
Oxidative nucleophilic alkoxylation of nitrobenzenes†
Viktor V.
Khutorianskyi
 ,
Norbert
Baris
and
Petr
Beier
*
,
Norbert
Baris
and
Petr
Beier
*
Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo náměstí 2, 166 10 Prague 6, Czech Republic. E-mail: beier@uochb.cas.cz
First published on 5th November 2020
Abstract
Oxidative nucleophilic aromatic substitution of hydrogen in nitroaromatics with potassium alkoxides derived from primary, secondary or tertiary alcohols in the presence of oxygen was achieved. Mechanistic investigations suggest the formation of a deprotonated σH adduct. The products showed good reactivity in nucleophilic aromatic substitution of the nitro group with oxygen or nitrogen nucleophiles to access selectively substituted aromatic compounds.
Introduction
Selectively substituted nitroaromatics are key intermediates in organic synthesis and important building blocks for the preparation of valuable chemicals such as pigments, agrochemicals, pharmaceuticals, explosives and electronic materials.1,2 Their synthesis is usually based on electrophilic aromatic nitration3–5 or nucleophilic aromatic substitution of nitro-substituted aryl halides.6 However, these methodologies are often limited by the availability of starting aromatics and aryl halides, by substrate compatibility with strongly acidic or basic reaction conditions and by reaction selectivity. A method which does not rely on aryl halides is nucleophilic displacement of aromatic hydrogens in nitroaromatics. It is well known that nitrobenzenes are prone to attack by strong nucleophiles at activated positions (typically ortho and para positions relative to the nitro group). However, the reversibly formed σH adduct cannot easily eliminate the hydride ion to complete the substitution7,8 (except for rare cases such as in the Chichibabin reaction).9 Several strategies have been invented to overcome this problem.10,11 In vicarious nucleophilic substitution (VNS), pioneered by Makosza and co-workers, the nucleophile contains a leaving group, which is able to depart together with the hydrogen atom bonded to the attacked aromatic carbon leading to substitution of aromatic hydrogen under mild conditions.12 In cine- or tele-substitutions, the auxiliary eliminated group is present in the substrate.13 In oxidative nucleophilic substitution of hydrogen (ONSH), the anionic σH adduct is oxidized to radical intermediates (using an internal or external oxidant, or electrochemically) facilitating hydrogen atom transfer.14–16 This process was used for amination,17–19 alkylation,20–23 or alkynylation24 of nitrobenzenes. The use of other nucleophiles including oxygen nucleophiles such as alkoxides is very limited. Guthrie studied the reaction of nitrobenzene with t-BuOK in THF and observed the formation of ortho- and para–tert-butoxynitrobenzene in low conversion.25 The study was focused on investigation of the reaction mechanism and kinetics. In the absence of an external oxidant, a mechanism involving dianion A was suggested (Scheme 1A).25 Later, Suzuki studied reactions of 1,3-dinitrobenzenes and nitronaphthalenes with potassium methoxide in dipolar aprotic DMI affording 2,4-dinitroanisoles in modest to good yields (Scheme 1B). Again, in the absence of any external oxidant, the starting 1,3-dinitrobenzene derivative acted as an oxidant of the σH adduct or the dianion intermediate.26 On the other hand, some nitrobenzenes are known to undergo nucleophilic substitution of the nitro group using excess of alkali metal alkoxides in the presence of a polar aprotic solvent at ambient temperature (Scheme 2, pathway a).27,28 However, in these reactions the primary attack of the alkoxide still occurs on the carbon atom at activated positions (ortho or para to the nitro group) and under thermodynamic conditions (excess of alkoxide, longer reaction time, and room temperature) an ipso attack takes place, followed by the irreversible departure of the nitrite leaving group. We envisioned that a more efficient and synthetically applicable ONSH process in substituted nitrobenzenes could be realized using alkali metal alkoxides under much milder conditions (low temperature) and in the presence of a suitable oxidant (Scheme 2, pathway b). This process is an alternative to the known SNAr of halonitrobenzenes (Scheme 2, pathway c).6,29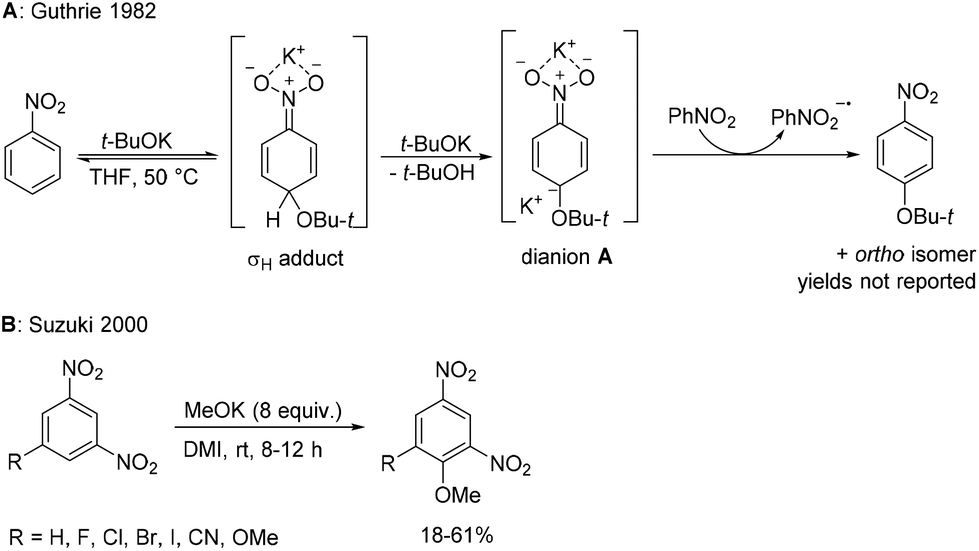 | ||
| Scheme 1 Reported nucleophilic substitutions of aromatic hydrogen for alkoxy groups in nitrobenzenes. | ||
 | ||
| Scheme 2 Nucleophilic aromatic substitution chemistry of nitrobenzenes. Pathway a – known nitro group substitution (SNAr); pathway b – proposed oxidative substitution of hydrogen (ONSH); and pathway c – known halogen substitution (SNAr). | ||
Results and discussion
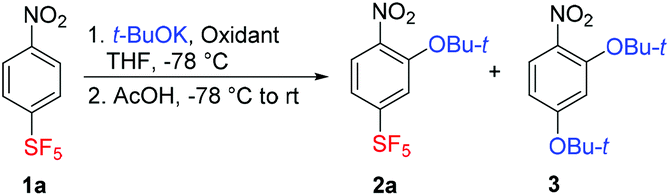
For initial investigations, the reaction of 4-nitro-1-(pentafluorosulfanyl)benzene (1a) with t-BuOK in THF at low temperature was selected. Nitrobenzene 1a was chosen as a model activated substrate for nucleophilic aromatic addition. The progress of the reaction was conveniently monitored by 19F NMR spectroscopy. Compounds with the SF5 group are intensively studied as drug candidates and agrochemicals, and in materials science.30 Potassium tert-butoxide was chosen as a stable, easily available and highly nucleophilic alkoxide soluble in THF. Furthermore, it is known that t-BuOK reacts with 1a only sluggishly in SNAr for nitro group even at room temperature.28 Performing the reaction at −78 °C using potassium permanganate dissolved in liquid ammonia (standard oxidant in ONSH with carbon nucleophiles or in aminations)19,22,23 afforded a low yield of the expected product 2a (Table 1, entry 1). Introducing oxygen in the reaction mixture increased the yield of 2a, especially when using an excess of t-BuOK (entries 2–6). The optimal conditions appeared to be 10 equiv. of t-BuOK introduced in 1 equiv. portions every 15 minutes (entry 7). Longer reaction time than 2.5 hours or lower temperature than −78 °C was less effective (entries 8 and 9). The optimized conditions shown in entry 7 were matched using di-tert-butyl peroxide as the oxidant (entry 10); however, in both the cases, about a third of starting 1a remained unreacted. For quenching the reaction mixture, acetic acid was found to be superior to aqueous ammonium chloride solution, since the latter caused partial deprotection of the tert-butyl group to the corresponding phenol derivative. With a large excess of t-BuOK or a long reaction time, previously unreported di-tert-butoxynitrobenzene 3 was formed via unusual aromatic substitution of the SF5 group.|
|
|||||
|---|---|---|---|---|---|
| Entry | t-BuOK (equiv.) | Time (h) | Oxidant (excess) | 2a, yieldb (%) | 3, yieldb (%) |
a
1a (1 mmol), t-BuOK (2.5–50 equiv.), and THF (20 mL) at −78 °C followed by AcOH (4.4–11 equiv.) and allowing to warm up to rt.
b ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19F NMR yield using an internal standard.
c KMnO4 and liquid ammonia added.
d
t-BuOK added in 1 equiv. portions every 15 min.
e Reaction performed at −100 °C. 19F NMR yield using an internal standard.
c KMnO4 and liquid ammonia added.
d
t-BuOK added in 1 equiv. portions every 15 min.
e Reaction performed at −100 °C.
|
|||||
| 1 | 2.5 | 1.5 | KMnO4c |
21 | 0 |
| 2 | 4 | 2.5 | O2 | 35 | 0 |
| 3 | 8 | 2.5 | O2 | 45 | 4 |
| 4 | 10 | 2.5 | O2 | 51 | 0 |
| 5 | 20 | 2.5 | O2 | 61 | 9 |
| 6 | 50 | 2.5 | O2 | 41 | 44 |
| 7 | 10d | 2.5 | O2 | 67 | 0 |
| 8 | 10 | 5 | O2 | 47 | 21 |
| 9 | 10e | 2.5 | O2 | 0 | 0 |
| 10 | 10d | 2.5 | (t-BuO)2 | 67 | 0 |
The scope of ONSH functionalization of nitroaromatics was studied using t-BuOK and optimized conditions (Table 2). Pentafluorosulfanyl- or trifluoromethyl-substituted benzenes at positions two, three or four were efficiently tert-butoxylated selectively at the position ortho to the nitro group. A similar reactivity profile was observed for nitrobenzonitriles. 1,3-Dinitrobenzene afforded 1-(tert-butoxy)-2,4-dinitrobenzene (2i) (position two is too crowded for substitution) and 1,2-dinitrobenzene gave 1-(tert-butoxy)-3,4-dinitrobenzene (2j), both in moderate yields. In the case of 1,2-dinitrobenzene, a side product (ca. 25%) of SNAr for the nitro group was also observed. A sulfonamide-containing nitrobenzene provided the tert-butoxy derivative 2k in good yield.

Protected formyl or oxo groups as dimethylacetal or ketal forms were also competent substrates in the reaction, and showed good reactivity also with moderately electron-deficient nitrobenzenes. 1-Nitronaphthalene afforded substitution at position two. The reaction was also possible with heterocyclic systems such as nitrothiophene; however, with 2-nitrofuran, no expected product could have been isolated. Some other substrates failed in the reaction. Nitrobenzene, ortho- meta- and para-anisole, 1-(tert-butyl)-4-nitrobenzene, 1-chloro-4-nitrobenzene, 1-bromo-4-nitrobenzene, 2-methyl-2-(4-nitrophenyl)-1,3-dithiane, quinoline, and isoquinoline were unreactive under the conditions. 4-Nitrotoluene afforded a product of methyl group oxidation rather than the expected aryl ether. 1,4-Dinitrobenzene gave a product of nitro group substitution and 1,3,5-trinitrobenzene was unreactive because of its poor solubility in THF at −78 °C. Prolonged reaction time can lead to the deprotection of the t-Bu group to the respective phenol. This was demonstrated on 1f, which after 2.5 hours gave a mixture of 2f and the corresponding phenol in 85% and 15% 19F NMR yields, respectively, while after 4 hours the yields changed to 33% and 66%, respectively.
Reaction regioselectivity is likely to be influenced by the chelation of the potassium ion of t-BuOK with the nitro group of the substrate. When the reaction with 1h was repeated in the presence of HMPA (10 equiv.), a mixture of 2h (50%) and regioisomers 2h′ (26%) and 2h′′ (24%) was formed (Scheme 3). HMPA solvated the potassium ion and converted t-BuOK into a loose ion pair, which may add to other electrophilic positions of the substrate with lower selectivity.
 | ||
| Scheme 3 Observation of decreased regioselectivity in the presence of HMPA, which solvates the potassium ion (1H NMR yields). | ||
Substrate 1a was then subjected to the reactions of various primary and secondary potassium alkoxides prepared in situ from the corresponding alcohols and potassium hydride. The corresponding 1-alkoxy-2-nitro-5-(pentafluorosulfanyl)-benzenes 4–7 were isolated in moderate to good yields (Table 3). Potassium phenolate did not react, presumably due to its insufficient nucleophilicity and soft character. The same result was observed with potassium benzylate, which was oxidized to benzaldehyde under the conditions.
| a Isolated yield. |
|---|

|
The mentioned SF5 group substitution of 2a in the presence of a large excess of t-BuOK was exploited in a two-step process leading to the new compound 3 in good yield (Scheme 4). Less bulky nucleophiles such as potassium methoxide or lithium morpholide afforded SF5-benzenes 8 and 9, respectively – products of the sequential substitution of aromatic hydrogen and the nitro group (Scheme 4). Apparently, the presence of the t-BuO group in 2a increased nucleofugacity of the proximal aromatic nitro group; however, the presence of two t-BuO groups at ortho positions was sterically unfavourable.
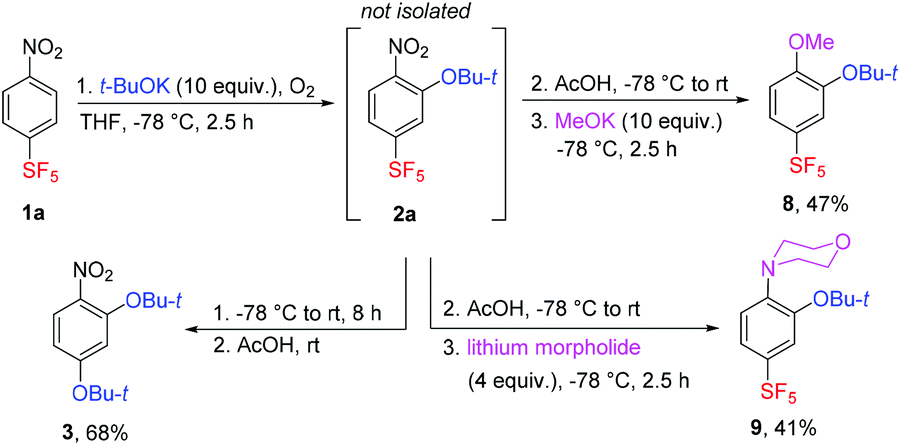 | ||
| Scheme 4 Sequential ONSH and SNAr of 1a. | ||
To probe the reaction mechanism, we used 1,3-dinitrobenzene-d4 (1i-d4). While 1i under standard conditions (Table 2) afforded the product of ONSH in 50% NMR or GC-MS yields (45% isolated yield), the perdeuterated substrate 1i-d4 was surprisingly largely unreactive. Furthermore, the use of a 1:1 mixture of 1i and 1i-d4 decreased dramatically the yield of 2i and the ratio of 2i to all deuterated 2i-dn observed by the GC-MS analysis was 4:1 (Table 4). This value does not represent a true isotope effect because an excess of both the nucleophile and oxidant had to be used to ensure some reactivity. Previously, a deuterium isotope effect of 9.8 was observed in the ONSH reaction of nitrobenzene with the anion of 2-phenylpropionitrile.31 Repeating the experiment in the absence of an oxidant led to the formation of a mixture of 1i, 1i-d, 1i-d3 and 1i-d4 (no 1i-d2) (Scheme 5). 1H NMR analysis revealed that H/D exchange took place at the most acidic position 2 of 1,3-dinitrobenzene.
 | ||
| Scheme 5 Proton/deuterium exchange in 1,3-dinitrobenzene. | ||
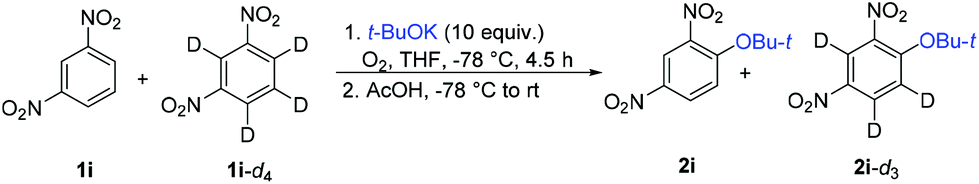
Based on these experiments and previous studies,25,30 the following reaction mechanism was proposed. Addition of t-BuOK takes place exclusively to position 4 of 1i to afford the σH adduct, which does not undergo electron transfer (oxidation by O2) but is deprotonated by a second equivalent of t-BuOK to dianion B. After hydrogen transfer, electron transfer takes place to form final 2i (Scheme 6). t-BuOK also causes deprotonation of 1i; however, the aryl anion does not act as a substrate for t-BuOK or O2. The reason why 1i becomes much less reactive in the presence of 1i-d4 is the diminished concentration of 1i by deuteration of position 2. Both deuterated substrates 1i-d and 1i-d4 are unreactive because of too slow CAr–D deprotonation to the dianion. If this is the case, then 1i-d3 should exhibit much better reactivity than 1i-d or 1i-d4. Indeed, 1i-d3 prepared by deprotonation and acid quenching from 1i-d4 afforded 2i-d2 (Scheme 7). However, more detailed mechanistic studies are required to understand the mechanism fully including the identification of the rate-determining step.
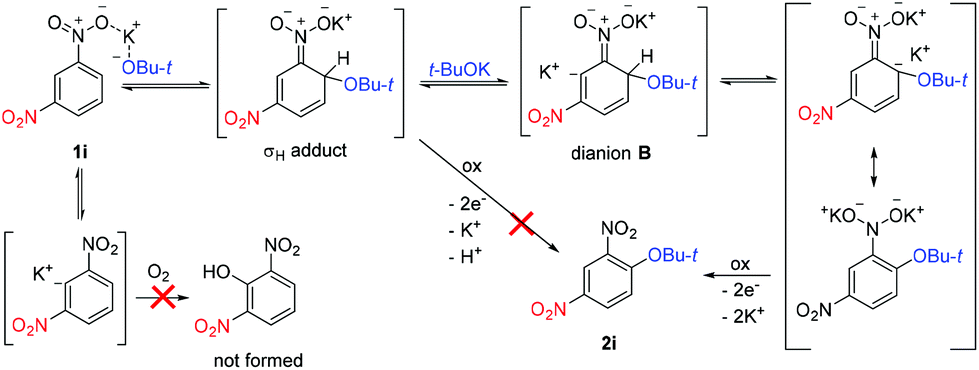 | ||
| Scheme 6 Proposed mechanism of the ONSH reaction of 1,3-dinitrobenzene with t-BuOK. | ||
 | ||
| Scheme 7 Demonstration of the reactivity of 1i-d3 in the ONSH reaction with t-BuOK. | ||
Experimental
Synthetic procedure for the preparation of 2a
To a solution of 1a (1 mmol, 249 mg) in THF (8 mL) at −78 °C (dry ice-acetone), a solution of t-BuOK (10 mmol, 1.12 g) in THF (10 mL) was added in portions (1 mL) every 15 minutes while dry oxygen was slowly bubbled through the apparatus. After 2.5 hours, AcOH (1 mL, 10 eq.) was added and the cooling bath was removed. After warming to room temperature, brine (30 mL) was added and the product was extracted with Et2O (3 × 50 mL). The combined organic phase was washed with brine (3 × 50 mL) and with water (3 × 50 mL), dried over magnesium sulfate, and the solvent was evaporated under reduced pressure. Purification was carried out by reverse phase chromatography (MeOH/H2O, 85:15) to afford pure 2a as a yellow oil (205 mg, 64% yield); 1H NMR (CDCl3, 400 MHz) δ 7.76 (1H, d, J = 8.9 Hz), 7.62 (1H, d, J = 2.2 Hz), 7.52 (1H, dd, J = 8.9, 2.3 Hz), 1.46 (9H, s); 19F NMR (CDCl3, 376 MHz) δ 82.10–80.35 (1F, m), 61.80 (4F, d, J = 150.5 Hz); 13C NMR (CDCl3, 100 MHz) δ 155.69 (quint, J = 19.5 Hz), 149.16, 146.63, 124.73, 122.33 (quint, J = 4.5 Hz), 120.41 (quint, J = 4.6 Hz), 84.71, 28.88; GCMS (EI) m/z 265 (1%), 248 (7), 127 (1), 63 (9), 57 (100), 41 (30), 39 (9); HRMS (EI+) m/z calcd for C10H12NO3SF5 [M]+: 321.0458; found: 321.0457.
Conclusions
In conclusion, we have demonstrated that structurally diverse nitrobenzenes and other nitroaromatics such as nitronaphthalene or nitrothiophene underwent oxidative nucleophilic aromatic substitution of hydrogen with potassium alkoxides (from primary, secondary and tertiary alcohols) in the presence of an oxidant (oxygen or di-tert-butyperoxide) to afford alkoxy-substituted nitroaromatics. The reaction took place at the position ortho to the nitro group of the substrate (with the exception of 1,2-dinitrobenzene) and the nucleophilic attack occurred at low temperature (−78 °C). Based on the mechanistic studies, a reaction mechanism was proposed involving the formation of a σH adduct, deprotonation to the dianion and electron transfer. Certain criteria must be met in order to observe efficient ONSH alkoxylation: (1) the aromatic substrate must be sufficiently electrophilic (i.e. mildly to highly electron-acceptor-substituted nitroaromatic or highly electron poor aromatic); (2) the formed σH adduct must have sufficient acidity for deprotonation with an excess of alkoxide; and (3) the formed deprotonated σH adduct must have sufficient electron-rich character for oxidation to take place.Primary products contained the nitro group of increased nucleofugacity. This feature was exploited in their derivatization by nucleophilic aromatic substitution of the nitro group with oxygen or nitrogen nucleophiles to afford selectively substituted benzene rings. Our new CAr–H functionalization method constitutes an important addition to the established nucleophilic aromatic substitution processes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was obtained from the Czech Academy of Sciences (RVO: 61388963) and the Czech Science Foundation (18-00215J).Notes and references
- K.-S. Ju and R. E. Parales, Nitroaromatic Compounds, from Synthesis to Biodegradation, Microbiol. Mol. Biol. Rev., 2010, 74, 250–272 CrossRef CAS.
- N. Ono, The Nitro Group in Organic Synthesis, Wiley-VCH, 2007 Search PubMed.
- J. G. Hoggett, R. B. Moodie, J. R. Penton and K. Schofield, Nitration and Aromatic Reactivity, Cambridge University Press, 1971 Search PubMed.
- G. A. Olah, S. C. Narang, J. A. Olah and K. Lammertsma, Recent aspects of nitration: New preparative methods and mechanistic studies (A Review), Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 4487–4494 CrossRef CAS.
- J. F. Queiroz, J. W. M. Carneiro, A. A. Sabino, R. Sparrapan, M. N. Eberlin and P. M. Esteves, Electrophilic Aromatic Nitration: Understanding Its Mechanism and Substituent Effects, J. Org. Chem., 2006, 71, 6192–6203 CrossRef.
- F. Terrier, Modern Nucleophilic Aromatic Substitution, Wiley-VCH, Weinheim, Germany, 2013 Search PubMed.
- K. Blaziak, W. Danikiewicz and M. Makosza, How Does Nucleophilic Aromatic Substitution Really Proceed in Nitroarenes? Computational Prediction and Experimental Verification, J. Am. Chem. Soc., 2016, 138, 7276–7281 CrossRef CAS.
- M. Makosza, Reactions of nucleophiles with nitroarenes: multifacial and versatile electrophiles, Chem. – Eur. J., 2014, 20, 5536–5545 CrossRef CAS.
- C. K. Mcgill and A. Rappa, Advances in the Chichibabin reaction, Adv. Heterocycl. Chem., 1988, 44, 1–79 CrossRef CAS.
- M. Makosza, Nucleophilic substitution of hydrogen in electron-deficient arenes, a general process of great practical value, Chem. Soc. Rev., 2010, 39, 2855–2868 RSC.
- M. Makosza, Nucleophilic Substitution of Hydrogen in Nitroarenes: A New Chapter of Aromatic Chemistry, Synthesis, 2011, 2341–2356 CrossRef CAS.
- M. Makosza and J. Winiarski, Vicarious nucleophilic substitution of hydrogen, Acc. Chem. Res., 1987, 20, 282–289 CrossRef CAS.
- J. Suwiński and K. Świerczek, cine- and tele-Subtitution reactions, Tetrahedron, 2001, 57, 1639 CrossRef.
- O. N. Chupakhin and V. N. Charushin, Recent advances in the field of nucleophilic aromatic substitution of hydrogen, Tetrahedron Lett., 2016, 57, 2665–2672 CrossRef CAS.
- M. Makosza and K. Staliński, Oxidative Nucleophilic Substitution of Hydrogen in Nitroarenes, Chem. – Eur. J., 1997, 3, 2025–2031 CrossRef CAS.
- M. Makosza and M. Paszewski, Oxidative Nucleophilic Substitution of Hydrogen in Nitroarenes. A short review, Pol. J. Chem., 2005, 79, 163–178 CAS.
- Z. Wróbel, C. Gulko, K. Plichta and A. Kwast, A two-step oxidative aromatic substitution of hydrogen as a convenient way to 2-nitrodiarylamines, Tetrahedron, 2016, 72, 8252–8260 CrossRef.
- Z. Wróbel and A. Kwast, Simple Synthesis of N-Aryl-2-nitrosoanilines in the Reaction of Nitroarenes with Aniline Anion Derivatives, Synthesis, 2010, 3865–3872 CrossRef.
- V. Khurorianskyi, M. Sonawane, M. Pošta, B. Klepetářová and P. Beier, Oxidative nucleophilic aromatic amination of nitrobenzenes, Chem. Commun., 2016, 52, 7237–7240 RSC.
- F. Kienzle, Nukleophile Alkylierung aromatischer Nitroverbindungen, Helv. Chim. Acta, 1978, 61, 449–452 CrossRef CAS.
- G. Iwasaki, S. Saeki and M. Hamana, Nucleophilic substitution of p-dinitrobenzene with some carbanions. Formation of p-substituted nitrobenzenes, Chem. Lett., 1986, 31–34 CrossRef CAS.
- M. Makosza, K. Staliński and C. Klępka, Oxidative nucleophilic substitution of hydrogen in nitrobenzene with 2-phenylpropionitrile carbanion and potassium permanganate oxidant, Chem. Commun., 1996, 837–838 RSC.
- N. Vida and P. Beier, Oxidative nucleophilic substitution of hydrogen in nitro(pentafluorosulfanyl)benzenes with alkyl Grignard and lithium reagents, J. Fluorine Chem., 2012, 143, 130–134 CrossRef CAS.
- R. Bujok and M. Makosza, Direct synthesis of nitroaryl acetylenes from acetylenes and nitroarenes via oxidative nucleophilic substitution of hydrogen, Chem. Commun., 2016, 52, 12650–12652 RSC.
- R. D. Guthrie and D. E. Nutter, Mechanism of the apparent electron-transfer reaction between tert-butoxide ion and nitrobenzene, J. Am. Chem. Soc., 1982, 104, 7478–7482 CrossRef CAS.
- T. Kawakami and H. Suzuki, Direct methoxylation of nitroarenes and nitroazaarenes with alkaline methoxides via nucleophilic displacement of an aromatic hydrogen atom, J. Chem. Soc., Perkin Trans. 1, 2000, 1259–1264 RSC.
- F. Effenberger, M. Koch and W. Streicher, Nucleophile Substitution von Nitrit in Nitrobenzolen, Nitrobiphenylen und Nitronaphthalinen, Chem. Ber., 1991, 124, 163–173 CrossRef CAS.
- P. Beier, T. Pastýříková, N. Vida and G. Iakobson, SNAr Reactions of Nitro-(pentafluorosulfanyl)benzenes To Generate SF5 Aryl Ethers and Sulfides, Org. Lett., 2011, 13, 1466–1469 CrossRef CAS.
- S. Ando, M. Tsuzaki and T. Ishizuka, Aryl ether syntheses via aromatic substitution proceeding under mild conditions, J. Org. Chem., 2020, 85, 11181–11189 CrossRef CAS.
- P. R. Savoie and J. T. Welch, Preparation and Utility of Organic Pentafluorosulfanyl-Containing Compounds, Chem. Rev., 2015, 115, 1130–1190 CrossRef CAS.
- M. Makosza and K. Staliński, Isotope effect in oxidative nucleophilic substitution of hydrogen in nitroarenes, Tetrahedron Lett., 1998, 39, 3575–3576 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental and synthetic procedures, characterization and copies of NMR spectra of all synthesized compounds. See DOI: 10.1039/d0qo01291b |
| This journal is © the Partner Organisations 2021 |