
Probing the ligand exchange kinetics of phenynyl-based ligands on colloidal Au nanoparticles†
Ting
Xiang
,
Jianpeng
Zong
,
Wenjia
Xu
,
Yuhua
Feng
* and
Hongyu
Chen
 *
*
Institute of Advanced Synthesis (IAS), School of Chemistry and Molecular Engineering, Jiangsu National Synergetic Innovation Centre for Advanced Materials, Nanjing Tech University, Nanjing 211816, P. R. China. E-mail: iashychen@njtech.edu.cn; ias_yhfeng@njtech.edu.cn
First published on 27th October 2020
Abstract
Ligands are the primary tool for stabilizing nanoparticles and surface treatments. Understanding their relative strength and modulating their exchange are the initial steps towards their application. By real-time monitoring of surface-enhanced Raman scattering (SERS), we show that phenynyl ligands could readily bind to colloidal Au nanoparticles giving strong SERS signals. On the basis of the relative exchange ratio, the phenynyl ligands are in general weaker than the strong thiol ligand, and stronger than the polymeric PVP. The method could also be applied to rank the relative strength of phenynyl ligands. We believe that our method provides a general platform for studying ligand affinity. Being a new class of ligands, the understanding of phenynyl ligands would promote their applications in SERS, nanosynthesis, surface treatment, and beyond.
Introduction
The main difference between nanoparticles and macroscopic objects is their size. The enormous surface of nanoparticles leads to high surface energy and instability, and thus ligands play an essential role in their surface passivation. Ligand exchange is also indispensible in order to modulate the physical and chemical properties of nanoparticles.1,2Despite the broad application of ligands in nanotechnology, there are few studies on ligand exchange.3,4 While it is obvious that strong ligands could exchange for weak ones, the exchange in reverse order, or the exchange between ligands of similar strength are so far poorly understood. Further understanding of ligand exchange and the detailed kinetics would assist future efforts in nanosynthesis, surface treatment, and beyond.
The commonly used ligands for nanosynthesis are rather limited in variety.5 They are nearly all weak ligands or surfactants, such as citrate, oleylamine, oleic acid, polyvinylpyrrolidone (PVP), cetyltrimethylammonium bromide (CTAB), etc. In recent years, strong thiol-based ligands have also been explored in controlling nanostructures.6–13 Given the critical significance of ligands, exploring new ligands, particularly new class of ligands, would set the first step in advancing synthetic control in nanosynthesis.
Phenynyl molecules are a class of small organic molecules containing the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C group. While it is frequently used in organic synthesis, it is rarely used in the field of nanosynthesis: Terminal phenynyl ligand (R–CCH) can adsorb on bulk Au surface and give Raman signals.14–17 Phenynyl-functionalized Au nanoclusters have been synthesized and characterized by X-ray crystallography.18–21 It is shown to exchange PVP and stabilize nanoparticles and nanoclusters.14,22–24 Hence, PhA appears to be a potential strong ligand, and understanding its relative strength to other common ligands would be helpful for future studies.
C group. While it is frequently used in organic synthesis, it is rarely used in the field of nanosynthesis: Terminal phenynyl ligand (R–CCH) can adsorb on bulk Au surface and give Raman signals.14–17 Phenynyl-functionalized Au nanoclusters have been synthesized and characterized by X-ray crystallography.18–21 It is shown to exchange PVP and stabilize nanoparticles and nanoclusters.14,22–24 Hence, PhA appears to be a potential strong ligand, and understanding its relative strength to other common ligands would be helpful for future studies.
Previously, we show that ligand exchange on colloidal Au nanoparticles could be monitored by its SERS signal in real time.25 In this work, we apply this method to study phenynyl ligands and use the rate of ligand exchange to reveal their relative strength. The results showed that phenylacetylene (PhA) is a weaker ligand than the standard thiol ligand, 2-naphthalenethiol (2-NT), but with a concentration bias it can still exchange the latter. Similarly, PhA is a stronger ligand than PVP, but ligand exchange could occur both ways. We believe that terminal phenynyl are a promising class of ligands for nanosynthesis and surface treatment, where their relative strength would offer important guidance for rational selection.
Results and discussion
For high SERS intensity, we choose large-size citrate-stabilized AuNPs as the substrate,6 with narrow size distribution of 60 ± 5 nm (Fig. S2, ESI†). Given the poor solubility of the non-polar ligands in water, the phenynyl-functionalized AuNPs would easily aggregate and interfere with SERS measurement. Hence, the AuNPs are first transferred into a DMF solution before applying the ligands. Specifically, the as-synthesized citrate–AuNPs were first isolated by centrifugation to remove the excess reactants; they are dispersed in DMF, and then centrifuged again to remove the remnant water, before finally dispersed in DMF. The two-step centrifugation led to about 8% decrease in UV-Vis extinction, likely due to the irreversible aggregation when the AuNPs are compressed together. But the normalized spectra showed little change before and after the process (Fig. 1a), except a 9.5 nm red shift that can be attributed to the higher refractive index of DMF.25,26 There was no noticeable peak at 600–800 nm, which is characteristic of aggregated AuNPs due to longitudinal absorption.27,28 Therefore, the AuNPs were free of aggregation after the solvent exchange. | ||
| Fig. 1 (a) UV-Vis spectra of citrate–AuNPs before and after solvent shifting from water to DMF. (b) UV-Vis spectra of citrate–AuNPs before and after incubation with PhA. (c) Ligand structures in the order of relative strength: 2-NT > PhA > PVP. SERS intensity of (d) 2-NT–AuNPs in DMF, (e) PhA–AuNPs in DMF, (f) PVP–AuNPs in DMF (no recognizable peak from PVP). | ||
The AuNPs are stable in the DMF solution, with no noticeable colour change for days. To ensure their stability, all of the AuNPs used in the following study were prepared freshly in the same day. As SERS intensity is extremely sensitive to the degree of aggregation, once ligand is added, all processing was avoided such as centrifugation and solvent change. All spectra were collected in real-time with minimal interference to the samples. Thus, strong DMF peaks could be observed at 657, 866, 1090, 1407, 1439, 1660 and 2932 cm−1 in all samples.
As the simplest phenynyl molecule, PhA is used for the model study. It is added to the solution of AuNPs, reaching 8.3 μM (final concentration, same below), followed by incubation for 3 h. The UV-Vis spectra before and after this ligand treatment (Fig. 1b) are exactly overlapping, indicating the absence of aggregation. The kinetics of PhA adsorption on the citrate–AuNPs was monitored by SERS at 5 min interval (Fig. 2a–e). In contrast to the unvarying DMF peaks, the PhA peaks showed obvious increase at 997, 1172, 1198, 1588 and 2013 cm−1. The enhancement factor (EF) at 1588 cm−1 is estimated to be 8.9 × 105 (see ESI†),29 meaning that the free ligand in the solution is negligible against the strong SERS signal.
 | ||
| Fig. 2 (a) Temporal evolution of the SERS intensity of citrate–AuNPs incubated with 8.3 μM PhA in DMF. (b–e) Expansion of the framed regions in a. (f) Intensity traces of the 4 peaks in a. (g) Normalized intensity traces of the 4 peaks in a. | ||
Most of the SERS peaks show little shift from those of the free PhA, except the 2108 cm−1 peak which is shifted to 2013 cm−1 (Fig. S6, ESI†). In most of our experiments with AuNPs, only the 2013 cm−1 peak is visible; with huge excess of PhA (200 mM), the 2108 cm−1 peak became visible in addition to the 2013 cm−1 peak (Fig. S7, ESI†). This peak is assigned as the CC vibration, and it is thus expected to decrease in energy/frequency when one end is bound the Au surface.30,31
The intensity traces of the 4 prominent peaks were plotted in Fig. 2f, where the small fluctuation demonstrates the reliability of the ensemble measurement of colloidal SERS substrates.32 After normalization (Fig. 2g), the 4 traces nearly overlap with each other, showing the strong correlation among them. The ligand adsorption mostly occurs before 40 min and gradually approaches a plateau.
The PhA adsorption experiment was carried out for 3 times. Their normalized traces at 2013 cm−1 are nearly overlapping (Fig. S8, ESI†), showing that the SERS kinetics is highly reproducible. With decreasing PhA concentration of 75, 25, 8.3, 2.8, and 0.9 μM, the rate of ligand adsorption decreases, with a lowering plateau (Fig. S9, ESI†). At these low concentrations and particularly the 8.3 μM condition, it appears that the Au surface has not been fully saturated. There should be a dynamic exchange between the free ligand and those adsorbed on the Au surface.
In the literature, reaction of PhA with Au was often carried out in inert atmosphere with a strong base.14,16,33 In our system, the strong base would destabilize the AuNPs and interfere with SERS measurements. Nonetheless, we carried out control experiments under Ar atmosphere, where the citrate–AuNPs were exchanged by PhA. The SERS peaks were only slightly weaker than the experiments under ambient environment (Fig. 3a). In addition, the ligand exchange under Ar atmosphere was monitored by SERS (Fig. 3b) and the trend was not particularly different (Fig. 3c).
 | ||
| Fig. 3 (a) SERS intensity at 2013 cm−1 of citrate–AuNPs after 3 h incubation with 8.3 μM PhA without (black) and with (purple) Ar gas bubbling (5 min). (b) Temporal evolution of the SERS intensity at 2013 cm−1 of citrate–AuNPs incubated with 8.3 μM PhA under Ar atmosphere in a cuvette which was sealed in a glove box. (c) Intensity traces of the peak at 2013 cm−1 of citrate–AuNPs incubated with 8.3 μM PhA under ambient (black) and Ar atmosphere (purple). (d) Raman spectra of pure DMF (black), pure THF (red), 60 nm citrate–AuNPs (blue) and PhA–AuNPs (purple) in THF. (e–g) Schematic diagram of the coordination configuration of PhA ligand on the Au surface. | ||
The SERS peak of C–Au σ bond is within 400–420 cm−1.16,33,34 As DMF shows overlapping peaks and THF does not, we use THF as solvent to check the 400 cm−1 region after the ligand exchange with PhA (Fig. 3d). No Raman band was observed, for both the solution sample and the dried samples on different substrates (Fig. S12 and S13, ESI†). We speculate that the PhA ligand may simply adsorb on the Au seeds, instead of forming the strong Au–C bond. It is reasonable considering that no base or catalyst was used in our experiments. Without prior deprotonation, the terminal PhA ligands may adsorb on the Au surface via flat-lying configuration through d–p π coordination bond of the alkene group (Fig. 3e).35–37
It is clear that PhA is a stronger ligand for Au than citrate ion. Thus, we compare it to a typical strong thiol-based ligand 2-NT,25 whose characteristic peaks occur at 515, 596, 767, 1578 and 1615 cm−1. The citrate–AuNPs were first treated with 8.3 μM PhA for 3 h, and then 8.2 μM 2-NT was added to start the exchange (3 h). As shown in Fig. 4a and d, the intensity of the 2013 cm−1 peak decreased by 25% at room temperature. In comparison, the same exchange experiment carried out at 60 °C led to 76% decrease of the peak (Fig. 4b–d), indicating a much more extensive exchange.
 | ||
| Fig. 4 (a) Temporal evolution of the SERS intensity at 2013 cm−1 of 8.3 μM PhA–AuNPs incubated with 8.2 μM 2-NT in DMF at room temperature. (b) Expansion of the framed region in c. (c) Temporal evolution of the SERS intensity of 8.3 μM PhA–AuNPs incubated with 8.2 μM 2-NT in DMF at 60 °C. (d) Intensity traces of the SERS peak at 2013 cm−1 of PhA in a (black), b (red), at 767 cm−1 of 2-NT in c (purple). (e) The SERS intensity at 767 cm−1 of citrate–AuNPs incubated with 8.3 μM and 0.41 μM 2-NT in DMF. (f) The SERS intensity of 0.41 μM 2-NT–AuNPs incubated with 20 mM PhA in DMF. (g) Expansion of the framed region in f. | ||
In the reverse exchange, the citrate–AuNPs were first treated with 8.3 μM 2-NT for 3 h and then 8.2 μM PhA for 3 h. There was no change of peak intensity for both room temperature and 60 °C experiments. When the 2-NT concentration was reduced to 1/20 (0.41 μM), the peak at 767 cm−1 decreased to 68% before the exchange (Fig. 4e). After treatment of 20 mM PhA at 60 °C for 3 h, new PhA peaks emerged but the 767 cm−1 peak of 2-NT showed only 26% decrease (Fig. 4g). It appears that the incoming PhA could easily occupy the empty sites, but the exchange for 2-NT was extremely difficult even when the former was 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times higher in concentration.
000 times higher in concentration.
Given the fact that the exchange of 2-NT for PhA is much easier than the reverse exchange, 2-NT should be a stronger ligand. In the reactions of organic or coordination compounds, exchange could occur via the SN2 mechanism, where the incoming and leaving groups could both form partial bonding at the diametric side of the central atom. For a ligand on a nearly flat metal surface, however, concerted ligand exchange is almost impossible. Instead, the leaving ligand is expected to first dissociate like in the SN1 mechanism, before the binding of the incoming ligand. From our results, it appears that the dynamic exchange between the free and bound PhA cannot be fast, otherwise the weaker PhA would be easily lost. At elevated temperature (60 °C), the higher kinetic energy is expected to promote both dissociation and association reactions, leading to a higher degree of exchange. The concentration bias is also expected to promote exchange, as the dissociated ligand would be statistically less probable to re-associate, given the overwhelming concentration of the incoming ligand.
XPS measurements were carried out for PhA and 2-NT before and after the ligand exchange. As shown in Fig. 5a–c, the presence of 2-NT was verified by the S 2p peak. Compared to the pure 2-NT power (Fig. 5a), the S 2p peak showed slight shift to lower binding energy (from 163.7 to 162.7 eV) due to their coordination on the Au surface (black line, Fig. 5b and c).38–42 The 162.7 eV peak was almost the same for the sample after exchanging 2-NT–AuNPs with PhA (purple line, Fig. 5b), or after exchanging PhA–AuNPs with 2-NT (purple line, Fig. 5c).
 | ||
| Fig. 5 The S 2p XPS of (a) 2-NT powder; (b) 2-NT–AuNPs (black), PhA exchange 2-NT–AuNPs (purple); (c) 2-NT–AuNPs (black), 2-NT exchange PhA–AuNPs (purple). (d) Temporal evolution of the SERS intensity of 8.3 μM PhA–AuNPs incubated with 2.5 mM PVP in DMF. (e) Expansion of the framed region in d. (f) Intensity traces of the peak in e. (g) Temporal evolution of the SERS intensity at 2013 cm−1 of 8.3 μM PVP–AuNPs incubated with 8.2 μM PhA in DMF. (h) Intensity traces of the peak in g. | ||
PVP is a non-ionic polymer with weak binding residues. Its multi-dentate binding mode is thought to be responsible for its relatively strong binding on metal surfaces, preserving the colloidal stability of metal nanoparticles. But its strength of binding is hard to evaluate. To understand its relative strength with PhA, PVP (equivalent monomer concentration of 2.5 mM) was used to exchange PhA–AuNPs (8.3 μM) at 60 °C for 2 h. As shown in Fig. 5d–f, the PhA signal quickly decreased, with only 34% left after 1 h. In the reverse exchange, 8.2 μM of PhA was used to exchange PVP–AuNPs (8.3 μM) at 60 °C for 2 h. As shown in Fig. 5g and h, the PhA peak quickly increased, reaching the plateau intensity in only 15 min. Judging from the relative concentration and the rate of the intensity trace, it is clear that PhA is a stronger ligand than PVP.
A series of phenynyl molecules were studied using this method of ligand exchange at 60 °C for 3 h. As shown in Table 1, the molecules 1–7 was first exchanged with PhA (purple letters), and the relative exchange ratio at 3 h was used to establish a rough trend (1 > 2 > 4 > 5 > 3 > PhA > 6 > 7). The relative strength among 4, 5, 3 and 6, 7 is not consistent from the values of the 2-way exchange. Thus, a second round of 2-way exchange was carried out for these ligand couples. The order of strength is corrected as 3 > 4 > 5 and 6 > 7.
|
|
|
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|---|---|
| Notes: (1) The ligand in each row was used to exchange the ligands in the corresponding column. (2) The data in the brackets is the ligand molar ratio. (3) All the ligand exchange were carried out at 60 °C for 3 h. | ||||||||
| 1 | — | 58% (1:1) |
68% (1:1) |
81% (1:1) |
— | 74% (1:1) |
— | — |
| 81% (10:1) |
86% (10:1) |
98% (10:1) |
||||||
| 2 | 45% (10:1) |
— | 50% (1:1) |
46% (1:1) |
— | 45% (1:1) |
— | — |
| 70% (10:1) |
67% (10:1) |
|||||||
| 3 | 4% (1:1) |
41% (1:1) |
— | 60% (1:1) |
86% (1:1) |
100% (1:1) |
— | — |
| 54% (10:1) |
68% (10:1) |
79% (10:1) |
97% (10:1) |
|||||
| 4 | 5% (1:1) |
23% (1:1) |
56% (1:1) |
— | 48% (1:1) |
56% (1:1) |
— | — |
| 48% (10:1) |
59% (10:1) |
75% (10:1) |
77% (10:1) |
|||||
| 5 | — | — | 64% (1:1) 78% (10:1) |
48% (1:1) |
— | 63% (1:1) |
— | — |
| 75% (10:1) |
||||||||
| PM | 20% (1:1) |
20% (1:1) |
72% (1:1) |
22% (1:1) |
38% (1:1) |
— | 100% (1:1) |
100% (1:1) |
| 38% (10:1) |
42% (10:1) |
72% (10:1) |
43% (10:1) |
73% (10:1) |
||||
| 6 | — | — | — | — | — | 71% (1:1) |
— | 100% (1:1) |
| 7 | — | — | — | — | — | 14% (1:1) |
38% (1:1) |
— |
| 45% (10:1) |
||||||||






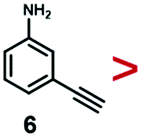

In the corrected series of 1 > 2 > 3 > 4 > 5 > PhA > 6 > 7, molecule 1 with two alkynyl groups is the strongest. As the two CC groups at the para-position cannot possibly bind simultaneously in upright configuration on the Au surface, its much stronger binding affinity than PhA shows that both alkynyl groups attached to the Au surface. It is consistent with the above hypothesis of flat-lying adsorption configuration. Moreover, it appears that the electron-donating (4, 6) or withdrawing groups (3, 5, 7) are not the dominant factor; neither are the polar (3, 4, 5, 6, 7) versus nonpolar molecules (1, 2, PhA).
Conclusions
In summary, we show that phenynyl ligands could readily bind to Au nanoparticles giving strong SERS signals, allowing the binding kinetics to be monitored in real time. Judging from the relative exchange ratio with PhA, the phenynyl ligands are in general weaker than thiol ligand, and stronger than PVP. Most importantly, we show that ligand exchange is not a black-and-white clean process: The exchange can occur both ways, that is, even a weak ligand could replace a strong one. More often than not, the exchange is not complete, even when a strong ligand exchanges for a weak one.43 Elevated temperature is recommended to promote both dissociation and association reactions, and thus the extent of the exchange reaction. Concentration bias is a convenient handle to modulate the exchange. It is recommended to thoroughly remove the initial ligand before applying the new one.We believe that understanding the relative affinity of phenynyl ligands would promote their applications in SERS, nanosynthesis, surface treatment, and beyond. The kinetic SERS monitoring provides a convenient platform for evaluating the relative strength of ligands, which is otherwise difficult to study.
Experimental
The synthesis and phase transfer of AuNPs
The 60 nm AuNPs were synthesized via seeded growth method.6 In a typical synthesis, 0.25 mL HAuCl4 solution (10 mg mL−1) was added to 25 mL H2O in a 500 ml round bottle flask equipped with a condenser. The mixture was refluxed for 30 min in an oil bath, followed by the addition of 0.375 mL sodium citrate solution (1 wt%). The color of the reaction solution changed quickly from light yellow to dark gray and black, then to purple. About 10 min later, the solution changed to red. Monodispersed AuNPs in 40 nm diameter formed at this stage, which were used as seed in later growth stage. After refluxed for another 15 min, 25 mL boiled water was added into the red AuNPs solution, followed by the dropwise addition of 50 μL 6.6 mg mL−1 NaOH solution and the quick addition of 0.25 mL sodium citrate solution (1 wt%) and 0.25 mL HAuCl4 solution (10 mg mL−1). The mixture was heated for 20 min to completely reduce the HAuCl4 and form Au layer on the seed NP surface. In the third cycle, 50 mL H2O, 0.1 mL NaOH, 0.5 mL sodium citrate and 0.5 mL HAuCl4 were added in sequence via the same way. The above cycle was repeated for twice times, after which the AuNPs in 60 nm diameter were obtained.For transferring the AuNPs into DMF solvent, 1.5 mL 60 nm citrate–AuNPs was centrifuged at 4000 rpm for 5 min. After removing the supernatant, the concentrated AuNPs collected at the bottom of centrifuge tube was dispersed in 1.5 mL DMF, centrifuged for the second time (3500 rpm, 10 min). The concentrated AuNPs were re-dispersed in 1.35 mL DMF for ligand exchange. The concentration of the AuNPs at this stage was calibrated by comparing its absorption intensity with that of the as-synthesized AuNPs solution.
Ligand exchange experiments
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21673117 and 91956109), Jiangsu Provincial Foundation for Specially-Appointed Professor, start-up fund at Nanjing Tech University (39837102 and 39837140) and SICAM Fellowship from Jiangsu National Synergetic Innovation Center for Advanced Materials.Notes and references
- L. Xu, H.-W. Liang, Y. Yang and S.-H. Yu, Stability and Reactivity: Positive and Negative Aspects for Nanoparticle Processing, Chem. Rev., 2018, 118, 3209–3250 CrossRef CAS.
- M. S. Inkpen, Z. F. Liu, H. Li, L. M. Campos, J. B. Neaton and L. Venkataraman, Non-chemisorbed gold–sulfur binding prevails in self-assembled monolayers, Nat. Chem., 2019, 11, 351–358 CrossRef CAS.
- B. Fritzinger, I. Moreels, P. Lommens, R. Koole, Z. Hens and J. C. Martins, In Situ Observation of Rapid Ligand Exchange in Colloidal Nanocrystal Suspensions Using Transfer NOE Nuclear Magnetic Resonance Spectroscopy, J. Am. Chem. Soc., 2009, 131, 3024–3032 CrossRef CAS.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Self-Assembled Monolayers of Thiolates on Metals as a Form of Nanotechnology, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS.
- A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak and A. G. Kanaras, The Role of Ligands in the Chemical Synthesis and Applications of Inorganic Nanoparticles, Chem. Rev., 2019, 119, 4819–4880 CrossRef CAS.
- Y. Feng, J. He, H. Wang, Y. Y. Tay, H. Sun, L. Zhu and H. Chen, An Unconventional Role of Ligand in Continuously Tuning of Metal-Metal Interfacial Strain, J. Am. Chem. Soc., 2012, 134, 2004–2007 CrossRef CAS.
- Y. Feng, Y. Wang, J. He, X. Song, Y. Y. Tay, H. H. Hng, X. Y. Ling and H. Chen, Achieving Site-Specificity in Multistep Colloidal Synthesis, J. Am. Chem. Soc., 2015, 137, 7624–7627 CrossRef CAS.
- Y. Feng, Y. Wang, X. Song, S. Xing and H. Chen, Depletion sphere: Explaining the number of Ag islands on Au nanoparticles, Chem. Sci., 2017, 8, 430–436 RSC.
- J. Huang, Y. Zhu, C. Liu, Z. Shi, A. Fratalocchi and Y. Han, Unravelling Thiol's Role in Directing Asymmetric Growth of Au Nanorod-Au Nanoparticle Dimers, Nano Lett., 2016, 16, 617–623 CrossRef CAS.
- L. Lin, Q. Zhang, X. Li, M. Qiu, X. Jiang, W. Jin, H. Gu, D. Y. Lei and J. Ye, Electron Transport Across Plasmonic Molecular Nanogaps Interrogated with Surface-Enhanced Raman Scattering, ACS Nano, 2018, 12, 6492–6503 CrossRef CAS.
- D.-K. Lim, K.-S. Jeon, J.-H. Hwang, H. Kim, S. Kwon, Y. D. Suh and J.-M. Nam, Highly uniform and reproducible surface-enhanced Raman scattering from DNA-tailorable nanoparticles with 1 nm interior gap, Nat. Nanotechnol., 2011, 6, 452–460 CrossRef CAS.
- J.-H. Lee, G.-H. Kim and J.-M. Nam, Directional Synthesis and Assembly of Bimetallic Nanosnowmen with DNA, J. Am. Chem. Soc., 2012, 134, 5456–5459 CrossRef CAS.
- J.-H. Lee, M.-H. You, G.-H. Kim and J.-M. Nam, Plasmonic Nanosnowmen with a Conductive Junction as Highly Tunable Nanoantenna Structures and Sensitive, Quantitative and Multiplexable Surface-Enhanced Raman Scattering Probes, Nano Lett., 2014, 14, 6217–6225 CrossRef CAS.
- S. Zhang, K. L. Chandra and C. B. Gorman, Self-Assembled Monolayers of Terminal Alkynes on Gold, J. Am. Chem. Soc., 2007, 129, 4876–4877 CrossRef CAS.
- Y. Chen, J.-Q. Ren, X.-G. Zhang, D.-Y. Wu, A.-G. Shen and J.-M. Hu, Alkyne-Modulated Surface-Enhanced Raman Scattering-Palette for Optical Interference-Free and Multiplex Cellular Imaging, Anal. Chem., 2016, 88, 6115–6119 CrossRef CAS.
- L. Herrer, A. González-Orive, S. Marqués-González, S. Martín, R. J. Nichols, J. L. Serrano, P. J. Low and P. Cea, Electrically transmissive alkyne-anchored monolayers on gold, Nanoscale, 2019, 11, 7976–7985 RSC.
- H. M. Osorio, P. Cea, L. M. Ballesteros, I. Gascón, S. Marqués-González, R. J. Nichols, F. Pérez-Murano, P. J. Low and S. Martín, Preparation of nascent molecular electronic devices from gold nanoparticles and terminal alkyne functionalised monolayer films, J. Mater. Chem. C, 2014, 2, 7348–7355 RSC.
- N. Kobayashi, Y. Kamei, Y. Shichibu and K. Konishi, Protonation-Induced Chromism of Pyridylethynyl-Appended [core + exo]-Type Au8 Clusters. Resonance-Coupled Electronic Perturbation through π-Conjugated Group, J. Am. Chem. Soc., 2013, 135, 16078–16081 CrossRef CAS.
- X.-K. Wan, Z.-J. Guan and Q.-M. Wang, Homoleptic Alkynyl-Protected Gold Nanoclusters: Au44(PhCC)28 and Au36(PhCC)24, Angew. Chem., Int. Ed., 2017, 56, 11494–11497 CrossRef CAS.
- Z. Lei, J.-J. Li, X.-K. Wan, W.-H. Zhang and Q.-M. Wang, Isolation and Total Structure Determination of an All-Alkynyl-Protected Gold Nanocluster Au144, Angew. Chem., Int. Ed., 2018, 57, 8639–8643 CrossRef CAS.
- J.-J. Li, Z.-J. Guan, Z. Lei, F. Hu and Q.-M. Wang, Same Magic Number but Different Arrangement: Alkynyl-Protected Au25 with D3 Symmetry, Angew. Chem., Int. Ed., 2019, 58, 1083–1087 CrossRef CAS.
- P. Maity, S. Takano, S. Yamazoe, T. Wakabayashi and T. Tsukuda, Binding Motif of Terminal Alkynes on Gold Clusters, J. Am. Chem. Soc., 2013, 135, 9450–9457 CrossRef CAS.
- P. Maity, H. Tsunoyama, M. Yamauchi, S. Xie and T. Tsukuda, Organogold Clusters Protected by Phenylacetylene, J. Am. Chem. Soc., 2011, 133, 20123–20125 CrossRef CAS.
- P. Maity, T. Wakabayashi, N. Ichikuni, H. Tsunoyama, S. Xie, M. Yamauchi and T. Tsukuda, Selective synthesis of organogold magic clusters Au54(CCPh)26, Chem. Commun., 2012, 48, 6085–6087 RSC.
- Y. Feng, S. Xing, J. Xu, H. Wang, J. W. Lim and H. Chen, Probing the kinetics of ligand exchange on colloidal gold nanoparticles by surface-enhanced Raman scattering, Dalton Trans., 2010, 39, 349–351 RSC.
- G. Zhao, Y. Zhou, J. Wang, Z. Wu, H. Wang and H. Chen, Self-Healing of Polarizing Films via the Synergy between Gold Nanorods and Vitrimer, Adv. Mater., 2019, 31, 1900363 CrossRef.
- G. Chen, Y. Wang, L. H. Tan, M. Yang, L. S. Tan, Y. Chen and H. Chen, High-Purity Separation of Gold Nanoparticle Dimers and Trimers, J. Am. Chem. Soc., 2009, 131, 4218–4219 CrossRef CAS.
- M. Yang, G. Chen, Y. Zhao, G. Silber, Y. Wang, S. Xing, Y. Han and H. Chen, Mechanistic investigation into the spontaneous linear assembly of gold nanospheres, Phys. Chem. Chem. Phys., 2010, 12, 11850–11860 RSC.
- Y. Feng, Y. Wang, H. Wang, T. Chen, Y. Y. Tay, L. Yao, Q. Yan, S. Li and H. Chen, Engineering “Hot” Nanoparticles for Surface-Enhanced Raman Scattering by Embedding Reporter Molecules in Metal Layers, Small, 2012, 8, 246–251 CrossRef CAS.
- W. K. H. Ho, Z. Y. Bao, X. Gan, K.-Y. Wong, J. Dai and D. Lei, Probing Conformation Change and Binding Mode of Metal Ion-Carboxyl Coordination Complex through Resonant Surface-Enhanced Raman Spectroscopy and Density Functional Theory, J. Phys. Chem. Lett., 2019, 10, 4692–4698 CrossRef CAS.
- H. Zhang, P. Yin, T. You, T. Sun, X. Lang, E. Tan, X. Liang and L. Guo, Au@Phenyacetylene organogold clusters: Direct spectroscopic evidence of gold-carbon covalent band, Spectrochim. Acta, Part A, 2015, 134, 96–100 CrossRef CAS.
- G. Chen, Y. Wang, M. Yang, J. Xu, S. J. Goh, M. Pan and H. Chen, Measuring Ensemble-Averaged Surface-Enhanced Raman Scattering in the Hotspots of Colloidal Nanoparticle Dimers and Trimers, J. Am. Chem. Soc., 2010, 132, 3644–3645 CrossRef CAS.
- A. Moneo, A. González-Orive, S. Bock, M. Fenero, I. L. Herrer, D. C. Milan, M. Lorenzoni, R. J. Nichols, P. Cea, F. Perez-Murano, P. J. Low and S. Martin, Towards molecular electronic devices based on ‘all-carbon’ wires, Nanoscale, 2018, 10, 14128–14138 RSC.
- L. Laurentius, S. R. Stoyanov, S. Gusarov, A. Kovalenko, R. Du, G. P. Lopinski and M. T. McDermott, Diazonium-Derived Aryl Films on Gold Nanoparticles: Evidence for a Carbon-Gold Covalent Bond, ACS Nano, 2011, 5, 4219–4227 CrossRef CAS.
- Q. Li, C. Han, M. Fuentes-Cabrera, H. Terrones, B. G. Sumpter, W. Lu, J. Bernholc, J. Yi, Z. Gai, A. P. Baddorf, P. Maksymovych and M. Pan, Electronic Control over Attachment and Self-Assembly of Alkyne Groups on Gold, ACS Nano, 2012, 6, 9267–9275 CrossRef CAS.
- M. L. Patterson and M. J. Weaver, Surface-enhanced Raman spectroscopy as a probe of adsorbate-surface bonding: simple alkenes and alkynes adsorbed at gold electrodes, J. Phys. Chem., 1985, 89, 5046–5051 CrossRef CAS.
- H. Feilchenfeld and M. J. Weaver, Binding of alkynes to silver, gold, and underpotential deposited silver electrodes as deduced by surface-enhanced Raman spectroscopy, J. Phys. Chem., 1989, 93, 4276–4282 CrossRef CAS.
- D. G. Castner, K. Hinds and D. W. Grainger, X-ray Photoelectron Spectroscopy Sulfur 2p Study of Organic Thiol and Disulfide Binding Interactions with Gold Surfaces, Langmuir, 1996, 12, 5083–5086 CrossRef CAS.
- E. Pensa, E. Cortés, G. Corthey, P. Carro, C. Vericat, M. H. Fonticelli, G. Benítez, A. A. Rubert and R. C. Salvarezza, The Chemistry of the Sulfur-Gold Interface: In Search of a Unified Model, Acc. Chem. Res., 2012, 45, 1183–1192 CrossRef CAS.
- C. Vericat, M. E. Vela, G. Benitez, P. Carro and R. C. Salvarezza, Self-assembled monolayers of thiols and dithiols on gold: new challenges for a well-known system, Chem. Soc. Rev., 2010, 39, 1805–1834 RSC.
- H. Wang, F. Cheng, M. Li, W. Peng and J. Qu, Reactivity and Kinetics of Vinyl Sulfone-Functionalized Self-Assembled Monolayers for Bioactive Ligand Immobilization, Langmuir, 2015, 31, 3413–3421 CrossRef CAS.
- A. W. Snow, E. E. Foos, M. M. Coble, G. G. Jernigan and M. G. Ancona, Fluorine-labeling as a diagnostic for thiol-ligand and gold nanocluster self-assembly, Analyst, 2009, 134, 1790–1801 RSC.
- J. B. Schlenoff, M. Li and H. Ly, Stability and Self-Exchange in Alkanethiol Monolayers, J. Am. Chem. Soc., 1995, 117, 12528–12536 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of characterizations, absorption and temporal evolution of the SERS spectra. See DOI: 10.1039/d0qm00612b |
| This journal is © the Partner Organisations 2021 |