
An electrochemically controlled release of NHCs using iron bis(dithiolene) N-heterocyclic carbene complexes†
Jayaraman
Selvakumar
 a,
Scott M.
Simpson
b,
Eva
Zurek
c and
Kuppuswamy
Arumugam
*a
a,
Scott M.
Simpson
b,
Eva
Zurek
c and
Kuppuswamy
Arumugam
*a
aDepartment of Chemistry, Wright State University, 3640 Colonel Glenn Hwy., Dayton, OH 45435, USA. E-mail: kuppuswamy.arumugam@wright.edu
bDepartment of Chemistry, St. Bonaventure University, 3261 West State Road, St. Bonaventure, NY 14778, USA
cDepartment of Chemistry, State University of New York at Buffalo, Buffalo, NY 14260, USA
First published on 23rd September 2020
Abstract
A series of five coordinated iron bis(dithiolene) complexes [Fe(NHC)(S2C2R2)2] (R = C6H5 or C6H4-p-OCH3) containing N-heterocyclic carbene (NHC) (NHC = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene or 1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene) were isolated in high yield (84–92%). The iron complexes were characterized by NMR spectroscopy and confirmed by single crystal X-ray diffraction studies. The combination of cyclic voltammetry and spectroelectrochemical analysis revealed that iron complexes undergo Fe–CNHC bond cleavage and release NHC upon subjection to electrochemical reduction. The electrochemically released NHC was trapped using 1-naphthylisothiocyanate and the adduct was isolated in nearly quantitative yield (∼99%). As a proof of concept, the electrochemically released NHC was subsequently used as a catalyst for synthesis of γ-butyrolactones from commercially available cinnamaldehydes.
Introduction
Since their discovery,1 transition metal dithiolene complexes have been an active area of research. Intriguing structural characteristics2 coupled with interesting electrochemical properties2 makes this class of metal complexes attractive for a variety of potential applications,3 including sensing,3h photophysical,3a,d superconducting,3c,f and hydrogen evolution catalysts.3b,4 In general, a dithiolene ligand can exist in three different oxidation states, a fully reduced ene-1,2-dithiolate (S2C2R2)2− or a radical monoanion (S2C2R2)˙− or a α-dithione (S2C2R2) (Fig. 1). When coordinated to a transition metal atom, the ligand may exist in either one of these oxidation states, thus leaving the metal oxidation state ambiguous.1b,2a The redox non-innocent character of the dithiolene ligand allows it to adopt multiple redox states without affecting the oxidation state of the central metal atom.2a,b This distinctive feature assists the metal complexes in ligand based reactivity, i.e., reactions with alkenes.3g,5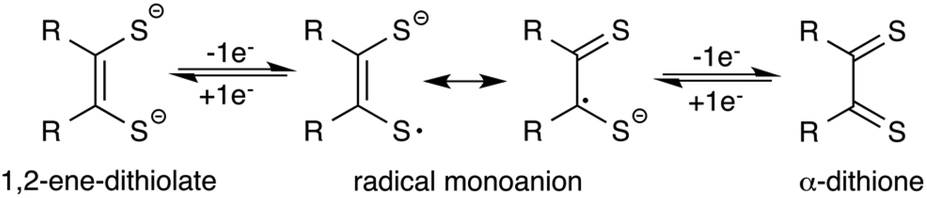 | ||
| Fig. 1 Redox states of dithiolene ligands. | ||
Of all metal dithiolene complexes, dimeric iron bis(dithiolene) complexes [Fe(S2C2R2)2]2 are among the most studied.2,6 Dimeric iron bis(dithiolene) complexes readily react with Lewis bases such as tri-alkyl/aryl phosphines,7 phosphites,8 nitrosyl,9 pyridines,10 and halides,11 to yield the corresponding five coordinate square pyramidal adducts [Fe(X)(S2C2R2)2] (X = PR3, P(OR)3, NO, pyridine, and I). The appealing feature of five coordinated metal complexes, especially, the phosphine and the phosphite adducts is that they undergo reversible release and binding of phosphine/phosphite when subject to an electrochemical reduction and oxidation cycle. Donahue and coworkers have elegantly demonstrated an electrochemical release and recovery of triphenylphosphine with [Fe(PPh3)(S2C2(C6H4-p-OCH3)2)2] (Fig. 2a).7b Similarly, Wieghardt and coworkers have also observed an electrochemical release of phosphite with five coordinated [Fe(P(OPh)3)(S2C2Ph2)2] when subjected to electrochemical reduction.8d The binding characteristics of an exogenous ligand (PPh3 or P(OPh)3) as a function of the redox state of the host ([Fe(S2C2R2)2]) could potentially be exploited to design an “on–off” switch. If the exogenous ligand is a catalyst, the strategy may be used to control the activity of an organic transformation. Relevant to this context, we envisioned to develop [Fe(NHC)(S2C2R2)2] type iron complexes to facilitate the release of NHC as a function of the redox state of the metal bis(dithiolene) unit (Fig. 2b).
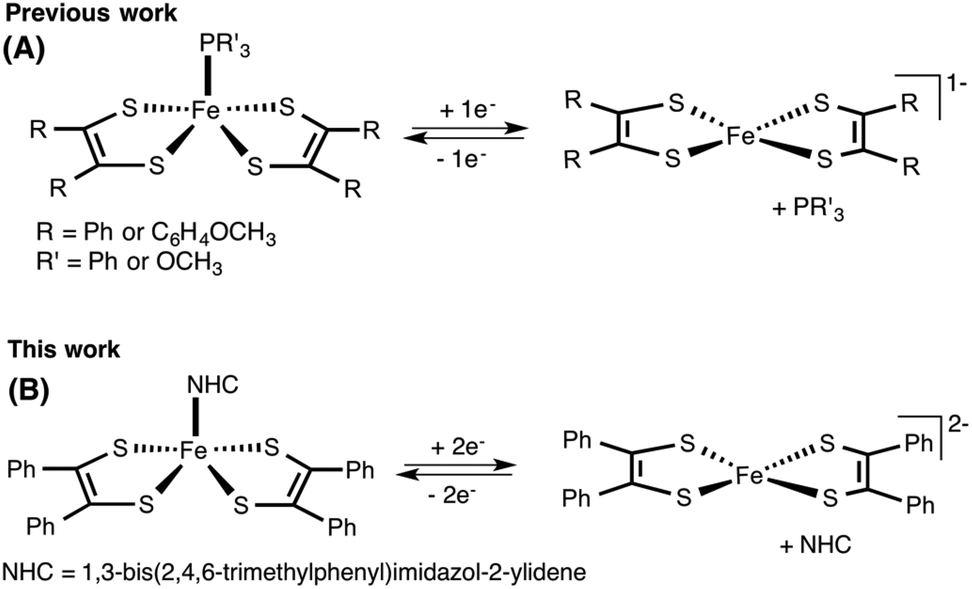 | ||
| Fig. 2 (A) Electrochemically controlled release and binding of phosphine and phosphite to iron bis(dithiolene) complexes. (B) Proposed electrochemically controlled release of NHC ligands to iron bis(dithiolene) complex. | ||
N-Heterocyclic carbenes12 are one of the most attractive classes of molecules with a variety of applications.13 The prime use of this ligand is in homogeneous catalysis, such as olefin metathesis,14 C–C cross-coupling reactions,15 transfer hydrogenation,16 and C–H activation reactions.17 A number of transition metal complexes containing NHC ligands also exhibit potential therapeutic value.18 In addition, free NHCs are also- used as organocatalysts for a variety of organic transformations,13a,19 including benzoin condensation reactions,20 Stetter reactions,21 umpolung acylanion catalysis,22 annulation reactions,23 catalysis involving acyl azolium intermediates,24 and zwitterionic ring-opening polymerization reactions.25
Free NHCs are usually generated in situ by treatment of imidazolium, pyrazolium or thiazolium salts with strong bases such as NaN(SiMe3)2, KNa(SiMe3)2, NaH, and KOtBu.13a Considerable developments have been made to generate NHCs from masked carbene sources such as NHC-carboxylates,26 imidazolidines-pentafluorophenyl,27 NHC-isothiocyanates,28 imidazolidines-trichloromethyl,29 NHC-alcohol adducts and NHC-metal systems.14d,30 Alternatively, NHCs can also be generated via electrochemical reduction of imidazolium salts.31,32 Careful analysis reveals that the usefulness of NHCs in organocatalysis is inherently tied to its ease of generation and recovery from the reaction media. Unfortunately, the methods that hinge on these principles are not common with NHCs. As a proof of concept, herein, we disclose a system that uses a less hazardous approach than traditional chemical methods for the release of NHC from [Fe(NHC)(S2C2Ph2)2] adducts and its subsequent use in organocatalysis.
Results and discussion
Synthesis and characterization
Since NHCs are excellent σ-donors with π-back bonding capability, the metal complexes they form are expected to be stable to oxygen and moisture.33 Hence, we envisaged that like PPh3, NHCs (3 and 4) would readily react with [Fe(S2C2R2)2]2 (1 and 2) to yield air and moisture stable square pyramidal adducts. The resulting adducts can be subsequently subjected to electrochemical release and recovery studies. The results of these studies are discussed below.The neutral iron bis(dithiolene) dimers (1 and 2)1b and NHCs (3 and 4)12,34 were readily prepared according to the known literature procedure. Using the dimers and the NHCs, metal complexes 5–8 were prepared (Scheme 1). Two equiv. of in situ generated 3 was treated with 1 equiv. of [Fe(S2C2Ph2)2]2 in THF. The resulting mixture was allowed to stir for 2 h at 25 °C to yield 5 in 90% yield. Similarly, two equiv. of 4 was treated with [Fe(S2C2Ph2)2]2 to yield 6 in 84% yield. By following the same procedure, 7 and 8 were isolated in 92% and 87% yield, respectively. Compounds 1 and 2 are sparingly soluble in polar organic solvents, while compounds 5–8 were readily soluble in common organic solvents such as THF and CH2Cl2.
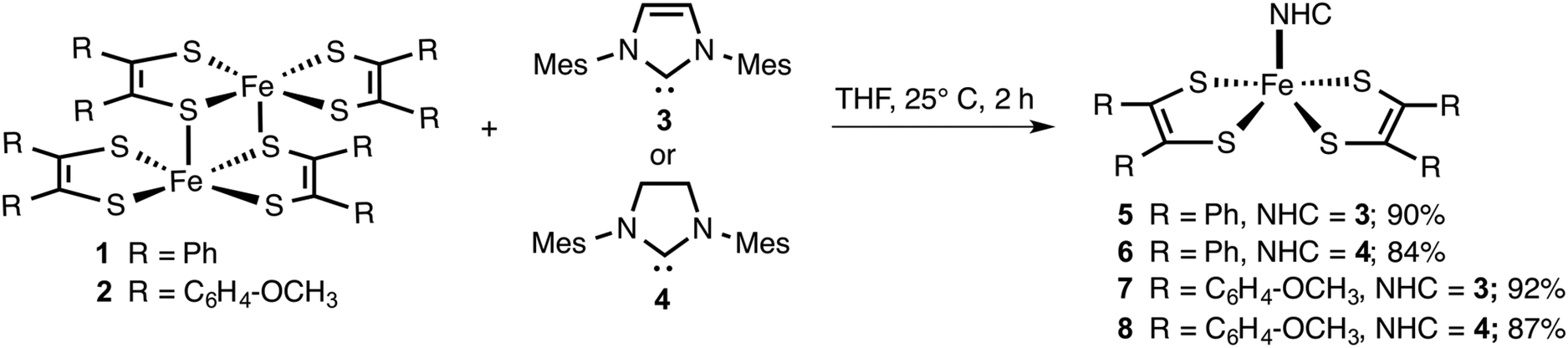 | ||
| Scheme 1 Synthesis of iron bis(dithiolene) N-heterocyclic carbene adducts 5–8. | ||
Compounds 5–8 displayed characteristic 1H and 13C signals corresponding to bis(dithiolene) and NHC units in the NMR spectroscopy. For example, in 1H NMR, 5 displayed characteristic phenyl hydrogens (20H) as a multiplet between 7.32–7.18 ppm, aromatic C–H (mesityl) hydrogens (4H) as a singlet at 6.96 ppm, and olefinic (imidazole) hydrogens (2H) as a singlet at 6.84 ppm. Two sets of singlets corresponding to mesityl-CH3 hydrogens (6H and 12H) were observed at 2.36 and 2.00 ppm, respectively. Likewise, compounds 6–8 were all characterized using 1H NMR spectroscopy. In 13C NMR, all four metal complexes displayed a characteristic CNHC peak between 180–183 ppm, confirming the presence of a Fe–CNHC bond.35
Crystal structures
Molecular structure of compounds 5–8 were unambiguously assigned using single-crystal X-ray diffraction studies. The thermal ellipsoid plots of 5–8 are presented in Fig. 3 and selected bond parameters are presented in Table 1. Compounds 5, 6 and 8 crystallized in a monoclinic (C2/c or P21/c) space group, while 7 crystallized in a triclinic (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) space group. All four compounds 5–8 displayed iron centered distorted square pyramidal geometries with four sulfur atoms occupying the base of a pyramid and the apex occupied by a CNHC atom. The average Fe–CNHC distance for 5–8 was observed to be 1.958[2]ave Å, which is consistent with values previously reported for Fe–CNHC (1.982–2.148 Å).35b,36 To better understand the nature of bonding in 5–8, the refinement parameters were compared with those of previously reported phosphine iron complexes, [Fe(X)(S2C2Ph2)2] (X = PPh3, P(OPh)3).7 The average C–S and S–C
) space group. All four compounds 5–8 displayed iron centered distorted square pyramidal geometries with four sulfur atoms occupying the base of a pyramid and the apex occupied by a CNHC atom. The average Fe–CNHC distance for 5–8 was observed to be 1.958[2]ave Å, which is consistent with values previously reported for Fe–CNHC (1.982–2.148 Å).35b,36 To better understand the nature of bonding in 5–8, the refinement parameters were compared with those of previously reported phosphine iron complexes, [Fe(X)(S2C2Ph2)2] (X = PPh3, P(OPh)3).7 The average C–S and S–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–S distances in 5–8 are 1.720[3]ave Å and 1.381[2]ave Å, respectively, which is consistent with the phosphine/phosphite iron complexes (C–S = 1.710[2]ave Å; S–CC–S = 1.3825[2]ave Å). Moreover, the direct comparison with the phosphine iron complex strongly suggests that the central iron is divalent and the electronic structure for dithiolene units can be best described as radical monoanion (S2C2Ar2)˙−.7b,8c,d,37
C–S distances in 5–8 are 1.720[3]ave Å and 1.381[2]ave Å, respectively, which is consistent with the phosphine/phosphite iron complexes (C–S = 1.710[2]ave Å; S–CC–S = 1.3825[2]ave Å). Moreover, the direct comparison with the phosphine iron complex strongly suggests that the central iron is divalent and the electronic structure for dithiolene units can be best described as radical monoanion (S2C2Ar2)˙−.7b,8c,d,37
 | ||
| Fig. 3 Thermal ellipsoid plots of 5–8 drawn at the 50% probability level, hydrogen atoms are omitted for clarity. The plots were rendered using POV-Ray. Selected distances and angles are presented in Table 1. | ||
| 5 | 6 | 7 | 8 | [Fe(PPh3)(S2C2(C6H4-p-OCH3)2)2] | [Fe(P(OPh)3)(S2C2Ph2)2] | |
|---|---|---|---|---|---|---|
| a Average values are reported when two or more identical bond parameters are present. b τ values are computed as described in ref. 38. | ||||||
| Fe–Ccarbene | 1.963(3) | 1.962(3) | 1.968(3) | 1.940(3) | ||
|
N–CC–Na |
1.327[8] | 1.512[6] | 1.333[4] | 1.496[6] | ||
|
S–CC–Sa |
1.381[3] | 1.383[3] | 1.3845[4] | 1.382[5] | 1.3825[2] | 1.3955[4] |
| C–Sa | 1.717[3] | 1.718[2] | 1.728[3] | 1.718[3] | 1.710[2] | 1.70475[3] |
| Fe–Sa | 2.1683[5] | 2.1688[5] | 2.1712[8] | 2.1650[9] | 2.1567[8] | 2.1576[11] |
| S–Fe–Sa | 88.25[2] | 87.025[19] | 87.825[3] | 87.81[3] | 88.13[10] | 88.46[4] |
| N–C–Na | 103.7[3] | 107.4[2] | 103.4[2] | 107.1[3] | ||
| τ | 0.22 | 0.41 | 0.63 | 0.60 | 0.19 | 0.11 |
For all four compounds 5–8, the iron atom was observed to be ∼0.46 Å above the mean plane defined by the four sulfur atoms and all four compounds displayed a distorted square pyramidal geometry. The magnitude of distortion from an ideal square pyramidal geometry can be deduced by computing Addison's τ value.38 These values are used to determine whether five-coordinate iron complexes adopt a perfect square pyramidal or a trigonal bipyramidal geometry. For example, for an ideal square pyramidal geometry τ = 0, while for an ideal trigonal bipyramidal geometry τ = 1. The calculated τ values for 5–8 are presented in Table 1.39 Of all the complexes, 5 was the least distorted with τ = 0.22 and compound 7 was the most distorted with τ = 0.63. This distortion may be attributed to steric and electronic effects exerted by an NHC unit.40
Electrochemistry
To assign formal reduction and oxidation potentials for compounds 5–8, cyclic voltammetry (CV) was recorded in THF with [N(nBu)4]PF6 as the supporting electrolyte at 25 °C using a platinum disc working electrode, platinum wire working electrode and silver wire quasi-reference electrode under N2 atmosphere. The CVs of 5–8 were very similar and the key electrochemical potentials are summarized in Table 2. As a representative example only compound 5 is discussed here. Compound 5 sustained one oxidation and two reduction waves in the cyclic voltammetry (Fig. 4). A reversible oxidation wave was observed at E11/2 = +0.51 V, yielding 51+.8d A reversible reduction wave was observed at E21/2 = −0.67 V, ascribed to 5 → 51−, and an irreversible reduction at E3 = −1.97 V attributed to 5 → 52−. The irreversibility of the second reduction (E3) signifies dissociation of compound 5, which is consistent with breaking of the Fe–CNHC bond and formation of [Fe(S2C2Ph2)2]2−.7b,8d,41 Similar behaviour was reported by Donahue and coworkers and Weighardt and coworkers with [Fe(PPh3)(S2C2(C6H4-p-OCH3)2)2] and [Fe(P(OPh)3)(S2C2Ph2)2] in CH2Cl2, respectively. Surprisingly for phosphine/phosphite iron complexes, the groups observed only one irreversible reduction wave in CH2Cl2. However, in a separate report, McCleverty has reported the existence of two reductions for [Fe(PPh3)(S2C2(C6H4-p-OCH3)2)2] in DMF solvent. This clearly suggests that the solvent plays a critical role in stabilizing the chemical species that are generated during the course of the reduction processes. Moreover, Donahue and Weighardt studies with [Fe(PPh3)(S2C2(C6H4-p-OCH3)2)2] and [Fe(P(OPh)3)(S2C2Ph2)2] suggests that the reduction processes is predominantly ligand based, rather metal based. A similar behaviour was observed with compounds 5–8. Molecular gas-phase calculations (vide infra) is in close accordance with ligand-based reduction processes.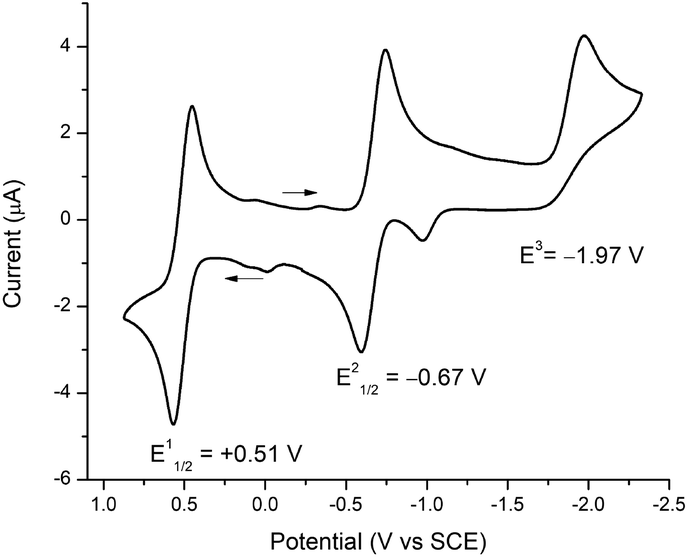 | ||
| Fig. 4 Cyclic voltammogram of 5 with 0.1 M [N(nBu)4]PF6 in THF (1 mM), decamethylferrocene was added as an internal standard and the potentials were adjusted to SCE. | ||
| Compound | E 11/2 (ox) | E 21/2 (red) | E 3 (red) |
|---|---|---|---|
| Potentials obtained from CV in THF with 0.10 M [N(nBu)4][PF6] and 0.1 mM analyte. The obtained redox potentials were referenced to SCE. See Fig. S6 and S9–S11† for corresponding cyclic voltammograms of 5–8. Abbreviations: r = reversible, ir = irreversible, ox = oxidation, red = reduction. | |||
| 5 | 0.51 (r) | −0.67 (r) | −1.97 (ir) |
| 6 | 0.55 (r) | −0.73 (r) | −1.96 (ir) |
| 7 | 0.38 (r) | −0.74 (r) | −2.04 (ir) |
| 8 | 0.43 (r) | −0.79 (r) | −2.02 (ir) |
Just like compound 5, compounds 6–8 displayed analogous electrochemical behaviour with [N(nBu)4]PF6 in THF. Close examination of the oxidation potentials for 5–8 revealed that 7 and 8 are easier to oxidize than 5 and 6 (Table 2). This characteristic is attributed to the electron rich nature of anisyl-substituted dithiolene ligands in 7 and 8. Subsequently, compounds 5 and 6 were easier to reduce relative to 7 and 8. Hence, these finding suggests that the electrochemical potentials are highly regulated by the nature of the ligand (Fig. S6 and S8–S10†). Again, these assignments are consistent with those reported for [Fe(PPh3)(S2C2(C6H4-p-OCH3)2)2] and [Fe(P(OPh)3)(S2C2Ph2)2] complexes.7b,8d
During the course of reduction, small peaks were observed upon scanning to negative potentials (around −0.1 and −1 V). These peaks may be attributed to the electrochemical response generated from the formation of [Fe(S2C2Ph2)2]2−, as a consequence of Fe–CNHC bond dissociation.41 To corroborate this, cyclic voltammetry was performed with a narrow electrochemical window that curtails the second reduction (Fig. S6†). The absence of small peaks with the newly defined window clearly suggests that these peaks were observed upon scanning to the potential pertaining to the second reduction. To probe the reversible nature of first reduction and confirm that the bond cleavage exclusively occurs after the second reduction, a scan rate dependent study was undertaken (see Fig. S7†). Either at low scan rate or at higher scan rate, the reversibility of E21/2 was maintained, indicative of stability of 5 after the first reduction.
Density functional theory calculations
Molecular gas phase density functional theory (DFT) calculations were performed on compound 5. The starting coordinates in each geometry optimization were taken from those experimentally determined for the crystalline compound, and all parameters were relaxed to the nearest stationary point. The experimental NMR spectrum of 5 clearly shows that the iron complex is diamagnetic (S = 0), however there are two ways in which such a state may be achieved: one in which the dithiolene ligand radicals are Pauli coupled, and another in which they are strongly antiferromagnetically coupled localized ligand radicals. We refer to the latter as the S = 0* state, and it was studied utilizing the broken-symmetry approach and methodology developed by Noodleman.41,42 We also considered the S = 1, 2 spin states, since their energies relative to the S = 0/0* states, may be able to help determine the most appropriate density functional to model our system. To these ends the ground state geometry of 5 was optimized with a variety of density functionals, and the resulting distances were compared with those measured for the crystal. Detailed results are provided in the ESI.† The functionals that were tested include those that employ the generalized gradient approximation, GGA, (BP86, PBE, PBEsol, revPBE), hybrid functionals (B3LYP, B3LYP*, PBE0), and the TPSS meta-GGA.43 Hartree–Fock type theories systematically favor the high-spin state in Fe(II) transition-metal complexes. As a result, the admixture of exact exchange in standard hybrid functionals, such as B3LYP, tends to over-stabilize the high-spin state. It has therefore been proposed that reducing the coefficient of exact exchange can yield more reliable results, and the resulting functional has been dubbed B3LYP*.43h Nonetheless, Jacobsen and Donahue have found that the BP86 functional provides a more appropriate description of iron bis(dithiolene) complexes than either B3LYP or B3LYP*.41 We have arrived to a similar conclusion based upon the rationalization contained in the ESI,† therefore, we report the results of our BP86 calculations. We also report our results for the B3LYP* and TPSS functionals, for comparison purposes.A symmetrized fragment orbital (SFO) analysis was carried out on the S = 0 state, using the BP86 functional, to determine the composition of the frontier molecular orbitals (Fig. 5). The highest occupied molecular orbital (HOMO) was composed primarily of the iron's dyz-orbital (∼59%) and occupied/unoccupied orbitals localized on the dithiolene ligand (31%). The major contribution to the lowest unoccupied molecular orbital (LUMO) of the iron complex was from the unoccupied LUMO+1 of the distorted dithiolene ligands (66%) and a contribution of 28% from the iron's dxz-orbital. In the previously reported phosphine compound, [Fe(P(CH3)3)(S2C2(C6H4-p-OCH3)2)2], the doubly degenerate π* orbitals of the dithiolene ligand contributed to the LUMO.7c However, in the case of 5, the degeneracy of the π* orbitals of the dithiolene is broken because of the distortion of the dithiolene ligand from an ideal square pyramidal geometry.
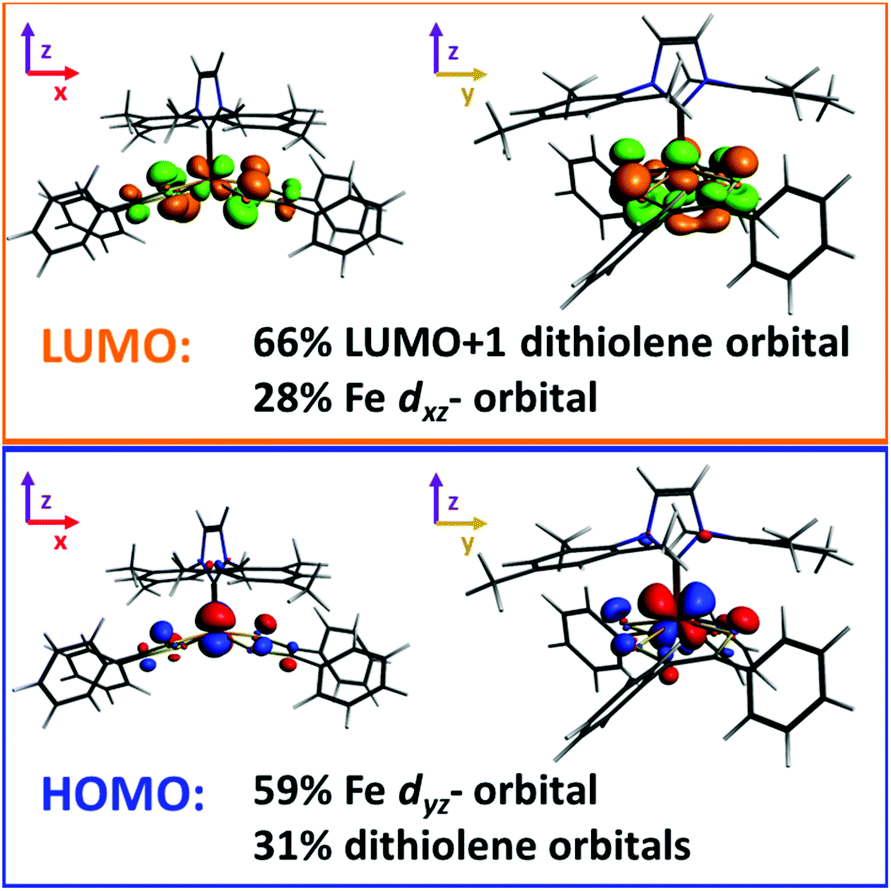 | ||
| Fig. 5 Stick models of the (bottom; blue/red) HOMO and the (top; orange/green) LUMO of compound 5. The views displayed on the right/left are aligned with the x-axis/y-axis, respectively. Isovalue of ±0.05 au. | ||
The Fe–CNHC binding energy (BE) of 5, along with the reduced forms of 5 ([5]1− & [5]2−) was calculated with the BP86, B3LYP*, and TPSS functionals. The BE was calculated by:
| BE=E(5−q) − E(3) − E(bis(dithiolene)−q) |
| Charge | BP86 | B3LYP* | TPSS | |||
|---|---|---|---|---|---|---|
| BE (kcal mol−1) | d(Fe–CNHC) (Å) | BE (kcal mol−1) | d(Fe–CNHC) (Å) | BE (kcal mol−1) | d(Fe–CNHC) (Å) | |
| 0 | −28.44 | 1.960 | −12.93 | 2.019 | −29.24 | 1.963 |
| −1 | −12.29 | 1.936 | −6.33 | 2.335 | −12.47 | 1.932 |
| −2 | −2.27 | 1.870 | −2.61 | 2.309 | −2.74 | 1.859 |
In order to investigate the nature of the reduction of 5 to [5]1− to [5]2−, we shall discuss their electronic structure in terms of atomic charges. We elect to use the Hirshfeld charges (q) as our charge models over the widely used Mulliken charge analysis as Mulliken charges have been shown to be highly basis set dependent. The results of our analysis are shown in Table S3.† The ESI† contains the iron spin state derived for Mulliken populations for 5, [5]1−, and [5]2− using the BP86, B3LYP*, and TPSS functionals.
It is apparent comparing the Hirshfeld charges of the reduction of 5 to [5]1− that the charge of the iron does not significantly change for the BP86 and TPSS functionals. While there is an appreciable amount of change in the charge for the Fe in the B3LYP* calculations, it is overmasked by the change in charge on the bis(dithiolene) ligand. The BP86 and TPSS functionals also find that the change in charge accumulation is localized on the sulfur containing ligand. This trend appears to continue for the dianionic species. It is clear from our calculations that it is not only the iron center that is involved in the reduction, but also the ligands surrounding the iron center. Predominantly, the bis(thiolene) ligand gains a substantial amount of charge during the reduction. One may conclude that this reduction is a ligand-based event.
Electronic absorption spectra
To better understand electronic excitations in compounds 5–8, UV-vis absorption measurements were carried out in THF. All four compounds 5–8 displayed distinctive absorptions at λ = 681, 672, 697 and 704 nm, respectively (Fig. S11†), these absorptions were attributed to intra ligand charge transfer transitions (ILCT).7b,8c Such absorption is completely absent for reduced iron bis(dithiolene) [Fe(S2C2R2)2]n− complexes (n = 1 or 2).7b Hence, electrochemically reducing compound 5 followed by concomitant monitoring of its electronic excitations in situ will help to verify the breaking of the Fe–CNHC bond. Independently, we treated compound 5 with 1 and 2 equiv. of cobaltocene and analysed its UV-vis absorption spectra (Fig. S13†). The spectra revealed the absence of characteristic ILCT transition at 681 nm for the doubly reduced compound ([Fe(S2C2R2)2]2−).To ascertain the electronic absorption properties of [5]2−, the adduct was subjected to UV-vis spectroelectrochemical measurements (Fig. 6). An electrochemical quartz cell fitted with Pt mesh working electrode, Pt wire counter electrode and quasi reference Ag wire was used for the measurement. The potentiostat was held at a constant potential (−2.25 V) and the absorption measurements (400–900 nm) were made using a UV-Vis absorption spectrometer. Compound 5 displayed a strong absorption feature at 681 nm attributed to ILCT. Upon electrochemical reduction, an exponential decrease in the concentration of the 681 nm peak was observed, consistent with a decrease in the concentration of 5. As evident from Fig. 6, during pre-electrolysis, the solution contained only 5, while during electrolysis, the concentration of 5 gets depleted and the concentration of [5]2− increases. After 300 seconds of electrolysis, significant suppression of the absorbance peak corresponding to ILCT was observed, this indicates disappearance of 5 and appearance of [5]2−.41
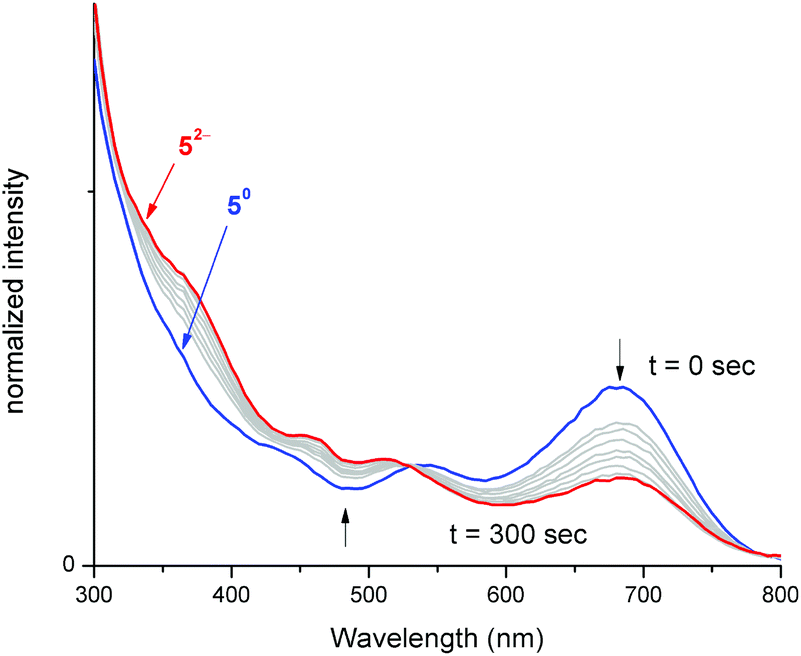 | ||
| Fig. 6 Electronic absorption spectra recorded during potential bulk electrolysis (−2.25 V vs. AgCl) of 5 (5 → 52−) in THF with 0.1 M [N(nBu)4][PF6] as a supporting electrolyte with platinum mesh working electrode. The arrows indicate spectra change during the process. | ||
Having documented spectroscopic evidence for two-electron reduction of 5 and concomitant dissociation of [5]2− to generate 3, we turned our focus towards trapping the generated carbene (3). We hypothesized that bulk electrolytic reduction of 5 → 52− is expected to release free carbenes that can be chemically trapped using isothiocyanates.28 By quantifying the amount of isothiocyanate-3 adduct formation, the amount of free carbene released during the process can be computed. Bulk electrolysis of 5 was conducted in a two-compartment cell separated by a glass frit at 25 °C in 20 mL of THF with 0.1 M [N(nBu)4]PF6 as the supporting electrolyte using a Pt mesh working electrode, Pt mesh counter electrode, and Ag/AgCl non-aqueous reference electrode (see the ESI† for more information). Adduct 5 (0.02 mmol) was taken in the working electrode compartment and the reference electrode and counter electrode were immersed in the other compartment. Bulk reduction was performed by holding a constant potential at −2.25 V (vs. Ag/AgCl). The initial current during the beginning of the experiment was read at 1 mA, as the experiment progressed, the current continue to steadily drop and read near zero as the concentration of 5 completely exhausted. After the passage of 3.81 coulombs of electrons (0.0395 mmol), the relative current read near zero, signifying the endpoint of electrolysis. The resulting electrolytic solution on the cathodic compartment was transferred to a 20 mL vial containing 2 equivalent of 1-naphthyl isothiocyanate and the resulting reaction mixture was stirred for 1 h. Further purification of the reaction mixture yielded the expected adduct 9 in near quantitative yield (Scheme 2).
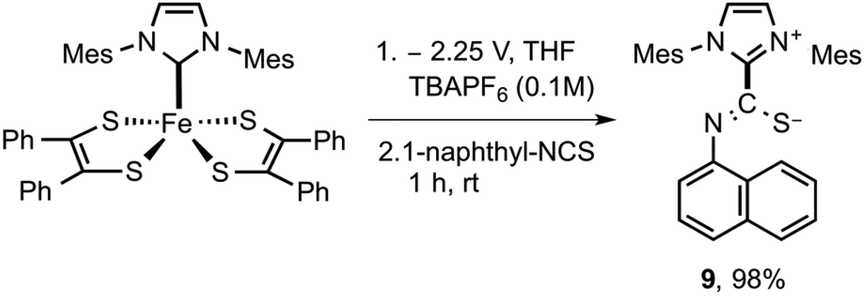 | ||
| Scheme 2 Trapping of electrochemically generated IMes carbene. | ||
Organocatalysis with electrochemically released NHC
After successfully trapping the electro-generated carbene, we turned our focus towards electrochemically-controlled organocatalysis. Carbene catalyzed conjugate umpolung reaction of an unsaturated aldehyde was recently disclosed by Glorius, such reaction has great utility in synthesizing γ-butyrolactones.23,44 Electrochemically released carbene (3) via bulk electrolysis of 5 was transferred into a 20 mL vial containing trans-cinnamaldehyde (0.4 mmol) and the resulting mixture was stirred at 25 °C for 24 h (Scheme 3). The progress of the reaction was monitored using thin layer chromatography. At the end of 24 h, the crude mixture was subjected to column chromatography to yield γ-butyrolactone 10 in 34% yield (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.15/cis:trans isomers). Control experiments were conducted without 5 and with 5 and no electrolysis, in either case, the formation of γ-butyrolactone was not observed. Moreover, heating 5 with trans-cinnamaldehyde to reflux temperature in THF resulted with no product. These control experiments further confirm that suitable electrochemical potential has to be applied for the release of 3.
:0.15/cis:trans isomers). Control experiments were conducted without 5 and with 5 and no electrolysis, in either case, the formation of γ-butyrolactone was not observed. Moreover, heating 5 with trans-cinnamaldehyde to reflux temperature in THF resulted with no product. These control experiments further confirm that suitable electrochemical potential has to be applied for the release of 3.
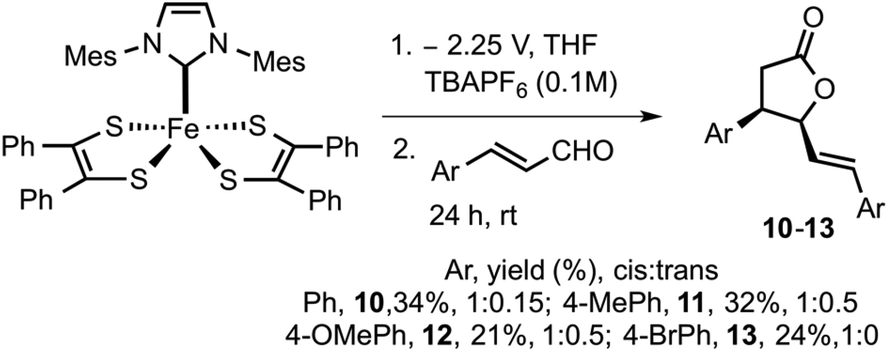 | ||
| Scheme 3 Electrochemically generated carbene catalyzed synthesis of γ-butyrolactones. | ||
To increase the scope of electro-released NHC catalysis, the methodology was expanded to other substituted cinnamaldehydes. Varying substituents on aryl include electron-donating functionalities such as methyl and methoxy and moderately electron withdrawing group such as bromine were subjected to similar catalytic conditions. Methyl and methoxy substituted aldehydes furnished the respective γ-butyrolactones 11 in 32% and 12 in 21% yield. Bromo substituted cinnamaldehyde yielded 13 in 24% yield. γ-Butyrolactones 10–12 were isolated as cis and trans mixture, while 13 was isolated in pure cis form. Electrochemical methods employed here use a less hazardous approach than traditional reagent-based chemistry. The benefits of such a system include mild reaction conditions, functional group tolerance and scalability. However, such systems also present challenges in product isolation and purification.
Conclusions
To the best of our knowledge, these are the first five coordinated [Fe(NHC)(S2C2R2)] systems that are prepared by cleaving the [Fe(S2C2Ar2)2]2 dimer with N-heterocyclic carbenes (3 and 4). The iron complexes 5–8 were characterized by NMR spectroscopy and confirmed by X-ray crystallography. DFT calculations showed that the reduction of 5 weakens the Fe–CNHC bond. The calculations of the neutral and reduced forms of the iron complex demonstrate that the reduction is primarily ligand based. Electrochemical and spectroelectrochemical measurements revealed that [Fe(NHC)(S2C2R2)] undergo facile electrochemical reduction that leads to dissociation of the adduct to release free NHCs. This behaviour was further confirmed by a trapping 3 with 1-naphthylisothiocyanate. The in situ electro-released free NHC (3) was used as a catalyst in organocatalysis to convert cinnamaldehydes to γ-butyrolactones. Efforts are underway to fine tune the electronics of the dithiolene system to achieve reversible binding of NHC so that activity of an organic transformation can be effectively controlled and the pre-catalyst can be actively restored. Furthermore, efforts are under way to explore a variety of redox-active NHC systems to expand the scope of [Fe(NHC)(S2C2R2)] systems for other potential applications, including electronic and material applications.Experimental section
Materials and methods
All synthetic manipulations were done in a nitrogen filled glove box (Inert Purelab) unless otherwise noted. All glassware were oven dried at 120 °C for 12 hours prior to use. The compounds 1,3-bis(2,4,6-trimethylphenyl)imidazolinium chloride ([3]·HCl) and 1,3-bis(2,4,6-trimethylphenyl)imidazolium chloride ([4]·HCl) were synthesized according to the literature procedure.12,34 Iron bis(dithiolene) dimers [Fe(S2C2Ph2)2]2 and [Fe(S2C2(C6H4-p-OCH3)2)]2 were prepared following the procedure reported by Schrauzer et al.1b All other reagents were purchased from commercial sources and used as received, such as sodium bis(trimethylsilyl)amide, trans-cinnamaldehyde, and substituted trans-cinnamaldehyde. [N(nBu)4][PF6] was recrystallized two times from ethanol and dried at 70 °C for 12 hours prior to use. Solvents were dried with a solvent purification system from Inert Pure Company (THF, CH2Cl2, Et2O and toluene) and degassed using three freeze–pump–thaw cycles prior to use.45 All solvents were stored over 4 Å molecular sieves in a nitrogen filled glove box. CDCl3 (99.9%) was purchased from Acros Laboratories and dried over 4 Å molecular sieves prior to use.Instrumentation
UV-Vis spectra were obtained at 25 °C with a Varian Cary 50 Bio UV-vis spectrophotometer and molar absorptivities were reported in M−1 cm−1. 1H and 13C NMR spectra were recorded on a Bruker 300 MHz NMR spectrometer. Spectra were referenced to the residual solvent as an internal standard, for 1H NMR: CDCl3, 7.26 ppm; for 13C NMR: CDCl3, 77.16. Coupling constants (J) are expressed in hertz (Hz). Elemental analyses were performed by Midwest Microlab, LLC in Indianapolis, IN. Electrochemical measurements were performed on a CHI620E electrochemical workstation using a silver wire quasi-reference electrode, a platinum disk working electrode, and a Pt wire auxiliary electrode in a gas tight three-electrode cell under nitrogen atmosphere. Unless specified otherwise, the measurements were performed using 1.0 mM solutions of the analyte in dry THF with 0.1 M [N(nBu)4][PF6] as the electrolyte and decamethylferrocene (Fc*) as the internal standard. Differential pulse voltammetry measurements were performed with 50 mV pulse amplitudes and 4 mV data intervals. All potentials listed herein were determined by cyclic voltammetry at 100 mV s−1 scan rates and referenced to a saturated calomel electrode (SCE) in decamethylferrocene 0/+ to 0.102 V (THF).46 Bulk electrolysis were conducted on two compartment cells separated by a glass frit, Pt mesh working electrode, Pt mesh counter electrode and Ag/Ag+ (in MeCN) non aqueous reference electrode.Synthesis of compound 5
A 10 mL scintillation vial with a stir bar was charged with [3]·HCl (99 mg, 0.29 mmol), NaN(Si(CH3)2)2 (58 mg mg, 0.31 mmol) and 4 mL of dry THF. The resulting mixture was stirred at 25 °C for 45 min, which resulted in a pale-yellow solution with white suspension. The heterogeneous mixture was filtered through a plug of Celite into a clean 20 mL scintillation vial containing compound 1 (78 mg, 0.145 mmol) in 2 mL THF. An immediate color change was noticed from black to black green solution. The resulting mixture was stirred at 25 °C for 2 h. The black green solution was filtered through a plug of Celite and the volatiles were removed under vacuum. The vacuum dried black green residue was dissolved in a minimum amount of dichloromethane and titrated with 15 mL of hexanes resulting with a black green precipitate, which was further washed with hexane and the volatiles were removed under vacuum. 110 mg, 90% yield; 1H-NMR (300 MHz, CDCl3): δ 7.31–7.28 (m, 8H), 7.22–7.18 (m, 12H), 6.96 (s, 4H), 6.84 (s, 2H), 2.36 (s, 6H), 2.00 (s, 12H). 13C-NMR (75 MHz, CDCl3): δ 182.18, 143.19, 139.13, 136.82, 135.80, 129.65, 129.31, 127.69, 127.00, 126.77, 21.41, 18.60. HRMS (ESI) for [C49H44FeN2S4]+[M]+ calcd 844.1737. Found 844.1732. Anal. calcd for: C49H44FeN2S4: C, 69.57, H, 5.36, N, 3.31; found: C, 69.50, H, 5.60, N, 3.46.Synthesis of compound 6
A 10 mL scintillation vial with stir bar was charged with [4]·HCl (100 mg, 0.29 mmol), NaN(Si(CH3)2)2 (58 mg mg, 0.31 mmol) and 4 mL of dry THF. The resulting mixture was stirred at 25 °C for 45 min, which resulted in a yellow solution with white suspension. The heterogeneous mixture was filtered through a plug of Celite into a clean 20 mL scintillation vial containing compound 1 (78 mg, 0.145 mmol) in 2 mL THF. An immediate color change was noticed from black to black green solution. The resulting mixture was stirred at 25 °C for 2 h. The black colored solution was filtered through a pad of Celite and dried. The dried black residue was dissolved in minimum amount of dichloromethane solvent and the compound was precipitated out using hexane 15 mL resulting in a black green precipitate, which was further washed with hexane and dried under vacuum for 24 h. 103 mg, 84% yield; 1H-NMR (300 MHz, CDCl3): δ 7.28–7.26 (m, 8H), 7.23–7.18 (m, 12H), 6.95 (s, 4H), 3.72 (s, 4H), 2.34 (s, 6H), 2.26 (s, 12H). 13C-NMR (75 MHz, CDCl3): δ 181.3, 143.06, 138.20, 137.26, 136.39, 129.62, 127.65, 127.02, 51.97, 21.35, 18.75. HRMS (ESI) for [C49H46FeN2S4]+[M]+ calcd 846.1893. Found 846.1888. Anal. calcd for: C49H46FeN2S4: C, 69.48, H, 5.47, N, 3.31; found: C, 68.75, H, 5.41, N, 3.22.Synthesis of compound 7
A 10 mL scintillation vial with stir bar was charged with [3]·HCl (99 mg, 0.29 mmol), NaN(Si(CH3)2)2 (58 mg mg, 0.31 mmol) and 4 mL of dry THF. The resulting mixture was stirred at 25 °C for 45 min, which resulted in a yellow solution with white suspension. The heterogeneous mixture was filtered through a plug of Celite into a clean 20 mL scintillation vial containing compound 2 (96 mg, 0.145 mmol) in 2 mL THF. An immediate generation of homogenous black solution occurred due to the breaking of dimer and adduct formation. The resulting mixture was stirred at 25 °C for 2 h. The black colored solution was filtered through a pad of Celite and dried. The dried dark grey residue was dissolved in a minimum amount of dichloromethane solvent and the compound was precipitated out using 15 mL diethyl ether resulting in a dark grey precipitate, which was further washed with diethyl ether and dried under vacuum for 24 h. 129 mg, 92% yield; 1H-NMR (300 MHz, CDCl3): δ 7.27–7.24 (m, 8H), 6.94 (s, 4H), 6.82 (s, 2H), 6.76–6.73 (m, 8H), 3.82 (s, 12 H), 2.36 (s, 6H), 2.00 (s, 12H); 13C-NMR (75 MHz, CDCl3): δ 181.05, 158.68, 138.94, 136.80, 136.51, 135.87, 130.73, 129.17, 126.63, 113.09, 55.33, 21.49, 18.62. HRMS (ESI) for [C53H52FeN2O4S4]+[M]+ calcd 964.2159. Found 964.2155. Anal. calcd for: C53H52FeN2O4S4: C, 65.96, H, 5.43, N, 2.90; found: C, 65.80, H, 5.46, N, 2.87.Synthesis of compound 8
A 10 mL scintillation vial with stir bar was charged with [4]·HCl (100 mg, 0.29 mmol), NaN(Si(CH3)2)2 (58 mg mg, 0.31 mmol) and 4 mL of dry THF. The resulting mixture was stirred at 25 °C for 45 min, which resulted in a yellow solution with white suspension. The heterogeneous mixture was filtered through a plug of Celite into a clean 20 mL scintillation vial containing compound 2 (96 mg, 0.145 mmol) in 2 mL THF. An immediate generation of homogenous black solution occurred due to the breaking of dimer and adduct formation. The resulting black mixture was stirred at 25 °C for 2 h. The black colored solution was filtered through a pad of Celite and dried. The dried black residue was dissolved in a minimum amount of dichloromethane solvent and the compound was precipitated out using diethyl ether 15 resulting black precipitate, which was further washed with diethyl ether and dried under vacuum for 24 h. 122 mg, 87% yield; 1H-NMR (300 MHz, CDCl3): δ 7.21–7.28 (m, 8H), 6.90 (s, 4H), 6.73–6.70 (m, 8H), 3.79 (s, 12H), 3.64 (s, 4 H), 2.33 (s, 6H), 2.22 (s, 12H). 13C-NMR (75 MHz, CDCl3): δ 180.32, 158.70, 138.03, 137.29, 136.48, 136.39, 130.72, 129.53, 113.06, 55.33, 51.94, 21.42, 18.77. HRMS (ESI) for [C53H54FeN2O4S4]+[M]+ calcd 966.2316. Found 966.2311. Anal. calcd for: C53H54FeN2O4S4: C, 65.82, H, 5.63, N, 2.90; found: C, 64.85, H, 5.26, N, 3.06.Computational details
Spin unrestricted and spin restricted Density Functional Theory (DFT) calculations were carried out using the Amsterdam Density Functional (ADF) software package.47 Triple-ζ Slater-type basis set with polarization functions (TZP) from the ADF basis-set library were employed on all atoms. Computations involving the metaGGA TPSS functional utilized the full electron basis sets. All other functionals considered in this study utilized basis functions where the core shells up to 1s for C, N, O, 2p for S, and 3p for Fe were kept frozen. The calculations were conducted without symmetry constraints on the geometry.DFT calculations must be able to predict the correct ground-state net total spin polarization in order to obtain reliable results. To this end, we have optimized the Fe(pdt)2(IMes) complex with different spin polarizations utilizing different functionals to confirm that our computational methodology matches the diamagnetic nature of the iron complex as confirmed by our 1H and 13C spectra. Besides iron complexes with ferromagnetically coupled density between the ligands and iron metal, we also considered complexes where the iron center and the ligands are spin-polarized but with opposing spin densities, i.e. anitferromagnetically coupled. This was done using the spin–flip method for broken symmetries implemented in ADF that was developed by Noodleman.41,42 The ESI† contains the relative energies (Erel) in kcal mol−1 for each of the tested functionals. Unsurprisingly, the hybrid functionals predicted the S = 1 spin state to be lowest in energy while the GGA functionals predicted the S = 0 state to be the energetic minimum. It was only for the Meta GGA TPSS functional that the antiferromagnetic S = 0 state (referred to as S = 0* in the text) was found to be slight lower in energy than the S = 0 by 0.3 kcal mol−1. Based upon a comparison of the results obtained for the iron bis(dithiolene) complexes using the BP86 GGA and the hybrid B3LYP, and B3LYP* functionals, Jacobsen et al. concluded that BP86 is the most appropriate method to describe these systems.41 Using the BP86 functional, we have determined that Fe(pdt)2(IMes) has a spin state of S = 0 and is Pauli coupled. Generally speaking, the mean absolute error (MAE) for the measured distances increased as the number of unpaired electrons in the calculation increased for all of the tested functionals (see the ESI†), further supporting the conclusion that the S = 0 state is the preferred spin state. A symmetrized fragment orbitals (SFO) analysis was utilized to investigate the percentage that the molecular orbitals of the constituent moieties contribute to the frontier molecular orbitals of the compound 5. The NHC molecule, iron atom, and bis(dithiolene) ligand were chosen to be the molecular fragments for the analysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. A. acknowledges support by funds from the Army Research Office (Department of Defense; W911NF-16-1-0197) and funds from The American Chemical Society Petroleum Research Fund (PRF-59893-UR7). Support by funds from Chemistry Department, Wright State University, College of Science and Mathematics for the purchase of the X-ray instrument is greatly acknowledged. The authors would like to acknowledge Dr Grossie, Wright State University and Dr Fratini, University of Dayton for help with low temperature single crystal X-ray diffraction measurements. S. S. acknowledges the donors of the American Chemical Society Petroleum Research Fund (PRF-58954-UNI5) and the National Science Foundation (Award #1904825) for support of this research.Notes and references
- (a) G. N. Schrauzer and V. Mayweg, Reaction of Diphenylacetylene with Nickel Sulfides, J. Am. Chem. Soc., 1962, 84, 3221–3221 CrossRef CAS; (b) G. N. Schrauzer, V. P. Mayweg, H. W. Finck and W. Heinrich, Coordination Compounds with Delocalized Ground States. Bisdithiodiketone Complexes of Iron and Cobalt1, J. Am. Chem. Soc., 1966, 88, 4604–4609 CrossRef CAS.
- (a) R. Eisenberg and H. B. Gray, Noninnocence in Metal Complexes: A Dithiolene Dawn, Inorg. Chem., 2011, 50, 9741–9751 CrossRef CAS PubMed; (b) S. Sproules and K. Wieghardt, Dithiolene radicals: Sulfur K-edge X-ray absorption spectroscopy and Harry's intuition, Coord. Chem. Rev., 2011, 255, 837–860 CrossRef CAS; (c) E. I. Stiefel, Dithiolene Chemistry: Progress in Inorganic Chemistry, Wiley, New York, 2004 Search PubMed.
- (a) A. P. Abbott, P. R. Jenkins and N. S. Khan, Novel complexes with new electro-optic properties, J. Chem. Soc., Chem. Commun., 1994, 1935–1936 RSC; (b) H. Lv, T. P. A. Ruberu, V. E. Fleischauer, W. W. Brennessel, M. L. Neidig and R. Eisenberg, Catalytic Light-Driven Generation of Hydrogen from Water by Iron Dithiolene Complexes, J. Am. Chem. Soc., 2016, 138, 11654–11663 CrossRef CAS PubMed; (c) K. L. Marshall, G. Painter, K. Lotito, A. G. Noto and P. Chang, Transition Metal Dithiolene Near-IR Dyes and Their Applications in Liquid Crystal Devices, Mol. Cryst. Liq. Cryst., 2006, 454, 47–79 Search PubMed; (d) U. T. Mueller-Westerhoff, B. Vance and D. Ihl Yoon, The synthesis of dithiolene dyes with strong near-IR absorption, Tetrahedron, 1991, 47, 909–932 CrossRef CAS; (e) T. Nakajima, Y. Yabushita and I. Tabushi, Amino acid synthesis through biogenetic-type CO2 fixation, Nature, 1975, 256, 60 CrossRef CAS PubMed; (f) N. Robertson and L. Cronin, Metal bis-1,2-dithiolene complexes in conducting or magnetic crystalline assemblies, Coord. Chem. Rev., 2002, 227, 93–127 CrossRef CAS; (g) K. Wang and E. I. Stiefel, Toward Separation and Purification of Olefins Using Dithiolene Complexes: An Electrochemical Approach, Science, 2001, 291, 106 CrossRef CAS PubMed; (h) H. Liu, X. Li, C. Shi, D. Wang, L. Chen, Y. He and J. Zhao, First-principles prediction of two-dimensional metal bis(dithiolene) complexes as promising gas sensors, Phys. Chem. Chem. Phys., 2018, 20, 16939–16948 RSC.
- W. R. McNamara, Z. Han, P. J. Alperin, W. W. Brennessel, P. L. Holland and R. Eisenberg, A Cobalt–Dithiolene Complex for the Photocatalytic and Electrocatalytic Reduction of Protons, J. Am. Chem. Soc., 2011, 133, 15368–15371 CrossRef CAS PubMed.
- (a) L. Dang, M. F. Shibl, X. Yang, A. Alak, D. J. Harrison, U. Fekl, E. N. Brothers and M. B. Hall, The Mechanism of Alkene Addition to a Nickel Bis(dithiolene) Complex: The Role of the Reduced Metal Complex, J. Am. Chem. Soc., 2012, 134, 4481–4484 CrossRef CAS PubMed; (b) D. J. Harrison, N. Nguyen, A. J. Lough and U. Fekl, New Insight into Reactions of Ni(S2C2(CF3)2)2 with Simple Alkenes: Alkene Adduct versus Dihydrodithiin Product Selectivity Is Controlled by [Ni(S2C2(CF3)2)2]- Anion, J. Am. Chem. Soc., 2006, 128, 11026–11027 CrossRef CAS PubMed.
- A. Zarkadoulas, E. Koutsouri and C. A. Mitsopoulou, A perspective on solar energy conversion and water photosplitting by dithiolene complexes, Coord. Chem. Rev., 2012, 256, 2424–2434 CrossRef CAS.
- (a) A. L. Balch, Cleavage reactions of cobalt and iron dithiolate compounds. Five-coordinate complexes, Inorg. Chem., 1967, 6, 2158–2162 CrossRef CAS; (b) R. Yu, K. Arumugam, A. Manepalli, Y. Tran, R. Schmehl, H. Jacobsen and J. P. Donahue, Reversible, Electrochemically Controlled Binding of Phosphine to Iron and Cobalt Bis(dithiolene) Complexes, Inorg. Chem., 2007, 46, 5131–5133 CrossRef CAS PubMed; (c) T. Selby-Karney, D. A. Grossie, K. Arumugam, E. Wright and P. Chandrasekaran, Structural and spectroscopic characterization of five coordinate iron and cobalt bis(dithiolene)-trimethylphosphine complexes, J. Mol. Struct., 2017, 1141, 477–483 CrossRef CAS.
- (a) J. A. McCleverty, N. M. Atherton, N. G. Connelly and C. J. Winscom, Transition-metal dithiolenes. Part VII. Five- and six-co-ordinate Lewis-base complexes of cobalt and iron bisdithiolenes, J. Chem. Soc. A, 1969, 2242–2257 RSC; (b) J. A. McCleverty and D. G. Orchard, Transition metal dithiolene complexes. Part XVIII. Adducts of cobalt and iron bis-1,2-dithiolenes containing ethyldiphenylphosphine and bis(1,2-diphenylphosphino)ethane, J. Chem. Soc. A, 1971, 626–631 RSC; (c) J. A. McCleverty and B. Ratcliff, Transtion-metal dithiolene complexes. Part XIV. Five-co-ordinate phosphine and phosphite adducts of cobalt and iron bis(diaryl-1,2-dithiolenes), J. Chem. Soc. A, 1970, 1631–1637 RSC; (d) A. K. Patra, E. Bill, E. Bothe, K. Chlopek, F. Neese, T. Weyhermüller, K. Stobie, M. D. Ward, J. A. McCleverty and K. Wieghardt, Electronic Structure of Mononuclear Bis(1,2-diaryl-1,2-ethylenedithiolato)iron Complexes Containing a Fifth Cyanide or Phosphite Ligand: A Combined Experimental and Computational Study, Inorg. Chem., 2006, 45, 7877–7890 CrossRef CAS PubMed.
- (a) J. Locke, J. A. McCleverty, E. J. Wharton and C. J. Winscom, Iron and cobalt nitrosyl 1,2-dithiolenes, Chem. Commun., 1966, 677–678 RSC; (b) J. A. McCleverty, N. M. Atherton, J. Locke, E. J. Wharton and C. J. Winscom, Transition metal-dithiolene complexes. III. Nitrosyl complexes of iron and cobalt, J. Am. Chem. Soc., 1967, 89, 6082–6092 CrossRef CAS; (c) J. A. McCleverty and B. Ratcliff, Transition-metal dithiolene complexes. Part XIII. Nitric oxide adducts of iron bis(diaryl-1,2-dithiolenes), J. Chem. Soc. A, 1970, 1627–1631 RSC; (d) P. Surawatanawong, S. Sproules, F. Neese and K. Wieghardt, Electronic Structures and Spectroscopy of the Electron Transfer Series [Fe(NO)L2]z (z = 1+, 0, 1–, 2–,3–; L = Dithiolene), Inorg. Chem., 2011, 50, 12064–12074 CrossRef CAS PubMed.
- K. Ray, E. Bill, T. Weyhermüller and K. Wieghardt, Redox-Noninnocence of the S,S‘-Coordinated Ligands in Bis(benzene-1,2-dithiolato)iron Complexes, J. Am. Chem. Soc., 2005, 127, 5641–5654 CrossRef CAS PubMed.
- S. Friedle, D. V. Partyka, M. V. Bennett and R. H. Holm, Synthesis of metal dithiolene complexes by Si–S bond cleavage of a bis(silanylsulfanyl)alkene, Inorg. Chim. Acta, 2006, 359, 1427–1434 CrossRef CAS.
- A. J. Arduengo, J. R. Goerlich and W. J. Marshall, A stable diaminocarbene, J. Am. Chem. Soc., 1995, 117, 11027–11028 CrossRef CAS.
- (a) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed; (b) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, An overview of N-heterocyclic carbenes, Nature, 2014, 510, 485 CrossRef CAS PubMed; (c) R. S. Menon, A. T. Biju and V. Nair, Recent advances in N-heterocyclic carbene (NHC)-catalysed benzoin reactions, Beilstein J. Org. Chem., 2016, 12, 444–461 CrossRef CAS PubMed; (d) E. Peris, Smart N-Heterocyclic Carbene Ligands in Catalysis, Chem. Rev., 2018, 118, 9988–10031 CrossRef CAS PubMed; (e) R. Singh and S. P. Nolan, N-Heterocyclic carbenes: Advances in transition metal and organic catalysis, Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 2006, 102, 168–196 RSC; A. Obanda, K. Valerius, J. T. Mague, S. Sproules and J. P. Donahue, Group 10 Metal Dithiolene Bis(isonitrile) Complexes: Synthesis, Structures, Properties, and Reactivity, Organometallics, 2020, 39, 2854–2870 Search PubMed.
- (a) T. Weskamp, F. J. Kohl, W. Hieringer, D. Gleich and W. A. Herrmann, Highly Active Ruthenium Catalysts for Olefin Metathesis: The Synergy of N-Heterocyclic Carbenes and Coordinatively Labile Ligands, Angew. Chem., Int. Ed., 1999, 38, 2416–2419 CrossRef CAS; (b) S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, Efficient and Recyclable Monomeric and Dendritic Ru-Based Metathesis Catalysts, J. Am. Chem. Soc., 2000, 122, 8168–8179 CrossRef CAS; (c) J. Huang, E. D. Stevens, S. P. Nolan and J. L. Petersen, Olefin Metathesis-Active Ruthenium Complexes Bearing a Nucleophilic Carbene Ligand, J. Am. Chem. Soc., 1999, 121, 2674–2678 CrossRef CAS; (d) M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Synthesis and Activity of a New Generation of Ruthenium-Based Olefin Metathesis Catalysts Coordinated with 1,3-Dimesityl-4,5-dihydroimidazol-2-ylidene Ligands, Org. Lett., 1999, 1, 953–956 CrossRef CAS PubMed; (e) M. Scholl, T. M. Trnka, J. P. Morgan and R. H. Grubbs, Increased ring closing metathesis activity of ruthenium-based olefin metathesis catalysts coordinated with imidazolin-2-ylidene ligands, Tetrahedron Lett., 1999, 40, 2247–2250 CrossRef CAS.
- (a) S. Díez-González, N. Marion and S. P. Nolan, N-Heterocyclic Carbenes in Late Transition Metal Catalysis, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed; (b) G. C. Fortman and S. P. Nolan, N-Heterocyclic carbene (NHC) ligands and palladium in homogeneous cross-coupling catalysis: a perfect union, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC; (c) E. A. B. Kantchev, C. J. O'Brien and M. G. Organ, Palladium Complexes of N-Heterocyclic Carbenes as Catalysts for Cross-Coupling Reactions—A Synthetic Chemist's Perspective, Angew. Chem., Int. Ed., 2007, 46, 2768–2813 CrossRef CAS PubMed; (d) N. Marion and S. P. Nolan, Well-Defined N-Heterocyclic Carbenes−Palladium(II) Precatalysts for Cross-Coupling Reactions, Acc. Chem. Res., 2008, 41, 1440–1449 CrossRef CAS PubMed.
- (a) M. Albrecht, J. R. Miecznikowski, A. Samuel, J. W. Faller and R. H. Crabtree, Chelated Iridium(III) Bis-carbene Complexes as Air-Stable Catalysts for Transfer Hydrogenation, Organometallics, 2002, 21, 3596–3604 CrossRef CAS; (b) A. C. Hillier, H. M. Lee, E. D. Stevens and S. P. Nolan, Cationic Iridium Complexes Bearing Imidazol-2-ylidene Ligands as Transfer Hydrogenation Catalysts, Organometallics, 2001, 20, 4246–4252 CrossRef CAS; (c) D. Wang and D. Astruc, The Golden Age of Transfer Hydrogenation, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- (a) S. Burling, B. M. Paine, D. Nama, V. S. Brown, M. F. Mahon, T. J. Prior, P. S. Pregosin, M. K. Whittlesey and J. M. J. Williams, CH Activation Reactions of Ruthenium N-Heterocyclic Carbene Complexes: Application in a Catalytic Tandem Reaction Involving CC Bond Formation from Alcohols, J. Am. Chem. Soc., 2007, 129, 1987–1995 CrossRef CAS PubMed; (b) S. Gaillard, C. S. J. Cazin and S. P. Nolan, N-Heterocyclic Carbene Gold(I) and Copper(I) Complexes in C–H Bond Activation, Acc. Chem. Res., 2012, 45, 778–787 CrossRef CAS PubMed; (c) Y. Wang, D. Wei, Y. Wang, W. Zhang and M. Tang, N-Heterocyclic Carbene (NHC)-Catalyzed sp3 β-C–H Activation of Saturated Carbonyl Compounds: Mechanism, Role of NHC, and Origin of Stereoselectivity, ACS Catal., 2016, 6, 279–289 CrossRef CAS.
- (a) F. Cisnetti and A. Gautier, Metal/N-Heterocyclic Carbene Complexes: Opportunities for the Development of Anticancer Metallodrugs, Angew. Chem., Int. Ed., 2013, 52, 11976–11978 CrossRef CAS PubMed; (b) W. Liu and R. Gust, Update on metal N-heterocyclic carbene complexes as potential anti-tumor metallodrugs, Coord. Chem. Rev., 2016, 329, 191–213 CrossRef CAS; (c) L. Oehninger, R. Rubbiani and I. Ott, N-Heterocyclic carbene metal complexes in medicinal chemistry, Dalton Trans., 2013, 42, 3269–3284 RSC; (d) M.-L. Teyssot, A.-S. Jarrousse, M. Manin, A. Chevry, S. Roche, F. Norre, C. Beaudoin, L. Morel, D. Boyer, R. Mahiou and A. Gautier, Metal-NHC complexes: a survey of anti-cancer properties, Dalton Trans., 2009, 6894–6902 RSC; (e) T. Zou, C.-N. Lok, P.-K. Wan, Z.-F. Zhang, S.-K. Fung and C.-M. Che, Anticancer metal-N-heterocyclic carbene complexes of gold, platinum and palladium, Curr. Opin. Chem. Biol., 2018, 43, 30–36 CrossRef CAS PubMed; (f) J. F. Arambula, R. McCall, K. J. Sidoran, D. Magda, N. A. Mitchell, C. W. Bielawski, V. M. Lynch, J. L. Sessler and K. Arumugam, Targeting antioxidant pathways with ferrocenylated N-heterocyclic carbene supported gold(i) complexes in A549 lung cancer cells, Chem. Sci., 2016, 7, 1245–1256 RSC; (g) P. J. Barnard, M. V. Baker, S. J. Berners-Price and D. A. Day, Mitochondrial permeability transition induced by dinuclear gold(I)–carbene complexes: potential new antimitochondrial antitumour agents, J. Inorg. Biochem., 2004, 98, 1642–1647 CrossRef CAS PubMed; (h) R. McCall, M. Miles, P. Lascuna, B. Burney, Z. Patel, K. J. Sidoran, V. Sittaramane, J. Kocerha, D. A. Grossie, J. L. Sessler, K. Arumugam and J. F. Arambula, Dual targeting of the cancer antioxidant network with 1,4-naphthoquinone fused Gold(I) N-heterocyclic carbene complexes, Chem. Sci., 2017, 8, 5918–5929 RSC; (i) J. Selvakumar, M. H. Miles, D. A. Grossie and K. Arumugam, Synthesis and molecular structure of biologically significant bis(1,3-dimesityl-4,5-naphthoquinoimidazol-2-ylidene)gold(I) complexes with chloride and dichloridoaurate counter-ions, Acta Crystallogr., Sect. C: Struct. Chem., 2019, 75, 462–468 CrossRef CAS PubMed.
- (a) P.-C. Chiang and J. W. Bode, On the Role of CO2 in NHC-Catalyzed Oxidation of Aldehydes, Org. Lett., 2011, 13, 2422–2425 CrossRef CAS PubMed; (b) W. Jeong, E. J. Shin, D. A. Culkin, J. L. Hedrick and R. M. Waymouth, Zwitterionic Polymerization: A Kinetic Strategy for the Controlled Synthesis of Cyclic Polylactide, J. Am. Chem. Soc., 2009, 131, 4884–4891 CrossRef CAS PubMed; (c) V. Nair, S. Vellalath, M. Poonoth and E. Suresh, N-Heterocyclic Carbene-Catalyzed Reaction of Chalcones and Enals via Homoenolate: an Efficient Synthesis of 1,3,4-Trisubstituted Cyclopentenes, J. Am. Chem. Soc., 2006, 128, 8736–8737 CrossRef CAS PubMed; (d) S. S. Sohn, E. L. Rosen and J. W. Bode, N-Heterocyclic Carbene-Catalyzed Generation of Homoenolates: γ-Butyrolactones by Direct Annulations of Enals and Aldehydes, J. Am. Chem. Soc., 2004, 126, 14370–14371 CrossRef CAS PubMed.
- D. Enders and J. Han, Synthesis of enantiopure triazolium salts from pyroglutamic acid and their evaluation in the benzoin condensation, Tetrahedron: Asymmetry, 2008, 19, 1367–1371 CrossRef CAS.
- C. S. P. McErlean and A. C. Willis, Application of an Intramolecular Stetter Reaction to Access trans,syn,trans-Fused Pyrans, Synlett, 2009, 233–236 CrossRef CAS.
- H. U. Vora and T. Rovis, Aldrichimica Acta, 2011, 44, 3 CAS.
- C. Burstein and F. Glorius, Organocatalyzed Conjugate Umpolung of α,β-Unsaturated Aldehydes for the Synthesis of γ-Butyrolactones, Angew. Chem., Int. Ed., 2004, 43, 6205–6208 CrossRef CAS PubMed.
- J. Mahatthananchai, P. Zheng and J. W. Bode, α,β-Unsaturated Acyl Azoliums from N-Heterocyclic Carbene Catalyzed Reactions: Observation and Mechanistic Investigation, Angew. Chem., Int. Ed., 2011, 50, 1673–1677 CrossRef CAS PubMed.
- (a) H. A. Brown, Y. A. Chang and R. M. Waymouth, Zwitterionic Polymerization to Generate High Molecular Weight Cyclic Poly(Carbosiloxane)s, J. Am. Chem. Soc., 2013, 135, 18738–18741 CrossRef CAS PubMed; (b) H. A. Brown and R. M. Waymouth, Zwitterionic Ring-Opening Polymerization for the Synthesis of High Molecular Weight Cyclic Polymers, Acc. Chem. Res., 2013, 46, 2585–2596 CrossRef CAS PubMed; (c) D. A. Culkin, W. Jeong, S. Csihony, E. D. Gomez, N. P. Balsara, J. L. Hedrick and R. M. Waymouth, Zwitterionic Polymerization of Lactide to Cyclic Poly(Lactide) by Using N-Heterocyclic Carbene Organocatalysts, Angew. Chem., Int. Ed., 2007, 46, 2627–2630 CrossRef CAS PubMed.
- M. Y. Lui, A. K. L. Yuen, A. F. Masters and T. Maschmeyer, Masked N-Heterocyclic Carbene-Catalyzed Alkylation of Phenols with Organic Carbonates, ChemSusChem, 2016, 9, 2312–2316 CrossRef CAS PubMed.
- (a) A. P. Blum, T. Ritter and R. H. Grubbs, Synthesis of N-Heterocylic Carbene-Containing Metal Complexes from 2-(Pentafluorophenyl)imidazolidines, Organometallics, 2007, 26, 2122–2124 CrossRef CAS; (b) G. W. Nyce, S. Csihony, R. M. Waymouth and J. L. Hedrick, A General and Versatile Approach to Thermally Generated N-Heterocyclic Carbenes, Chem. – Eur. J., 2004, 10, 4073–4079 CrossRef CAS PubMed.
- B. C. Norris, D. G. Sheppard, G. Henkelman and C. W. Bielawski, Kinetic and Thermodynamic Evaluation of the Reversible N-Heterocyclic Carbene−Isothiocyanate Coupling Reaction: Applications in Latent Catalysis, J. Org. Chem., 2011, 76, 301–304 CrossRef CAS PubMed.
- D. J. Cardin, B. Cetinkaya, E. Cetinkaya and M. F. Lappert, Carbene complexes. Part I. Electron-rich olefins as a source of carbene complexes of platinum(II) and palladium(II); and some experiments with (CF3)2CN2, J. Chem. Soc., Dalton Trans., 1973, 514–522 RSC.
- T. M. Trnka, J. P. Morgan, M. S. Sanford, T. E. Wilhelm, M. Scholl, T.-L. Choi, S. Ding, M. W. Day and R. H. Grubbs, Synthesis and Activity of Ruthenium Alkylidene Complexes Coordinated with Phosphine and N-Heterocyclic Carbene Ligands, J. Am. Chem. Soc., 2003, 125, 2546–2558 CrossRef CAS PubMed; S. Naumann and M. Buchmeiser, Liberation of N-heterocyclic carbenes (NHCs) from thermally labile progenitors: protected NHCs as versatile tools in organo- and polymerization catalysis, Catal. Sci. Technol., 2014, 4, 2466 RSC.
- (a) E. K. Bullough, M. A. Little and C. E. Williams, Organometallics, 2013, 32, 570 CrossRef CAS; (b) B. Gorodetsky, T. Ramnial, N. R. Branda and J. A. C. Clyburne, Electrochemical reduction of an imidazolium cation: a convenient preparation of imidazol-2-ylidenes and their observation in an ionic liquid, Chem. Commun., 2004, 1972–1973 RSC; (c) G. D. Robillard, C. H. Devillers, D. Kunz, H. E. Cattey, E. Digard and J. Andrieu, J. Org. Lett., 2013, 15, 4410 CrossRef PubMed.
- (a) J. P. Canal, T. Ramnial, D. A. Dickie and J. A. C. Clyburne, From the reactivity of N-heterocyclic carbenes to new chemistry in ionic liquids, Chem. Commun., 2006, 1809–1818 RSC; (b) M. Feroci, I. Chiarotto, M. Orsini and A. Inesi, Electrogenerated NHC as an organocatalyst in the Staudinger reaction, Chem. Commun., 2010, 46, 4121–4123 RSC; (c) M. Feroci, I. Chiarotto, M. Orsini, G. Sotgiu and A. Inesi, Reactivity of Electrogenerated N-Heterocyclic Carbenes in Room-Temperature Ionic Liquids. Cyclization to 2-Azetidinone Ring via C-3/C-4 Bond Formation, Adv. Synth. Catal., 2008, 350, 1355–1359 CrossRef CAS.
- F. E. Hahn and M. C. Jahnke, Heterocyclic Carbenes: Synthesis and Coordination Chemistry, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed.
- K. M. Kuhn and R. H. Grubbs, A Facile Preparation of Imidazolinium Chlorides, Org. Lett., 2008, 10, 2075–2077 CrossRef CAS PubMed.
- (a) S. Jiang, T. Zhang, X. Zhang, G. Zhang and B. Li, Nitrogen heterocyclic carbene containing pentacoordinate iron dicarbonyl as a [Fe]-hydrogenase active site model, Dalton Trans., 2015, 44, 16708–16712 RSC; (b) K. Riener, S. Haslinger, A. Raba, M. P. Högerl, M. Cokoja, W. A. Herrmann and F. E. Kühn, Chemistry of Iron N-Heterocyclic Carbene Complexes: Syntheses, Structures, Reactivities, and Catalytic Applications, Chem. Rev., 2014, 114, 5215–5272 CrossRef CAS PubMed.
- (a) T. Hashimoto, S. Urban, R. Hoshino, Y. Ohki, K. Tatsumi and F. Glorius, Synthesis of Bis(N-heterocyclic carbene) Complexes of Iron(II) and Their Application in Hydrosilylation and Transfer Hydrogenation, Organometallics, 2012, 31, 4474–4479 CrossRef CAS; (b) B. Li, T. Liu, C. V. Popescu, A. Bilko and M. Y. Darensbourg, Synthesis and Mössbauer Characterization of Octahedral Iron(II) Carbonyl Complexes FeI2(CO)3L and FeI2(CO)2L2: Developing Models of the [Fe]-H2ase Active Site, Inorg. Chem., 2009, 48, 11283–11289 CrossRef CAS PubMed.
- K. Arumugam, M. Selvachandran, A. Obanda, M. C. Shaw, P. Chandrasekaran, S. L. Caston Good, J. T. Mague, S. Sproules and J. P. Donahue, Redox-Active Metallodithiolene Groups Separated by Insulating Tetraphosphinobenzene Spacers, Inorg. Chem., 2018, 57, 4023–4038 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- The distortion from an ideal square pyramidal geometry can be computed into an equation to analyze structural fluctuations for compounds 5–8. The following formula is used to compute τ, τ = (β − α)/60. For an ideal square pyramidal geometry, α = β = 180° (α is defined as ∠S1–Fe–S3; β is defined as ∠S2–Fe–S4) and the apex position is occupied by CNHC atom. Subsequently, for an ideal trigonal bipyramidal geometry, α = 120° (α is defined as ∠S1–Fe–S3 or ∠S1–Fe–CNHC or ∠S3–Fe–CNHC) and β = 180° (β is defined as ∠S2–Fe–S4). For an ideal square pyramidal geometry, τ = 0, while for an ideal trigonal bipyramidal τ = 1. By computing τ, the degree of deviation from an ideal square pyramidal geometry can be deduced for compounds for 5–8.
- The manuscript is currently under preperation that details the extent of distortion for a variety of iron bis(dithiolene) complexes bearing an NHC ligand. Preliminary screening reveals both steric and electronic effects contribute to the distortion excerted by an NHC unit.
- H. Jacobsen and J. P. Donahue, Computational Study of Iron Bis(dithiolene) Complexes: Redox Non-Innocent Ligands and Antiferromagnetic Coupling, Inorg. Chem., 2008, 47, 10037–10045 CrossRef CAS PubMed.
- (a) L. Noodleman, Valence bond description of antiferromagnetic coupling in transition metal dimers, J. Chem. Phys., 1981, 74, 5737–5743 CrossRef CAS; (b) L. Noodleman and E. J. Baerends, Electronic structure, magnetic properties, ESR, and optical spectra for 2-iron ferredoxin models by LCAO-X.alpha. valence bond theory, J. Am. Chem. Soc., 1984, 106, 2316–2327 CrossRef CAS.
- (a) A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed; (b) M. Ernzerhof and G. E. Scuseria, Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS; (c) S. Grimme, Accurate description of van der Waals complexes by density functional theory including empirical corrections, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed; (d) J. P. Perdew, Density-functional approximation for the correlation energy of the inhomogeneous electron gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed; (e) J. P. Perdew, Erratum: Density-functional approximation for the correlation energy of the inhomogeneous electron gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 7406–7406 CrossRef; (f) J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed; (g) J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed; (h) M. Reiher, O. Salomon and B. A. Hess, Reparameterization of hybrid functionals based on energy differences of states of different multiplicity, Theor. Chem. Acc., 2001, 107, 48–55 Search PubMed; (i) V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS; (j) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS; (k) J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed; (l) Y. Zhang and W. Yang, Comment on ``Generalized Gradient Approximation Made Simple’‘, Phys. Rev. Lett., 1998, 80, 890–890 CrossRef CAS.
- M. M. D. Wilde and M. Gravel, Bis(amino)cyclopropenylidenes as Organocatalysts for Acyl Anion and Extended Umpolung Reactions, Angew. Chem., Int. Ed., 2013, 52, 12651–12654 CrossRef CAS PubMed.
- W. L. F. Armarego and C. L. L. Chai, Purification of laboratory chemicals, Butterworth-Heinemann, Amsterdam, 2003 Search PubMed.
- J. R. Aranzaes, M.-C. Daniel and D. Astruc, Metallocenes as references for the determination of redox potentials by cyclic voltammetry — Permethylated iron and cobalt sandwich complexes, inhibition by polyamine dendrimers, and the role of hydroxy-containing ferrocenes, Can. J. Chem., 2006, 84, 288–299 CrossRef.
- (a) C. F. Guerra, J. G. Snijders, G. t. Velde and E. J. Baerends, Towards an order-N DFT method, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed; (b) G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, Chemistry with ADF, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS; (c) E. J. Baerends, T. Ziegler, A. J. Atkins, J. Autschbach, O. Baseggio, D. Bashford, A. Bérces, F. M. Bickelhaupt, C. Bo, P. M. Boerrigter, L. Cavallo, C. Daul, D. P. Chong, D. V. Chulhai, L. Deng, R. M. Dickson, J. M. Dieterich, D. E. Ellis, M. van Faassen, L. Fan, T. H. Fischer, C. Fonseca Guerra, M. Franchini, A. Ghysels, A. Giammona, S. J. A. van Gisbergen, A. Goez, A. W. Götz, J. A. Groeneveld, O. V. Gritsenko, M. Grüning, S. Gusarov, F. E. Harris, P. van den Hoek, Z. Hu, C. R. Jacob, H. Jacobsen, L. Jensen, L. Joubert, J. W. Kaminski, G. van Kessel, C. König, F. Kootstra, A. Kovalenko, M. V. Krykunov, E. van Lenthe, D. A. McCormack, A. Michalak, M. Mitoraj, S. M. Morton, J. Neugebauer, V. P. Nicu, L. Noodleman, V. P. Osinga, S. Patchkovskii, M. Pavanello, C. A. Peeples, P. H. T. Philipsen, D. Post, C. C. Pye, H. Ramanantoanina, P. Ramos, W. Ravenek, J. I. Rodríguez, P. Ros, R. Rüger, P. R. T. Schipper, D. Schlüns, H. van Schoot, G. Schreckenbach, J. S. Seldenthuis, M. Seth, J. G. Snijders, M. Solà, M. Stener, M. Swart, D. Swerhone, V. Tognetti, G. te Velde, P. Vernooijs, L. Versluis, L. Visscher, O. Visser, F. Wang, T. A. Wesolowski, E. M. van Wezenbeek, G. Wiesenekker, S. K. Wolff, T. K. Woo and A. L. Yakovlev, ADF2017, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1946471–1946474. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0qi00638f |
| This journal is © the Partner Organisations 2021 |