
Sendai virus acts as a nano-booster to excite dendritic cells for enhancing the efficacy of CD47-directed immune checkpoint inhibitors against breast carcinoma†
Yanan
Xu‡
a,
Bin
Zheng‡
 *bc,
Mengqian
Huang
a,
Xianhuang
Li
a,
Zhiyun
Wang
d,
Jin
Chang
a and
Tao
Wang
*a
*bc,
Mengqian
Huang
a,
Xianhuang
Li
a,
Zhiyun
Wang
d,
Jin
Chang
a and
Tao
Wang
*a
aSchool of Life Sciences, Tianjin University, 92 Weijin Road, Nankai District, Tianjin 300072, China. E-mail: wangtaobio@tju.edu.cn
bAcademy of Medical Engineering and Translational Medicine, Tianjin University, 300072, Tianjin, China. E-mail: binzheng@tju.edu.cn
cTianjin Key Laboratory of Brain Science and Neural Engineering, Tianjin University, 300072, Tianjin, China
dSchool of Environmental Science and Engineering, Tianjin University, 92 Weijin Road, Nankai District, Tianjin 300072, China
First published on 18th September 2020
Abstract
Dendritic cells (DCs) are vital hubs for exciting systemic adaptive immune responses. But the uppermost antigen presenting cells are in a state of severe suppression in breast carcinoma for making tumor cells more resistant and viable. In order to reverse this immunosuppressive state in the tumor microenvironment, here we showed that non-pathogenic Sendai virus (SeV) is employed to execute multi-channel recruitment and excitation of DCs. Simultaneously, the signal of “don't eat me” on the surface of tumor cells is blocked with a CD47 blocker to greatly enhance the phagocytosis and antigen presentation of macrophages to the tumor cells. With the excitation of these two cells by SeV and CD47 blocker nanocomposites, breast carcinoma antigens are presented to the lymph nodes in large numbers to mature T lymphocytes for specifically killing tumor cells. Our findings indicate that the nanohybrids can successfully excite DCs and macrophages in vitro and in vivo, and then systemically arouse cytotoxic T lymphocytes and helper T lymphocytes, and further motivate the formation of humoral immunity and immune memory. Therefore, the oncolytic nanocomposite strategy combining non-genetically modified viruses and immune checkpoint inhibitors might serve as the next generation of personalized anti-tumor immunotherapy agents, representing an alternative way to suppress tumor metastasis and recurrence.
1. Introduction
Immunotherapy is a novel treatment method to control and clear tumors by restarting and maintaining the immune system's ability to recognize and eliminate tumor cells1–3 as well as restoring the systemic anti-tumor immune response.4–6 This treatment has better specificity and efficiency and avoids bodily damage compared to existing methods for surgical treatment, radiotherapy and chemotherapy7,8 and has been the subject of study in recent years.6,9 However, current immunotherapy methods still have numerous shortcomings, especially free therapeutic antibodies based on immune checkpoint, such as programmed death-1 (PD-1/PD-L1), cytotoxic T lymphocyte antigen 4 (CTLA-4), lymphocyte activation gene-3 (LAG-3), T cell immunoglobulin-3 (TIM-3), T cell immunoglobulin and ITIM domain protein (TIGIT), and V-domain immunoglobulin-containing suppressor of T cell activation (VISTA), are easily removed by the body,10–13 and can also cause many side effects, such as diarrhea and colitis, skin rash and mucous membrane irritation, liver toxicity, endocrine disorders, nervous system syndrome, hematopoietic system syndrome, infection, etc.14–18 Moreover, these methods have a short effect time and a single mode of action, causing inefficient clearance of tumors and, ultimately, tumor recurrence and metastasis.19–21 Therefore, there is an urgent need to introduce novel strategies to overcome these shortcomings faced by conventional immune checkpoint inhibitors in order to improve the safety and effectiveness of anti-tumor immunotherapy.The immune system is a self-protection mechanism formed by organisms in the long-term evolution against microbial invasion. In view of the complex structures of microorganisms, which can excite the body's immune protection mechanism from multiple signaling pathways,22–24 microorganisms have become a “booster” for tumor immunotherapies.25,26 Abundant non-pathogenic viruses, such as deactivated or non-infectious viruses,18,27–29 can strongly stimulate host innate and adaptive immune systems in vivo by a variety of pathogen-associated molecular patterns (PAMPs) to recruit and arouse immunocytes including macrophages, dendritic cells (DCs),30 natural killer (NK) cells and cytotoxic T lymphocytes (CTLs).31,32 Interestingly, these innate and adaptive immunocytes are also effector cells for eliminating tumor cells.33–35 These immune cells can also secrete a wide variety of immune-activating cytokines upon maturation including interleukins (IL), tumor necrosis factors (TNF) and interferons (IFN) after being stimulated by non-pathogenic viruses.36,37 These cytokines in turn enhance the phagocytosis of immunocytes towards tumor cells, the presentation of tumor antigens, the activation of immune cells, the killing effect of immune cells on tumors, and the formation of anti-tumor immune memory.38–42 Therefore, non-pathogenic viruses may serve as a powerful tool for stimulating the systemic immune system to induce durable tumor regression and prevent tumor metastasis.
Integrin-associated protein (IAP), also known as CD47, is abundantly expressed on the surface of several malignant cell types, allowing tumor cells to escape recognition by innate immunocytes by acting as an anti-phagocytic “don’t eat me” signal.43,44 This is a major cause of the failure of most tumor treatment methods. Hence, blocking the interaction of CD47 with its ligand signal regulatory protein-α (SIRPα),45 which is expressed on macrophages, DCs and neutrophils, will arouse innate immunocytes and promote the capacity of these immunocytes to phagocytose tumor cells. Recent studies have demonstrated that CD47-blockade not only increases the phagocytosis of tumor cells but also promotes cross-presentation of tumor antigens by the STING-pathway of DCs to enhance priming of anti-tumor effector T cells.46,47 Therefore, CD47-directed immune checkpoint inhibitors have an important role in anti-tumor immunotherapy by awaking phagocytosis towards tumor cells and tumor antigen presentation by macrophages and DCs. However, as demonstrated in both preclinical models and human trials, systemic CD47 blockade often causes anemia and thrombocytopenia due to the high expression of CD47 on red blood cells and platelets, respectively.48 Hence, efforts to avoid these severe side effects are essential to make CD47-blocking immunotherapies clinically applicable. In addition, the excitation of Toll-like receptor (TLR) pathways in DCs by some classic viral stimuli, which is the most important pathway for arousing DCs, is likely to improve the efficacy of anti-tumor immunotherapy for CD47-directing immune checkpoint inhibitors.
Herein, we report a novel strategy of non-pathogenic viruses to boost anti-tumor immunotherapy based on immune checkpoint. To efficiently stimulate the innate immune pathway in the tumor microenvironment, we employed non-pathogenic Sendai virus (SeV),49 a commonly used innate immune activator,50,51 which can directly actuate DCs by combining its multiple complex structures with different cellular pattern recognition receptors (PRRs) based on the TLR-PAMP immune activation pathway, and promote the expression of CD11c, MHC II, CD80 and CD86 on the surface of DCs, and stimulate DCs and tumor cells to secrete IFN-β, IL-6 and other immune enhancing cytokines,52,53 among which IFN-β can fully arouse NK cells, enhance the killing capability of activated NK cells, and secrete IFN-γ, which in turn can further promote the excitation of DCs and macrophages for improving the level of innate immunity and cellular immunity. Besides, SeV can stimulate DCs to produce IL-6, which can inhibit the vitality of regulatory T cells (Tregs), and promote the proliferation of CD4+ and CD8+ T cells. Hence, SeV plays an important role in the treatment of tumors, allergic diseases, infectious diseases and patients with low immune function. In addition, to prevent tumor cells from escaping surveillance and elimination by the immune system, the anti-CD47 antibody (aCD47) was employed to block CD47 molecules on the surface of malignant cells from interacting with SIRPα. The biocompatible poly(lactic-co-glycolic acid)-b-poly(ethylene glycol) (PLGA–PEG) was used as a carrier to assemble SeV and aCD47 to serve as a release reservoir of immunomodulatory therapeutics.54–57 To trace the immunotherapeutic agents in real-time, a fluorescent molecule of N,N′-((6,7-diphenyl-[1,2,5]thiadiazolo[3,4-g]quinoxaline-4,9-diyl)bis(4,1-phenylene))bis(N-phenylnaphthalen-1-amine) (TQ-BPN), owing to the aggregation-induced emission (AIE) effect in the near-infrared region,58,59 was selectively embedded in the hydrophobic shell of the amphiphilic PLGA–PEG to form (SeV + aCD47)@PLGA nanohybrids (Scheme 1). The SeV from the nanocomposites can efficiently recruit and stimulate DCs to the periphery of the tumor, while the CD47 blocker can specifically shield the “don't eat me” signal on the surface of breast cancer cells, which in turn causes the tumor cells to be effectively phagocytosed and tumor antigens are presented to lymph nodes to mature T lymphocytes. Therefore, the oncolytic nanocomposite strategy combining non-genetically modified viruses and CD47 blocker might serve as the next generation of personalized anti-tumor immunotherapy agents with enhanced features over standard free immune checkpoint inhibitor regimens, representing an alternative way to suppress tumor metastasis and recurrence.
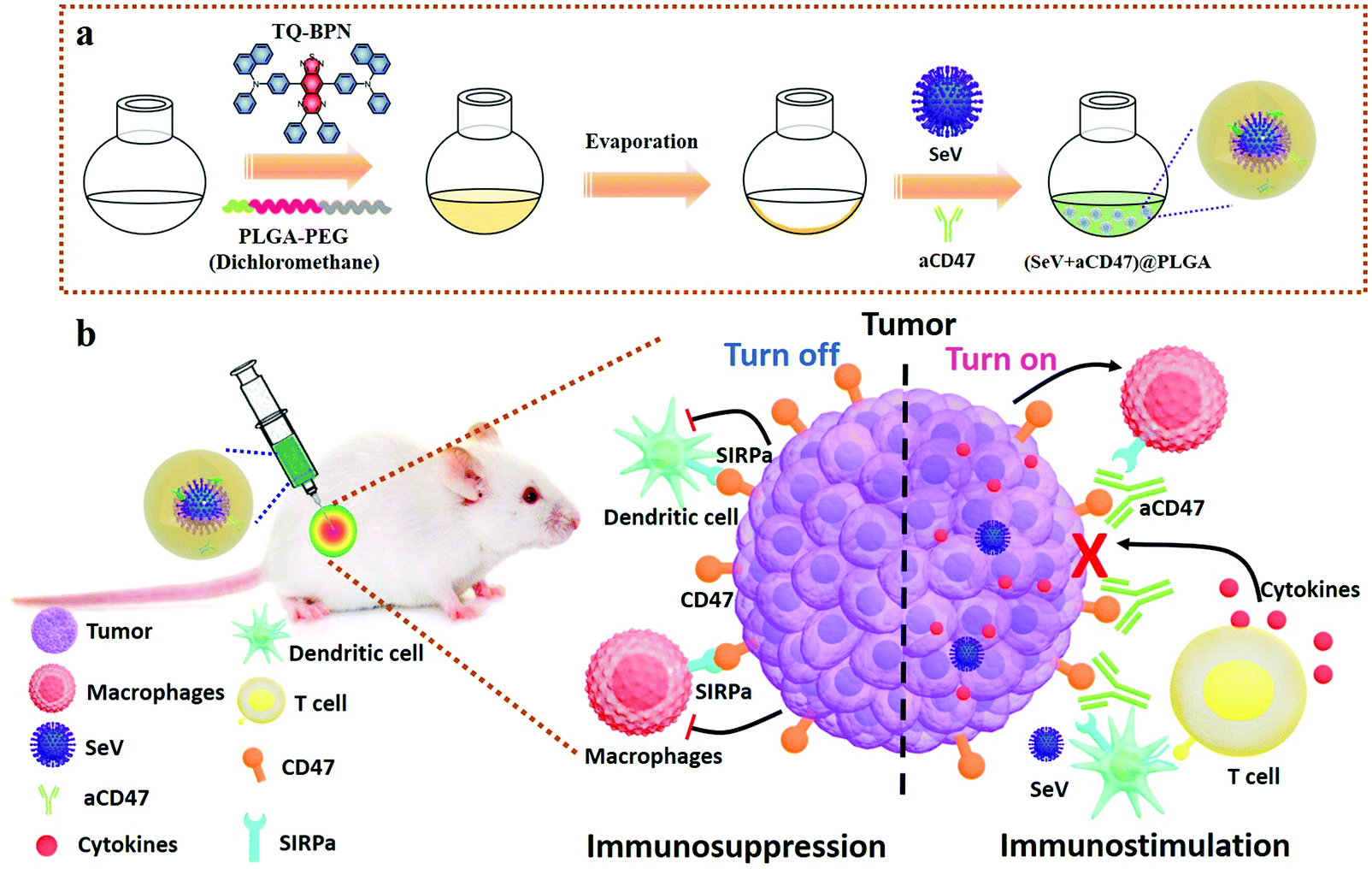 | ||
| Scheme 1 Schematic diagram of oncolytic nanohybrid synthesis and excitation of immune responses. (a) The fabrication process of oncolytic nanocomposites by poly(lactic-co-glycolic acid)-b-poly(ethylene glycol) (PLGA–PEG) block co-polymers self-assembling with Sendai virus (SeV) and aCD47 ((SeV + aCD47)@PLGA). (b) Schematic showing that the oncolytic nanocomposites turn on the anti-tumor response. The SeV from the nanohybrids can efficiently recruit and stimulate DCs to the periphery of the tumor, while the CD47 blocker can specifically shield the “don't eat me” signal on the surface of breast cancer cells, which in turn causes the tumor cells to be effectively phagocytosed and tumor antigens are presented to lymph nodes to mature T lymphocytes for awakening the systemic specific anti-tumor immune killing. Besides, the nanocomposites stimulate a large number of cytokines secreted by immune cells, which in turn can further enhance the phagocytosis of immunocytes towards tumor cells, the presentation of tumor antigens, the activation of immune cells, the killing effect of immune cells on tumors, and the formation of anti-tumor immune memory. | ||
2. Materials and methods
2.1 Materials, cell lines and animals
Poly(lactic-co-glycolic acid)-b-poly(ethylene glycol) (PLGA–PEG) di-block copolymers were synthesized by the following method. First, PLGA-COOH was dissolved in N,N-dimethylformamide, and EDC and sulfo-NHS were added for 10 min to activate the carboxylic group. Then PEG-NH2 was added and allowed to react for 3 h under stirring. Finally, PLGA–PEG was obtained by dialyzing the reaction products overnight. Anti-mouse CD47 was purchased from Biolegend (cat. no. 127518). Sendai virus (SeV) was provided by Professor Lishu Zheng from Chinese Center for Disease Control and Prevention; dendritic cells 2.4 (immature immune cells) (DC 2.4) were provided by Professor Shaokai Sun from Tianjin Medical University (China). 4T1 (mouse breast cancer cells) and macrophages (RAW 264.7 cells) were purchased from Cell Bank of the Chinese Academy of Sciences. Female BALB/c mice (6–8 weeks) were purchased from the Beijing HFK Bioscience Co, Ltd. All mouse studies were carried out following protocols approved by Tianjin Medical University Animal Care and Use Committee and complied with all relevant ethical regulations.2.2 Nanoparticle preparation and characterization
PLGA, aCD47@PLGA, SeV@PLGA, and (SeV + aCD47)@PLGA nanoparticles were prepared by the thin film dispersion method. First, an appropriate amount of PLGA–PEG was dissolved in dichloromethane to prepare a solution of 0.5 mg mL−1. The AIE agent was dissolved in dichloromethane to prepare a solution of 100 mg mL−1, and PVA was dissolved in distilled water to prepare a solution of 6 mg mL−1. Next, 2 mL of PLGA–PEG solution was pipetted into a bottle for 10 min; 100 μL of N,N′-((6,7-diphenyl-[1,2,5]thiadiazolo[3,4-g]quinoxaline-4,9-diyl)bis(4,1-phenylene))bis(N-phenylnaphthalen-1-amine) (TQ-BPN) and 1 mL of PVA solution were slowly added during ultrasonication. Following ultrasonication completion, the liquid was transferred to a round bottom bottle and placed on a rotary evaporator until all the dichloromethane and water were evaporated and a film was formed on the bottom of the bottle. Then, 1 mL SeV was added to a round bottom flask containing the copolymer, to which an appropriate amount of diluted anti-CD47 antibody was added; this mixture was stirred until uniformly mixed at 4 °C and then transferred into an EP tube for use.The prepared (SeV + aCD47)@PLGA nanoparticles were studied by dynamic light scattering (laser particle size analyzer, LITESIZER 500), and the nanoparticle morphology was observed with a transmission electron microscope (Hitachi HT7700). The fluorescence emission spectrum and the fluorescence excitation spectrum were measured with a fluorophotometer (Edinburgh Instruments Ltd, Spectrofluometer FS5), and the ultraviolet absorption spectrum was measured with a multi-plate reader (EnSpire Multilabel Reader). Fluorescence was detected with a small animal in vivo imaging system (IVIS Spectrum).
2.3 Dual luciferase reporter assay
HEK293T cells were grown in the log phase, and the cell concentration was adjusted for plating on a 24-well plate. The cells were maintained in a 37 °C, 5% CO2 incubator. After 24 hours of culture, each well was transfected with 200 ng of IFN-β-luc reporter plasmid and 1 ng of pRL-SV40 plasmid or the indicated amount of empty plasmid for 24 h. Subsequently, fresh medium containing the respective materials was added separately to each well. After 16 hours of culture, the supernatant was aspirated and the cells were washed 3 times with PBS. One hundred microliters of 1× lysis buffer PLB was added to each well, and then the 24-well plate was shaken for 20 min (to ensure total cell lysis), centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min, and then processed with a Centro XS3 LB 960 (Berthold Technologies).
000 rpm for 10 min, and then processed with a Centro XS3 LB 960 (Berthold Technologies).
2.4 Cytokine measurements
DC 2.4 cells and RAW 264.7 cells were grown in the log phase and the concentration was adjusted prior to being grown on a 6-well plate. After 24 hours of culture, fresh medium containing different materials was added and the culture was continued for 48 hours. The culture supernatant was then collected for cytokine detection, and the precipitate was removed by centrifugation for 10 min. The cells were incubated with the diluted antibody for 1.5 h. After washing the plate, the cells were incubated with horseradish peroxidase for 30 min, and then the color developing solution was added. The stop solution was added at the end of the color development, and the absorbance was measured at 450 nm. This study was performed using an ELISA kit of IL-6 (MULTISCIENCES, EK206/3), TNF-α (MULTISCIENCES, EK282/3) and INF-γ (MULTISCIENCES, EK280/3).2.5 In vitro flow cytometry analysis of activated APCs
DC 2.4 cells and RAW 264.7 cells were grown in the log phase, and the cell concentration was adjusted for plating on a 6-well plate. After 24 hours of culture, fresh medium containing different materials was separately added to each well, and the culture was then maintained for 48 hours. The culture supernatant was aspirated and washed twice with PBS prior to cell collection. For overnight permeabilization, 70% ethanol solution was added. The cells were labeled with fluorescent anti-mouse CD80 (Biolegend), and rat mAb to CD83 (Abcam) following the manufacturer's instructions for staining. The labeled cells were studied on a cell expansion flow cytometer (BD FACSCalibur) and analyzed using FlowJo software. The numbers displayed in the flow cytometry analysis image are based on percentages.2.6 Endocytosis
After incubating the nanocomplexes with DC 2.4 cells and RAW 264.7 cells for 4 h, the cells were washed with PBS and then fixed with a 4% paraformaldehyde solution for 15 min. After thorough washing, the cells were permeabilized with 0.1% Triton X-100 for 10 min. Subsequently, the cells were stained with a 50 mg mL−1 fluorescent phalloidin conjugate solution for 40 min at room temperature. The unbound phalloidin conjugate was removed through several washes with PBS. The cells were stained with 4,6-diamino-2-phenylhydrazine hydrochloride (DAPI, Solarbio Life Sciences) for 10 min at room temperature. This was followed by observation with a laser confocal microscope (PerkinElmer, UltraView Vox).2.7 In vitro phagocytosis experiments
4T1 mouse breast cancer cells were grown in the log phase, and then the cell concentration was adjusted for plating on a 6-well plate. The culture medium was changed for medium containing different samples after 24 hours, and the culture was continued for an additional 48 hours. Simultaneously, mouse macrophages were grown in the log phase, and the cellular concentration was adjusted for plating on a 6-well plate for 24 hours. Following medium removal, the macrophages were stained with CellTracker™ Green CMFDA dye (Thermo-Fisher Scientific) for 30 min and the 4T1 cells were stained with CellTracker™ Dark Red (Thermo-Fisher Scientific) for 30 min. The 4T1 cells were co-cultured with the macrophages in serum-free medium and incubated for 3 h at 37 °C. Phagocytosis was observed by confocal microscopy (PerkinElmer, UltraView Vox).2.8 Animal experiments
To investigate the therapeutic effect of (SeV + aCD47)@PLGA, 1 × 106 4T1 cells were injected into the left and right sides of mice. When the tumors reached 5–7 mm in diameter, these mice were randomized into 5 groups (n = 5 per group) for different treatments. The tumors were injected with 50 μL of the control or nanocomposite, depending on these groups (i.e., PBS, PLGA, aCD47@PLGA, SeV@PLGA and (SeV + aCD47)@PLGA).The tumors were monitored daily to measure changes in the tumor volume and mouse body weight. After various treatments, in vivo fluorescence images of tumors and major organs were obtained using a small animal in vivo imaging system (IVIS SPECTRUM). Mice were anesthetized with isoflurane in an induction chamber (up to 5%; 1–3% maintenance). Anesthesia was maintained through the nose cone, and the mice were subsequently photographed. All the animal experiments involved in this work were approved by the Animal Ethics Committee of Tianjin University.
2.9 In vivo fluorescence detection
Tumor sections were prepared with a cryostat (CM1950, Germany). The sections were placed with fixative for 15 min, and then washed and incubated with 0.1% Triton X-100 for 10 min using antibodies for CD3+ (Biolegend) and CD8+ (Biolegend) staining overnight at 4 °C according to the manufacturer's instructions. The next day, they were stained with DAPI for 10 min. After mounting the film, the slides were observed with an inverted fluorescence microscope (Olympus, IX73) and confocal microscopy (PerkinElmer, UltraView Vox).2.10 In vivo measurement of immune activation
In order to study the in vivo immunotherapy effect of the oncolytic nanocomposites, 4T1 cells were injected into mice. After tumor formation, the mice were grouped and treated. Seven days after the treatment, the mice were euthanized, and the tumor infiltration fluid, spleen, lymph nodes, blood, and abdominal fluid of the mice were collected. The cells were stained with fluorescence-labelled antibodies CD45, CD11b, CD44, CD62L, CD138, F4/80, CD80, CD83, CD3, CD4, CD8, Foxp3, CD11c, CD103, Ly-6Chi and Gr-1 following the manufacturer's instructions (all antibodies above are from Biolegend). The stained cells were measured on a flow cytometer and analysed using FlowJo software.3. Results and discussion
3.1 Synthesis and characterization of oncolytic nanocomposites
Oncolytic nanocomposites were prepared from PLGA–PEG block copolymers self-assembling with SeV and aCD47. Monodisperse oncolytic nanocomposites were obtained, and were found by transmission electron microscopy (TEM) and dynamic light scattering (DLS) to have a diameter of approximately 170 nm (Fig. 1a–c); the size was controlled by the agitation speed. The optical properties of the AIE from the TQ-BPN molecule, embedded in the hydrophobic shell of the amphiphilic PLGA–PEG from the (SeV + aCD47)@PLGA nanoparticles, were tested by UV-vis spectrophotometry and fluorescence spectroscopy. The results showed that the largest absorption peak was at 625 nm and the largest emission peak was at 810 nm for the nanoparticles (Fig. 1e and f and Fig. S1, ESI†). Therefore, the TQ-BPN molecule is a near-infrared fluorescent probe with high penetrability. The nanoparticles were added to a 96-well plate at different concentrations, and the fluorescence from TQ-BPN was monitored using an in vivo imaging system (IVIS) (Fig. 1d and h), while the fluorescence of free TQ-BPN molecules was weak. Hence, these results also suggested that the polymer was assembled into nanoparticles, and the TQ-BPN molecule was successfully embedded in the hydrophobic shell of the polymer vesicle, which could be used as a molecular probe for tracing SeV and aCD47 in real time. In short, due to the particular aggregation-induced emission effect of the TQ-BPN molecule, it has better tracking ability than conventional near-infrared dyes.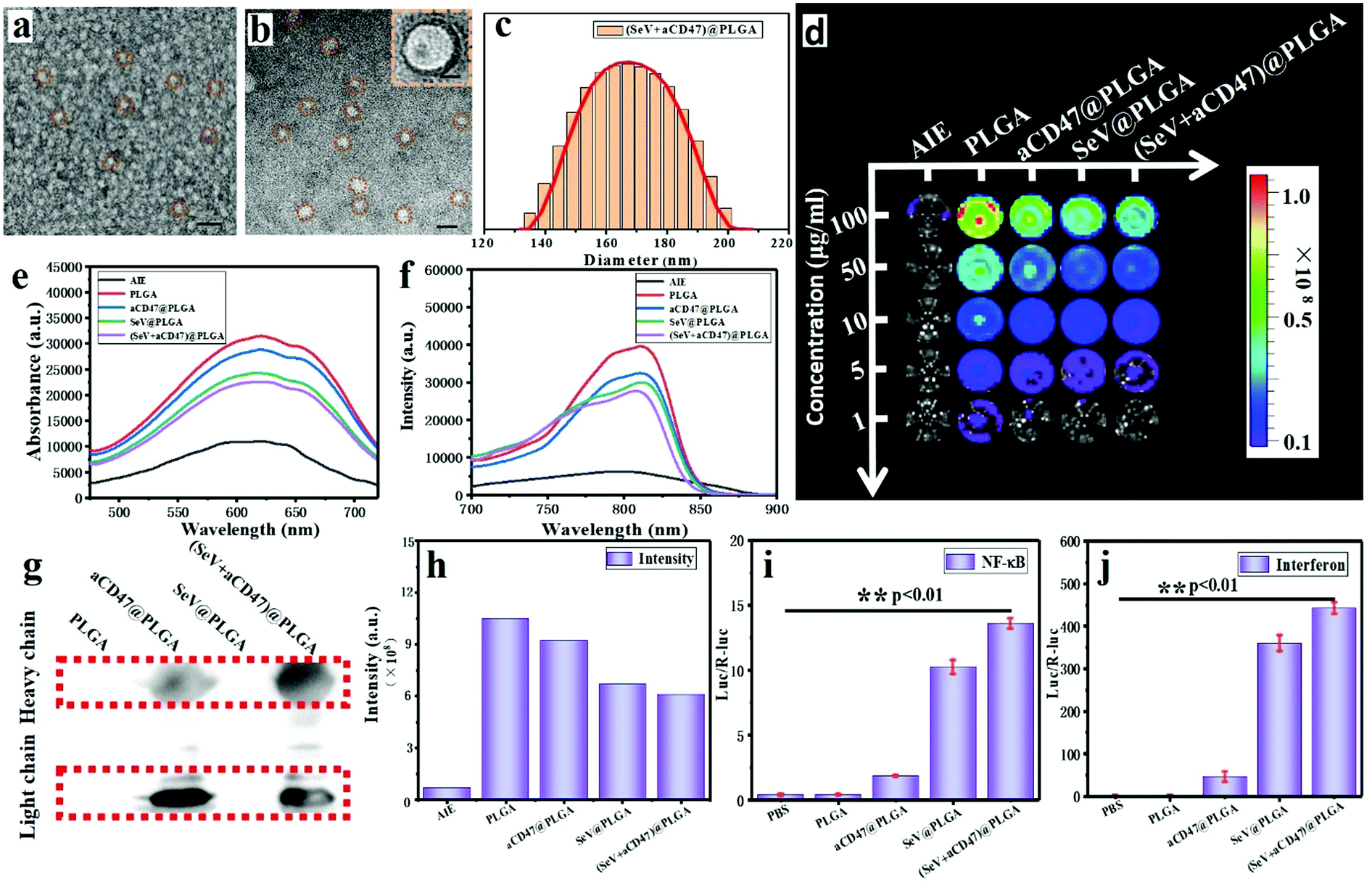 | ||
| Fig. 1 Schematic and characterization of the oncolytic nanohybrids. (a) Transmission electron microscopy (TEM) image of SeV with negative staining using sodium phosphotungstate. Scale bar, 250 nm. (b) Transmission electron microscopy (TEM) image of (SeV + aCD47)@PLGA nanoparticles with negative staining using sodium phosphotungstate. Scale bar, 250 nm. The insert shows an enlarged image of the nanoparticle. The scale bar represents 50 nm. Experiments were repeated three times; a representative image is shown. (c) Average hydrodynamic size of (SeV + aCD47)@PLGA nanoparticles determined by dynamic light scattering. (d) The fluorescence properties of different samples after TQ-BPN was embedded in the hydrophobic shell of PLGA–PEG were monitored using an in vivo imaging system (IVIS). (e) UV-vis absorption spectra. (f) Fluorescence emission spectra. (g) Detection of anti-CD47 antibodies in nanoparticles by western blotting. (h) Quantification of 100 μg ml−1 different sample solutions by IVIS. a.u., arbitrary unit. The activity of double-reporter genes containing (i) NF-κB and (j) interferon β (IFN-β) promoters was detected respectively by firefly luciferase and renilla luciferase. n = 5, mean ± SD. Statistical significance was calculated via one-way analysis of variance (ANOVA) with a Tukey post hoc test. **P < 0.01. | ||
In order to demonstrate that the anti-CD47 antibody was successfully encapsulated into the nanoparticles, we detected the presence of light chains and heavy chains from antibodies in the nanoparticles by western blotting (Fig. 1g). To determine whether the nanoparticles contained functional SeV capable of activating the natural immune response pathway, we tested the activity of double-reporter genes containing interferon-β (IFN-β) and NF-κB promoters driving firefly luciferase and renilla luciferase, respectively. The results exhibited that SeV could still strongly activate the expression of IFN-β and NF-κB after being embedded into nanoparticles with PLGA–PEG (Fig. 1i and j). These results indicated that the viruses were successfully encapsulated in the nanoparticles and still had corresponding immune activation properties. According to the above measurements, Sendai virus and CD47-directed immune checkpoint inhibitor nanohybrids were successfully prepared.
3.2 Oncolytic nanocomposites triggered innate immune cells in vitro
Dendritic cells (DCs) are the most powerful professional antigen presenting cells (APCs) in vivo, efficiently absorbing, processing and presenting antigens.60 Excitation of DCs by exogenous foreign bodies leads to maturation of initial T cells—the central link in initiating, regulating and maintaining the anti-neoplastic immune response.61 The increased expression of CD80, which recognizes and binds to B7 molecules on T cells to provide the T cell maturation signal, as well as CD83, which modulates the maturity of DCs and excites T cells and B cells, on the cytomembrane was recognized as an important marker for DC maturation.To demonstrate that (SeV + aCD47)@PLGA nanoparticles could excite DCs successfully in vitro, first, the biocompatibility and endocytosis of the nanoparticles were detected by live–dead cell staining, the MTT method, hemolysis tests and fluorescence imaging (Fig. S2–S6, ESI†). Immediately thereafter, CD80 and CD83 markers on the surface of DC 2.4 cells were quantitatively detected by flow cytometry and laser scanning confocal microscopy after co-incubation of oncolytic nanocomposites and cells for 48 h. SeV@PLGA and (SeV + aCD47)@PLGA both resulted in remarkably higher expression of CD80 and CD83 (Fig. 2a–d and Fig. S7, S8, ESI†). These results speculated that the synthesized oncolytic nanocomposites could effectively stimulate DCs in vitro. Notably, similar to the expression of CD molecules on the DC 2.4 cell surface, the SeV@PLGA and (SeV + aCD47)@PLGA nanoparticles induced the mass secretion of tumor necrosis factor alpha (TNF-α) and interleukin 6 (IL-6) (Fig. 2e and f). Immunofluorescence staining visually also indicated the remarkable increase in DC 2.4 cells after incubation with the (SeV + aCD47)@PLGA nanohybrids (Fig. 2g and Fig. S9, ESI†). Therefore, these results demonstrated that the oncolytic nanocomposites were efficiently phagocytosed by DC 2.4 cells and promote APC maturity for triggering the anti-tumor immune response.
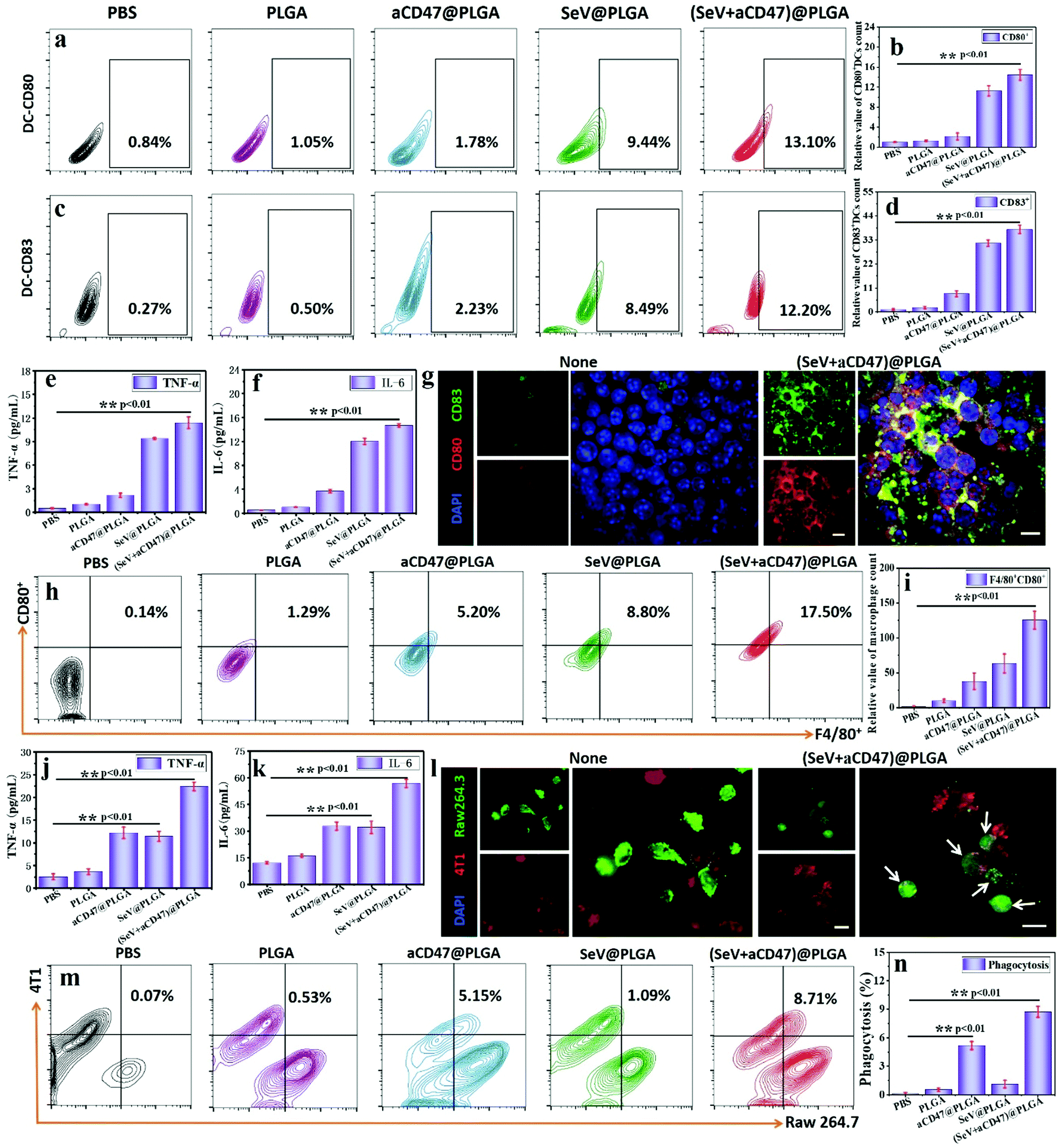 | ||
| Fig. 2 Innate immune response induced by oncolytic nanohybrids in vitro. DC 2.4 cells were incubated with oncolytic nanocomposites for 48 h. (a) Expression levels of surface molecules (CD80) on DC 2.4 cells after incubation with PBS, PLGA, aCD47@PLGA, SeV@PLGA and (SeV + aCD47)@PLGA analyzed by flow cytometry. (b) The relative quantification of DC 2.4 cells (CD80 molecules). n = 5, mean ± SD. (c) Expression levels of surface molecules (CD83) on DC 2.4 cells after incubation with PBS, PLGA, aCD47@PLGA, SeV@PLGA and (SeV + aCD47)@PLGA analyzed by flow cytometry. (d) The relative quantification of DC 2.4 cells (CD83 molecules). n = 5, mean ± SD. Secretion levels of (e) TNF-α and (f) IL-6 in the DC 2.4 cell culture supernatant after incubation with PBS, PLGA, aCD47@PLGA, SeV@PLGA and (SeV + aCD47)@PLGA for 48 h. n = 5, mean ± SD. (g) Representative immunofluorescence images of DC 2.4 cells showing CD80+ and CD83+ for the groups with or without (SeV + aCD47)@PLGA. Scale bars, 25 μm. Experiments were repeated three times. (h) Expression levels of surface molecules (F4/80 and CD80) on RAW 264.7 cells after treatment with PBS, PLGA, aCD47@PLGA, SeV@PLGA and (SeV + aCD47)@PLGA analyzed by flow cytometry. (i) The relative quantification of DC RAW 264.7 cells (F4/80 and CD80 molecules). n = 5, mean ± SD. Secretion levels of (j) TNF-α and (k) IL-6 in the RAW 264.7 cell culture supernatant after incubation with PBS, PLGA, aCD47@PLGA, SeV@PLGA and (SeV + aCD47)@PLGA for 48 h. n = 5, mean ± SD. (l) Representative fluorescence images of RAW 264.7 cells and phagocytic 4T1 cells following treatment with oncolytic nanocomposites. Scale bars, 50 μm. Experiments were repeated three times. (m) Quantitative analysis of macrophage phagocytic tumor cells by flow cytometry after 4T1 cell treatment with PBS, PLGA, aCD47@PLGA, SeV@PLGA and (SeV + aCD47)@PLGA. (n) The relative quantification of macrophage phagocytic tumor cells. n = 5, mean ± SD. Statistical significance was calculated via one-way analysis of variance (ANOVA) with a Tukey post hoc test. **P < 0.01. | ||
In order to verify that the (SeV + aCD47)@PLGA nanoparticles could successfully actuate macrophages and enhance the ability to phagocytose cancer cells, flow cytometry, ELISA and confocal imaging were employed to detect the specific activation situation after the nanoparticles were incubated with the macrophages and cancer cells, respectively. The expression of F4/80 and CD80, a well-characterized and extensively referenced marker after mouse macrophage maturation,31 was significantly increased in RAW 264.7 cells detected by flow cytometry and confocal imaging (Fig. 2h and i and Fig. S10–S14, ESI†). A large amount of TNF-α and IL-6 was detected in the cell culture supernatants by ELISA after 48 hours of co-incubation of RAW 264.7 cells and (SeV + aCD47)@PLGA nanoparticles (Fig. 2j and k). In addition, the CD47 blockade increased the phagocytosis of cancer cells by macrophages in vitro, as shown by confocal imaging and flow cytometry (Fig. 2l–n). Thus, these results demonstrated that the (SeV + aCD47)@PLGA oncolytic nanocomposites enhanced macrophage and dendritic cell phagocytosis of cancer cells, thereby promoting APC maturity for triggering the anti-tumor immune response.
3.3 Oncolytic nanocomposites stimulated immune responses in vivo
To validate that the oncolytic nanocomposites could excite immune responses in vivo, 4T1-bearing mice were intratumorally injected with PLGA, aCD47@PLGA, SeV@PLGA and (SeV + aCD47)@PLGA individually. After TQ-BPN molecules were embedded in the hydrophobic layer of the nanoparticles, strong fluorescence could be observed in the tumor site by IVIS, while other organs were almost without any fluorescence (Fig. 3a and b and Fig. S15, ESI†). After euthanasia, the tumors were isolated and dissected; a large amount of red AIE fluorescence could be observed in the tumor tissue (Fig. 3i). These results imply that intratumoral injection has a very high delivery efficiency for nanocomposites and a very low systemic distribution, which can not only improve the oncolytic effect, but also reduce the systemic toxicity of nanocomposites.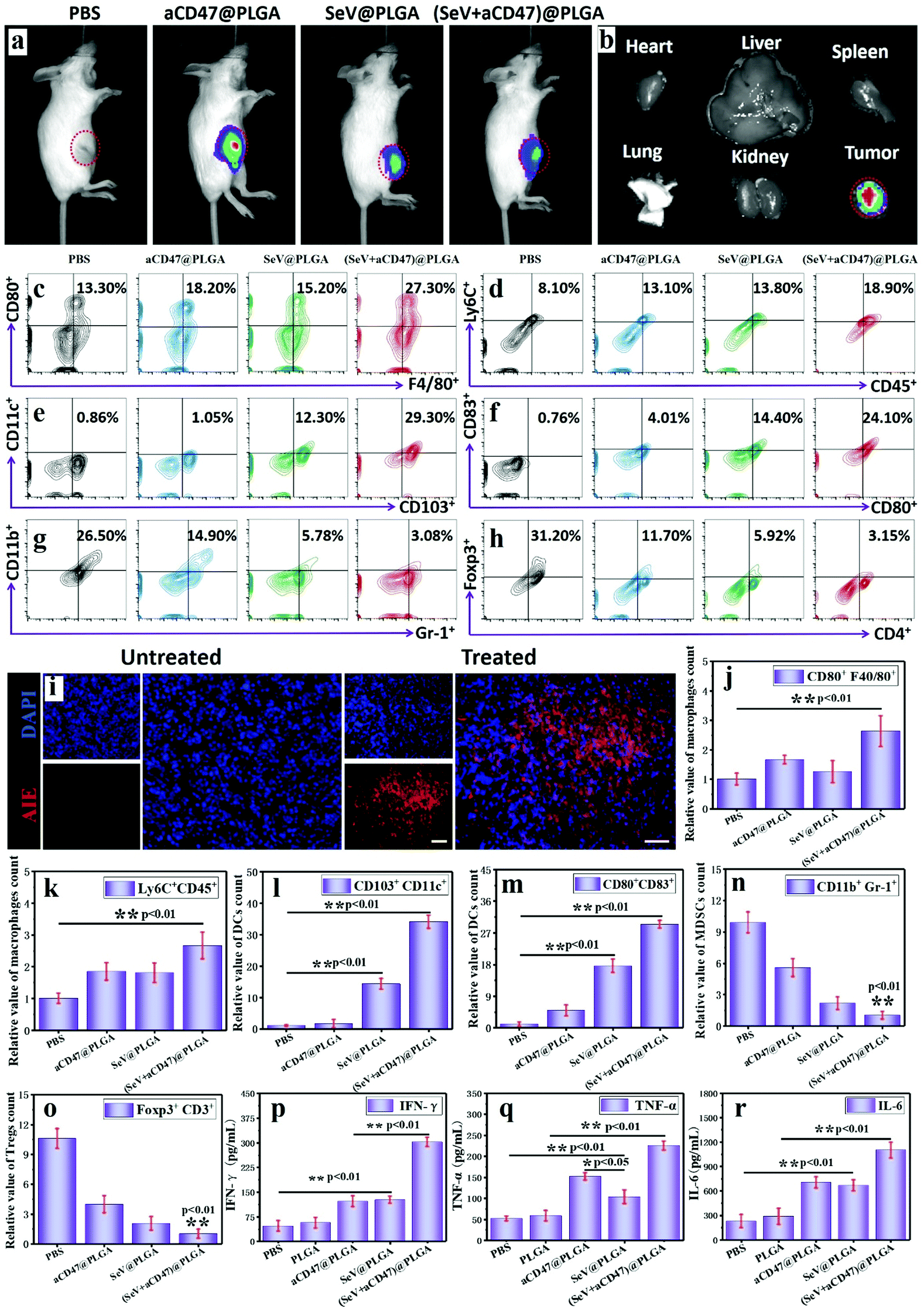 | ||
| Fig. 3 Immune response induced by oncolytic nanohybrids in vivo. (a) Representative fluorescence images using an in vivo imaging system (IVIS) depicting in vivo oncolytic nanocomposites after intratumoral injection in different samples for 2 h. Experiments were repeated in triplicate. (b) Representative fluorescence IVIS imaging of the heart, liver, spleen, lungs, kidneys and tumor 2 h after intratumoral injection of oncolytic nanocomposites. Representative flow cytometric analysis images of (c) CD80+ F4/80+ and (d) Ly6Chi macrophages gating on CD45+ cells. Representative flow cytometric analysis images of (e) CD80+CD86+ and (f) CD103+ dendritic cells gating on CD45+CD11c+ cells. (g) Representative flow cytometric analysis images of MDSCs (CD11b+Gr-1+) gating on CD45+ cells. (h) Representative flow cytometric analysis images of Tregs (Foxp3+CD4+) gating on CD3+ cells. (i) Representative fluorescence images of tumor sections after intratumoral injection in different samples for detecting the oncolytic nanocomposites. Scale bars, 200 μm. Relative quantitative analysis of the (j) CD80+ F4/80+ macrophages, (k) Ly6Chi macrophages, (l) CD80+CD86+ dendritic cells, (m) CD103+ dendritic cells, (n) MDSCs cells and (o) Tregs cells. Secretion levels of (p) IFN-γ, (q) TNF-α and (r) IL-6 in tumor infiltration by ELISA after intratumoral injection in different samples for 7 days. n = 5, mean ± SD. Statistical significance was calculated via one-way analysis of variance (ANOVA) with a Tukey post hoc test. **P < 0.05; **P < 0.01. | ||
The efficacy of nanocomposites containing Sendai virus and CD47 blocker in exciting dendritic cells and macrophages was first tested to indicate that the anti-tumor immune response was initiated. This process is also the key to arousing the organism's adaptive immune response. It has been demonstrated above that CD47 blockers can increase the phagocytosis of tumor cells by macrophages in vitro. Here we further verify the performance of this nanocomposite to actuate macrophages in tumors. The flow cytometry results display that 48 h after the nanocomposite injection, the anti-tumor M1 (CD80+F4/80+) macrophages and Ly6Chi monocytes had more than twice more than the control group (Fig. 3c, d, j and k). At the same time, the proportion of CD11c+ dendritic cells expressing mature surface molecules of CD80, CD86 and CD103 also increased significantly in the tumor, blood, spleen and lymph nodes after nanocomposite stimulation (Fig. 3e, f, l, m and Fig. S16, S17, ESI†). Among them, we can also find another interesting phenomenon that the number of mature DCs after SeV@PLGA treatment is significantly higher than that after aCD47@PLGA treatment, indicating that SeV has a strong ability to recruit and activate DCs. In addition, the number of immunosuppressive cells represented by MDSCs (CD11b+Gr-1+) and Tregs (Foxp3+CD4+) in the tumor microenvironment decreased by nearly 90% after being treated with the nanocomposites (Fig. 3g, h, n, o and Fig. S18, S19, ESI†). These results suggest from different levels and areas that under the combined action of SeV and CD47 blocker, macrophages and DCs can be efficiently excited to release breast cancer antigens, and the released antigens can be efficiently presented to lymph nodes by DCs around the tumor recruited by SeV. The secretion of anti-tumor cytokines in tumors including IFN-γ, TNF-α and IL-6 further confirmed the effective immune responses induced by the nanocomposites containing Sendai virus and CD47 blocker (Fig. 3p–r). These cytokines, in turn, can inhibit tumor cell growth, increase phagocytosis of tumor cells, enhance tumor antigen presentation, and activate adaptive immune responses.
In the process of tumor cell killing, cellular immunity plays a vital role and CD8+ T cells are the most important adaptive immune cells for tumor immune clearance in vivo.6,62 To demonstrate that CD8+ T cells were activated by APCs after treatment with oncolytic nanocomposites, tumors were harvested and analyzed using flow cytometry and immunofluorescence staining 7 days after intratumoral injection. Treatment with (SeV + aCD47)@PLGA caused an observable increase in tumor-infiltrating lymphocytes (TILs, CD3+), cytotoxic T lymphocytes (CTLs, CD3+CD8+), and helper T cells (Th, CD3+CD4+) within the tumor by flow cytometry (Fig. 4a, b, e and f). Immunofluorescence staining indicated an increase in CTLs in the tumors after (SeV + aCD47)@PLGA treatment (Fig. 4i and Fig. S20, ESI†). These results indicated that our oncolytic nanocomposites after intratumoral injection could effectively recruit and mature T cells to the tumor area for eliminating tumors.
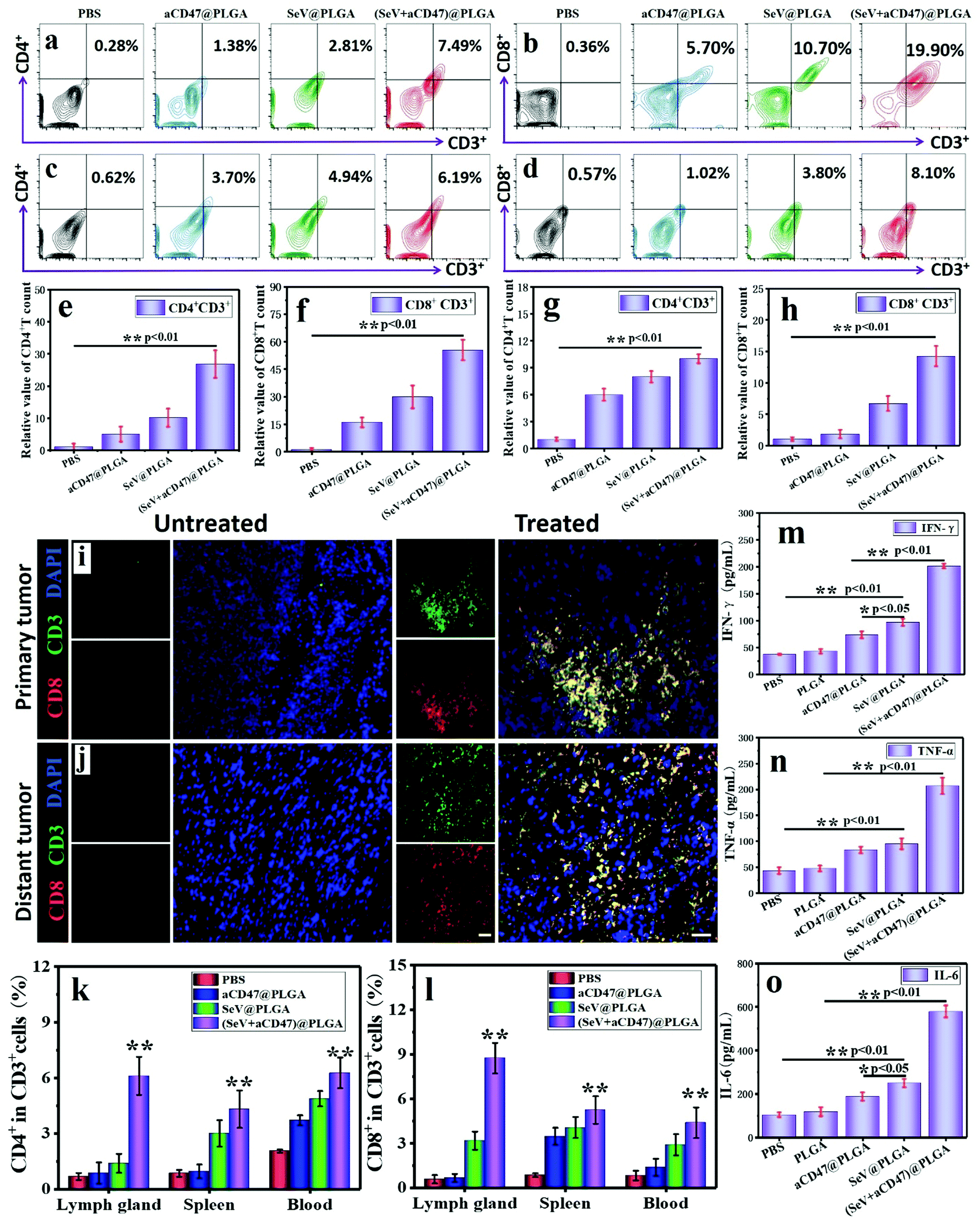 | ||
| Fig. 4 Systemic immune response in distant tumors induced by oncolytic nanohybrids in vivo. (a) Representative flow cytometric analysis images of Th cells (CD4+CD3+) gating on CD3+ cells in primary tumors after intratumoral injection in different samples for 7 days. (b) Representative flow cytometric analysis images of CTL cells (CD8+CD3+) gating on CD3+ cells in primary tumors. (c) Representative flow cytometric analysis images of Th cells (CD4+CD3+) gating on CD3+ cells in distant tumors. (d) Representative flow cytometric analysis images of CTL cells (CD8+CD3+) gating on CD3+ cells in distant tumors. Relative quantitative analysis of the (e) Th cells and (f) CTL cells in primary tumors. Relative quantitative analysis of the (g) Th cells and (h) CTL cells in distant tumors. (e) Representative immunofluorescent staining of tissue sections in (i) primary tumors and (j) distant tumors showing CD3+ and CD8+ in the groups with or without (SeV + aCD47)@PLGA. Scale bars, 200 μm. The absolute percentage of (k) Th cells and (l) CTL cells in the tumor-draining lymph node, spleen, and blood for different treatment groups by flow cytometry gating on CD3+ cells. Secretion levels of (m) IFN-γ, (n) TNF-α and (o) IL-6 in distant tumor infiltration by ELISA after intratumoral injection in different samples for 7 days. Experiments were repeated in triplicate. n = 5, mean ± SD. Statistical significance was calculated via one-way analysis of variance (ANOVA) with a Tukey post hoc test. **P < 0.05; **P < 0.01. | ||
With confirmation that oncolytic nanocomposites aroused local antitumor immunity in vivo, we investigated whether local treatment with (SeV + aCD47)@PLGA triggered systemic immune responses. 4T1 tumor cells were inoculated in the opposite flank of the primary tumor to mimic tumor metastasis. Seven days after the primary tumors were injected with (SeV + aCD47)@PLGA nanoparticles, distal tumors were isolated and CD4+ and CD8+ T cells were examined by flow cytometry and immunofluorescence staining. Similar to the results of primary tumors, the CTL and Th content in distal tumors increased significantly for the (SeV + aCD47)@PLGA treatment group as evidenced by flow cytometry (Fig. 4c, d, g and h). The immunofluorescence of CTLs in distant tumors was also detected, and the results showed that CTLs were also present in the distal tumor (Fig. 4j and Fig. S21, ESI†). In addition, CD4+ T cells and CD8+ T cells were detected by flow cytometry in different regions of the body. The results showed that the anti-tumor T lymphocytes in the lymph nodes, spleen and blood were also significantly increased after the nanocomposite treatment (Fig. 4k, l and Fig. S22, S23, ESI†). The secretion of cytokines in the distant tumor, including IFN-γ, TNF-α and IL-6, further confirmed the effective innate and adaptive immune responses induced by (SeV + aCD47)@PLGA treatment (Fig. 4m–o). The results might be due to the cross-presentation of tumor antigens by macrophages and dendritic cells excited by oncolytic nanocomposites that trigger systemic anti-tumor immunity. These results reveal that intratumoral injection with high drug delivery efficiency and low systemic distribution can not only excite the immune activation in the injection area, but also energize the systemic anti-tumor immune function to prevent tumor metastasis.
3.4 Oncolytic nanocomposites stimulated the systemic immune response for tumor elimination and immune memory formation
We described the excitation of the systemic anti-tumor immune system by oncolytic nanocomposites. On this basis, we verified the tumor growth and mouse survival time. 4T1-bearing mice were injected with PBS, PLGA, aCD47@PLGA, SeV@PLGA and (SeV + aCD47)@PLGA nanoparticles individually in the left tumor (Fig. 5a). The tumor sizes on both sides were measured daily using calipers. After treatment with (SeV + aCD47)@PLGA, the growth of the tumors was significantly inhibited in the left tumor. The growth of the tumors on the opposite flank of the primary tumor was similar because the systemic immune system had been activated by (SeV + aCD47)@PLGA (Fig. 5b). After aCD47@PLGA treatment, the survival time of the mice was extended from 16 days in the untreated group to about 30 days, which indicated that CD47 blocker also has a certain anti-tumor effect. When treated with nanohybrids containing SeV and CD47 blocker, 60% of mice survived longer than 36 days, revealing that SeV could improve the therapeutic effect of CD47-directed immune checkpoint inhibitors against breast cancer (Fig. 5c).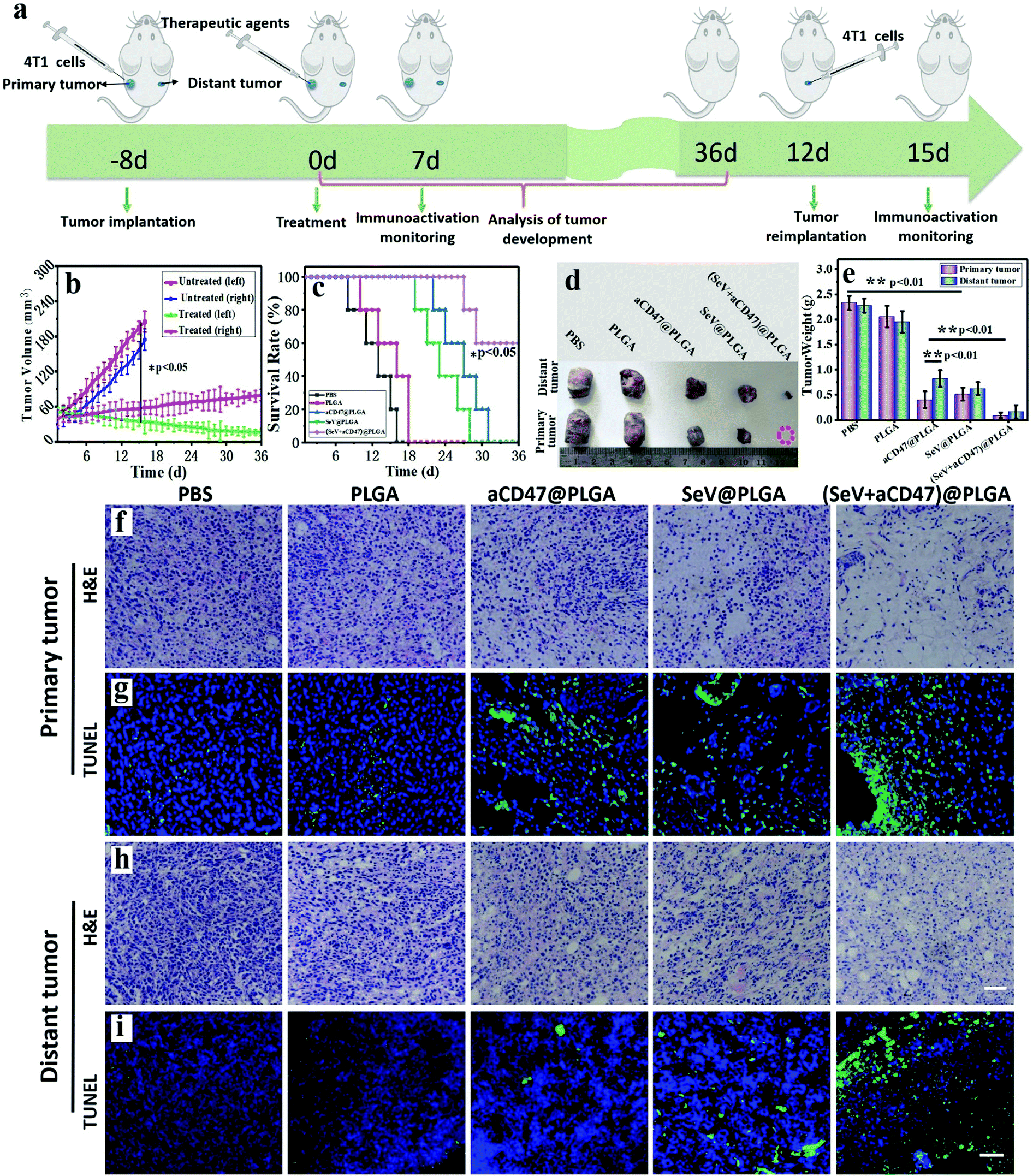 | ||
| Fig. 5 Evaluation of the therapeutic effect on tumors in mice treated with oncolytic nanohybrids. (a) Schematic illustrating (SeV + aCD47)@PLGA therapy in a mouse model of primary tumors and distant tumors. Tumors on the left side were designated as ‘primary tumors’ with (SeV + aCD47)@PLGA intra-tumor injection and those on the right side were designated as ‘distant tumors’ without any treatment. (b) Both sides of the tumor growth kinetics in the oncolytic nanocomposite-treated group. (c) Survival corresponding to the tumor size of mice after different treatments as indicated (n = 5). Statistical significance was calculated via the log-rank (Mantel–Cox) test. (d) Representative size of the excised tumor on both sides after 16 days with different treatments. (e) The excised tumor weight on both sides after 16 days with different treatments. (f) Representative H&E staining images of primary tumor sections after different treatments. (g) Representative TUNEL staining images of primary tumor sections after different treatments. (h) Representative H&E staining images of distant tumor sections after different treatments. (i) Representative TUNEL staining images of distant tumor sections after different treatments. Scale bars, 200 μm. n = 5, mean ± SD. Statistical significance was calculated via one-way ANOVA with a Tukey post hoc test for multiple comparisons. **P < 0.05; **P < 0.01. | ||
After oncolytic nanocomposite treatment, representative mice were euthanized, and then the tumors were isolated, and the size and weight of the tumors were measured (Fig. 5d and e). Just like conventional immunocheckpoint therapy, aCD47@PLGA alone has a certain inhibitory effect on tumor growth. Compared with the aCD47@PLGA group, tumors were difficult to identify in the tumor-bearing mice treated with the (SeV + aCD47)@PLGA nanohybrids. There was no significant change in the body weight of mice treated with the oncolytic nanocomposites compared to the other groups (Fig. S24, ESI†). Moreover, haematoxylin and eosin (H&E) and TUNEL staining of the tumor tissues on both sides showed extensive necrosis and apoptosis after the oncolytic nanocomposite treatment (Fig. 5f–i and Fig. S25, S26, ESI†). We speculate that this result is caused by the stimulation of cellular immunity by the synergistic therapy of the virus and immune checkpoint. Meanwhile, histology analysis of major organs collected from mice 16 days after oncolytic nanocomposite treatment indicated that local delivery of the nanocomposites did not induce significant side effects in the mice (Fig. S27, ESI†). However, even more surprising was the fact that the free anti-CD47 antibodies induced hemolysis of a large amount of mouse red blood cells and caused them to rupture to form a crescent shape, but it was perfectly avoided after the antibody was encapsulated into the PLGA–PEG polymer vesicles (Fig. S28, ESI†). Besides, the clotting agglutination caused by Sendai virus was also significantly alleviated after surface camouflage (Fig. S29, ESI†). These phenomena indicated that our (SeV + aCD47)@PLGA oncolytic nanocomposites could efficiently induce tumor regression and had good biocompatibility.
One of the main functions of Th is to induce humoral immunity, so mature plasma cells in the spleen and lymph nodes are detected by flow cytometry. The B lymphocytes expressing CD138+ surface molecules increased significantly after treatment with the (SeV + aCD47)@PLGA nanocomposites (Fig. 6a, b and f and Fig. S30, ESI†). At the same time, the antibodies produced by plasma cells in different parts of the body were detected by ELISA. These results showed that the number of antibodies against 4T1 tumor cell surface antigen in the lymph nodes, tumor infiltration fluid, and blood increased significantly (Fig. 6e), and the resulting trends are similar with plasma cells. This phenomenon implies that the (SeV + aCD47)@PLGA nanohybrids can indirectly stimulate anti-tumor humoral immunity by exciting Th cells. The formation of memory T lymphocytes is an important symbol of the success of cellular immunity, and it is also a long-term immune protection mechanism formed by the body in order to resist the same antigen from invading again. Flow cytometry results showed that the number of central immune memory T cells (CD44+CD62L+) in the lymph nodes, spleen, and blood of mice increased significantly after (SeV + aCD47)@PLGA nanohybrid treatment (Fig. 6c, d and g and Fig. S31, ESI†). These results illustrate that the CD47-directed immune checkpoint inhibitor and Sendai virus nanohybrids can induce the formation of memory T cells.
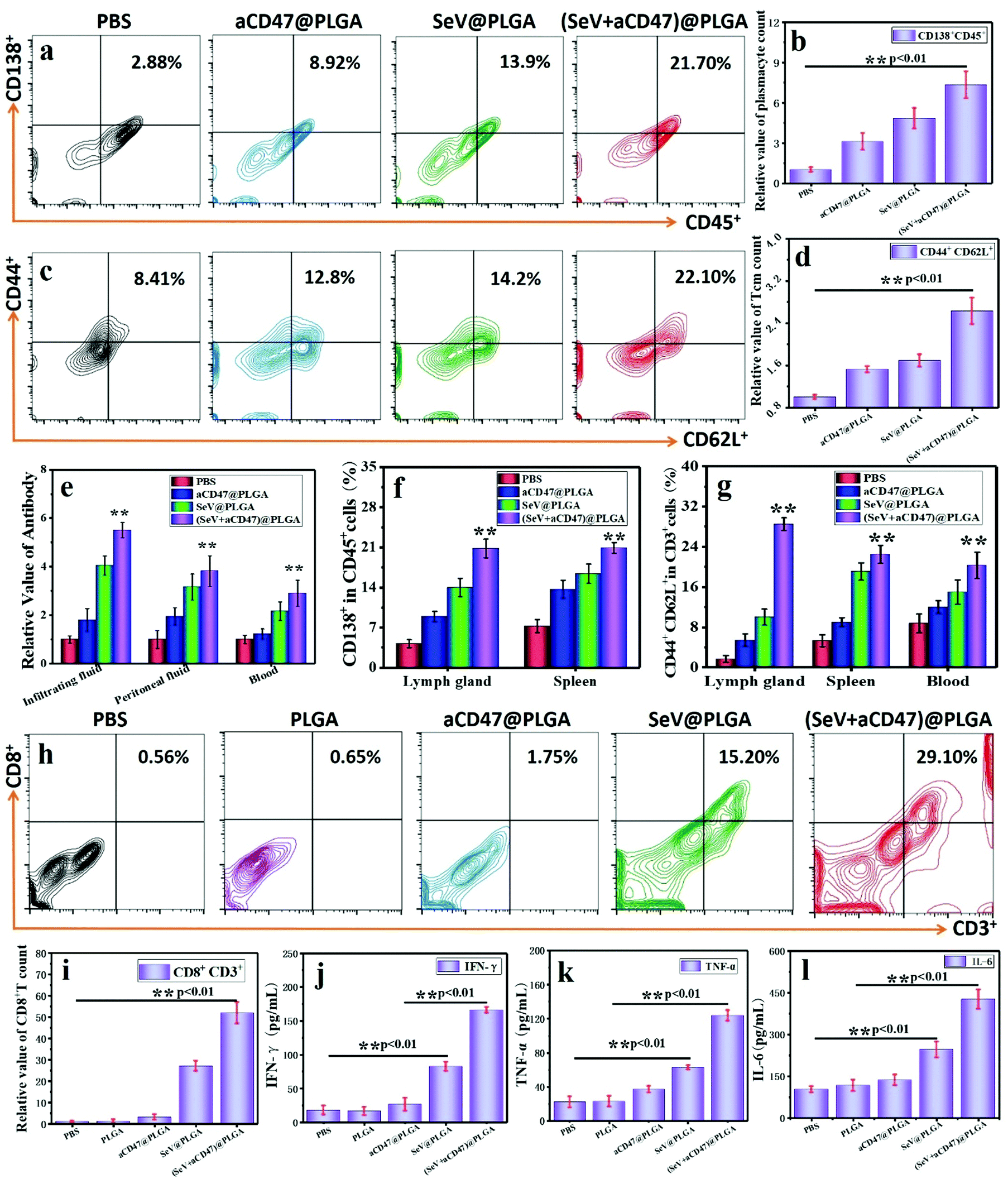 | ||
| Fig. 6 Evaluation of immune memory for tumors in mice treated with oncolytic nanohybrids. (a) Representative flow cytometric analysis images and (b) relative quantification of CD138+ B cells gating on CD45+ cells in the spleen. (c) Representative flow cytometric analysis images and (d) relative quantification of CD44+CD62L+ memory T cells gating on CD3+ cells in the lymph nodes. (e) Relative quantification of the antibodies against 4T1 cell antigen by ELISA in the interstitial fluid, peritoneal fluid and blood. (f) The absolute percentage of CD138+ B cells in the tumor-draining lymph node and spleen for different treatment groups by flow cytometry gating on CD45+ cells. (g) The absolute percentage of CD44+CD62L+ memory T cells in the tumor-draining lymph node, spleen and blood for different treatment groups by flow cytometry gating on CD3+ cells. (h) Quantitative analysis of the maturation of CD8+ T cells in the new tumor infiltration by flow cytometry after subcutaneous re-injection of 4T1 cells after 3 days. (i) The relative quantification of CD8+ T cells in the new tumor infiltration after subcutaneous re-injection of the same tumor cells. Secretion levels of (j) IFN-γ, (k) TNF-α and (l) IL-6 in tumor infiltration by ELISA after intratumoral re-injection of the same tumor cells after 3 days. n = 5, mean ± SD. Statistical significance was calculated via one-way analysis of variance (ANOVA) with a Tukey post hoc test. **P < 0.01. | ||
To verify the formation of systemic immune memory against tumors, a new subcutaneous area was re-injected with the same tumor cells after 12 days of oncolytic nanocomposite treatment. The tumor-infiltrating CTLs and cytokines were collected and then detected by flow cytometry and ELISA after 3 days of subcutaneous injection of 4T1 cells. The results showed that the content of CTLs in the injected area was significant increased for the oncolytic nanocomposite treated group compared to that in the PBS, PLGA, and aCD47@PLGA groups injected with the 4T1 cells again (Fig. 6h and i). Similarly, IFN-γ, TNF-α and IL-6 were also significantly higher than those in the virus-free treatment groups (Fig. 6j–l). These results suggest that memory T cells induced by immunocheckpoint inhibitors combined with Sendai virus can stimulate immune protection rapidly after exposure to the same tumor antigen.
4. Conclusion
In summary, we engineered Sendai virus as a nano-booster to excite dendritic cells for enhancing the anti-tumor therapeutic effect of immune checkpoint. In the cancer collaborative therapy strategy, the non-genetically modified Sendai virus can significantly energize dendritic cells in vivo and in vitro. Simultaneously, the signal of “don't eat me” on the surface of tumor cells is blocked with a CD47 blocker to greatly enhance the phagocytosis and antigen presentation of macrophages to the tumor cells. When CD47-directed immune checkpoint inhibitors and Sendai virus are camouflaged into the copolymer, hemolysis caused by anti-CD47 antibody and coagulation caused by the virus were successfully avoided. After intratumoral injection with high drug delivery efficiency and low systemic distribution, the oncolytic nanohybrids can induce the production of a large number of cytotoxic T lymphocytes and helper T lymphocytes and secrete a lot of anti-tumor cytokines throughout the body. In addition, this immunotherapy strategy can also induce humoral immunity and form immune memory with the help of Th cells induced by oncolytic nanocomposites. Therefore, the oncolytic nanohybrid strategy combining non-genetically modified viruses and immune checkpoint inhibitors might serve as the next generation of personalized anti-tumor immunotherapy agents with enhanced features over standard single immunotherapeutic agents, representing an alternative way to suppress tumor metastasis and recurrence.Author contributions
Bin Zheng conceived and designed the experiments. Yanan Xu, Xianhuang Li and Mengqian Huang performed the experiments and analysed the data. Bin Zheng wrote the paper. Bin Zheng and Tao Wang were in charge of the overall direction and planning of this study. Zhiyun Wang and Jin Chang provided instrumental, technical and financial assistance for the smooth running of the experiment. All authors discussed the results and implications and edited the manuscript at all stages.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by grants from the National Key Research and Development Program of China (2017YFA0205102), National Natural Science Foundation of China (32000999), and Key Project of Tianjin Natural Science Foundation (19JCZDJC34100). The authors thank Shao-Kai Sun for providing the DC 2.4 cells and Xuanyuan Zheng for providing the transmission electron microscopy (TEM) and in vivo imaging system (IVIS) at Tianjin Medical University.References
- X. Li, C. Shao, Y. Shi and W. Han, Lessons learned from the blockade of immune checkpoints in cancer immunotherapy, J. Hematol. Oncol., 2018, 11, 31 CrossRef.
- J. Gong, A. Chehrazi-Raffle, S. Reddi and R. Salgia, Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations, J. Immunother. Cancer, 2018, 6, 8 CrossRef.
- T. A. Yap, J. G. Aerts, S. Popat and D. A. Fennell, Novel insights into mesothelioma biology and implications for therapy, Nat. Rev. Cancer, 2017, 17, 475–488 CrossRef CAS.
- C. H. June and M. Sadelain, Chimeric Antigen Receptor Therapy, N. Engl. J. Med., 2018, 379, 64–73 CrossRef CAS.
- M. Lim, Y. Xia, C. Bettegowda and M. Weller, Current state of immunotherapy for glioblastoma, Nat. Rev. Clin. Oncol., 2018, 15, 422–442 CrossRef CAS.
- J. S. O'Donnell, M. W. L. Teng and M. J. Smyth, Cancer immunoediting and resistance to T cell-based immunotherapy, Nat. Rev. Clin. Oncol., 2019, 16, 151–167 CrossRef.
- W. Ngwa, O. C. Irabor, J. D. Schoenfeld, J. Hesser, S. Demaria and S. C. Formenti, Using immunotherapy to boost the abscopal effect, Nat. Rev. Cancer, 2018, 18, 313–322 CrossRef CAS.
- P. A. Mayes, K. W. Hance and A. Hoos, The promise and challenges of immune agonist antibody development in cancer, Nat. Rev. Drug Discovery, 2018, 17, 509–527 CrossRef CAS.
- W. H. Fridman, L. Zitvogel, C. Sautès-Fridman and G. Kroemer, The immune contexture in cancer prognosis and treatment, Nat. Rev. Clin. Oncol., 2017, 14, 717–734 CrossRef CAS.
- Y. K. Chae, A. Arya, W. Iams, M. R. Cruz, S. Chandra, J. Choi and F. Giles, Current landscape and future of dual anti-CTLA4 and PD-1/PD-L1 blockade immunotherapy in cancer; lessons learned from clinical trials with melanoma and non-small cell lung cancer (NSCLC), J. Immunother. Cancer, 2018, 6, 39 CrossRef.
- C. Sun, R. Mezzadra and T. N. Schumacher, Regulation and Function of the PD-L1 Checkpoint, Immunity, 2018, 48, 434–452 CrossRef CAS.
- J. Bellmunt, T. Powles and N. J. Vogelzang, A review on the evolution of PD-1/PD-L1 immunotherapy for bladder cancer: The future is now, Cancer Treat. Rev., 2017, 54, 58–67 CrossRef CAS.
- F. Teng, X. Meng, L. Kong and J. Yu, Progress and challenges of predictive biomarkers of anti PD-1/PD-L1 immunotherapy: A systematic review, Cancer Lett., 2018, 414, 166–173 CrossRef CAS.
- R. Mandal, R. M. Samstein, K.-W. Lee, J. J. Havel, H. Wang, C. Krishna, E. Y. Sabio, V. Makarov, F. Kuo, P. Blecua, A. T. Ramaswamy, J. N. Durham, B. Bartlett, X. Ma, R. Srivastava, S. Middha, A. Zehir, J. F. Hechtman, L. G. T. Morris, N. Weinhold, N. Riaz, D. T. Le, L. A. Diaz, Jr. and T. A. Chan, CANCER Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response, Science, 2019, 364, 485 CrossRef.
- J. A. du Payrat, C. Cugnet-Anceau, D. Maillet, M. Levy, G. Raverot, E. Disse and F. Borson-Chazot, Checkpoint inhibitors-induced hypophysitis, Bull. Cancer, 2020, 107, 490–498 CrossRef.
- R. Cohen, M. Svrcek, A. Duval, Y. Parc, P. P. Osterlund and T. Andre, Immune checkpoint inhibitors for patients with colorectal cancer: mismatch repair deficiency and perspectives, Colorectal Cancer, 2017, 6, 23–31 CrossRef.
- M. Chen, S. Yang, L. Fan, L. Wu, R. Chen, J. Chang and J. Hu, Combined Antiangiogenic Therapy and Immunotherapy Is Effective for Pancreatic Cancer With Mismatch Repair Proficiency but High Tumor Mutation Burden A Case Report, Pancreas, 2019, 48, 1232–1236 CrossRef.
- M. F. Sanmamed and L. Chen, A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization, Cell, 2018, 175, 313–326 CrossRef CAS.
- R. W. Jenkins, D. A. Barbie and K. T. Flaherty, Mechanisms of resistance to immune checkpoint inhibitors, Br. J. Cancer, 2018, 118, 9–16 CrossRef CAS.
- H. M. Knochelmann, A. S. Smith, C. J. Dwyer, M. M. Wyatt, S. Mehrotra and C. M. Paulos, CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies, Front. Immunol., 2018, 9, 1740 CrossRef.
- R. Zappasodi, T. Merghoub and J. D. Wolchok, Emerging Concepts for Immune Checkpoint Blockade-Based Combination Therapies, Cancer Cell, 2018, 33, 581–598 CrossRef CAS.
- A. M. Skoda, D. Simovic, V. Karin, V. Kardum, S. Vranic and L. Serman, The role of the Hedgehog signaling pathway in cancer: A comprehensive review, Bosnian J. Basic Med. Sci., 2018, 18, 8–20 CrossRef CAS.
- Y. Sun, W.-Z. Liu, T. Liu, X. Feng, N. Yang and H.-F. Zhou, Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis, J. Recept. Signal Transduction, 2015, 35, 600–604 CrossRef CAS.
- W. J. Leonard and J. X. Lin, Cytokine receptor signaling pathways, J. Allergy Clin. Immunol., 2000, 105, 877–888 CrossRef CAS.
- J. Nam, S. Son, K. S. Park, W. Zou, L. D. Shea and J. J. Moon, Cancer nanomedicine for combination cancer immunotherapy, Nat. Rev. Mater., 2019, 4, 398–414 CrossRef.
- J. Galon and D. Bruni, Approaches to treat immune hot, altered and cold tumours with combination immunotherapies, Nat. Rev. Drug Discovery, 2019, 18, 197–218 CrossRef CAS.
- J. A. Marin-Acevedo, A. E. Soyano, B. Dholaria, K. L. Knutson and Y. Lou, Cancer immunotherapy beyond immune checkpoint inhibitors, J. Hematol. Oncol., 2018, 11, 8 CrossRef.
- L. Dyck and K. H. G. Mills, Immune checkpoints and their inhibition in cancer and infectious diseases, Eur. J. Immunol., 2017, 47, 765–779 CrossRef CAS.
- P. K. Bommareddy, M. Shettigar and H. L. Kaufman, Integrating oncolytic viruses in combination cancer immunotherapy, Nat. Rev. Immunol., 2018, 18, 498–513 CrossRef CAS.
- F. Veglia and D. I. Gabrilovich, Dendritic cells in cancer: the role revisited, Curr. Opin. Immunol., 2017, 45, 43–51 CrossRef CAS.
- K. Twumasi-Boateng, J. L. Pettigrew, Y. Y. E. Kwok, J. C. Bell and B. H. Nelson, Oncolytic viruses as engineering platforms for combination immunotherapy, Nat. Rev. Cancer, 2018, 18, 419–432 CrossRef CAS.
- M. Hashimoto, A. O. Kamphorst, S. J. Im, H. T. Kissick, R. N. Pillai, S. S. Ramalingam, K. Araki and R. Ahmed, CD8 T Cell Exhaustion in Chronic Infection and Cancer: Opportunities for Interventions, Annu. Rev. Med., 2018, 69, 301–318 CrossRef CAS.
- H. Liu, L. Chen, Y. Peng, S. Yu, J. Liu, L. Wu, L. Zhang, Q. Wu, X. Chang, X. Yu and T. Liu, Dendritic cells loaded with tumor derived exosomes for cancer immunotherapy, Oncotarget, 2018, 9, 2887–2894 CrossRef.
- C. E. Bryant, S. Sutherland, B. Kong, M. S. Papadimitrious, P. D. Fromm and D. N. J. Hart, Dendritic cells as cancer therapeutics, Semin. Cell Dev. Biol., 2019, 86, 77–88 CrossRef CAS.
- L. Chiossone, P.-Y. Dumas, M. Vienne and E. Vivier, Natural killer cells and other innate lymphoid cells in cancer, Nat. Rev. Immunol., 2018, 18, 671–688 CrossRef CAS.
- J. Weiden, J. Tel and C. G. Figdor, Synthetic immune niches for cancer immunotherapy, Nat. Rev. Immunol., 2018, 18, 212–219 CrossRef CAS.
- D. G. Fisher, G. M. Coppock and C. B. Lopez, Virus-derived immunostimulatory RNA induces type I IFN-dependent antibodies and T-cell responses during vaccination, Vaccine, 2018, 36, 4039–4045 CrossRef CAS.
- I. Raphael, S. Nalawade, T. N. Eagar and T. G. Forsthuber, T cell subsets and their signature cytokines in autoimmune and inflammatory diseases, Cytokine, 2015, 74, 5–17 CrossRef CAS.
- W. Ouyang, S. Rutz, N. K. Crellin, P. A. Valdez and S. G. Hymowitz, Regulation and Functions of the IL-10 Family of Cytokines in Inflammation and Disease, Annu. Rev. Immunol., 2011, 29, 71–109 CrossRef CAS.
- W. Ouyang and A. O'Garra, IL-10 Family Cytokines IL-10 and IL-22: from Basic Science to Clinical Translation, Immunity, 2019, 50, 871–891 CrossRef CAS.
- G. A. Duque and A. Descoteaux, Macrophage cytokines: involvement in immunity and infectious diseases, Front. Immunol., 2014, 5, 1–12 CAS.
- D. M. Schwartz, M. Bonelli, M. Gadina and J. J. O'Shea, Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases, Nat. Rev. Rheumatol., 2016, 12, 25–36 CrossRef CAS.
- Q. Chen, C. Wang, X. Zhang, G. Chen, Q. Hu, H. Li, J. Wang, D. Wen, Y. Zhang, Y. Lu, G. Yang, C. Jiang, J. Wang, G. Dotti and Z. Gu, In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment, Nat. Nanotechnol., 2019, 14, 89–97 CrossRef CAS.
- M. Mohme, S. Riethdorf and K. Pantel, Circulating and disseminated tumour cells – mechanisms of immune surveillance and escape, Nat. Rev. Clin. Oncol., 2017, 14, 155–167 CrossRef CAS.
- K. Weiskopf, Cancer immunotherapy targeting the CD47/SIRP alpha axis, Eur. J. Cancer, 2017, 76, 100–109 CrossRef CAS.
- A. A. Barkal, K. Weiskopf, K. S. Kao, S. R. Gordon, B. Rosental, Y. Y. Yiu, B. M. George, M. Markovic, N. G. Ring, J. M. Tsai, K. M. McKenna, P. Y. Ho, R. Z. Cheng, J. Y. Chen, L. J. Barkal, A. M. Ring, I. L. Weissman and R. L. Maute, Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy, Nat. Immunol., 2018, 19, 76–84 CrossRef CAS.
- X. Liu, Y. Pu, K. Cron, L. Deng, J. Kline, W. A. Frazier, H. Xu, H. Peng, Y. X. Fu and M. M. Xu, CD47 blockade triggers T cell-mediated destruction of immunogenic tumors, Nat. Med., 2015, 21, 1209–1215 CrossRef CAS.
- S. Chowdhury, S. Castro, C. Coker, T. E. Hinchliffe, N. Arpaia and T. Danino, Programmable bacteria induce durable tumor regression and systemic antitumor immunity, Nat. Med., 2019, 25, 1057–1063 CrossRef CAS.
- S. Li, M. Zhu, R. Pan, T. Fang, Y.-Y. Cao, S. Chen, X. Zhao, C.-Q. Lei, L. Guo, Y. Chen, C.-M. Li, E. Jokitalo, Y. Yin, H.-B. Shu and D. Guo, The tumor suppressor PTEN has a critical role in antiviral innate immunity, Nat. Immunol., 2016, 17, 241–249 CrossRef CAS.
- F. Hou, L. Sun, H. Zheng, B. Skaug, Q. X. Jiang and Z. J. Chen, MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response, Cell, 2011, 146, 448–461 CrossRef CAS.
- O. V. Matveeva, Z. S. Guo, S. A. Shabalina and P. M. Chumakov, Oncolysis by paramyxoviruses: multiple mechanisms contribute to therapeutic efficiency, Mol. Ther.–Oncolytics, 2015, 2, 15011 CrossRef CAS.
- X. Liu, Y. Pu, K. Cron, L. Deng, J. Kline, W. A. Frazier, H. Xu, H. Peng, Y.-X. Fu and M. M. Xu, CD47 blockade triggers T cell-mediated destruction of immunogenic tumors, Nat. Med., 2015, 21, 1209–1215 CrossRef CAS.
- R. H. Vonderheide, CD47 blockade as another immune checkpoint therapy for cancer, Nat. Med., 2015, 21, 1122–1123 CrossRef CAS.
- X. Duan, C. Chan and W. Lin, Nanoparticle-Mediated Immunogenic Cell Death Enables and Potentiates Cancer Immunotherapy, Angew. Chem., Int. Ed., 2019, 58, 670–680 CrossRef CAS.
- O. S. Fenton, K. N. Olafson, P. S. Pillai, M. J. Mitchell and R. Langer, Advances in Biomaterials for Drug Delivery, Adv. Mater., 2018, e1705328, DOI:10.1002/adma.201705328.
- C. M. Hartshorn, M. S. Bradbury, G. M. Lanza, A. E. Nel, J. Rao, A. Z. Wang, U. B. Wiesner, L. Yang and P. Grodzinski, Nanotechnology Strategies To Advance Outcomes in Clinical Cancer Care, ACS Nano, 2018, 12, 24–43 CrossRef CAS.
- S. Sau, H. O. Alsaab, K. Bhise, R. Alzhrani, G. Nabil and A. K. Iyer, Multifunctional nanoparticles for cancer immunotherapy: a groundbreaking approach for reprogramming malfunctioned tumor environment, J. Controlled Release, 2018, 274, 24–34 CrossRef CAS.
- J. Qi, C. Sun, A. Zebibula, H. Zhang, R. T. K. Kwok, X. Zhao, W. Xi, J. W. Y. Lam, J. Qian and B. Z. Tang, Real-Time and High-Resolution Bioimaging with Bright Aggregation-Induced Emission Dots in Short-Wave Infrared Region, Adv. Mater., 2018, 30, e1706856 CrossRef.
- J. Qi, C. Sun, D. Li, H. Zhang, W. Yu, A. Zebibula, J. W. Y. Lam, W. Xi, L. Zhu, F. Cai, P. Wei, C. Zhu, R. T. K. Kwok, L. L. Streich, R. Prevedel, J. Qian and B. Z. Tang, Aggregation-Induced Emission Luminogen with Near-Infrared-II Excitation and Near-Infrared-I Emission for Ultradeep Intravital Two-Photon Microscopy, ACS Nano, 2018, 12, 7936–7945 CrossRef CAS.
- J. Pan, Y. Wang, C. Zhang, X. Wang, H. Wang, J. Wang, Y. Yuan, X. Wang, X. Zhang and C. Yu, Antigen-Directed Fabrication of a Multifunctional Nanovaccine with Ultrahigh Antigen Loading Efficiency for Tumor Photothermal-Immunotherapy, Adv. Mater., 2018, 30, 1704408 CrossRef.
- W. Wang, M. Green, J. E. Choi, M. Gijón, P. D. Kennedy, J. K. Johnson, P. Liao, X. Lang, I. Kryczek, A. Sell, H. Xia, J. Zhou, G. Li, J. Li, W. Li, S. Wei, L. Vatan, H. Zhang, W. Szeliga, W. Gu, R. Liu, T. S. Lawrence, C. Lamb, Y. Tanno, M. Cieslik, E. Stone, G. Georgiou, T. A. Chan, A. Chinnaiyan and W. Zou, CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy, Nature, 2019, 569, 270–274 CrossRef CAS.
- S. L. Park, A. Buzzai, J. Rautela, J. L. Hor, K. Hochheiser, M. Effern, N. McBain, T. Wagner, J. Edwards, R. McConville, J. S. Wilmott, R. A. Scolyer, T. Tüting, U. Palendira, D. Gyorki, S. N. Mueller, N. D. Huntington, S. Bedoui, M. Hölzel, L. K. Mackay, J. Waithman and T. Gebhardt, Tissue-resident memory CD8(+) T cells promote melanoma-immune equilibrium in skin, Nature, 2019, 565, 366–371 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0qm00393j |
| ‡ These author contributed equally to this work. |
| This journal is © the Partner Organisations 2021 |