
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal–organic frameworks vs. buffers: case study of UiO-66 stability†
Daniel
Bůžek
 *ab,
Slavomír
Adamec
b,
Kamil
Lang
a and
Jan
Demel
a
*ab,
Slavomír
Adamec
b,
Kamil
Lang
a and
Jan
Demel
a
aInstitute of Inorganic Chemistry of the Czech Academy of Sciences, 250 68 Husinec-Řež, Czech Republic. E-mail: buzek@iic.cas.cz
bFaculty of Environment, Jan Evangelista Purkyně University, 400 96 Ústí nad Labem, Czech Republic
First published on 7th December 2020
Abstract
UiO-66 is a zirconium-based metal–organic framework (MOF) that has numerous applications. Our group recently determined that UiO-66 is not as inert in aqueous dispersions as previously reported in the literature. The present work therefore assessed the behaviour of UiO-66 in buffers: 2-amino-2-(hydroxymethyl)-1,3-propanediol (TRIS), 4-(2-hydroxyethyl)piperazine-1-ethane sulfonic acid (HEPES), N-ethylmorpholine (NEM) and phosphate buffer (PB), all of which are commonly used in many UiO-66 applications. High performance liquid chromatography and inductively coupled plasma mass spectrometry were used to monitor degradation of the MOF at 25 °C. In each buffer, the terephthalate linker was released to some extent. The chemical nature of the buffer media played a decisive role in the stability with a more pronounced leaching effect in the saline forms of these buffers. The HEPES buffer was found to be the most benign, whereas NEM and PB should be avoided at any concentration as they were shown to rapidly degrade the UiO-66 framework. Low concentration TRIS buffers are also recommended, although these offer minimal buffer capacity to adjust pH. Regardless of the buffer used, rapid terephthalate release was observed, indicating that the UiO-66 was attacked immediately after mixing with the buffer. This process was even more pronounced at 37 °C, i.e., at typical temperature in biological and medical applications. In addition, the dissolution of zirconium, observed in some cases, intensified the UiO-66 decomposition process. These results demonstrate that sensitive analytical techniques have to be used to monitor the release of MOF components so as to quantify the stabilities of these materials in liquid environments.
Introduction
Metal–organic frameworks (MOFs) are coordination networks consisting of inorganic nodes (referred to as secondary building units or SBUs) joined together by organic linkers giving structure with potential voids.1,2 The use of different metals together with the wide range of potential linkers allows for the synthesis of MOFs having numerous different structures, topologies, properties and functionalizations.3,4 MOFs typically exhibit significant porosity and high surface areas often exceeding 1000 m2 g−1.5 Owing to their potential variability, MOFs are promising materials for many applications, such as gas storage and separation,6,7 adsorption of pollutants from liquid media,8,9 drug delivery and biomedicine,10,11 catalysis,12 energy storage13 and sensing.14At present, the poor stability of MOFs precludes their use in many applications, partly because MOFs are less stable than purely inorganic materials such as zeolites.15,16 Mechanical stability is important when shaping MOFs into pellets or when these materials are used for mechanical operations,17 while thermal and hydrothermal stability are vital in industrial processes that are associated with high temperatures or the presence of water vapour and steam.18–20 Finally, chemical stability is required when employing MOFs in liquid-based applications, including drug delivery, biomedicine, sensing and water treatment.15,18 Aqueous environments represent especially challenging conditions for MOFs because of potential for the hydrolytic cleavage of the coordination bonds constituting the MOF backbone.21,22 The stability of MOFs in water is determined by the strength of the metal-linker coordination bonds,23 and so metal ions that are trivalent (such as CrIII, AlIII and FeIII) or primarily tetravalent (such as TiIV, ZrIV and HfIV) form the most water-stable MOFs when employing carboxylate-type linkers.24–26
Zirconium-based MOFs (Zr-MOFs) represent a large family of water-stable structures. The most common Zr-MOFs, UiO-66, UiO-67 and UiO-68, are composed of [Zr6O6(OH)4]12+ SBUs connected by terephthalate, 4,4′-biphenyldicarboxylate and 4,4′-terphenyldicarboxylate linkers, respectively, to form 12-connected structures having an fcu-a-topology.27 Since the initial discovery of Zr-MOFs, many researchers has focused on the use of polytopic or functional linkers as well as different connective patterns among the Zr-SBUs. Such research has led to the design of dozens of new structures, including those of the MOF-808, NU-1000, PCN-222/MOF-545, MOF-525 and PCN-224 materials.28,29 Even so, the most commonly utilized Zr-MOF is UiO-66, because this MOF is robust and easy to synthesize and has numerous potential applications (Fig. 1).30
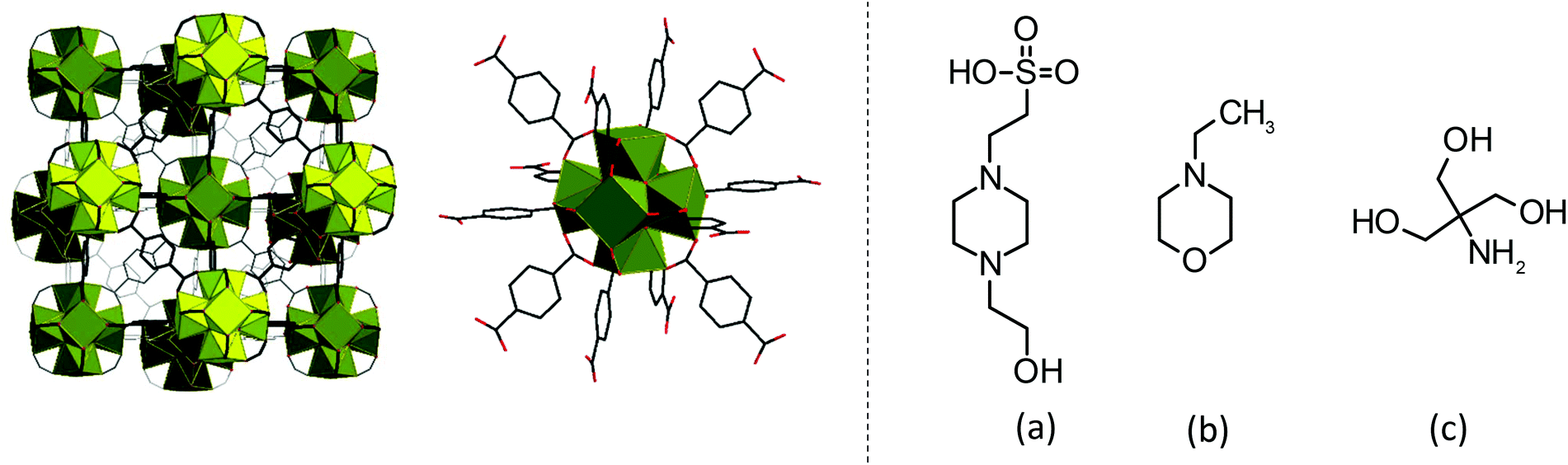 | ||
| Fig. 1 From left: The theoretical structure of UiO-66, a detailed view of the 12-connected [Zr6O6(OH)4]12+ SBU of UiO-66, and the molecular structures of the (a) HEPES, (b) NEM and (c) TRIS buffer constituents. | ||
Zr-MOFs are widely used in water-based applications such as catalysis, sensing, water treatment and drug delivery.11,31,32 In many of these applications, the Zr-MOFs are not utilized in neat water, but rather are dispersed in aqueous media with specific pH values, often achieved by the addition of buffers. Despite the common use of buffers, thus far there have been no detailed studies assessing the stability of Zr-MOFs in such solutions. Those studies that have examined the stability of MOFs have assessed post-exposed solids retrieved from various dispersions using powder X-ray diffraction (PXRD), gas adsorption analyses and scanning electron microscopy (SEM).18,33–39 Recently, our own group demonstrated that these methods are not able to conclusively assess structural changes induced in MOFs by exposure to aqueous media having various pH values.40 Indeed, even though the UiO-66 structure is quite stable in pure water, leaching of the terephthalate linkers has been shown to occur after adjustment to a pH of 6.0 while, at higher pH values, the UiO-66 framework is completely degraded.
The present work investigated the stability of UiO-66 in the most commonly utilized buffers, namely 2-amino-2-(hydroxymethyl)-1,3-propanediol (TRIS), (4-(2-hydroxyethyl)piperazine-1-ethane sulfonic acid (HEPES), phosphate buffer (PB) and N-ethylmorpholine (NEM), and in their saline forms (Fig. 1). The stability of this material was assessed by monitoring the release of the terephthalate linker and zirconium, as this approach has been shown to be superior when characterizing MOFs after exposure to water.40–42 The high performance liquid chromatography (HPLC) analysis of released linker also allowed the kinetics of the linker release to be studied. The results, together with data regarding the release of zirconium, were compared with data obtained using the PXRD, N2 adsorption and SEM techniques more commonly applied during stability characterizations.
Results and discussion
Synthesis and characterization of UiO-66
Zirconium-based UiO-66 was prepared according to a previously published procedure.40 Briefly, zirconium chloride, terephthalic acid and acetic acid were combined in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:95 and allowed to react in N,N-dimethylformamide (DMF) at 120 °C to generate UiO-66. This product was thoroughly washed with DMF and acetone then activated by heating at 100 °C under a dynamic vacuum. Water was not used for washing so as to eliminate any possibility of hydrolytic reactions. The parent UiO-66 prepared in this manner was characterized by PXRD, N2 adsorption analyses, SEM, 1H nuclear magnetic resonance (NMR) spectroscopy after dissolution in deuterated sodium hydroxide (see discussion and Fig. S1 in the ESI†); and by thermogravimetric analysis (TGA, Fig. S2, ESI†). The latter analysis showed an initial endothermic process with an associated mass loss of 10% between room temperature and 230 °C that was attributed to the release of water. This was followed by an exothermic peak at approximately 400 °C ascribed to the decomposition of monocarboxylates originating from the initial synthesis. The complete thermal decomposition of the UiO-66 between 450 and 560 °C was accompanied by the release of large amounts of CO2 and H2O. The TGA data showed an overall mass loss of 55% which is sum of 10% of water and approximately 45% was attributed to the degradation of the material (primarily the terephthalate linkers) yielding to ZrO2.
:1:95 and allowed to react in N,N-dimethylformamide (DMF) at 120 °C to generate UiO-66. This product was thoroughly washed with DMF and acetone then activated by heating at 100 °C under a dynamic vacuum. Water was not used for washing so as to eliminate any possibility of hydrolytic reactions. The parent UiO-66 prepared in this manner was characterized by PXRD, N2 adsorption analyses, SEM, 1H nuclear magnetic resonance (NMR) spectroscopy after dissolution in deuterated sodium hydroxide (see discussion and Fig. S1 in the ESI†); and by thermogravimetric analysis (TGA, Fig. S2, ESI†). The latter analysis showed an initial endothermic process with an associated mass loss of 10% between room temperature and 230 °C that was attributed to the release of water. This was followed by an exothermic peak at approximately 400 °C ascribed to the decomposition of monocarboxylates originating from the initial synthesis. The complete thermal decomposition of the UiO-66 between 450 and 560 °C was accompanied by the release of large amounts of CO2 and H2O. The TGA data showed an overall mass loss of 55% which is sum of 10% of water and approximately 45% was attributed to the degradation of the material (primarily the terephthalate linkers) yielding to ZrO2.
This material was found to have a Brunauer–Emmett–Teller (BET) specific surface area of 1395 m2 g−1 and a terephthalate:monocarboxylates molar ratio of 1.00:0.31. Details of these analyses are provided in Fig. S1 and S2 in the ESI† and in our previous paper.40 The results of the above analyses demonstrated that the UiO-66 was defective, primarily due to missing clusters, similar to the UiO-66 described by Lillerud et al.43
Stability of UiO-66 in buffer solutions
Many potential applications of MOFs require buffered media having a specific pH value. In the present work, the hydrolytic stability of UiO-66 was therefore assessed in TRIS, HEPES, PB and NEM buffers, all of which are commonly used in biomedicine for in vivo or in vitro applications such as drug delivery, and also for catalytic or sensing purposes. These buffers were prepared by carefully combining their acidic and basic components so as to avoid the requirement to add sodium hydroxide or acid for the purpose of pH adjustment, and thus to reduce variations in the ionic strength between buffers (see details in Table S2, ESI†).In each trial, 50 mg of the UiO-66 was combined with 50 mL of a buffer incorporating the buffering compound in the concentration range between 0.01 and 1.0 M (with the exception of PB), after which the kinetics of the terephthalate release were monitored for 4 h using HPLC. In selected cases, the kinetics was followed for 24 h. The concentrations and pH values of the various dispersions are summarized in Table 1. We also examined the effects of the saline forms of TRIS, HEPES and phosphate-buffered saline (PBS) along with those of NaCl solutions, acting as references. The dissolved zirconium concentrations were determined using inductively coupled plasma mass spectrometry (ICP-MS), also after 4 h (Table S3, ESI†). Following each trial in a given solution, the solid was separated, washed and characterized by PXRD, N2 adsorption and SEM.
| Buffer | C buffer/mol L−1 | pHa | pHb | S BET /m2 g−1 | S BET decreased/% | Linker release 15 mine/% | Linker release 4 he/% | Time of total linker releasef/min |
|---|---|---|---|---|---|---|---|---|
| a The pH of the as-prepared buffer. b The pH after a 4 h trial. c The specific surface area calculated by the BET method from N2 adsorption isotherms after a 4 h trial. d The decrease in SBET in comparison to that of the parent UiO-66 (SBET = 1395 m2 g−1) in %. e The amount (%) of the linker released after 15 min and 4 h, respectively, with respect to the total amount of the linker in parent UiO-66 (100% = 446 mg L−1 terephthalic acid). f The time required for the total release of all linker in the material (446 mg L−1 terephthalic acid determined by HPLC). g Complete linker release was not observed. h A colloidal system was formed. <LOD indicates that the concentration of terephthalic acid was below the HPLC detection limit (0.01 mg L−1). | ||||||||
| H2O | n. a. | 5.5 | 3.8 | 1331 | 5 | <LOD | <LOD | n. a.g |
| TRIS pH 7.5 | 0.01 | 7.5 | 7.0 | 1222 | 12 | 5.6 | 10 | n. a.g |
| 0.05 | 7.5 | 7.4 | 1107 | 21 | 8.0 | 16 | n. a.g | |
| 0.1 | 7.5 | 7.5 | 1100 | 21 | 10 | 26 | n. a.g | |
| 0.5 | 7.5 | 7.5 | 785 | 44 | 46 | 96 | n. a.g | |
| 1.0 | 7.5 | 7.5 | n. a.h | n. a. | 81 | 100 | 120 | |
| TRIS pH 9.0 | 0.01 | 9.0 | 8.2 | 849 | 39 | 33 | 49 | n. a.g |
| 0.05 | 9.0 | 8.5 | n. a.h | n. a. | 92 | 100 | 60 | |
| 0.1 | 9.0 | 8.8 | n. a.h | n. a. | 96 | 100 | 60 | |
| 0.5 | 9.0 | 9.0 | n. a.h | n. a. | 99 | 100 | 30 | |
| 1.0 | 9.0 | 9.0 | n. a.h | n. a. | 100 | 100 | 10 | |
| HEPES | 0.01 | 7.5 | 7.2 | 1319 | 5 | 4.2 | 9.4 | n. a.g |
| 0.05 | 7.5 | 7.3 | 1308 | 6 | 5.0 | 12 | n. a.g | |
| 0.1 | 7.5 | 7.5 | 1289 | 8 | 5.2 | 13 | n. a.g | |
| 0.5 | 7.5 | 7.5 | 1254 | 10 | 5.8 | 15 | n. a.g | |
| 1.0 | 7.5 | 7.5 | 1122 | 20 | 5.3 | 17 | n. a.g | |
| PB | 0.01 | 7.5 | 7.0 | 128 | 91 | 16 | 99 | n. a.g |
| 0.05 | 7.5 | 7.3 | 38 | 97 | 83 | 100 | 180 | |
| 0.1 | 7.5 | 7.5 | 224 | 84 | 93 | 100 | 120 | |
| 0.2 | 7.5 | 7.5 | 248 | 82 | 99 | 100 | 60 | |
| NEM | 0.01 | 8.2 | 8.1 | 952 | 32 | 16 | 23 | n. a.g |
| 0.05 | 9.2 | 8.8 | 493 | 65 | 44 | 61 | n. a.g | |
| 0.1 | 9.6 | 9.1 | 370 | 73 | 72 | 93 | n. a.g | |
| 0.5 | 10.1 | 9.6 | 10 | 99 | 91 | 100 | 120 | |
| 1.0 | 10.2 | 10.1 | 5 | 99 | 97 | 100 | 120 | |
| NaCl | 0.01 | 6.0 | 4.4 | 1373 | 2 | <LOD | <LOD | n. a.g |
| 0.05 | 6.0 | 4.8 | 1356 | 3 | <LOD | 0.1 | n. a.g | |
| 0.1 | 6.0 | 4.7 | 1323 | 5 | 0.1 | 0.1 | n. a.g | |
| 0.5 | 6.0 | 5.0 | 1319 | 5 | 0.7 | 0.8 | n. a.g | |
| 1.0 | 6.0 | 4.9 | 1322 | 5 | 1.3 | 1.6 | n. a.g | |
In a blank experiment, UiO-66 was also immersed in neat water. In this case, the pH of the aqueous medium was 3.8 due to the release of formate and acetate anions originally bound at defect sites in the UiO-66.43 Prior work has shown that both these species (although primarily the acetates) are leached out from the MOF, resulting in an acidic pH.40 In this trial, the concentration of released terephthalic acid in the water was found to be below the detection limit of the HPLC (<0.01 mg L−1) and subsequent PXRD, N2 adsorption and SEM assessments of the solid did not indicate any changes relative to the parent UiO-66 (Fig. S3, ESI†). These results established the structural and compositional stability of UiO-66 in water after a 4 h treatment.
The total amount of terephthalate linker in the parent UiO-66 was determined by dissolving 50 mg of UiO-66 in 50 mL of 1.0 M sodium hydroxide.43 The resulting solution contained 446 ± 16 mg L−1 terephthalic acid, indicating that the original material contained approximately 45% by mass of the linkers, in good agreement with the TGA results. The stability of a MOF is typically also determined by the solubility of the linker in a given medium, and so the solubility of terephthalic acid in the various solutions was examined. This solubility was found to vary greatly depending on pH and ionic strength. In neat water, the solubility was quite low at approximately 20 mg L−1 while solubilities in the buffer solutions were more than an order of magnitude greater, as can be seen from the data in Table S4 (ESI†). For each buffer solution assessed, the solubility of terephthalic acid was much greater than the terephthalate concentration that could be leached from the UiO-66 (compare Table 1 and Table S4, ESI†), demonstrating that the release of terephthalate was never limited by its solubility.
Stability of UiO-66 in TRIS buffers
TRIS buffers are made by combining 2-amino-2-(hydroxymethyl)-1,3-propanediol (the alkaline form of the compound) and its hydrochloride (the acidic form) in various ratios to obtain pH values ranging between 7 and 9. These buffers are often used as a medium for cell cultivation and molecular biochemistry, with MOFs having applications in drug delivery and as biosensors; TRIS buffers are also utilized as an environment for the post-synthetic modification of MOFs.44–46 In all these cases, the buffer concentrations are between 0.01 and 1.0 M with a typical working pH of 7.5. Therefore, we concentrated on ascertaining the effects of a TRIS buffer having a pH 7.5 on the stability of UiO-66, by assessing the kinetics of terephthalate release from the UiO-66 framework in detail (Table 1). To delineate the effect of pH, we also performed the same set of experiments at pH 9.0.We initially analysed the effect of the TRIS concentration on the extent of terephthalate release (Fig. 2). The exposure of the UiO-66 to 0.01 and 0.05 M TRIS solutions at pH 7.5 resulted in 10 and 16% linker release after 4 h, respectively. These values are similar to the 12% terephthalate release in water adjusted to pH 7.0 using a sodium hydroxide solution.40 It is important to keep in mind that the release in these trials were not limited by the terephthalate solubility. As can be seen from Table S4 (ESI†), the solubility of terephthalate in 0.01 M TRIS is much greater (302 mg L−1) than the concentration that was leached from the UiO-66 (45 mg L−1, equivalent to 10% release). However, the buffer capacity of the 0.01 M TRIS solution was insufficient to maintain a constant pH in conjunction with the release of terephthalate and monocarboxylic acids occupying defect sites in the UiO-66.40 The resulting variation in the pH could be important in certain biological applications. Higher TRIS concentrations were found to significantly enhance the terephthalate release, such that complete loss of the terephthalate linker occurred within 120 min in 1.0 M TRIS, at which point the solid MOF transitioned to a colloid dispersion and recovery of the solid was impossible. These observations indicate that the TRIS buffer constituents coordinated to zirconium ions (see the section entitled “Zirconium in buffers” below). Regardless of the TRIS concentration, the majority of the terephthalate release was also found to occur during the first 30 min of the treatment.
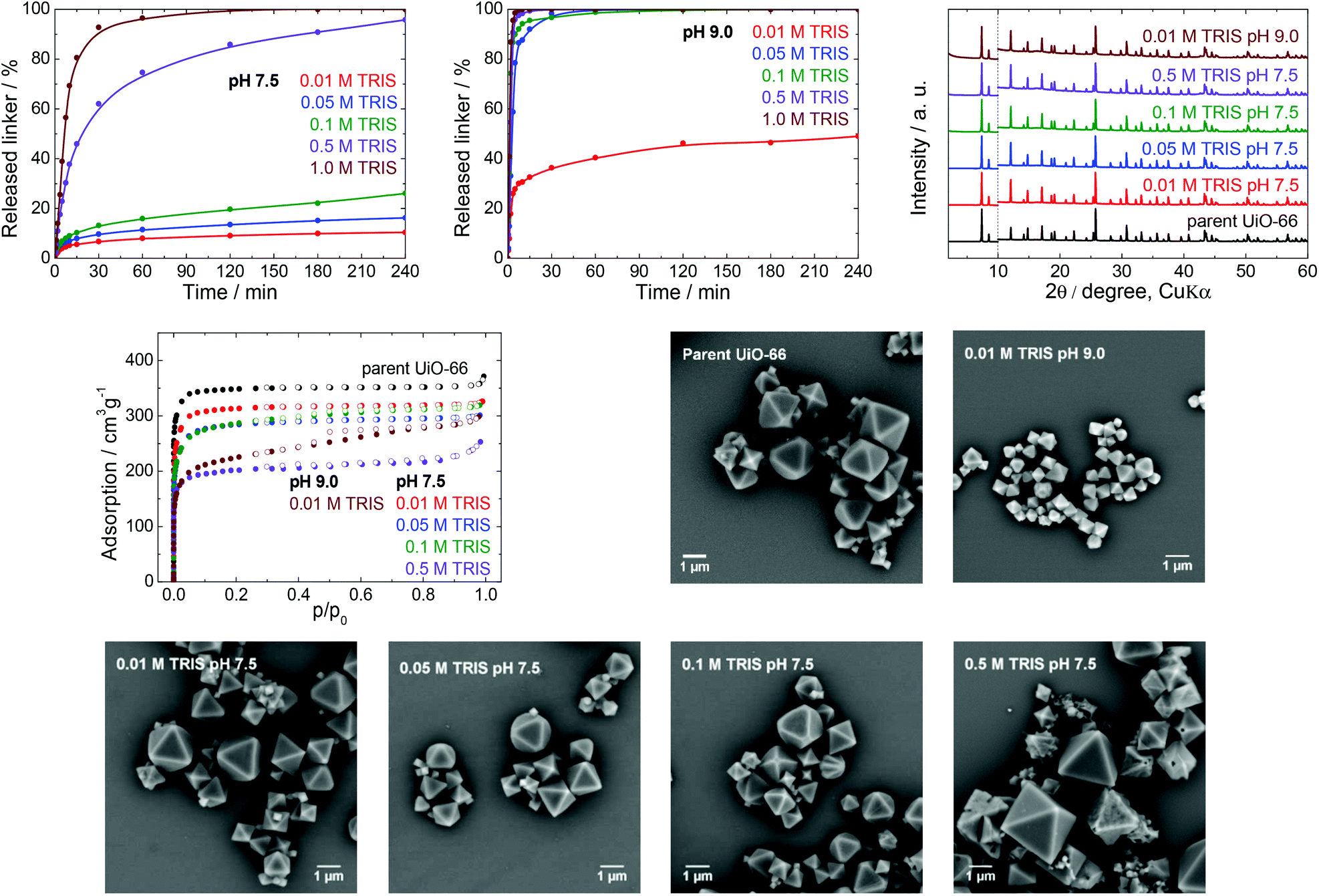 | ||
| Fig. 2 The stability of UiO-66 in TRIS buffers, a 4 h treatment. Upper row from left: Kinetics of the terephthalate release in TRIS buffers of different concentrations and pH 7.5 or 9.0, and a comparison of the PXRD pattern of the parent UiO-66 with those of post-exposed specimens. These patterns have been normalized and shifted vertically to avoid overlaps and the vertical dashed line divides the diffractograms at 2θ = 10° so that the less intensive diffractions above 10° can be enlarged. Middle row from left: N2 adsorption isotherms and SEM images of the parent UiO-66 and a sample after immersion in 0.01 M TRIS at pH 9.0. Bottom row: SEM images of UiO-66 samples after immersion in TRIS solutions of varying concentrations and pH values. | ||
The prolongation of the release experiments in 0.01, 0.05 and 0.1 M TRIS to 24 h showed that the linker release occurs slowly even in a longer times scale. The most pronounced effect was observed in 0.1 M TRIS solution (Fig. S4, ESI†).
PXRD, N2 adsorption isotherms and SEM were all used to characterize the MOF samples after buffer exposure for 4 h, with the exception of the sample immersed in the 1.0 M TRIS solution at pH 7.5, because no solid could be recovered (Fig. 2). The positions of diffraction lines suggested that the UiO-66 phase was retained even after immersion in 0.5 M TRIS, during which the MOF lost 96% of its terephthalate linker content. These observations are consistent with the results of our previous work,40 in which PXRD patterns recorded only total decomposition of the UiO-66. In the case of the parent UiO-66, the diffraction line at a 2θ value of approximately 12.1° was more intense than the two lines at 14.2° and 14.8° and this intensity ratio was even more evident in the patterns of the post-exposed UiO-66. This behaviour was common to all the buffers assessed in this work (see PXRD patterns in Fig. 3–6) and can be ascribed to the degree of hydroxylation of the UiO-66 structure, which increased the intensity of the line at 12.1° after contact of the UiO-66 with water.47
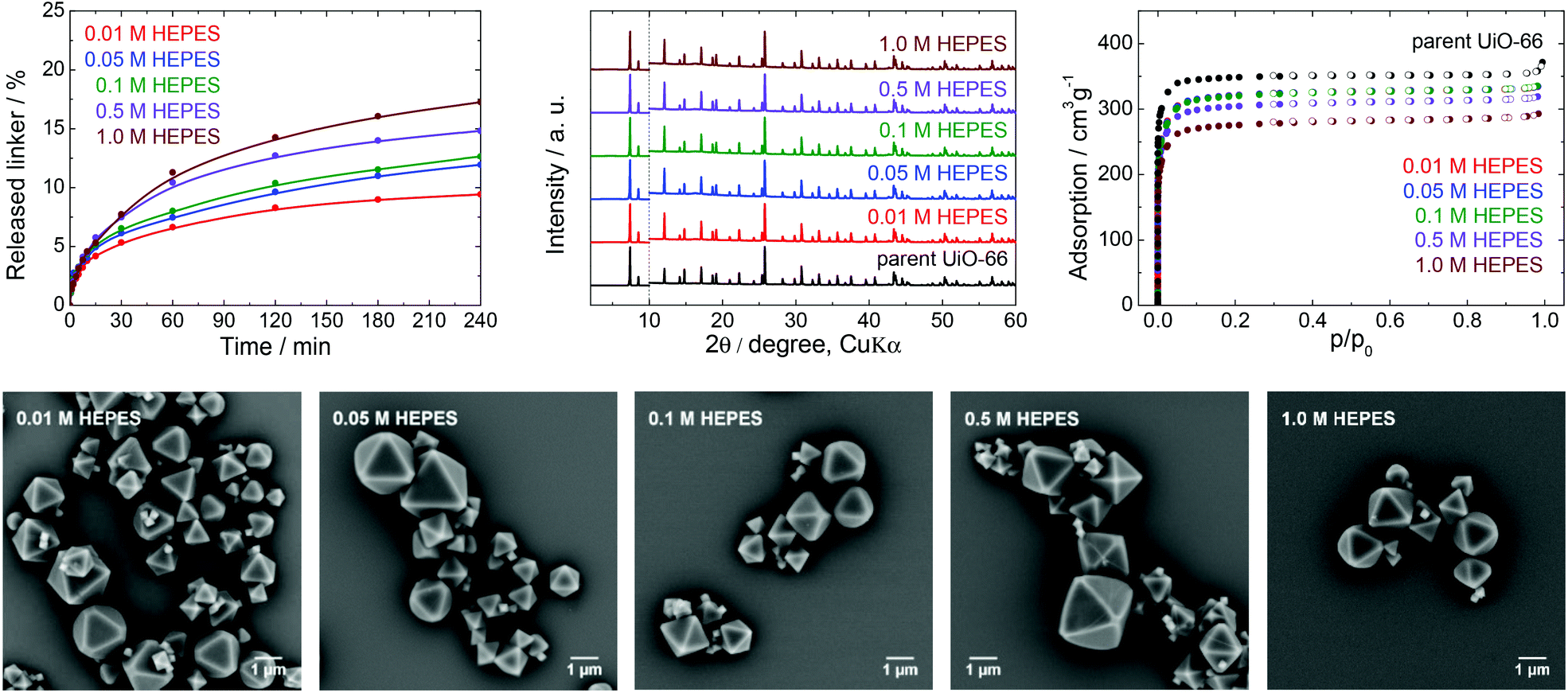 | ||
| Fig. 3 The stability of UiO-66 in HEPES buffers at pH 7.5, a 4 h treatment. Upper row from left: Kinetics of the terephthalate release in HEPES buffers of different concentrations, comparison of the PXRD pattern of the parent UiO-66 with those of post-exposed specimens, and N2 adsorption isotherms of the parent UiO-66 and of post-exposed specimens. The PXRD patterns have been normalized and shifted vertically to avoid overlaps and the vertical dashed line divides the diffractograms at 2θ = 10° so that the less intensive diffractions above 10° can be enlarged. Bottom row: SEM images of UiO-66 samples after immersion in HEPES solutions of varying concentrations (see Fig. 2 for an SEM image of the parent UiO-66). | ||
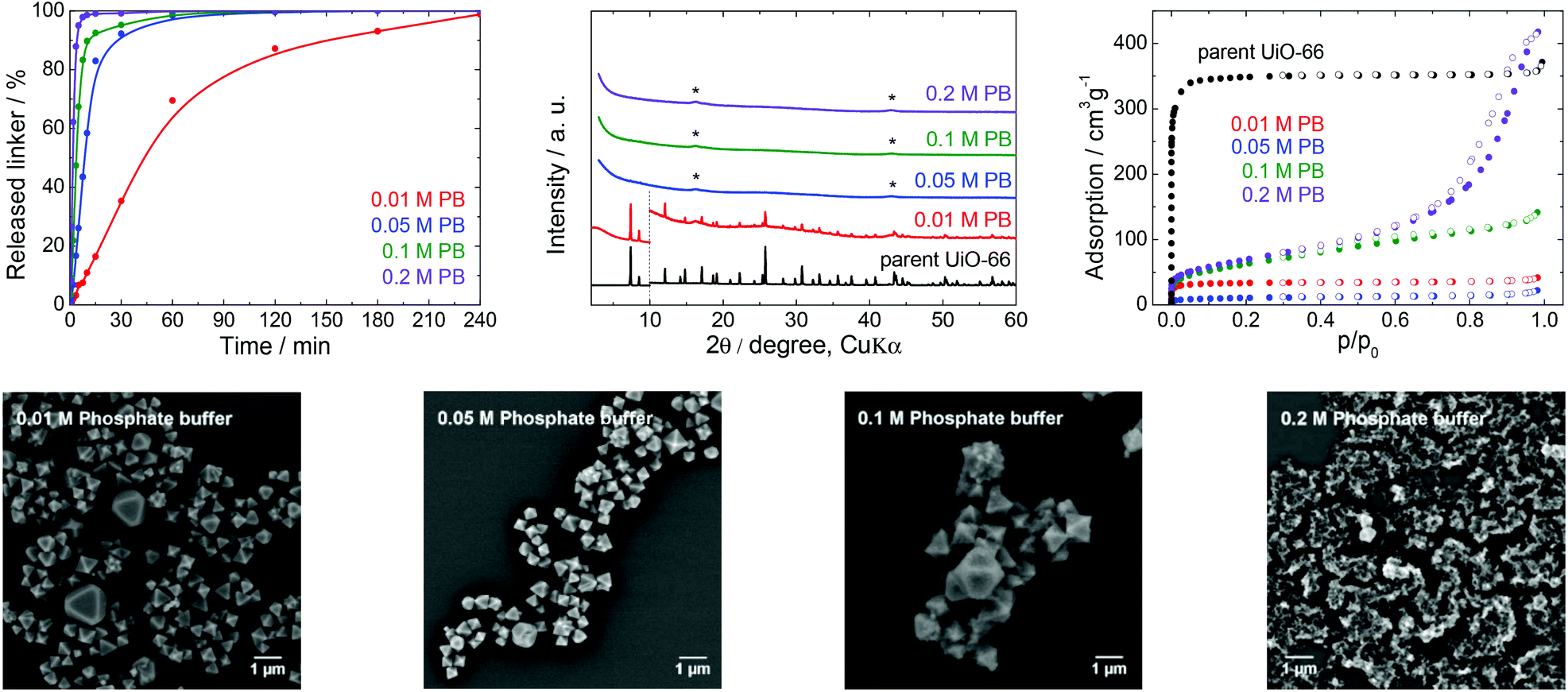 | ||
| Fig. 4 The stability of UiO-66 in PB solutions at pH 7.5, a 4 h treatment. Upper row from left: Kinetics of the terephthalate release in PB of different concentrations, comparison of the PXRD pattern of the parent UiO-66 with those of post-exposed specimens, and N2 adsorption isotherms of the parent UiO-66 and of post-exposed specimens. The PXRD patterns have been normalized and shifted vertically to avoid overlaps and the vertical dashed line divides the diffractograms at 2θ = 10° so that the less intensive diffractions above 10° can be enlarged. The diffraction lines marked with an asterisk resulted from the Mylar foil support. Bottom row: SEM images of UiO-66 samples after immersion in PB solutions of varying concentrations (see Fig. 2 for an SEM image of the parent UiO-66). | ||
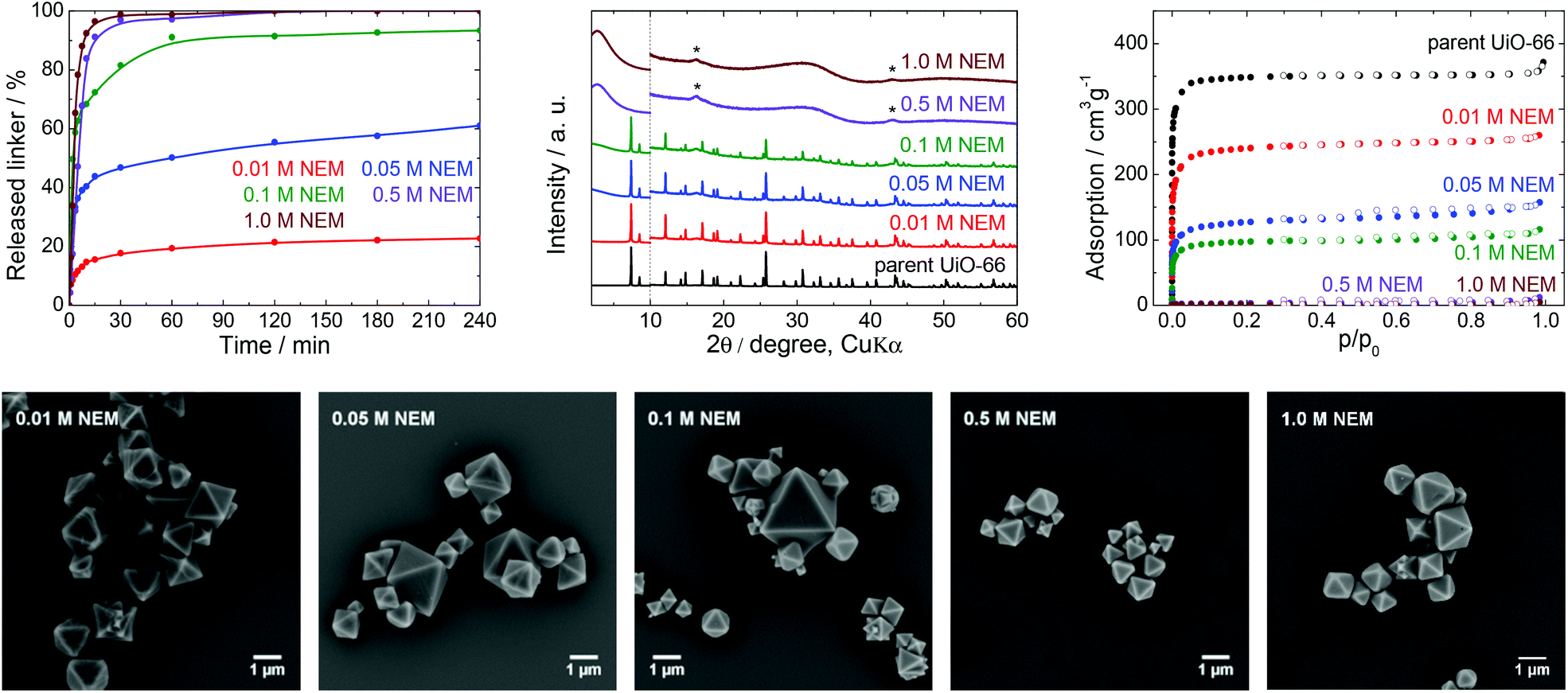 | ||
| Fig. 5 The stability of UiO-66 in NEM solutions, a 4 h treatment. Upper row from left: Kinetics of the terephthalate release in NEM solutions at different pH, comparison of the PXRD pattern of the parent UiO-66 with those of post-exposed specimens, and N2 adsorption isotherms of the parent UiO-66 and of post-exposed specimens. The PXRD patterns have been normalized and shifted vertically to avoid overlaps and the vertical dashed line divides the diffractograms at 2θ = 10° so that the less intensive diffractions above 10° can be enlarged. The diffraction lines marked with an asterisk resulted from the Mylar foil support. Bottom row: SEM images of UiO-66 samples after immersion in NEM solutions of varying concentrations (see Fig. 2 for an SEM image of the parent UiO-66). | ||
 | ||
| Fig. 6 The stability of UiO-66 in NaCl solutions, a 4 h treatment. Upper row from left: Kinetics of the terephthalate release in NaCl solutions of different concentrations, comparison of the PXRD pattern of the parent UiO-66 with those of post-exposed specimens, and N2 adsorption isotherms of the parent UiO-66 and of post-exposed specimens. The PXRD patterns have been normalized and shifted vertically to avoid overlaps and the vertical dashed line divides the diffractograms at 2θ = 10° so that the less intensive diffractions above 10° can be enlarged. Bottom row: SEM images of UiO-66 samples after immersion in NaCl solutions of varying concentrations (see Fig. 2 for an SEM image of the parent UiO-66). | ||
Contrary to PXRD, the N2 adsorption isotherms were found to be more sensitive to changes in the UiO-66 network, and indicated decreasing specific surface areas but with preservation of the microporous nature of the material at increasing TRIS concentrations (Table 1, Fig. 2). These decreases in specific surface area did not correlate with the percentage of the released linker, and so this parameter does not reflect the degree of degradation of the UiO-66. In addition, the SEM images did not show any changes in the morphology over the concentration range of 0.01–0.1 M TRIS compared with the parent UiO-66. Interestingly, exposure to the 0.5 M TRIS disrupted the surfaces of microcrystals even though their shapes were preserved. This behaviour is likely connected to the behaviour of the zirconium ions, which partially dissolved in the solution and were partly hydrolysed and precipitated to form an amorphous solid on the microcrystal surfaces (see the section titled “Zirconium in buffers” below).
To better understand the effect of pH on the UiO-66 stability, analogous experiments were repeated in a TRIS solution at pH 9.0, which is the upper limit for a TRIS buffer. At this pH, a large amount of terephthalate was released even at low TRIS concentrations such as 0.01 and 0.05 M, accompanied by a lowering of the final pH value (Table 1). Except for the 0.01 M TRIS trial, complete release occurred within 60 min or less. We recovered a small amount of the solid only from the 0.01 M TRIS, while at higher TRIS concentrations the solid formed colloids. Similar to the effects observed at pH 7.5, the positions of diffraction lines were unaffected. However, the specific surface areas were considerably decreased, the shapes of the adsorption isotherms indicated the loss of microporosity, and the SEM analyses revealed significant decreases in the particle sizes, although with preservation of the shape of the microcrystals (Fig. 2).
In general, the TRIS buffers at pH 7.5 represented a destructive environment for UiO-66, especially at concentrations above 0.05 M. The main reason was that the buffer components coordinated to zirconium ions and the transfer of these ions into solution was accompanied by the release of a large portion of the linker and destruction of the UiO-66 framework. The retrieved solid, even after leaching of 96% of the terephthalate linker, was found to generate the same diffraction lines. Therefore, PXRD is suitable only for the detection of the total destruction of the UiO-66 framework. The specific surface area decreased when the TRIS molarity increased, although the microporous nature of the UiO-66 was maintained (with the exception of the 0.01 M TRIS solution at pH 9.0). Evidently, UiO-66 should be used only in 0.01 or 0.05 M TRIS buffers at pH 7.5, and the associated small release of the linker should still be considered. TRIS buffers at pH 9.0 should be excluded completely.
Stability of UiO-66 in HEPES buffer
HEPES buffers are a mixture of 4-(2-hydroxyethyl)piperazine-1-ethane sulfonic acid and its sodium salt and are used in the pH range from 7.0 to 8.0 (Fig. 1). There are reports on applications of UiO-66 in HEPES in areas such as drug delivery, cell imaging, luminescence (chemodosimetry), biomedicine applications and catalysis,48–50 including the post-synthetic modification of Zr-MOFs.51 HEPES buffers are commonly used at concentrations between 0.01 and 0.1 M with a pH 7.5.From Fig. 3 and Table 1, it is apparent that the extent of terephthalate leaching from the UiO-66 HEPES was comparable to those in water adjusted to pH 7.0.40 In contrast to the results obtained with TRIS, the release was quite limited even at high HEPES concentrations. As an example, immersion in 0.01 and 1.0 M HEPES led to the release of 9.4 and 17% of the terephthalate linker from the UiO-66 network after 4 h, respectively, indicating that the dependence of the linker release on the HEPES concentrations was significantly lower. The buffer capacity of the 0.01 or 0.05 M HEPES was evidently not high enough to hold the pH constant, although the final pH was not greatly affected. Clearly, because the solubility of terephthalic acid in HEPES solutions was much greater than the concentration of the leachates, the terephthalate release was not affected by the terephthalate solubility limit (Table S4, ESI†). Similarly to TRIS buffers, the slow release of terephthalate continued at longer treatment times (Fig. S4, ESI†).
The PXRD patterns did not indicate changes in the UiO-66 structure after the treatment, whereas the N2 adsorption isotherms showed that the specific surface area decreased by approximately 20% after immersion in the 1.0 M HEPES, with the preservation of the microporous structure of the UiO-66. In addition, SEM images of the post-exposed solids showed the same morphology as that of the parent UiO-66.
These results demonstrate that HEPES buffers are relatively benign to the UiO-66 framework, the effect of the HEPES concentration on linker release is very low, and the terephthalate release is predominantly controlled by the hydrolytic activity of the water itself. Highly concentrated HEPES buffers are therefore much more compatible with UiO-66 and more suitable for UiO-66-based applications than TRIS buffers. However, the partial terephthalate release from UiO-66 connected with the formation of additional structural defects cannot be ignored in some cases.
Stability of UiO-66 in phosphate buffer
PB buffers and PBS are the most widely utilized media for biological and medical applications and for cell cultivations because of their good compatibility with living tissues and microorganisms. For these reasons, these buffers are often combined with bulk or nanosized MOFs for drug delivery or photodynamic therapy, or as contrast agents.42,52,53Fig. 4 and the data in Table 1 establish that the UiO-66 rapidly lost the terephthalate linker at all PB concentrations. Thus, the degradation was complete shortly after 180 min in 0.05 M PB and was greatly accelerated in 0.2 M PB to just 60 min. With the exception of 0.01 M PB, the majority of the terephthalate was released within the first 15 min. The increase in the buffer molarity accelerated the UiO-66 decomposition. Similar to the results obtained with the low concentration TRIS and HEPES solutions, the PB did not maintain a constant pH at concentrations below 0.05 M due to the large release of the terephthalate linker and/or monocarboxylates.
The PXRD pattern of the solid retrieved from 0.01 M PB after 4 h contained diffraction lines attributable to the UiO-66 phase, even though 99% of the terephthalate linker was released. At higher PB concentrations, the UiO-66 was transformed to an amorphous solid phase. The PB also had a significant effect on the N2 adsorption isotherms and BET specific surface areas (Fig. 4). The BET specific surface area of the solid immersed in 0.05 M PB was significantly decreased to 38 m2 g−1 even though the area was increased considerably to 224 and 248 m2 g−1 after treatment in 0.1 and 0.2 M PB, respectively. Most likely, the amorphous solid examined by PXRD was an amorphous zirconium–phosphate coordination polymer generated in the highly concentrated PB and having an increased specific surface area.54,55 In agreement with the above results, SEM images demonstrated a significant decrease in particle size at lower PB concentrations (0.01 or 0.05 M) and complete disintegration at concentrations above 0.1 M PB.
In general, PB buffers provide a highly destructive environment for UiO-66 even at very low concentrations. The effect of PB is so strong that more than 80% of the terephthalate linkers are released within 15 min of immersion in 0.05 M PB and higher concentrations. Clearly, PB buffers are not suitable for any application of UiO-66 that take advantage of its porous structure. This would also be the case in drug delivery formulations, which are generally based on a slow release of active compounds from carrier structures.
Stability of UiO-66 in N-ethylmorpholine solution
NEM solutions (Fig. 1) were selected for analysis based on the frequent usage of these media in catalysis primarily for the degradation of organophosphates such as methyl paraoxon and chemical warfare agents together with Zr-MOFs.56–58 NEM solutions are typically used at concentrations in the vicinity of 0.5 M with pH values varying between 8.0 and 10.2.NEM solutions were found to induce the release of terephthalate from UiO-66 even at low concentrations. The extent of the release was also significantly increased with increases in the NEM concentration and at higher pH values up to pH 10.2 in 1.0 M NEM (Table 1, Fig. 5). The leaching kinetics were similar to those observed in trials with the other buffers, meaning that the linker release was very fast during the first 15 min of the treatment, followed by a considerable deceleration. The terephthalate linker was completely released from the UiO-66 in the 0.5 and 1.0 M NEM within 120 min.
Diffraction patterns confirming the UiO-66 structure were obtained after immersion of the material in solutions having low NEM concentrations (Fig. 5). The PXRD data for the solid retrieved after treatment in 0.1 M NEM did not exhibit changes in the positions of the diffraction lines of UiO-66 or the formation of a new phase, even after 93% of terephthalate had been leached. The dissolution of the terephthalate was correlated with a decrease in the specific surface area to 370 m2 g−1 together with retention of microporosity. After the complete release of terephthalate in 0.5 or 1.0 M NEM, the diffraction patterns no longer indicated the presence of UiO-66 and the porosity of the material disappeared. Surprisingly, even the solid recovered from the 1.0 M NEM contained some particles (albeit with reduced sizes) having the original morphology as shows SEM images (Fig. 5).
NEM solutions greatly affected the UiO-66 framework in a short time frame, such that the UiO-66 structure was completely destroyed at higher NEM concentrations. These results suggest that NEM buffers are not suitable in cases where UiO-66 is utilized as a catalyst. In addition, the reuse of this material after contact with NEM would evidently be challenging or even impossible because of the extensive degradation of the material.
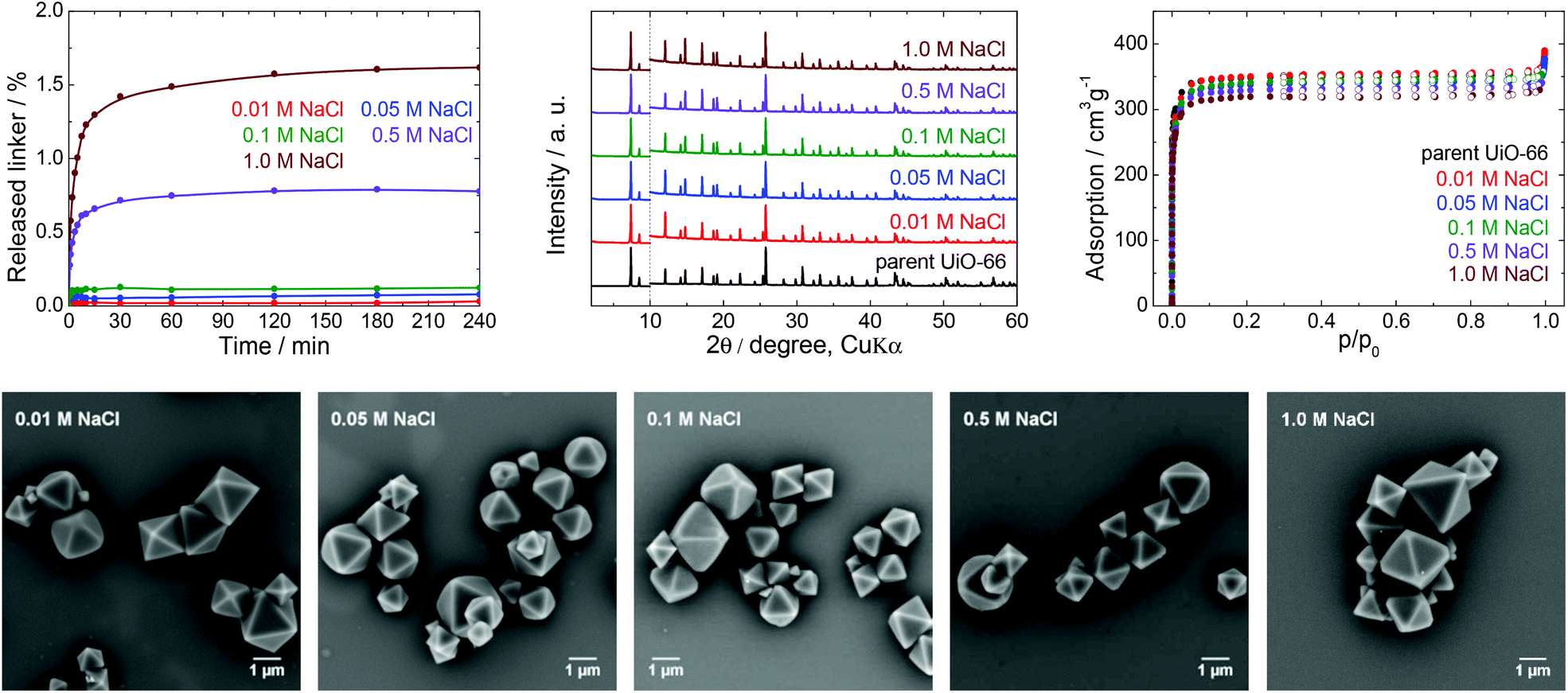
Stability of UiO-66 in saline buffers
Many physiological buffers contain sodium chloride (or potassium chloride or their combination) as an essential component. Thus, PBS, TRIS saline and HEPES saline are widely used in biological and medical applications. The effect of saline on UiO-66 stability was assessed by immersing UiO-66 in sodium chloride solutions having concentrations ranging from 0.01 to 1.0 M without pH adjustment (Table 1, Fig. 6). The results were compared with those observed after exposure to PBS, TRIS saline and HEPES saline (Fig. 7), all of which comprised 0.01 M of the buffering compound together with 0.15 M NaCl (the concentration commonly used in saline buffers).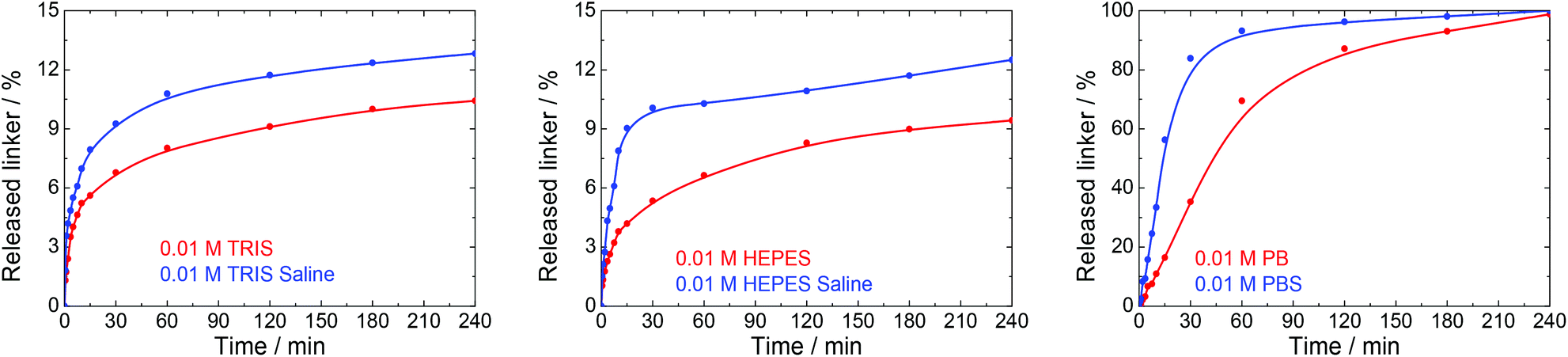 | ||
| Fig. 7 The release kinetics of terephthalate from UiO-66 in 0.01 M TRIS, HEPES and PB buffers at pH 7.5 and their saline forms containing 0.15 M NaCl. | ||
First, we studied the effect of NaCl solutions on UiO-66. The initial pH values of the sodium chloride solutions decreased from 6.0 to pH 4.4–5.0 after mixing with the parent UiO-66. HPLC analyses established that the amount of released terephthalate was negligible, with a maximum value of approximately 1.6 and 2.1% in 1.0 M NaCl after 4 and 24 h, respectively, and that the majority of the terephthalate was released during the first 30 min (Fig. S4, ESI†). PXRD patterns and SEM images of the material after these trials did not indicate any changes, and specific surface areas obtained from N2 adsorption isotherms showed a maximum decrease of only 5% which is within the experimental error.
Fig. 7 depicts the effects of salinity on the terephthalate release from UiO-66 in TRIS saline, HEPES saline and PBS buffers. Importantly, the presence of NaCl had a negative effect on the UiO-66 stability. Specifically, the total amount of released terephthalate was increased by 20–30% in the saline forms of the TRIS and HEPES buffers compared with the corresponding pure buffers. In addition, the decomposition of UiO-66 was considerably accelerated in the PBS solution as compared with the PB solution.
Data resulting from saline solutions indicate that the presence of sodium chloride in water has a negligible, if any, effect on UiO-66 stability (Fig. 6). In contrast, the presence of saline in buffers generates a synergistic effect leading to a higher degree of UiO-66 decomposition. Therefore, saline buffers should be used with care in UiO-66-based applications.
Effect of temperature on the UiO-66 stability
Many biological and medical applications of MOFs require temperature of 37 °C. We selected 0.01 M TRIS, HEPES and PB buffers and 0.01 M NaCl solution, the most often utilized environments in these applications, to evaluate the effects of temperature on the stability of UiO-66. These trials were only done at concentration of buffers of 0.01 M for 4 h.A blank experiment in water showed no effect of 37 °C treatment temperature on the UiO-66 stability. The same result was obtained in 0.01 M NaCl. In contrast, the kinetics of the linker release was significantly accelerated in buffer solutions reaching similar terephthalate concentrations after 4 h of treatment as observed at 25 °C (Fig. 8). These results document that the damage to the UiO-66 structure is much faster at 37 °C than at 25 °C and is completed within 10–30 min after the mixing of UiO-66 with the buffer solutions.
 | ||
| Fig. 8 The effect of temperature on the release kinetics of terephthalate from UiO-66 in 0.01 M TRIS, HEPES, and PB. Blank experiments in water and NaCl solutions are not shown since terephthalate concentrations were below limit of detection. | ||
Zirconium in buffers
Dissolved Zr4+ ions exist only in strongly acidic environments, whereas in neutral or basic solutions Zr4+ immediately hydrolyses to form amorphous zirconium oxide/hydroxide species.59,60 In addition to this process which was anticipated in the buffers in the present work, the mass losses exhibited by the UiO-66 during trials in the TRIS buffers were higher than could be explained by the amount of terephthalate released and, in some cases, no solid was even recovered. These results indicate the solubilization of zirconium and the formation of colloidal dispersions, and so the concentrations of dissolved zirconium were evaluated using ICP-MS (Table S3, ESI†).Total amount of zirconium in parent UiO-66 was determined using ICP-MS after microwave-assisted total dissolution of the MOF in mixture of nitric, hydrochloric and hydrofluoric acids. Resulting data showed that content of zirconium in parent UiO-66 was 33.5 ± 1.2%, i.e. maximum release of zirconium can lead to 335 ± 12 mg L−1 in our experimental setup. This content of zirconium is in good agreement with similar defective UiO-66 described in literature.43,61 Moreover, amount of zirconium was confirmed by TGA measurement which showed 333 mg L−1 (for details see Table S3, ESI†) and which is well comparable with data resulting from ICP-MS.
Fig. 9 shows the percentages of zirconium released into the TRIS (pH 7.5 and 9.0), PB and NEM buffers after 4 h. Data for the HEPES buffer and NaCl solution are not presented here because the zirconium concentrations were below the detection limit of the analytical method (0.001 mg L−1, representing 3 × 10−4% of total zirconium) in these cases.
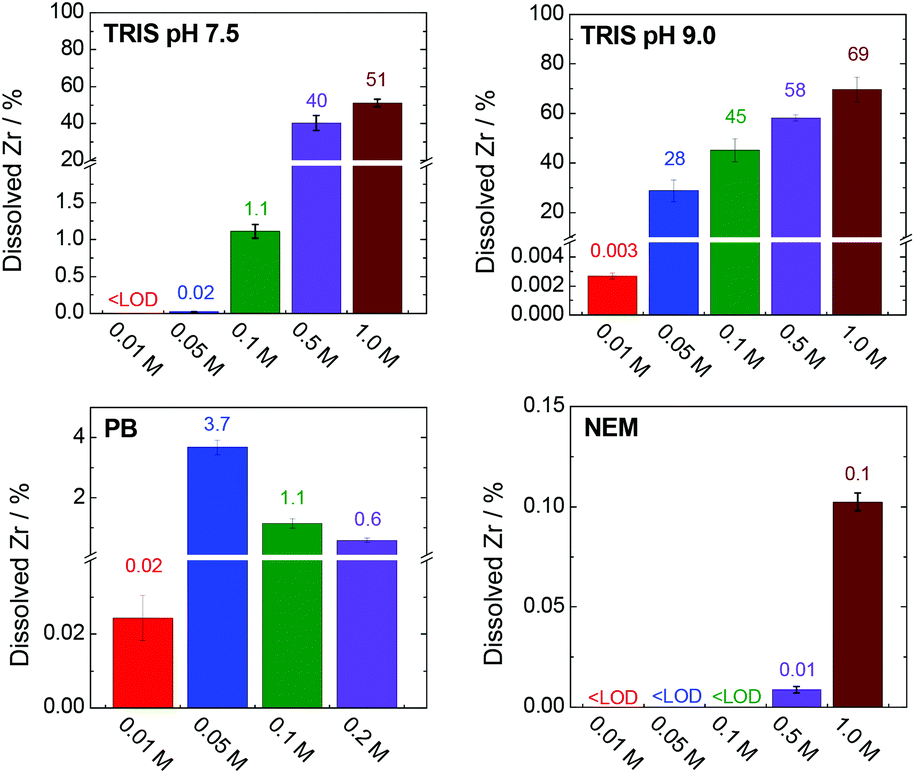 | ||
| Fig. 9 Percentages of dissolved and stabilized zirconium after a 4 h treatment in various buffers. The corresponding concentrations and experimental errors are given in Table S3, ESI.† Total amount of zirconium (100%) corresponds to 335 mg L−1. | ||
The TRIS buffers had the strongest ability to stabilize zirconium in solution via coordination with 2-amino-2-(hydroxymethyl)-1,3-propanediol. The amount of dissolved zirconium was correlated with both the TRIS concentration and pH (Fig. 9). Indeed, the zirconium concentrations in the separated liquid phases after the treatment of UiO-66 in 0.01–0.1 M TRIS at pH 7.5 were below the detection limit or very low. Under these conditions, the extent of terephthalate leaching was also minor. Increasing the TRIS concentration to 0.5 or 1.0 M greatly increased the concentration of dissolved zirconium, which was consistent with the increased release of the terephthalate linker. Even so, the zirconium concentration in 1.0 M TRIS at pH 7.5 (171 mg L−1i.e. 51% of total zirconium content) was still less than the total amount of zirconium in the UiO-66. The treatment was accompanied by visible changes in the transparency of the mixture and recovery of the post-exposed solid was not possible due to formations of colloid. In the case of TRIS buffers at pH 9.0 (i.e., above the pKa of the TRIS conjugated acid), zirconium was stabilized in the solutions, such that the suspensions became transparent above 0.05 M TRIS. The concentration of dissolved zirconium increased to 233 mg L−1 (equivalent to 69% of the total zirconium content) in 1.0 M TRIS, which was still less than the total content of 335 mg L−1. This discrepancy can be attributed to the hydrolysis of the non-complexed or partially-complexed zirconium by 2-amino-2(hydroxymethyl)-1,3-propandiol to form colloidal particles.
The use of the HEPES buffers did not result in any zirconium dissolution because the zirconium did not coordinate with the 4-(2-hydroxyethyl)piperazine-1-ethanesulfonate. As was discussed in section entitled “Stability of UiO-66 in HEPES buffer” above, the amount of linker released in HEPES solutions was significantly less affected by the buffer concentration compared with trials using TRIS. In both cases, the linker releases were comparable at concentrations in the range of 0.01–0.05 M. However, increasing the TRIS concentration was found to promote linker release, evidently because of the partial dissolution of zirconium, which was not observed in highly concentrated HEPES solutions. The solubilization of zirconium therefore seems to have promoted decomposition of the MOF.
In agreement with the rapid disintegration of UiO-66 in the PB solutions (Fig. 4), the zirconium concentration was found to increase in the trials up to 0.05 M PB (Fig. 9, Table S3, ESI†). At higher PB concentrations the zirconium concentrations decreased, evidently because of the formation of zirconium phosphate coordination polymer (based on the PXRD patterns, N2 adsorption isotherms and SEM images of the materials (Fig. 4) that incorporated dissolved zirconium. NEM has minimal ability to coordinate with zirconium and so there was little stabilization of the metal in the 0.5 and 1.0 M NEM buffers and the majority of the zirconium was hydrolysed, accompanied by the total release of the terephthalate in the MOF (Fig. 5 and 8).
These data indicate that the constituents of some buffers can strongly coordinate with zirconium and that this effect directly modifies the stability of UiO-66. Indeed, the decomposition of UiO-66, as indicated by the release of the linker, appears to be initiated by the solubilization of zirconium. Clearly, pre- and post-exposure weighing of an MOF cannot conclusively show the stability of the material in a buffered solution or be used to quantify the formation of missing-linker defects. The observations provided in this section may also serve to elucidate the initial stages of the degradation of a zirconium-based MOF.
Conclusions
This work examined the stability of UiO-66 in various buffers to illustrate the limitations and possible failure mechanisms of this material. None of the tested buffers were found to be harmless to UiO-66. Low TRIS concentrations at pH 7.5 induced the release of the terephthalate linker, but this was primarily because of the hydrolytic effect of the water in the buffer at given pH. Thus, 0.01 and 0.05 M TRIS buffers with a pH of 7.5 are the most suitable UiO-66 media as they induce only partial UiO-66 decomposition. However, these buffers allow pH variability. At higher TRIS concentrations or pH values, the leaching of terephthalate and zirconium is enhanced considerably and eventually leads to the complete destruction of the UiO-66 framework. In contrast, the HEPES buffers were found to have a relatively small effect on the UiO-66 framework and could be suitable for UiO-66 applications, similar to TRIS at low concentrations, but again with partial release of the linker. PB and NEM buffers, even at low concentrations, significantly degrade the MOF framework and should be avoided in all UiO-66-based applications. We also found that saline buffers, commonly utilized in biological and medical applications, support the terephthalate release when compared with corresponding pure buffers. In addition, raising temperature from 25 °C to 37 °C accelerates the kinetics of the terephthalate release considerably.Regardless the type and concentration of the buffer, a common feature was the rapid release of terephthalate within the first 15–30 min of exposure, indicating immediate attack of the UiO-66 by the buffer constituents. When saline buffers were used, both the rate and magnitude of the linker leaching was increased. The present results confirm that standard techniques such as PXRD, N2 adsorption, SEM, and weighing before and after the treatment are not conclusive as they do not provide quantitative data regarding changes in the UiO-66. Thus, more sensitive analytical approaches based on HPLC or ICP-MS will be important to increasing our understanding of the stabilities of various MOFs in different environments, including neat water, aqueous solutions and organic solvents.
Moreover, this work determined that the fate of zirconium is also a very important issue in UiO-66 stability. Primarily TRIS and PB can act as ligands to coordinate with zirconium and promote its dissolution. The degree of UiO-66 decomposition also appears to be highly correlated with the ability of the buffering compound (and the concentration of the buffer) to dissolve zirconium, as shown by a comparison between the results obtained using TRIS and HEPES. This phenomenon is likely to be common to all zirconium-based MOFs, evidence for which is provided by our previous work with the Zr-porphyrin-MOF PCN-222.62
Experimental
Materials and methods
The materials and chemicals used in this work are summarized in Table S1 (ESI†). Buffer solutions were prepared by mixing the acidic and basic forms of respective components in ratios required to give the desired pH value. To eliminate ionic strength changes, we avoided the addition of sodium hydroxide or acids for the purpose of pH adjustment. Detailed descriptions of buffer preparation procedures are provided in Table S2 (ESI†).Powder X-ray diffraction patterns were acquired using a PANalytical X'Pert PRO diffractometer in the transmission mode with a Cu X-ray tube (40 kV, 30 mA). Qualitative data analysis was performed with the HighScorePlus software package (PANalytical, Almelo, version 3.0) and the JCPDS PDF-2 database.63 Nitrogen adsorption isotherms were obtained at −196.2 °C with a Belsorp max II instrument (Microtrac Bel). Prior to each trial, the sample was heated under vacuum at 90 °C for at least 24 h. Sample surface areas were calculated using the Brunauer–Emmett–Teller (BET) method. High-resolution scanning electron microscopy (SEM) was performed using a FEI Nova NanoSEM microscope equipped with a circular backscatter detector in the backscattered electron mode, operating at an accelerating voltage of 5 kV. The samples were prepared by deposition onto a silicon wafer chip followed by drying in air overnight.
Concentrations of terephthalic acid released from the UiO-66 were determined by high performance liquid chromatography using a DIONEX UltiMate 3000 instrument equipped with a diode array detector (operating at 240 nm), a 20 μL sampling loop and a Hydrosphere 5 μm C18 column (YMC Co. Ltd, Japan, 150 mm × 4.6 mm). The mobile phase was acetonitrile/water (v/v = 30/70, both acidified with 0.1% formic acid) with an isocratic elution having a run time of 4 min at a flow rate of 1 mL min−1. The separation was performed at a constant temperature of 30 °C. Terephthalic acid concentrations were determined using a calibration curve with a relative standard deviation of less than 5% based on replicate analyses of standard solutions.
Dissolved zirconium was quantified using an Agilent 7900 ICP-MS instrument equipped with an Ar burner, an ORS 4 collision reaction cell and a hyperbolic quadrupole mass analyser with an orthogonal detection system. Samples were filtered through 0.1 μm PTFE microfilters (Whatman) and analysed in the no-gas and He modes. The m/z signal at 90 (90Zr) was used for analysis and a 20 ppb Indium solution was continuously added as an internal standard. The zirconium concentration was determined using a calibration curve and the relative standard deviation based on repeated analyses of the same sample was less than 5%. Total content of zirconium was measured after microwave-assisted decomposition of parent UiO-66 (10 mg) in a mixture of nitric acid (4 mL), hydrochloric acid (12 mL) and hydrofluoric acid (4 mL).
Synthesis of UiO-66
The synthesis of UiO-66 was performed using a solvothermal procedure described previously.40 Briefly, zirconium chloride (106 mg, 0.455 mmol) was dissolved in N,N-dimethylformamide (DMF; 20 mL) in a 40 mL Wheaton vial, followed by 20 min of sonication to assure complete dissolution of the salt. Terephthalic acid (75.6 mg, 0.455 mmol) was subsequently added and the mixture was sonicated for a further 10 min, after which acetic acid (99.8%, 2.5 mL) was added as a modulator. The reaction solution was subsequently sealed and crystallization was carried out in a preheated oven (Memmert UF30 Plus) at 120 °C for 24 h. The resulting white solid was separated by centrifugation (Hettich Rotina 380 R) at 10000 rpm for 5 min, then washed four times with DMF and five times with acetone. Importantly, water was not used for washing to avoid hydrolysis of the product. After air-drying at room temperature, the UiO-66 powder was activated by heating at 100 °C under a dynamic vacuum for 24 h.
Stability of UiO-66
In each trial, UiO-66 (50 mg) and water (25 mL) were transferred into a SIMAX glass bottle followed by 1 min sonication. Following this, a stock buffer solution (25 mL) was added to give a final UiO-66 concentration of 1 mg mL−1, the bottle was capped and the suspension was magnetically stirred at 450 rpm for 4 h. In experiments using the 0.2 M PB and 1.0 M HEPES buffers, the UiO-66 was instead mixed directly with 50 mL of the stock buffer solution because of the limited solubility of the buffer components. In trials with the saline buffers, the 25 mL stock buffer also contained 0.3 M NaCl. During each trial, the pH of the solution was continually monitored using a pH electrode and 0.2 mL aliquots were extracted at predefined time intervals and passed through PTFE microfilters (Whatman, 0.1 μm). The concentration of leached terephthalic acid in each aliquot was determined by HPLC. The residual solid after each experiment was separated by centrifugation, washed three times with water to remove traces of the buffer solution and then washed four times with acetone. Finally, the solid was air-dried and activated by heating for 24 h at 100 °C under a dynamic vacuum before being assessed by PXRD, N2 adsorption and SEM. In selected cases, the kinetics of the terephthalate release was followed for 24 h.Blank experiments were performed based on the same procedure. In this case, the 25 mL of the buffer solution was replaced with pure water. These blank trials confirmed that the UiO-66 was stable in water during the experimental time span of 4 h and that the concentration of leached terephthalic acid was below the detection limit of the HPLC analysis.
The zirconium released into each solution in analogous experiments was determined using ICP-MS. In these experiments, the suspensions were prepared in 50 mL plastic centrifugation tubes and all sample manipulations were performed in plastic vials, to avoid interactions between zirconium and glass surfaces.
Each experiment was repeated three times in an air-conditioned laboratory with a constant temperature of 25 ± 1 °C. The errors in the concentrations of terephthalic acid (determined by HPLC) and zirconium (determined by ICP-MS) were below 7% and 20%, respectively. The error in the zirconium concentration was relatively high as a result of distribution of the zirconium between the solution and amorphous and colloidal phases.
Terephthalate release experiments were also repeated in water, 0.01 M NaCl solution and 0.01 M TRIS, HEPES and PB buffers at 37 ± 1 °C. A mixture of UiO-66 in water was preheated to 37 °C in a SIMAX glass bottle and then mixed with a preheated stock solution.
The total amount of terephthalic acid in the parent UiO-66 was determined by dissolving UiO-66 (50 mg) in 1.0 M sodium hydroxide (50 mL) with stirring overnight. The same procedure was used to dissolve UiO-66 in deuterated sodium hydroxide in preparation for 1H NMR analysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Czech Science Foundation (No. 20-04408S). The authors acknowledge the assistance provided by the Research Infrastructure NanoEnviCz, supported by the Ministry of Education, Youth and Sports of the Czech Republic under Project No. LM2018124. The authors are grateful to the working group Interactions of Inorganic Clusters, Cages, and Containers with Light within the AV21 Strategy of the Czech Academy of Science. Authors thanks Petr Bezdička and Jitka Bezdičková for PXRD and TGA measurements, respectively.Notes and references
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. P. Suh and J. Reedijk, Terminology of Metal-Organic Frameworks and Coordination Polymers (IUPAC Recommendations 2013), Pure Appl. Chem., 2013, 85, 1715–1724 CAS.
- J. L. C. Rowsell and O. M. Yaghi, Metal–Organic Frameworks: A New Class of Porous Materials, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS.
- M. J. Kalmutzki, N. Hanikel and O. M. Yaghi, Secondary Building Units as the Turning Points in the Development of the Reticular Chemistry of MOFs, Sci. Adv., 2018, 4, 1–16 Search PubMed.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H.-C. Zhou, Tuning the Structure and Function of Metal-Organic Frameworks via Linker Design, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, Ch. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydin and J. T. Hupp, Metal–Organic Framework Materials with Ultrahigh Surface Areas: Is the Sky the Limit?, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS.
- Y. He, W. Zhou, G. Qian and B. Chen, Methane Storage in Metal-Organic Frameworks, Chem. Soc. Rev., 2014, 43, 5657–5678 RSC.
- F.-J. Zhao, Y.-X. Tan, W. Wang, Z. Ju and D. Yuan, Optimizing H2, D2, and C2H2 Sorption Properties by Tuning the Pore Apertures in Metal–Organic Frameworks, Inorg. Chem., 2018, 57, 13312–13317 CrossRef CAS.
- M. Feng, P. Zhang, H.-C. Zhou and V. K. Sharma, Water-Stable Metal-Organic Frameworks for Aqueous Removal of Heavy Metals and Radionuclides: A Review, Chemosphere, 2018, 209, 783–800 CrossRef CAS.
- X.-Q. Zhan, F.-Ch. Tsai, L. Xie, K.-D. Zhang, H.-L. Liu, N. Ma, D. Shi and T. Jiang, Ligands-Coordinated Zr-Based MOF for Wastewater Treatment, Nanomaterials, 2018, 8, 655 CrossRef.
- S. Rojas, A. Arenas-Vivo and P. Horcajada, Metal-Organic Frameworks: A novel Platform for Combined Advanced Therapies, Coord. Chem. Rev., 2019, 388, 202–226 CrossRef CAS.
- I. A. Lázaro and R. S. Forgan, Application of Zirconium MOFs in Drug Delivery and Biomedicine, Coord. Chem. Rev., 2019, 380, 230–259 CrossRef.
- A. Karmakar and A. J. L. Pombeiro, Recent Advances in Amide Functionalized Metal Organic Frameworks for Heterogeneous Catalytic Applications, Coord. Chem. Rev., 2019, 395, 86–129 CrossRef CAS.
- T. Mehtab, G. Yasin, M. Arif, M. Shakeel, R. M. Korai, M. Nadeem, N. Muhammad and X. Lu, Metal-Organic Frameworks for Energy Storage Devices: Batteries and Supercapacitors, J. Energy Storage, 2019, 21, 632–646 CrossRef.
- J. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Metal-Organic Framework Materials as Chemical Sensors, Chem. Rev., 2012, 112, 1105–1125 CrossRef.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Metal–Organic Frameworks as Heterogeneous Catalysts for Oxidation Reactions, Catal. Sci. Technol., 2011, 1, 856–867 RSC.
- N. C. Burtch, H. Jasuja and K. S. Walton, Water Stability and Adsorption in Metal-organic Frameworks, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS.
- J. Ch. Tan and A. K. Cheetham, Mechanical Properties of Hybrid Inorganic–Organic Framework Materials: Establishing Fundamental Structure–property Relationships, Chem. Soc. Rev., 2011, 40, 1059–1080 RSC.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Chemical, Thermal and Mechanical Stabilities of Metal-Organic Frameworks, Nat. Rev. Mater., 2016, 1, 1–15 Search PubMed.
- J. Canivet, A. Fateeva, Y. Guo, B. Coasne and D. Farrusseng, Water Adsorption in MOFs: Fundamentals and Applications, Chem. Soc. Rev., 2014, 43, 5594–5617 RSC.
- N. T. T. Nguyen, H. Furukawa, F. Gándara, H. T. Nguyen, K. E. Cordova and O. M. Yaghi, Selective Capture of Carbon Dioxide under Humid Conditions by Hydrophobic Chabazite-Type Zeolitic Imidazolate Frameworks, Angew. Chem., Int. Ed., 2014, 53, 10645–10648 CrossRef CAS.
- K. Tan, N. Nijem, P. Canepa, Q. Gong, J. Li, T. Thonhauser and Y. J. Chabal, Stability and Hydrolyzation of Metal Organic Frameworks with Paddle-Wheel SBUs upon Hydration, Chem. Mater., 2012, 24, 3153–3167 CrossRef CAS.
- J. B. DeCoste, G. W. Peterson, H. Jasuja, T. G. Glover, Y. G. Huang and K. S. Walton, Stability and Degradation Mechanisms of Metal–Organic Frameworks Containing the Zr6O4(OH)4 Secondary Building Unit, J. Mater. Chem. A, 2013, 1, 5642–5650 RSC.
- J. Hynek, P. Brázda, J. Rohlíček, M. G. S. Londesborough and J. Demel, Phosphinic Acid Based Linkers: Building Blocks in Metal–Organic Framework Chemistry, Angew. Chem., Int. Ed., 2018, 57, 5116–5119 CrossRef.
- S. Yuan, J.-S. Qin, Ch. T. Lollar and H.-C. Zhou, Stable Metal-Organic Frameworks with Group 4 Metals: Current Status and Trends, ACS Cent. Sci., 2018, 4, 440–450 CrossRef CAS.
- D. Lv, J. Chen, Y. Chen, Z. Liu, Y. Xu, Ch. Duan, H. Wu, Y. Wu, J. Xiao, H. Xi, Z. Li and Q. Xia, Moisture Stability of Ethane-Selective Ni(II), Fe(III), Zr(IV)-Based Metal–Organic Frameworks, AIChE J., 2019, 65, e16616 CrossRef.
- M. Ding, X. Cai and H.-L. Jiang, Improving MOF Stability: Approaches and Applications, Chem. Sci., 2019, 10, 10209–10230 RSC.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, New Zirconium Inorganic Building Brick Forming Metal Organic Frameworks with Exceptional Stability, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef.
- Z. Chen, S. L. Hanna, L. R. Redfern, D. Alezi, T. Islamoglu and O. K. Farha, Reticular Chemistry in the Rational Synthesis of Functional Zirconium Cluster-Based MOFs, Coord. Chem. Rev., 2019, 386, 32–49 CrossRef CAS.
- M. Taddei, When Defects Turn into Virtues: The Curious Case of Zirconium-Based Metal-Organic Frameworks, Coord. Chem. Rev., 2017, 343, 1–24 CrossRef CAS.
- J. Winarta, B. Shan, S. M. McIntyre, L. Ye, Ch. Wang, J. Liu and B. Mu, Decade of UiO-66 Research: A Historic Review of Dynamic Structure, Synthesis Mechanisms, and Characterization Techniques of an Archetypal Metal-Organic Framework, Cryst. Growth Des., 2020, 20, 1347–1362 CrossRef CAS.
- Ch. Wang, X. Liu, N. K. Demir, J. P. Chen and K. Li, Applications of Water Stable Metal-Organic Frameworks, Chem. Soc. Rev., 2016, 45, 5107–5134 RSC.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Zr-Based Metal–Organic Frameworks: Design, Synthesis, Structure, and Applications, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- I. J. Kang, N. A. Khan, E. Haque and S. H. Jhung, Chemical and Thermal Stability of Isotypic Metal–Organic Frameworks: Effect of Metal Ions, Chem. – Eur. J., 2011, 17, 6437–6442 CrossRef CAS.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, Ch. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Synthesis and Stability of Tagged UiO-66 Zr-MOFs, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- K. Leus, T. Bogaerts, J. D. Decker, H. Depauw, K. Hendrickx, H. Vrielinck, V. V. Speybroeck and P. V. D. Voort, Systematic Study of the Chemical and Hydrothermal Stability of Selected “Stable” Metal Organic Frameworks, Microporous Mesoporous Mater., 2016, 226, 110–116 CrossRef CAS.
- M. A. Luzuriaga, C. E. Benjamin, M. W. Gaertner, H. Lee, F. C. Herbert, S. Mallick and J. J. Gassensmith, ZIF-8 Degrades in Cell Media, Serum, and Some - but not All - Common Laboratory Buffers, Supramol. Chem., 2019, 31, 485–490 CrossRef CAS.
- X. Liu, N. K. Demir, Z. Wu and K. Li, Highly Water-Stable Zirconium Metal-Organic Framework UiO-66 Membranes Supported on Alumina Hollow Fibers for Desalination, J. Am. Chem. Soc., 2015, 137, 6999–7002 CrossRef CAS.
- C. G. Piscopo, A. Polyzoidis, M. Schwarzer and S. Loebbecke, Stability of UiO-66 under Acidic Treatment: Opportunities and Limitations for Post-Synthetic Modifications, Microporous Mesoporous Mater., 2015, 208, 30–35 CrossRef CAS.
- B. Liu, K. Vikrant, K.-H. Kim, V. Kumar and S. K. Kailasa, Critical Role of Water Stability in Metal–Organic Frameworks and Advanced Modification Strategies for the Extension of their Applicability, Environ. Sci.: Nano, 2020, 7, 1319–1347 RSC.
- D. Bůžek, J. Demel and K. Lang, Zirconium Metal–Organic Framework UiO-66: Stability in an Aqueous Environment and Its Relevance for Organophosphate Degradation, Inorg. Chem., 2018, 57, 14290–14297 CrossRef.
- I. A. Lázaro, S. Haddad, S. Sacca, C. Orellana-Tavra, D. Fairen-Jimenez and R. S. Forgan, Selective Surface PEGylation of UiO-66 Nanoparticles for Enhanced Stability, Cell Uptake, and pH-Responsive Drug Delivery, Chem, 2017, 2, 561–578 Search PubMed.
- E. Bellido, M. Guillevic, T. Hildago, M. J. Santander-Ortega, Ch. Serre and P. Horcajada, Understanding the Colloidal Stability of the Mesoporous MIL-100(Fe) Nanoparticles in Physiological Media, Langmuir, 2014, 30, 5911–5920 CrossRef CAS.
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Defect Engineering: Tuning the Porosity and Composition of the Metal−Organic Framework UiO-66 via Modulated Synthesis, Chem. Mater., 2016, 28, 3749–3761 CrossRef CAS.
- K. A. Mocniak, I. Kubajewska, D. E. M. Spillane, G. R. Williams and R. E. Morris, Incorporation of Cisplatin into the Metal–Organic Frameworks UiO66-NH2 and UiO66 – Encapsulation vs. Conjugation, RSC Adv., 2015, 5, 83648–83656 RSC.
- P. Xu and G. Liao, Novel Fluorescent Biosensor for Adenosine Triphosphate Detection Based on a Metal–Organic Framework Coating Polydopamine Layer, Materials, 2018, 11, 1616 CrossRef.
- G. Ji, X. Gao, T. Zheng, W. Guan, H. Liu and Z. Liu, Postsynthetic Metalation Metal−Organic Framework as a Fluorescent Probe for the Ultrasensitive and Reversible Detection of PO43− Ions, Inorg. Chem., 2018, 57, 10525–10532 CrossRef CAS.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Disclosing the Complex Structure of UiO-66 Metal Organic Framework: A Synergic Combination of Experiment and Theory, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- M. Sk, S. Nandi, R. K. Singh, V. Trivedi and S. Biswas, Selective Sensing of Peroxynitrite by Hf-Based UiO-66-B(OH)2 Metal−Organic Framework: Applicability to Cell Imaging, Inorg. Chem., 2018, 57, 10128–10136 CrossRef CAS.
- M. Sk, M. R. U. Z. Khan, A. Das, S. Nandi, V. Trivedi and S. Biswas, A Phthalimide-functionalized UiO-66 Metal–organic Framework for the Fluorogenic Detection of Hydrazine in Live Cells, Dalton Trans., 2019, 48, 12615–12621 RSC.
- Y. Hao, S. Chen, Y. Zhou, Y. Zhang and M. Xu, Recent Progress in Metal-Organic Framework (MOF) Based Luminescent Chemodosimeters, Nanomaterials, 2019, 9, 974 CrossRef CAS.
- M. Xu, L. Feng, L.-N. Yan, S.-S. Meng, S. Yuan, M.-J. He, H. Liang, X.-Y. Chen, H.-Y. Wei, Z.-Y. Gu and H.-C. Zhou, Discovery of Precise pH-Controlled Biomimetic Catalysts: Defective Zirconium Metal-Organic Frameworks as Alkaline Phosphatase Mimics, Nanoscale, 2019, 11, 11270–11278 RSC.
- K. E. deKrafft, W. S. Boyle, L. M. Burk, O. Z. Zhou and W. Lin, Zr- and Hf-based Nanoscale Metal–Organic Frameworks as Contrast Agents for Computed Tomography, J. Mater. Chem., 2012, 22, 18139–18144 RSC.
- C. Orellana-Tavra, R. J. Marshall, E. F. Baxter, I. A. Lázaro, A. Tao, A. K. Cheetham, R. S. Forgan and D. Fairen-Jimenez, Drug Delivery and Controlled Release from Biocompatible Metal–Organic Frameworks Using Mechanical Amorphization, J. Mater. Chem. B, 2016, 4, 7697–7707 RSC.
- C. W. Abney, K. M. L. Taylor-Pashow, S. R. Russell, Y. Chen, R. Samantaray, J. V. Lockard and W. Lin, Topotactic Transformations of Metal−Organic Frameworks to Highly Porous and Stable Inorganic Sorbents for Efficient Radionuclide Sequestration, Chem. Mater., 2014, 26, 5231–5243 CrossRef CAS.
- Ch.-Y. Wang, Y.-J. Lee, J.-P. Hsu and D.-J. Lee, Phosphate or Arsenate Modified UiO-66-NO2: Amorphous Mesoporous Matrix, J. Taiwan Inst. Chem. Eng., 2020, 108, 129–133 CrossRef CAS.
- A. M. Ploskonska and J. B. DeCoste, Insight into Organophosphate Chemical Warfare Agent Simulant Hydrolysis in Metal-Organic Frameworks, J. Hazard. Mater., 2019, 375, 191–197 CrossRef.
- H. Wang, J. J. Mahle, T. M. Tovar, G. W. Peterson, M. G. Hall, J. B. DeCoste, J. H. Buchanan and Ch. J. Karwacki, Solid-Phase Detoxification of Chemical Warfare Agents using Zirconium-Based Metal Organic Frameworks and the Moisture Effects: Analyze via Digestion, ACS Appl. Mater. Interfaces, 2019, 11, 21109–21116 CrossRef CAS.
- M. J. Katz, R. C. Klet, S. Y. Moon, J. E. Mondloch, J. T. Hupp and O. K. Farha, One Step Backward Is Two Steps Forward: Enhancing the Hydrolysis Rate of UiO-66 by Decreasing [OH−], ACS Catal., 2015, 5, 4637–4642 CrossRef CAS.
- P. Brown, E. Curti and B. Grambow, et al., in Chemical Thermodynamics of Zirconium, Chemical Thermodynamics, ed. F. J. Mompean, et al., Elsevier, North-Holland, Amsterdam, 2005, vol. 8 Search PubMed.
- T. Kobayashi, T. Sasaki, I. Takagi and H. Moriyama, Solubility of Zirconium(IV) Hydrous Oxides, J. Nucl. Sci. Technol., 2007, 44, 90–94 CrossRef CAS.
- Y. Jiao, Y. Liu, G. Zhu, J. T. Hungerford, S. Bhattacharyyam, R. P. Lively, D. S. Sholl and K. S. Walton, Heat-Treatment of Defective UiO-66 from Modulated Synthesis: Adsorption and Stability Studies, J. Phys. Chem. C, 2017, 121, 23471–23479 CrossRef CAS.
- D. Bůžek, J. Zelenka, P. Ulbrich, T. Ruml, I. Křížová, J. Lang, P. Kubát, J. Demel, K. Kirakci and K. Lang, Nanoscaled Porphyrinic Metal-Organic Frameworks: Photosensitizer Delivery Systems for Photodynamic Therapy, J. Mater. Chem. B, 2017, 5, 1815–1821 RSC.
- JCPDS PDF-2 database, release 54, International Centre for Diffraction Data: Newtown Square, PA, USA, 2004.
Footnote |
| † Electronic supplementary information (ESI) available: List of used materials, preparation of buffer solutions, dissolved zirconium, solubility of terephthalic acid in buffers, 1H NMR spectra of dissolved parent UiO-66 with description, DTA/TGA curves including experiment setup, blank experiment – stability of parent UiO-66 in neat water. Prolonged experiments in buffers. See DOI: 10.1039/d0qi00973c |
| This journal is © the Partner Organisations 2021 |