DOI:
10.1039/D0QI01303J
(Research Article)
Inorg. Chem. Front., 2021,
8, 711-719
Aluminium(III) and zinc(II) complexes of azobenzene-containing ligands for ring-opening polymerisation of ε-caprolactone and rac-lactide†
Received
2nd November 2020
, Accepted 22nd November 2020
First published on 23rd November 2020
Abstract
The ability to control the outcome of polymerisations using an external stimulus remains a formidable challenge. Herein, we report a series of photoactive Schiff base ligands bearing azobenzene moieties, as well as seven Al(III) and Zn(II) complexes. Trans–cis isomerisation of the ligands and complexes occurred by exposure to UV light. Photoisomerisation was investigated using spectroscopic techniques and real-time reaction monitoring was conducted using FlowNMR. The activity of the complexes was tested in ring-opening polymerisation (ROP) of ε-caprolactone and rac-lactide under ambient and UV light conditions, with the isomers of the Al(III) complexes displaying marked differences in activity in the ROP of ε-caprolactone.
Introduction
The proliferation of plastic usage since its commercial development in the 1930s has led to increasing concerns regarding its environmental impact. In 2018, 359 million tonnes (MT) of plastic was produced globally,1 compared to 1.5 MT in 1950.2 Biodegradable polymers are deemed to be a suitable alternative to oil-derived polymers with polylactic acid (PLA), an aliphatic polyester, being one of the most well-known of the renewable polymers. One advantage of PLA, besides being degradable under compostable conditions, is that it can be chemically recycled to form other useful derivatives such as methyl lactate.3 Prices of PLA have dropped significantly over the last 20 years which has expanded its use to many areas, including biomedical and packaging applications.4–8 Research in this area is becoming highly sophisticated and its continuous evolution is reliant on new highly capable catalytic systems.
The use of an external stimulus such as light,9 redox processes,10 or mechanical force11 to control polymerisations is a very promising area of research. Light has many advantages over other external triggers, such as its non-invasive nature, easy handling, and its ability to exert a high level of control by tuning the wavelength or intensity of the light source.12 The use of light to control molecular weight and gain sequence control is therefore highly attractive in the area of polymer chemistry.13 Hawker and co-workers have demonstrated the effective use of light in both iridium based and metal-free atom transfer radical polymerisations (ATRP), which operate via a redox equilibrium process. The use of light in the activation and deactivation of the copolymerisation allowed a high level of sequence control as well as accurate control over molecular weight and chain end groups.14 However, the main drawback of photoredox catalysis is the need for continuous irradiation to maintain population of the excited states. In contrast, photoswitchable catalysis involves a photochromic moiety being incorporated into the catalyst. This form of light controlled catalysis relies on a difference in activity or selectivity for the “on” and “off” catalytic species. For example, a catalyst in its initial “off” state could proceed slowly, or not at all, and once irradiated with light of an appropriate wavelength it could lead to an increase in activity or selectivity towards a single monomer, allowing incorporation of that monomer into the chain. In order for the catalyst to be effective, a significant geometrical change must be induced upon irradiation, which can result in a cooperative,15,16 steric17–19 or electronic effect.20,21
Azobenzene is one of the most widely used photochromic units for this purpose,22 and irradiation of the trans isomer with light of an appropriate wavelength results in trans–cis isomerisation. The reverse cis–trans isomerisation can be induced thermally or by the use of visible light.23–26 One of the earliest examples of photoswitchable catalysis based on a conformational change induced by trans–cis isomerisation of azobenzene was reported by Ueno, Takahashi and Osa in 1981, whereby capping a β-cyclodextrin with an azobenzene unit allowed modulation of the rate of ester hydrolysis.17 In the area of polymer chemistry, Hecht and co–workers have reported a highly robust organocatalyst whose catalytic activity towards ring-opening polymerisation (ROP) of cyclic monomers could be turned on and off by light-induced keto–enol tautomerism. One of the most remarkable aspects of this work was the ability to control the monomer sequence of copolymerisations by a single catalytic system.27 Chen et al. demonstrated the use of salicylaldimine Zn(II) catalysts bearing azobenzene ligands for the ROP of various monomers, with a remarkable 6-fold reactivity difference reported for ε-caprolactone upon irradiation with light.28 However, reports of metal catalysts bearing azobenzene units for photocontrolled ROP are generally rare, as most of the photoswitchable catalysis to date has focused on the transformation of small molecules using organocatalysts.16,29,30
Herein, we report the successful synthesis of five Al(III) and two Zn(II) based complexes bearing ligands containing azobenzene units. The extent of their trans–cis isomerisation has been investigated using UV-Vis absorption spectroscopy, 1H NMR spectroscopy and FlowNMR. The activity of the complexes has been tested in the application of ROP of rac-lactide (LA) and ε-caprolactone (ε-CL) under ambient and UV light conditions.
Results
Ligand and complex preparation
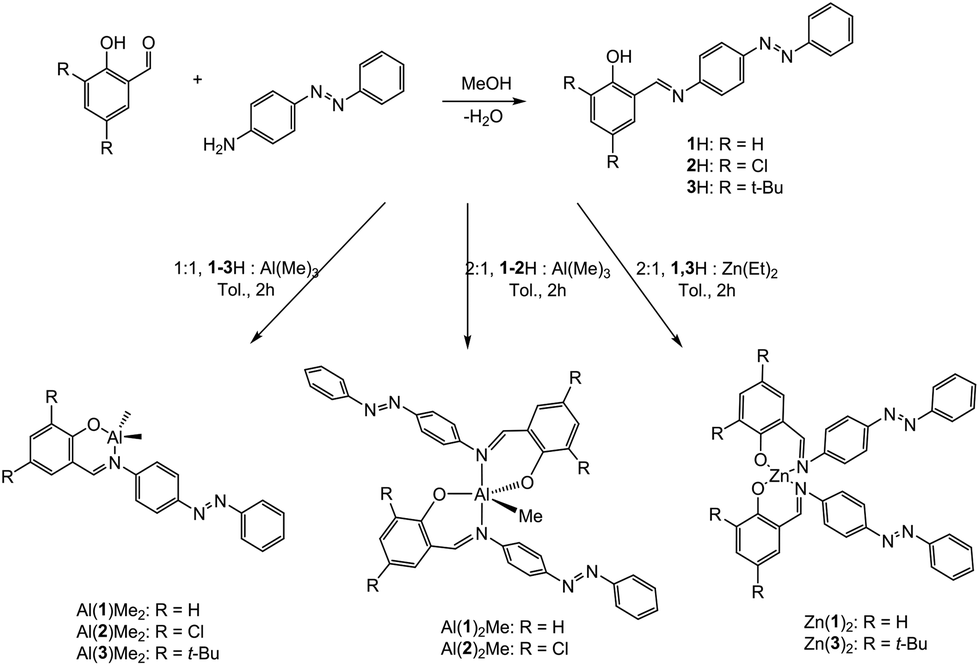
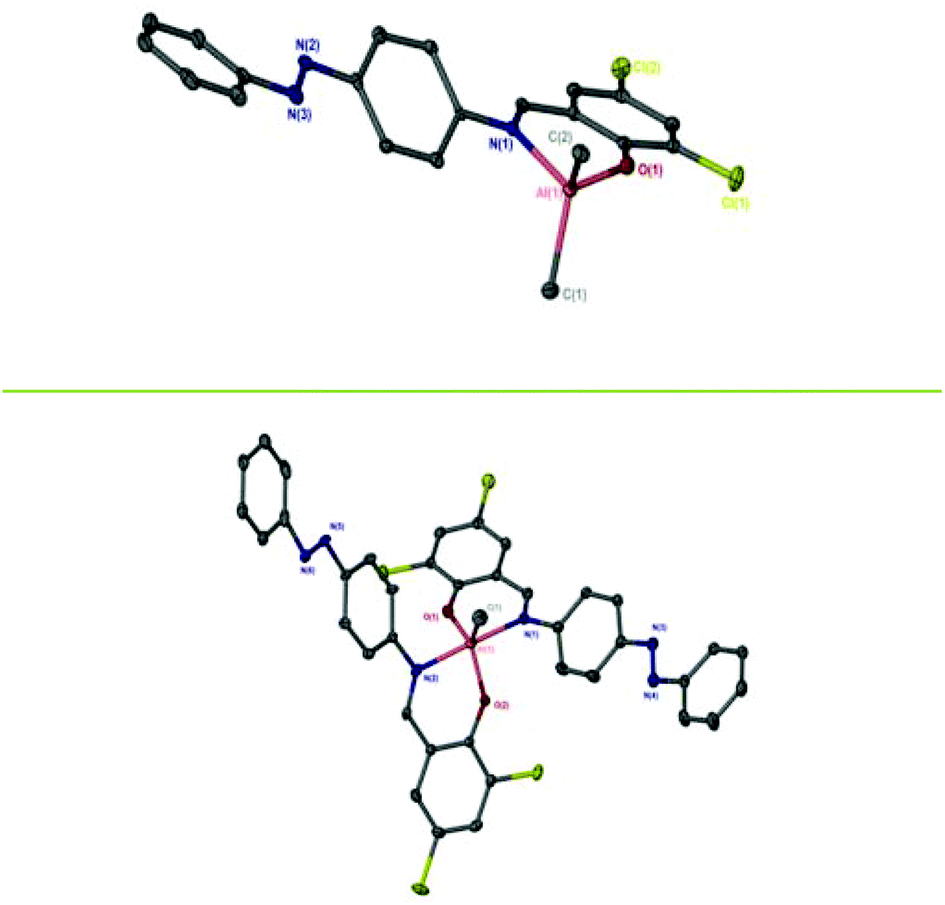
A series of three Schiff-base ligands containing photoactive azobenzene moieties were easily synthesised in high yields of ∼90% by a condensation reaction of the substituted salicylaldehyde and 4-phenyl(diazenyl)aniline (Scheme 1). 1H has been reported previously by methods similar to those reported here.31–33 The formation of the ligands was confirmed by NMR spectroscopy, with primarily (>97%) trans-azo species being present. The 1H NMR spectra of 1–3H exhibit a sharp singlet resonance in the region δ = 8.50–8.70 ppm, corresponding to imine environments, with the concurrent 13C{1H} NMR signal at ca. 165 ppm. Subsequently, five Al(III) complexes were successfully prepared by complexation of the pro-ligands (1–3H) to Al(Me)3 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 2:1 molar ratio (M:XH) in anhydrous toluene. Solid-state structures of Al(III) complexes were obtained by single crystal X-ray diffraction (Fig. 1). Elemental analysis for some complexes, particularly Al(1)Me2, showed small deviations from the expected values, however further analysis by NMR spectroscopy, along with the solid-state structures confirmed formation of the pure complexes.
:1 or 2:1 molar ratio (M:XH) in anhydrous toluene. Solid-state structures of Al(III) complexes were obtained by single crystal X-ray diffraction (Fig. 1). Elemental analysis for some complexes, particularly Al(1)Me2, showed small deviations from the expected values, however further analysis by NMR spectroscopy, along with the solid-state structures confirmed formation of the pure complexes.
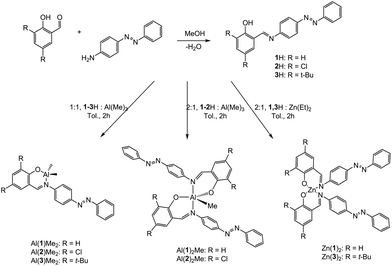 |
| | Scheme 1 Preparation of the ligands 1–3H and Al(III) and Zn(II) complexes. | |
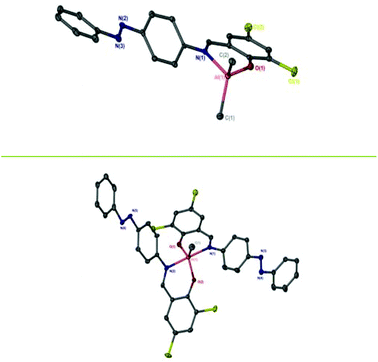 |
| | Fig. 1 Solid-state structures of Al(2)Me2 (top) and Al(2)2Me (bottom). Hydrogen atoms have been omitted for clarity. Ellipsoids are shown at 50% probability level. | |
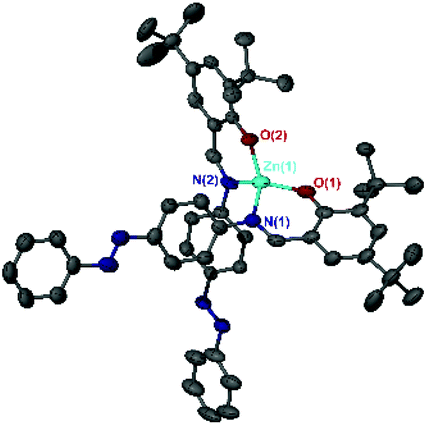
Two Zn(II) complexes are reported here by complexation of the pro-ligands with Zn(Et)2 in a procedure similar to the Al(III) complexes. Zn(1)2 has been reported previously by Markiewicz et al. via a subcomponent self-assembly procedure, where the ligand was not isolated prior to complexation with the metal.31 A single crystal suitable for X-ray diffraction of Zn(3)2 was obtained. Following numerous attempts at forming a single crystal of Zn(1)2, the characterisation of Zn(1)2 was based on NMR spectroscopy and elemental analysis, which both supported formation of the complex. Despite numerous attempts, Al(3)2Me and Zn(2)2 could not be successfully isolated. 1H NMR spectroscopy of the complexes revealed the imine proton to be upfield shifted with reference to the free ligands. For the Al(III) complexes, the metal-alkyl signals δ = −0.15 to −0.97 ppm integrated to 3 and 6 for 2:1 and 1:1 complexes, respectively.
Crystal structure analysis of Al(III) and Zn(II) complexes
Al(1)Me2 exhibited a four-coordinate structure with the Al(III) centre tended towards a tetrahedral geometry and a τ4 index of 0.9, calculated using Houser's method.34Table 1 summarises selected bond lengths and angles for each of the complexes. The complexes prepared by a 1:1 molar ratio have near identical geometries and τ4 values were in agreement with the tetrahedral geometries displayed in the solid-state structures obtained for Al(2)Me2 and Al(3)Me2 (τ4 = 0.9 in both cases). The complexes prepared by a 2:1 molar ratio were penta-coordinate and the calculated τ5 values for Al(1–2)2Me demonstrate a distorted trigonal bipyramidal geometry, with the perfect trigonal bipyramidal index being equal to 1.35 The solid state-structure of Zn(3)2 revealed a four-coordinate complex with a τ4 value of 0.9 suggestive of a distorted tetrahedral geometry (Table 2, Fig. 2).
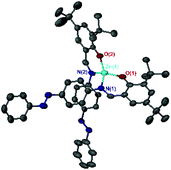 |
| | Fig. 2 Solid-state structure of Zn(3)2. Hydrogen atoms have been omitted for clarity. Ellipsoids are shown at 50% probability level. | |
Table 1 Selected bond lengths (Å) and angles (°) for Al complexes Al(1–3)Me2 and Al(1–2)2Me
| |
Al(1)Me2 |
Al(1)2Me |
Al(2)Me2 |
Al(2)2Me |
Al(3)Me2 |
| Al–O(1) |
1.780(3) |
1.775(2) |
1.771(5) |
1.777(2) |
1.779(9) |
| Al–C(1) |
1.956(4) |
1.981(4) |
1.969(7) |
1.975(3) |
1.957(15) |
| Al–N(1) |
1.979(3) |
2.130 (3) |
1.979(5) |
2.138(3) |
1.981(11) |
| Al–N(2) |
— |
2.082(3) |
— |
2.119(3) |
— |
| O(1)–Al–N(1) |
94.79(12) |
86.57(11) |
94.8(2) |
88.60(10) |
93.5(4) |
| N(1)–Al–C(1) |
113.13(16) |
92.91(13) |
108.9(3) |
93.37(15) |
110.1(5) |
| O(1)–Al–C(1) |
111.35(17) |
122.37(13) |
109.1(3) |
123.96(15) |
111.9(6) |
| C(1)–Al–C(2) |
117.0(2) |
— |
119.4(3) |
— |
119.6(7) |
| O(2)–Al–N(1) |
— |
88.17(10) |
— |
87.35(10) |
— |
| N(2)–Al–N(2) |
— |
167.84 |
— |
173.01(10) |
— |
|
τ
4
|
0.92 |
— |
0.91 |
— |
0.91 |
|
τ
5
|
— |
0.75 |
— |
0.82 |
— |
Table 2 Selected bond lengths (Å) and angles (°) for Zn(3)2
| |
Zn(3)2 |
| N(1)–Zn |
1.996(3) |
| N(2)–Zn |
1.983(3) |
| O(1)–Zn |
1.903(3) |
| O(2)–Zn |
1.983(3) |
| O(1)–Zn–N(2) |
121.01(12) |
| O(1)–Zn–N(1) |
117.81(11) |
| O(1)–Zn–N(1) |
95.14(12) |
| N(1)–Zn–N(2) |
109.82(13) |
|
τ
4
|
0.9 |
Photoisomerisation behaviour of the ligands and complexes
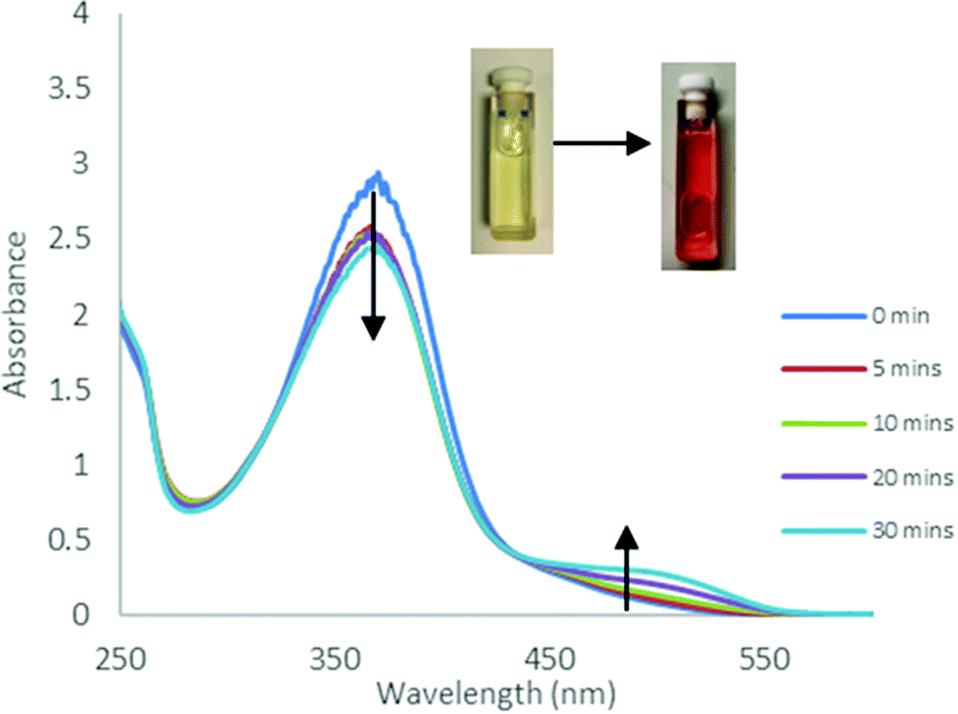
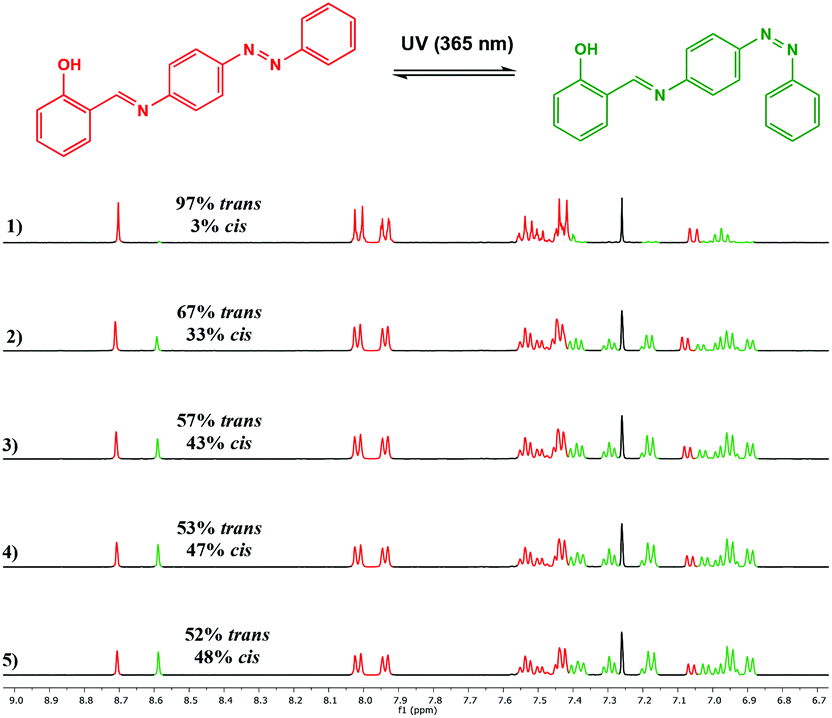
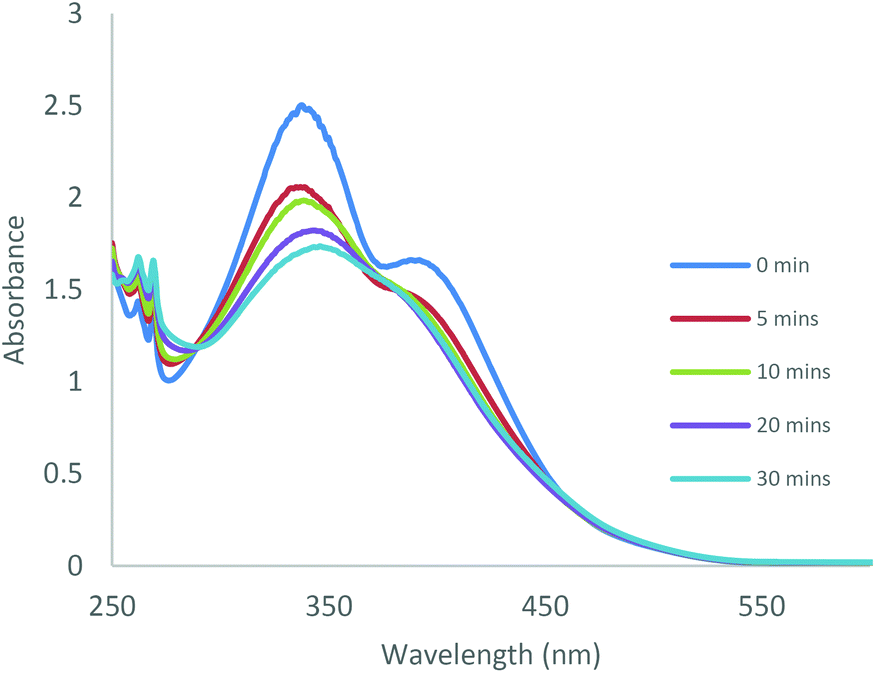
The photoisomerisation behaviour of the ligands and complexes was studied initially by UV-Vis and 1H NMR spectroscopies. The UV-Vis absorption spectra of a CHCl3 solution of the free ligand 1H displayed a maximum absorption at around 365 nm corresponding to the π–π* transition (ε ∼ 14400 L mol−1 cm−1) of the trans isomer and a weaker band at around 500 nm (ε ∼ 750 L mol−1 cm−1) due to the n–π* transition of the cis azobenzene unit (Fig. 3).22 Irradiation with 365 nm light led to a decrease in the absorbance of the π–π* band and an increase in the n–π* band, accompanied by a colour change. The 1H NMR spectrum of 1H prior to irradiation exhibited a set of imine signals which supported formation of 97% trans isomer in solution. Upon irradiation of the CDCl3 solutions in an NMR tube with 365 nm light, a set of new signals became progressively apparent in the aromatic region of the spectrum, corresponding to cis-1H (Fig. 4). Trans–cis isomerisation could be quantified by integration of the imine signal. The original signal at δ = 8.71 ppm decreased in intensity, whilst a signal at δ = 8.59 ppm increased. After 30 minutes of irradiation a photostationary state (PSS) was reached with a mixture of 48% cis and 52% trans isomers present. 2H and 3H also demonstrated photoresponsive behaviour when exposed to UV light (ESI Fig. 21†). Of all the ligands synthesised 1H contained the highest abundance of cis isomer at the PSS.
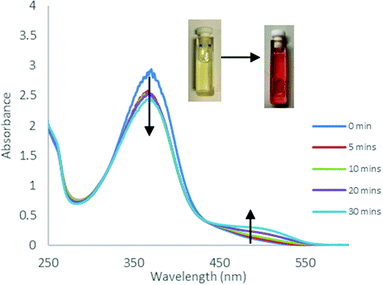 |
| | Fig. 3 UV-Vis absorption spectra obtained during UV irradiation (365 nm) of 1H in CHCl3 (C = 2 × 10–4 M) at various time intervals. | |
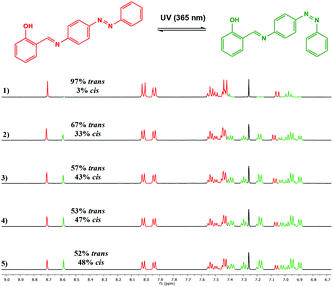 |
| | Fig. 4
1H NMR spectrum (500 MHz, CDCl3) of 1H showing spectral changes during UV irradiation: (1) prior to irradiation; (2) after 5 min; (3) after 10 min; (4) after 20 min and (5) after 30 min. | |
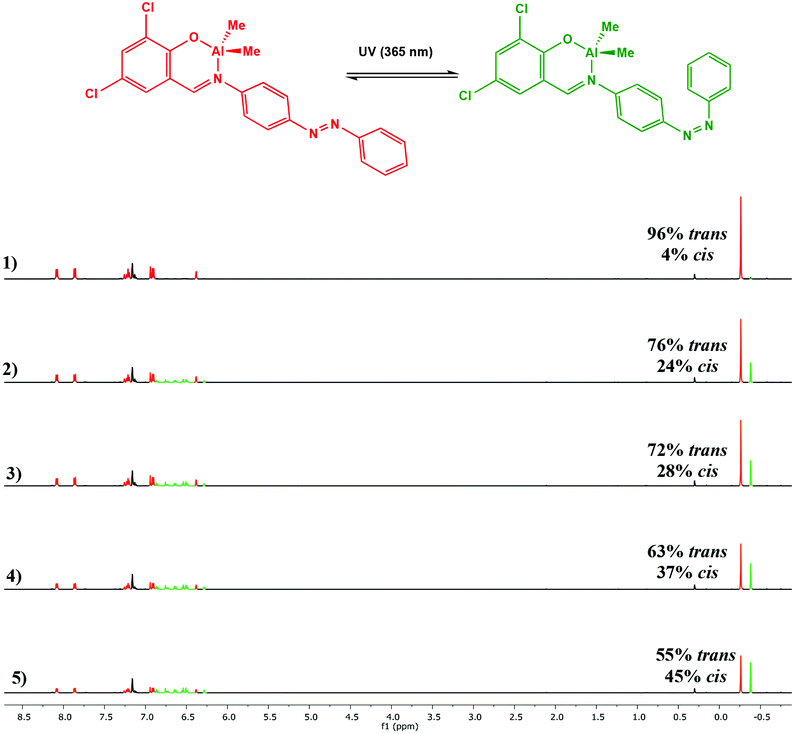
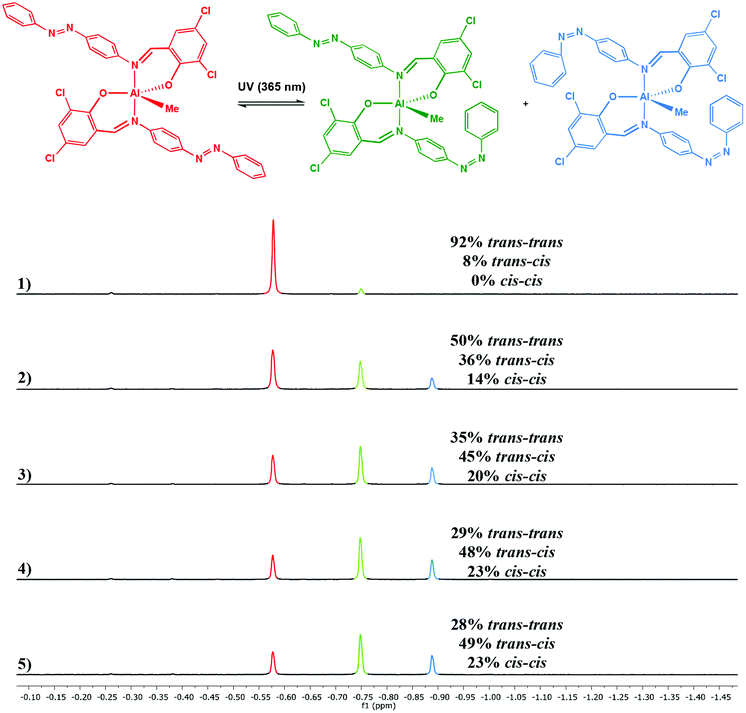
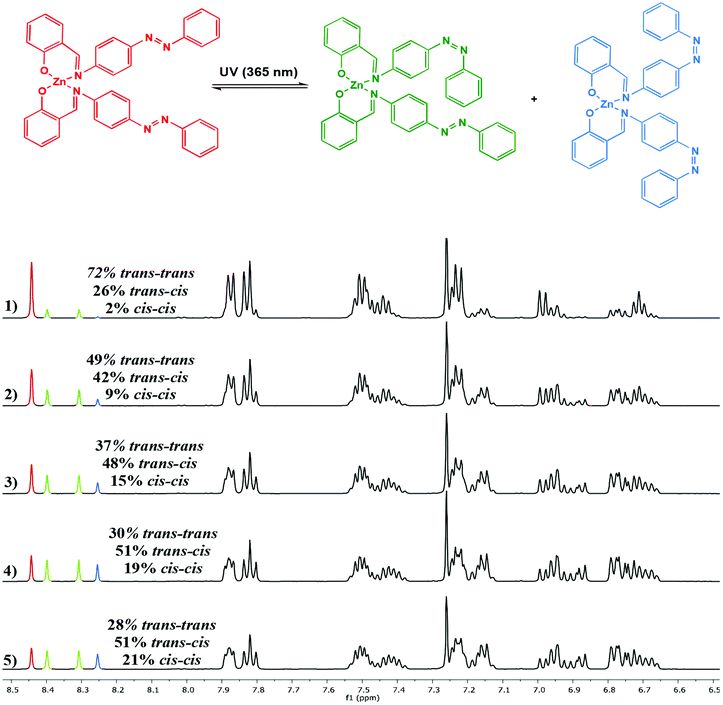
Photoisomerisation of the complexes was studied in a similar manner to the ligands to investigate the effects of complexation to Al(III) and Zn(II) on the extent of cis isomer formation. UV-Vis absorption spectra of Al(1)Me2 revealed a hypsochromic shift of the maximum absorption to around 340 nm (ε ∼ 12500 L mol−1 cm−1) (Fig. 5). Upon irradiation with UV light, the π–π* transition was observed to decrease as a function of time. 1H NMR spectroscopy was used to further support this by irradiation of a C6D6 sample within an NMR tube. The spectrum for Al(2)Me2 displayed a sharp singlet for the expected aluminium alkyl signal at δ = −0.27 ppm, which could be assigned to the trans configuration, with 4% cis isomer detected at δ = −0.37 ppm (Fig. 6). Upon irradiation, the signal at δ = −0.37 ppm increased in intensity suggestive of increasing formation of cis-Al(2)Me2. A set of new resonances in the aromatic region could also be observed and integration of the signals indicated the presence of two species with identical numbers of protons. In comparison, 1H NMR spectra of the complexes Al(1–2)2Me showed singlets relating to three possible configurations due to the presence of two ligand arms on the metal centre. For example, the spectrum for Al(2)2Me (Fig. 7) suggested a mixture of two isomers present in solution prior to irradiation, with the azobenzene units in either the trans–trans or trans–cis configuration. Spectral changes across the whole 1H NMR spectrum could be observed upon irradiation; however, the most easily quantifiable change was observed in the intensities of the Al-Me signals. A third signal of increasing intensity appeared upfield with increasing irradiation, indicating formation of the cis–cis isomer.
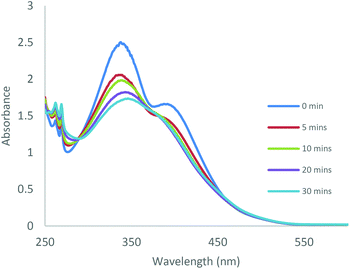 |
| | Fig. 5 UV-Vis absorption spectra obtained during UV irradiation of Al(1)Me2 in CDCl3 (C = 2 × 10−4 M) at various time intervals. | |
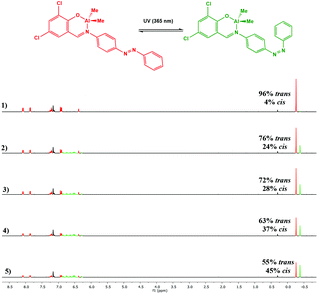 |
| | Fig. 6
1H NMR spectrum (500 MHz, C6D6) of Al(2)Me2 showing spectral changes during UV irradiation: (1) prior to irradiation; (2) after 5 min; (3) after 10 min; (4) after 20 min (5) after 30 min. | |
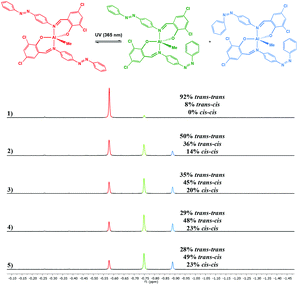 |
| | Fig. 7
1H NMR spectrum (500 MHz, CDCl3) of Al(2)2Me showing spectral changes during UV irradiation: (1) prior to irradiation; (2) after 5 min; (3) after 10 min; (4) after 20 min and (5) after 30 min. | |
Zn(II) complexes, Zn(1,3)2, also exhibited trans–cis isomerisation upon exposure to UV light, with the clearest spectral changes observed for the imine signal. An increase in irradiation time resulted in four well-separated resonances corresponding to the imine protons (Fig. 8). The signal furthest downfield corresponded to both ligand arms in the trans–trans configuration. A further signal upfield to the major trans isomer was assigned to a ligand in the trans configuration, with a further peak of identical intensity slightly upfield being attributed to the neighbouring ligand in the cis configuration. Finally, the signal furthest upfield was that of the imine protons in the cis–cis configuration. Similar to results obtained by Markiewicz et al., we observed that the extent of photoisomerisation of the Zn(II) complexes was in closest agreement with the free ligand, compared to the Al(III) complexes, with nearly 40% cis–cis isomer present in Zn(3)2.31 The thermal cis–trans isomerisation was also investigated by heating a solution of sample Al(2)Me2 which had been irradiated for 30 min (λmax = 365 nm) to reach a PSS. The sample was heated at 80 °C for one hour and monitored via1H NMR spectroscopy. The 1H NMR spectrum after heat treatment resulted in restoration of the original signals prior to irradiation with no cis isomer detected at this point and no photodecomposition of the complexes was detected.
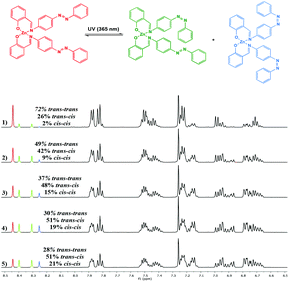 |
| | Fig. 8
1H NMR spectrum (500 MHz, C6D6) of Zn(1)2 showing spectral changes during UV irradiation: (1) prior to irradiation; (2) after 5 min; (3) after 10 min; (4) after 20 min (5) after 30 min. | |
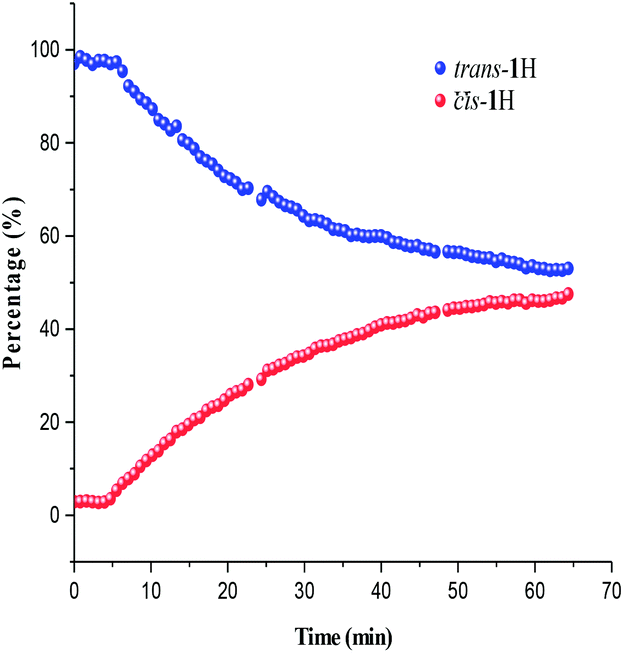
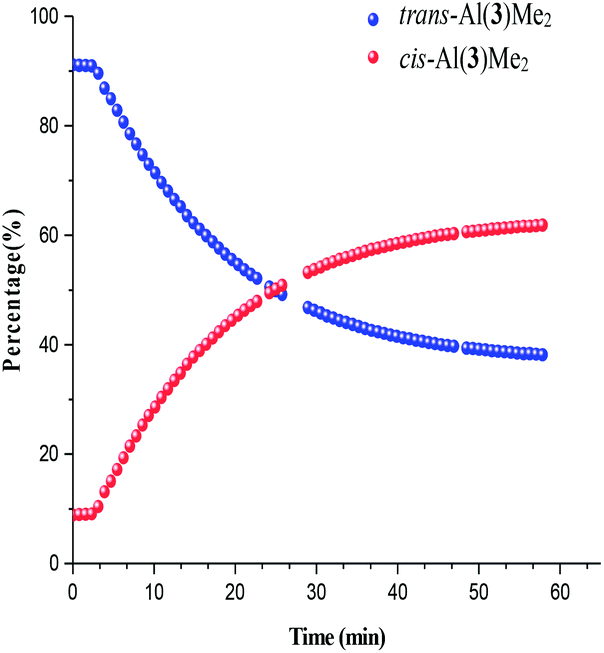
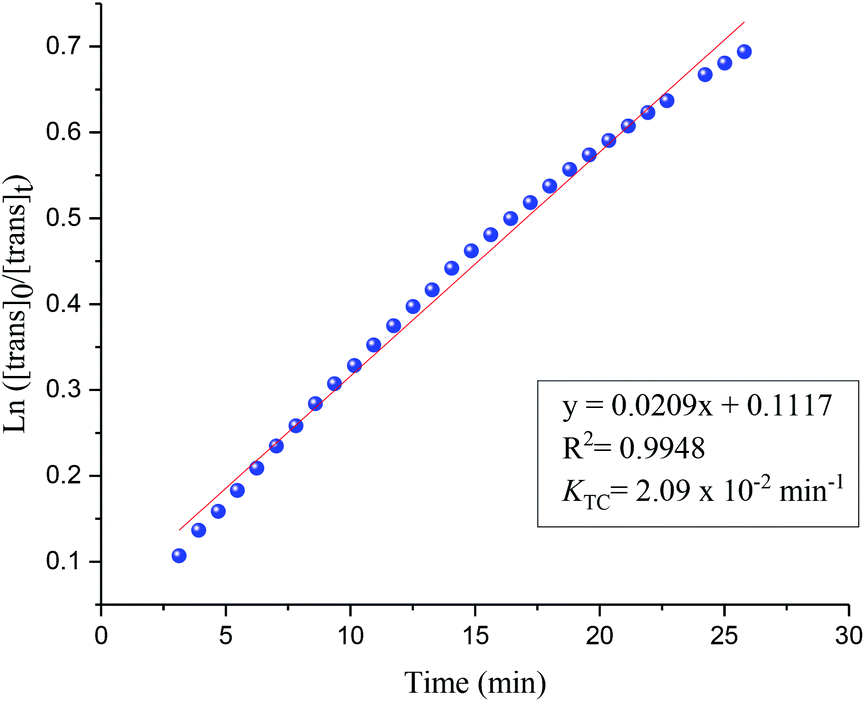
Photoisomerisation investigations of the ligands and complexes was further complimented by FlowNMR which allows monitoring of the catalytic species in real-time and is especially useful for identification of changes in photochemical reactions.36 On-line FlowNMR studies were carried out by continuous irradiation of the reaction vessel containing a solution of the complex which was flowed into the NMR spectrometer. Monitoring the reaction in continuous flow allowed 1H NMR measurements to be recorded every 60 seconds, however a two- fold delay in reaching a PSS was noted, presumably due to the quantity of the solution in the flow tube not being illuminated (Fig. 9 and 10). The change in concentration of the trans and cis isomers could be easily calculated from integration of the imine signals from each spectrum obtained (ESI Fig. 36 and 37†). The results obtained were in good agreement with the static NMR experiments. Fig. 11 shows a kinetics plot of the trans–cis isomerisation in Al(3)Me2 fitted from results obtained by FlowNMR experiments. The rate of change of concentration of trans isomer to form cis isomer indicates first-order kinetics with a rate constant of kTC = 2.09 × 10−2 min−1.
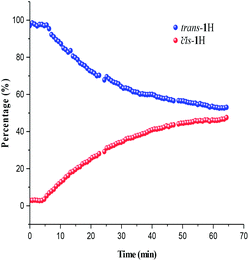 |
| | Fig. 9
Trans–cis isomerisation of ligand 1H calculated from FlowNMR spectroscopy. | |
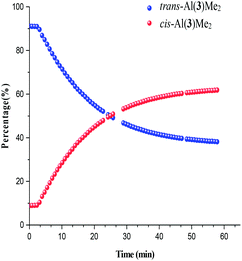 |
| | Fig. 10
Trans–cis isomerisation of Al(3)Me2. Note: blanks in data due to spectrometer shimming. | |
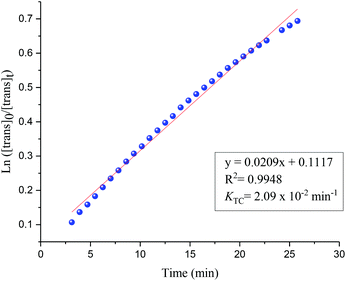 |
| | Fig. 11 First-order plot for trans–cis isomerisation (UV = 365 nm) fitted with results obtained from FlowNMR. [trans]0 = 0.0123 mol dm−3. | |
Photo-controlled ring-opening polymerisation
Following investigation of the photoswitching capabilities of the free ligands and their complexes, we envisaged that they could be tested as initiators in the ROP of rac-LA and ε-CL, with the aim of controlling the outcome of the reactions using light to exert some steric and electronic effects around the metal centre. The polymerisations of rac-LA were conducted in anhydrous toluene at 80 °C for 4 h, and monitored by 1H NMR spectroscopy, whereby the conversion was calculated by analysis of the methine region. Generally, the Al(III) dimethyl complexes, Al(1–3)Me2, displayed superior activity compared to their monomethyl counterparts, for example a comparison of Al(1)Me2 and Al(1)2Me displayed a two-fold increase in activity. Increasing steric congestion around the Al(III) centre with the presence of two ligand arms could affect coordination of the monomer, making it less favourable. The effect of modifying the substituents on the ligand also appeared to play a role in the activity of the complexes. The increasing steric bulk on the phenoxy moiety resulted in lower conversions, with the tert-butyl substituted Al(3)Me2 achieving a conversion of 18% (Table 3, entry 9). Modification of the two R groups effects both the accessibility and Lewis acidity of the metal centre, with bulky tert-butyl electron donating groups resulting in a sterically hindered and less Lewis acidic Al(III) centre. The Zn(II) complexes displayed a similar pattern with regards to activity to the Al(III) complexes, however the disparity between activities achieved was less pronounced upon switching from a H-substituted phenoxy fragment to a more sterically hindered one (Table 4).
Table 3 ROP of rac-LA initiated by Al(III) complexes under ambient and UV light conditionsa
| Entry |
Initiator |
Conv.b/% |
M
nGPC
|
M
ntheo
|
Đ
|
P
re |
Light |
|
General conditions: toluene (10 mL), 100:1:1 {[LA]/[Cat.]/[I]}, 80 °C/4 h.
Obtained from 1H NMR analysis.
Determined via GPC analysis in THF.
M
n,theo = {Mw(lactide) × conv.(%) + Mw(BnOH)}.
Determined via homonuclear decoupled NMR spectroscopy.
|
| 1 |
Al(1)Me2 |
92 |
9450 |
13369 |
1.17 |
0.42 |
Ambient |
| 2 |
Al(1)Me2 |
85 |
11850 |
12360 |
1.34 |
0.44 |
UV |
| 3 |
Al(1)2Me |
45 |
10100 |
6593 |
1.10 |
0.42 |
Ambient |
| 4 |
Al(1)2Me |
45 |
7700 |
6593 |
1.12 |
0.43 |
UV |
| 5 |
Al(2)Me2 |
80 |
13550 |
11639 |
1.12 |
0.47 |
Ambient |
| 6 |
Al(2)Me2 |
80 |
4100 |
11639 |
1.11 |
0.48 |
UV |
| 7 |
Al(2)2Me |
72 |
6950 |
10486 |
1.10 |
0.45 |
Ambient |
| 8 |
Al(2)2Me |
71 |
11800 |
10342 |
1.10 |
0.44 |
UV |
| 9 |
Al(3)Me2 |
18 |
1300 |
2702 |
1.15 |
0.43 |
Ambient |
| 10 |
Al(3)Me2 |
31 |
5200 |
4576 |
1.12 |
0.42 |
UV |
Table 4 ROP of rac-LA initiated by Zn(II) complexes under ambient and UV light conditionsa
| Entry. |
Initiator |
Conv. (%)b |
M
n,GPCc |
M
n,theo
|
Đ
|
P
re |
Light |
|
General conditions: toluene (10 mL), 100:1:1 ([LA]/[Cat]/[I]), 80 °C/4 h.
Obtained from 1H NMR analysis.
Determined via GPC analysis in THF.
M
n,theo = {(Mw(rac-LA) × conv.(%) + Mw(BnOH)}.
Determined via homonuclear decoupled NMR spectroscopy.
|
| 1 |
Zn(1)2 |
78 |
10650 |
11351 |
1.18 |
0.40 |
Ambient |
| 2 |
Zn(1)2 |
88 |
8000 |
12792 |
1.42 |
0.51 |
UV |
| 3 |
Zn(3)2 |
79 |
7400 |
11495 |
1.16 |
0.47 |
Ambient |
| 4 |
Zn(3)2 |
74 |
7750 |
10774 |
1.13 |
0.49 |
UV |
All PLA samples were analysed by GPC and demonstrated moderate number average molecular weights and narrow dispersities (1.10–1.42), indicating a controlled polymerisation process. The tacticity of the polymers produced was analysed using homonuclear decoupled 1H NMR and all initiators were atactic (Prca. 0.5). End group determination was carried out using MALDI-ToF (ESI Fig. 32 and 33†) and confirmed the polymers bear –H and –OBn end groups. Two series were observed with a repeating unit of 72 Da, indicating some transesterification. Upon switching to UV light, a third minor series was also observed, particularly in the low mass region with no end groups, suggesting formation of some cyclic oligomers under UV irradiation. The polymerisations conducted under UV conditions were carried out under continuous in situ irradiation to maintain population of the excited states and prevent reverse cis–trans isomerisation. Similar activities were observed for the rac-LA monomer under ambient and UV light conditions, with the activities remaining near identical. This could be explained by the application of heat which could facilitate the reverse isomerisation from the meta-stable cis isomer to the more thermodynamically favourable trans isomer. Generally, good Mn control was still observed under UV conditions and narrow dispersity values were retained. Following this, we attempted to alter the reaction conditions to eliminate the requirement for heat, and so a second series of experiments were conducted in CH2Cl2 at room temperature, however the Al(III) complexes were inactive under these conditions. Since several aluminium based complexes have been reported for the ROP of ε-CL,37–40 we postulated that the complexes prepared here could effectively catalyse the ROP of ε-CL, with the added benefit of using light to exert a higher degree of control where required. The ROP experiments of ε-CL were carried out at room temperature in anhydrous toluene (Table 5). All Al(III) complexes were active toward the ROP of ε-CL, however complexes Al(1–3)Me2 displayed superior activities under these conditions. Al(2)Me2 afforded the highest conversions (Table 5, entry 5), presumably due to the presence of electron withdrawing groups present on the phenoxy unit of the ligand. All PCL samples (exception Table 5, entries 3 and 4) were analysed by GPC and afforded narrow dispersity values (1.10–1.27), however experimental Mn values were marginally higher than those calculated, indicating the possibility of a higher concentration of inactive initiator present. Switching the polymerisations to UV conditions presented some exciting results with higher activities under UV radiation. For example, polymerisations initiated by Al(2)Me2 under UV light afforded near quantitative conversion compared to the same experiment under ambient light (Table 5, entries 5 and 6). It is also worth noting that a series of ε-CL polymerisations were also conducted at 80 °C to determine whether the thermal cis–trans isomerisation prevented any distinct differences in activity between polymerisations conducted under varying light conditions, however at 80 °C the reactions went to completion in <15minutes, preventing any meaningful comparison between UV and ambient light conditions. For example, an ε-CL polymerisation with Al(2)Me2 for 15 minutes afforded 100% conversion (Mn = 26800 g mol−1, Đ = 1.34). The MALDI-ToF spectrum of PCL obtained under ambient and UV light (Fig. ESI 34 and S35†) confirmed the polymer to be –OBn and –H group capped. A single series of peaks was observed, separated by intervals of 114 Da, corresponding to the repeating unit mass. The polymerisation activities of the complexes probably originate from a mixture of steric and electronic effects, dependent upon substitution of the ligand and the monomer being polymerised. A steric effect seemed to be dominating for rac-LA polymerisations, whereas the complexes bearing electron withdrawing groups [Al(2)Me2 and Al(2)2Me] displayed superior activity in ROP of ε-CL.
Table 5 ROP of ε-CL initiated by Al(III) complexes Al(1–3)Me2 under ambient and light conditionsa
| Entry |
Initiator |
Conv.b (%) |
M
n,GPC
|
M
n,theod |
Đ
|
Light |
|
General conditions: toluene (10 mL), 100:1:1 ([ε-CL]/[Cat.]/[I]), rt/12 h.
Obtained from 1H NMR analysis.
Determined via GPC analysis in THF.
M
n,theo = {(Mw(ε-CL) × conv.(%) + Mw(BnOH). GPC values not obtained for entries 3 and 4 due to low conversions. ND, not determined.
|
| 1 |
Al(1)Me2 |
33 |
8000 |
3874 |
1.17 |
Ambient |
| 2 |
Al(1)Me2 |
56 |
13100 |
6500 |
1.20 |
UV |
| 3 |
Al(1)2Me |
11 |
ND |
1363 |
ND |
Ambient |
| 4 |
Al(1)2Me |
19 |
ND |
2276 |
ND |
UV |
| 5 |
Al(2)Me2 |
75 |
21350 |
8668 |
1.23 |
Ambient |
| 6 |
Al(2)Me2 |
97 |
22200 |
11179 |
1.27 |
UV |
| 7 |
Al(2)2Me |
47 |
8900 |
5472 |
1.10 |
Ambient |
| 8 |
Al(2)2Me |
59 |
16700 |
6842 |
1.10 |
UV |
| 9 |
Al(3)Me2 |
57 |
7650 |
6614 |
1.12 |
Ambient |
| 10 |
Al(3)Me2 |
65 |
13900 |
7527 |
1.15 |
UV |
Conclusions
In summary, we have successfully demonstrated the photoswitching behaviour of a range of azobenzene containing ligands with varying substituents, and subsequently their Al(III) and Zn(II) complexes. The extent of cis isomerisation formation was altered depending on the substitution of the ligands. The aluminium complex Al(1)Me2 exhibited a lower abundance of cis isomer, compared to its free ligand, however a reverse trend was observed for Al(3)Me2 and Al(2)Me2, where an enhancement in trans–cis isomerisation was observed. The photoswitching of the complexes was exploited in the ROP of rac-LA and ε-CL. There were no distinguishable differences in activity for rac-LA polymerisation upon switching from ambient to UV light conditions. The application of heat during polymerisations of rac-LA is believed to play a role in this, however further investigation is required. For ε-CL, a clear increase in catalytic activity was observed upon trans–cis isomerisation under UV light with up to 23% increase in conversion observed. The switching in activity using light described herein should allow for the selective incorporation of monomers under variable light conditions, and therefore allow access to polymers with varying structures. Further challenges will include the careful design of highly active initiators towards ROP that also offer highly efficient and quantitative conversion during trans–cis isomerisation.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
We would like to thank the EPSRC for funding a PhD studentship to SK (EP/L016443/1) and for funding for PM (EP/P016405/1). The University of Bath and MC2 are acknowledged for their use of facilities, including FlowNMR (EP/P001475/1). We also thank Alejandro Bara-Estaún and Dr Uli Hintermair for useful discussions regarding FlowNMR measurements.
References
- Plastics-the Facts 2019, https://www.plasticseurope.org/application/files/1115/7236/4388/FINAL_web_version_Plastics_the_facts2019_14102019.pdf, (accessed 25 March 2020).
- World Plastics Production 1950–2015, https://committee.iso.org/files/live/sites/tc61/files/The_Plastic_Industry_Berlin_Aug_2016_-_Copy.pdf, (accessed 25 March 2020).
- L. A. Román-Ramírez, P. Mckeown, M. D. Jones and J. Wood, Poly(lactic acid) Degradation into Methyl Lactate Catalyzed by a Well-Defined Zn(II) Complex, ACS Catal., 2019, 9, 409–416 CrossRef.
- V. Siracusa, P. Rocculi, S. Romani and M. D. Rosa, Biodegradable polymers for food packaging: a review, Trends Food Sci. Technol., 2008, 19, 634–643 CrossRef CAS.
- I. Manavitehrani, A. Fathi, H. Badr, S. Daly, A. N. Shirazi and F. Dehghani, Biomedical Applications of Biodegradable Polyester, Polymers, 2016, 8, 20 CrossRef.
- Y. Lu and S. C. Chen, Micro and nano-fabrication of biodegradable polymers for drug delivery, Adv. Drug Delivery Rev., 2004, 56, 1621–1633 CrossRef CAS.
- A. Ammala, Biodegradable polymers as encapsulation materials for cosmetics and personal care markets, Int. J. Cosmet. Sci., 2013, 35, 113–124 CrossRef CAS.
- W. Amass, A. Amass and B. Tighe, A review of biodegradable polymers: Uses, current developments in the synthesis and characterization of biodegradable polyesters, blends of biodegradable polymers and recent advances in biodegradation studies, Polym. Int., 1998, 47, 89–144 CrossRef CAS.
- S. Shankar, M. Peters, K. Steinborn, B. Krahwinkel, F. D. Sönnichsen, D. Grote, W. Sander, T. Lohmiller, O. Rüdiger and R. Herges, Light-controlled switching of the spin state of iron(III), Nat. Commun., 2018, 9, 4750 CrossRef.
- C. Chen, Redox-Controlled Polymerization and Copolymerization, ACS Catal., 2018, 8, 5506–5514 CrossRef CAS.
- A. Piermattei, S. Karthikeyan and R. P. Sijbesma, Activating catalysts with mechanical force, Nat. Chem., 2009, 1, 133–137 CrossRef CAS.
- B. M. Neilson and C. W. Bielawski, Illuminating Photoswitchable Catalysis, ACS Catal., 2013, 3, 1874–1885 CrossRef CAS.
- B. P. Fors and C. J. Hawker, Control of a Living Radical Polymerization of Methacrylates by Light, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS.
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. Read de Alaniz, B. P. Fors and C. J. Hawker, Metal-Free Atom Transfer Radical Polymerization, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS.
- R. Cacciapaglia, S. Di Stefano and L. Mandolini, The bis-barium complex of a butterfly crown ether as a phototunable supramolecular catalyst, J. Am. Chem. Soc., 2003, 125, 2224–2227 CrossRef CAS.
- F. Würthner and J. Rebek, Light-switchable catalysis in synthetic receptors, Angew. Chem., Int. Ed., 1995, 34, 446–448 CrossRef.
- A. Ueno, K. Takahashi and T. Osa, Photocontrol of catalytic activity of capped cyclodextrin, J. Chem. Soc., Chem. Commun., 1981, 94 RSC.
- H. Sugimoto, T. Kimura and S. Inoue, Photoresponsive molecular switch to control chemical fixation of CO2, J. Am. Chem. Soc., 1999, 121, 2325–2326 CrossRef CAS.
- L. Osorio-Planes, C. Rodríguez-Escrich and M. A. Pericàs, Photoswitchable Thioureas for the external manipulation of catalytic activity, Org. Lett., 2014, 16, 1704–1707 CrossRef CAS.
- D. Wilson and N. R. Branda, Turning “On” and “Off” a pyridoxal 5′-Phosphate mimic using light, Angew. Chem., Int. Ed., 2012, 51, 5431–5434 CrossRef CAS.
- B. M. Neilson and C. W. Bielawski, Photoswitchable Organocatalysis: Using Light To Modulate the Catalytic Activities of N-Heterocyclic Carbenes, J. Am. Chem. Soc., 2012, 134, 12693–12699 CrossRef CAS.
- H. M. D. Bandara and S. C. Burdette, Photoisomerization in different classes of azobenzene, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- G. S. Hartley, The cis-form of azobenzene, Nature, 1937, 140, 281–281 CrossRef CAS.
- H. Rau and E. Lueddecke, On the Rotation-Inversion Controversy on Photoisomerization of Azobenzenes. Experimental Proof of Inversion, J. Am. Chem. Soc., 1982, 104, 1616–1620 CrossRef CAS.
- C. R. Crecca and A. E. Roitberg, Theoretical study of the isomerization mechanism of azobenzene and disubstituted azobenzene derivatives, J. Phys. Chem. A, 2006, 110, 8188–8203 CrossRef CAS.
- V. Blanco, D. A. Leigh and V. Marcos, Artificial switchable catalysts, Chem. Soc. Rev., 2015, 44, 5341–5370 RSC.
- F. Eisenreich, M. Kathan, A. Dallmann, S. P. Ihrig, T. Schwaar, B. M. Schmidt and S. Hecht, A photoswitchable catalyst system for remote-controlled (co)polymerization in situ, Nat. Catal., 2018, 1, 516–522 CrossRef CAS.
- M. Li, P. Zhang and C. Chen, Light-Controlled Switchable Ring Opening Polymerization, Macromolecules, 2019, 52, 5646–5651 CrossRef CAS.
- T. Imahori, R. Yamaguchi and S. Kurihara, Azobenzene-Tethered Bis(Trityl Alcohol) as a Photoswitchable Cooperative Acid Catalyst for Morita-Baylis-Hillman Reactions, Chem. – Eur. J., 2012, 18, 10802–10807 CrossRef CAS.
- M. Samanta, V. S. R. Krishna and S. Bandyopadhyay, A photoresponsive glycosidase mimic, Chem. Commun., 2014, 50, 10577 RSC.
- G. Markiewicz, A. Walczak, F. Perlitius, M. Piasecka, J. M. Harrowfield and A. R. Stefankiewicz, Photoswitchable transition metal complexes with azobenzene-functionalized imine-based ligands: structural and kinetic analysis, Dalton Trans., 2018, 47, 14254–14262 RSC.
- L. Fan, L. Wu and M. Ke, Synthesis of novel phenylazo-substituted salicylaldimine-based boron difluoride complexes, J. Chem. Res., 2015, 39, 442–444 CrossRef CAS.
- S. Suganya and S. Velmathi, Simple azo-based salicylaldimine as colorimetric and fluorescent probe for detecting anions in semi-aqueous medium, J. Mol. Recognit., 2013, 26, 259–267 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Structural variation in copper(I) complexes with pyridylmethylamide ligands: structural analysis with a new four-coordinate geometry index, tau(4), Dalton Trans., 2007, 955–964 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- A. M. R. Hall, R. Broomfield-Tagg, M. Camilleri, D. R. Carbery, A. Codina, D. T. E. Whittaker, S. Coombes, J. P. Lowe and U. Hintermair, Online monitoring of a photocatalytic reaction by real-time high resolution FlowNMR spectroscopy, Chem. Commun., 2018, 54, 30–33 RSC.
- C.-T. Chen, C.-A. Huang and B.-H. Huang, Aluminium metal complexes supported by amine bis-phenolate ligands as catalysts for ring-opening polymerization of epsilon-caprolactone, Dalton Trans., 2003, 3799–3803 RSC.
- W. Yao, Y. Mu, A. Gao, Q. Su, Y. Liu and Y. Zhang, Efficient ring-opening polymerization of epsilon-caprolactone using anilido-imine-aluminum complexes in the presence of benzyl alcohol, Polymer, 2008, 49, 2486–2491 CrossRef CAS.
- N. Nomura, T. Aoyama, R. Ishii and T. Kondo, Salicylaldimine-aluminum complexes for the facile and efficient ring-opening polymerization of epsilon-caprolactone, Macromolecules, 2005, 38, 5363–5366 CrossRef CAS.
- L. Qin, Y. Zhang, J. Chao, J. Cheng and X. Chen, Four- and five-coordinate aluminum complexes supported by N,O-bidentate
beta-pyrazylenolate ligands: synthesis, structure and application in ROP of epsilon-caprolactone and lactide, Dalton Trans., 2019, 48, 12315–12325 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental protocols and data. CCDC 2041481–2041486. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0qi01303j |
|
| This journal is © the Partner Organisations 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Benjamin D.
Ward
a,
Benjamin D.
Ward