
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into the co-assemblies formed by different aromatic short-peptide amphiphiles†
Cristina
Gila-Vilchez
 a,
Mari C.
Mañas-Torres
b,
Juan A.
González-Vera
c,
Francisco
Franco-Montalban
d,
Juan A.
Tamayo
d,
Francisco
Conejero-Lara
e,
Juan Manuel
Cuerva
b,
Modesto T.
Lopez-Lopez
*af,
Angel
Orte
*c and
Luis
Álvarez de Cienfuegos
*bf
a,
Mari C.
Mañas-Torres
b,
Juan A.
González-Vera
c,
Francisco
Franco-Montalban
d,
Juan A.
Tamayo
d,
Francisco
Conejero-Lara
e,
Juan Manuel
Cuerva
b,
Modesto T.
Lopez-Lopez
*af,
Angel
Orte
*c and
Luis
Álvarez de Cienfuegos
*bf
aUniversidad de Granada, Departamento de Física Aplicada, Facultad de Ciencias, 18071 Granada, Spain. E-mail: modesto@ugr.es
bUniversidad de Granada, Dpto de Química Orgánica, Facultad de Ciencias, Unidad de Excelencia Química Aplicada a Biomedicina y Medioambiente (UEQ), 18071-Granada, Spain. E-mail: lac@ugr.es
cUniversidad de Granada, Nanoscopy-UGR Laboratory. Dpto de FisicoQuímica, Facultad de Farmacia, UEQ, 18072-Granada, Spain. E-mail: angelort@ugr.es
dUniversidad de Granada, Dpto de Química Farmacéutica y Orgánica, Facultad de Farmacia, 18072-Granada, Spain
eUniversidad de Granada, Dpto de Química Física, Facultad de Ciencias, UEQ, Spain
fInstituto de Investigación Biosanitaria ibs.GRANADA, Spain
First published on 12th November 2021
Abstract
We have investigated the co-self-assembly, in water and at room temperature, of different aromatic short peptides containing Fmoc- (fluorenylmethyloxycarbonyl-) and Nap- (2-(naphthalen-2-yloxy)acetyl) groups having also different chirality. Using a combination of spectroscopy and microscopy techniques we have shown that mixtures of peptides have a stronger preference to form co-assemblies giving rise to different types of fibrils of well-defined morphology. Kinetic analysis of fluorescence resonance energy transfer (FRET) between Fmoc- and Nap- side groups reported more information about the process of self-assembly between different dipeptides. We have shown that when peptides are mixed in an equimolar ratio, the kinetics of co-aggregation is faster than that occurring when the proportion is unbalanced. Moreover, following the emission band of Nap-excimers we have shown that these peptides form co-assemblies in an alternate fashion at an equimolar ratio. The mechanism of self-assembly has been studied by molecular dynamics and monitored by differential scanning calorimetry. The mechanical properties of the resulting composite hydrogels have been evaluated by rheology. These results show that the formation of co-assemblies is promoted by π–π interactions between the different aromatic groups resulting in accelerating polymerization due to destabilization of the intermediates.
Introduction
Over the last two decades, the field of supramolecular polymers has experienced a major advance leading to the development of soft functional materials with potential applications ranging from optoelectronics to biotechnology and medicine.1–9 Based on the seminal works of Meijer et al., supramolecular polymers can be classified alluding to their mechanism of growth, correlating with different covalent polymerization models. The three different mechanisms for supramolecular polymerization are isodesmic (analogue of step growth polymerization), ring-chain (similar to ring opening polymerization) and cooperative or nucleation–elongation (analogue of chain growth polymerization).5 Despite certain similitudes between covalent and supramolecular polymerization, the latter occurs via noncovalent interactions between monomers, making these processes reversible and more susceptible to environmental changes. In fact, self-assembled systems, particularly those governed by a cooperative mechanism, can often give rise to more than one polymorph, which suggests that metastable intermediates can evolve and be trapped under kinetic conditions.10–12 Thus, parameters such as concentration, temperature, solvent composition and the application of external stimuli have a major impact on the final outcome of the corresponding supramolecular polymer. These factors make the process of supramolecular polymerization very difficult to control being particularly challenging to obtain copolymers with a well-defined composition.13 Although different strategies have been implemented to exert a control over monomers’ relative distribution, very few reports have shown strong evidence about the type of copolymer formed.13 Some of these strategies rely on different types of molecular recognition (complementary H bonds, donor–acceptor, electrostatic interactions, etc.) to control the copolymer composition.14–16 Recently, intensive efforts have been directed to exert a kinetic control of pathway selectivity, which has allowed the development of seed-induced and seed-induced living supramolecular polymerizations, and the possibility to obtain copolymers with a better control over monomers’ distribution.12,13,17 While most of these recent polymerization strategies have been explored in organic solvents using molecules such as perylene bisimide dyes, porphyrins, etc.,12,18–20 less work has been conducted in biomimetic molecules that self-assemble in water at room temperature.21,22 In particular, amphiphilic short peptides such as those having Fmoc- (fluorenylmethyloxycarbonyl) and Nap- (2-(naphthalen-2-yloxy)acetyl) groups are able to self-assemble by a cooperative process in which, starting from an initial nucleation phase, the promotion of the polymerization usually happens under kinetic control triggered by the application of different stimuli.23–27 The copolymerization of these aromatic peptides28–30 has been mainly investigated by the groups of Gazit,31,32 Ulijn33,34 and Adams,35–40 respectively. Ulijn et al. have described three different types of co-assemblies (orthogonal, cooperative and disruptive) based on the structural similarities of the peptides.34 On the other hand, Adams et al. have shown that these peptides are able to undergo a self-sorting process based on the differences between the apparent pKa of the peptides.35 The assembly of different monomers has several advantages; for instance, it allows the production of novel polymers having different morphologies and materials, such as gels, with a broad control over their mechanical properties.36,38–40 Moreover, the ability to control the assembly process allows obtaining more sophisticated materials with specific functions, such as energy transport41,42 and biomaterials.43 Nevertheless, the mechanism of copolymerization of these types of peptides has not been studied in detail, justifying the formation of co-assemblies based on the structural similarities between co-monomers and/or the resulting morphological differences between polymers. Moreover, strong evidence about monomers’ distribution within copolymers is also scarce.It has been recently demonstrated that the mechanism of polymerization of these amphiphilic short peptides involves a nucleation–elongation or cooperative process.44–46 This nucleation phase is a metastable state whose persistence in time depends on the hydrophobicity and concentration of the peptide. Supramolecular fibril formation only happens when a change in the conditions allows the system to overcome the free energy barrier of polymerization.
Considering this, herein, we have studied in detail the mechanism of co-assembly of slightly different aromatic short peptides, containing Fmoc- or Nap-groups, with the intention to understand the co-assembly process and shed light on monomer's relative distribution within the supramolecular polymer (Fig. 1). We have been able to prove that both monomers have a stronger tendency to form co-assemblies giving rise to alternate copolymers. We have studied the co-assemblies formed by the combination of Fmoc-diphenylalanine (Fmoc-FF, L) with Fmoc-dialanine (Fmoc-AA, L and D), Fmoc-diglycine (Fmoc-GG), Nap-dialanine (Nap-AA, L and D) and Nap-diglycine (Nap-GG). The self-assembly process has been studied by transmission electron microscopy (TEM), Fourier transform infrared spectroscopy (FTIR), and circular dichroism (CD). To gain insights into the copolymerization process, fluorescence resonance energy transfer (FRET) between Fmoc- and Nap-dipeptides has been performed. Additional experiments of FTIR, differential scanning calorimetry (DSC), all-atom molecular dynamics (AAMD) simulations, FRET kinetic studies and fluorescence lifetime imaging microscopy (FLIM) have been carried out with the particular mixture of Fmoc-FF(L) and Nap-GG. Finally, the mechanical properties of the resulting copolymers have been analyzed by shear rheology.
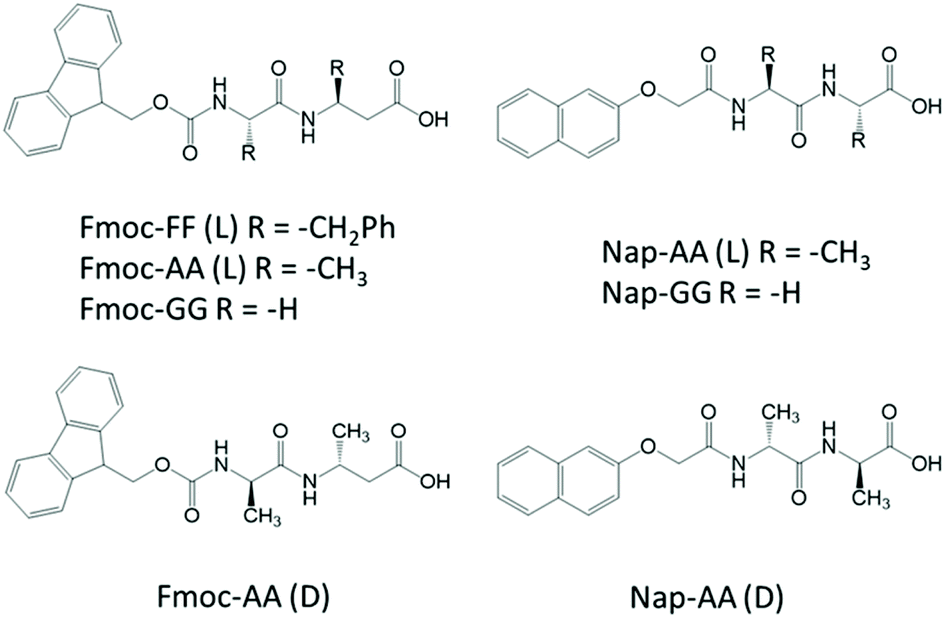 | ||
| Fig. 1 Peptide structures studied in this work. | ||
Experimental section
Reagents and materials
N-Fluorenylmethoxycarbonyl-diphenylalanine (Fmoc-FF), N-fluorenylmethoxycarbonyl-dialanine (Fmoc-AA) and N-fluorenylmethoxycarbonyl-diglycine (Fmoc-GG) were purchased from Bachem Co., Switzerland and were used without further purification. Sodium hydroxide (NaOH) and δ-gluconolactone were provided by Sigma Aldrich, USA. AQui fluorophore (9-(azetidin-1-yl)-5-butyl-4H-benzo[de][2,6]naphthyridine-4,6(5H)-dione47 was provided by Dr Rosario Herranz and Dr Francisco Fueyo-Gonzalez at Instituto de Química Médica-CSIC (IQM-CSIC). The synthesis of Nap-dipeptides was performed following a described protocol;48 characterization of Nap-GG is also described.49 Full details of the synthesis and characterization of the intermediates as well as copies of the 1H- and 13C-NMR of the final compounds can be found in the ESI.†Hydrogel preparation
In all cases, the peptide hydrogels were prepared following a previously reported protocol.50 Peptides were simultaneously weighed in the desired proportion and MilliQ water was added to obtain a final concentration of 10 mM. This suspension was sonicated in a cold AL04-03-230 ultrasonic bath for 25 minutes, until peptides were totally dispersed and a homogeneous mixture was obtained. Then, a solution 0.5 M of NaOH was added in drops of 10 in 10 μL using a micropipette, sonicating for 2 minutes when each drop was added, until a clear solution was obtained. In order to initiate the gelation process, 5 μL of a concentrated solution of δ-gluconolactone = 200 mg mL−1 was added per mL of basic solution of peptides and homogenized by vortex stirring. Finally, peptide hydrogels were rested overnight under a water-saturated atmosphere at room temperature to ensure a complete gelation.For FLIM studies, the hydrogels were prepared adding the AQui fluorophore to a clear peptide solution for a final fluorophore concentration of 12.5 μM. The mixture was vortexed and gelation was induced by adding the δ-gluconolactone solution.
Transmission electron microscopy (TEM)
The hydrogel internal structure was studied using a LIBRA 120 PLUS Carl Zeiss. Hydrogels were dispersed in ultrapure water at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 proportion and sonicated for 3 minutes. A drop of this suspension was placed on a 300-mesh copper grid and uranyl acetate 2% was used to perform a negative staining. After 2 minutes, the sample was washed 3 times with water and dried for 30 minutes at room temperature.
:5 proportion and sonicated for 3 minutes. A drop of this suspension was placed on a 300-mesh copper grid and uranyl acetate 2% was used to perform a negative staining. After 2 minutes, the sample was washed 3 times with water and dried for 30 minutes at room temperature.
Circular dichroism (CD)
The hydrogels were gelified into a 0.1 mm quartz cell (Hellma 0.1 mm quartz Suprasil) and their CD spectra were measured using a Jasco J-815 spectropolarimeter equipped with an air cooled 150 W Xenon lamp. A constant temperature of 20 °C was maintained during the measurements using a PFD-425 Peltier controller. The spectra were recorded from 200 to 350 nm with a step of 1 nm and 0.5 s of integration time per step. For each experimental condition, we took the average value of 100 measurements. CD was performed at 5 mM final dipeptide concentration to avoid artefacts derived from the samples.Fourier-transform infrared (FTIR) spectroscopy
A PerkinElmer Spectrum Two FTIR ATR spectrometer was used to carry out the spectra measurements. The hydrogels were compressed onto the diamond crystal and the spectra were scanned over the wavenumber range from 4000 to 450 cm−1. The time evolution of the FTIR spectra of peptide mixtures was studied using a Bruker IFS-66 spectrometer (Bruker, Ettlingen, Germany) equipped with a liquid nitrogen-cooled MCT detector and an attenuated total reflection (BioATR-II) accessory thermostated at 25 °C. FTIR spectra were first acquired with freshly prepared peptide mixtures dissolved in water/NaOH as described above. Then, δ-gluconolactone was added and the mixtures were immediately deposited on the ATR cell and spectra were scanned every 5 min over the range between 4000 and 900 cm−1 with a 2 cm−1 resolution and 128 accumulations. Background spectra previously measured with water/NaOH solution were subtracted from the sample spectra to remove the contribution of the strong water bands. Spectral contributions from residual water vapour were reduced using the atmospheric compensation filter built in the Bruker OPUS software. The final spectra were smoothed using a FFT filter with a 10-point window.Fluorescence
Fluorescence emission spectra and kinetics were obtained on a Jasco FP-8300 spectrofluorimeter (Jasco, Tokyo, Japan), at the excitation wavelength λex of 280 nm.Rheology
The shear rheological properties of the peptide hydrogels were determined using a Haake MARS III controlled-stress rheometer (Thermo Fisher Scientific, Waltham, MA, USA) with a plate–plate geometry of 35 mm of diameter (sensor P35 Ti L S serrated) and at a constant temperature of 25.0 ± 0.1 °C. First, the hydrogels were subjected to deformation amplitude sweep tests at a constant frequency of 1 Hz and stepwise increasing shear strain amplitude in order to determine the linear viscoelastic region (LVR). From these measurements, the values of the storage (G′) and loss (G′′) moduli were obtained as a function of γ0. From the resulting curves, the characteristic G′ and G′′ values within the LVR were calculated by averaging for the total extension of the LVR. Afterwards, the hydrogels were subjected to frequency sweep tests at a fixed shear strain amplitude of 0.02% within the LVR, and increasing frequency in the range 0.1 to 16 Hz. From these measurements we obtained the values of G′ and G′′ as a function of frequency. For each set of experimental conditions we measured at least 3 different samples. In the case of deformation amplitude sweep tests we performed 3 different repetitions for each sample. Thus, in total we have at least 9 values in deformation amplitude sweep tests and 3 in the rest of experiments for each set of experimental conditions. In this manuscript we provide the corresponding mean values and standard deviations of the performed rheological measurements.Gelation kinetics
The gelation kinetics of the different hydrogels was studied using a Haake MARS III controlled-stress rheometer (Thermo Fisher Scientific, Waltham, MA, USA) with a cone-plate geometry of 60 mm in diameter and an angle of 2° (sensor DC60/2° Ti L) and at a constant temperature of 25.0 ± 0.1 °C. The hydrogel solution was poured in the geometry plate immediately after the addition of the δ-gluconolactone and the mixture was left for gelation for 2 hours at fixed parameters of 1 Hz of frequency and 0.02% of shear strain amplitude.Differential scanning calorimetry (DSC)
DSC experiments were performed in a DASM-4 microcalorimeter51 equipped with capillary platinum cells. The reference cell was filled with volume of a salt solution in water, and the sample cell was filled with the hydrogel-precursor mixture (without δ-gluconolactone) prepared immediately prior to its introduction into the instrument. The samples were scanned up to 100 °C at a scan rate of °C min−1, cooled and rescanned again to check the reversibility of the thermograms. Instrumental baselines were recorded with both cells filled with salt solution in water and subtracted from the experimental thermograms. The corrected thermograms were normalized to heat capacity relative to the water using the Fmoc-FF molar concentrations.Multidimensional FLIM microscopy
These experiments were carried out on a MicroTime 200 instrument (PicoQuant GmbH, Berlin, Germany), previously described in detail.47 The excitation source was a pulsed, 470 nm diode laser (LDH-P-C-470, PicoQuant), operated at a repetition rate of 20 MHz. The excitation beam was focused on the sample through a ×100, 1.4NA oil immersion objective. Fluorescence emission was collected back, filtered through the main dichroic mirror and a cleanup cutoff filter (500LP) and focused onto the 75 μm confocal aperture. The emitted light was split in an orange channel and a red channel using a 600DCXR dichroic mirror and appropriate bandpass filters to define I550 (Thorlabs 550/40 bandpass filter) and I630 (Chroma 630/60 bandpass filter). Two avalanche single photon detectors (PerkinElmer) were used to detect individual photons and a TimeHarp 200 photon-counting module (PicoQuant) was used for individual photon time tagging. FLIM images were obtained with a spatial resolution of 19.5 nm per pixel and a temporal resolution of 29 ps per channel in the microtime scale. Images were analyzed using SymPhoTime 64 (PicoQuant) and home-coded scripts in FIJI (distribution of ImageJ).52 FLIM images were obtained by fitting all pixels of the image to a monoexponential decay function, using a reconstructed instrument response function, after applying a 5-pixel spatial binning and an 8-channel temporal binning. Segmentation was performed by subtracting the background defined by a 50-pixel median filter53 and applying the robust automatic threshold selection tool in FIJI.Results and discussion
Hydrogel formation of different dipeptides
Mixtures between Fmoc-FF and other dipeptides [Fmoc-AA(L and D), Fmoc-GG, Nap-AA(L and D), Nap-GG] at a ratio of 1:1 and 1:3 were dissolved in water adjusting the pH to 10.35 using NaOH. Peptide mixtures formed clear solutions at a final total concentration of 10 mM. Gelification was triggered by pH switch adding δ-gluconolactone to a final gel concentration of 1 mg mL−1.48,54 In all cases, gels of different consistency were formed after 24 hours [Fig. 2O–R, see Fig. S1† for the results using Fmoc-AA (D) and Nap-AA (D)].55 It is important to note that Fmoc-FF at 2.5 mM and the other dipeptides at 7.5 mM were unable to form gels by themselves, but only when mixed at this 1:3 ratio, the mixture of dipeptides yielded gels. At 1:4 ratio, none of the mixture samples formed gels. At a final concentration of 10 mM only Fmoc-FF and Fmoc-AA were able to form gels by themselves, although Fmoc-AA formed weaker gels.55 To study the capacity of peptide's self-assembly, we analysed peptide solutions at 10 mM, before and after adding δ-gluconolactone, by TEM. It is important to highlight that, in the absence of δ-gluconolactone, the predominant aggregates found in samples of individuals Fmoc-(AA and GG) and Nap-AA sodium salt were small nanospheres except in the case of Nap-GG sodium salt that also showed small fibrils (Fig. 2B–E and Fig. S1†). In contrast, Fmoc-FF sodium salt, at the same concentration, showed exclusively the presence of thin fibrils (Fig. 2A). As expected, these images showed the higher propensity of Fmoc-FF to aggregate into fibrillar assemblies promoted by the presence of metal cations, in agreement with the data reported by other groups56–60 and us.46 Surprisingly, TEM images of samples containing a mixture of Fmoc-FF:other dipeptides in a 1:3 ratio showed the presence of small fibrils in all cases (Fig. 2F–I and Fig. S1†). Each individual peptide mixture showed the predominance of one type of fibril with a well-defined morphology. In addition, fibril morphology was different between the different mixtures, pointing out the preferential formation of co-assemblies (see Fig. S2† for fibril width). The presence of nanospheres in most of the cases was residual, and, in some images, it could be seen that these fibrils were formed by the aggregation and transformation of the pre-existing nanospheres (Fig. 2F, see Fig. S3† for a magnified image). These results showed that Fmoc-(AA and GG) and Nap-(AA and GG) sodium salts were able to form fibrils in the presence of Fmoc-FF sodium salt. It is known that these types of peptides form micellar aggregates in the presence of metal salts; aggregates that can evolve into fibrils by an increase of peptide concentration or by lowering the pH of the medium.46,56–59,61 In this case, the formation of fibrils when Fmoc-FF is combined with other peptides suggests that mixed micellar aggregates responsible for fibril co-assemblies have had to form at high pH. Adams et al. have shown similar results in mixtures of Na-dipeptides.37 The different density of nanospheres and fibrils suggests that both peptides are involved in the co-assembly in different mixtures. After the addition of δ-gluconolactone, TEM images of peptide mixtures showed, in all cases, fibers and ribbons of higher aspect-ratio (Fig. 2J–N and Fig. S1†). In this case, the morphology of the aggregates was more similar between each other and similar to the fibers formed by Fmoc-FF alone.
 | ||
| Fig. 2 TEM images of 10 mM samples diluted in water at 1:5 proportion (A)–(E) sodium salt solution of each of the individual dipeptides. Sodium salt solution of Fmoc-FF and: Fmoc-GG (F), Fmoc-AA(L) (G), Nap-GG (H), Nap-AA(L) (I) at 1:3 proportion without δ-gluconolactone. Hydrogels, formed by the addition of δ-gluconolactone, of Fmoc-FF (J) and: Fmoc-GG (K), Fmoc-AA(L) (L), Nap-GG (M), and Nap-AA(L) (N) at 1:3 proportion; and macroscopic images of hydrogels formed by Fmoc-FF and Fmoc-GG (O), Fmoc-AA(L) (P), Nap-GG (Q), and Nap-AA(L) (R) at a final concentration of 10 mM. | ||
In order to gain more knowledge on the intra-fibrillar environment under more native-like conditions, we employed an environment-sensitive fluorophore, a 9-azetidinyl quinolimide (AQui, 9-(azetidin-1-yl)-5-butyl-4H-benzo[de][2,6] naphthyri-dine-4,6(5H)-dione), to image fibrils using fluorescence lifetime imaging microscopy (FLIM).46,47 AQui has the ability of interacting with fibrils, undergoing a drastic enhancement in fluorescence emission, and reporting on different environments through the fluorescence lifetime value, τ. Fig. 3 shows the fibrillar structure of gels formed with Fmoc-FF or Fmoc-FF:Nap-GG 1:1 mixtures. Fmoc-FF:Nap-GG mixtures exhibited fibrils of similar morphology to those of Fmoc-FF only. Nevertheless, the AQui reporter displayed slightly higher τ values in the mixtures, with average τ values of 2.5 ± 0.5 ns (s.d., n = 4) and 4.4 ± 0.1 ns (s.d., n = 4) for hydrogels of Fmoc-FF only or Fmoc-FF:Nap-GG 1:1 mixture, respectively. This significative difference can be also seen in the τ distributions of AQui from several FLIM images from the two types of hydrogels (Fig. S4†). This suggests that the effective co-aggregation of the two peptides resulted in changes in the fibrillar microenvironment. In some fibrils, the FLIM images showed the presence of repetitive patterns of high-lifetime values (indicated as arrows in Fig. 3). This may be due to the helical structure of the fibril as well as the presence of repeated hydrophobic motifs that are more noticeable in the Fmoc-FF:Nap-GG mixtures.
 | ||
| Fig. 3 FLIM images of AQui (12.5 uM) interacting with Fmoc-FF (10 mM) (A) or Fmoc-FF:Nap-GG 1:1 (5 mM:5 mM) (B) hydrogels. Arrows indicate repetitive sections of high lifetime of the dye within the hydrogel fibrils. | ||
Spectroscopy studies of co-assemblies of different dipeptides
Further analysis of the secondary structure of these peptide mixtures as well as the capacity to interact with each other to form co-assemblies was studied by FTIR (Fig. 4). First, the process of self-assembly was monitored by time dependent FTIR of Fmoc-FF and Nap-GG alone and its mixture at 1:3 ratio, before and after the addition of δ-gluconolactone. Before addition of δ-gluconolactone, Fmoc-FF (10 mM) showed the presence of two strong bands in the amide I region at 1638 cm−1 and 1690 cm−1 (Fig. 4A). Many authors attribute these two bands to formation of an antiparallel β structure based on the typical spectra of polypeptides and proteins, but a study shows that the band at 1690 cm−1 could be attributed to the stacking of the carbamate group.62 In contrast, Nap-GG did not show those two intense peaks, but a wider one at 1652 cm−1 and another more acute but less intense at 1629.5 cm−1. The spectrum of the 1:3 Fmoc-FF:Nap-GG mixture shows characteristics of both peptides. In fact, the theoretically calculated spectrum of the 1:3 mixture is similar, although not identical to the observed spectrum. This suggests that the two peptides mutually influence their structure in solution. Subsequently, the evolution of the spectra (for about 5 hours) was followed after adding δ-gluconolactone, trying to observe the changes that occur as the gels form (Fig. 4B). In the case of Fmoc-FF alone, the spectral changes show the development of a broad band centered at about 1633 cm−1, attributable to the formation of a β-sheet structure. In contrast, Nap-GG showed different spectral changes, presenting an increase in the intensity of the band at 1662 cm−1. This is consistent with a lack of capacity of this peptide to self-assemble into a β-sheet fibrillar structure.
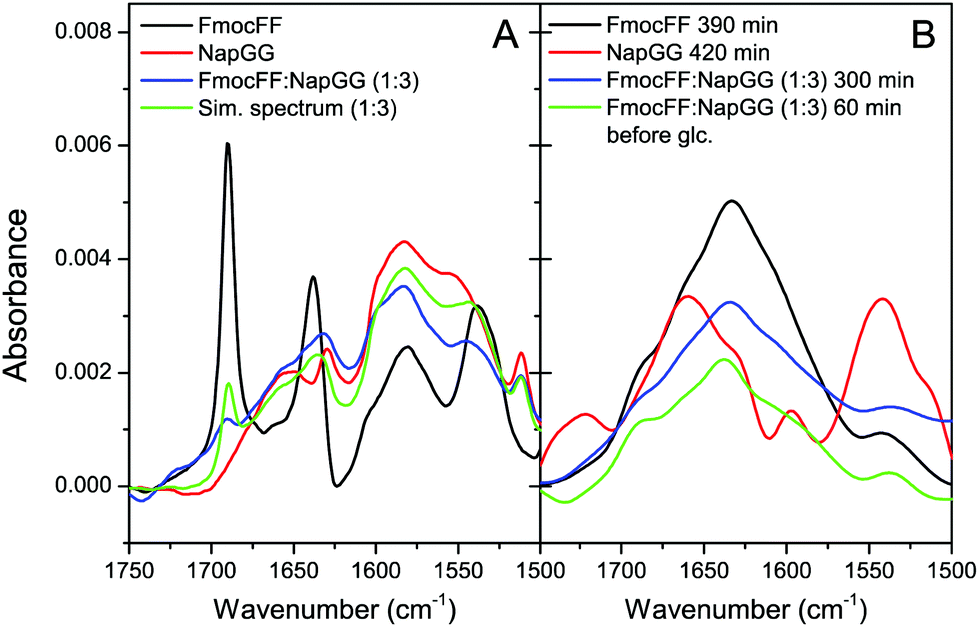 | ||
| Fig. 4 FTIR spectra of peptides. (A) Spectra of fresh solutions of Fmoc-FF, Nap-GG (10 mM) and 1:3 (2.5 mM:7.5 mM) Fmoc-FF:Nap-GG mixture. The simulated spectrum of the mixture is also shown for comparison. (B) Difference spectra obtained by subtracting the initial spectrum measured immediately after δ-gluconolactone addition from the spectrum measured after incubation of each peptide at the indicated times; the difference spectrum of the 1:3 mixture after incubation without δ-gluconolactone is also shown in green. | ||
In the spectra of the 1:3 Fmoc-FF:Nap-GG mixture, a band centered at about 1635 cm−1, very similar to the spectra of the Fmoc-FF gel alone, evolved with the incubation time. This result, consistent with those observed by TEM, suggests the formation of co-assemblies in which the resulting aggregates rearrange into β-sheets. Finally, FTIR spectroscopy of the rest of the peptide mixtures in the form of gels at 1:1 and 1:3 ratios showed practically superimposed spectra for all the mixtures and almost identical to the Fmoc-FF spectra alone (Fig. S5†), presenting again a broad band centered at about 1635 cm−1, in agreement with the results observed for the mixture Fmoc-FF:Nap-GG.
Next, we studied the supramolecular chirality of the dipeptide aggregates at 1:1 and 1:3 ratios in the gel state using CD spectroscopy (Fig. 5A and B, see Fig. S6† for CD of individual peptides and HT spectra). Most of these supramolecular amphiphilic peptides, similar to proteins and polypeptides, have CD bands in the far ultraviolet region (210–240 nm) derived mainly from the amide groups and are related to the secondary structure of the aggregates, and in the near ultraviolet region (250–320 nm) derived from aromatic groups giving information about the environment and interaction between aromatic groups.63
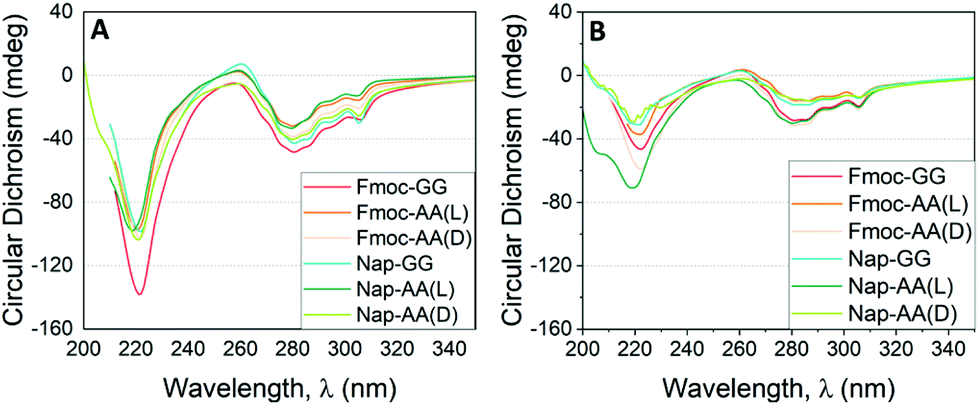 | ||
| Fig. 5 CD results as a function of the incident wavelength for hydrogels formed by Fmoc-FF and other dipeptides at (A) 1:1 (2.5 mM:2.5 mM) and (B) 1:3 (1.25 mM:3.75 mM) proportion. | ||
As commented previously, Fmoc-GG, Nap-AA and Nap-GG did not form gels and were CD silent. Fmoc-AA and Fmoc-FF gels showed CD spectra matching with those already reported.64–66 Fmoc-AA gel showed a negative peak at 228 nm, a positive peak ∼260 nm and an intense negative peak ∼280 nm. A previous report has shown that this peptide tends to adopt an extended polyproline type II rather than a β-sheet-type conformation.67 Similarly, Fmoc-FF showed an intense negative peak at 220 nm and a less intense negative peak at 280 nm. For this peptide an antiparallel β-sheet conformation has been proposed.68 The CD spectra of peptide mixtures looked all similar to the spectra of the Fmoc-FF alone. In all cases, the spectra presented two main negative bands, one in the far ultraviolet region around 220 nm and another in the near ultraviolet region around 280 nm. This result goes in line with that observed by TEM and FTIR, in which the secondary structure of β-sheets formed by Fmoc-FF alone is preserved in the mixtures. This result is quite interesting considering that dipeptides of different chirality or achiral did not modify the supramolecular chirality of the copolymer. It is also significant that the CD values found for the mixtures at 1:1 ratio were higher than those found at 1:3 ratio. This result is also coherent since the secondary structure is defined by Fmoc-FF thus, the greater proportion of Fmoc-FF, the higher ellipticity values. This effect is particularly relevant in the mixtures derived from achiral dipeptides (Fmoc-GG and Nap-GG). In this case, the preservation of the chirality supposes a process of supramolecular chiral amplification by a sergeants-and-soldiers principle.37,69,70 The retention of the handedness in mixtures containing dipeptides of opposite chirality (Fmoc-AA(D) and Nap-AA(D) showed again the predominant role of Fmoc-FF in locking and directing the supramolecular arrangement of the dipeptides. This result also shows that supramolecular copolymers and gels can be formed with peptides of different chirality, as previously shown by Adams et al.,71 and implies that the major driving force that promotes aggregation is π–π interaction between aromatic groups enhanced in water by the hydrophobic effect.72–74
As commented above, the analysis of all the data obtained by TEM, FTIR and CD is in agreement if we consider that mixtures of peptides are interacting in a cooperative way, giving rise to copolymers of similar morphological characteristics. Our results show that mixtures of peptides are interacting at high pH probably forming mixed micellar aggregates that evolve to mixed fibrillar structures upon increasing peptide concentration and/or pH reduction. Adams et al. have shown that, to a certain extent, the co-assembly of two dipeptides can be controlled toward self-sorting by lowering the pH of the media and if the pKa of the two gelators is sufficiently different.35 In this case, all the mixtures tested showed the formation of co-assemblies, reasons behind this result are commented below and it might be due to the strong hydrophobic character of Fmoc-FF that forms stable micelles that can accommodate other dipeptides, as suggested by Adams et al. for Nap-FF in ref. 37. Additionally, we used approx. 20 times more δ-gluconolactone than Adams’ experiments; therefore, in our case, the fast drop of pH (less than 5 min) does not allow a sequential self-assembly of the different dipeptides based on their relative pKa, see Fig. S7† for the variations of pH in time of the peptides.
Next, taking advantage of the fluorescence properties of the Fmoc- and Nap-groups, we performed fluorescence studies to gain insights about the type of copolymer that these peptides were forming. In particular, the Fmoc-group emits fluorescence at 325 nm, whereas Nap-monomers emit at around 350 nm. Moreover, Nap-groups have the ability to form dynamic and static excimers,75,76 exhibiting red-shifted luminescence, characterized by a broad band at >400 nm. Likewise, fluorescence resonance energy transfer (FRET) would be feasible from Fmoc- to Nap-groups when in close proximity, due to a favorable spectral overlap of the emission of Fmoc, acting as a donor, with the absorption of Nap, as an energy acceptor. Hence, by investigating the emission properties and the efficacy of the FRET process one can infer on the co-aggregation ability of Fmoc-FF with Nap-AA(D), Nap-AA(L) or Nap-GG, as it was previously demonstrated for similar dipeptides carrying Nap and pyrene units.77 To unequivocally confirm that FRET occurs from Fmoc to Nap in co-aggregated dipeptides, first, we focused on the emission spectra of the individual incubated dipeptides without δ-gluconolactone (Fig. 6A). Fmoc-FF showed the typical emission due to the Fmoc aromatic ring, centered at 325 nm. Certain contribution of the emission of phenylalanine may also be present, though this would be usually centered around 280 nm, so that its effect would be negligible. Interestingly, for Nap-AA(D), Nap-AA(L) and Nap-GG, the formation of Nap-static excimers within the individual peptides provides a measurement of the extent of self-interaction in solution. In all three emission spectra (Fig. 6A), the emission of both Nap-monomers (∼350 nm) and static excimers (∼400 nm) was present. Nevertheless, there were notable differences between the different peptides. Nap-GG showed an almost negligible contribution of monomers, and the largest excimer formation was found. This is in agreement with the largest extent of fibrillization of this peptide compared to Nap-AA (Fig. 2). In contrast, Nap-AA(L) still exhibited a notable emission of the monomer at ∼350 nm, indicating a lower extent of self-interaction compared to Nap-AA(D), as it can be also confirmed in TEM images (Fig. 2).
 | ||
| Fig. 6 (A) Fluorescence emission spectra (λex = 280 nm) of Fmoc-FF (black), Nap-AA(L) (blue), Nap-AA(D) (red), and Nap-GG (green). (B) Time evolution of the fluorescence emission spectra (λex = 280 nm) from Fmoc-FF:Nap-GG mixtures at 1:1 (5 mM:5 mM, i), 1:3 (2.5 mM:7.5 mM, ii) and 3:1 (7.5 mM:2.5 mM, iii) ratios. Arrows indicate increasing time from 0 min to 60 min. (C) Aggregation kinetics followed using the I340/I400 intensity ratio from spectra in panel (B) from Fmoc-FF:Nap-GG mixtures at 1:1 (5 mM:5 mM, black symbols), 1:3 (2.5 mM:7.5 mM, red symbols) and 3:1 (7.5 mM:2.5 mM, blue symbols) ratios. Curves represent non-linear fittings to an exponential growth equation. (D) Illustration representing how effective co-aggregation of Nap-GG with Fmoc-FF results in a disruption of Nap excimers, increasing the emission of Nap monomers, as well as FRET Fmoc → Nap. | ||
We then measured the emission spectra of incubated 1:3 ratio mixtures of Fmoc-FF:Nap-dipeptides and detected FRET from Fmoc to Nap as identified by the almost total quenching of the Fmoc emission and the subsequent enhancement of the Nap emission, confirming the effective co-aggregation of the dipeptides especially in the Fmoc-FF:Nap-AA(D) and Fmoc-FF:Nap-GG mixtures (Fig. S8†), with Nap-AA(D) or Nap-GG well integrated within the Fmoc-FF network. Interestingly, this effect was far less notable in the Fmoc-FF:Nap-AA(L) mixture (Fig. S8†). The emission of Fmoc at 325 nm was effectively quenched, but the increase in the emission of the Nap excimer was low. This indicated a lower degree of interaction and co-aggregation of Nap-AA(L) with Fmoc-FF. TEM images showed effective fibrillization for the Fmoc-FF:Nap-AA(L) mixture (Fig. 2). This supports that Nap-AA(L) may still maintain certain self-aggregation, independent of Fmoc-FF. Indeed, by inspecting the corresponding CD spectra (Fig. 5B) the mixture Fmoc-FF:Nap-AA(L) exhibited a distinctive negative band around 205 nm, which can be assigned to the unordered structure,47,78 resulting in less effective Nap excimer formation and energy transfer Fmoc → Nap.
Importantly, we also obtained quantitative information on the co-aggregation kinetics by fluorescence spectrometry. Co-aggregation of Fmoc-FF:Nap-GG at 1:1, 1:3, and 3:1 ratios was investigated by the time evolution of the emission spectra (Fig. 6B). In all these spectra, Fmoc fluorescence is practically fully quenched supporting a rapid interaction between the two peptides, Fmoc-FF and Nap-GG, in agreement with DSC experiments (vide infra), so that FRET indicates the formation of packed co-aggregates yet in the early stages of gelification. The main temporal changes were detected in a decrease in the band centered around 400 nm, where Nap excimers emit, with a concomitant increase in a band centered around 340 nm, in the region of Nap monomer emission. Additional effects, such as light scattering by fibrillar nanostructures of the hydrogel of the shorter wavelength emission of Fmoc, may contribute to the disappearance of Fmoc emission. Therefore we studied the aggregation kinetics by plotting the intensity ratio at 340 nm and 400 nm (I340/I400), as a measurement of the ratio monomer:excimer of Nap (Fig. 6C). In all the hydrogels studied, the I340/I400 ratio exhibited an increase with time, thus a transformation from Nap-excimers to monomers. Indeed, whereas Nap-excimers are formed in self-aggregation of Nap-GG, the loss of excimer and increase of Nap-monomer indicate disruption of self-aggregation. Given there is a correlation between the breakup of Nap-GG aggregates and the actual macroscopic formation of the hydrogel, the underlying cause must be the effective co-aggregation with Fmoc-FF (Fig. 6D). Then, we fitted the time-dependent I340/I400 ratio to an exponential growth function:
| I340/I400 = p·e−kobst + y0 | (1) |
:excimer ratio. We also obtained the initial velocity as: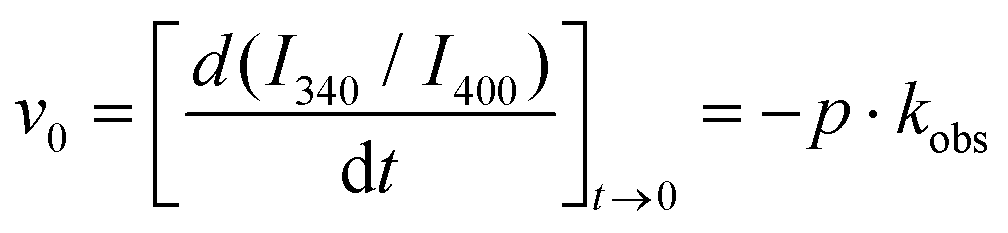 | (2) |
The kinetics of aggregation provided very interesting features. The fastest kinetics of aggregation were found for the Fmoc-FF:Nap-GG 1:1 mixture, with an average kobs of (3.5 ± 0.1) × 10−3 s−1 (s.d.) and average initial velocity of (5.6 ± 0.4) × 10−3 s−1 (s.d.). Likewise, the reached plateau at (I340/I400) = 1.64 was the largest, indicating the most effective disruption of Nap-excimers into monomers. These results contrast with the average kobs values of (1.6 ± 0.4) × 10−3 s−1 and (1.0 ± 0.1) × 10−3 s−1 and average initial velocity values of (1.2 ± 0.1) × 10−3 s−1 and (0.22 ± 0.01) × 10−3 s−1 for the Fmoc-FF:Nap-GG 1:3 and 3:1 mixtures, respectively. The fact that the 1:1 mixture exhibited the most effective and fastest co-aggregation clearly supports that Fmoc-FF and Nap-GG may be aggregating in an alternating fashion, since Nap-GG self-contacts are highly disrupted. Accordingly, an excess of Nap-GG did show remaining Nap-GG excimers that were not efficiently incorporated into the polymer. In contrast, the large excess of Fmoc-FF in the 3:1 mixture resulted in the slowest co-aggregation and less disruption of Nap-GG contacts, indicating that the molar ratio of the two peptides did not favor alternating arrangement. The slowest kinetics of co-aggregation in this last proportion suggests also that the initial metastable phase is more stable due to a larger proportion of the more hydrophobic Fmoc-FF, as we have previously shown.46
Intermediate states define co-aggregation properties
To investigate the energy exchanges and intermediate states involved in gel formation, we carried out differential scanning calorimetry (DSC) experiments with freshly prepared peptide solutions without δ-gluconolactone (Fig. 7). We reported previously that DSC can provide information about the thermal properties of the precursor intermediates and energy exchanges during heat-induced gel formation.46 Freshly prepared peptide mixtures at different molar ratios were scanned at 2 °C min−1. The thermogram of individual Fmoc-FF shows an initial strong endothermic peak near ∼50 °C attributable to disaggregation of metastable nanospheres that then evolve to fibrillar gel in an exothermic transition at higher temperature (∼75 °C). In the case of Fmoc-GG both endothermic and exothermic peaks are much weaker, consistently with a lower tendency to form gel (Fig. 7A). The Fmoc-FF:Fmoc-GG mixtures show different and more complex profiles with two sharp endothermic peaks and a more intense exotherm, consistent with more extensive gel formation. The sharp endothermic peaks at ∼44 °C and ∼50 °C could be attributed to the small amorphous aggregates. The observations that the peak ∼44 °C appears only in the mixtures and that its relative intensity with the other peak varies with the relative proportion of Fmoc-FF to Fmoc-GG suggest the formation of mixed aggregates. Interestingly, the intensity of the exotherm is in line with the presence of the peak at ∼44 °C.
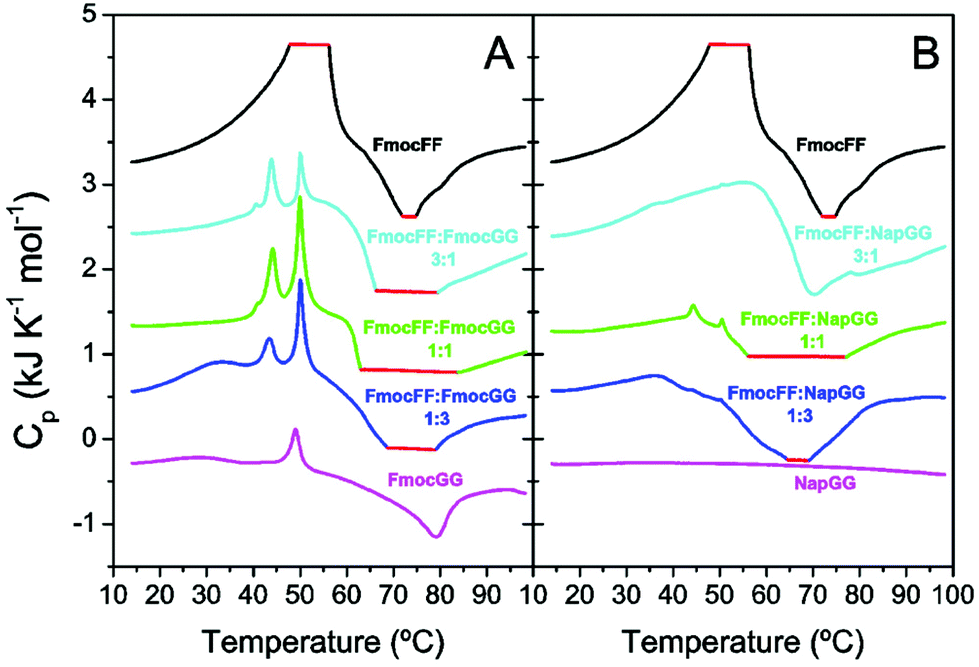 | ||
| Fig. 7 DSC scans of fresh solutions of the peptides at a total concentration of 10 mM. (A) FmocFF:Fmoc-peptide mixtures; (B) FmocFF:Nap-peptide mixtures. Scans were carried out at 2 °C min−1. The red flat regions indicate saturation of the calorimeter's signal due to strong peaks. The thermograms have been displaced vertically by 1 kJ K−1 mol−1 intervals for the sake of clarity. | ||
With regard to the Fmoc-FF:Nap-GG mixtures (Fig. 7B), while the individual Nap-GG peptide does not show any transition, in agreement with its weak-aggregating character, the mixtures show significant exothermic peaks indicating gel formation. However, sharp endothermic peaks only appear faintly for the 1:1 mixture, which is actually the one showing a stronger gel formation. These results support that peptide co-assembly into mixed gels is promoted by the formation of mixed aggregate precursors and is in agreement with the fluorescence experiments (Fig. 6), which revealed fastest and most efficient kinetics for the 1:1 Fmoc-FF:Nap-GG mixture.
Next, we explored the molecular organization of Fmoc-FF, Nap-GG and the mixtures of both monomers at 1:1 ratio by tracking the dynamic evolution of self-assemblies through AAMD simulations (see the ESI† for more details). After 300 ns AAMD simulations, the most populated cluster of Fmoc-FF showed a β-sheet-like distribution of the monomeric subunits, characterized by Fmoc-Fmoc stacking and H-bonding interactions.32 The size of the cluster showed average distances of 66.2 Å, 46.4 Å and 34.7 Å (Fig. 8A). The Nap-GG major cluster, however, developed into a more sphere-like distribution of its monomers predominantly controlled by stacking interactions within naphthyl groups; in this case the distances were 39.5 Å; 36.2 Å, and 37.9 Å (Fig. 8B). Clusters formed by mixtures of Fmoc-FF and Nap-GG at 1:1 ratio showed again the formation of β-sheet-type aggregates but, in this case, distances between monomers were smaller, revealing a tightly molecular stacking network (cluster average distances of 56.19 Å; 20.5 Å; 35.71 Å) (Fig. 8C). The co-assembly showed the preference for an alternate disposition of both monomers. This tight arrangement between both monomers might justify the preferred co-assembled aggregates, in which the average number of intramolecular H bonds (19) did not reduce significantly compared to the cluster of Fmoc-FF (22).
 | ||
| Fig. 8 Most representative assembly cluster of Fmoc-FF (A) and Nap-GG (B) and Fmoc-FF:Nap-GG 1:1 ratio (C) (60 molecules) of the 300 ns MD simulations, side view. Fmoc-and Nap-groups are represented as yellow and cyan van der Waals spheres, respectively. FF and GG peptides are represented as grey wires and benzyl groups in FF as orange sticks. H-Bonds are shown as dashed purple lines. | ||
Rheological properties of the hydrogels
From the rheological experiments we obtained the values of the storage (G′) and loss (G′′) moduli of the hydrogels at different Fmoc-FF:other dipeptide proportions, for a total peptide concentration of 10 mM (Fig. 9A and Fig. S9†). First, it should be noticed that only Fmoc-FF and Fmoc-AA were able to form gels by themselves, other single dipeptide samples behaving as solutions. Taking as the reference sample a single Fmoc-FF hydrogel, at a dipeptide concentration of 10 mM, by partial substitution of the Fmoc-FF by other dipeptides (i.e., samples Fmoc-FF:other dipeptides) we obtained a strengthening in most of the cases (Fig. 9A). For a ratio of Fmoc-FF:other dipeptides = 3:1, all compositions resulted in an increase in G′—this increase was maximum for sample Fmoc-FF:Nap-AA(D). For a ratio 1:1 sample Fmoc-FF:Fmoc-GG resulted in a weaker gel, while other 1:1 compositions resulted in stronger hydrogels—remark the one-order of magnitude increase for Fmoc-FF:Nap-AA(D). Finally, for composition 1:3 the largest enhancement took place for Fmoc-FF:Fmoc-AA(L), followed by Fmoc-FF:Fmoc-GG and Fmoc-FF:Fmoc-AA(D), whereas a decrease was obtained for Fmoc-FF:Nap-GG. These results are very interesting, since they demonstrate not only that a mixture of dipeptides made possible the generation of gels at dipeptide concentrations that did not result in gels separately39 but, more interestingly, that strong strengthening of the gel network was achieved by partial substitution of Fmoc-FF by other dipeptides.
 | ||
| Fig. 9 Rheological characterization of hydrogels at a total concentration of 10 mM formed by Fmoc-FF individual dipeptides and those formed by Fmoc-FF and other dipeptides. (A) Average storage modulus (G′) in the LVR depending on the dipeptide proportion. (B) Representative gelation kinetics at 1:1 ratio—sol–gel transitions are indicated with circles. | ||
We also investigated the kinetics of sol–gel transition by monitoring the viscoelastic moduli of the samples as a function of time starting right after addition of δ-gluconolactone (Fig. S10†). A first, rough idea of the kinetics is given by the trend of the loss tangent tanδ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) G′′/G′ (Fig. 9B). At low time, tanδ values were noisy and fluctuated around a value of 1, which is typical of the sol region. At longer times, oscillations of tanδ progressively disappeared and its values presented a marked decrease, which defines the sol–gel transition. Finally, at the longest times, tanδ tended to constant values, as the gelation process was completed. From these curves, the values of gelation time, defined as the time for which tanδ decreases below 1 in a stable manner, can be obtained (Fig. S11†). We can also infer about the gel-like character of the samples at the end of the gelation process by the corresponding values of tanδ (Fig. S12†). As observed, samples showed values in the range of 0.2–0.6, which are typical of weak gels.79 An exception is sample Fmoc-FF:Nap-AA(L) that can be defined as a strong gel (tanδ < 0.1), despite its relatively low value of storage modulus.
G′′/G′ (Fig. 9B). At low time, tanδ values were noisy and fluctuated around a value of 1, which is typical of the sol region. At longer times, oscillations of tanδ progressively disappeared and its values presented a marked decrease, which defines the sol–gel transition. Finally, at the longest times, tanδ tended to constant values, as the gelation process was completed. From these curves, the values of gelation time, defined as the time for which tanδ decreases below 1 in a stable manner, can be obtained (Fig. S11†). We can also infer about the gel-like character of the samples at the end of the gelation process by the corresponding values of tanδ (Fig. S12†). As observed, samples showed values in the range of 0.2–0.6, which are typical of weak gels.79 An exception is sample Fmoc-FF:Nap-AA(L) that can be defined as a strong gel (tanδ < 0.1), despite its relatively low value of storage modulus.
The absence of a linear relationship between the kinetics of gel formation and the final mechanical properties has already been observed by Adams et al.37 revealing that in co-assembled systems the ratio of both monomers and the kinetics of gelation has a major impact on the final gel's mechanical values.
To gain more information on the kinetics of the gelation, we analysed the rheological data based on Avrami's equation.80 Since we can consider that gelation occurred via a nucleation process in which a sol phase was progressively transformed in a more stable gel phase, we could assume that the following relation held during the gelation process:
| ln{−ln[(G′∞ − G′t)/(G′∞ − G′0)]} = lnK + nlnt | (3) |
Here G′0, G′t, and G′∞ are the values of the storage modulus at time 0, given time, t, and at the end of the gelation process, respectively. K is a temperature dependent parameter and n is the Avrami exponent, which indicates the type of growth (see scheme in Fig. 10), and that can be expressed as:80
| n = n| + d·nG | (4) |
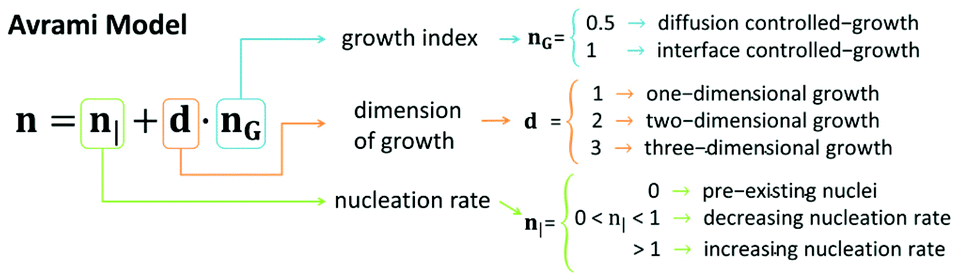 | ||
| Fig. 10 Avrami model of kinetics of nucleation and growth. | ||
We obtained the n exponent as the fitting factor for the three regions: sol, transition, and gel (Table S1†). Roughly, we observe low n values for the sol region, which can be justified by pre-existing nuclei (n| = 0), in agreement with TEM observations prior to the addition of δ-gluconolactone (Fig. 2). Furthermore, nG = 0.5 (controlled-diffusion growth) and d ≈ 1 (1-dimensional growth) would be required for the obtained values of n in the sol region. Note, nevertheless, that the value n = 1.04 ± 0.03 for Fmoc-FF:Nap-GG at 1:1 proportion would require a dimensional growth larger than 1 and/or interface-controlled growth (nG = 1) and/or an increasing nucleation rate (n| > 1) in the sol region that could explain the faster kinetics of aggregation observed for that particular ratio. In contrast, n values considerably smaller than 0.5 (samples Fmoc-FF, Fmoc-FF:Fmoc-GG, and Fmoc-FF:Nap-AA(L and D)) can only be explained by a decreasing nucleation rate in the sol region. In the transition region all samples demonstrated large n values (n considerably larger than 1) that can be explained by a combination of increasing nucleation rate (n > 1) and growth in more than 1 dimension, irrespective of the value of nG. Note that indeed 2-dimensional growth is expected for peptide gels, since fibrils tend to aggregate laterally giving rise to ribbons.66 In fact, TEM images of peptide mixtures (Fig. 2K–N) show the presence of ribbons in all cases. Finally, in the gel region, the values of n decreased with respect to those of the transition region. In general, we could explain these n values by a similar reasoning to that in the sol region but considering a 2-dimensional growth instead of 1-dimensional growth.
Conclusions
The self-assembly behavior of Fmoc-FF, in water and at room temperature, in the presence of different Fmoc- and Nap-dipeptides has been evaluated using a combination of microscopy and spectroscopy techniques. Starting from the corresponding sodium salt solutions of the dipeptides at different ratios, the self-assembly process has been studied before and after the addition of δ-gluconolactone from the initial solution state to the gel phase. As previously reported by other authors and us,44–46 the self-assembly of these peptides follows a nucleation–elongation mechanism, in which, starting from an initial nucleation phase, fibril formation is triggered by a change in the initial conditions. Herein we have shown that these peptides have a strong tendency to interact with each other in solution at high pH to form initial hybrid intermediates that are responsible for the final composition of the fibrils. Considering that the major driving force of interaction between aromatic molecules in water is the hydrophobic effect,72–74 molecules having a high percentage of aromatic groups would interact with each other to form hybrid aggregates. The polymerization of the different intermediate species only occurs when these intermediates are able to overcome the free energy barrier of polymerization. In this case, DSC studies showed that the copolymerization of the hybrid intermediates is thermodynamically favoured; therefore, the copolymer is the species that predominates in the final composition. This could be explained by analysing the results obtained from AAMD, in which a mixture of Fmoc-FF and Nap-GG is capable of forming more hydrophobic well-ordered β-sheet aggregates. Kinetic analysis of fluorescence resonance energy transfer (FRET) between Fmoc-FF and Nap-GG dipeptides has shown that when the proportion of both monomers is equimolar, the copolymerization process is faster pointing out the co-assembly of both monomers in an alternate fashion, in agreement with the results obtained following the emission band of Nap-excimers. Finally, the rheological analysis demonstrated that partial substitution of Fmoc-FF by other dipeptides resulted in strong enhancement of the storage modulus of the hydrogels. Analysis of the kinetics of sol–gel transition based on rheological measurements was consistent with the pre-existence of nuclei in peptide solutions, which assembled at an increasing rate into structures of more than one dimension (e.g., ribbons), under pH switch following addition of δ-gluconolactone.The present work provides insights into the mechanism of co-self-assembly between different aromatic short-peptides. We have shown that the ratio between both monomers determines the self-assembly kinetics and the arrangement of both monomers within the copolymer. The great tendency of these dipeptides to form co-assemblies opens the door to develop a variety of supramolecular copolymers having different characteristics and gels having also novel properties, expanding the scope of these soft interesting materials.
Author contributions
C.G.-V.: Conceptualization, formal analysis, investigation, and validation. M.C.M.-T.: Formal analysis, investigation, validation, and visualization. J.A.G.-V.: Conceptualization, formal analysis, investigation, and supervision. F.F.: Formal analysis, methodology, and investigation. J.T.: Formal analysis, methodology, and investigation. F.C.-L: Formal analysis, methodology, and writing – original draft. J.M.C.: Conceptualization, formal analysis, and writing – original draft. M.T.L.-L.: Conceptualization, methodology, funding acquisition, formal analysis, writing – original draft, and writing – review. A.O.: Conceptualization, formal analysis, funding acquisition, methodology, supervision, writing – original draft, and writing – review. L.A.C: Conceptualization, funding acquisition, methodology, project administration, supervision, writing – original draft, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by project PID2020-118498GB-I00 funded by MCIN/AEI/10.13039/501100011033, Spain and by project CTQ2017-85658-R funded by MCIN/AEI/10.13039/501100011033/FEDER “Una manera de hacer Europa”, Spain, and by FEDER/Junta de Andalucía-Consejería de Transformación Económica, Industria, Conocimiento y Universidades (Spain) project P18-FR-3533. CGV and MCMT acknowledge respectively grants FPU17/00491 and PRE2018-083773 funded by MCIN/AEI/10.13039/501100011033 and FSE “El FSE invierte en tu futuro”, Spain. Thanks go to the CIC personnel of the University of Granada for technical assistance. We thank the Centro de Servicios de Informática y Redes de Comunicaciones (CSIRC), Universidad de Granada, for providing the computing time. The authors acknowledge Dr Rosario Herranz and Dr Francisco Fueyo-Gonzalez for the fluorophore 9-azetidinyl-5-butyl-quinolimide (AQui) used in this study, which was synthesized at Instituto de Química Médica-CSIC (IQM-CSIC) with the support of the Ministerio de Ciencia e Innovación/Agencia Estatal de Investigación grant FU2015-67284-R.References
- J. R. Nitschke, Systems chemistry: molecular networks come of age, Nature, 2009, 462, 736–738 CrossRef CAS PubMed.
- E. Mattia and S. Otto, Supramolecular systems chemistry, Nat. Nanotechnol., 2015, 10, 111–119 CrossRef CAS PubMed.
- F. S. Kim, G. Ren and S. A. Jenekhe, One-dimensional nanostructures of π-conjugated molecular systems: Assembly, properties, and applications from photovoltaics, sensors, and nanophotonics to nanoelectronics, Chem. Mater., 2011, 23, 682–732 CrossRef CAS.
- L. Adler-Abramovich and E. Gazit, The physical properties of supramolecular peptide assemblies: From building block association to technological applications, Chem. Soc. Rev., 2014, 43, 6881–6893 RSC.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Supramolecular polymers, Chem. Rev., 2001, 101, 4071–4097 CrossRef CAS PubMed.
- S. Zhang, Fabrication of novel biomaterials through molecular self-assembly, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- S. Mann, Self-assembly and transformation of hybrid nano-objects and nanostructures under equilibrium and non-equilibrium conditions, Nat. Mater., 2009, 8, 781–792 CrossRef CAS PubMed.
- Y. Liu, Z. Wang and X. Zhang, Characterization of supramolecular polymers, Chem. Soc. Rev., 2012, 41, 5922–5932 RSC.
- E. Busseron, Y. Ruff, E. Moulin and N. Giuseppone, Supramolecular self-assemblies as functional nanomaterials, Nanoscale, 2013, 5, 7098–7140 RSC.
- S. H. Jung, M. Takeuchi and K. Sugiyasu, Molecular self-assembly under kinetic control, Elsevier, 2018 Search PubMed.
- J. Matern, Y. Dorca, L. Sánchez and G. Fernández, Revising Complex Supramolecular Polymerization under Kinetic and Thermodynamic Control, Angew. Chem., Int. Ed., 2019, 58, 16730–16740 CrossRef CAS.
- M. Wehner and F. Würthner, Supramolecular polymerization through kinetic pathway control and living chain growth, Nat. Rev. Chem., 2020, 4, 38–53 CrossRef CAS.
- B. Adelizzi, N. J. Van Zee, L. N. J. De Windt, A. R. A. Palmans and E. W. Meijer, Future of Supramolecular Copolymers Unveiled by Reflecting on Covalent Copolymerization, J. Am. Chem. Soc., 2019, 141, 6110–6121 CrossRef CAS PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. Ky Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Reversible polymers formed from self-complementary monomers using quadruple hydrogen bonding, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- S. Sivakova and S. J. Rowan, Nucleobases as supramolecular motifs, Chem. Soc. Rev., 2005, 34, 9–21 RSC.
- A. Das and S. Ghosh, Supramolecular assemblies by charge-transfer interactions between donor and acceptor chromophores, Angew. Chem., Int. Ed., 2014, 53, 2038–2054 CrossRef CAS PubMed.
- J. Kang, D. Miyajima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, A rational strategy for the realization of chain-growth supramolecular polymerization, Science, 2015, 347, 646–651 CrossRef CAS PubMed.
- S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Living supramolecular polymerization realized through a biomimetic approach, Nat. Chem., 2014, 6, 188–195 CrossRef CAS PubMed.
- S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, Mechanism of self-assembly process and seeded supramolecular polymerization of perylene bisimide organogelator, J. Am. Chem. Soc., 2015, 137, 3300–3307 CrossRef CAS PubMed.
- D. Görl, X. Zhang, V. Stepanenko and F. Würthner, Supramolecular block copolymers by kinetically controlled co-self-assembly of planar and core-twisted perylene bisimides, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- F. Tantakitti, J. Boekhoven, X. Wang, R. V. Kazantsev, T. Yu, J. Li, E. Zhuang, R. Zandi, J. H. Ortony, C. J. Newcomb, L. C. Palmer, G. S. Shekhawat, M. O. De La Cruz, G. C. Schatz and S. I. Stupp, Energy landscapes and functions of supramolecular systems, Nat. Mater., 2016, 15, 469–476 CrossRef CAS PubMed.
- S. Wang, F. Liu, N. Ma, Y. Li, Q. Jing, X. Zhou and Y. Xia, Mechanistic process understanding of the self-assembling behaviour of asymmetric bolaamphiphilic short-peptides and their templating for silica and titania nanomaterials, Nanoscale, 2021, 13, 13318–13327 RSC.
- G. Wei, Z. Su, N. P. Reynolds, P. Arosio, I. W. Hamley, E. Gazit and R. Mezzenga, Self-assembling peptide and protein amyloids: From structure to tailored function in nanotechnology, Chem. Soc. Rev., 2017, 46, 4661–4708 RSC.
- E. R. Draper and D. J. Adams, Low-Molecular-Weight Gels: The State of the Art, Chem, 2017, 3, 390–410 CAS.
- C. Yuan, W. Ji, R. Xing, J. Li, E. Gazit and X. Yan, Hierarchically oriented organization in supramolecular peptide crystals, Nat. Rev. Chem., 2019, 3, 567–588 CrossRef CAS.
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Biomimetic peptide self-assembly for functional materials, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS.
- A. Lampel, Biology-Inspired Supramolecular Peptide Systems, Chem, 2020, 6, 1222–1236 CAS.
- D. M. Ryan, T. M. Doran and B. L. Nilsson, Complementary π-π Interactions induce multicomponent coassembly into functional fibrils, Langmuir, 2011, 27, 11145–11156 CrossRef CAS PubMed.
- D. M. Raymond and B. L. Nilsson, Multicomponent peptide assemblies, Chem. Soc. Rev., 2018, 47, 3659–3720 RSC.
- Y. Tang, Y. Yao and G. Wei, Expanding the structural diversity of peptide assemblies by coassembling dipeptides with diphenylalanine, Nanoscale, 2020, 12, 3038–3049 RSC.
- P. Chakraborty, Y. Tang, T. Guterman, Z. A. Arnon, Y. Yao, G. Wei and E. Gazit, Co-Assembly between Fmoc Diphenylalanine and Diphenylalanine within a 3D Fibrous Viscous Network Confers Atypical Curvature and Branching, Angew. Chem., Int. Ed., 2020, 59, 23731–23739 CrossRef CAS PubMed.
- W. Ji, C. Yuan, P. Chakraborty, P. Makam, S. Bera, S. Rencus-Lazar, J. Li, X. Yan, E. Gazit and E. Gazit, Coassembly-Induced Transformation of Dipeptide Amyloid-Like Structures into Stimuli-Responsive Supramolecular Materials, ACS Nano, 2020, 14, 7181–7190 CrossRef CAS.
- S. Fleming and R. V. Ulijn, Design of nanostructures based on aromatic peptide amphiphiles, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- S. Fleming, S. Debnath, P. W. J. M. Frederix, N. T. Hunt and R. V. Ulijn, Insights into the coassembly of hydrogelators and surfactants based on aromatic peptide amphiphiles, Biomacromolecules, 2014, 15, 1171–1184 CrossRef CAS PubMed.
- K. L. Morris, L. Chen, J. Raeburn, O. R. Sellick, P. Cotanda, A. Paul, P. C. Griffiths, S. M. King, R. K. O'Reilly, L. C. Serpell and D. J. Adams, Chemically programmed self-sorting of gelator networks, Nat. Commun., 2013, 4, 1–6 Search PubMed.
- C. Colquhoun, E. R. Draper, E. G. B. Eden, B. N. Cattoz, K. L. Morris, L. Chen, T. O. McDonald, A. E. Terry, P. C. Griffiths, L. C. Serpell and D. J. Adams, The effect of self-sorting and co-assembly on the mechanical properties of low molecular weight hydrogels, Nanoscale, 2014, 6, 13719–13725 RSC.
- E. R. Draper, M. Wallace, R. Schweins, R. J. Poole and D. J. Adams, Nonlinear Effects in Multicomponent Supramolecular Hydrogels, Langmuir, 2017, 33, 2387–2395 CrossRef CAS PubMed.
- S. Panja, B. Dietrich, O. Shebanova, A. J. Smith and D. J. Adams, Programming Gels Over a Wide pH Range Using Multicomponent Systems, Angew. Chem., Int. Ed., 2021, 60, 9973–9977 CrossRef CAS PubMed.
- S. Panja, A. Seddon and D. J. Adams, Controlling hydrogel properties by tuning non-covalent interactions in a charge complementary multicomponent system, Chem. Sci., 2021, 12, 11197–11203 RSC.
- D. Giuri, L. J. Marshall, B. Dietrich, D. McDowall, L. Thomson, J. Y. Newton, C. Wilson, R. Schweins and D. J. Adams, Exploiting and controlling gel-to-crystal transitions in multicomponent supramolecular gels, Chem. Sci., 2021, 12, 9720–9725 RSC.
- H. A. M. Ardona, E. R. Draper, F. Citossi, M. Wallace, L. C. Serpell, D. J. Adams and J. D. Tovar, Kinetically Controlled Coassembly of Multichromophoric Peptide Hydrogelators and the Impacts on Energy Transport, J. Am. Chem. Soc., 2017, 139, 8685–8692 CrossRef CAS PubMed.
- N. K. Wijerathne, M. Kumar and R. V. Ulijn, Fmoc-Dipeptide/Porphyrin Molar Ratio Dictates Energy Transfer Efficiency in Nanostructures Produced by Biocatalytic Co-Assembly, Chem. – Eur. J., 2019, 25, 11847–11851 CrossRef CAS PubMed.
- A. Brito, Y. M. Abul-Haija, D. S. Da Costa, R. Novoa-Carballal, R. L. Reis, R. V. Ulijn, R. A. Pires and I. Pashkuleva, Minimalistic supramolecular proteoglycan mimics by co-assembly of aromatic peptide and carbohydrate amphiphiles, Chem. Sci., 2019, 10, 2385–2390 RSC.
- A. Levin, T. O. Mason, L. Adler-Abramovich, A. K. Buell, G. Meisl, C. Galvagnion, Y. Bram, S. A. Stratford, C. M. Dobson, T. P. J. Knowles and E. Gazit, Ostwalds rule of stages governs structural transitions and morphology of dipeptide supramolecular polymers, Nat. Commun., 2014, 5, 5219 CrossRef CAS PubMed.
- C. Yuan, A. Levin, W. Chen, R. Xing, Q. Zou, T. W. Herling, P. K. Challa, T. P. J. Knowles and X. Yan, Nucleation and Growth of Amino Acid and Peptide Supramolecular Polymers through Liquid–Liquid Phase Separation, Angew. Chem., Int. Ed., 2019, 58, 18116–18123 CrossRef CAS PubMed.
- M. C. Mañas-Torres, C. Gila-Vilchez, J. A. González-Vera, F. Conejero-Lara, V. Blanco, J. M. Cuerva, M. T. Lopez-Lopez, A. Orte and L. Álvarez de Cienfuegos, In situ real-time monitoring of the mechanism of self-assembly of short peptide supramolecular polymers, Mater. Chem. Front., 2021, 5, 5452–5462 RSC.
- F. Fueyo-González, J. A. González-Vera, I. Alkorta, L. Infantes, M. L. Jimeno, P. Aranda, D. Acuña-Castroviejo, A. Ruiz-Arias, A. Orte and R. Herranz, Environment-Sensitive Probes for Illuminating Amyloid Aggregation in Vitro and in Zebrafish, ACS Sens., 2020, 5, 2792–2799 CrossRef.
- L. Chen, K. Morris, A. Laybourn, D. Elias, M. R. Hicks, A. Rodger, L. Serpell and D. J. Adams, Self-assembly mechanism for a naphthalene-dipeptide leading to hydrogelation, Langmuir, 2010, 26, 5232–5242 CrossRef CAS PubMed.
- Z. Yang, G. Liang, M. Ma, Y. Gao and B. Xu, Conjugates of naphthalene and dipeptides produce molecular hydrogelators with high efficiency of hydrogelation and superhelical nanofibers, J. Mater. Chem., 2007, 17, 850–854 RSC.
- R. Contreras-Montoya, A. B. Bonhome-Espinosa, A. Orte, D. Miguel, J. M. Delgado-López, J. D. G. Duran, J. M. Cuerva, M. T. Lopez-Lopez and L. Álvarez de Cienfuegos, Iron nanoparticles-based supramolecular hydrogels to originate anisotropic hybrid materials with enhanced mechanical strength, Mater. Chem. Front., 2018, 2, 686–699 RSC.
- P. L. Privalov and V. V. Plotnikov, Three generations of scanning microcalorimeters for liquids, Thermochim. Acta, 1989, 139, 257–277 CrossRef CAS.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Fiji: An open-source platform for biological-image analysis, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- S. Rüttinger, P. Kapusta, M. Patting, M. Wahl and R. Macdonald, On the resolution capabilities and limits of fluorescence lifetime correlation spectroscopy (FLCS) measurements., J. Fluoresc., 2010, 20, 105–114 CrossRef.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, A new method for maintaining homogeneity during liquid-hydrogel transitions using low molecular weight hydrogelators, Soft Matter, 2009, 5, 1856–1862 RSC.
- D. J. Adams, L. M. Mullen, M. Berta, L. Chen and W. J. Frith, Relationship between molecular structure, gelation behaviour and gel properties of Fmoc-dipeptides, Soft Matter, 2010, 6, 1971–1980 RSC.
- L. Chen, G. Pont, K. Morris, G. Lotze, A. Squires, L. C. Serpell and D. J. Adams, Salt-induced hydrogelation of functionalised-dipeptides at high pH, Chem. Commun., 2011, 47, 12071–12073 RSC.
- L. Chen, T. O. McDonald and D. J. Adams, Salt-induced hydrogels from functionalised-dipeptides, RSC Adv., 2013, 3, 8714–8720 RSC.
- A. Z. Cardoso, L. L. E. Mears, B. N. Cattoz, P. C. Griffiths, R. Schweins and D. J. Adams, Linking micellar structures to hydrogelation for salt-triggered dipeptide gelators, Soft Matter, 2016, 12, 3612–3621 RSC.
- K. McAulay, P. A. Ucha, H. Wang, A. M. Fuentes-Caparrós, L. Thomson, O. Maklad, N. Khunti, N. Cowieson, M. Wallace, H. Cui, R. J. Poole, A. Seddon and D. J. Adams, Controlling the properties of the micellar and gel phase by varying the counterion in functionalised-dipeptide systems, Chem. Commun., 2020, 56, 4094–4097 RSC.
- W. Ji, C. Yuan, S. Zilberzwige-Tal, R. Xing, P. Chakraborty, K. Tao, S. Gilead, X. Yan and E. Gazit, Metal-Ion Modulated Structural Transformation of Amyloid-Like Dipeptide Supramolecular Self-Assembly, ACS Nano, 2019, 13, 7309 Search PubMed.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Fmoc-Diphenylalanine Self-assembly Mechanism Induces Apparent pK a Shifts, Langmuir, 2009, 25, 9447–9453 CrossRef CAS PubMed.
- S. Fleming, P. W. J. M. Frederix, I. Ramos Sasselli, N. T. Hunt, R. V. Ulijn and T. Tuttle, Assessing the utility of infrared spectroscopy as a structural diagnostic tool for β-sheets in self-assembling aromatic peptide amphiphiles, Langmuir, 2013, 29, 9510–9515 CrossRef CAS PubMed.
- P. G. Argudo, R. Contreras-Montoya, L. Álvarez de Cienfuegos, J. M. Cuerva, M. Cano, D. Alba-Molina, M. T. Martín-Romero, L. Camacho, J. J. Giner-Casares, L. Á. de Cienfuegos, J. M. Cuerva, M. Cano, D. Alba-Molina, M. T. Martín-Romero, L. Camacho and J. J. Giner-Casares, Unravelling the 2D self-assembly of Fmoc-dipeptides at fluid interfaces, Soft Matter, 2018, 14, 9343–9350 RSC.
- Y. Zhang, H. Gu, Z. Yang and B. Xu, Supramolecular Hydrogels Respond to Ligand-Receptor Interaction, J. Am. Chem. Soc., 2003, 125, 13680–13681 CrossRef CAS PubMed.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Nanostructured hydrogels for three-dimensional cell culture through self-assembly of fluorenylmethoxycarbonyl-dipeptides, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Fmoc-diphenylalanine self assembles to a hydrogel via a novel architecture based on π-π interlocked β-sheets, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- X. Mu, K. M. Eckes, M. M. Nguyen, L. J. Suggs and P. Ren, Experimental and computational studies reveal an alternative supramolecular structure for Fmoc-dipeptide self-assembly, Biomacromolecules, 2012, 13, 3562–3571 CrossRef CAS PubMed.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Fmoc-diphenylalanine self assembles to a hydrogel via a novel architecture based on π-π interlocked β-sheets, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- F. García and L. Sánchez, Structural rules for the chiral supramolecular organization of OPE-based discotics: Induction of helicity and amplification of chirality, J. Am. Chem. Soc., 2012, 134, 734–742 CrossRef PubMed.
- S. Arias, R. Rodríguez, E. Quiñoá, R. Riguera and F. Freire, Chiral Coalition in Helical Sense Enhancement of Copolymers: The Role of the Absolute Configuration of Comonomers, J. Am. Chem. Soc., 2018, 140, 667–674 CrossRef CAS PubMed.
- K. McAulay, B. Dietrich, H. Su, M. T. Scott, S. Rogers, Y. K. Al-Hilaly, H. Cui, L. C. Serpell, A. M. Seddon, E. R. Draper and D. J. Adams, Using chirality to influence supramolecular gelation, Chem. Sci., 2019, 10, 7801–7806 RSC.
- H. Weissman and B. Rybtchinski, Noncovalent self-assembly in aqueous medium: Mechanistic insights from time-resolved cryogenic electron microscopy, Curr. Opin. Colloid Interface Sci., 2012, 17, 330–342 CrossRef CAS.
- E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Supramolecular polymers in aqueous media, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS PubMed.
- M. F. J. Mabesoone, A. R. A. Palmans and E. W. Meijer, Solute-Solvent Interactions in Modern Physical Organic Chemistry: Supramolecular Polymers as a Muse, J. Am. Chem. Soc., 2020, 142, 19781–19798 CrossRef CAS PubMed.
- H. Zhang, L. Xie, W. Yan, C. He, X. Cao and C. Duan, Conformational switching fluorescent chemodosimeter for the selective detection of silver(I) ions, New J. Chem., 2009, 33, 1478–1481 RSC.
- P. K. Lekha, T. Ghosh and E. Prasad, Utilizing dendritic scaffold for feasible formation of naphthalene excimer, J. Chem. Sci., 2011, 123, 919–926 CrossRef CAS.
- L. Chen, S. Revel, K. Morris and D. J. Adams, Energy transfer in self-assembled dipeptide hydrogels, Chem. Commun., 2010, 46, 4267–4269 RSC.
- V. V. Andrushchenko, H. J. Vogel and E. J. Prenner, Solvent-dependent structure of two tryptophan-rich antimicrobial peptides and their analogs studied by FTIR and CD spectroscopy, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1596–1608 CrossRef CAS PubMed.
- A. Borzacchiello, F. Della Sala and L. A. Ambrosio, Rheometry of polymeric biomaterials, Woodhead Publishing, 2017, pp. 233–253 Search PubMed.
- R. Laishram and U. Maitra, Supramolecular Gelation of Europium and Calcium Cholates through the Nucleation-Elongation Growth Mechanism, ChemPlusChem, 2019, 84, 853–861 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Protocol of the synthesis of Nap-derivatives and characterization, molecular dynamics calculations and supplementary figures. See DOI: 10.1039/d1py01366a |
| This journal is © The Royal Society of Chemistry 2021 |