1 | ∼213 | ∼79 | 165 |
| Doped graphene with carbon nitride | ||||
| C3N4@N-doped graphene hybrid | 33 wt% g-C3N4 | ∼240 | ∼52 | 170 |
| Porous C3N4 nanolayers@N-doped graphene films after 750 CV cycles | 4.6 at% N, 9.1% PCN nanolayers, 58 m2 g−1 | ∼80 | ∼49 | 171 |
| N,P-doped nanoporous graphene/g-C3N4 hybrid | 2.14 at% P, 42.08 at% N (from C3N4 and N-graphene), 119 m2 g−1 | ∼340 | ∼90 | 166 |
| Doped graphene with metal-based materials | ||||
| Molybdenum sulfide clusters-N-doped graphene hybrid hydrogel film | 2.1 wt% MoSx | ∼140 | ∼105 | 173 |
| N-doped graphene decorated by few-layer MoS2 | 45.1 wt% MoSx, 2.01 at% N | ∼160b | ∼45 | 178 |
| WS2 nanolayers@heteroatom-doped graphene | 3.36 at% N, 9.73 at% O, 0.63 at% P, 20 wt% WS2 | ∼125 | ∼53 | 167 |
| Nanostructured SnS-N-doped graphene | 2.15 at% N, 17.31 at% S, 17.28 at% Sn | ∼125 | ∼38 | 180 |
| Atomic cobalt on N-doped graphene | 8.5 at% N, 0.57 at% Co | ∼147 | ∼82 | 181 |
| N-doped carbon-wrapped Co nanoparticles on N-doped graphene nanosheets | 7.5 at% N, 0.48 at% Co | ∼190b | ∼80 | 184 |
| Co embedded in N-doped carbon | 2.8 at% N, nanoparticle size: <10 nm | ∼265 | ∼98 | 182 |
| N-doped graphene/Co-embedded porous carbon polyhedron hybrid | 3.5 at% N, ∼0.5 at% Co on surface, 375 m2 g−1 | ∼229 | ∼126 | 183 |
| S-doped graphene with carbon black and Ru nanoparticles | 4.45 at% S, 0.75 at% Ru, 20% carbon black | ∼70b | ∼61 | 169 |
| 3D N-doped graphene supported MoS2 nanoparticles | 5.4 wt% N, 3.53 wt% S, 5.39 wt% Mo, 1066.6 m2 g−1, graphene thickness: ∼0.34 nm, MoS2 lateral size: ∼35 nm | ∼290b | ∼44 | 179 |
| Co nanoparticles at N-doped graphene with porous structure | 4.9 at% N, 0.8 at% Co, 104.5 m2 g−1, pore size: 10 to >100 nm | ∼125 | ∼94 | 185 |
| Ultrathin N-doped graphene shells encapsulate CoNi nanoalloya | 1.8 at% N, 12.2 at% Co, 9.9 at% Ni, 1 to 3 graphene layers per shell, nanoalloy size: 4 to 7 nm, 5 wt equivalent mass loadings used | ∼142 | ∼105 | 186 |
| Under basic conditions (0.1 M KOH) | ||||
| P,N-codoped graphene | 4.60 at% N, 1.63 at% P | ∼585c | ∼145 | 158 |
| Co embedded in N-doped carbon | 2.8 at% N, nanoparticle size: <10 nm | ∼337 | Not mentioned | 182 |
Even though p-type doping of graphene is usually not preferred for HER catalysis, Asefa and co-workers reported the use of B-doped graphene (p-type doping) material for this application.159 They explored different borylating agents (i.e., carborane, B(OH)3, NaBH4, and BH3-THF) on defective graphene and found that BH3-THF-treated graphene (1.85 at% B) achieved HER performance with a Tafel slope of ∼99 mV dec−1, indicating a Volmer-limiting step, and an overpotential of ∼440 mV.159 Compared to its undoped counterpart, there was ∼60 mV upshift in overpotential. Such observation, where p-type doping shows a similar catalytic effect to n-type doping, could be attributed to variations in the doping mechanism (i.e., addition instead of substitution) as suggested from computational studies by Lazar et al.161
Similar to ORR, the strategy of dual-doping was investigated to further enhance the catalytic performance of doped graphene. From DFT calculations of various single- and dual-doped graphenes, Qiao and co-workers predicted that dual-doped graphene would show higher HER activity compared to single-doped graphene. Their calculations revealed pyridinic-N and P dual-doped graphene to have the best HER performance among the different possible electrocatalysts studied, as it yielded the lowest ΔGH* value of 0.08 eV.158 This was further supported experimentally with N,P-codoped graphene (N,P-graphene-1), containing 4.60 at% N and 1.63 at% P, showing improved HER catalytic performance as compared to its single-doped graphene counterparts (Fig. 13). Additionally, the electrocatalyst was tested under alkaline conditions (0.1 M KOH) to demonstrate the stability of doped graphene materials over a wide range of pH. By qualitatively evaluating the number of active sites derived from Tafel plots, the group found that the enhancement seen in N,P-graphene was not merely due to a simple increase in the number of active sites. Instead, it was evidence for a synergistic coupling effect arising from the dual-doping of N and P. As suggested from a study by Woo and co-workers,163 the co-doping of P could promote preferential bonding of pyridinic-N to induce this synergistic effect.164 This finding has also been experimentally observed by several other studies.165–167 Interestingly, Qiao's group also found that random coupling of heteroatoms (i.e., B,N-graphene) could not produce such an effect. This highlights the importance of N and P co-doping in graphene for improved HER catalysis.
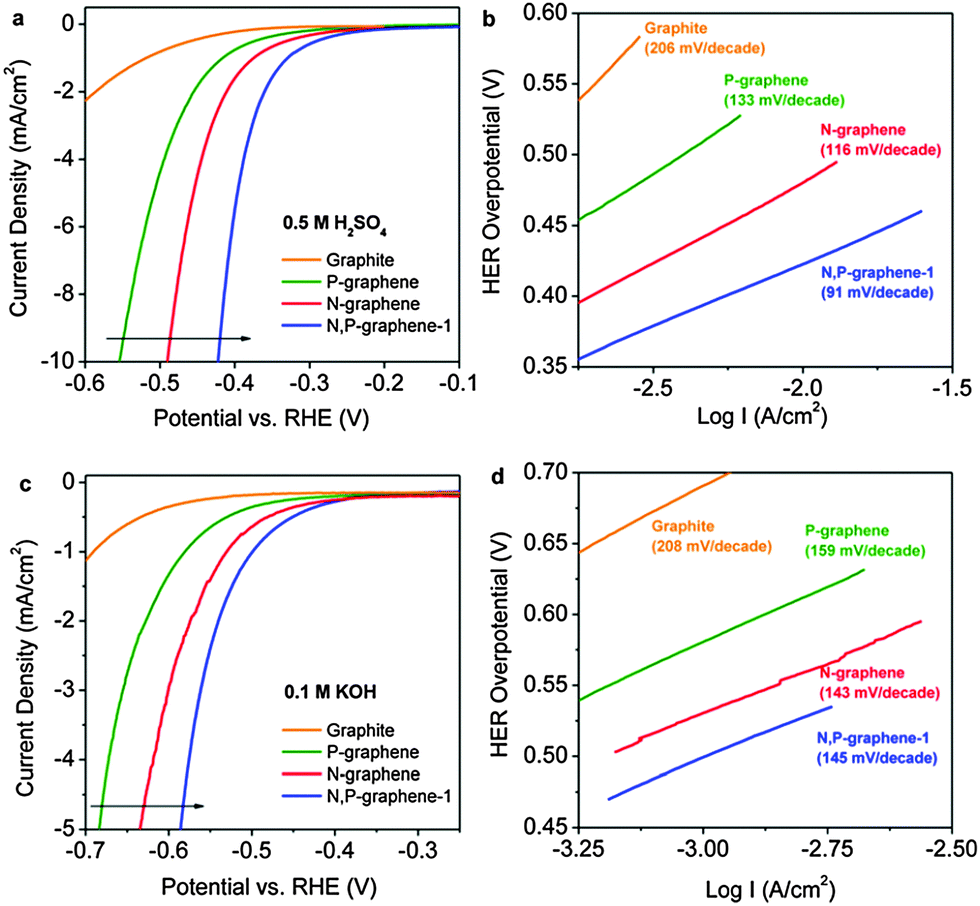 | ||
| Fig. 13 (a and c) HER polarization curves and (b and d) the corresponding Tafel slopes of N,P-graphene and its single-doped counterparts under (a and b) acidic and (c and d) alkaline conditions. Adapted with permission from ref. 158. Copyright (2014) American Chemical Society. | ||
From the predicted value of ΔGH* of the co-doped graphene material, one would expect a better HER performance for the material. However, in experimental studies, this could have been impeded due to strong π–π interactions and/or hydrophobic affinities between graphene layers leading to restacking and agglomeration during the electrode drying process. To overcome this, porous structures of doped graphene have been investigated as one possible solution.
Wang's group developed N-doped mesoporous graphene using micelle-template synthesis and compared its HER performance with its 2D graphene counterpart.162 The group found that porous architecture greatly increased the specific surface area (by over five times), thereby effectively accelerating the HER catalysis to achieve ∼245 mV overpotential from over 400 mV in N-doped graphene.
Chen and co-workers combined these strategies of dual-doping and porous structures to further enhance the HER performances of doped graphene materials.160 N and S co-doped graphene was grown on nanoporous Ni by the chemical vapour deposition method. The Ni template was then etched away to yield nanoporous dual-doped graphene. With an overpotential of ∼280 mV and a Tafel slope of ∼80.5 mV dec−1, the electrocatalyst displayed superior HER activity over its undoped and single-doped counterparts. The co-doping of S could have promoted the formation of graphitic-N to result in the observed synergistic effect.164 The group also studied the effect of different CVD synthesis temperatures (500 and 800 °C) and found that a lower temperature produced more favourable bonding configurations (i.e., C–S–C–, –CS–) for HER. From XPS analysis, the group found that 4 to 6 at% of –C–S–C– and –CS– were converted to –C–SO2– upon 1000 HER cycles, suggesting that these bonding types could be responsible for the observed HER catalysis. Moreover, the group found residual amounts of Ni (<0.07 at%) which could have also contributed to its HER activity.
Besides designing porous structures, inserting carbon black particles in between graphene layers was successfully investigated as an alternative solution to prevent restacking of graphene.168 Shervedani and Amini reported S-doped graphene showing improvements in HER performance after the treatment with carbon black (20%).169
5.3.2.1 Doped graphene with carbon nitride. Qiao's group coupled graphitic-carbon nitride (g-C3N4) with N-doped graphene (N-graphene; NG) to form a hybrid (C3N4@NG).170 Various proportions of C3N4 were investigated and 33 wt% was found to produce the optimum HER performance. C3N4@NG showed an overpotential of ∼240 mV and a Tafel slope of ∼51.5 mV dec−1, outperforming the control g-C3N4, N-graphene and C3N4/NG (physically mixed) samples. The authors credited this high HER activity to a synergistic coupling effect between N-graphene and g-C3N4. Electrical impedance spectroscopy (EIS) data revealed an increased electrical conductivity through the coupling effect, which could possibly have resulted in the improved HER performances observed. DFT calculations further supported the experimental results with C3N4@NG showing a favourable ΔGH* value close to zero (0.19 eV) as compared to g-C3N4 (ΔGH* = −0.54 eV) and N-graphene (ΔGH* = 0.57 eV).
In an effort to improve the HER performance, the group later reported a 3D porous structure of C3N4 nanolayers in N-graphene films, which contained more exposed active sites. Interestingly, they found that the HER activity greatly improved after an optimized electrochemical treatment of 750 cyclic voltammetry (CV) cycles (denoted as PCN@N-graphene-750), with remarkable overpotential observed at 80 mV and a Tafel slope of 49.1 mV dec−1 (Fig. 14).171 The HER mechanism shifted from the Volmer step being the RDS prior to the CV cycles, to become the desorption controlled step after the electrochemical treatment. Besides N-doped graphene, Lee and co-workers explored the HER activity of g-C3N4 with P-doped graphene hybrid material.166 They found that the P-doped graphene hybrid material performed better than the N-doped counterpart. However, one must always be cautious in evaluating the performance of the material and consider the possible effects of metallic impurities present in such systems.
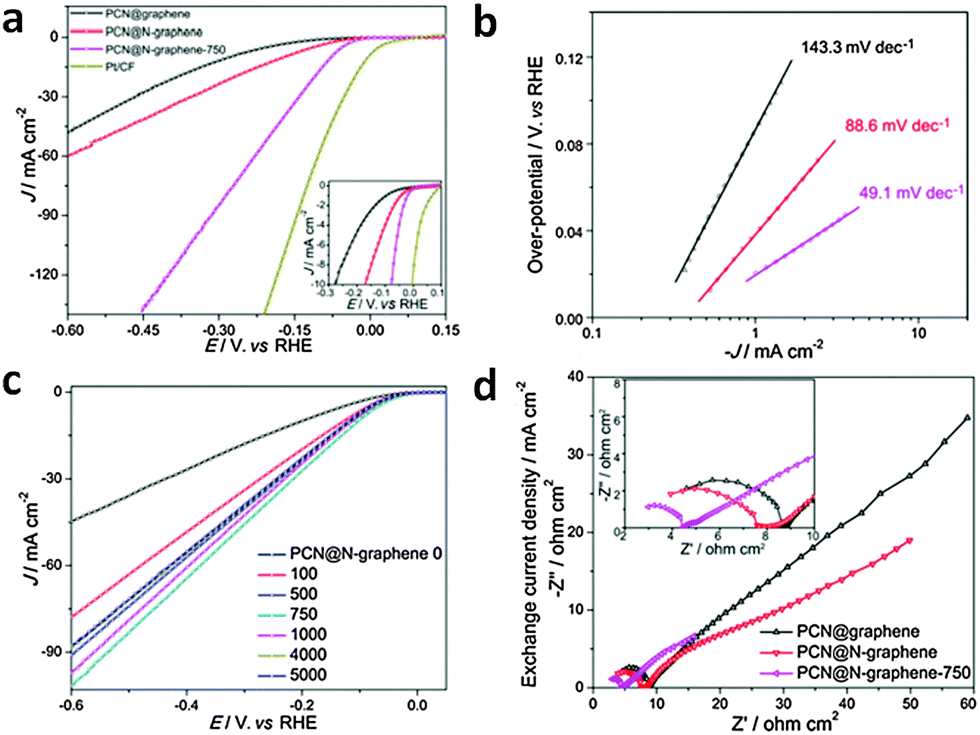 | ||
| Fig. 14 (a) HER polarization curves and (b) the corresponding Tafel plots for PCN@N-graphene-750 and its control samples. (c) HER polarization curves upon various CV cycles. (d) EIS data collected at −200 mV vs. reversible hydrogen electrode (RHE). Adapted with permission from ref. 171. Copyright (2015) American Chemical Society. | ||
5.3.2.2 Doped graphene with metal-based materials. Besides carbon-based materials, doped graphene has also been successfully coupled to metal-based materials for enhanced HER activity. These include transition metal dichalcogenides (TMDs) such as MoS2 and WS2 as well as metallic atoms/nanoparticles/nanoalloys. Here, doped graphene function as a support material which provides efficient electrical conductivity, high stability and large surface area for improved catalysis.155,172 Among various doped graphene materials, N-doped graphene is especially preferred due to its enhanced electrical conductivity, which has been proven both theoretically and experimentally.164 When coupled with metal-based materials, which have long been identified to be HER active, these hybrids can offer dual active centres arising from the metals as well as the pyridinic-N/pyrrolic-N structures of N-doped graphene.173 Alternatively, these metal-based materials may also act as dopants to tailor the electronic and electrochemical properties of neighbouring carbon atoms on graphene to be catalytically active sites for HER. In either case, these doped graphenes with metal-based hybrid materials have great potential to act as excellent HER electrocatalysts.
MoS2 and WS2 are well-studied examples of metal-based materials reported with promising HER activities.153,174,175 They have been coupled with graphene related materials to give enhanced HER performances. Starting from hybrid materials with undoped graphene,176,177 the trend then shifted towards coupling with N-doped graphene. Dai et al. demonstrated a facile synthesis of few layer MoS2 decorated on N-doped graphene displaying improved HER performance compared to bulk MoS2.178 Taking a step further, Qiao's group combined this strategy with 3D structures when reporting molybdenum sulfide clusters (MoSx; due to the presence of HER active MoS3 found with MoS2) with N-doped graphene hybrid hydrogel film, and showed that the 3D hybrid performed better than its 2D counterpart.173 Similarly, Zhang and co-workers reported MoS2 nanoparticles supported on 3D-N-doped graphene with good HER activity.179 Qiao's group also explored 3D WS2 nanolayers on tri-doped graphene (P,N,O) and found that they performed better relative to the non-P-doped film, highlighting the synergistic effect between N and P co-doping.167 Alongside MoSx and WS2, SnS has also been coupled with N-doped graphene to investigate its HER performance. This hybrid (SnS–rGr) presented by Lee and co-workers clearly showed improvements in HER activity through the coupling and doping strategies, by comparison with control samples (Fig. 15).180
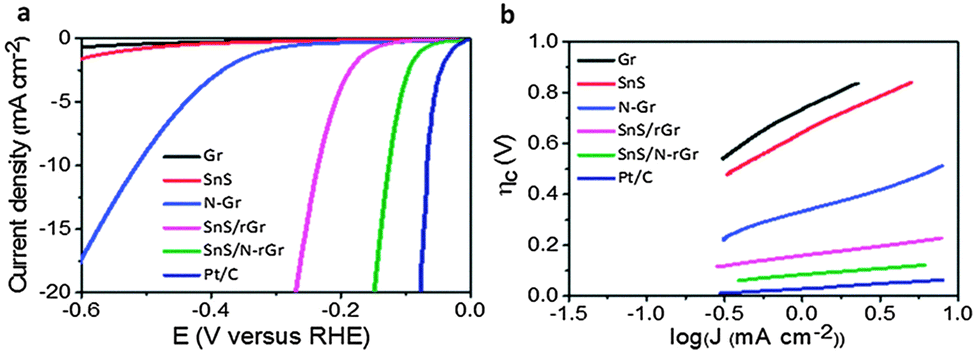 | ||
| Fig. 15 (a) HER polarization curves and (b) the corresponding Tafel plots for SnS–rGr and its control samples. Adapted with permission from ref. 180. Copyright (2015) Royal Society of Chemistry. | ||
Apart from TMDs, metallic atoms, nanoparticles and nanoalloys have also been coupled with doped graphene for HER applications. Recently, Fei et al. reported small amounts of atomic Co dispersed on N-doped graphene with good HER catalytic activity (∼147 mV overpotential).181 The amount of Co was optimized to be 0.57 at%, beyond which the HER performance deteriorated. They found that the presence of N-dopants (8.5 at%) greatly enhanced the HER activity of the hybrid (∼200 mV upshift in overpotential) compared to the control sample without N-dopants. The group also investigated the role of Co by replacing Co with Fe in the catalyst, in another study.182 They observed poorer HER performance when Fe was used, which highlights an important synergistic effect of Co–N interactions in enhancing the HER catalysis. This synergistic effect was further supported by other studies.183,184 Besides Co atoms, Co nanoparticles (∼15 nm) wrapped by N-doped carbon on N-doped graphene nanosheets as HER electrocatalysts have been reported by Zhou et al.184 The group later expanded this strategy to 3D porous structures with improved HER activity.185 In addition to Co atoms and nanoparticles, a Co nanoalloy was also investigated. Bao's group reported a CoNi nanoalloy encapsulated by ultrathin graphene shells (1 to 3 layers) with good HER activity.186 They found that thinner graphene shells and higher amounts of nitrogen dopants can significantly increase the electron density in the graphene shells to enhance HER activities. Besides cobalt, ruthenium nanoparticles have also been reported to improve the HER performance of S-doped graphene materials by Shervedani and Amini.169
6. Electrochemistry of graphene modified with d-block elements
Whilst there is interest in doping graphene with metals (d-block elements), one should not overlook the point that the only difference between dopants and impurities is semantic. Generally, dopants are introduced into the system on purpose, while impurities tend to be inherently present or introduced unintentionally. Graphene materials are known to host a wide range of impurities, which can either be inherent in the graphitic precursors used to synthesize graphene materials, or the result of unintentional contamination of the material at various stages of the synthesis process.136 Metallic impurities thus introduced are often electrochemically active, and have been shown to be responsible for the apparent electrocatalytic behavior of graphene materials towards an array of analytes (e.g. hydrazine, peroxides, sulfides) as well as the oxygen reduction reaction.187–192 For top-down approaches to the synthesis of graphene materials, graphite or carbon nanotube (CNT) precursors contain metallic impurities that are extremely challenging to remove, even after exposure to strong oxidative or reductive conditions, ultrasonication, and Cl2 at 1000 °C.135,193 Synthetic procedures and CVD graphene transfer methods involving metal-based reagents (e.g. KMnO4, FeCl3) often contaminate the end materials with significant amounts of metallic impurities, despite many washing steps taken to maximize the removal of excess reagents.8,194 Extended exposure of CVD graphene to ammonium persulfate (APS) as the chemical etchant was able to significantly decrease (but not eliminate) the amount of residual metallic impurities (Fig. 16A).195 Unfortunately, Raman mapping (Fig. 16B) as well as optical microscopy (Fig. 16C and D) of the graphene sheets showed that this reduction of impurities came at the expense of graphene layer integrity. To complicate matter further, inadvertent contamination can occur due to metallic impurities within the synthetic reagents themselves, making it difficult to anticipate the type and amount of contamination.196 This issue was exemplified in the fabrication of graphene screen-printed electrodes using commercially available graphene inks, where the resulting devices exhibited electrocatalytic responses for the oxidation of hydrazine that was dominated by the presence of metallic impurities.197 As the compositions and production methods of such inks are proprietary information of the manufacturer, the exact stage at which metallic impurities were introduced into the graphene could not be ascertained. Since these metallic impurities exert a dramatic influence on the electrochemical properties of graphene materials, recent research efforts have attempted to tackle this issue via purification methods. It was shown that redox-accessible metallic impurities such as Fe could be partially removed from chemically reduced graphene oxide via an electrochemical purification process by cycling the electrode in nitric acid. Successive repetitions of the purification step led to an increasing suppression of the electrocatalytic activity of the material towards the reduction of cumene hydroperoxide (CHP), paving the way for the extension of this method to other types of graphene materials. A similar electrochemical etching method was also demonstrated to remove residual metallic impurities on CVD graphene using a solution of HCl/H2O2; 20 cycles of this electrochemical etching was sufficient to completely remove any parasitic electrochemical signals arising from Cu and its oxides, as well as the disappearance of cathodic and anodic peaks corresponding to the electrocatalytic reduction and oxidation of CHP and L-glutathione, respectively (Fig. 16E and F).198 Another strategy to exclude the influence of electrochemically active metallic impurities in graphene is to pursue alternative fabrication procedures that explicitly do not involve the use of such metals, thus minimizing the possibility of such contamination. Using this line of reasoning, pure elemental lithium was combusted in carbon dioxide (in the form of dry ice), yielding graphene powder and lithium oxide.199 Conceptually, the use of high purity starting materials (Li and CO2) should have led to impurity-free graphene; however, ultralow traces of transition metals were still detected in the graphene sample. These levels of metallic impurities were found to be electrochemically benign toward the oxidation of NaHS, presumably because they were present at levels many factors less than conventional chemically reduced graphene. | ||
| Fig. 16 Removal of metallic impurities from CVD graphene materials. (A) ToF-SIMS mass spectra in the 63Cu region for CVD graphene samples etched for 8 h and 72 h in APS. (B) Mapping of Raman 2D peak intensity on CVD graphene etched for 72 h, mapped area shown is 20 × 20 μm2. (C) Optical microscopy images of CVD graphene on SiO2 substrates after 8 h and (D) 72 h etching. Reproduced with permission from ref. 195. Copyright (2015) American Chemical Society. (E) CVs of CVD graphene before and after electrochemical etching in the presence of 5 mmol L−1 CHP (inset shows a zoomed-in region) as well as (F) 5 mmol L−1L-glutathione. Reproduced with permission from ref. 198. Copyright (2014) John Wiley and Sons. | ||
While the profound effect of metallic impurities on the electrochemical properties of graphene materials has been reasonably studied, another major impurity that cannot be overlooked is amorphous carbon. These carbonaceous impurities, which are common in CNT samples, are known to be highly active in the electrocatalytic responses of CNTs towards many compounds.200 The possibility of amorphous carbon impurities making their way into graphene materials should be immediately obvious if they are derived from the unzipping of CNTs, but can also occur as a side-product when graphitic materials are exposed to strong oxidative conditions.201 It was also recently shown that even under well-controlled CVD graphene growth on Pt, amorphous carbon impurity islands can form in between the catalyst surface and monolayer graphene, and is often misidentified as areas of few-layer graphene.202 Unsurprisingly, amorphous carbon impurities were found to be responsible for the supposed electrocatalytic responses of graphene materials toward NADH, acetaminophen, as well as hydroquinone.201
7. Recent electrochemical applications of graphene and its derivatives: towards the fabrication of sensors and biosensors
Owing to the extraordinary characteristics of graphene and its analogues, these materials have been widely employed to fabricate electrochemical sensors and biosensors. Here we wish to focus our attention only on the past year 2014–2015, reviewing studies on the detection of selected examples of small molecules followed by the description of studies on the detection of biomolecules.7.1 Electrochemical sensing: direct detection of target molecules
The electrochemical sensing approach is exemplified by a direct electron transfer between the sensing platform (graphene and its derivatives) and an electroactive species, which can either be the target molecule itself or a molecule in which its produced electrochemical signal can be correlated to the target molecule. For such an approach, graphene and its derivatives are mainly utilized due to their high surface area and electron conductivity, leading to enhanced heterogeneous electron transfer and thus greater output signal intensity. In some cases, detection of target molecules can be achieved at a lower applied potential. In addition, functionalities on the surface of graphene and its derivatives can also be exploited to confer a greater affinity for the target molecules, thereby reducing the effects from interferences. Hence, with the utilization of graphene and its derivatives, greater sensitivity of the electrochemical sensor and its applicability for real sample matrices can be achieved.Chemically modified graphene (CMG) materials203 have been successfully used as transducers for the analysis of electroactive probes in food samples. Given the multitude of CMGs prepared by following various protocols and possessing different structural features such as the amount of disorders and oxygen functionalities, comparisons have been performed in order to identify the material that could be better suited to detect a specific analyte. Detection of nitrite, which is commonly used as a food additive but can be hazardous to humans, has been performed on both chemically reduced graphene oxide (CRGO) and electrochemically reduced graphene oxide (ERGO).204 The former showed higher sensitivity, lower potential for nitrite oxidation and a better selectivity in the presence of interferences. The authors attribute this behavior to the presence of unreduced oxygen moieties on ERGO, which were absent on the CRGO surface, as shown also by the FTIR study. The transducer based on CRGO was finally employed for the selective detection of nitrite in water samples.
Several CMG platforms namely graphite oxide (GPO), graphene oxide (GO) and ERGO were used for a comparative study on the detection of caffeine in coffee, tea and energetic drink samples.205 It was found that ERGO, due to the lower amount of oxygen functionalities, provided the best analytical performance for the detection of caffeine based on sensitivity, linearity and reproducibility of response. The same authors also demonstrated that depending on the graphite oxide preparation protocol, different intrinsic electrochemistry can be shown by the electrochemically reduced graphene materials.206 The authors used electrochemically reduced graphene nanoribbons in which the corresponding graphite oxide was obtained by using permanganate oxidants. They observed that the oxidation peak of caffeine on such a platform was strongly influenced by the inherent background signal of the reduced graphene nanoribbons. This showed that the potential window of these materials should be carefully taken into account when detecting analytes which are electroactive at positive potentials, such as most of the biological and clinical probes. A comparative study on how the structural properties of the graphene material could influence the assessment of antioxidant capacity in wine samples was also performed.207 The authors employed three CMG platforms namely GO, CRGO and ERGO, possessing different amounts of oxygen-containing groups. They demonstrated that the latter could favour the interaction between the graphene surface and the analyte of interest, and at the same time be detrimental to the heterogeneous charge transfer. The best electrochemical performance in terms of calibration sensitivity, selectivity, and linearity of response was shown by ERGO, due to the presence of non-electrochemically reducible oxygen functionalities which played a major role in promoting the interactions between the graphene surface and the analyte. Therefore, the same group was able to tune the analytical performance of the graphene oxide platform by carrying out the electrochemical reduction of the starting material at increasing negative potentials from −0.25 V to −1.50 V.208 The eight ERGO platforms, carrying a decreasing amount of oxygen-containing groups, were used for the detection of standard gallic acid, a standard polyphenol which is commonly used as an index of the antioxidant capacity of food and beverages. ERGO obtained after reduction at −0.90 V, which provided the best electroanalytical performance, was then employed for the assessment of the antioxidant capacity of fruit juice samples. Lately, several nanomaterials have been used to fabricate functional graphene nanocomposites which show improved performances when used as sensing and biosensing platforms. This behavior has been attributed to various reasons, including the increased surface area and the synergistic effect derived from the combination of the materials. Long et al.209 developed a novel imprinted electrochemical sensor based on a composite including graphene nanosheets and cobalt–nickel bimetallic nanoparticles for the detection of octylphenol, a compound known to be involved in the disruption of the mammalian endocrine system. The electrochemical detection of the analyte in plastic bottles, metal bottles and food packaging bags was performed and the achieved detection limit was in the pM range. An electrodeposited bi-enzymatic biosensor based on graphene and gold nanoparticles was employed for the detection of carbamates, a major class of pesticides with very severe adverse effects on both humans and animals.210 The proposed biosensor exhibited an improved Michaelis–Menten kinetic constant and a low detection limit for the detection of several carbamates in citrus fruit samples, without significant interferences from ascorbic and citric acid and glucose.
Electrochemical detection of nitrite was carried out on reduced graphene oxide and dendritic copper nanoclusters electrodeposited on a glassy carbon electrode.211 The developed sensor showed improved electrocatalytic activity for nitrite detection as well as a low detection limit of 0.4 μM, due to the enlarged surface-to-volume ratio including more electroactive sites for the reduction of nitrite. The same analyte was detected on a biosensor based bifunctionalized graphene–gold nanoparticle hybrid, obtained by the in situ growth of gold nanoparticles. Haemoglobin was then employed as a biorecognition element and the developed biosensor allowed the detection of nitrite in pickled radish by achieving a detection limit of 0.01 μM.
A novel electrochemical sensor for the detection of fructose was developed by immobilizing a highly dispersed CuO–Cu nanocomposite on graphene, which was previously non-covalently functionalized with sodium dodecyl benzene sulfonate (SDBS).212 The improved electrochemical performance of the composite material for fructose detection was mainly due to the exceptional cation anchoring ability of SDBS, which prevented the aggregation among Cu-based nanoparticles during the nanocomposite synthesis.
The detection of colorants in food has also been successfully performed on graphene-composite materials. For safety issues, either the amount of dyes used in the food industry as colouring agents or the trace colorants present as food contaminants have to be strictly controlled, those compounds being harmful for humans at high doses. Gan et al. used graphene sheets decorated with highly-ordered mesoporous TiO2 nanoparticles to detect Sudan I, one of the most used azo-dyes in petroleum and textile industries which can be found as trace in animal tissues.213 The low detection limit achieved with the composite material was due to its high specific area, strong adsorptive capacity and excellent electrocatalytic properties. The same authors used the same transducer to analyse chilli products and ketchup samples for the presence of Orange II, an azo-colorant which is not permitted in food preparation due to its high toxicity.214 In addition, the graphene–TiO2 composite was also used by the same group for the simultaneous detection of sunset yellow and tartrazine,215 two very common colorants used as food additives. Well separated voltammetric peaks were obtained by using the composite materials, thus allowing the determination of both compounds in several food sample extracts. The simultaneous detection of the same colorants in commercial soft drinks was also performed by Ye et al.216 on a beta-cyclodextrin-coated poly(diallyldimethylammonium chloride)-functionalized graphene composite film, obtained by using L-ascorbic acid as the reducing agent. Finally, a graphene quantum dot-gold nanoparticle multi-layered material was employed by Hou et al. for the determination of malachite green, a dye generally used in materials such as silk, leather and paper.217 The developed transducer presented good recoveries and a detection limit of 1 × 10−7 M for the detection of malachite green in fish samples.
Doped graphene platforms have also been recently used for the assessment of food quality and safety. Recent reports have shown that p- and n-type doped graphene materials obtained by performing doping with either electron donating or electron withdrawing species yield graphene materials possessing different electronic properties.218 Bonanni and coworkers compared boron-doped graphene and nitrogen-doped graphene platforms containing different amounts of heteroatoms for the antioxidant activity quantification of tea samples.219 They demonstrated that for this purpose, the type and amount of dopant have a significant influence on the electrochemical detection of gallic acid rather than the structural properties of the materials. In a different study performed by the same group, the electrochemical detection of catechin was carried out on boron-doped graphene, nitrogen-doped graphene and undoped graphene platforms.220 The authors found out that the undoped graphene, possessing a lower amount of oxygen functionalities, a higher density of defects and a larger electroactive surface area provided the best electroanalytical performance for the determination of catechin in commercial beer samples.
7.2 Electrochemical biosensing: detection of target molecules via the bio-recognition process
The electrochemical biosensing approach shows that detection of the target molecules is achieved through a specific bio-recognition process between the target molecules and their respective bio-recognition elements. The main aspect of this approach lies in the employment of a bio-recognition layer and this layer is responsible for conferring selectivity and specificity to the respective biosensors. The bio-recognition layer is fabricated on the sensing platform with biological recognition structures such as nucleic acids, antibodies and enzymes, as depicted in Fig. 17. Similarly, for the electrochemical biosensing approach, graphene and its derivatives are generally adopted because of their large surface area and excellent electron conductivity. The large surface area displayed by these materials is beneficial for enhancing the amount of bio-recognition elements that can be immobilized during the fabrication of a bio-recognition layer. In addition, they can also provide a greater available surface area for heterogeneous electron transfer. For achieving excellent electron conductivity, they aid in faster heterogeneous electron transfer. The properties of both graphene and its derivatives can contribute greatly to the enhancement of the analytical signal intensity, therefore improving the sensitivity of the electrochemical biosensor. | ||
| Fig. 17 The different classes of bio-recognition elements utilized in the fabrication of electrochemical biosensors. | ||
• Physical adsorption – in physical adsorption, the immobilization of nucleic acid probes onto graphene materials exploits the hydrophobic and π–π interactions between the aromatic rings of nucleobases in nucleic acids and the hexagonal cells of graphene materials. The resulting orientation of the immobilized nucleic acid probes is random.
• Covalent attachment – in covalent attachment, the immobilization process involves the formation of a covalent bond, usually an amide bond, between the graphene materials and the nucleic acid probes. To achieve an amide bond, the inherent carboxylic functional groups on the surface of the materials are often exploited. Upon activation by carbodiimide/N-hydroxysuccinimide (EDC/NHS) chemistry, nucleic acid with a NH2 terminal can subsequently be covalently attached to the surface of graphene materials.
• Cross-linking –the cross-linking method often involves an organic linker molecule which facilitates the covalent immobilization of nucleic acid probes onto the surface of graphene materials. A linker molecule is usually employed when the graphene materials do not have sufficient inherent carboxylic functional groups on the surface for direct covalent attachment with the nucleic acid probes. Hence, linker molecules generally have dual properties; containing hydrophobic regions for conjugation to the basal plane of graphene materials by hydrophobic and π–π interactions, and containing carboxylic functional groups for formation of amide linkages with nucleic acid probes with NH2 terminal.
• Biotin–avidin interaction – the biotin–avidin immobilization technique is a form of bio-recognition process on its own. Avidin is a protein which has the ability to bind up to four biotin molecules. Interesting to note, the biotin–avidin complex is the strongest known non-covalent interaction between a protein and a ligand. For this technique, prior modification of the surface of graphene materials with a layer of avidin molecules is required before the introduction of biotinylated nucleic acid probes. Upon introduction, biotinylated nucleic acid probes will bind to the avidin molecules by affinity, resulting in the immobilization of nucleic acid probes on graphene materials.
• Electrostatic attraction – in electrostatic attraction, immobilization of nucleic acid probes is achieved through the negatively charged phosphate backbone of nucleic acids, thereby exposing the nucleobases for efficient hybridization. Hence, this suggests that the surface of graphene materials has to be modified in order to confer a certain degree of positive charge to it. Similar to the cross-linking method, an organic molecule with dual properties may be exploited.
With nucleic acids functioning as the bio-recognition elements, electrochemical biosensors for the detection of various target molecules have been developed, with their concepts elaborated in the subsequent subsections, according to the class of target molecules.
7.2.1.1 Nucleic acid analysis. For the purpose of detecting nucleic acids with nucleic acids also serving as the recognition elements, various forms of graphene materials have been employed in recent years, with the most common being chemically reduced graphene oxide (CRGO). Mukherjee et al.221 designed a biosensor for the detection of Mycobacterium tuberculosis gene, with CRGO functioning as the sensing platform. A single-stranded nucleic acid probe was first covalently immobilized onto the CRGO surface, with methylene blue (MB) serving as an electroactive hybridization indicator. Due to the greater affinity which MB has for single-stranded nucleic acid as compared to double-stranded nucleic acid, a smaller differential pulse voltammetry (DPV) peak current was derived after hybridization with the target nucleic acid. A detection range of 2.872 × 10−3–2.872 × 105 ng μL−1 was obtained for the fabricated biosensor, with good selectivity against single-base mismatched and non-complementary nucleic acids. In another report, Benvidi et al.222 demonstrated an impedimetric biosensor for the sensing of amelogenin gene, also using CRGO as the transducing platform, and covalent nucleic acid probe immobilization (Fig. 18A). The proposed biosensor presented a wide detection range of 1 × 10−20–1 × 10−14 M, with a limit of detection (LOD) of 3.2 × 10−21 M. In addition, the biosensor also displayed good specificity against non-complementary nucleic acid. Subsequently, the same group attempted to improve the analytical performance of their biosensor in another study.223 Employing CRGO as the sensing interface, Zhang et al.224 reported on an impedimetric biosensor for the detection of hepatitis B virus. In this study, the nucleic acid probe was covalently immobilized onto CRGO by the cross-linking technique. In order to facilitate the cross-linking process, CRGO was first functionalized with tryptamine and then glutaraldehyde. The sensing platform exhibited a detection range of 1 × 10−12–1 × 10−7 M, with a LOD of 5.2 × 10−13 M. Furthermore, the biosensor also showed high selectivity against single-base mismatched and non-complementary nucleic acids.
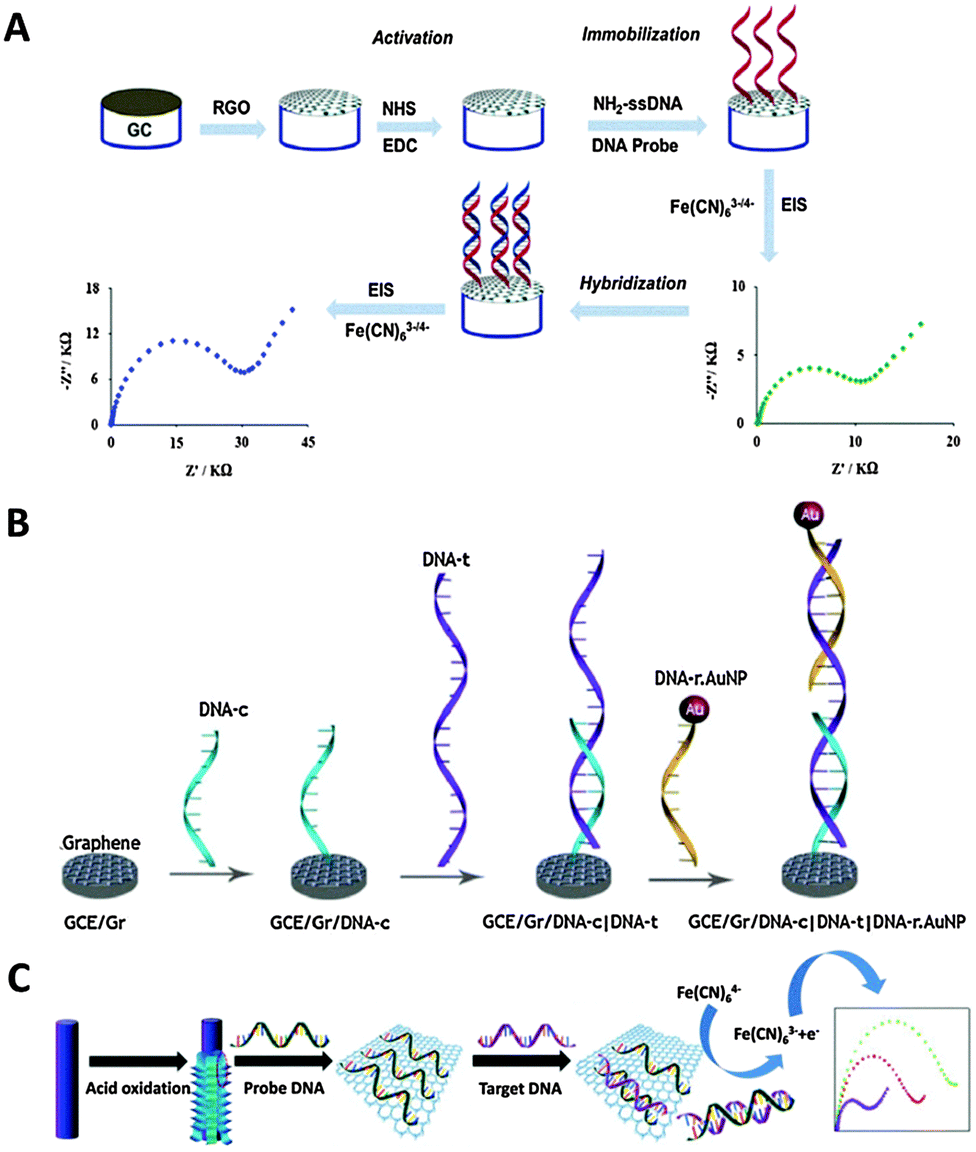 | ||
| Fig. 18 (A) Illustration of impedimetric biosensor for the sensing of amelogenin gene, based on CRGO as the transducing platform and covalent immobilization of a single-stranded nucleic acid probe. Upon hybridization with the amelogenin gene, charge transfer resistance (Rct) between [Fe(CN)6]3−/4− and the electrode surface increased. Reproduced with permission from ref. 222. Copyright (2014) Elsevier. (B) Schematic representation of the various fabrication stages of the biosensor designed for BRCA1 gene detection. Reproduced with permission from ref. 225. Copyright (2014) Elsevier. (C) Illustration of a scaly GO/graphite fiber hybrid electrode employed for impedimetric DNA sensing. Upon hybridization with a complementary DNA target, the partial release of the double-stranded DNA resulted in a lower Rct. Reproduced with permission from ref. 230. Copyright (2015) John Wiley and Sons. | ||
Apart from chemical reduction, graphene oxide (GO) can be reduced by thermal methods as well. In 2014, Rasheed and Sandhyarani225 published a work on employing thermally reduced graphene oxide (TRGO) as the sensing platform for the detection of breast cancer 1 (BRCA1) gene. The proposed sensor used a sandwich detection strategy, as illustrated in Fig. 18B. The capture probe (DNA-c) underwent physical adsorption onto TRGO while gold nanoparticle (AuNP) were covalently conjugated to the reporter probe (DNA-r) to form DNA-r.AuNP. Upon hybridization with the BRCA1 gene, a sandwich complex was formed and the oxidation signal of AuNPs was utilized for BRCA1 gene detection by chronoamperometry. A detection range of 1 fM–1 nM was attained and selectivity was achieved with three-base mismatched and non-complementary nucleic acids.
Following CRGO and TRGO, electrochemically reduced graphene oxide (ERGO) was employed for the purpose of nucleic acid detection. Li et al.226 presented a human immunodeficiency virus 1 (HIV1) gene biosensor based on an ERGO modified graphene surface. The nucleic acid probe was physically immobilized onto the sensing platform, before undergoing hybridization with HIV1 gene. Herein, the analytical signal was derived from the DPV peak current of ferricyanide. The sensing interface achieved a detection range of 10−12–10−7 M, with a LOD of 1.58 × 10−13 M and selectivity against non-complementary nucleic acid.
Moving on, in addition to reduced analogues of GO, other types of graphene materials have also been applied to nucleic acid sensing. These materials include thiofluorographene,227 GO228 and GO/graphite hybrid.229,230 Urbanova et al.227 demonstrated the proof-of-concept of using thiofluorographene as a platform for impedimetric nucleic acid sensing. Thiofluorographene was obtained by substituting fluorine atoms in fluorographene with nucleophilic sulfhydryl groups. Physical adsorption was adopted to immobilize the nucleic acid probe before hybridization reaction. It was shown that thiofluorographene could distinguish between complementary, single-base mismatched and non-complementary nucleic acids, while fluorographene did not have such capability. On another note, Sun et al.228 fabricated a biosensor for the sensing of miRNA-21, a diagnostic tool for lung cancer. In this report, GO was utilized as the transducing platform with a silver nanoparticle (AgNP)-labelled nucleic acid probe physically adsorbed on the surface. In the presence of miRNA-21, hybridization occurred, forming a duplex structure which was then released from GO. Hence, the stripping current of AgNPs can be exploited as the analytical signal and a detection range of 100 fM–1 nM was acquired, with a LOD of 60 fM. Selectivity of the biosensor was also displayed against up to three mismatched sites. For the application of the GO/graphite hybrid, two separate studies have been published. Firstly, Congur et al.229 presented an impedimetric biosensor for detecting miRNA-34a, which is related to Alzheimer's disease and cancers. In this work, a pencil graphite electrode modified with GO was adopted as the sensing surface. Interesting to note, their sensing approach was to hybridize the nucleic acid probes with the miRNA-34a targets before exposing the sensing surface to the formed duplex. Due to the NH2 terminal of the nucleic acid probe, the duplex underwent covalent bonding with the GO/graphite surface. The fabricated sensor achieved a detection range of 0–10 μg mL−1, with a LOD of 1.9 μg mL−1 and selectivity against other miRNAs. In the second study conducted by Zhang et al.,230 a scaly GO/graphite fiber hybrid electrodes were utilized as the sensing platform for impedimetric detection of nucleic acid (Fig. 18C). GO sheets were synthesized in situ at the surface of graphite fibers by acid oxidation to yield scaly GO/graphite fiber hybrid electrodes. The partially peeled GO sheets provided numerous binding sites for nucleic acid probe immobilization and thus enhanced electron transfer. Herein, the nucleic acid probe underwent physical adsorption on a GO/graphite hybrid prior to hybridizing with its complementary nucleic acid target. Due to the lower affinity towards double-stranded nucleic acid as compared to single-stranded nucleic acid, detection of the nucleic acid target was achieved. The range of detection was determined to be 0.01–1 nM, with a LOD of 5.6 pM and selectivity demonstrated against single-base mismatched and non-complementary nucleic acids.
7.2.1.2 Biomolecule analysis. In addition to the detection of nucleic acids, sensing of smaller biological molecules is also achievable with nucleic acids functioning as the recognition elements. Wu et al.231 reported an aptasensor for the detection of adenosine triphosphate (ATP) using the ATP aptamer as the recognition element (Fig. 19). Aptamers are synthetic single-stranded nucleic acids which are in vitro selected through the systematic evolution of ligands by exponential enrichment (SELEX) process to bind to specific target biomolecules. They exhibit several advantages, including high specificity, simple structure, easy synthesis and long-term stability. Herein, CRGO was utilized as a label and its electrocatalytic property towards ascorbic acid (AA) oxidation, derived from its defective sites, was exploited. For the fabrication of the aptasensor, the ATP aptamer with a SH terminal was first covalently linked to the gold electrode. In the absence of ATP, CRGO can bind to the ATP aptamer through π–π stacking interactions, leading to a greater DPV oxidation current and a lower oxidation potential of AA. On the other hand, in the presence of ATP, the immobilized aptamer formed a complex with ATP and CRGO was released from the biosensing interface, resulting in a sharp decrease in oxidation current and an increase in the oxidation potential of AA. The changes in the oxidation current and the oxidation potential of AA were both employed as the analytical signals for the detection of ATP. With oxidation current as the analytical signal, a low LOD of 11.7 pM and a wide detection range of 0.05 nM–1.0 μM were attained. Whereas, when using oxidation potential as the analytical signal, a LOD of 25.0 pM with a detection range of 0.05–10.0 nM was acquired. In addition, the aptasensor also displayed excellent selectivity for ATP over the other analogues.
 | ||
| Fig. 19 Schematic representation of an electrochemical aptasensor for ATP. Reproduced with permission from ref. 231. Copyright (2015) Elsevier. | ||
7.2.1.3 Heavy metal analysis. Nucleic acids playing the role of a recognition element can contribute to the monitoring of environmental safety by detecting for pollution by heavy metals. Zhang et al.232 demonstrated a biosensor for mercury (Hg2+) detection based on thymine–mercury–thymine (T–Hg2+–T) interactions and CRGO as the transducing platform. As shown in Fig. 20A, a single-stranded, ferrocene-tagged T-rich nucleic acid probe was first physically adsorbed onto the surface of CRGO, with the redox peaks of ferrocene utilized as the analytical signal with DPV. In the presence of Hg2+, the ferrocene-labelled T-rich probe could hybridize with another single-stranded nucleic acid, forming a double-stranded nucleic acid complex through Hg2+-mediated coordination of T–Hg2+–T base pairs. Owing to the weaker affinity between CRGO and a double-stranded nucleic acid complex, ferrocene drew away from the surface of CRGO and the resulting redox peaks were substantially diminished. The proposed biosensor displayed a LOD of 5 pM, with a detection range of 25 pM–10 μM. Furthermore, high selectivity of the biosensor was also reported.
 | ||
| Fig. 20 (A) Schematic illustration of an electrochemical Hg2+ biosensor based on T–Hg2+–T interactions and CRGO as the transducing platform. Reproduced with permission from ref. 232. Copyright (2015) Elsevier. (B) Representation of an electrochemical biosensor for Hg2+ based on the catalytic formation of AuNPs. Reproduced with permission from ref. 233. Copyright (2014) Elsevier. | ||
In another study, Tang et al.233 also reported a biosensor for Hg2+ sensing, exploiting T–Hg2+–T interactions and ERGO as the sensing platform. As seen in Fig. 20B, a single-stranded nucleic acid was first covalently immobilized onto the sensing platform by cross-linking. Upon exposure to Hg2+, stable duplex complexes were formed through strong T–Hg2+–T bonding with two immobilized nucleic acid strands in close proximity. The captured Hg2+ subsequently catalyzed the formation of AuNPs. Since the extent of AuNPs formation was proportional to the concentration of Hg2+, Hg2+ concentration was directly quantified by stripping voltammetry of the formed AuNPs. The proposed biosensor demonstrated a range of detection of 0.5–120 nM, with a LOD of 0.06 nM. Similar to the previous work, excellent selectivity against other metal ions was also obtained.
• Physical adsorption – in physical adsorption, antibodies are conjugated onto the surface of graphene materials through mainly hydrophobic interactions. Physical adsorption is the simplest and most straightforward immobilization method as compared to the other strategies. However, such a method results in the random orientation of the immobilized antibodies.
• Covalent attachment – based on the covalent attachment technique, antibodies are linked to the surface of graphene materials through the formation of covalent bonds, usually an amide bond. To form an amide bond, antibodies with a NH2 terminal and inherent carboxylic functional groups on the surface of graphene materials are often exploited. Upon activation by EDC/NHS chemistry, amide linkages are generated and antibodies are covalently attached to the surface of graphene materials. Apart from amide bonds, other types of covalent bonds are also plausible, depending on the type of functional groups available on the antibodies and graphene materials. As compared to physical adsorption, covalent attachment allows control over the orientation of the immobilized antibodies.
• Cross-linking – the cross-linking immobilization approach makes use of linker molecules to facilitate the covalent attachment of antibodies to the surface of graphene materials. This approach is generally employed in cases where there are no or insufficient quantity of the desired functional groups present on the surface of graphene materials. In addition, cross-linking can also be employed to create spacing, thereby enhancing the accessibility of the antibodies to the target molecules.
• Biotin–avidin interaction – the principle behind this immobilization technique lies in the exceptional affinity between biotin and avidin molecules. Each avidin molecule is capable of binding to four molecules of biotin. To adopt this immobilization strategy, pre-modification of graphene materials and antibodies is crucial. Graphene materials have to be modified with avidin molecules while antibodies have to be functionalized with a biotin moiety each. Upon exposure, the biotinylated antibodies will bind to the avidin molecules by affinity, thereby immobilizing the antibodies onto graphene materials.
The following subsections will demonstrate how electrochemical biosensors exploiting antibodies as the bio-recognition elements can be used for the detection of various target molecules.
7.2.2.1 Disease biomarker analysis. The detection of disease biomarkers using antibodies as the recognition elements represents a popular and effective clinical diagnostic tool. Among the various measurement techniques, electrochemical immunoassays are the most ideal owing to their advantages of affordability, rapid analysis, sensitivity and portability. To date, graphene materials have been extensively involved in the development of electrochemical immunosensors for the detection of biomarkers and other target molecules.
Chemical vapor deposition grown graphene represents an example of graphene materials, which has been gaining research attention in recent years due to the demand for high quality graphene materials. Eissa et al.234 reported on the utilization of CVD graphene for the development of an impedimetric immunosensor for ovalbumin. The ovalbumin antibody was covalently immobilized on CVD graphene using the cross-linking approach, where CVD graphene underwent prior functionalization with carboxylic groups. The reported immunosensor displayed a detection range of 1 pg mL−1–100 ng mL−1, with a LOD of 0.9 pg mL−1 and selectivity against non-specific proteins. In another study adopting CVD graphene as the transducing platform, Jin et al.235 designed a voltammetric immunosensor for carcinoembryonic antigen (CEA). Interesting to note, the capture antibodies were first covalently immobilized onto magnetic beads before getting attached onto a CVD graphene platform by an external magnetic field. A sandwich strategy was also employed, using AuNPs conjugated with detection antibodies and horseradish peroxidase (HRP) enzymes. The sensing approach is presented in Fig. 21A and the HRP/H2O2 catalytic system provided an analytical signal which was monitored by cyclic voltammetry. Based on the sensing approach, the detection range was concluded to be 5–60 ng mL−1, with a LOD of 5 ng mL−1 and superior specificity. CVD grown graphene was also exploited as the transducing material for the detection of immunoglobulin G (IgG) in a report by Loo et al.236 Loo et al. demonstrated impedimetric immunosensing of IgG, with the recognition element physically immobilized on CVD graphene. The fabricated immunosensor displayed a range of detection of 0.1–100 μg mL−1, a LOD of 0.136 μg mL−1 and selectivity against other proteins.
 | ||
| Fig. 21 (A) Preparation of a magnetic field-controlled voltammetric CEA immunosensor with CVD graphene functioning as the sensing platform. Reproduced with permission from ref. 235. Copyright (2014) Elsevier. (B) Schematic representation of the fabrication process and electrochemical response of the PSA immunosensor. Reproduced with permission from ref. 237. Copyright (2015) Springer. (C) Preparation of CRGO conjugated with respective metal ions and antibodies to function as labels for simultaneous detection of CRP and CD40L. Schematic illustration of the electrochemical immunoassay employed. After CRP and CD40L had been captured by their respective antibodies on the sensing interface, CRGO hybrid labels selectively bound to CRP and CD40L, forming a sandwich complex. Reproduced with permission from ref. 238. Copyright (2015) Elsevier. | ||
Apart from CVD grown graphene, reduced analogues of GO, such as CRGO, have also been widely used for immunosensing. Wang et al.237 fabricated an impedimetric immunosensor for the detection of prostate specific antigen (PSA). In their work, CRGO was employed as the transducing material and it was functionalized with silk peptide and glutaraldehyde to facilitate the covalent immobilization of anti-PSA via the cross-linking technique. The immunosensing approach is shown in Fig. 21B. Based on the fabricated sensor, 0.1–5.0 ng mL−1 and 5.0–80.0 ng mL−1 were determined as the detection ranges, with a LOD of 0.053 ng mL−1. Finally, selectivity against other proteins was also observed. In another two studies, CRGO functionalized with tetraethylene pentamine (CRGO-TEPA) was utilized. Firstly, Yuan et al.238 reported a voltammetric immunosensor for simultaneous detection of C-reactive protein (CRP) and CD40 ligand (CD40L) based on a sandwich assay (Fig. 21C). Herein, CRGO-TEPA was employed as a label, with metal ions conjugated to it and detection antibodies covalently immobilized on it through cross-linking with glutaraldehyde. Detection of CRP and CD40L was achieved by detecting their respective metal ions on the CRGO-TEPA labels with DPV. With the proposed sensing interface, the detection range was assessed to be 0.05–100 ng mL−1, with a LOD of 16.7 pg mL−1 and 13.1 pg mL−1 for CRP and CD40L, respectively. Furthermore, specificity was also shown by the immunosensor. In the second study, Wu et al.239 used CRGO-TEPA as the platform to design a voltammetric immunosensor for the simultaneous detection of two cervical cancer biomarkers, CEA and squamous cell carcinoma antigen (SCCA). The respective capture antibodies were covalently immobilized on CRGO-TEPA and a sandwich assay was employed. Two different redox mediators, neutral red and thionine, were utilized as labels to produce the analytical signals which were then monitored by DPV. Based on the immunosensing approach, CEA and SCCA could be detected in the range of 0.05–20 ng mL−1 and 0.03–20 ng mL−1 accordingly. In addition, LOD of CEA and SCCA was found to be 0.013 ng mL−1 and 0.010 ng mL−1, respectively. Acceptable selectivity parameters were also displayed by the immunosensor. As an extension of CRGO, CRGO nanoribbons have also been demonstrated by Shi et al.240 to be an useful platform for the concurrent detection of interleukin-6 (IL-6) and matrix metallopeptidase-9 (MMP-9). In their work, IL-6 and MMP-9 capture antibodies were immobilized onto CRGO nanoribbons by physical adsorption to fabricate the sensing interface. A sandwich assay was also employed with two different metals acting as labels for detection by stripping voltammetry. The developed immunosensor exhibited a detection range of 10 fg mL−1–1 μg mL−1 and 1 pg mL−1–1 μg mL−1, with a LOD of 5 fg mL−1 and 0.1 pg mL−1, for MMP-9 and IL-6, respectively. In addition, good specificity was also displayed by the immunosensor.
Moving on from CRGO, other types of graphene materials have also been used in recent years. For example, Kailashiya et al.241 presented a GO-based impedimetric immunosensor for platelet-derived microparticles (PMPs). In their work, the antibody for PMPs was covalently immobilized on the GO surface and the sensing platform attained a detection range of 100–7000 counts per μL with specificity. In another study, Lim et al.242 reported on a graphene-based immunosensor for human chorionic gonadotropin (hCG), where AuNPs were utilized as labels. The capture antibody was first physically adsorbed onto graphene, before exposing to hCG and sandwiched with a detection antibody conjugated with AuNPs. DPV was subsequently employed to measure the analytical signal derived from AuNPs and the detection range was found to be 0–500 pg mL−1, with a LOD of 5 pg mL−1.
7.2.2.2 Nucleic acid analysis. Apart from biomarkers, the application of antibodies as recognition elements can be extended to nucleic acid analysis. Tran et al.243 demonstrated the use of antibodies in the fabrication of a voltammetric immunosensor for the detection of miRNA-141. Herein, a CRGO and carbon nanotube composite was employed as the transducing platform and a single-stranded DNA probe was first covalently immobilized onto the surface. Upon exposure to the target miRNA, hybridization occurred and a DNA/RNA hybrid was formed. Subsequently, an antibody bound selectively to the DNA/RNA hybrid before a secondary antibody, which was conjugated to HRP, bound to the first antibody. The HRP/H2O2 catalytic system oxidized hydroquinone to benzoquinone and the subsequent reduction of benzoquinone back to hydroquinone at the electrode was exploited as the analytical signal using square wave voltammetry (SWV). With this immunosensing approach, the LOD was calculated to be 30 fM, with selectivity shown against non-complementary miRNA.
7.2.2.3 Relevant biological molecule analysis. For the monitoring of food safety, Narayanan et al.244 designed a voltammetric immunosensor for the detection of botulinum neurotoxin (BoNT). The proposed sensor utilized carboxylic groups functionalized CRGO as the sensing platform, with the BoNT antibody covalently immobilized on it. Fig. 22 briefly illustrates the immunosensing protocol, ending with the deposition of AgNPs. As such, the stripping current of AgNPs was used for the quantification of BoNT. The developed immunosensor could selectively detect BoNT in the range of 10 pg mL−1–10 ng mL−1, with the LOD at 5 pg mL−1.
 | ||
| Fig. 22 Fabrication steps for sandwich immunoassay-based detection of BoNT. Upon specific binding with BoNT, a series of recognition events followed and ended with deposition of AgNPs generated by the reaction between MIgG-ALP/AuNPs and 3-IP. Reproduced with permission from ref. 244. Copyright (2015) Elsevier. | ||
Other than food toxins, antibodies can also be applied in the sensing of important biological molecules such as cholesterol. Ali et al.245 demonstrated an impedimetric lipid cholesterol immunosensor, with the antibody covalently immobilized onto aminated CRGO as the sensing interface. Based on the sensing interface, a detection range of 5–120 mg dL−1 and a LOD of 5 mg dL−1 were acquired.
• Physical adsorption – physical adsorption is a fast and simple method, utilizing hydrophobic interactions to immobilize enzymes onto the surface of graphene materials. Based on this method, there is little or no control over the orientation of the resulting immobilized enzymes.
• Covalent attachment – in covalent attachment, inherent functional groups present on the surface of graphene materials are exploited to form covalent bonds with the exposed side chains of amino acids used to build the enzymes. Based on the type of covalent bonding formed, it may be possible to influence the final orientation of the immobilized enzymes. In addition, this method allows direct anchoring of the enzyme onto the graphene material modified electrode surface. Hence, direct transfer of electrons to the enzyme's active site is enabled.
• Cross-linking – the cross-linking approach involves the use of linker molecules to assist in immobilizing enzymes onto graphene materials through covalent bonds. Such an approach is generally adopted in cases where the surface of graphene materials does not contain or contain insufficient moieties of the desired functional groups.
• Electropolymerization – for this technique, the enzyme is mixed with a monomer molecule which can undergo polymerization before applying an electrochemical procedure to initiate electropolymerization. Incorporation of the enzymes into the polymer matrix is mainly achieved by electrostatic interactions. Using this technique, thin layers can be produced with superior control.
• Layer by layer – the principle behind the layer by layer immobilization approach is based on the attraction between oppositely charged ionic layers on the electrode's surface. By combining layer by layer immobilization and graphene materials, the response of enzymatic biosensors has demonstrated improvements.
Employing an enzymatic bio-recognition layer, electrochemical detection of small molecular targets has been reported.
7.2.3.1 Small molecule analysis. Baptista-Pires et al.246 presented an enzymatic biosensor for catechol using tyrosinase enzyme as a proof-of-concept receptor and graphene materials as the transducing platforms. Tyrosinase enzyme was immobilized onto GO and CRGO by physical adsorption and the biosensing performance of these two materials was compared. The principle of detection lies in tyrosinase enzyme catalyzing the oxidation of catechol to o-quinone. The electrochemical reduction of o-quinone to catechol was then exploited as the analytical signal using chronoamperometry (Fig. 23). The coupling between catalytic oxidation (catechol to o-quinone) and electrochemical reduction (o-quinone to catechol) shuttled catechol into a repetitive cycle, leading to potential amplification of the signal. In this work, it was demonstrated that CRGO had better analytical performance than GO. CRGO exhibited a LOD of 0.0103 nM and a sensitivity of 0.0898 μA μM−1 while GO displayed a LOD of 0.0711 nM and a sensitivity of 0.0679 μA μM−1. The enhanced analytical performance by CRGO was attributed to its higher conductivity and its mode of interactions with the tyrosinase enzyme. Tyrosinase enzyme was physically immobilized onto CRGO via hydrophobic interactions while GO interacted with tyrosinase through electrostatic attractions. Hydrophobic interactions for tyrosinase enzyme immobilization were more efficient than electrostatic attraction as the latter adversely affected the activity of the immobilized enzyme.247 Nevertheless, both types of graphene materials led to better analytical response for the detection of catechol than the unmodified bare electrode.
 | ||
| Fig. 23 Schematic diagram displaying tyrosinase and the reactions involved in the enzymatic detection of catechol on GO and CRGO surfaces. Reproduced with permission from ref. 246. Copyright (2014) Elsevier. | ||
8. Conclusions
Graphene and its derivatives, such as graphene oxide and heteroatom-doped graphene, proved to have rich electrochemistry. This rich electrochemistry stems from the fact that practically any graphene materials applied for electrochemical measurements do not have an ideal structure as per the IUPAC definition of graphene, but in contrast, they contain defects, amorphous carbon-based impurities, and heteroatoms, among which the most common is oxygen-containing groups, as well as metals either intentionally or unintentionally introduced into the graphene materials. This rich electrochemistry is not only reflected in the observed “electrocatalysis”, but also in the inherent ability of graphene oxide to be oxidized or reduced electrochemically. While there has been tremendous progress in the investigation of graphene since 2010, when our Chem. Soc. Rev.3 tutorial review was published, there is still more room for further research on the determination of underlying reasons for the catalytic activity of graphene materials, such as the recent insight which established that ORR catalysis on N-doped graphene may stem from the electron-deficient carbon neighbouring pyridinic nitrogen.248 Future avenues are expected to take advantage of developments in materials chemistry, specifically in the chemistry on graphene with inclination towards well-defined graphene structures of nearly stoichiometric composition (i.e., fluorographene, hydrogenated graphene, and graphene oxide), in order to attain both reproducibility and well-understood mechanisms for electrocatalysis.Acknowledgements
We acknowledge a Tier 2 grant (MOE2013-T2-1-056; ARC 35/13) from the Ministry of Education, Singapore.References
- R. K. Joshi, S. Alwarappan, M. Yoshimura, V. Sahajwalla and Y. Nishina, Appl. Mater. Today, 2015, 1, 1–12 CrossRef.
- A. C. Ferrari, et al. , Nanoscale, 2015, 7, 4598–4810 RSC.
- M. Pumera, Chem. Soc. Rev., 2010, 39, 4146–4157 RSC.
- J. N. Coleman, Acc. Chem. Res., 2013, 46, 14–22 CrossRef CAS PubMed.
- A. Ambrosi, C. K. Chua, A. Bonanni and M. Pumera, Chem. Rev., 2014, 114, 7150–7188 CrossRef CAS PubMed.
- A. V. Krasheninnikov and R. M. Nieminen, Theor. Chem. Acc., 2011, 129, 625–630 CrossRef CAS.
- F. M. Hu, T. X. Ma, H. Q. Lin and J. E. Gubernatis, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 075414 CrossRef.
- A. Ambrosi and M. Pumera, Nanoscale, 2014, 6, 472–476 RSC.
- Y. Takada and R. Fujii, Tanso, 1985, 1985, 110–113 CrossRef.
- M. Noel, R. Santhanam and M. F. Flora, J. Appl. Electrochem., 1994, 24, 455–459 CrossRef CAS.
- H. Takenaka, M. Kawaguchi, M. Lerner and N. Bartlett, J. Chem. Soc., Chem. Commun., 1987, 1431–1432, 10.1039/c39870001431.
- M. Inagaki, N. Iwashita, Z. D. Wang and Y. Maeda, Synth. Met., 1988, 26, 41–47 CrossRef CAS.
- A. Jnioui, A. Metrot and A. Storck, Electrochim. Acta, 1982, 27, 1247–1252 CrossRef CAS.
- G. Wang, B. Wang, J. Park, Y. Wang, B. Sun and J. Yao, Carbon, 2009, 47, 3242–3246 CrossRef CAS.
- G. M. Morales, P. Schifani, G. Ellis, C. Ballesteros, G. Martinez, C. Barbero and H. J. Salavagione, Carbon, 2011, 49, 2809–2816 CrossRef CAS.
- J. Wang, K. K. Manga, Q. Bao and K. P. Loh, J. Am. Chem. Soc., 2011, 133, 8888–8891 CrossRef CAS PubMed.
- Y. L. Zhong and T. M. Swager, J. Am. Chem. Soc., 2012, 134, 17896–17899 CrossRef CAS PubMed.
- A. J. Cooper, N. R. Wilson, I. A. Kinloch and R. A. W. Dryfe, Carbon, 2014, 66, 340–350 CrossRef CAS.
- M. Alanyalioglu, J. Jose Segura, J. Oro-Sole and N. Casan-Pastor, Carbon, 2012, 50, 142–152 CrossRef CAS.
- V. V. Singh, et al. , Adv. Funct. Mater., 2012, 22, 2352–2362 CrossRef CAS.
- J. Wang, J. Huang, R. Yan, F. Wang, W. Cheng, Q. Guo and J. Wang, J. Mater. Chem. A, 2015, 3, 3144–3150 CAS.
- C.-Y. Su, A.-Y. Lu, Y. Xu, F.-R. Chen, A. N. Khlobystov and L.-J. Li, ACS Nano, 2011, 5, 2332–2339 CrossRef CAS PubMed.
- K. Parvez, et al. , ACS Nano, 2013, 7, 3598–3606 CrossRef CAS PubMed.
- K. Parvez, Z.-S. Wu, R. Li, X. Liu, R. Graf, X. Feng and K. Müllen, J. Am. Chem. Soc., 2014, 136, 6083–6091 CrossRef CAS PubMed.
- T. J. Davies, M. E. Hyde and R. G. Compton, Angew. Chem., Int. Ed., 2005, 44, 5121–5126 CrossRef CAS PubMed.
- A. Ambrosi and M. Pumera, Chem. – Eur. J., 2016, 22, 153–159 CrossRef CAS PubMed.
- J. Liu, et al. , Nano Energy, 2013, 2, 377–386 CrossRef CAS.
- K. S. Rao, J. Senthilnathan, Y.-F. Liu and M. Yoshimura, Sci. Rep., 2014, 4, 4237 Search PubMed.
- K. S. Rao, J. Sentilnathan, H.-W. Cho, J.-J. Wu and M. Yoshimura, Adv. Funct. Mater., 2015, 25, 298–305 CrossRef CAS.
- S. Yang, et al. , J. Am. Chem. Soc., 2015, 137, 13927–13932 CrossRef CAS PubMed.
- M. Hofmann, C. Wan-Yu, T. D. Nguyễn and Y.-P. Hsieh, Nanotechnology, 2015, 26, 335607 CrossRef PubMed.
- Y. Wang, Y. Zheng, X. F. Xu, E. Dubuisson, Q. L. Bao, J. Lu and K. P. Loh, ACS Nano, 2011, 5, 9927–9933 CrossRef CAS PubMed.
- L. B. Gao, et al. , Nat. Commun., 2012, 3, 699 CrossRef PubMed.
- X. H. Wang, et al. , Small, 2014, 10, 694–698 CrossRef CAS PubMed.
- C. T. Cherian, F. Giustiniano, I. Martin-Fernandez, H. Andersen, J. Balakrishnan and B. Ozyilmaz, Small, 2015, 11, 189–194 CrossRef CAS PubMed.
- F. Pizzocchero, et al. , Carbon, 2015, 85, 397–405 CrossRef CAS.
- X. W. Yang, H. L. Peng, Q. Xie, Y. Zhou and Z. F. Liu, J. Electroanal. Chem., 2013, 688, 243–248 CrossRef CAS.
- L. J. Shi, Y. Q. Liu, F. Yang, L. Gao and J. Sun, Nanotechnology, 2014, 25, 145704 CrossRef PubMed.
- M. Zhou, Y. Wang, Y. Zhai, J. Zhai, W. Ren, F. Wang and S. Dong, Chem. – Eur. J., 2009, 15, 6116–6120 CrossRef CAS PubMed.
- A. Y. S. Eng, A. Ambrosi, C. K. Chua, F. Šaněk, Z. Sofer and M. Pumera, Chem. – Eur. J., 2013, 19, 12673–12683 CrossRef CAS PubMed.
- A. Bonanni, A. Ambrosi, C. K. Chua and M. Pumera, ACS Nano, 2014, 8, 4197–4204 CrossRef CAS PubMed.
- L. Chen, Y. Tang, K. Wang, C. Liu and S. Luo, Electrochem. Commun., 2011, 13, 133–137 CrossRef CAS.
- A. H. Loo, A. Bonanni and M. Pumera, Nanoscale, 2013, 5, 4758–4762 RSC.
- C. S. Lim, C. K. Chua and M. Pumera, J. Phys. Chem. C, 2014, 118, 23368–23375 CAS.
- A. Ambrosi and M. Pumera, Chem. – Eur. J., 2013, 19, 4748–4753 CrossRef CAS PubMed.
- D. A. C. Brownson, D. K. Kampouris and C. E. Banks, Chem. Soc. Rev., 2012, 41, 6944–6976 RSC.
- J. G. S. Moo, A. Ambrosi, A. Bonanni and M. Pumera, Chem. – Asian J., 2012, 7, 759–770 CrossRef CAS PubMed.
- M. M. Hantel, T. Kaspar, R. Nesper, A. Wokaun and R. Kötz, J. Electrochem. Soc., 2013, 160, A747–A750 CrossRef CAS.
- M. S. Goh and M. Pumera, Electrochem. Commun., 2010, 12, 1375–1377 CrossRef CAS.
- M. D. Stoller, C. W. Magnuson, Y. Zhu, S. Murali, J. W. Suk, R. Piner and R. S. Ruoff, Energy Environ. Sci., 2011, 4, 4685–4689 CAS.
- J. Xia, F. Chen, J. Li and N. Tao, Nat. Nanotechnol., 2009, 4, 505–509 CrossRef CAS PubMed.
- H. Marsh and F. Rodríguez Reinoso, Activated carbon, Elsiever Ltd., 2006 Search PubMed.
- Y. Zhu, et al. , Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- Y. Chen, X. Zhang, H. Zhang, X. Sun, D. Zhang and Y. Ma, RSC Adv., 2012, 2, 7747–7753 RSC.
- L. Zhang, et al. , Sci. Rep., 2013, 3, 1408 Search PubMed.
- X. Sun, P. Cheng, H. Wang, H. Xu, L. Dang, Z. Liu and Z. Lei, Carbon, 2015, 92, 1–10 CrossRef CAS.
- S. Yun, S.-O. Kang, S. Park and H. S. Park, Nanoscale, 2014, 6, 5296–5302 RSC.
- Y. Xu, et al. , Nano Lett., 2015, 15, 4605–4610 CrossRef CAS PubMed.
- L. Buglione, A. Bonanni, A. Ambrosi and M. Pumera, ChemPlusChem, 2012, 77, 71–73 CrossRef CAS.
- H. R. Byon, S. W. Lee, S. Chen, P. T. Hammond and Y. Shao-Horn, Carbon, 2011, 49, 457–467 CrossRef CAS.
- T. Lu, L. Pan, H. Li, C. Nie, M. Zhu and Z. Sun, J. Electroanal. Chem., 2011, 661, 270–273 CrossRef CAS.
- L. Qiu, X. Yang, X. Gou, W. Yang, Z.-F. Ma, G. G. Wallace and D. Li, Chem. – Eur. J., 2010, 16, 10653–10658 CrossRef CAS PubMed.
- Q. Cheng, J. Tang, J. Ma, H. Zhang, N. Shinya and L.-C. Qin, Phys. Chem. Chem. Phys., 2011, 13, 17615–17624 RSC.
- K.-S. Kim and S.-J. Park, Electrochim. Acta, 2011, 56, 1629–1635 CrossRef CAS.
- S.-Y. Yang, et al. , J. Mater. Chem., 2011, 21, 2374–2380 RSC.
- L. Buglione and M. Pumera, Electrochem. Commun., 2012, 17, 45–47 CrossRef CAS.
- L. Buglione, E. L. K. Chng, A. Ambrosi, Z. Sofer and M. Pumera, Electrochem. Commun., 2012, 14, 5–8 CrossRef CAS.
- A. Bonanni and M. Pumera, Electrochem. Commun., 2013, 26, 52–54 CrossRef CAS.
- W. Song, X. Ji, W. Deng, Q. Chen, C. Shen and C. E. Banks, Phys. Chem. Chem. Phys., 2013, 15, 4799–4803 RSC.
- K. Chen, F. Liu, S. Song and D. Xue, CrystEngComm, 2014, 16, 7771–7776 RSC.
- F. Liu, S. Song, D. Xue and H. Zhang, Adv. Mater., 2012, 24, 1089–1094 CrossRef CAS PubMed.
- M. Seredych, D. Hulicova-Jurcakova, G. Q. Lu and T. J. Bandosz, Carbon, 2008, 46, 1475–1488 CrossRef CAS.
- W. Kim, J. B. Joo, N. Kim, S. Oh, P. Kim and J. Yi, Carbon, 2009, 47, 1407–1411 CrossRef CAS.
- L. Zhao, L.-Z. Fan, M.-Q. Zhou, H. Guan, S. Qiao, M. Antonietti and M.-M. Titirici, Adv. Mater., 2010, 22, 5202–5206 CrossRef CAS PubMed.
- J. P. Paraknowitsch, J. Zhang, D. Su, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 87–92 CrossRef CAS PubMed.
- J. Han, et al. , ACS Nano, 2013, 7, 19–26 CrossRef CAS PubMed.
- L. L. Zhang, et al. , Energy Environ. Sci., 2012, 5, 9618–9625 CAS.
- A. Ambrosi, H. L. Poh, L. Wang, Z. Sofer and M. Pumera, ChemSusChem, 2014, 7, 1102–1106 CrossRef CAS PubMed.
- M. Seredych and T. J. Bandosz, J. Mater. Chem. A, 2013, 1, 11717–11727 CAS.
- D. Hulicova-Jurcakova, A. M. Puziy, O. I. Poddubnaya, F. Suárez-García, J. M. D. Tascón and G. Q. Lu, J. Am. Chem. Soc., 2009, 131, 5026–5027 CrossRef CAS PubMed.
- V. Thirumal, A. Pandurangan, R. Jayavel, K. S. Venkatesh, N. S. Palani, R. Ragavan and R. Ilangovan, J. Mater. Sci.: Mater. Electron., 2015, 26, 6319–6328 CrossRef CAS.
- Y. Wen, B. Wang, C. Huang, L. Wang and D. Hulicova-Jurcakova, Chem. – Eur. J., 2015, 21, 80–85 CrossRef CAS PubMed.
- Y. Xu, X. Huang, Z. Lin, X. Zhong, Y. Huang and X. Duan, Nano Res., 2013, 6, 65–76 CrossRef CAS.
- H. Wang, H. S. Casalongue, Y. Liang and H. Dai, J. Am. Chem. Soc., 2010, 132, 7472–7477 CrossRef CAS PubMed.
- W. Zhou, et al. , Phys. Chem. Chem. Phys., 2011, 13, 14462–14465 RSC.
- L. Xie, et al. , ChemSusChem, 2015, 8, 2917–2926 CrossRef CAS PubMed.
- F. Yang, et al. , ACS Appl. Mater. Interfaces, 2015, 7, 20513–20519 CAS.
- K. K. Lee, et al. , Nanoscale, 2012, 4, 2958–2961 RSC.
- B. G. Choi, M. Yang, W. H. Hong, J. W. Choi and Y. S. Huh, ACS Nano, 2012, 6, 4020–4028 CrossRef CAS PubMed.
- J. Yan, Z. Fan, T. Wei, W. Qian, M. Zhang and F. Wei, Carbon, 2010, 48, 3825–3833 CrossRef CAS.
- S. D. Perera, A. D. Liyanage, N. Nijem, J. P. Ferraris, Y. J. Chabal and K. J. Balkus Jr, J. Power Sources, 2013, 230, 130–137 CrossRef CAS.
- W. Shi, et al. , J. Mater. Chem., 2011, 21, 3422–3427 RSC.
- R. B. Rakhi, W. Chen, D. Cha and H. N. Alshareef, J. Mater. Chem., 2011, 21, 16197–16204 RSC.
- R. B. Rakhi and H. N. Alshareef, J. Power Sources, 2011, 196, 8858–8865 CrossRef CAS.
- K. Zhang, L. L. Zhang, X. S. Zhao and J. Wu, Chem. Mater., 2010, 22, 1392–1401 CrossRef CAS.
- D.-W. Wang, et al. , ACS Nano, 2009, 3, 1745–1752 CrossRef CAS PubMed.
- R. R. Salunkhe, S.-H. Hsu, K. C. W. Wu and Y. Yamauchi, ChemSusChem, 2014, 7, 1551–1556 CrossRef CAS PubMed.
- E. G. da Silveira Firmiano, A. C. Rabelo, C. J. Dalmaschio, A. N. Pinheiro, E. C. Pereira, W. H. Schreiner and E. R. Leite, Adv. Energy Mater., 2014, 4, 1301380 Search PubMed.
- M. A. Bissett, I. A. Kinloch and R. A. W. Dryfe, ACS Appl. Mater. Interfaces, 2015, 7, 17388–17398 CAS.
- K. S. Novoselov, et al. , Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- K. S. Novoselov, et al. , Nature, 2005, 438, 197–200 CrossRef CAS PubMed.
- F. Schedin, A. K. Geim, S. V. Morozov, E. W. Hill, P. Blake, M. I. Katsnelson and K. S. Novoselov, Nat. Mater., 2007, 6, 652–655 CrossRef CAS PubMed.
- K. S. Novoselov, V. I. Fal'ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- M. Pumera, J. Mater. Chem. C, 2014, 2, 6454–6461 RSC.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- M. Pumera and C. H. A. Wong, Chem. Soc. Rev., 2013, 42, 5987–5995 RSC.
- F. Karlicky, K. K. R. Datta, M. Otyepka and R. Zboril, ACS Nano, 2013, 7, 6434–6464 CrossRef CAS PubMed.
- J. J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Catal., 2015, 5, 5207–5234 CrossRef CAS.
- R. L. McCreery, Chem. Rev., 2008, 108, 2646–2687 CrossRef CAS PubMed.
- A. Lherbier, X. Blase, Y. M. Niquet, F. Triozon and S. Roche, Phys. Rev. Lett., 2008, 101, 036808 CrossRef PubMed.
- P. Lazar, R. Zboril, M. Pumera and M. Otyepka, Phys. Chem. Chem. Phys., 2014, 16, 14231–14235 RSC.
- Y. Wang, Y. Shao, D. W. Matson, J. Li and Y. Lin, ACS Nano, 2010, 4, 1790–1798 CrossRef CAS PubMed.
- M. J. Ju, et al. , ACS Nano, 2013, 7, 5243–5250 CrossRef CAS PubMed.
- H. L. Poh, P. Simek, Z. Sofer, I. Tomandl and M. Pumera, J. Mater. Chem. A, 2013, 1, 13146–13153 CAS.
- T. Lin, F. Huang, J. Liang and Y. Wang, Energy Environ. Sci., 2011, 4, 862–865 Search PubMed.
- S. M. Tan, H. L. Poh, Z. Sofer and M. Pumera, Analyst, 2013, 138, 4885–4891 RSC.
- A. G. Garcia, S. E. Baltazar, A. H. R. Castro, J. F. P. Robles and A. Rubio, J. Comput. Theor. Nanosci., 2008, 5, 2221–2229 CrossRef CAS.
- J. Dai, J. Yuan and P. Giannozzi, Appl. Phys. Lett., 2009, 95, 232105 CrossRef.
- P. A. Denis, R. Faccio and A. W. Mombru, ChemPhysChem, 2009, 10, 715–722 CrossRef CAS PubMed.
- P. A. Denis, Chem. Phys. Lett., 2010, 492, 251–257 CrossRef CAS.
- Z. Yang, et al. , ACS Nano, 2011, 6, 205–211 CrossRef CAS PubMed.
- H. L. Poh, P. Šimek, Z. Sofer and M. Pumera, ACS Nano, 2013, 7, 5262–5272 CrossRef CAS PubMed.
- Y. Liu, Y. Ma, Y. Jin, G. Chen and X. Zhang, J. Electroanal. Chem., 2015, 739, 172–177 CrossRef CAS.
- M. Li, C. Liu, H. Zhao, H. An, H. Cao, Y. Zhang and Z. Fan, Carbon, 2015, 86, 197–206 CrossRef CAS.
- J. O. Sofo, A. S. Chaudhari and G. D. Barber, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 153401 CrossRef.
- X. Feng, et al. , J. Mater. Chem. A, 2013, 1, 12818 CAS.
- S. M. Tan, Z. Sofer and M. Pumera, Electroanalysis, 2013, 25, 703–705 CrossRef CAS.
- T. H. Seah, H. L. Poh, C. K. Chua, Z. Sofer and M. Pumera, Electroanalysis, 2014, 26, 62–68 CrossRef CAS.
- R. R. Nair, et al. , Small, 2010, 6, 2877–2884 CrossRef CAS PubMed.
- H. L. Poh, Z. Sofer, K. Klimova and M. Pumera, J. Mater. Chem. C, 2014, 2, 5198–5207 RSC.
- S. Boopathi, T. N. Narayanan and S. Senthil Kumar, Nanoscale, 2014, 6, 10140–10146 RSC.
- P. W. Gong, et al. , Nanoscale, 2014, 6, 3316–3324 RSC.
- H. L. Poh, Z. Sofer, P. Šimek, I. Tomandl and M. Pumera, Chem. – Eur. J., 2015, 21, 8130–8136 CrossRef CAS PubMed.
- X. Ji, X. Zhang and X. Zhang, J. Nanomater., 2015, 2015, 9 Search PubMed.
- A. Ambrosi, C. K. Chua, B. Khezri, Z. Sofer, R. D. Webster and M. Pumera, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12899–12904 CrossRef CAS PubMed.
- C. K. Chua, A. Ambrosi, Z. Sofer, A. Mackova, V. Havranek, I. Tomandl and M. Pumera, Chem. – Eur. J., 2014, 20, 15760–15767 CrossRef CAS PubMed.
- C. H. A. Wong, Z. Sofer, M. Kubešová, J. Kučera, S. Matějková and M. Pumera, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13774–13779 CrossRef CAS PubMed.
- L. Wang, A. Ambrosi and M. Pumera, Angew. Chem., Int. Ed., 2013, 52, 13818–13821 CrossRef CAS PubMed.
- D. S. Geng, et al. , Energy Environ. Sci., 2011, 4, 760–764 CAS.
- Y. Okamoto, Appl. Surf. Sci., 2009, 256, 335–341 CrossRef CAS.
- L. P. Zhang and Z. H. Xia, J. Phys. Chem. C, 2011, 115, 11170–11176 CAS.
- H. Kim, K. Lee, S. I. Woo and Y. Jung, Phys. Chem. Chem. Phys., 2011, 13, 17505–17510 RSC.
- L. Lai, et al. , Energy Environ. Sci., 2012, 5, 7936–7942 CAS.
- D. W. Chang, H.-J. Choi and J.-B. Baek, J. Mater. Chem. A, 2015, 3, 7659–7665 CAS.
- J. Wu, et al. , ACS Appl. Mater. Interfaces, 2015, 7, 14763–14769 CAS.
- S. Yasuda, L. Yu, J. Kim and K. Murakoshi, Chem. Commun., 2013, 49, 9627–9629 RSC.
- X. Kong, Q. Chen and Z. Sun, ChemPhysChem, 2013, 14, 514–519 CrossRef CAS PubMed.
- L. Kavan, Chem. Rec., 2012, 12, 131–142 CrossRef CAS PubMed.
- L. Wang, Z. Sofer, P. Šimek, I. Tomandl and M. Pumera, J. Phys. Chem. C, 2013, 117, 23251–23257 CAS.
- L. Zhang, J. Niu, M. Li and Z. Xia, J. Phys. Chem. C, 2014, 118, 3545–3553 CAS.
- J. Wang, R. Ma, Z. Zhou, G. Liu and Q. Liu, Sci. Rep., 2015, 5, 9304 CrossRef PubMed.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, ACS Catal., 2014, 4, 3957–3971 CrossRef CAS.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Catal., 2015, 5, 5207–5234 CrossRef CAS.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS PubMed.
- B. E. Conway and B. V. Tilak, Electrochim. Acta, 2002, 47, 3571–3594 CrossRef CAS.
- Y. Zheng, Y. Jiao, L. H. Li, T. Xing, Y. Chen, M. Jaroniec and S. Z. Qiao, ACS Nano, 2014, 8, 5290–5296 CrossRef CAS PubMed.
- B. R. Sathe, X. Zou and T. Asefa, Catal. Sci. Technol., 2014, 4, 2023–2030 Search PubMed.
- Y. Ito, W. Cong, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed., 2015, 54, 2131–2136 CrossRef CAS PubMed.
- P. Lazar, R. Zboril, M. Pumera and M. Otyepka, Phys. Chem. Chem. Phys., 2014, 16, 14231–14235 RSC.
- X. Huang, Y. Zhao, Z. Ao and G. Wang, Sci. Rep., 2014, 4, 7557 CrossRef CAS PubMed.
- C. H. Choi, M. W. Chung, H. C. Kwon, S. H. Park and S. I. Woo, J. Mater. Chem. A, 2013, 1, 3694–3699 CAS.
- X. Wang, G. Sun, P. Routh, D.-H. Kim, W. Huang and P. Chen, Chem. Soc. Rev., 2014, 43, 7067–7098 RSC.
- H. Jiang, Y. Zhu, Y. Su, Y. Yao, Y. Liu, X. Yang and C. Li, J. Mater. Chem. A, 2015, 3, 12642–12645 CAS.
- S. S. Shinde, A. Sami and J.-H. Lee, ChemCatChem, 2015, 7, 3873–3880 CrossRef CAS.
- J. Duan, S. Chen, B. A. Chambers, G. G. Andersson and S. Z. Qiao, Adv. Mater., 2015, 27, 4234–4241 CrossRef CAS PubMed.
- Y. Li, Y. Li, E. Zhu, T. McLouth, C.-Y. Chiu, X. Huang and Y. Huang, J. Am. Chem. Soc., 2012, 134, 12326–12329 CrossRef CAS PubMed.
- R. K. Shervedani and A. Amini, Carbon, 2015, 93, 762–773 CrossRef CAS.
- Y. Zheng, et al. , Nat. Commun., 2014, 5, 3783 Search PubMed.
- J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Nano, 2015, 9, 931–940 CrossRef CAS PubMed.
- N. M. Julkapli and S. Bagheri, Int. J. Hydrogen Energy, 2015, 40, 948–979 CrossRef CAS.
- S. Chen, J. Duan, Y. Tang, B. Jin and S. Z. Qiao, Nano Energy, 2015, 11, 11–18 CrossRef CAS.
- J. Yang and H. S. Shin, J. Mater. Chem. A, 2014, 2, 5979–5985 CAS.
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- L. Liao, J. Zhu, X. Bian, L. Zhu, M. D. Scanlon, H. H. Girault and B. Liu, Adv. Funct. Mater., 2013, 23, 5326–5333 CrossRef CAS.
- X. Dai, et al. , Electrochim. Acta, 2015, 171, 72–80 CrossRef CAS.
- H. Dong, et al. , Sci. Rep., 2015, 5, 17542 CrossRef CAS PubMed.
- S. S. Shinde, A. Sami, D.-H. Kim and J.-H. Lee, Chem. Commun., 2015, 51, 15716–15719 RSC.
- H. Fei, et al. , Nat. Commun., 2015, 6, 8668 CrossRef CAS PubMed.
- H. Fei, et al. , ACS Appl. Mater. Interfaces, 2015, 7, 8083–8087 CAS.
- Y. Hou, Z. Wen, S. Cui, S. Ci, S. Mao and J. Chen, Adv. Funct. Mater., 2015, 25, 872–882 CrossRef CAS.
- W. Zhou, et al. , Chem. Mater., 2015, 27, 2026–2032 CrossRef CAS.
- D. Hou, et al. , J. Mater. Chem. A, 2015, 3, 15962–15968 CAS.
- J. Deng, P. Ren, D. Deng and X. Bao, Angew. Chem., Int. Ed., 2015, 54, 2100–2104 CrossRef CAS PubMed.
- A. Ambrosi, S. Y. Chee, B. Khezri, R. D. Webster, Z. Sofer and M. Pumera, Angew. Chem., Int. Ed., 2012, 51, 500–503 CrossRef CAS PubMed.
- A. Ambrosi, C. K. Chua, B. Khezri, Z. Sofer, R. D. Webster and M. Pumera, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12899–12904 CrossRef CAS PubMed.
- M. Pumera, A. Ambrosi and E. L. K. Chng, Chem. Sci., 2012, 3, 3347–3355 RSC.
- L. Wang, A. Ambrosi and M. Pumera, Angew. Chem., Int. Ed., 2013, 52, 13818–13821 CrossRef CAS PubMed.
- S. Y. Chee and M. Pumera, Analyst, 2012, 137, 2039–2041 RSC.
- R. J. Toh and M. Pumera, Faraday Discuss., 2013, 164, 275–282 RSC.
- C. H. A. Wong, C. K. Chua, B. Khezri, R. D. Webster and M. Pumera, Angew. Chem., Int. Ed., 2013, 52, 8685–8688 CrossRef CAS PubMed.
- L. Wang, C. H. A. Wong, B. Khezri, R. D. Webster and M. Pumera, ChemCatChem, 2015, 7, 1650–1654 CrossRef CAS.
- G. Lupina, et al. , ACS Nano, 2015, 9, 4776–4785 CrossRef CAS PubMed.
- C. H. A. Wong, Z. Sofer, M. Kubesova, J. Kucera, S. Matejkova and M. Pumera, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13774–13779 CrossRef CAS PubMed.
- J. P. Smith, C. W. Foster, J. P. Metters, O. B. Sutcliffe and C. E. Banks, Electroanalysis, 2014, 26, 2429–2433 CrossRef CAS.
- R. M. Iost, F. N. Crespilho, L. Zuccaro, H. K. Yu, A. M. Wodtke, K. Kern and K. Balasubramanian, ChemElectroChem, 2014, 1, 2070–2074 CrossRef CAS.
- H. L. Poh, Z. Sofer, J. Luxa and M. Pumera, Small, 2014, 10, 1529–1535 CrossRef CAS PubMed.
- A. Ambrosi and M. Pumera, J. Phys. Chem. C, 2011, 115, 25281–25284 CAS.
- L. Wang, A. Ambrosi and M. Pumera, Chem. – Asian J., 2013, 8, 1200–1204 CrossRef CAS PubMed.
- J. L. Ping and M. S. Fuhrer, J. Appl. Phys., 2014, 116, 044303 CrossRef.
- D. R. Dreyer, R. S. Ruoff and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 9336–9344 CrossRef CAS PubMed.
- V. Mani, A. P. Periasamy and S. M. Chen, Electrochem. Commun., 2012, 17, 75–78 CrossRef CAS.
- W. Y. H. Khoo, M. Pumera and A. Bonanni, Anal. Chim. Acta, 2013, 804, 92–97 CrossRef CAS PubMed.
- C. E. Chng, M. Pumera and A. Bonanni, Electrochem. Commun., 2014, 46, 137–139 CrossRef CAS.
- K. H. Hui, M. Pumera and A. Bonanni, Chem. – Eur. J., 2015, 21, 11793–11798 CrossRef CAS PubMed.
- K. H. Hui, A. Ambrosi, M. Pumera and A. Bonanni, Chem. – Eur. J., 2016, 22, 3830–3834 CrossRef CAS PubMed.
- F. Long, Z. H. Zhang, J. Wang, L. Yan and B. W. Zhou, Electrochim. Acta, 2015, 168, 337–345 CrossRef CAS.
- T. M. B. F. Oliveira, et al. , Bioelectrochemistry, 2014, 98, 20–29 CrossRef CAS PubMed.
- D. Zhang, et al. , Electrochim. Acta, 2013, 107, 656–663 CrossRef CAS.
- S. Zhou, D. Wei, H. Shi, X. Feng, K. Xue, F. Zhang and W. Song, Talanta, 2013, 107, 349–355 CrossRef CAS PubMed.
- T. Gan, J. Y. Sun, M. M. He and L. L. Wang, Ionics, 2014, 20, 89–95 CrossRef CAS.
- T. Gan, J. Y. Sun, Z. M. Lin and Y. L. Li, Anal. Methods, 2013, 5, 2964–2970 RSC.
- T. Gan, J. Y. Sun, W. Meng, L. Song and Y. X. Zhang, Food Chem., 2013, 141, 3731–3737 CrossRef CAS PubMed.
- X. Ye, Y. Du, D. Lu and C. Wang, Anal. Chim. Acta, 2013, 779, 22–34 CrossRef CAS PubMed.
- J. Hou, F. Bei, M. Wang and S. Ai, J. Appl. Electrochem., 2013, 43, 689–696 CrossRef CAS.
- H. T. Liu, Y. Q. Liu and D. B. Zhu, J. Mater. Chem., 2011, 21, 3335–3345 RSC.
- K. H. Hui, A. Ambrosi, Z. Sofer, M. Pumera and A. Bonanni, Nanoscale, 2015, 7, 9040–9045 RSC.
- C. E. Chng, Z. Sofer, M. Pumera and A. Bonanni, Sci. Rep., 2016, 6, 20673 CrossRef CAS PubMed.
- M. Das Mukherjee, et al. , Sens. Actuators, B, 2015, 210, 281–289 CrossRef.
- A. Benvidi, N. Rajabzadeh, M. Mazloum-Ardakani, M. M. Heidari and A. Mulchandani, Biosens. Bioelectron., 2014, 58, 145–152 CrossRef CAS PubMed.
- A. Benvidi, N. Rajabzadeh, H. M. Zahedi, M. Mazloum-Ardakani, M. M. Heidari and L. Hosseinzadeh, Talanta, 2015, 137, 80–86 CrossRef CAS PubMed.
- Z. Zhang, L. Q. Luo, G. F. Chen, Y. P. Ding, D. M. Deng and C. H. Fan, Biosens. Bioelectron., 2014, 60, 161–166 CrossRef CAS PubMed.
- P. A. Rasheed and N. Sandhyarani, Sens. Actuators, B, 2014, 204, 777–782 CrossRef CAS.
- B. Li, G. H. Pan, N. D. Avent, R. B. Lowry, T. E. Madgett and P. L. Waines, Biosens. Bioelectron., 2015, 72, 313–319 CrossRef CAS PubMed.
- V. Urbanova, et al. , Adv. Mater., 2015, 27, 2305–2310 CrossRef CAS PubMed.
- E. L. Sun, et al. , RSC Adv., 2015, 5, 69334–69338 RSC.
- G. Congur, E. Eksin and A. Erdem, Electrochim. Acta, 2015, 172, 20–27 CrossRef CAS.
- J. Zhang, et al. , Adv. Mater. Interfaces, 2015, 2, 1500072 Search PubMed.
- L. A. Wu, E. H. Xiong, Y. Yao, X. Zhang, X. H. Zhang and J. H. Chen, Talanta, 2015, 134, 699–704 CrossRef CAS PubMed.
- Y. L. Zhang, et al. , Electrochim. Acta, 2015, 170, 210–217 CrossRef CAS.
- S. R. Tang, P. Tong, W. Lu, J. F. Chen, Z. M. Yan and L. Zhang, Biosens. Bioelectron., 2014, 59, 1–5 CrossRef CAS PubMed.
- S. Eissa, et al. , Nano Res., 2015, 8, 1698–1709 CrossRef CAS.
- B. Jin, et al. , Biosens. Bioelectron., 2014, 55, 464–469 CrossRef CAS PubMed.
- A. H. Loo, A. Ambrosi, A. Bonanni and M. Pumera, RSC Adv., 2014, 4, 23952–23956 RSC.
- Y. Y. Wang, et al. , Microchim. Acta, 2015, 182, 2061–2067 CrossRef CAS.
- G. L. Yuan, C. Yu, C. Y. Xia, L. L. Gao, W. L. Xu, W. J. Li and J. L. He, Biosens. Bioelectron., 2015, 72, 237–246 CrossRef CAS PubMed.
- D. Wu, A. P. Guo, Z. K. Guo, L. L. Xie, Q. Wei and B. Du, Biosens. Bioelectron., 2014, 54, 634–639 CrossRef CAS PubMed.
- J. J. Shi, T. T. He, F. Jiang, E. S. Abdel-Halim and J. J. Zhu, Biosens. Bioelectron., 2014, 55, 51–56 CrossRef CAS PubMed.
- J. Kailashiya, N. Singh, S. K. Singh, V. Agrawal and D. Dash, Biosens. Bioelectron., 2015, 65, 274–280 CrossRef CAS PubMed.
- S. A. Lim, H. Yoshikawa, E. Tamiya, H. M. Yasin and M. U. Ahmed, RSC Adv., 2014, 4, 58460–58466 RSC.
- H. V. Tran, B. Piro, S. Reisberg, L. H. Nguyen, T. D. Nguyen, H. T. Duc and M. C. Pham, Biosens. Bioelectron., 2014, 62, 25–30 CrossRef CAS PubMed.
- J. Narayanan, M. K. Sharma, S. Ponmariappan, S. M. Shaik and S. Upadhyay, Biosens. Bioelectron., 2015, 69, 249–256 CrossRef CAS PubMed.
- M. A. Ali, K. K. Reza, S. Srivastava, V. V. Agrawal, R. John and B. D. Malhotra, Langmuir, 2014, 30, 4192–4201 CrossRef CAS PubMed.
- L. Baptista-Pires, et al. , Biosens. Bioelectron., 2014, 61, 655–662 CrossRef CAS PubMed.
- Y. Zhang, J. Zhang, X. Huang, X. Zhou, H. Wu and S. Guo, Small, 2012, 8, 154–159 CrossRef CAS PubMed.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |