
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vivo degeneration and the fate of inorganic nanoparticles
Neus
Feliu
ab,
Dominic
Docter
c,
Markus
Heine
d,
Pablo
del Pino
bef,
Sumaira
Ashraf
b,
Jelena
Kolosnjaj-Tabi
g,
Paolo
Macchiarini
a,
Peter
Nielsen
*d,
Damien
Alloyeau
*h,
Florence
Gazeau
*g,
Roland H.
Stauber
*c and
Wolfgang J.
Parak
*bf
aAdvanced Center for Translational Regenerative Medicine (ACTREM), Department of Clinical Science, Intervention and Technology (CLINTEC), Division of Ear, Nose and Throat, Karolinska Institutet, Stockholm, Sweden
bFachbereich Physik, Philipps Universität Marburg, Marburg, Germany. E-mail: wolfgang.parak@physik.uni-marburg.de
cDepartment of Nanobiomedicine, ENT/University Medical Center of Mainz, Mainz, Germany. E-mail: roland.stauber@unimedizin-mainz.de
dUniversitätsklinikum Hamburg-Eppendorf, Hamburg, Germany. E-mail: nielsen@uke.uni-hamburg.de
eCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CIQUS) and Departamento de Física de la Materia Condensada, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain
fCIC biomaGUNE, 20009 Donostia-San Sebastián, Spain
gLaboratoire Matière et Systèmes Complexes, UMR 7057 CNRS/Université Paris Diderot, Paris, France. E-mail: florence.gazeau@univ-paris-diderot.fr
hLaboratoire Matériaux et Phénomènes Quantiques, UMR 7162 CNRS/Université Paris Diderot, Paris, France. E-mail: damien.alloyeau@univ-paris-diderot.fr
First published on 10th February 2016
Abstract
What happens to inorganic nanoparticles (NPs), such as plasmonic gold or silver, superparamagnetic iron oxide, or fluorescent quantum dot NPs after they have been administrated to a living being? This review discusses the integrity, biodistribution, and fate of NPs after in vivo administration. The hybrid nature of the NPs is described, conceptually divided into the inorganic core, the engineered surface coating comprising of the ligand shell and optionally also bio-conjugates, and the corona of adsorbed biological molecules. Empirical evidence shows that all of these three compounds may degrade individually in vivo and can drastically modify the life cycle and biodistribution of the whole heterostructure. Thus, the NPs may be decomposed into different parts, whose biodistribution and fate would need to be analyzed individually. Multiple labeling and quantification strategies for such a purpose will be discussed. All reviewed data indicate that NPs in vivo should no longer be considered as homogeneous entities, but should be seen as inorganic/organic/biological nano-hybrids with complex and intricately linked distribution and degradation pathways.
Neus Feliu graduated in Chemistry from Universitat de Barcelona (UB) in 2007 and obtained her Master Science degree in Biomedical Materials from the Royal Institute of Technology (KTH) in Stockholm, in 2009. She obtained her PhD degree in Medical Science from Karolinska Institutet, Stockholm in the field of Engineered Nanomaterials for Biomedical Applications in 2014. Since then, she is postdoctoral researcher at the Advanced Centre of Translational and Regenerative Medicine (ACTREM), Karolinska Institutet, Sweden. Her research interest is focus on understanding the interactions between nanoparticles and biological systems and to explore their use in medical applications. |
Dominic Docter received his diploma degree in biology in 2008 and his PhD degree at the Johannes Gutenberg University Mainz in 2014 after participating in the “Schwerpunktprogramm: Biological response to Nanoscale Particles” of the German Research Foundation. He has received the NMFZ-research award from the University Medical Center of Mainz. He is currently a junior group leader in the Department of Nanobiomedicine at the ENT University Medical Center of Mainz. His research is focused on understanding events at the host-nano-bio interface to optimize and explore possible nano-diagnostic and -therapeutic applications. |
Markus Heine studied Biology at the Free University Berlin and obtained his PhD at the Humboldt University Berlin. After a short stay at the German Institute of Human Nutrition Potsdam Rehbruecke in the group of Prof. Regina Brigelius-Flohé he became Postdoc in the group of Prof. Udo Schumacher at the University Medical Center Hamburg-Eppendorf (UKE). Since 2013, he is Postdoc in the group of Prof. Jörg Heeren at the UKE. His research interest is focused on the regulation of triglyceride-rich lipoprotein uptake in to adipose tissue and visualization of uptake processes with nanoparticles using intravital microscopy and magnetic resonance imaging (MRI). |
Pablo del Pino studied Physics at the Universidad de Sevilla, Spain and obtained his PhD degree from Technische Universität München, in 2007. He has been a researcher at the Ludwig-Maximilians-Universität München (2007–2009), Institute of Nanoscience of Universidad de Zaragoza (2009–2013) and CIC biomaGUNE (2013–2015). In 2015 he was awarded with a Ramon y Cajal grant. His current research interests focus on the development of materials for applications in life science. |
Sumaira Ashraf obtained her Masters degree in Chemistry from Institute of Chemistry, University of the Punjab, Lahore and her PhD (Nano-biotechnology) from Quaid-i-Azam University Islamabad and in the Biophotonics group, Philipps University of Marburg. She joined Pakistan Institute of Engineering and Applied Sciences (PIEAS) as Assistant Professor in 2013. Since then, she is a postdoctoral fellow supported by the Alexander von Humboldt (AvH). Her research interest is focused on the synthesis and characterization of colloidal nano- and microparticles and their interaction with cells. |
Jelena Kolosnjaj-Tabi received her undergraduate degree in pharmacy from the University of Ljubljana, Slovenia and earned her MSc and PhD in nano toxicology form the University Paris 11. She is an awardee of the Chancellery of the Universities of Paris and spent four years as a postdoctoral fellow at the Paris Cardiovascular Research Center (University Paris Descartes) and University Paris Diderot. Her main research interest concerns the evaluation of the in vivo behavior of nanoparticles, their use for cell-based therapies and the application of nanomaterials for improving the therapy and diagnosis of cancer. |
Paolo Macchiarini obtained his Medical Degree in 1986 from the University of Pisa. He obtained his MSc and PhD degree in Organ and Tissue Transplantation at the University of Franche-Cômpte. He became Chairman of the Department of General Thoracic and Vascular Surgery at the Heidehaus Hospital at the Medical School Hannover. Since 2010, he is Director of the Advanced Center of Translational Regenerative Medicine (ACTREM) at Karolinska Institutet, Stockholm, Sweden. |
Peter Nielsen obtained his PhD in Organic Chemistry and Biochemistry and his MD at the Technische Universität München. He is currently an assistant professor and group leader at the Department of Biochemistry and Molecular Biology at the University Medical Centre Hamburg-Eppendorf. He works also as physician in a special ambulance on diseases of iron metabolism. His research interest spans across metabolism of iron and other trace elements and is focussed in the last year on the synthesis of radiolabelled nanoparticles and their biodistribution and degradation in animal models. |
Damien Alloyeau obtained his PhD thesis on the thermodynamic properties of magnetic nanoalloys performed at the French Aerospace Lab (ONERA). He was postdoctoral fellow at the Lawrence Berkeley National Laboratory. He joined the Materials and Quantum Phenomena laboratory at the National Research Center for Scientific Research (CNRS) as a permanent researcher in 2010. He is a CNRS scientist at the Paris Diderot University. His research interests focus on the fabrication of nanostructured materials, and to explore the life cycle of nanostructures in their application media. Transmission electron microscopy still serve as a cornerstone of his researches. |
Florence Gazeau obtained her PhD from the University Paris 7-Diderot, in 1997, focusing on the magnetic and hydrodynamic properties of ferrofluids. She joined the National Research Center for Scientific Research (CNRS) as a staff scientist in 1998, when she broadened her research on biomedical applications of magnetic nanoparticles. Her current research interests focus on the physics of nanomagnetism applied to nanomedicine, cell–nanoparticles interactions, cellular MRI, nanoparticles-mediated hyperthermia, magnetic targeting, nanoparticles behavior in vivo and nanotoxicology. She is a senior CNRS scientist since 2009, and she works in the laboratory MSC at the University Paris Diderot. |
Roland H. Stauber received his PhD degree from Würzburg University in 1994 and did his post-doctoral training at the National Cancer Institute (USA). He has received the Alexander Karl Award for Cancer Research and a President's Fellowship of the Chinese Academy of Sciences. He is currently a Professor at the Molecular and Cellular Oncology/Nanobiomedicine department at the ENT Medical University Mainz. His research interests focuses on the understanding of molecular mechanisms at the nano-bio interface, the development of nano- and -microtechnology for cancer treatment and diagnosis, as well as drug development. |
Wolfgang Parak studied Physics at the Technische Universität München and obtained his PhD at the Ludwig Maximilians Universität Münche. After a PostDoctoral stay at the University of California, Berkeley he became Assistant Professor at the Center for Nanoscience in Munich. Since 2007 he is Full Professor for Physics at the Philipps Universität Marburg and since 2013 in addition group leader at CIC biomaGUNE in San Sebastian. Since 2010 he is also Associate Editor for ACS Nano. His research interest is focussed on the synthesis and characterization of colloidal nano- and microparticles and on their interaction with cells. |
Nanoparticles as tunable tools towards application in nanobiomedicine
The applications of engineered nanomaterials (NMs) are not only increasing in technical products, but are also more and more common in biotechnology and biomedicine.1–4 The intersection of nanotechnology and biomedicine defines one of the most exciting and cross-disciplinary developments over the last decade.1,2,4,5 NMs and in particular, colloidal inorganic nanoparticles (NPs), are increasingly considered as novel, promising tools with improved therapeutic efficacy, biodistribution, and pharmacokinetics.3,6–9 Recent advancements in synthesis and the ability to rationally manipulate NM and NP features, such as their physical, chemical, and biological properties open up additional possibilities in designing a new generation of nanoprobes for theranostic applications.10–18 For example, considerable progress in the development of magnetic NPs with engineered physicochemistry and tailored surfaces properties19 has opened up a variety of clinically relevant applications, such as magnetic resonance imaging (MRI), drug delivery, magnetic hyperthermia, and low cost in vitro diagnostics.3,6,8,9,20–24 Also, plasmonic NPs, in particular Au NPs, are used for similar purposes,25 ranging from plasmonic sensing, photoacoustic imaging, drug delivery, photothermal therapy (PTT), photodynamic therapy (PDT), and many others.26–29 Fluorescent semiconductor NPs, the so-called quantum dots (QDs),30 have been proposed as contrast agents for fluorescence imaging, guided surgery,31 PDT, etc., though their clinical in vivo use is still under debate due to their potential toxicity. Moreover, with the advent of the concept of so-called ‘personalized medicine’, the field of nanobiomedicine has started to grow, producing a huge variety of different multi-functional NMs. However, despite the increasing production of new nano-tools, to date only a few of them have reached the clinics.32 Yet, some formulations based on gold and iron oxide NPs have already been approved or are in phase 3 of clinical trials. One of the most challenging technical difficulties that NMs are facing in biomedicine, is to successfully cross biological barriers and specifically recognize their targets while they circulate through the body5 (cf.Fig. 1). Moreover, the use of NMs may pose unknown risks to patients, thus current enthusiasm for nanotechnology might shift towards safer approaches.5,33–37 This review focuses on NMs and NPs designed for biomedical application. However, as these materials are also used in industrial and technological sectors, from which they may be released into the biological environment at a certain period of their life cycle, the arguments on degradation of NPs given below also apply to materials involved in non-intentional exposure of living organisms.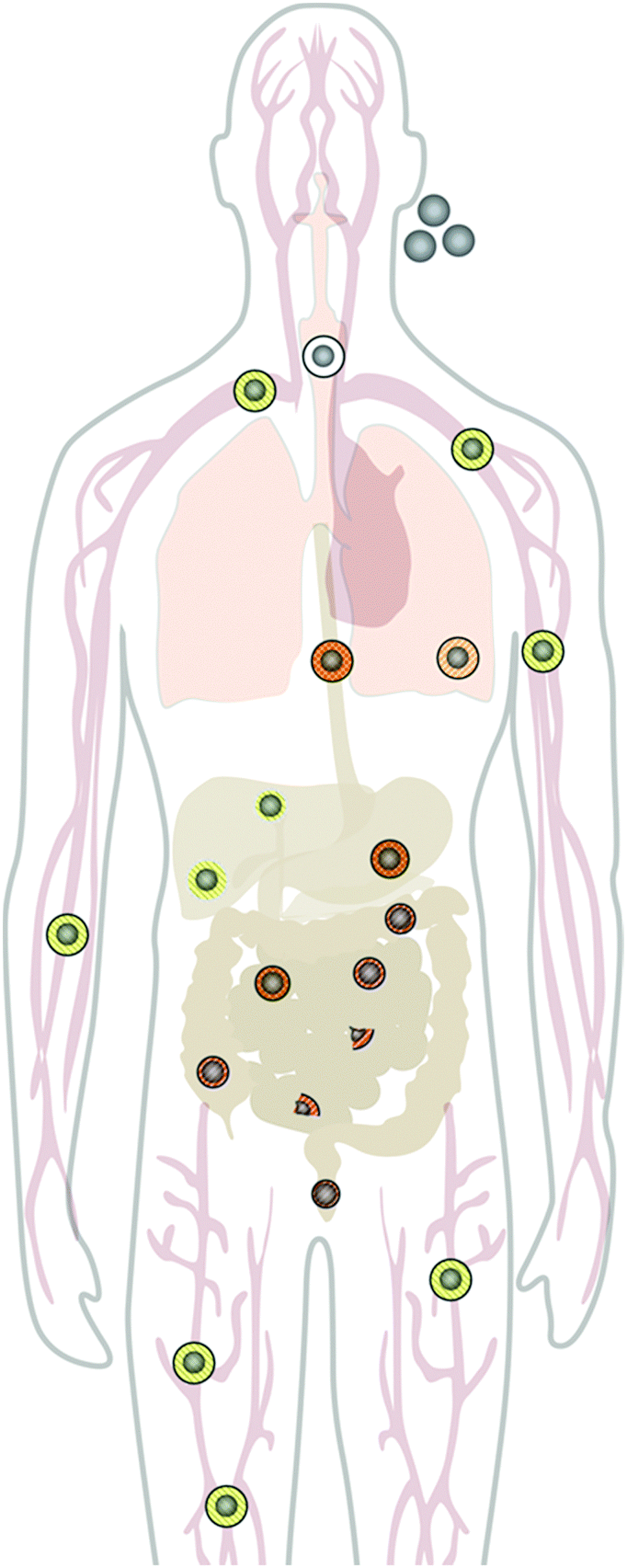 | ||
| Fig. 1 The in vivo pathway and fate of NPs through the human body. While accidental exposure of humans to NPs and their entry into the body most likely occur through inhalation and the oral route, biomedical applications mainly focus on intravenous injection or oral application routes. Upon inhalation, ingestion, or injection of pristine NPs, they come into contact with various complex physiological environments, leading to the covering of NPs with various biomolecules. Upon the formation of such a corona made of biomolecules, the in vivo transformation of pristine NPs is rapidly initiated. During their passage through different compartments of the body, the in vivo degeneration and fate of NPs differ significantly, which is indicated by the different symbols for the NPs. While NP–biomolecule corona complexes are quite stable in plasma, their uptake into organs, such as the liver can trigger corona and NP degradation. Likewise, the conditions in the digestive tract, such as low pH and the presence of surfactants, may lead to similar degradation processes. Such in vivo transformation of nanomaterials should be considered in nanotoxicology and nanomedicine. | ||
Nanoparticles in the biological environment transform into composites
What happens to NPs once they have been administered in vivo? Though the in vivo biodistribution of inorganic NPs (e.g., plasmonic NPs, superparamagnetic NPs, and quantum dots) is relatively well investigated, the appraisal mainly concerns the inorganic NP cores. However, the plain inorganic cores would not be stable in biological environments. Without an organic surface coating, either obtained by the chemical design or due to adsorbed proteins, the NPs would agglomerate. Thus, NPs within the in vivo environment are complex hybrids with an inorganic core and an organic/biologic surface coating,38,39cf.Fig. 2. Conceptually, we will describe each NP as a hybrid object composed of three different entities: the inorganic core, the engineered surface coating, and compounds adsorbed from the biological environment.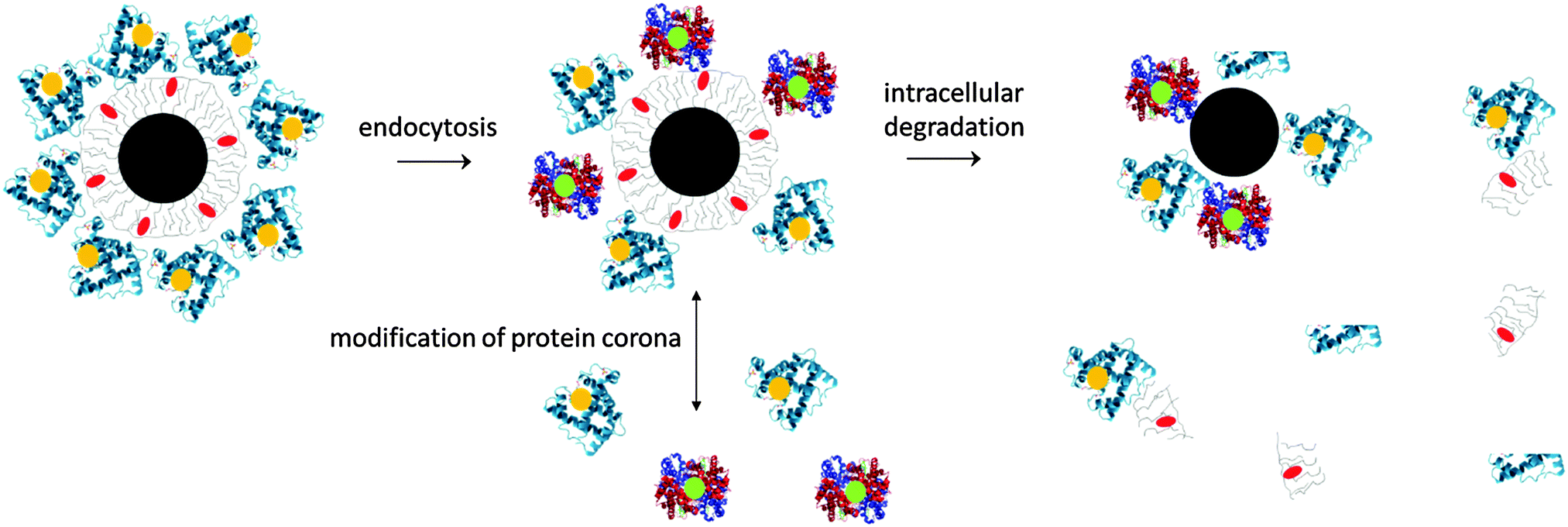 | ||
| Fig. 2 A typical inorganic NP within an in vivo environment comprises of an inorganic core (drawn in black), a capping organic surface, which provides colloidal stability (drawn in grey) and a shell of adsorbed proteins (drawn in blue). The NPs can change their physicochemical properties under in vivo conditions. This may involve dynamic exchange of the protein corona, depending on the variations in the biological environment and/or the activity of “cellular degradation machineries”. Degradation may decompose the NPs into individual parts. Depending on the NP core material, the inorganic core may start to decompose, and thereby change their physical and morphological properties. The organic coating around the inorganic NP core could also be partly removed, as adsorbed proteins may be digested. A possible dissolution of the NP core is not depicted in the sketch. | ||
The NP core defines the “physical identity” or, in other words, the basic functional physical properties of NPs, such as being plasmonic, superparamagnetic, or fluorescent.40 In this way, the core may provide the contrast for several imaging/detection modalities or create heat upon excitation for hyperthermia treatment, etc. The persistence of the physical properties of the core in biological environment is critical for theranostic efficiency of NPs. However, biotransformation of the NPs (aggregation, dissolution, and degradation) might jeopardize such properties over time, depending on the NP environment.
The engineered surface coating determines the intrinsic physico-chemical properties of the NPs, sometimes also termed “synthetic identity”.41 Typical surface coatings for inorganic NPs involve short ligand molecules such as lipoic acid42,43 or peptides,44 silica shells,45,46 polymer micelles,47 or lipid micelles.48,49 The resulting intrinsic physico-chemical properties, such as surface charge, hydrophilicity/hydrophobicity, etc., play an important role in the colloidal stability of the NPs. Appropriate surface coatings prevent NPs from agglomeration and ensure dispersion of the NPs in complex environments. The coatings also determine how the NPs interact with biological environments, i.e., how molecules from these environments adsorb to the NP surface. Distinct surface coatings were shown to have a profound impact on the biocompatibility and fate of NPs, including cell viability or cell adhesion, their cellular uptake, lifetime in the blood system, and the biopersistence in tissues belonging to the mononuclear phagocyte system (MPS; formerly referred to as the reticuloendothelial system, RES).50–56 In addition, coatings may involve functionalization via conjugation with targeting ligands and/or bioactive molecules for obtaining multifunctional ‘intelligent’ NPs.57 In this way, the engineered coating plays an important role in active targeting schemes.
It is often neglected that in complex physiological environments, such as blood, a certain degree of in situ biotransformation will most likely occur for all NPs. For the majority of in vivo applications, NPs will be intravenously injected and immediately exposed to a highly complex biological environment. There, a plethora of ions58,59 and biomolecules, such as lipids, metabolites, sugars, and especially proteins,60 will adsorb onto the surface of the NPs, mediated by van der Waals, electrostatic, hydrogen bonding, and hydrophilic/hydrophobic interactions.38,61–66 The sum of all adsorption processes will result in the formation of the so-called ‘biomolecule corona’, of which, so far, the protein corona (PC) has mostly been studied. It is now accepted, but far from being understood in detail, that the formation of a protein/biomolecule corona critically affects not only the physico-chemical characteristics of NPs,67 but also the (patho-) physiological and biomedical identity, often simply referred to as “biological identity”41,68 of NPs in general.5,69–71 Hence, the properties of corona-covered NPs differ (in most cases significantly) from the intrinsic physico-chemical properties of the NPs, before their exposure to biological environments.5,33,64,71 In the area of corona research, the term ‘hard corona’ was coined to define a protein adsorption signature of a NP, but is sometimes also used to describe the ‘long-lived’ equilibrium protein signature of a NP, e.g., the plasma protein signature of a NP in the blood.4,62,64,71–73 On top of this ‘hard corona’, some models also suggested the existence of a ‘soft corona’, which can be conceptualized as a putative, loosely associated, and rapidly transient layer of biomolecules.62,72,74–78 However, since such a ‘soft corona’ seems to desorb during current purification processes, its existence, (patho)biological, and medical relevance still remain vague.3,5 Here, a standardized definition would be very helpful. In the following, only the analytically accessible proteins associated as NP–protein complexes will be referred to as ‘protein corona’ (PC). Notably, the PC not only (co)defines interaction interfaces between NPs and biological environments, but may also additionally trigger the NP transformation by altering their colloidal stability. The PC can either have a stabilizing effect by inducing steric stabilization or have a destabilizing impact caused by protein mediated bridging, charge compensation and/or introduction of charge inhomogeneity onto the NP surface.63,79–82 Upon aggregation, multiple interactions may result in stronger affinities compared to proteins binding to single NPs, which is likely to occur in a biological solution, in which NPs are highly diluted. Moreover, there could even be a trapping of proteins in such aggregates, with otherwise low or no affinity for single NPs. Depending on the type of NP, such aggregation may also require a certain time, and may thus additionally impact the evolution of the PC.79–82 As protein adsorption is a dynamic process depending on the affinity of individual components for each other, it also varies with the ratio of NPs over available proteins. An important consequence is that the PC differs in a medium with 10% serum, which is currently used in cell culture in vitro, in comparison to in vivo conditions under which the protein ratio is much higher.68
Taking together the statements from the last paragraphs, NPs should be seen as complex hybrids formed via their interaction with biological environments. The interplay between molecular constituents of the biological medium in which the NPs are dispersed and the synthetic NP surface determines the surface properties of the NPs and their colloidal stability. Being colloids, the “synthetic identity”41 of NPs as well as their physical properties need to be characterized in solution (ad minima in water), which defines their surface charge, hydrophilicity/hydrophobicity, and aggregation state. In the next step, the “biological identity”41,68 also involves molecules from the biological medium which adsorb to the NP surface. In this way, for a full characterization, NPs dispersed in the actual biological medium are required. The final step of characterization is to follow the processing of NPs within an in vivo system.
Application routes and biodistribution of nanoparticles
Upon administration, NPs will interact with cells. It is well recognized from cell-culture studies that almost all mammalian cells can, in principle, incorporate NPs to some extent, due to a variety of non-specific uptake mechanisms.83 Many studies have explored the NP properties which influence the efficiency of cell uptake and also determine their intracellular processing, resulting in complete or partial degradation, or storage of biopersistent NPs in unchanged form in cells.84 However, how relevant are these in vitro studies for the in vivo situation? Mammalian cells can clearly process and degrade larger molecules and NPs. Yet, this is not routinely necessary for all cells. Cells in culture are also either dividing rapidly (cancer lines) or are restricted in their metabolism (primary culture) and interactions. Contrariwise, a close interaction between tissues exists in vivo, in which certain cell types, such as the professional macrophages in the mononuclear phagocyte system (MPS), are very potent in fast clearance and processing of larger NPs from the blood flow, whereas other cells may be very limited in their capacity to uptake and process NPs. Importantly, in vivo, the NP clearance directly depends on the global status of the immune system.85 This implicates that, besides the physico-chemical properties of the individual NP, the cell type, which is primarily exposed to the NPs, and the model system used, will also influence the biodistribution, intracellular transfer, and degradation of NPs in vivo.Bolus injection into a peripheral vein seems to be the most recurrent application route for most nanomedical applications. Following intravenous (i.v.) injection, the blood flow would spread the NPs into the right heart, through the lung capillaries, back to the left heart and then into the arterial system supplying each organ with the respective NPs. Under these conditions, a main blood fraction will enter the liver and spleen, which have a huge capacity to remove xenobiotic NPs from the blood stream. A few studies have quantified the distribution of labeled NPs,86,87 and it can be expected that a large fraction of most protein-covered NPs will be taken up by these organs within minutes after injection (cf.Fig. 3). Another cell type, involved in the efficient uptake of NPs in vivo are endothelial cells, which line veins, arteries, and capillaries throughout the body, and thus come in direct contact with the injected NPs.4 Evidence indicates that there could be a difference between endothelia in the liver (liver sinus endothelial cells (LSECs) and peripheral endothelial cells (PECs)), which differentially uptake NPs with respect to their surface charge, with anionic NPs taken up by the liver4,80,88,89 and cationic NPs preferentially binding to PECs.90
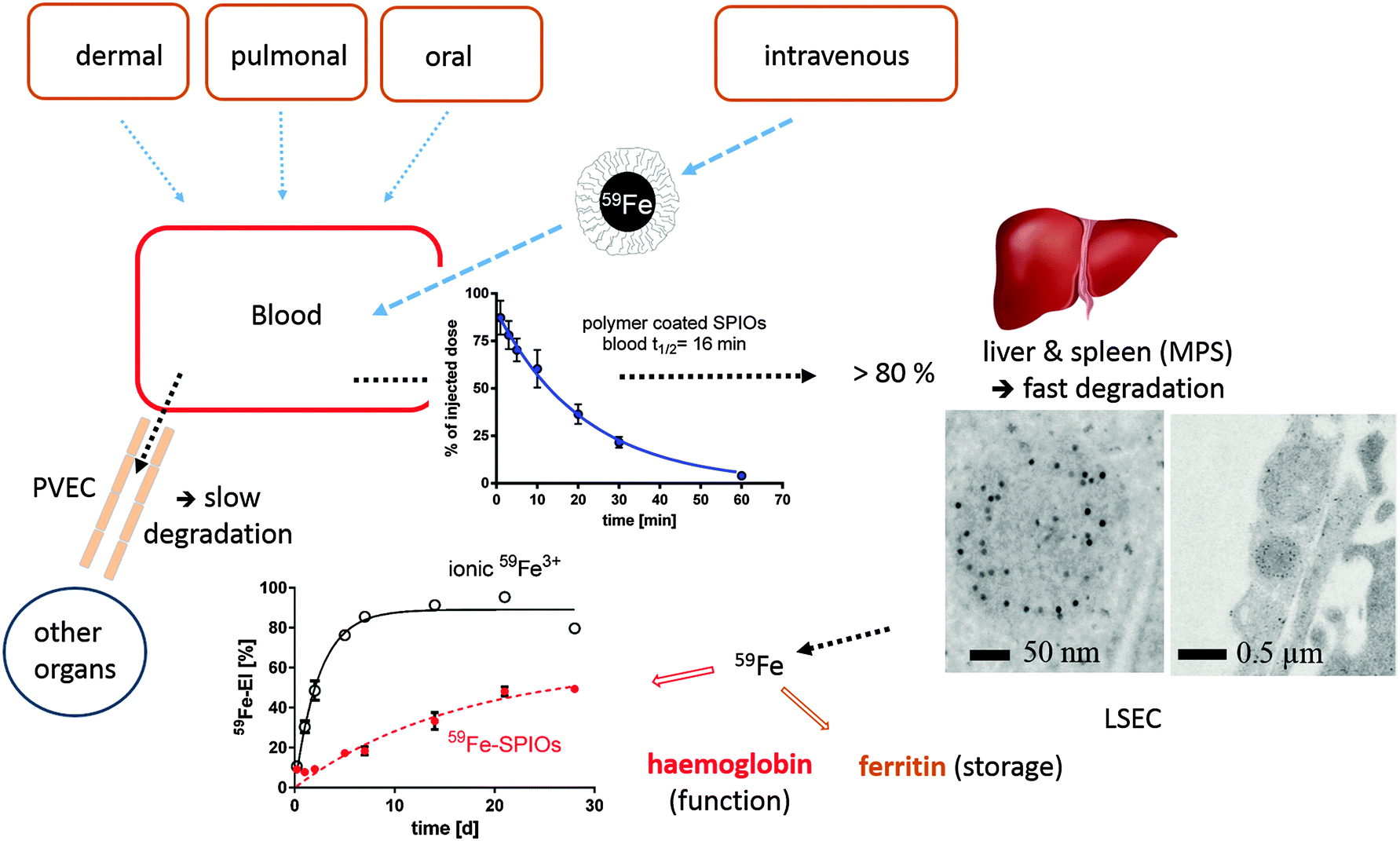 | ||
| Fig. 3 Distribution and degradation of intravenously injected 59Fe-labeled FeOx NPs in mice. The FeOx NPs consisted of a monodisperse iron oxide core (11 nm diameter) coated with an amphiphilic polymer, poly(maleic anhydride-alt-1-octadecene), resulting in 25 nm negatively charged NPs in aqueous solution. A large fraction of the NPs is taken up within minutes by liver cells (Kupffer cells and liver sinusoidal endothelial cells (LSECs)). The degradation of the FeOx core of the NPs can be monitored and quantified by measuring the amount of 59Fe incorporated into the hemoglobin of newly formed erythrocytes. The FeOx NPs are also incorporated in peripheral vascular endothelial cells (PVECs), which represent a large surface area in veins and capillaries. Differences in the degradation efficiency among different cells indicate that FeOx NPs or NP remnants might remain in some cells, and thus elicit a cell-specific chronic toxicity, which is hard to measure in vivo by standard toxicity tests. MPS = mononuclear phagocyte system (mainly liver and spleen). The figure represents a compilation of images from several publications.80,86,88,89 | ||
The situation is different after injection of NPs into the peritoneal cavity or directly into a tumoral lesion,91 which is also relevant for nanomedicines.92 From the peritoneal cavity, the NPs have to pass the visceral peritoneum first, a monolayer of mesothelial cells, and then enter the interstitium, with its lymph or blood vessels, which can transport them into the blood pool. Likewise, pulmonary,93 dermal,94 and oral uptake routes95,96 also have to be taken into account, in particular in the case of environmental NP exposure due to air pollution or spills. Very little quantitative information is available so far on cell processing efficiency after in vivo administration through abovementioned routes. Lademann et al. investigated the NP–skin interactions for silica, titanium dioxide and silver NPs. The vast amount of topically applied solid NPs stays on the skin surface, deeper penetration of single NPs was seen by X-ray microscopy, Raman spectroscopy and flow cytometric studies, with hair follicles representing an important storage and putative entry site.97,98 Kreyling et al. found that inert gold NPs, administered intratracheally are phagocytosed mainly by lung macrophages and only a tiny fraction of gold NPs translocated into systemic circulation.99,100 Enterocytes in the gastrointestinal tract seem to represent an effective barrier against NP uptake. So far, experiments with gold NPs,95 carboxylate-functionalized polystyrene NPs,101 and iron oxide NPs88 have been performed to elucidate the oral application route. Bargheer et al. studied the intestinal absorption of iron from 59Fe or 51Cr-labeled cores of FeOx NPs. After oral application of labelled NPs in mice, a significant absorption of 59Fe but not of 51Cr was observed by sensitive whole-body-counting. As ionic Cr3+ is known to be almost not absorbed in rodents, the results show a partial degradation of the iron oxide cores in the acidic stomach, followed by the physiological absorption or released ionic Fe2+, and almost exclude a relevant intestinal absorption of intact NPs. It should be emphasized that we know far too little about the accurate distribution of a given NP composite in vivo and reliable techniques are urgently needed to document which cells take up the respective NPs and to what extent.
Biological environments may impose hostile conditions to nanoparticles
As outlined in the case of intended or accidental in vivo exposure, NPs take a complex route through the body. From administration until eventual intracellular long-term deposition or alternatively excretion, the NPs are exposed to a variety of different biological environments. The hostile conditions in those environments can vary significantly, for example in pH or local protein (i.e., enzyme) species composition, etc. Highly acidic compartments can be either found at the organ level, inside the stomach or tumors, or at the cell organelle level, in endosomes/lysosomes. Low pH and protein adsorption can affect colloidal stability and favor NP aggregation inducing detrimental modifications of their magnetic or optical properties, when the NPs are clustered inside intra-cellular compartments.102–104 Acidic media also facilitate the corrosion of inorganic NP cores,105 such as in the case of plasmonic Ag NPs,106–108 superparamagnetic FeOx NPs,109,110 or fluorescent CdSe NPs.111 Silica NPs can be, for example, completely dissolved by hydrolysis.112 Enzymes and reactive oxygen species on the other hand have been demonstrated, to be able under certain conditions, to digest carbonic parts of NPs, as demonstrated for example for carbonic, purely inorganic carbon nanotubes (CNTs),113 the strongest and stiffest materials in terms of tensile strength and elastic modulus.114 Thus, it is likely that enzymes can also digest parts of organic surface coatings around inorganic NPs. Indeed, the first evidence has been reported.115–117 Enzymes may also attack proteins adsorbed onto the NP surface,118 and the PC has been demonstrated to dynamically evolve.119Thus, an important question to be addressed is the in vivo integrity of the NPs. Will the organic surface coating and the PC of internalized NPs remain unmodified, or will they be (partly) degraded in vivo? Continuing with this train of thought leads to many potential implications. If the ligand shell of organic NPs is degraded after internalization, how much can fancy surface chemistries performed on NPs direct their biodistribution? This would have an impact on the ongoing discussion of passive versus active tumor targeting. Can organic surface coatings possibly resist enzymatic degradation? On the contrary, while for some applications NP degradation is unwanted, other applications might require it. For example, can inorganic NPs be designed to be degradable into fragments that can be cleared from the body by renal excretion? An improved NP design could accordingly allow for a new generation of inorganic NPs tailored for medical applications with controlled degradability.
Since this kind of research is still in its infancy, this review aims at providing an overview about what is already known about the in vivo integrity of inorganic NPs and how their in vivo physico-chemical and biomedical properties evolve. As in this review NPs are considered as hybrids, including the inorganic core, engineered surface coating, and adsorbed biological molecules (cf.Fig. 2), the fate of all these compounds needs to be investigated for a detailed analysis. The fate of a given hybrid NP in vivo depends on various factors such as the entrance route, PC formation in blood, distribution in the cardiovascular system, etc., uptake into different cell types, intracellular degradation and release, processing of the inorganic core or organic materials, etc. Even if uniformity and full colloidal stability of the NPs are provided during intravenous injection, it cannot be expected that all NP components end up in the same functional pool in vivo. Therefore, multiple labeling strategies are needed by which the different components can be observed independently.120 These would involve reliable quantification techniques for the inorganic core material as well as for attached organic molecules, in order to balance at least the main distribution paths for a given NP. Radioactive labeling is a historical tool with great value to monitor the transport and metabolism pathways of biomolecules. This technique has already been used to trace the kinetics of Au,121 Cd, and Fe-based NPs in vivo.86 In addition, MRI and fluorescence imaging can be used to record biodistributions,87,122,123 but are incapable of detecting absolute amounts as the fluorescence of molecules and magnetic resonance relaxivities may depend on the local environment and aggregation state of NPs. It is also of utmost importance to involve all the controls which are necessary to prove that the labels are attached permanently to the respective NP compound.124,125 Detachment of the label would lead to the determination of the biodistribution of the free label, and not of the labeled NPs. In the following text, the in vivo fate of the three different NP compounds, i.e. the core, engineered coating, and adsorbed biomolecules will be discussed individually. We will hereby start from the outside to the inner parts of the NPs, i.e., from the PC to engineered organic coatings to the inorganic core.
Degradation/evolution of the corona of surface-adsorbed biomolecules
The first part of NPs, which is likely to interact with the environment, is the PC, being the outermost entity. Having pointed out that the PC clearly (co)defines the “biological identity”41,68 and fate of NPs in biological environments, it is clear that understanding its evolution and exchange processes in vivo becomes crucial to produce nontoxic and effective NPs.3,62,71,126In vitro experiments have shown that the formation of the PC influences cellular uptake of NPs, which appears to be dependent on the nature of the adsorbed proteins.3,61,62,64,71,126 Elucidating the fate of the PC during the delivery of the NPs in vivo is also a key determinant for developing efficient nanomedicines.3 The complexity of the biological environment can strongly alter the composition of PCs during their systemic route through the body with possible biological implications (cf.Fig. 3). The specific fate of NPs mostly depends on the entry or chosen administration route (cf.Fig. 3). For example, an intravenously injected NP formulation will incur the following biological processes: vascular transport, extravasation, interstitial passage (extra-cellular matrix), cellular uptake, and clearance. Thus, NPs and more realistically cognate PCs will need to overcome different biological barriers before reaching their final intended or unintended destination.Numerous studies have shown that the physicochemical properties of pristine NPs, such as size, shape, and surface chemistry (termed the 3 ‘S’) can influence the amount, composition and in situ evolution of the PC, which in turn can (co)determine the NP bioactivity.33,63,64,72,127–129 For example, there is evidence that the PC is capable of regulating various cell–NP interactions,64,126,130–132 blood residence time,133,134 (tumor) cell targeting activity and pharmacokinetic profiles,134 albeit the underlying molecular mechanisms are not yet fully resolved. A variety of ex situ and some in situ studies have been conducted to dissect and mechanistically understand the biomolecule corona on the nanoscale, its dependence on the physico-chemical properties of NPs and its impact on the biotransformation and fate of NMs in the human body and environmental systems.33,63,64,72,128,129,135 Typically, corona profiles differ significantly from the protein composition of the investigated (biological) fluid.62–64,82,126 Distinct proteins will either enrich or display only weak affinity for the NP surface. Despite significant work concerning the important relationship between the original surface functionality of the NPs and the nature of the corona, it currently still remains impossible to predict or simulate these interactions in complex physiological environments.5,63,64,72,126
Despite the complexity and analytical challenges already occurring in ex situ characterization of the PC, additional challenges are faced during its in situ analysis. In particular, when NPs move from one physiological (micro)environment of the body to another, e.g., from the circulation via different cellular uptake mechanisms into cells and different organs, such as the liver or spleen, a key question is whether the original corona remains stable or is subjected to substantial changes, which again adds an additional level of complexity.62–64,71,72,86,92,105,125,126,129,136,137 So far, it is assumed that even after passing through several “physiological (micro)environments”, the final corona would still contain a fingerprint of its history and keep a memory of its prior passage through the body,63,105,138 which is in line with recent reports showing the stability of PC signatures ex situ.5,64
Though other studies suggest that PCs may be subject to modifications in their composition while they reside in the body, when they encounter different biological environments, as it occurs for NPs conceived to target tumors.3,63,105,139,140 A study on silica NPs indicated that PCs formed in serum and then transferred to cytosolic extracts experienced qualitative changes in the composition. Yet, these results should be carefully interpreted, as analytical methods may not detect low corona protein concentrations.138
However, the majority of the studies conducted so far demonstrated a stable PC fingerprint, originating from the biological fluid the NPs encounter first, unless processing is performed by enzymatic cellular machineries.63,125,138,141 Here, dissolution processes have been recognized as being essential for the fate, biodistribution and toxicity, particularly for metal and metal oxide NPs.126,142–146 Even while investigating the intracellular fate of silica-coated magnetite NPs by recovering NP-containing cellular organelles, employing magnetic separation techniques, studies demonstrated that PCs, associated with NPs extracted from different cellular compartments, still retained a cytoplasmic fingerprint, albeit additional proteins adsorbed to the “precoated” NPs.147 Radioactive double-labeling was a convenient analytical technique for investigating the stability of a preformed protein corona in vivo. For example, adsorbed 121I-labelled transferrin was transported efficiently into the liver.80 Collectively, the conservation of specific protein signatures in the PCs from different physiological compartments encountered by the NPs may potentially allow reconstructing the NP history through the body. The extent of rearrangements/evolution in PC fingerprints are related to the 3 ‘S’ of NPs, and additional factors such as exposure time,64 temperature,148 and composition of the encountered physiological environments. As the PC remains an unpredictable complex factor, there are currently numerous attempts to chemically prevent and/or modulate protein adsorption.62,63,134,149–151 Such chemical strategies include hydrophilic oligomeric or polymeric PEGylation152 and zwitterionic low molecular weight and polymeric coatings which reduce PC formation.
Again, there are many issues that are yet to be unraveled. What role does enzymatic degradation play in addition to composition exchange for the PC? In vitro experiments have suggested that after intracellular incorporation, lysosomal enzymes may digest parts of the original PC.118 Advanced techniques are required to detect the modifications occurring in situ. These techniques would complement classical methods, which allow detecting the composition of the PC, but require extractions steps, which might be responsible for the formation of a new equilibrium after each purification procedure, and might significantly change the PC composition.153 Standard in situ techniques, such as dynamic light scattering (DLS) and fluorescence correlation spectroscopy (FCS), which can be conveniently applied to PC characterization in sample solutions of biological liquids148,154,155 are unlikely to be suitable for in vivo analysis. Instead, spectroscopy techniques in which the read-out signal depends on the adsorption of proteins, such as surface enhanced Raman scattering (SERS), may offer good possibilities.156–158
Degradation of engineered surface coatings of nanoparticles
The engineered organic surface coating providing the “synthetic identity”41 of NPs lies beneath the PC. However, concerning the in vivo stability of engineered surface coatings, there is not much work reported in the literature. Clearly there is an indication that in vitro, inside endosomes/lysosomes, part of the surface coating may be released from the NP core. The endo/lysosomal enzyme cathepsin L, for example, potentially cleaves a third of the human proteome and the degradation of peptides conjugated to the surface of NPs has been shown within endosomal compartments.115 The enzyme α-glucosidase has also been demonstrated to degrade the carboxydextran shell around the NP core.116Degradation has been shown quantitatively by using a double labeling technique. A 14C-labeled peptide or a 125I-labeled protein was covalently bound to 59Fe-labeled iron oxide cores, and the radioactivity of the labels was followed in the blood and in ex vivo samples of organs after two hours. From both NPs, no separation between core and shell molecules was found while in blood circulation (cf.Fig. 4). A covalently bound 11-As myelin basic protein (MBP) was transported efficiently into LSECs, where the immune competent peptide must have been separated from the core and was presented on the surface of the cells, because the onset of experimental autoimmune encephalomyelitis was completely prevented in a mouse model of multiple sclerosis89 (cf.Fig. 4, upper lane). Likewise, covalently bound or in vitro adsorbed 125I-labeled mouse-transferrin was not separated from the 59Fe-label in blood and was transported into the liver, where however, a complex metabolism and re-distribution of this physiologically relevant plasma protein occurred (cf.Fig. 4, lower lane).80,88
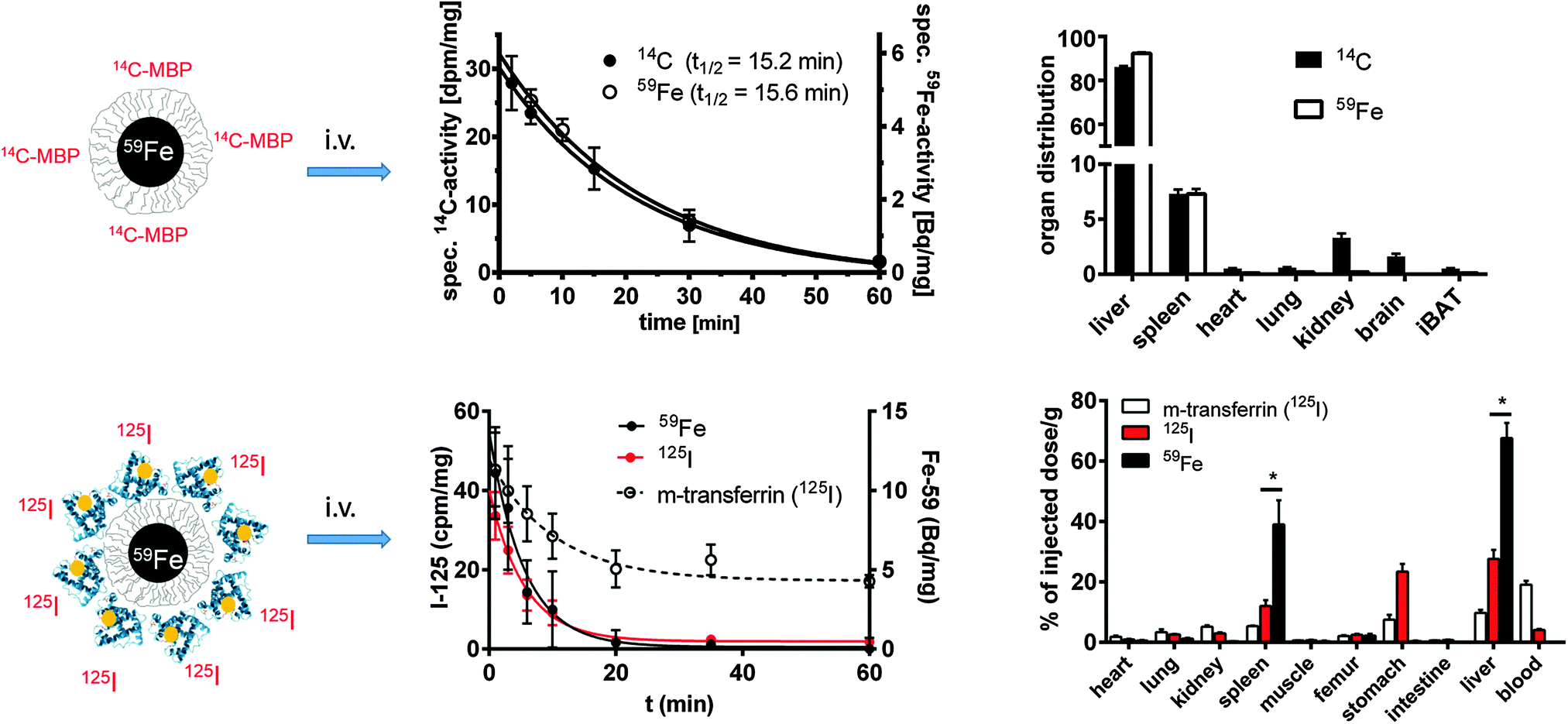 | ||
| Fig. 4 Fate of two double-labeled NPs in vivo. The upper lane displays polymer-coated 59Fe-labeled FeOx NPs. The carboxyl groups of the surface were covalently coupled to a 14C-labeled peptide (11-As, myelin basic protein). After i.v. injection in groups of mice (n = 3–4), the 59Fe and 14C-labels disappeared synchronously from blood following a first order kinetics due to the fast uptake mainly into liver and spleen.89 In the bottom lane, polymer-coated 59Fe-labeled FeOx NPs were covalently labeled with 125I-labeled mouse-transferrin. The blood half-life and organ distribution were followed in groups of mice after i.v. injection. Again, a synchronous removal of both labels from the blood was found, probably indicating the uptake of intact NPs into the liver, without separation of the protein covered shell/polymer from the core already in the bloodstream.80 | ||
The use of radiolabeled NPs thus clearly provides a powerful tool, especially for the sensitive and reliable quantification of the distribution of the different parts of NPs. However, this requires special equipment and the selection of appropriate isotopes is critical. For each isotope, the individual transport mechanisms must be carefully taken into account by including a control group, in which an ionic probe of the element under study is handled in the same way as the NPs.
In yet another study, partial separation of the engineered polymer shell around the Au core was demonstrated.125 Here the polymer shell was labeled with 111In, and the Au NPs with 198Au via neutron activation, cf.Fig. 5. After intravenous application to rats, biodistributions of 111In and 198Au were recorded after one hour and 24 hour retention. Controls ensured that the labels were not lost and in fact corresponded to the locations of the core and polymer shell. The data presented in Fig. 5 indicate that in particular in urine, more polymer shells than NP cores were found. Therefore, it was concluded that after intravenous injection, the polymer coated Au NPs are cleared by the immune system and are transported to the liver, where they are endocytosed. While in this way the majority of the NPs (comprising core and engineered polymer shell) are trapped in the cells of the liver, this also initiates their degradation. Proteases in the endosomes/lysosomes of the cells where the NPs are located may start cleaving the polymer shell. In the particular case, the polymer shell comprising peptide bonds could be readily cut by proteases present in endosomes/lysosomes. The liberated polymer fragments were small enough to be exocytosed leading to final renal excretion.
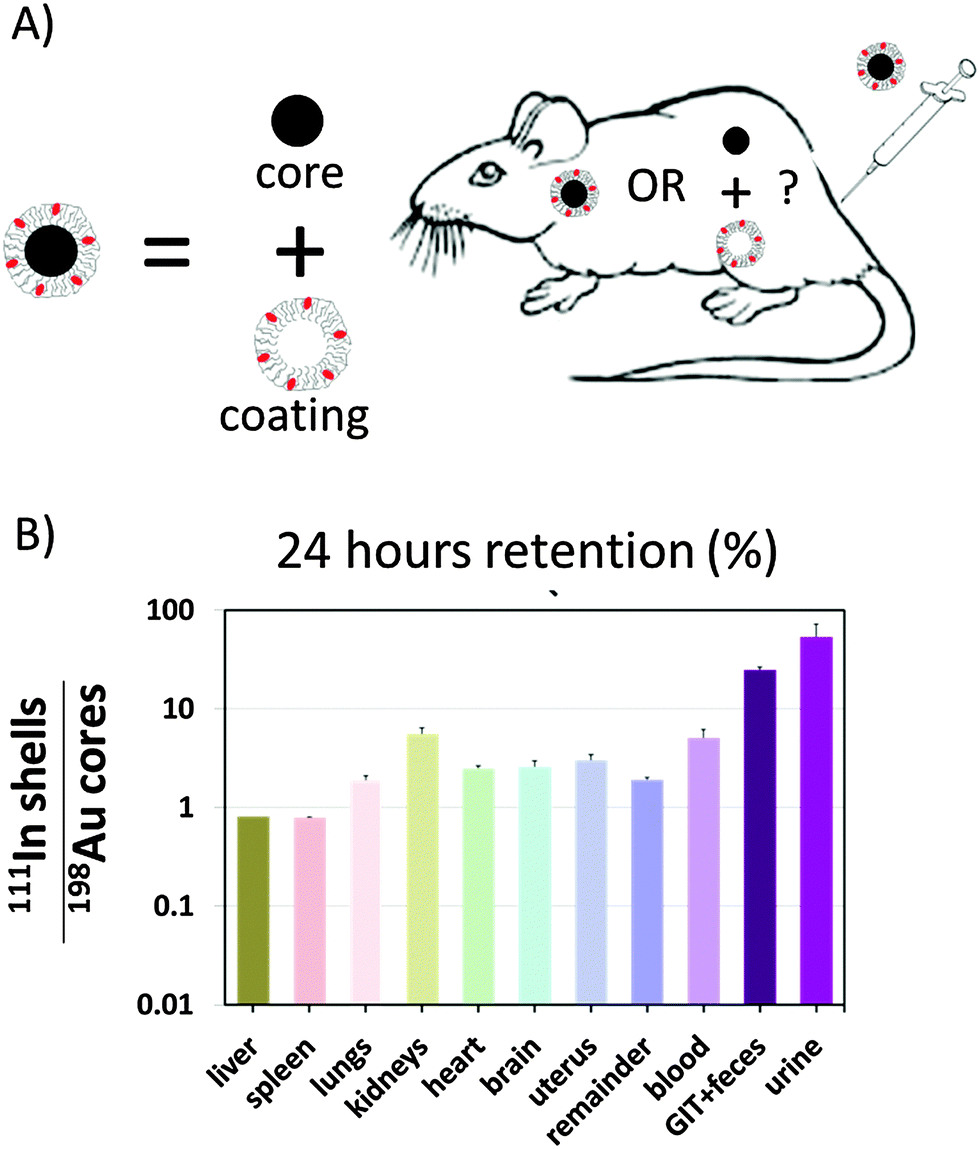 | ||
| Fig. 5 (A) Au cores were coated with an amphiphilic polymer. Cores and polymer shells were labelled with 198Au and 111In, respectively. The question was whether in vivo the polymer shells would remain around the cores or whether they would separate. (B) The ratio of radioactivities originating from 111In and 198Au is shown for different organs 24 hours after retention. In case this ratio is higher than one, the polymer must have come off the NP cores. A figure adopted from Kreyling et al.,125 GIT = gastrointestinal tract. | ||
While these findings offer a first indication that engineered surface coatings can be degraded, there is not much quantitative information to be found in the literature. It is clear that in endosomes/lysosome enzymes exist, which can cleave organic surface coatings. These involve proteases, designed to cut proteins or polypeptides into peptide fragments, which can cut peptide bonds (such as cathepsin L or trypsin), and enzymes, designed to cut polysaccharides into saccharides (such as α-glucosidase), which can cut sugar bonds. However, this does not exclude the existence of engineered NP coatings, which could withstand enzymatic degradation. In addition to intracellular enzymes, degradation may occur in blood by esterases,159 which could, for example, cut coatings containing ester bonds. In this way, for each engineered NP coating one would need to take into account which enzyme could degrade this coating, and what the biodistribution of this enzyme is. Double labeling of the core and shell and double detection schemes, such as correlative microscopy (scanning electron microscopy/fluorescence microscopy), will surely be a useful tool for the investigation of particle and coating distribution.
Degradation of the inorganic nanoparticle core
Ultimately, the inorganic core, as the innermost part of NPs may also be degraded. Obviously the degradation of the inorganic NP core will be highly dependent on the composition of the NP material. While for example NPs made of Ag, ZnO, CdSe, and FeOx are known to corrode, and thus release metal ions,105 other materials such as NPs made from Au are typically considered to be inert and thus stable against degradation. However, as will be discussed in this section, such NPs can also undergo structural alterations. This can be assumed from different points of view. First, ligands such as thiols can strongly bind to the Au surface, and under certain conditions, this may lead to pulling-out Au atoms via the ligand from the Au surface.160,161 Thiols are available in cells, as for example in glutathione, an antioxidant produced by the cells. Thus, in principle, it is possible that Au NPs (very slowly) dissolve. Second, Au NPs can be made with different shapes. The surface A and volume V of a cubic NP with side lengths d are A = 6d2 and V = d3, respectively, leading to a unity surface A* = 6 for a NP with unity volume V* = 1. In contrast, for the NP sphere with diameter d the surface and volume are A = 4π(d/2)2 = π·d2 and V = (4π/3)(d/2)3 = (π/6)d3, respectively, leading to a unity surface A* ≈ 4.8 for a NP with unity volume V* = 1. Thus, for the same volume, a cubic NP has a significantly bigger surface than a spherical NP. The most stable and energetically favorable configuration is the one with the smallest surface and thus NPs with spherical geometries. Thermodynamic laws might therefore govern shape transitions of internalized NPs. In addition, some crystal facets may be more stable than others, stabilizing different shapes or favoring degradation on preferential faces. Thus, even for “inert” materials such as Au, degradation or crystalline reorganization might occur to some extent.The case of superparamagnetic iron oxide (FeOx) NPs, which are known to be chemically reactive is discussed. In order to quantify the inorganic core of NPs and its degradation residues over time in the body, elemental analysis such as inductively coupled plasma mass spectrometry (ICP-MS) is often used. The difficulty is however in distinguishing between the original NPs and their products of degradation. For FeOx NPs, elemental analysis is inappropriate due to the high amount of endogenous iron forms. However, FeOx can be quantified and distinguished from endogenous iron and non-magnetic residues by following their magnetic properties by nanomagnetism methods such as electron paramagnetic resonance (EPR) and temperature-dependent susceptibility measurements.110,162 These magnetic characterizations give information about the biotransformation of the superparamagnetic iron core at short and long terms. Soon after engulfment of FeOx NPs by macrophages, their intracellular confinement impacts their magnetic dynamics due to impaired rotational and translational mobility as well as magnetic interactions. The high local density of FeOx NPs in lysosomes results on one hand in a decrease of magnetic susceptibility and, on the other hand, in an increase of the temperature of transition between superparamagnetic and ferromagnetic regimes, affecting in turn the MRI relaxivity and heating capacity of the FeOx NPs under alternating magnetic fields.102–104 At longer terms the evolution of superparamagnetic properties of NPs may reflect the degradation of their iron oxide core.109,110,163 MRI provides a non-invasive means to detect the distribution and integrity of FeOx NPs over time in the same animal, although quantification remains challenging since MRI relaxivity significantly depends on the local environment and physical state of NPs. EPR was used to quantify the dissolution or elimination of superparamagnetic iron oxide from liver and spleen over one year after intravenous administration of FeOx NPs at the relevant dose for MRI application (2.5 mg kg−1 body weight). This included 7–8 nm spherical FeOx NPs with a hydrophilic glucose-derivative coating, proposed by Guerbet et al. as contrast agents for MRI,110 20 nm FeOx nanocubes, coated with polyethylene glycol (PEG),91,163 and 13 nm iron oxide/5 nm gold dimer heterostructures, coated with an amphiphilic polymer or PEG.50 As expected, the nature of the coating of the FeOx NPs determined their initial uptake in the organs of the mononuclear phagocyte system,50 the PEG-coated NPs being less accumulated in spleen and liver than the NPs coated with the amphiphilic polymer. More surprisingly, the difference of the initial coating has a long-lasting effect on the degradation/elimination of FeOx NPs, which persisted longer in spleen and liver (more than one year), when coated with the amphiphilic polymer. Regardless of the NPs, the elimination of magnetic iron was almost complete in liver after a few months, while 10 to 30% of the initial amount of magnetic iron still persisted in spleen six months after administration. It is worth noting that the total uptake of NPs is much higher in liver than in spleen (15 to 80% of the injected dose in liver depending on NPs versus 2 to 6% in spleen), but the concentration per gram of organ is larger in the spleen. Thereby, the degradative capacity of the organ could be saturated by a high local concentration of the material in the spleen.
Apart from MRI, the in vivo degradation of FeOx NPs can be also followed via radioactive labeling. The core material, i.e., the inorganic core of the FeOx NPs can be labeled either during the synthesis or by neutron activation of the already synthesized material. Recently, a fast, gentle, and quasi on-demand method for post-synthetic labeling of monodisperse iron oxide cores with 59Fe has been developed, which allows for studying the distribution and metabolism of these cores in detail.86 After intravenous injection in mice, the fate of FeOx NPs was studied, cf.Fig. 3. After a lag phase of 3–7 days, 59Fe from the administered FeOx NPs appeared in the hemoglobin of newly formed erythrocytes, indicating the intracellular degradation of the FeOx NPs. 59Fe was released from the cores and channeled into the physiological transport ways for iron. However, a substantial part of the label from the NPs was obviously retained in organs and tissues, probably also indicating storage of intact or only partially degraded FeOx NPs, presumably in cells other than macrophages.
It should be noted that the so-called 59Fe-erythrocyte incorporation rate (59Fe-EI) is a long-known and very unique parameter for following the in vivo processing of iron from any given iron supplement, including NPs. This has been monitored by 59Fe-labeled dextran or carboxydextran coated FeOx NPs used as MRI contrast agents (Endorem®, Resovist®), during studies involved in the registration procedure.164,165 The information obtained from 59Fe-EI in living animals is whether iron is released from the core of a NP at all, to what extent, and at which time scale. In comparison to a test substance (e.g., ionic Fe-salt), the degradation of a given FeOx core and the incorporation of released iron in erythrocytes can be quantified. This technique, if available, is an optimal complement to EPR, which quantifies the persistent FeOx core through its magnetic properties and to multiscale imaging techniques including TEM in ex vivo samples, which can give more insights into the mechanisms of uptake and intracellular processing. However, without a reliable quantification technique, these techniques always lack the information about how relevant the collected data is for the studied in vivo system. Highly fluorescent QDs with the same surface modification as FeOx NPs can be alternatively used, to follow the cellular distribution and uptake kinetics in vivo with intra-vital fluorescence microscopy. This technique shows for example the special role of vascular endothelial cells in the liver or outside the liver for the uptake of different NPs.166
Together with the quantification at the organ level (i.e., the recording of biodistributions), following inorganic cores at the nanoscale in biological environments, is essential for evaluating the degree of degradation of the nanostructures over time and possibly, demonstrating the recycling of degradation products in proteins. In this regard, the multi-functionalities of TEM are of great interest because on the one hand, analytical methods (energy dispersive X-ray (EDX) and electron energy loss spectroscopies) allow studying the local biodistribution of exogenous materials (from cellular up to the single NP or protein levels) and on the other hand, high-resolution imaging techniques provide the unique opportunity to probe the atomic-structure of NP cores in the organism. Although the morphological degradation of polydisperse 7–8 nm spherical FeOx NPs is difficult to ascertain in vivo, FeOx nanocubes and gold/iron oxide heterostructures show evident features of erosion in the liver and spleen seven days after administration, cf.Fig. 6. This is particularly conclusive for heterostructures, because iron oxide crystals degrade around the gold cores, leaving the less reactive gold remnants as long-lived witnesses of iron oxide dissolution in lysosomes. Nanoscale observations also show that FeOx NPs become increasingly surrounded by monodisperse (6 nm) iron-storing ferritin proteins, which are rich in iron, but present different atomic structures than the NPs and can thus be identified by high resolution TEM (Fig. 6, day (D)14).50,110,163 The presence of ferritins proximally to FeOx NPs suggests that iron released from degraded FeOx NPs could be locally transferred to endogenous ferritins through a process regulated by iron homeostasis.167 Consistently, an increase of non-magnetic iron in spleens was observed, confirming the local transformation of superparamagnetic FeOx NPs into non-magnetic iron species. Such a scenario has a perfect plot: the cell firstly confines the NPs within the lysosomes, where iron remains bound in crystals until the proteins are synthesized and recruited in the vicinity of NPs. Secondarily, in order to allow a close approach of endogenous proteins, which tightly and safely unload the iron cargo, while avoiding the cell deleterious Fenton's reaction, the cell progressively isolates the NPs within the lysosomes, as observed by TEM.163 If confirmed, the mechanisms of metal transfer from NPs to endogenous proteins would exemplify a quintessential process in which biomolecules and homeostasis regulate the local degradation of NPs and recycle their by-products.
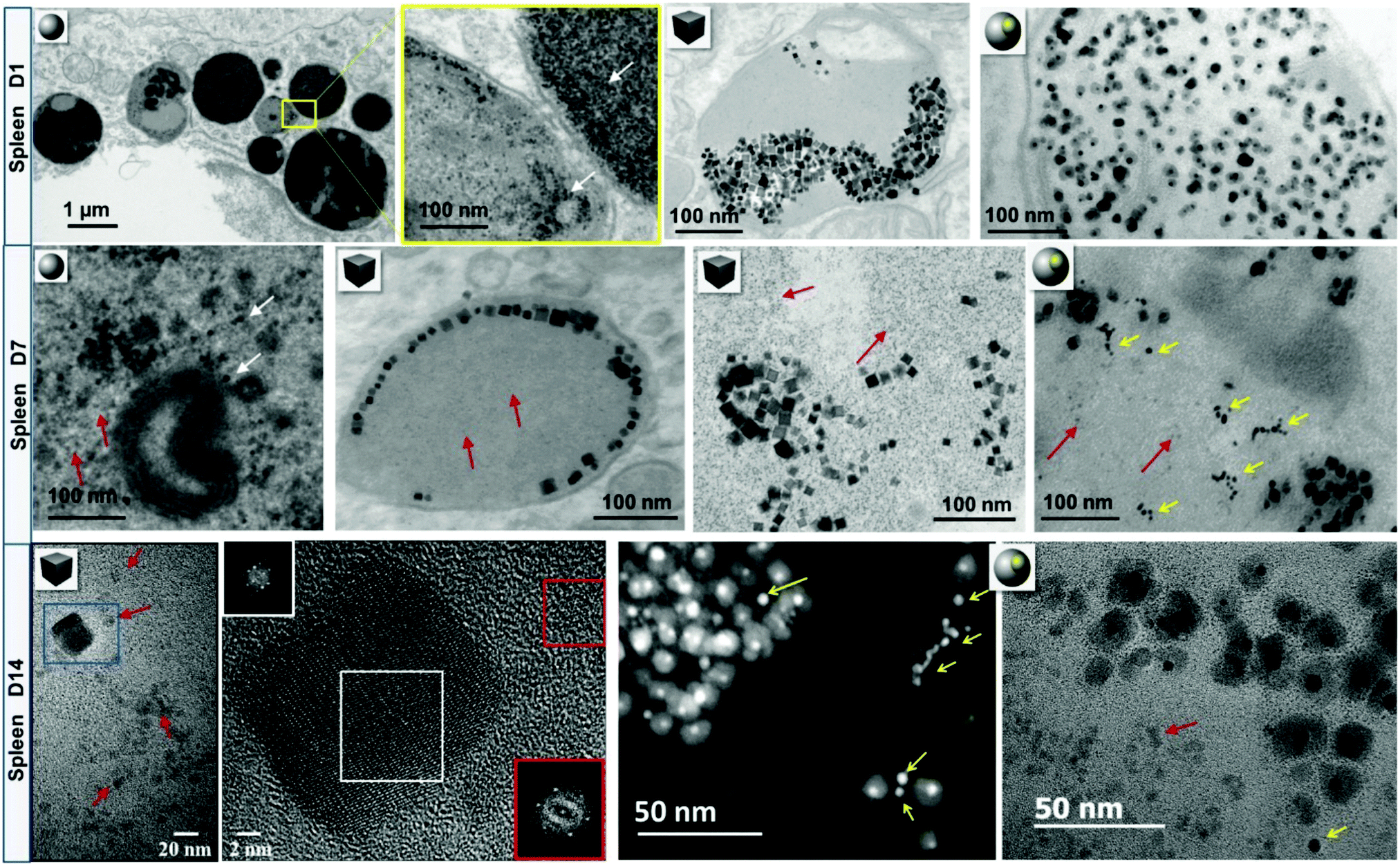 | ||
| Fig. 6 Intralysosomal degradation of FeOx nanospheres, nanocubes and gold/FeOx heterostructures in murine splenic macrophages. At day 1 (D1) post-injection, FeOx NPs are concentrated within lysosomes of macrophages. After one week (D7), a fair share of NPs arranges on the margins of lysosomes or appears to be more isolated. Red arrows point to NP-adjacent to monodisperse iron-rich ferritin proteins. Some parts of gold/FeOx heterostructures have been locally degraded leaving gold remnants (yellow arrows). Scanning transmission electron microscopy (STEM) – high-angle annular dark field (HAADF) and elemental nanoanalysis by STEM-EDX confirm the partial or total disappearance of iron oxidesurrounding the resilient gold core. The Fourier transformation (FT) of the degraded and resilient nanocubes at day 14 (D14, white square on the high resolution TEM micrograph) shows that nanocubes maintain their initial lattice structure (spinel inverse or vacancy-ordered γ-Fe2O3 structures). The FT of the ferritin protein core (red square) shows a hematite structure, suggesting local transfer of iron from degraded NPs to the storage protein. An image adapted from a previously published work.50,110,163 | ||
An advanced NP design could allow for a new generation of inorganic NPs with controlled degradability of the inorganic cores. For screening the factors that impact the lifecycle of NPs in the body, one could track the aging of single cores in a medium that mimics some features of the lysosomal environment, i.e., acidic pH (4.7), and the presence of iron chelators,109,168cf.Fig. 7. Consistent with in vivo observations, the surface coating raises a more or less efficient shield to the effect of the microenvironment and clearly governs the kinetics of FeOx NP dissolution. At the single NP level, the areas of FeOx NPs with less dense polymer coverage (e.g., the vertex of nanocubes) are the most prone to degradation,163cf.Fig. 8. In addition to the above mentioned argument of the increased surface-to-volume ratio present at edges, the degradation may be sterically facilitated at these positions. As observed in vivo for gold/iron oxide dimers, the hydrophilic PEG coating is less effective in retarding the FeOx NP degradation than a double chain amphiphilic polymer.50 In the case of Ag NPs, this effect has not been observed in the same way.107
 | ||
| Fig. 7 Progressive erosion of different FeOx NPs in lysosome-like medium. An image adopted from a previously published work.50,163,169 | ||
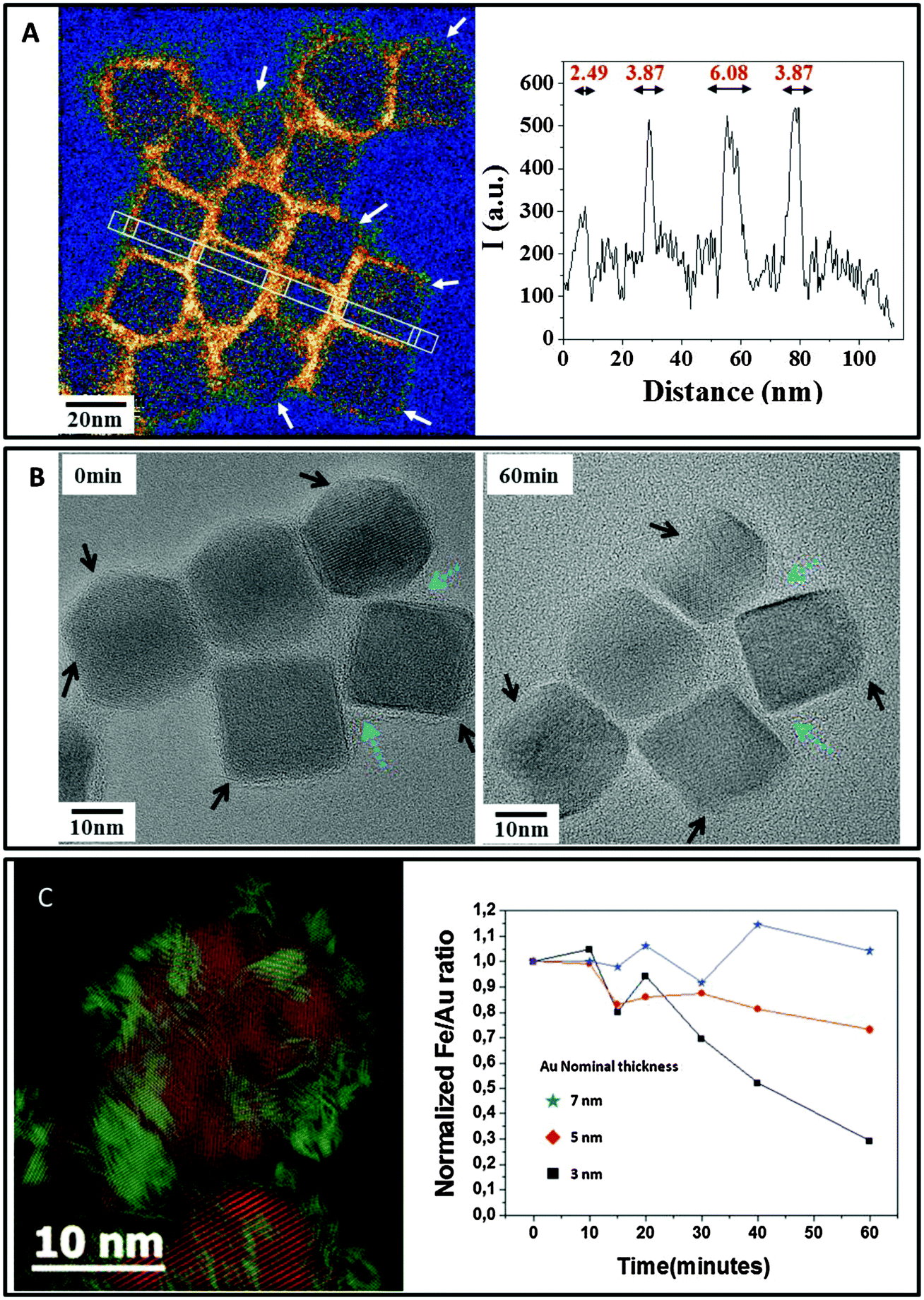 | ||
| Fig. 8 Protective role of coating on NP degradation. (A) Carbon mapping obtained by energy filtered TEM (EFTEM) evidences the uneven coverage of the amphiphilic polymer layer surrounding the cubes. White arrows point to polymer-poor regions where the degradation begins. An image adapted from Lartigue et al.163 (B) Step-by-step degradation of nanocubes showing faster degradation in zones with poor polymer coating (black arrows) and the protective effect of dense coating areas (blue arrows). (C) The coverage of multicore FeOx NPs with a 3 nm gold layer (iron featured in red and gold in green on the Bragg filtered high-resolution micrograph on the left) protects the NPs from degradation. The EDX quantitative measurement of the Fe/Au ratio shows that the thickest gold layer efficiently delays NP degradation and iron release in lysosome-like medium. | ||
Concerning NP geometry, the NP architecture also appears to be a major factor that impacts the NP bio-persistence. Multicore flower-shaped FeOx NPs, formed by the coalescence of magnetically oriented iron oxide seeds, rapidly disintegrate in lysosome-like medium, losing the outstanding properties that they had owing to their cooperative structure.169 Importantly, the junctions in the multicore structures are the most vulnerable sites. Apart from organic coatings, the association of different materials allows for modulating the biopersistence of NPs. For example, the disintegration of multicore FeOx nanoflowers is more or less delayed when they are covered by a layer of gold, depending on the thickness and porosity of the gold shell.169 In a general manner, the degradation process of inorganic cores is a step-by-step corrosion governed by surface reaction mechanisms. Therefore, the efficiency of organic or inorganic engineered coatings relies on their ability to prevent the access of the cellular medium to the core surfaces. In vivo, the 5 nm gold seeds associated with iron oxide spheres persisted much longer than the iron oxide crystals in splenic and hepatic macrophages, but also showed reorganization (as chains and assemblies) as well as degradation into smaller structures one year after injection, once the iron oxide part had been dissolved, cf.Fig. 6.50 Importantly, the size diminution of poorly reactive NPs could enable size-dependent elimination processes such as renal clearance, which could not occur in the case of originally injected NPs. In addition, the variation in the state of aggregation of the NPs in the lysosomes also plays an important role in the possibility of degradation and clearance. The way by which NPs could be excreted from macrophages or translocated and cleared in other organs is another issue. It has been shown recently that macrophages, endothelial cells or mesenchymal stem cells that have first internalized FeOx NPs170,171 Au NPs, QDs,172 or CNTs,173 can expulse NPs in the extracellular medium within microvesicles when the cells are stressed by starvation. Microvesicles are constitutively released by virtually all cell types in body fluids and are considered as potent vectors of intercellular communication in vivo. Such vesicles can spread NPs across the body and transfer these nanomaterials to distal cells. This propagation process, mediated by underestimated vectors of NP dissemination, additionally increases the importance of the in vivo fate of NPs. Despite the non-ambiguous local degradation processes of NPs, it must be noted that a few resilient intact NPs could be observed even one year after injection, regardless of the NP nature, even in the case of highly biodegradable iron oxide. As observed in vitro during single NP tracking, evidence shows that the degradation process is a non-linear and uneven process, which integrally dissolves some NPs, while other particles remain unmodified. The exact mechanisms of degradation, enzymatic attack and involvement of cell metabolism in such a process still remain open questions. Ideally the design of complex nanostructures should help modulating both the time frame of NP activity and the duration of degradation/excretion processes. Interestingly, the recent advances of TEM in liquid environments could also help contemplating nanomaterials at the nanoscale within biological environments, with the possibility of observing the interaction between NPs and wet-cell cultures in situ with unprecedented resolution.174,175 In addition, TEM in liquid media opens many avenues for studying oxidative transformations of NMs, by directly observing the effects of reactive-oxygen-species (ROS) created by the electron beam by radiolysis processes, on the atomic structure of NMs. As oxidative stress plays a critical role in the cellular processing of NMs,176 such dynamic nanoscale investigations allow recapitulating the ROS-induced aging of NMs in cellular media. For example, the in situ monitoring of CNT degradation induced by hydroxyl radicals provided a mechanistic understanding of the stigmata of degradation observed on nanotubes after aging into macrophages.177
The examples listed in this review clearly demonstrate that inorganic NP cores can be degraded in vivo. While this conclusion is commonly reached in the literature for different materials,105 such as Ag, FeOx, ZnO, the discussion about degradation of other materials, such as Au, has just begun. While some cores degrade, the exact mechanisms of degradation have not been fully understood yet. Can NPs be completely dissolved and how these products are excreted from the body? For NPs made of “inert” materials such as Au, what are the detailed mechanisms of intracellular gold dissolution? What is the role of the PC in core degradation, following the observation that the PC changes over time, in particular after NP internalization in lysosomes,118 and what is the role of intra-lysosomal proteins? In case NP concentrations in one organ are reduced over time, how to distinguish between translocation of intact NPs (with or without coating) from organs where local degradation occurs? If degradation happens, are the byproducts less toxic than the original non-reactive persistent NPs? Does degradation depend on NP concentration, i.e., will lysosome overloaded with NPs lead to impairment or acceleration of the degradative capacity of cells and autophagy? Importantly, corroborating nanoscale information (e.g., by TEM) with tissue level investigations (e.g., by ICP-MS, magnetic and optical techniques) and biological studies (genetic and proteomic techniques) is a cornerstone for addressing the many open questions on the degradation of NP cores.
Conclusions
As interactions of NPs with their environments are dominated by their surface and in this way by their engineered surface coating together with adsorbed biomolecules, the biodistribution and fate of NPs need to be correlated to their physicochemical properties as well as their biocoating. While the physicochemical properties and biocoating of NPs can be measured in different biological fluids, there are only few appropriate techniques to (kinetically) determine NPs in vivo and evaluate their evolution over time through their route in the body. The most drastic change in NP properties may involve in vivo degradation, and in this way the fate of all NP components – the inorganic core, the engineered surface coating, and the adsorbed biological molecules – need to be analyzed. Assessing biodistribution and clearance would involve multiple labeling strategies, in which all different components can be traced and analyzed separately. There is increasing experimental evidence that all of these compounds may degrade in vivo. Thus, the hybrid nature of NPs eventually transforms when they lose their integrity during their voyage through the human body. While detailed extracorporeal NP characterization provides information on the products we put “in” and take “out” of the body, the intracorporeal processes are still shrouded in mystery.Acknowledgements
This work was partly funded by the European Commission (grant FutureNanoNeeds to WJP), the MINECO (MAT2013-48169-R, NanoFATE, to WJP and PdP), BMBF-MRCyte/NanoBEL, Zeiss-ChemBioMed, Stiftung Rheinland-Pfalz (NanoScreen), Peter und Traudl Engelhorn foundation, BIOMATICS (grants to RS, DD), DFG SPP1313 (grants to RS, PN, WJP), the CNRS (Centre National de la Recherche Scientifique, Defi Nano program), the ANR (Agence Nationale de la Recherche) and CGI (Commissariat à l'Investissement d'Avenir) through the LabEx SEAM (Science and Engineering for Advanced Materials and devices; ANR 11 LABX 086, ANR 11 IDEX 05 02) (grants to DA and FG). SA acknowledges the Alexander von Humboldt Foundation for a PostDoc fellowship and NF the Lars Hierta Memorial Foundation.References
- T. J. Webster, Nanomedicine, 2013, 8, 525 CrossRef CAS PubMed.
- M. Reese, Health Matrix Clevel, 2013, 23, 537 Search PubMed.
- D. Docter, S. Strieth, D. Westmeier, O. Hayden, M. Y. Gao, S. K. Knauer and R. H. Stauber, Nanomedicine, 2015, 10, 503 CrossRef CAS PubMed.
- M. I. Setyawati, C. Y. Tay, D. Docter, R. H. Stauber and D. T. Leong, Chem. Soc. Rev., 2015, 44, 8174 RSC.
- D. Docter, D. Westmeier, M. Markiewicz, S. Stolte, S. K. Knauer and R. H. Stauber, Chem. Soc. Rev., 2015, 44, 6094 RSC.
- M. Pautler and S. Brenner, Int. J. Nanomed., 2010, 5, 803 Search PubMed.
- S. E. McNeil, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2009, 1, 264 CrossRef CAS PubMed.
- K. Riehemann, S. W. Schneider, T. A. Luger, B. Godin, M. Ferrari and H. Fuchs, Angew. Chem., Int. Ed., 2009, 48, 872 CrossRef CAS PubMed.
- M. Wahajuddin and S. Arora, Int. J. Nanomed., 2012, 7, 3445 CrossRef PubMed.
- L. Y. Rizzo, B. Theek, G. Storm, F. Kiessling and T. Lammers, Curr. Opin. Biotechnol., 2013, 24, 1159 CrossRef CAS PubMed.
- C. Seeney, J. O. Ojwang, R. D. Weiss and J. Klostergaard, Nanomedicine, 2012, 7, 289 CrossRef CAS PubMed.
- P. Prabhu and V. Patravale, J. Biomed. Nanotechnol., 2012, 8, 859 CrossRef CAS PubMed.
- J. S. Murday, R. W. Siegel, J. Stein and J. F. Wright, Nanomedicine, 2009, 5, 251 CAS.
- M. Ferrari, M. A. Philibert and W. R. Sanhai, Clin. Pharmacol. Ther., 2009, 85, 466 CrossRef CAS PubMed.
- X. M. Qian, X. H. Peng, D. O. Ansari, Q. Yin-Goen, G. Z. Chen, D. M. Shin, L. Yang, A. N. Young, M. D. Wang and S. M. Nie, Nat. Biotechnol., 2008, 26, 83 CrossRef CAS PubMed.
- C. L. Ventola, P T., 2012, 37, 582 Search PubMed.
- Y. Hou, M. Lai, X. Chen, J. Li, Y. Hu, Z. Luo, X. Ding and K. Cai, J. Biomed. Mater. Res., Part A, 2014, 102, 1726 CrossRef PubMed.
- P. d. Pino, J. Biomed. Opt., 2014, 19, 101507 CrossRef PubMed.
- M. Colombo, S. Carregal-Romero, M. F. Casula, L. Gutiérrez, M. P. Morales, I. B. Böhm, J. T. Heverhagen, D. Prosperi and W. J. Parak, Chem. Soc. Rev., 2012, 41, 4306 RSC.
- M. Helou, M. Reisbeck, S. F. Tedde, L. Richter, L. Bar, J. J. Bosch, R. H. Stauber, E. Quandt and O. Hayden, Lab Chip, 2013, 13, 1035 RSC.
- H. M. Ding and Y. Q. Ma, Biomaterials, 2014, 35, 8703 CrossRef CAS PubMed.
- J. T. Dias, M. Moros, P. del Pino, S. Rivera, V. Grazú and J. M. de la Fuente, Angew. Chem., Int. Ed., 2013, 52, 11526–11529 CrossRef CAS PubMed.
- P. del Pino, A. Munoz-Javier, D. Vlaskou, P. Rivera Gil, C. Plank and W. J. Parak, Nano Lett., 2010, 10, 3914 CrossRef CAS PubMed.
- H. W. Child, P. A. Del Pino, J. M. De La Fuente, A. S. Hursthouse, D. Stirling, M. Mullen, G. M. McPhee, C. Nixon, V. Jayawarna and C. C. Berry, ACS Nano, 2011, 5, 7910 CrossRef CAS PubMed.
- R. A. Sperling, P. Rivera Gil, F. Zhang, M. Zanella and W. J. Parak, Chem. Soc. Rev., 2008, 37, 1896 RSC.
- M. Perez-Hernandez, P. Del Pino, S. G. Mitchell, M. Moros, G. Stepien, B. Pelaz, W. J. Parak, E. M. Galvez, J. Pardo and J. M. de la Fuente, ACS Nano, 2015, 9, 52 CrossRef CAS PubMed.
- C. Bao, N. Beziere, P. del Pino, B. Pelaz, G. Estrada, F. Tian, V. Ntziachristos, J. M. de la Fuente and D. Cui, Small, 2013, 9, 68 CrossRef CAS PubMed.
- E. Polo, P. del Pino, B. Pelaz, V. Grazu and J. M. de la Fuente, Chem. Commun., 2013, 49, 3676 RSC.
- A. Ambrosone, P. D. Pino, V. Marchesano, W. J. Parak, J. M. d. l. Fuente and C. Tortiglione, Nanomedicine, 2014, 9, 1913 CrossRef CAS PubMed.
- W. J. Parak, T. Pellegrino and C. Plank, Nanotechnology, 2005, 16, R5 CrossRef PubMed.
- S. Kim, Y. T. Lim, E. G. Soltesz, A. M. D. Grand, J. Lee, A. Nakayama, J. A. Parker, T. Mihaljevic, R. G. Laurence, D. M. Dor, L. H. Cohn, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2004, 22, 93 CrossRef CAS PubMed.
- C. Bao, J. Conde, E. Polo, P. del Pino, M. Moros, P. Baptista, V. Grazu, D. Cui and J. de la Fuente, Nanomedicine, 2014, 9, 2353 CrossRef CAS PubMed.
- A. M. Nystrom and B. Fadeel, J. Controlled Release, 2012, 161, 403 CrossRef PubMed.
- R. Bawa, Curr. Drug Delivery, 2011, 8, 227 CrossRef CAS.
- A. Baun and S. F. Hansen, Nanomedicine, 2008, 3, 605 CrossRef PubMed.
- M. Rahman, M. Z. Ahmad, I. Kazmi, S. Akhter, M. Afzal, G. Gupta and V. R. Sinha, Curr. Drug Discovery Technol., 2012, 9, 319 CrossRef CAS.
- C. L. Ventola, P T., 2012, 37, 631 Search PubMed.
- P. Rivera Gil, D. Jimenez de Aberasturi, V. Wulf, B. Pelaz, P. del Pino, Y. Zhao, J. de la Fuente, I. Ruiz de Larramendi, T. Rojo, X.-J. Liang and W. J. Parak, Acc. Chem. Res., 2013, 46, 743 CrossRef CAS PubMed.
- B. Pelaz, G. Charron, C. Pfeiffer, Y. L. Zhao, J. M. de la Fuente, X. J. Liang, W. J. Parak and P. del Pino, Small, 2013, 9, 1573 CrossRef CAS PubMed.
- W. J. Parak, D. Gerion, T. Pellegrino, D. Zanchet, C. Micheel, S. C. Williams, R. Boudreau, M. A. L. Gros, C. A. Larabell and A. P. Alivisatos, Nanotechnology, 2003, 14, R15 CrossRef CAS.
- B. Fadeel, N. Feliu, C. Vogt, A. M. Abdelmonem and W. J. Parak, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 111 CrossRef CAS PubMed.
- C.-A. J. Lin, T.-Y. Yang, C.-H. Lee, S. H. Huang, R. A. Sperling, M. Zanella, J. K. Li, J.-L. Shen, H.-H. Wang, H.-I. Yeh, W. J. Parak and W. H. Chang, ACS Nano, 2009, 3, 395 CrossRef CAS PubMed.
- L. Shang, N. Azadfar, F. Stockmar, W. Send, V. Trouillet, M. Bruns, D. Gerthsen and G. U. Nienhaus, Small, 2011, 7, 2614 CrossRef CAS PubMed.
- S. Clarke, F. Pinaud, O. Beutel, C. J. You, J. Piehler and M. Dahan, Nano Lett., 2010, 10, 2147 CrossRef CAS PubMed.
- G. Gerion, F. Pinaud, S. C. Williams, W. J. Parak, D. Zanchet, S. Weiss and A. P. Alivisatos, J. Phys. Chem. B, 2001, 105, 8861 CrossRef.
- S. T. Selvan, P. K. Patra, C. Y. Ang and J. Y. Ying, Angew. Chem., Int. Ed., 2007, 46, 2448 CrossRef CAS PubMed.
- F. Zhang, E. Lees, F. Amin, P. Rivera_Gil, F. Yang, P. Mulvaney and W. J. Parak, Small, 2011, 7, 3113 CrossRef CAS PubMed.
- Y. Klapper, P. Maffre, L. Shang, K. N. Ekdahl, B. Nilsson, S. Hettler, M. Dries, D. Gerthsen and G. U. Nienhaus, Nanoscale, 2015, 7, 9980 RSC.
- B. Dubertret, P. Skourides, D. J. Norris, V. Noireaux, A. H. Brivanlou and A. Libchaber, Science, 2002, 298, 1759 CrossRef CAS PubMed.
- J. Kolosnjaj-Tabi, Y. Javed, L. Lartigue, J. Volatron, D. Elgrabli, I. Marangon, G. Pugliese, B. Caron, A. Figuerola, N. Luciani, T. Pellegrino, D. Alloyeau and F. Gazeau, ACS Nano, 2015, 9, 7925 CrossRef CAS PubMed.
- M. Levy, F. Lagarde, V. A. Maraloiu, M. G. Blanchin, F. Gendron, C. Wilhelm and F. Gazeau, Nanotechnology, 2010, 21, 395103 CrossRef PubMed.
- H. W. Yang, M. Y. Hua, H. L. Liu, R. Y. Tsai, C. K. Chuang, P. C. Chu, P. Y. Wu, Y. H. Chang, H. C. Chuang, K. J. Yu and S. T. Pang, ACS Nano, 2012, 6, 1795 CrossRef CAS PubMed.
- Y. Zhao, S. C. Burkert, Y. F. Tang, D. C. Sorescu, A. A. Kapralov, G. V. Shurin, M. R. Shurin, V. E. Kagan and A. Star, J. Am. Chem. Soc., 2015, 137, 675 CrossRef CAS PubMed.
- M. Branca, M. Marciello, D. Ciuculescu-Pradines, M. Respaud, M. D. Morales, R. Serra, M. J. Casanove and C. Amiens, J. Magn. Magn. Mater., 2015, 377, 348 CrossRef CAS.
- Y. Wang and H. C. Gu, Adv. Mater., 2015, 27, 576 CrossRef CAS PubMed.
- J. F. Zeng, L. H. Jing, Y. Hou, M. X. Jiao, R. R. Qiao, Q. J. Jia, C. Y. Liu, F. Fang, H. Lei and M. Y. Gao, Adv. Mater., 2014, 26, 2694 CrossRef CAS PubMed.
- J.-M. Montenegro, V. Grazu, A. Sukhanova, S. Agarwal, J. M. d. l. Fuente, I. Nabiev, A. Greiner and W. J. Parak, Adv. Drug Delivery Rev., 2013, 65, 677–688 CrossRef CAS PubMed.
- F. Zhang, Z. Ali, F. Amin, A. Feltz, M. Oheim and W. J. Parak, ChemPhysChem, 2010, 11, 730 CrossRef CAS PubMed.
- C. Carrillo-Carrion, M. Nazarenus, S. Sánchez Paradinas, S. Carregal-Romero, M. J. Almendral, M. Fuentes, B. Pelaz, P. del Pino, I. Hussain, M. J. D. Clift, B. Rothen-Rutishauser, X.-J. Liang and W. J. Parak, Curr. Opin. Chem. Eng., 2014, 4, 88 CrossRef.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050 CrossRef CAS PubMed.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543 CrossRef CAS PubMed.
- C. D. Walkey and W. C. W. Chan, Chem. Soc. Rev., 2012, 41, 2780 RSC.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Nat. Nanotechnol., 2013, 8, 772 CrossRef CAS PubMed.
- X. R. Xia, N. A. Monteiro-Riviere and J. E. Riviere, Nat. Nanotechnol., 2010, 5, 671 CrossRef CAS PubMed.
- S. Wan, P. M. Kelly, E. Mahon, H. Stockmann, P. M. Rudd, F. Caruso, K. A. Dawson, Y. Yan and M. P. Monopoli, ACS Nano, 2015, 9, 2157 CrossRef CAS PubMed.
- T. L. Moore, L. Rodriguez-Lorenzo, V. Hirsch, S. Balog, D. Urban, C. Jud, B. Rothen-Rutishauser, M. Lattuada and A. Petri-Fink, Chem. Soc. Rev., 2015, 44, 6287 RSC.
- L. Lartigue, C. Wilhelm, J. Servais, C. Factor, A. Dencausse, J. C. Bacri, N. Luciani and F. Gazeau, ACS Nano, 2012, 6, 2665 CrossRef CAS PubMed.
- P. D. Howes, R. Chandrawati and M. M. Stevens, Science, 2014, 346, 53 CrossRef CAS PubMed.
- A. Jedlovszky-Hajdu, F. B. Bombelli, M. P. Monopoli, E. Tombacz and K. A. Dawson, Langmuir, 2012, 28, 14983 CrossRef CAS PubMed.
- M. P. Monopoli, F. B. Bombelli and K. A. Dawson, Nat. Nanotechnol., 2011, 6, 11 CrossRef CAS PubMed.
- P. M. Kelly, C. Aberg, E. Polo, A. O'Connell, J. Cookman, J. Fallon, Z. Krpetic and K. A. Dawson, Nat. Nanotechnol., 2015, 10, 472 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. B. Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525 CrossRef CAS PubMed.
- S. Milani, F. B. Bombelli, A. S. Pitek, K. A. Dawson and J. Radler, ACS Nano, 2012, 6, 2532 CrossRef CAS PubMed.
- D. Walczyk, F. B. Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, J. Am. Chem. Soc., 2010, 132, 5761 CrossRef CAS PubMed.
- I. Lynch, T. Cedervall, M. Lundqvist, C. Cabaleiro-Lago, S. Linse and K. A. Dawson, Adv. Colloid Interface Sci., 2007, 134–135, 167 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, M. Foy, T. Berggad, S. C. Donnelly, G. Cagney, S. Linse and K. A. Dawson, Angew. Chem., Int. Ed., 2007, 46, 5754 CrossRef CAS PubMed.
- C. Bantz, O. Koshkina, T. Lang, H. J. Galla, C. J. Kirkpatrick, R. H. Stauber and M. Maskos, Beilstein J. Nanotechnol., 2014, 5, 1774 CrossRef CAS PubMed.
- D. Bargheer, J. Nielsen, G. Gebel, M. Heine, S. C. Salmen, R. Stauber, H. Weller, J. Heeren and P. Nielsen, Beilstein J. Nanotechnol., 2015, 6, 36 CrossRef CAS PubMed.
- L. Treuel, D. Docter, M. Maskos and R. H. Stauber, Beilstein J. Nanotechnol., 2015, 6, 857 CrossRef CAS PubMed.
- J. S. Gebauer, M. Malissek, S. Simon, S. K. Knauer, M. Maskos, R. H. Stauber, W. Peukert and L. Treuel, Langmuir, 2012, 28, 9673 CrossRef CAS PubMed.
- H. Hillaireu and P. Couvreur, Cell. Mol. Life Sci., 2009, 66, 2873 CrossRef PubMed.
- M. Nazarenus, Q. Zhang, M. G. Soliman, P. del Pino, B. Pelaz, S. Carregal_Romero, J. Rejman, B. Rothen-Ruthishauser, M. J. D. Clift, R. Zellner, G. U. Nienhaus, J. B. Delehanty, I. L. Medintz and W. J. Parak, Beilstein J. Nanotechnol., 2014, 5, 1477 CrossRef PubMed.
- S. W. Jones, R. A. Roberts, G. R. Robbins, J. L. Perry, M. P. Kai, K. Chen, T. Bo, M. E. Napier, J. P. Y. Ting, J. M. DeSimone and J. E. Bear, J. Clin. Invest., 2013, 123, 3061 CAS.
- B. Freund, U. I. Tromsdorf, O. T. Bruns, M. Heine, A. Giemsa, A. Bartelt, S. C. Salmen, N. Raabe, J. Heeren, H. Ittrich, R. Reimer, H. Hohenberg, U. Schumacher, H. Weller and P. Nielsen, ACS Nano, 2012, 6, 7318 CrossRef CAS PubMed.
- A. Moore, E. Marecos, J. Alexei Bogdanov and R. Weissleder, Radiology, 2000, 214, 568 CrossRef CAS PubMed.
- D. Bargheer, A. Giemsa, B. Freund, M. Heine, C. Waurich, G. Stachowski, S. Hickey, A. Eychmüller, J. Heeren and P. Nielsen, Beilstein J. Nanotechnol., 2015, 6, 111 CrossRef CAS PubMed.
- A. Carambia, B. Freund, D. Schwinge, O. T. Bruns, S. C. Salmen, H. Ittrich, R. Reimer, M. Heine, S. Huber, C. Waurisch, A. Eychmuller, D. C. Wraith, T. Korn, P. Nielsen, H. Weller, C. Schramm, S. Luth, A. W. Lohse, J. Heeren and J. Herkel, J. Hepatol., 2015, 62, 1349 CrossRef CAS PubMed.
- M. Rehberg, C. F. Leite, K. Mildner, J. Horstkotte, D. Zeuschner and F. Krombach, ACS Nano, 2012, 6, 1370 CrossRef CAS PubMed.
- J. Kolosnjaj-Tabi, R. Di Corato, L. Lartigue, I. Marangon, P. Guardia, A. K. A. Silva, N. Luciani, O. Clement, P. Flaud, J. V. Singh, P. Decuzzi, T. Pellegrino, C. Wilhelm and F. Gazeau, ACS Nano, 2014, 8, 4268 CrossRef CAS PubMed.
- C. Jung, M. G. Kaul, O. T. Bruns, T. Ducic, B. Freund, M. Heine, R. Reimer, A. Meents, S. C. Salmen, H. Weller, P. Nielsen, G. Adam, J. Heeren and H. Ittrich, Circ. Cardiovasc. Imaging, 2014, 7, 303 CrossRef PubMed.
- M. Lipka, M. Semmler-Behnke, R. A. Sperling, A. Wenk, S. Takenaka, C. Schleh, T. Kissel, W. J. Parak and W. G. Kreyling, Biomaterials, 2010, 31, 6574 CrossRef PubMed.
- G. J. Nohynek, J. Lademann, C. Ribaud and M. S. Roberts, Crit. Rev. Toxicol., 2007, 37, 251 CrossRef CAS PubMed.
- C. Schleh, M. Semmler-Behnke, J. Lipka, A. Wenk, S. Hirn, M. Schaeffler, G. Schmid, U. Simon and W. G. Kreyling, Nanotoxicology, 2012, 6, 36 CrossRef CAS PubMed.
- A. Frey, M. R. Neutra and F. A. Robey, Bioconjugate Chem., 1997, 8, 424 CrossRef CAS PubMed.
- F. Rancan, Q. Gao, C. Graf, S. Troppens, S. Hadam, S. Hackbarth, C. Kembuan, U. Blume-Peytavi, E. Ruehl, J. Lademann and A. Vogt, ACS Nano, 2012, 6, 6829 CrossRef CAS PubMed.
- A. Vogt, F. Rancan, S. Ahlberg, B. Nazemi, C. S. Choe, M. E. Darvin, S. Hadam, U. Blume-Peytavi, K. Loza, J. Diendorf, M. Epple, C. Graf, E. Ruehl, M. C. Meinke and J. Lademann, Beilstein J. Nanotechnol., 2014, 5, 2363 CrossRef CAS PubMed.
- M. Semmler-Behnke, W. G. Kreyling, J. Lipka, S. Fertsch, A. Wenk, S. Takenaka, G. Schmid and W. Brandau, Small, 2008, 4, 2108 CrossRef CAS PubMed.
- E. Sadauskas, N. R. Jacobsen, G. Danscher, M. Stoltenberg, U. Vogel, A. Larsen, W. Kreyling and H. Wallin, Chem. Cent. J., 2009, 3, 16 CrossRef PubMed.
- H. Sinnecker, T. Krause, S. Koelling, I. Lautenschlager and A. Frey, Beilstein J. Nanotechnol., 2014, 5, 2092 CrossRef CAS PubMed.
- M. Levy, C. Wilhelm, N. Luciani, V. Deveaux, F. Gendron, A. Luciani, M. Devaud and F. Gazeau, Nanoscale, 2011, 3, 4402 RSC.
- R. Di Corato, A. Espinosa, L. Lartigue, M. Tharaud, S. Chat, T. Pellegrino, C. Menager, F. Gazeau and C. Wilhelm, Biomaterials, 2014, 35, 6400 CrossRef CAS PubMed.
- M. Levy, C. Wilhelm, M. Devaud, P. Levitz and F. Gazeau, Contrast Media Mol. Imaging, 2012, 7, 373 CrossRef CAS PubMed.
- S. J. Soenen, W. J. Parak, J. Rejman and B. Manshian, Chem. Rev., 2015, 115, 2109 CrossRef CAS PubMed.
- S. Kittler, C. Greulich, J. Diendorf, M. Koller and M. Epple, Chem. Mater., 2010, 22, 4548 CrossRef CAS.
- E. Caballero-Díaz, C. Pfeiffer, L. Kastl, P. Rivera-Gil, B. Simonet, M. Valcárcel, J. Jiménez-Lamana, F. Laborda and W. J. Parak, Part. Part. Syst. Charact., 2013, 30, 1079 CrossRef.
- K. Loza, J. Diendorf, C. Greulich, L. Ruiz-Gonzales, J. M. Gonzalez-Calbet, M. Vallet-Regi, M. Koeller and M. Epple, J. Mater. Chem. B, 2014, 2, 1634 RSC.
- M. Lévy, F. Lagarde, V. A. Maraloiu, M. G. Blanchin, F. Gendron, C. Wilhelm and F. Gazeau, Nanotechnology, 2010, 21, 395103 CrossRef PubMed.
- M. Levy, N. Luciani, D. Alloyeau, D. Elgrabli, V. Deveaux, C. Pechoux, S. Chat, G. Wang, N. Vats, F. Gendron, C. Factor, S. Lotersztajn, A. Luciani, C. Wilhelm and F. Gazeau, Biomaterials, 2011, 32, 3988 CrossRef CAS PubMed.
- C. Kirchner, T. Liedl, S. Kudera, T. Pellegrino, A. Muñoz Javier, H. E. Gaub, S. Stölzle, N. Fertig and W. J. Parak, Nano Lett., 2005, 5, 331 CrossRef CAS PubMed.
- E. Mahon, D. R. Hristov and K. A. Dawson, Chem. Commun., 2012, 48, 7970 RSC.
- V. E. Kagan, N. V. Konduru, W. H. Feng, B. L. Allen, J. Conroy, Y. Volkov, I. I. Vlasova, N. A. Belikova, N. Yanamala, A. Kapralov, Y. Y. Tyurina, J. W. Shi, E. R. Kisin, A. R. Murray, J. Franks, D. Stolz, P. P. Gou, J. Klein-Seetharaman, B. Fadeel, A. Star and A. A. Shvedova, Nat. Nanotechnol., 2010, 5, 354 CrossRef CAS PubMed.
- M.-F. Yu, O. Lourie, M. J. Dyer, K. Moloni, T. F. Kelly and R. S. Ruoff, Science, 2000, 287, 637 CrossRef CAS PubMed.
- V. Sée, P. Free, Y. Cesbron, P. Nativo, U. Shaheen, D. Rigden, D. G. Spiller, D. G. Fernig, M. R. H. White, I. A. Prior, M. Brust, B. Lounis and R. Lévy, ACS Nano, 2009, 3, 2461 CrossRef PubMed.
- O. Lunov, T. Syrovets, C. Rocker, K. Tron, G. Nienhaus, V. Rasche, V. Mailander, K. Landfester and T. Simmet, Biomaterials, 2010, 31, 9015 CrossRef CAS PubMed.
- K. Bose, M. Koch, C. Cavelius, A. K. Kiemer and A. Kraegeloh, Part. Part. Syst. Charact., 2014, 31, 439 CrossRef.
- M. Chanana, P. Rivera Gil, M. A. Correa-Duarte, W. J. Parak and L. M. Liz-Marzán, Angew. Chem., Int. Ed., 2013, 52, 4179 CrossRef CAS PubMed.
- E. Casals, T. Pfaller, A. Duschl, G. J. Oostingh and V. F. Puntes, ACS Nano, 2010, 4, 3623 CrossRef CAS PubMed.
- Z. Ali, A. Z. Abbasi, F. Zhang, P. Arosio, A. Lascialfari, M. F. Casula, A. Wenk, W. Kreyling, R. Plapper, M. Seidel, R. Niessner, J. Knoll, A. Seubert and W. J. Parak, Anal. Chem., 2011, 83, 2877 CrossRef CAS PubMed.
- W. G. Kreyling, S. Hirn, W. Müller, C. Schleh, A. Wenk, G. Celik, J. Lipka, M. Schäffler, N. Haberl, B. D. Johnston, R. Sperling, G. Schmid, U. Simon, W. J. Parak and M. Semmler-Behnke, ACS Nano, 2014, 8, 222 CrossRef CAS PubMed.
- J. R. McCarthy and R. Weissleder, Adv. Drug Delivery Rev., 2008, 60, 1241 CrossRef CAS PubMed.
- M. G. Harisinghani, K. S. Jhaveri, R. Weissleder, W. Schima, S. Saini, P. F. Hahn and P. R. Mueller, Clin. Radiol., 2001, 56, 714 CrossRef CAS PubMed.
- A. Salvati, C. Aberg, T. dos Santos, J. Varela, P. Pinto, I. Lynch and K. A. Dawson, Nanomedicine, 2011, 7, 818 CAS.
- W. G. Kreyling, A. M. Abdelmonem, Z. Ali, F. Alves, M. Geiser, N. Haberl, R. Hartmann, S. Hirn, D. J. de Aberasturi, K. Kantner, G. Khadem-Saba, J. M. Montenegro, J. Rejman, T. Rojo, I. R. de Larramendi, R. Ufartes, A. Wenk and W. J. Parak, Nat. Nanotechnol., 2015, 10, 619 CrossRef CAS PubMed.
- C. D. Walkey, J. B. Olsen, F. Y. Song, R. Liu, H. B. Guo, D. W. H. Olsen, Y. Cohen, A. Emili and W. C. W. Chan, ACS Nano, 2014, 8, 2439 CrossRef CAS PubMed.
- P. P. Adiseshaiah, J. B. Hall and S. E. McNeil, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2010, 2, 99 CrossRef CAS PubMed.
- J. Leszczynski, Nat. Nanotechnol., 2010, 5, 633 CrossRef CAS PubMed.
- M. A. Dobrovolskaia, B. W. Neun, S. Man, X. Ye, M. Hansen, A. K. Patri, R. M. Crist and S. E. McNeil, Nanomedicine, 2014, 10, 1453 CAS.
- A. Lesniak, F. Fenaroli, M. R. Monopoli, C. Aberg, K. A. Dawson and A. Salvati, ACS Nano, 2012, 6, 5845 CrossRef CAS PubMed.
- C. C. Fleischer and C. K. Payne, Acc. Chem. Res., 2014, 47, 2651 CrossRef CAS PubMed.
- C. Y. Tay, M. I. Setyawati, J. Xie, W. J. Parak and D. T. Leong, Adv. Funct. Mater., 2014, 24, 5936–5955 CrossRef CAS.
- U. Sakulkhu, M. Mahmoudi, L. Maurizi, J. Salaklang and H. Hofmann, Sci. Rep., 2014, 4, 5020 CAS.
- C. Sacchetti, K. Motamedchaboki, A. Magrini, G. Palmieri, M. Mattei, S. Bernardini, N. Rosato, N. Bottini and M. Bottini, ACS Nano, 2013, 7, 1974 CrossRef CAS PubMed.
- A. Nel, Y. L. Zhao and L. Madler, Acc. Chem. Res., 2013, 46, 605 CrossRef CAS PubMed.
- F. J. Wang, L. Yu, M. P. Monopoli, P. Sandin, E. Mahon, A. Salvati and K. A. Dawson, Nanomedicine, 2013, 9, 1159 CAS.
- G. Caracciolo, D. Pozzi, A. L. Capriotti, C. Cavaliere, P. Foglia, H. Amenitsch and A. Lagana, Langmuir, 2011, 27, 15048 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, T. Cedervall, T. Berggard, M. B. Flanagan, I. Lynch, G. Elia and K. Dawson, ACS Nano, 2011, 5, 7503 CrossRef CAS PubMed.
- A. Syed and W. C. Chan, Cancer Treat. Res., 2015, 166, 227 Search PubMed.
- R. Liu, W. Jiang, C. D. Walkey, W. C. W. Chan and Y. Cohen, Nanoscale, 2015, 7, 9664 RSC.
- S. J. Lin, X. Wang, Z. X. Ji, C. H. Chang, Y. Dong, H. Meng, Y. P. Liao, M. Y. Wang, T. B. Song, S. Kohan, T. Xia, J. I. Zink, S. Lin and A. E. Nel, ACS Nano, 2014, 8, 4450 CrossRef CAS PubMed.
- R. Li, Z. Ji, C. H. Chang, D. R. Dunphy, X. Cai, H. Meng, H. Zhang, B. Sun, X. Wang, J. Dong, S. Lin, M. Wang, Y. P. Liao, C. J. Brinker, A. Nel and T. Xia, ACS Nano, 2014, 8, 1771 CrossRef CAS PubMed.
- N. Konduru, J. Keller, L. Ma-Hock, S. Gröters, R. Landsiedel, T. C. Donaghey, J. D. Brain, W. Wohlleben and R. M. Molina, Part. Fibre Toxicol., 2014, 11, 55 CrossRef PubMed.
- B. Gilbert, S. C. Fakra, T. Xia, S. Pokhrel, L. Madler and A. E. Nel, ACS Nano, 2012, 6, 4921 CrossRef CAS PubMed.
- X. Wang, Z. X. Ji, C. H. Chang, H. Y. Zhang, M. Y. Wang, Y. P. Liao, S. J. Lin, H. Meng, R. B. Li, B. B. Sun, L. V. Winkle, K. E. Pinkerton, J. I. Zink, T. Xia and A. E. Nel, Small, 2014, 10, 385 CrossRef CAS PubMed.
- H. Y. Zhang, S. Pokhrel, Z. X. Ji, H. Meng, X. Wang, S. J. Lin, C. H. Chang, L. J. Li, R. B. Li, B. B. Sun, M. Y. Wang, Y. P. Liao, R. Liu, T. Xia, L. Madler and A. E. Nel, J. Am. Chem. Soc., 2014, 136, 6406 CrossRef CAS PubMed.
- F. Bertoli, G. L. Davies, M. P. Monopoli, M. Moloney, Y. K. Gun'ko, A. Salvati and K. A. Dawson, Small, 2014, 10, 3307 CrossRef CAS PubMed.
- M. Mahmoudi, A. M. Abdelmonem, S. Behzadi, J. H. Clement, S. Dutz, M. R. Ejtehadi, R. Hartmann, K. Kantner, U. Linne, P. Maffre, S. Metzler, M. K. Moghadam, C. Pfeiffer, M. Rezaei, P. Ruiz-Lozano, V. Serpooshan, M. A. Shokrgozar, G. U. Nienhaus and W. J. Parak, ACS Nano, 2013, 7, 6555 CrossRef CAS PubMed.
- A. K. Murthy, R. J. Stover, W. G. Hardin, R. Schramm, G. D. Nie, S. Gourisankar, T. M. Truskett, K. V. Sokolov and K. P. Johnston, J. Am. Chem. Soc., 2013, 135, 7799 CrossRef CAS PubMed.
- K. Natte, J. F. Friedrich, S. Wohlrab, J. Lutzki, R. von Klitzing, W. Osterle and G. Orts-Gil, Colloids Surf., B, 2013, 104, 213 CrossRef CAS PubMed.
- D. Pozzi, V. Colapicchioni, G. Caracciolo, S. Piovesana, A. L. Capriotti, S. Palchetti, S. De Grossi, A. Riccioli, H. Amenitsch and A. Lagana, Nanoscale, 2014, 6, 2782 RSC.
- B. Pelaz, P. Del Pino, P. Maffre, R. Hartmann, M. Gallego, S. Rivera-Fernandez, J. M. de la Fuente, G. U. Nienhaus and W. J. Parak, ACS Nano, 2015, 9, 6996 CrossRef CAS PubMed.
- P. del_Pino, B. Pelaz, Q. Zhang, P. Maffre, G. U. Nienhaus and W. J. Parak, Mater. Horiz., 2014, 1, 301 RSC.
- C. Röcker, M. Pötzl, F. Zhang, W. J. Parak and G. U. Nienhaus, Nat. Nanotechnol., 2009, 4, 577 CrossRef PubMed.
- D. Hühn, K. Kantner, C. Geidel, S. Brandholt, I. De Cock, S. J. H. Soenen, P. Rivera Gil, J.-M. Montenegro, K. Braeckmans, K. Müllen, G. U. Nienhaus, M. Klapper and W. J. Parak, ACS Nano, 2013, 7, 3253 CrossRef PubMed.
- R. A. Alvarez-Puebla, A. Agarwal, P. Manna, B. P. Khanal, P. Aldeanueva-Potel, E. Carbo-Argibay, N. Pazos-Perez, L. Vigderman, E. R. Zubarev, N. A. Kotov and L. M. Liz-Marzan, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8157 CrossRef CAS PubMed.
- L. Guerrini, R. Arenal, B. Mannini, F. Chiti, R. Pini, P. Matteini and R. A. Alvarez-Puebla, ACS Appl. Mater. Interfaces, 2015, 7, 9420 CAS.
- L. Guerrini, E. Pazos, C. Penas, M. E. Vazquez, J. L. Mascarenas and R. A. Alvarez-Puebla, J. Am. Chem. Soc., 2013, 135, 10314 CrossRef CAS PubMed.
- D. Richter and P. G. Croft, Biochem. J., 1942, 36, 746 CrossRef CAS PubMed.
- M. Paulsson, C. Krag, T. Frederiksen and M. Brandbyge, Nano Lett., 2009, 9, 117 CrossRef CAS PubMed.
- D. Kruger, R. Rousseau, H. Fuchs and D. Marx, Angew. Chem., Int. Ed., 2003, 42, 2251 CrossRef CAS PubMed.
- R. Mejias, L. Gutierrez, G. Salas, S. Perez-Yague, T. M. Zotes, F. J. Lazaro, M. P. Morales and D. F. Barber, J. Controlled Release, 2013, 171, 225 CrossRef CAS PubMed.
- L. Lartigue, D. Alloyeau, J. Kolosnjaj-Tabi, Y. Javed, P. Guardia, A. Riedinger, C. Pechoux, T. Pellegrino, C. Wilhelm and F. Gazeaut, ACS Nano, 2013, 7, 3939 CrossRef CAS PubMed.
- R. Weissleder, D. Stark, B. L. Engelstad, B. R. Bacon, C. C. Compton, D. L. White, P. Jacobs and J. Lewis, Am. J. Roentgenol., 1989, 152, 167 CrossRef CAS PubMed.
- S. Majumdar, S. S. Zoghbi and J. C. Gore, Invest. Radiol., 1990, 25, 771 CrossRef CAS PubMed.
- M. Heine, A. Bartelt, O. T. Bruns, D. Bargheer, A. Giemsa, B. Freund, L. Scheja, C. Waurisch, A. Eychmuller, R. Reimer, H. Weller, P. Nielsen and J. Heeren, Beilstein J. Nanotechnol., 2014, 5, 1432 CrossRef PubMed.
- C. Beaumont and C. Delaby, Semin. Hematol., 2009, 46, 328 CrossRef CAS PubMed.
- A. S. Arbab, L. B. Wilson, P. Ashari, E. K. Jordan, B. K. Lewis and J. A. Frank, NMR Biomed., 2005, 18, 383 CrossRef CAS PubMed.
- Y. Javed, L. Lartigue, P. Hugounenq, V. Quoc Lam, Y. Gossuin, R. Bazzi, C. Wilhelm, C. Ricolleau, F. Gazeau and D. Alloyeau, Small, 2014, 10, 3325 CrossRef CAS PubMed.
- N. Luciani, C. Wilhelm and F. Gazeau, Biomaterials, 2010, 31, 7061 CrossRef CAS PubMed.
- A. K. Silva, C. Wilhelm, J. Kolosnjaj-Tabi, N. Luciani and F. Gazeau, Pharm. Res., 2012, 29, 1392 CrossRef CAS PubMed.
- A. K. A. Silva, R. Di Corato, T. Pellegrino, S. Chat, G. Pugliese, N. Luciani, F. Gazeau and C. Wilhelm, Nanoscale, 2013, 5, 11374 RSC.
- I. Marangon, N. Boggetto, C. Menard-Moyon, E. Venturelli, M. L. Beoutis, C. Pechoux, N. Luciani, C. Wilhelm, A. Bianco and F. Gazeau, Nano Lett., 2012, 12, 4830 CrossRef CAS PubMed.
- N. de Jonge, D. B. Peckys, G. J. Kremers and D. W. Piston, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2159 CrossRef CAS PubMed.
- E. S. Pohlmann, K. Patel, S. J. Guo, M. J. Dukes, Z. Sheng and D. F. Kelly, Nano Lett., 2015, 15, 2329 CrossRef CAS PubMed.
- V. E. Kagan, A. A. Kapralov, C. M. St Croix, S. C. Watkins, E. R. Kisin, G. P. Kotchey, K. Balasubramanian, I. I. Vlasova, J. Yu, K. Kim, W. Seo, R. K. Mallampalli, A. Star and A. A. Shvedova, ACS Nano, 2014, 8, 5610 CrossRef CAS PubMed.
- D. Elgrabli, W. Dachraoui, C. Ménard-Moyon, X. J. Liu, D. Bégin, S. Bégin-Colin, A. Bianco, F. Gazeau and D. Alloyeau, ACS Nano, 2015, 9, 10113 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |