
Application of palladium-catalyzed aryl C–H alkylation in total synthesis of (−)-berkelic acid†
Hui-Hong
Wang‡
a,
Xiao-Dong
Wang‡
b,
Fei
Cao
a,
Wei-Wei
Gao
a,
Shu-Meng
Ma
b,
Zhao
Li
b,
Xue-Mei
Deng
b,
Tao
Shi
*b and
Zhen
Wang
 *ab
*ab
aState Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, Gansu, China. E-mail: zhenw@lzu.edu.cn
bSchool of Pharmacy, Lanzhou University, West Donggang Road. No. 199, Lanzhou 730000, Gansu, China. E-mail: shit18@lzu.edu.cn
First published on 11th November 2020
Abstract
An unprecedented palladium(II)-catalyzed ortho-alkylation of N-methoxybenzamide with epoxides has been applied to the total synthesis of the isochroman natural product (–)-berkelic acid. Combining this strategy with a well documented spiroacetalization cascade reaction, (–)-berkelic acid is obtained in 13.9% overall yield with the longest linear sequence of 11 steps. The results of preliminary biological studies on the synthesized natural product and its analogues are also reported.
Over the past decades, transition metal-promoted C–H activation has brought new insights owing to its higher atom and step economies over conventional cross coupling reactions.1,2 Applications of C–H activation in the preparation of simple substrates are in blossom.3 Nevertheless, its implementation in total syntheses of complex natural products is largely hampered by the lack of site selectivity in molecules that have multiple similar C–H bonds,1a,2d by the issues of functional group tolerance in complex architectures,1a and particularly, by the difficulty in removing directing groups.1c
In 2015, the groups of Kanai4 and Yu5 reported Pd-catalyzed C–H alkylation reactions of N-methoxybenzamides and benzoic acids with epoxides, respectively. In these reactions, the directing group was converted directly into a part of the product via a subsequent intramolecular alcoholysis reaction, avoiding the removal of directing groups and providing a labor-saving alternative for the construction of isochromans and related natural products.
(–)-Berkelic acid (1), a novel tetracyclic natural product with highly substituted chroman and spiroketal structures, was isolated from an extremophilic Penicillium species by Stierle and co-workers in 2006.6 Since the pioneering synthesis-driven structure revision works on berkelic acid performed by the Fürstner,7 Snider8 and De Brabander9 groups, four total syntheses7–10 and several formal syntheses have been reported.11,12 In these syntheses, we note that access to the isochroman core of berkelic acid often needed multiple steps (Scheme 1), and that the efforts to develop modular approaches to the tetracyclic chroman spiroketal core did not lead to the total synthesis of berkelic acid, thus limiting the synthetic efficiency. In this context, we herein report a concise strategy of Pd(II)-catalyzed C–H alkylation providing rapid access to the isochroman skeleton of berkelic acid. This highly efficient C–H activation strategy, in combination with the well-documented deprotection/spiroacetalization cascade reaction,11 enabled an expeditious total synthesis of (–)-berkelic acid. In addition, preliminary studies on the biological activity of the synthesized natural product and its analogues have been performed.
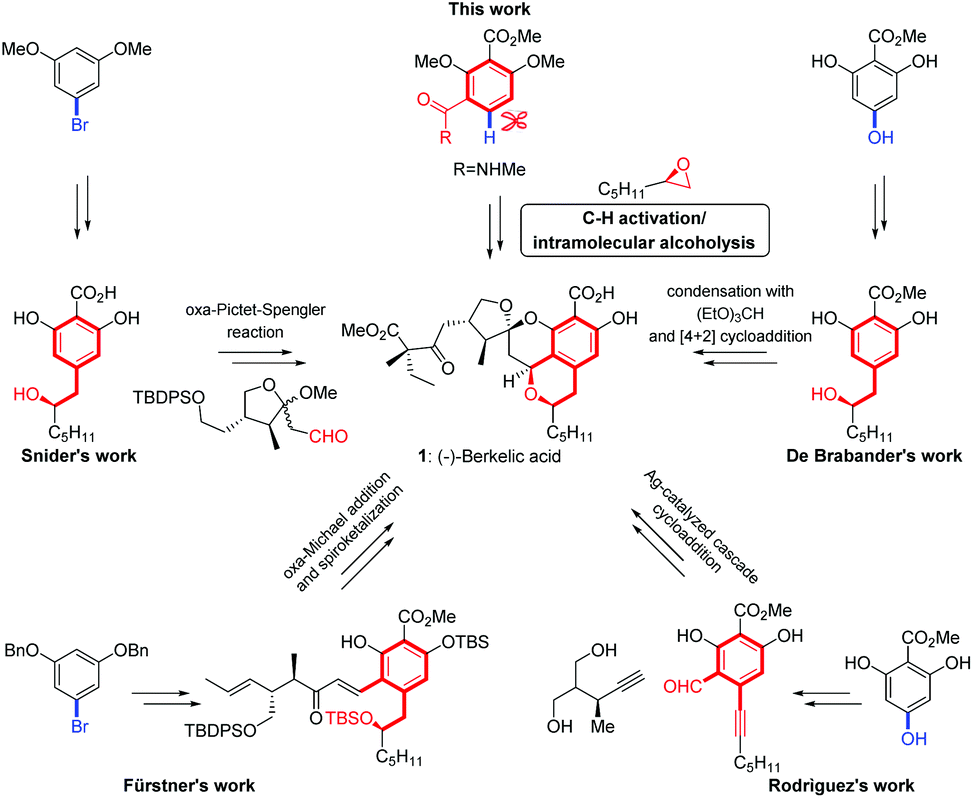 | ||
| Scheme 1 Previous approaches to construct the isochroman core in total syntheses of (−)-berkelic acid (1). | ||
With the aim to synthesize berkelic acid via an efficient approach that would allow the scalable synthesis of its analogues and biological evaluations, we adopted a redox-neutral umpolung alkylation strategy disconnecting 1 into cyanohydrin 2 and iodide 3. Following the work of Fañanás and Rodríguez,10 iodide 3 could be assembled from bis-silyl ether 4 by the well-established spontaneous spiroketalization upon deprotion that constructs the challenging spirocenter. In turn, bia-silyl ether 4 could be traced back to lactol 5 and silyl enol ether 6 with reference to Brimble's strategy.11 Finally, lactol 5 could arise from the Pd-catalyzed C–H alkylation of arene 8 with oxirane 7 (Scheme 2), which might provide one of the most efficient routes to this kind of highly substituted isochroman skeleton.
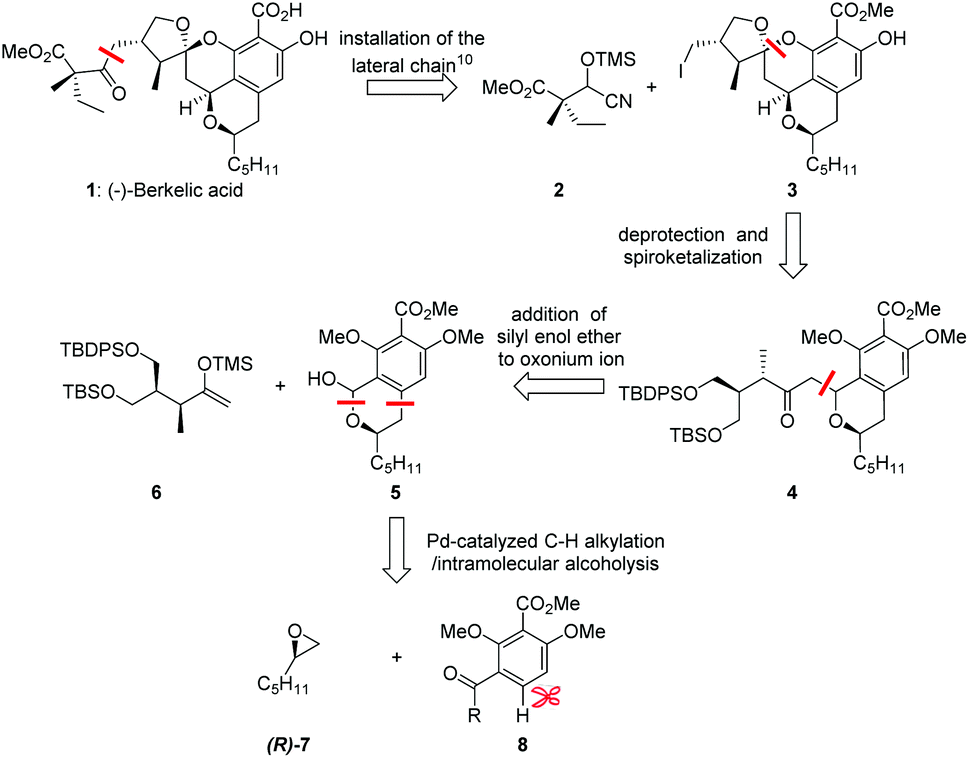 | ||
| Scheme 2 Retrosynthetic analysis of (–)-berkelic acid (1). | ||
With the above retrosynthetic analysis in mind, we started our synthesis by exploring the key Pd-catalyzed C–H activation/(R)-pentyloxirane opening reaction to build the dihydroisochroman precursor of 5. In view of the high efficiency of carbonyl-derived directing groups in isocoumarins synthesis as stated earlier,4,5 we initially employed 8a and (rac)-7 as the model substrates under Yu's conditions.5 While 9a was obtained with a yield of 15%, and efforts to increase the yield by modification of the reaction conditions proved unsuccessful (Scheme 3). We hypothesized that the presence of an electron-withdrawing group at the para position of the target C–H bond might reduce the nucleophilicity of arylpalladium intermediates and that multiple oxygen functional groups might attenuate the activity of the palladium catalyst by coordination. Thus, a stronger coordinating directing group among the carbonyl derivatives appeared to have to be selected. Considering that the N-methoxyamide was beneficial to this type of reaction4 and the reaction could be conducted under slightly basic conditions,13N-methoxybenzamide 8a′, in place of carboxylic acid 8a, was utilized to optimize this reaction under basic conditions. Gratifyingly, the yield of 9a substantially increased to 47% under the Pd(OAc)2/KOAc conditions (Table 1, entry 1), without requiring an adjacent coordinating group in the oxirane which was a restriction in the original report.4 Subsequently, we studied the effects of different ligands.
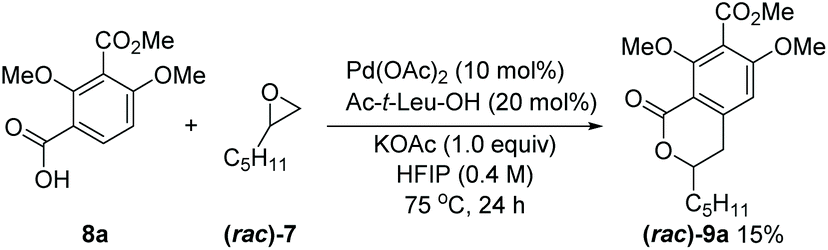 | ||
| Scheme 3 Exploration of the Pd(II)-catalyzed oxirane ring opening under Yu's conditions. | ||
|
|
||
|---|---|---|
| Entry | Change from the standard conditions | Yieldb % |
| a Standard reaction conditions: 8a′ (0.1 mmol, 1.0 equiv.), 7 (0.2 mmol, 2.0 equiv.), KOAc (0.1 mmol, 1.0 equiv.), Pd(OAc)2 (10 mol%) and HFIP (0.4 M), 75 °C, 24 h. b Isolated yields. c MPAA = mono-N-protected amino acid ligands. d 95 °C. | ||
| 1 | None | 47 |
| 2 | MPAAc | 15–30 |
| 3 | XPhos instead of MPAA | 0 |
| 4 | PPh3 instead of MPAA | 0 |
| 5 | PivOK + Pd(OPiv)2 | 32 |
| 6 | K2HPO4 instead of KOAc | 63 |
| 7 | K2HPO4 + 20 mol% CuCl2 | 72 (77)d |
| 8 | 20 mol% Cu(OAc)2 | 76 (84)d |
| 9 | 20 mol% CuCl2 | 79 (86)d |

However, the addition of several mono-N-protected amino acid ligands (MPAA, see the ESI† for details) led to 15–30% yields (entry 2), and phosphine ligands such as X-Phos and PPh3 significantly diminished the reactivity resulting in no formation of the desired product (entries 3 and 4). The Pd(OPiv)2/PivOK also promoted the transformation, but did not afford a better yield (entry 5). Replacing KOAc with K2HPO4 improved the yield to 63% (entry 6). Much to our pleasure, the addition of CuCl2 could enhance the yield, which was further improved to 77% upon raising the temperature to 95 °C (entry 7). As the poor solubility of K2HPO4 in HFIP caused inconvenience in the process of post treatment, KOAc instead of K2HPO4 was investigated and found to be beneficial to this reaction (up to 86%, entry 9), while replacing CuCl2 with Cu(OAc)2 showed no improvement on the yield (entry 8). Remarkably, the coupling reactions proceeded with a high degree of stereoretention (>98% ee) when (R)-7 was exploited, and a gram-scale reaction also gave 9a in comparable yield (1.23 g of 8a′ gave 1.15 g of 9a in 75% yield).
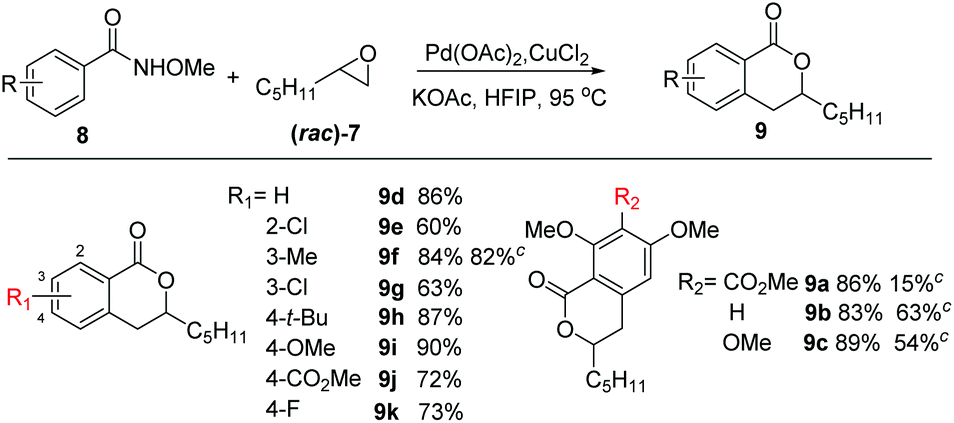
With the optimized reaction conditions in hand, we examined the scope of N-methoxybenzamide 8 using 2-pentyl oxirane (7) as the alkylating reagent (Table 2). Electron-donating groups on the aryl ring were well tolerated in general (9d, 9f, 9h, 9i, 84%–90%), while substrates with electron-withdrawing chloro (9e and 9g), ester (9j) and fluoro (9k) groups gave lower yields (60%–73%). In addition, substrates bearing substituents of different electronic properties at the meta position of the amide were also investigated. The electron-rich substrate 8c having 2,3,4-trimethoxy groups gave 9c in a yield of 89%. Similar yields were obtained from the reactions of the non-meta-substituted 2,4-dimethoxy amide 9b (83%) and electron-withdrawing ester-substituted 9a (86%), while the yields of 9a–9c were lower under Yu's conditions (15%–63%).
Having the optimal reaction conditions established, we turned our attention to the asymmetric synthesis of berkelic acid, first preparing silyl enol ether 6 (Scheme 4). The synthesis started from the known (R)-paraconyl alcohol (10), which was firstly subjected to silyl protection to synthesize TBDPS ether 11 in 92% yield. The subsequent methylation of 11 using NaHMDS and MeI at −78 °C gave α-methylated lactone 12 in high diastereoselectivity (>95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 dr). Addition of methyllithium to lactone 12 afforded lactol 13, which was trapped by TBS to render 14 in an overall yield of 80% for two steps. Then, treatment of ketone 14 with TMSOTf in the presence of DIPEA in DCM afforded silyl enol ether 6, which was directly used without further purification.
:5 dr). Addition of methyllithium to lactone 12 afforded lactol 13, which was trapped by TBS to render 14 in an overall yield of 80% for two steps. Then, treatment of ketone 14 with TMSOTf in the presence of DIPEA in DCM afforded silyl enol ether 6, which was directly used without further purification.
 | ||
| Scheme 4 Synthesis of silyl enol ether 6. | ||
With dihydroisochroman (R)-9a and silyl enol ether 6 in hand, we set out to combine them into Berkelic acid (Scheme 5). Lactone (R)-9a was reduced to lactol 5 by DIBAL-H in 72% yield. After treatment of 5 with BF3·OEt2, addition of silyl enol ether 6 at −78 °C rendered a diastereomeric mixture of lactol 15. Previous experience7,11 with the spiroketal moiety indicated that deprotection of 15 would form thermodynamically stable 16via equilibration under acidic conditions. Thus, a one-pot cascade sequence was orchestrated, which involved removal of the methyl protecting groups with BBr3 at low temperature and the subsequent spiroketalization in acidic methanol (5 eq. p-TsOH). As a result, spiroketal 16 with 53% yield from 14 was obtained as a single diastereoisomer, in which the TBDPS group was also removed smoothly in this acid condition. Iodination of alcohol 16 gave Fürstner's7 intermediate 3, which was characterized by single crystal X-ray analysis. Subsequently, installation of the lateral chain gave diester 19 in 86% yield. As for the synthesis of cyanohydrin 2, a four-step sequence from commercially available dimethyl-D-malate (17) and subsequent silylcyanation of the resulting aldehyde 18, referring to the reported method,10 produced cyanohydrin 2. Finally, selective hydrolysis of the methyl benzoate with (Bu3Sn)2O completed the asymmetric total synthesis of berkelic acid (1) in 55% yield.10 The 1H and 13C NMR spectra and optical rotation of the synthetic (–)-berkelic acid (1) were identical with the previously reported data.6
 | ||
| Scheme 5 Assembling of three fragments into (–)-berkelic acid (1). [a]Non-hydrogens atoms are shown as 30% ellipsoids. DMPU = 1,3-dimethyl-3,4,5,6-tetrahydropyrimidin-2(1H)-one; PNPCl = bis-(triphenylphosphoranylidene)ammonium. | ||
Since the β-keto carboxylic acid in berkelic acid (1) might serve as a potential site for further decoration,14 hydrolysis of the methyl ester of 1 was next tried (Scheme 6). Although the yield of the desired hydrolyzed product 20 was low, the yield of the decarboxylated product 21 (2 diastereoisomers, ∼1:1) was high. In the process of purifying diacid 20 with MeOH as the mobile phase, an aliphatic acid esterification product, berkelic acid, and the decarboxylated product 21 were also detected in LC-MS and 1H-NMR spectra. Considering that the esterification rate of the acid would be decreased by the steric hinderance of α-substituents,15 especially that of the α-quaternary carbon, the fromation of an esterified product even without any catalyst was puzzling. We surmised that the counter-intuitive result of the esterification that happened during the purification process might be attributed to the existence of a β-ketone. To verify this intriguing hypothesis, esterification and decarboxylation of two carboxylic acids with or without a β-ketone were tested in methanol in the presence of differential amounts of HCl (Table 3). From acid 22 devoid of a β-ketone, neither esterified nor decarboxylated products were detected, even with the addition of HCl. In the case of β-ketoacid 25, both reactions proceed, with decarboxylation more apt to occur than esterification, producing ketone 27 in a yield up to 87.6%. While the facile decarboxylation of β-ketoacids is well established,16 it is intriguing that esterification of an acid could also be assisted, albeit to a small degree, by a β-keto group.
 | ||
| Scheme 6 Hydrolysis of the aliphatic methyl ester in (–)-berkelic acid (1). | ||
|
|
||||
|---|---|---|---|---|
| Solvent (mmol% of HCl) | Esterificated producta | Decarboxylated producta | ||
| 23 | 26 | 24 | 27 | |
| a Yields were determined by HPLC-DAD-MS analysis of the crude product and quantified with external standard method after 24 h. b 48 h. ND = No detected (starting material was recovered). | ||||
| MeOH (1 mmol%) | ND | ND | ND | 54.31% |
| MeOH (10 mmol%) | ND | 1.07% | ND | 67.49% |
| MeOH (100 mmol%) | ND | 2.12% | ND | 72.47% |
| MeOH (100 mmol%)b | ND | 2.66% | ND | 87.60% |
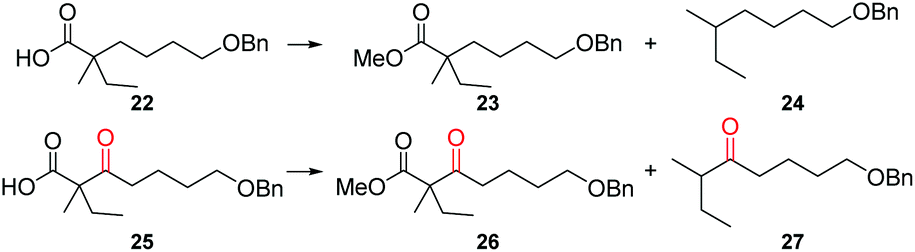
There is a pending controversy on the cytotoxicity of (–)-berkelic acid; the natural sample was reported to be active against ovarian cancer OVCAR-3 whereas the synthetic material did not show any significant activity.6,9,10 In light of the finding that the methyl ester product (berkelic acid) was formed in the process of purifying diacid 20 with MeOH (the natural berkelic acid was isolated by killing the fungus with MeOH),7 we then performed a biological evaluation of compounds 1, 19, and 21. However, at a range of indicated concentrations (1.56, 3.13, 6.25, 12.5, 25, 50 μM), none of these compounds exhibited any appreciable cytotoxicity against HCT-116, MGC-803, HuH-7 and SGC-7901 cancer cell lines (see the ESI†).
Conclusions
In conclusion, we have accomplished the asymmetric total synthesis of berkelic acid (1), which features a Pd-catalyzed ortho-alkylation of N-methoxybenzamides with epoxides for the construction of the isochroman core of the natural product. This novel strategy was channeled with the known deprotection-induced spontaneous spiroacetalization cascade reaction to establish the spiroketal carbon center and ultimately afforded berkelic acid in 13.9% overall yield with the longest linear sequence of 11 steps. Our synthetic strategy provides a new approach for concise and efficient synthesis of isochromans. Preliminary biological studies on (–)-berkelic acid and its analogues revealed low cytotoxicity and safety profiles associated with these compounds, which merit further research on their bioactivities. By taking advantage of this powerful Pd-catalyzed C–H activation/oxirane ring opening reaction, total synthesis investgations on other isochroman natural products are currently underway, which, together with this work, would accelerate further biological evaluations of these fascinating natural products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the Recruitment Program of Global Experts (1000 Talents Plan), Gansu Province Science Foundation for Distinguished Young Scholars (20JR5RA304), and Fundamental Research Funds for the Central Universities (lzujbky-2019-ct08).Notes and references
- (a) E. J. E. Caro-Diaz, M. Urbano, D. J. Buzard and R. M. Jones, C-H Activation Reactions as Useful Tools for Medicinal Chemists, Bioorg. Med. Chem. Lett., 2016, 26, 5378 CrossRef CAS; (b) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, 3d Transition Metals for C–H Activation, Chem. Rev., 2019, 119, 2192 CrossRef CAS; (c) K. M. Engle, T. S. Mei, M. Wasa and J. Q. Yu, Weak Coordination as a Powerful Means for Developing Broadly Useful C-H Functionalization Reactions, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS.
- (a) W. R. Gutekunst and P. S. Baran, C–H functionalization logic in total synthesis, Chem. Soc. Rev., 2011, 40, 1976 RSC; (b) B. Hong, C. Li, Z. Wang, J. Chen, H. Li and X. Lei, Enantioselective Total Synthesis of (−)-Incarviatone A, J. Am. Chem. Soc., 2015, 137, 11946 CrossRef CAS; (c) P. Tao and Y. Jia, C–H bond activation in the total syntheses of natural products, Sci. China: Chem., 2016, 59, 1109 CrossRef CAS; (d) P. B. Brady and V. Bhat, Recent Applications of Rh and Pd-Catalyzed C(sp3)-H Functionalization in Natural Product Total Synthesis, Eur. J. Org. Chem., 2017, 5179 CrossRef CAS; (e) Z. Zhang, J. Wang, J. Li, F. Yang, G. Liu, W. Tang, W. He, J. J. Fu, Y. H. Shen, A. Li and W. D. Zhang, Total Synthesis and Stereochemical Assignment of Delavatine A: Rh-Catalyzed Asymmetric Hydrogenation of Indene-Type Tetrasubstituted Olefins and Kinetic Resolution through Pd-Catalyzed Triflamide-Directed C−H Olefination, J. Am. Chem. Soc., 2017, 139, 5558 CrossRef CAS.
- (a) J. Jayakumar, K. Parthasarathy and C. Cheng, One-Pot Synthesis of Isoquinolinium Salts by Rhodium-Catalyzed C-H Bond Activation: Application to the Total Synthesis of Oxychelerythrine, Angew. Chem., Int. Ed., 2012, 51, 197 CrossRef CAS; (b) D. Y. K. Chen and S. W. Youn, C-H Activation: A Com-plementary Tool in the Total Synthesis of Complex Natural Products, Chem. – Eur. J., 2012, 18, 9452 CrossRef CAS.
- Z. Wang, Y. Kuninobu and M. Kanai, Palladium-Catalyzed Oxirane-Opening Reaction with Arenes via C–H Bond Activation, J. Am. Chem. Soc., 2015, 137, 6140 CrossRef CAS.
- G. Cheng, T. Li and J. Q. Yu, Practical Pd(II)-Catalyzed C–H Alkylation with Epoxides: One-Step Syntheses of 3,4-Dihydroisocoumarins, J. Am. Chem. Soc., 2015, 137, 10950 CrossRef CAS.
- A. A. Stierle, D. B. Stierle and K. Kelly, Berkelic Acid, A Novel Spiroketal with Selective Anticancer Activity from an Acid Mine Waste Fungal Extremophile, J. Org. Chem., 2006, 71, 5357 CrossRef CAS.
- (a) P. Buchgraber, T. N. Snaddon, C. Wirtz, R. Mynott, R. Goddard and A. Fürstner, A Synthesis-Driven Structure Revision of Berkelic Acid Methyl Ester, Angew. Chem., Int. Ed., 2008, 47, 8450 CrossRef CAS; (b) T. N. Snaddon, P. Buchgraber, S. Schulthoff, C. Wirtz, R. Mynott and A. Fürstner, Total Synthesis of Berkelic Acid, Chem. – Eur. J., 2010, 16, 12133 CrossRef CAS.
- (a) J. Zhou and B. B. Snider, Biomimetic Synthesis of the Tetra-cyclic Core of Berkelic Acid, Org. Lett., 2007, 9, 2071 CrossRef CAS; (b) X. Wu, J. Zhou and B. B. Snider, Synthesis of (−)-Berkelic Acid, Angew. Chem., Int. Ed., 2009, 48, 1283 CrossRef CAS.
- (a) C. F. Bender, F. K. Yoshimoto, C. L. Paradise and J. K. De Bra-bander, A Concise Synthesis of Berkelic Acid Inspired by Combining the Natural Products Spicifernin and Pulvilloric Acid, J. Am. Chem. Soc., 2009, 131, 11350 CrossRef CAS; (b) C. F. Bender, C. L. Paradise, V. M. Lynch, F. K. Yoshimoto and J. K. De Brabander, A bio-synthetically inspired synthesis of (−)-berkelic acid and analogs, Tetrahedron, 2018, 74, 909 CrossRef CAS.
- Fañanás's/Rodríguez's studies toward berkelic acid and simplified model compounds: (a) F. J. Fañanás, A. Mendoza, T. Arto, B. Temelli and F. Rodríguez, Scalable Total Synthesis of (−)-Berkelic Acid by Using a Protecting-Group-Free Strategy, Angew. Chem., Int. Ed., 2012, 51, 4930 CrossRef; (b) T. Arto, I. S. Santa-María, M. Chiara, F. Fañanás and J. F. Rodríguez, Berkelic Acid: Straightforward Analogue Synthesis and Studies of Activity Against Selected Cancer Cell Lines, Eur. J. Org. Chem., 2016, 5876 CrossRef CAS.
- Brimble's studies toward berkelic acid and simplified model compounds: (a) Z. E. Wilson and M. A. Brimble, A flexible asymmetric synthesis of the tetracyclic core of berkelic acid using a Horner–Wadsworth–Emmons/oxa-Michael cascade, Org. Biomol. Chem., 2010, 8, 1284 RSC; (b) M. C. McLeod, Z. E. Wilson and M. A. Brimble, An Enantioselective Formal Synthesis of Berkelic Acid, Org. Lett., 2011, 13, 5382 CrossRef CAS; (c) M. C. McLeod, Z. E. Wilson and M. A. Brimble, Formal Synthesis of Berkelic Acid: A Lesson in α-Alkylation Chemistry, J. Org. Chem., 2012, 77, 400 CrossRef CAS; (d) M. A. Brimble, I. Haym, J. Sperry and D. P. Furkert, Synthesis of the Tetracyclic Core of Berkelic Acid Using Gold(I)-Catalyzed Hydroarylation and Oxidative Radical Cyclizations, Org. Lett., 2012, 14, 5820 CrossRef CAS.
- Pettus's studies toward berkelic acid and simplified model compounds: (a) K. L. Wu, S. Wilkinson, N. O. Reich and T. R. R. Pettus, Facile Synthesis of Naphthoquinone Spiroketals by Diastereoselective Oxidative [3+2] Cycloaddition, Org. Lett., 2007, 9, 5537 CrossRef CAS; (b) M. A. Marsini, Y. Huang, C. C. Lindsey, K.-L. Wu and T. R. R. Pettus, Org. Lett., 2008, 10, 1477 CrossRef CAS; (c) Y. Huang and T. R. R. Pettus, A Cycloaddition Strategy for Use toward Berkelic Acid, an MMP Inhibitor and Potent Anticancer Agent Displaying a Unique Chroman Spiroketal Motif, Synlett, 2008, 1353 CAS; (d) T. A. Wenderski, M. A. Marsini and T. R. R. Pettus, A Diastereoselective Formal Synthesis of Berkelic Acid, Org. Lett., 2011, 13, 118 CrossRef CAS.
- R. Li and G. Dong, Direct Annulation between Aryl Iodides and Epoxides through Palladium/Norbornene Cooperative Catalysis, Angew. Chem., Int. Ed., 2018, 57, 1697 CrossRef CAS.
- M. Kimura, Recent topics in the syntheses of β-keto car-boxylic acids and the derivatives, Tetrahedron Lett., 2018, 59, 1295 CrossRef CAS.
- Y. Liu, E. Lotero and J. G. Goodwin Jr., Effect of carbon chain length on esterification of carboxylic acids with methanol using acid catalysis, J. Catal., 2006, 243, 221 CrossRef CAS.
- (a) D. B. Bigley and J. C. Thurman, Studies in decarboxylation. Part VI. A comparison of the transition states for the decarboxyla-tion of β-keto- and βγ-unsaturated acids, J. Chem. Soc. B, 1968, 436 RSC; (b) R. D. Bach and C. Canepa, Electronic Factors Influencing the Decarboxylation of β-Keto Acids. A Model Enzyme Study, J. Org. Chem., 1996, 61, 6346 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2004145. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0qo01003k |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2021 |