
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePost-polymerization ‘click’ end-capping of polyglyoxylate self-immolative polymers†
Peter G.
Maschmeyer
 a,
Xiaoli
Liang
b,
Allison
Hung
b,
Oksana
Ahmadzai
a,
Annmaree L.
Kenny
a,
Yuan C.
Luong
a,
Timothy N.
Forder
a,
Haoxiang
Zeng
a,
Elizabeth R.
Gillies
*bc and
Derrick A.
Roberts
*ad
a,
Xiaoli
Liang
b,
Allison
Hung
b,
Oksana
Ahmadzai
a,
Annmaree L.
Kenny
a,
Yuan C.
Luong
a,
Timothy N.
Forder
a,
Haoxiang
Zeng
a,
Elizabeth R.
Gillies
*bc and
Derrick A.
Roberts
*ad
aKey Centre for Polymers and Colloids, School of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia. E-mail: derrick.roberts@sydney.edu.au
bDepartment of Chemistry and the Centre for Advanced Materials and Biomaterials Research. The University of Western Ontario, 1151 Richmond St., London, Canada N6A 5B7. E-mail: egillie@uwo.ca
cDepartment of Chemical and Biochemical Engineering, The University of Western Ontario, 1151 Richmond St., London, Ontario, Canada N6A 5B9
dSydney Nano Institute, The University of Sydney, Sydney, NSW 2006, Australia
First published on 26th October 2021
Abstract
Post-polymerization modification is a powerful tool for expanding the functionality and tuning the properties of self-immolative polymers (SIPs), typically through reactions of the polymer repeat units. Herein, we investigate the use of post-polymerization ‘click’ reactions to attach stimuli-responsive end-caps to poly(ethyl glyoxylate) (PEtG) SIPs. Two classes of alkyne-terminated polymers were synthesized, with the alkyne attached to the PEtG either by an acetal or carbonate bridge. These PEtGs were treated with azide-functionalized self-immolative linker precursors via the copper(I) catalyzed azide–alkyne cycloaddition reaction to afford PEtGs end-capped with cleavable self-immolative triazole (SIT) linkers. Our results demonstrate that degradation of the PEtGs can be initiated by cleavage of the SIT end-caps, and that successful cleavage depends strongly on the moiety that connects the SIT linker to the PEtG backbone. Through depolymerization studies on the polymers and a series of small molecule model compounds, we examined in detail the mechanistic features of this system. Additionally, we demonstrate that this modular chemistry is versatile for introducing different stimuli-responsive end-caps to a single pre-synthesized PEtG.
Introduction
Linear self-immolative polymers (SIPs) are degradable macromolecules that undergo complete head-to-tail depolymerization following the cleavage of the backbone or a single stimuli-responsive end-cap (designated as the ‘trigger’ group).1 This contrasts with the behavior of most conventional degradable polymers, whereby the cleavage of one bond does not typically drive the cleavage of adjacent bonds (Scheme 1). SIPs naturally function as chemical amplifiers of weak triggering signals, with a single bond cleavage event releasing multiple degradation products.2 Additionally, a wide range of trigger end-caps have been reported to date, allowing SIPs to degrade in response to various environmental, chemical, and biological stimuli.3 These unique properties of SIPs have fueled interest for their use in controlled release systems,4–10 stimuli-responsive coatings,11,12 chemical sensors,13–15 and degradable bulk materials.16–18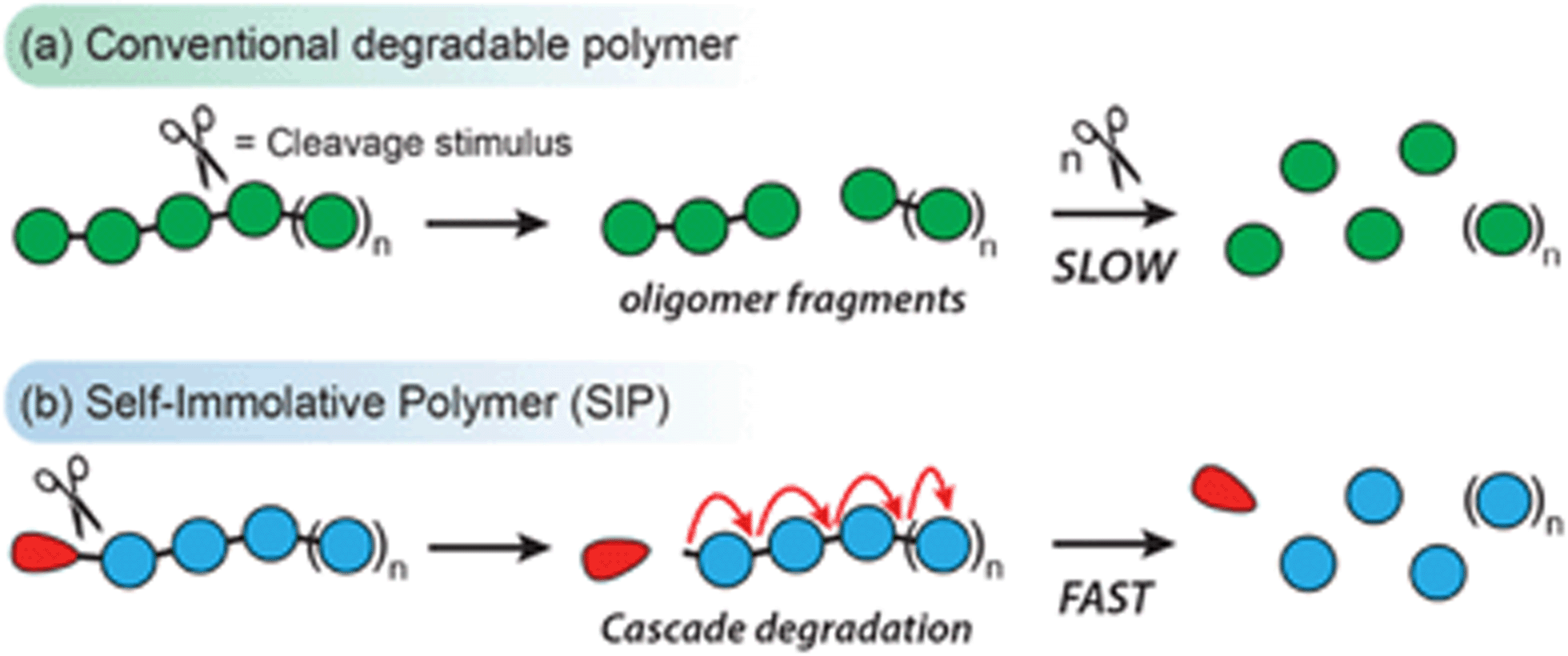 | ||
| Scheme 1 Comparison of (a) conventional degradable polymers, which tend to undergo stepwise fragmentation through random bond scission events; and (b) a linear end-capped SIP, which undergoes a head-to-tail depolymerization cascade upon removal of a single ‘trigger’ end-cap (red circle). | ||
Since their debut in 2008,19 a handful of SIP designs have been reported with degradation mechanisms based on 1,4- and 1,6-elimination reactions of aromatic compounds,20–22 heterocyclization reactions,23,24 olefin sulfone elimination reactions,25–27 and the depolymerization of polyacetals.28,29 While each of these designs have reliable polymerization methods, functional group intolerances preclude the direct polymerization of certain monomers. To circumvent this issue, post-polymerization modification (PPM) has become an important tool for introducing functional groups to SIPs that would otherwise interfere with their polymerization reactions. For example, Shabat and co-workers have shown that protecting groups can mask nucleophilic functionalities en route to water soluble poly(benzyl urethanes).30 Zhang and co-workers have used post-synthetic disulfide metathesis31 and ‘click’-type reactions32 to prepare SIP bottlebrushes and organogels. Gillies and co-workers have developed PPM strategies for preparing a range of functionalized polyglyoxylate and polyglyoxylamide SIPs via transesterification33 and amidation reactions,34 which significantly expand the functional scope of these polymers. Furthermore, PPM has been used to prepare block copolymers from both linear and branched SIPs.28,35–37
In addition to avoiding functional group incompatibilities, PPM also enables rapid derivatization of a single parent macromolecule, which can be useful for tuning chemical functionality and physical properties.38–43 While strategies for modifying the repeat units of SIPs have been explored previously, there are no reported approaches for introducing triggerable end-caps to an existing SIP backbone via PPM. In addition to being a fundamentally interesting pursuit, such a capability could also provide a modular route for varying the stimuli-responsivity of a pre-formed SIP through late-stage introduction of different trigger end-caps.
Recently, Roberts and co-workers developed a method for preparing self-immolative triazole (SIT) linkers using the copper(I) catalyzed azide–alkyne cycloaddition (CuAAC) ‘click’ reaction,44 and have demonstrated its suitability for the post-synthetic modification of RAFT polymers.45 A key feature of this linker design is that the triazole group plays an active role in the self-immolation cascade and allows rate-tuning of the degradation kinetics through substituent effects. These features led us to hypothesize that a SIT linker, if attached to the end of a SIP, could offer a convenient way to introduce different trigger end-caps through PPM. To test this hypothesis, we investigated the post-synthetic ‘click’ activation of alkyne-terminated poly(ethyl glyoxylate)s (PEtGs) using two exemplar azide capping groups, and studied their degradation behavior by in situ1H NMR spectroscopy (Scheme 2). Through this investigation, we show that a SIT cap can be successfully ‘clicked’ onto the terminus of a PEtG without causing incipient depolymerization. Additionally, self-immolation can successfully proceed via the degradation of the SIT linker for certain end-cap structures. Furthermore, we explore the interesting finding that the rate of SIT end-cap self-immolation is not sensitive to substituent effects when attached to the terminus of PEtG, contrary to the behavior of previously reported SIT linker systems.
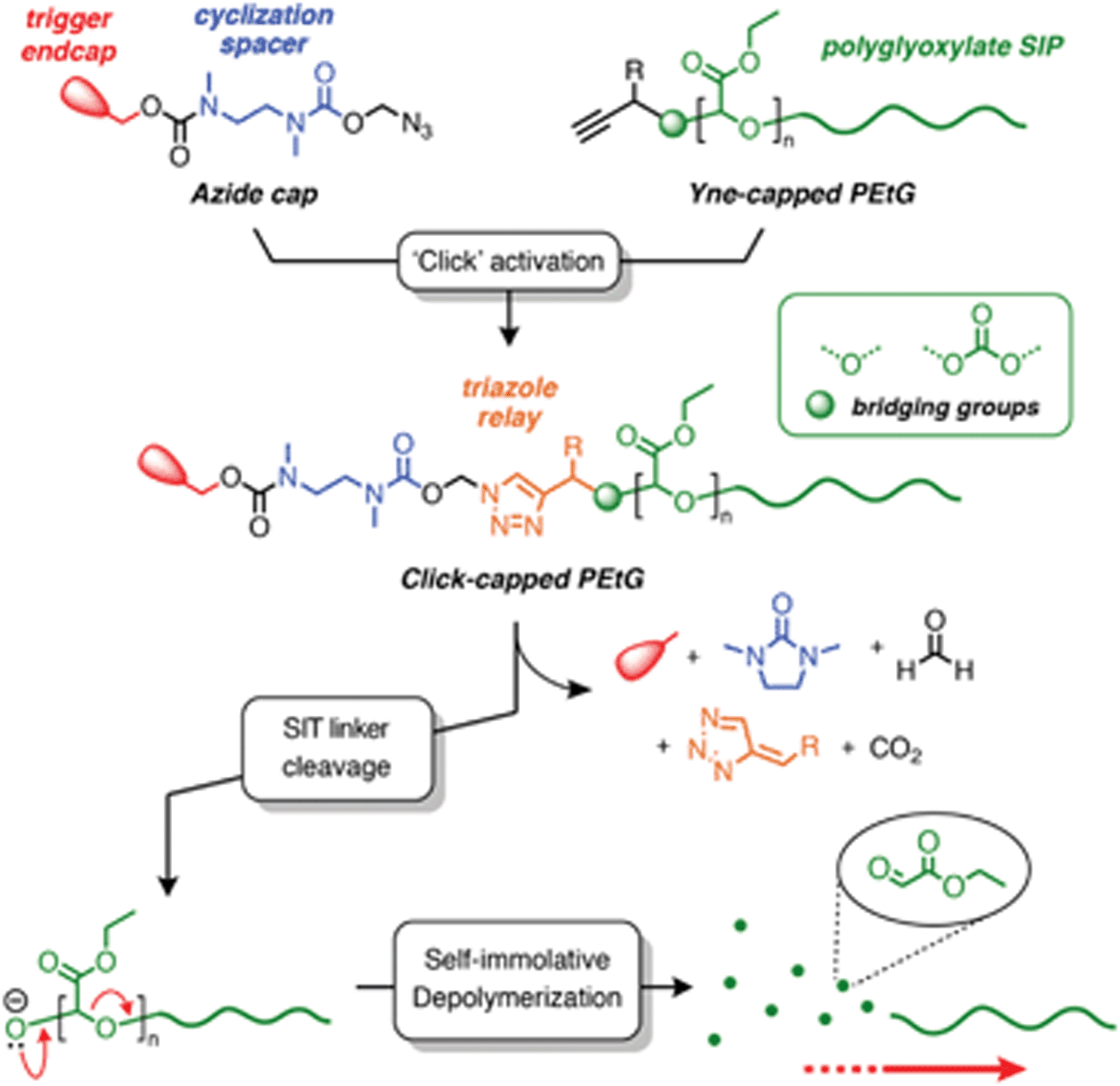 | ||
| Scheme 2 Overview scheme summarizing our approach for the post-synthetic ‘click’ activation of PEtG SIPs with self-immolative triazole (SIT) linkers. Structure–property relationships are explored by varying the bridging group (acetal or carbonate) and triazole Cα-substituent (R) then examining the effect these changes have on the kinetics of SIT linker degradation and overall depolymerization. | ||
Results and discussion
Polymer synthesis and ‘click’ activation
Alkyne-terminated PEtGs P1a–b and P2a–b (Scheme 3a and Table 1) were prepared by the anionic polymerization of ethyl glyoxylate.46 To prepare P1a–b, the polymerization reactions were initiated with the corresponding alkoxides of propargyl alcohol or but-3-yn-2-ol after their deprotonation by n-butyllithium (n-BuLi), and the resulting PEtGs were end-capped with benzyloxymethyl chloride (Scheme 3a). P2a–b were synthesized by direct initiation of the polymerization with n-BuLi and end-capping with either propargyl chloroformate or the chloroformate prepared from 3-butyn-2-ol (Scheme 3b). All PEtGs had peaks corresponding to their expected termini in their 1H NMR spectra (ESI, section S3†). Their number average molar masses (Mn) ranged from 4–10 kg mol−1 based on size exclusion chromatography (SEC) and their dispersities (Đ) ranged from 1.2–1.3 (Table 1). As a result of their polymerization mechanisms, each set of polymers has a different bridging moiety linking the propargyl end-group to the polymer backbone; P1a–b are acetal-bridged, while P2a–b are carbonate-bridged. Additionally, the use of propargyl versus but-3-yn-2-ol allows us to investigate how substitution at the carbon atom adjacent to the triazole 4-position (henceforth referred to as the Cα-position) influences the rate of depolymerization, since this has been shown to influence 1,4-elimination rates in related SIT linkers.44,47 Syntheses of alkyne-capped PEtGs with geminal methyl groups at the propargylic position were attempted, but failed to give the desired polymers, most likely due to a combination of steric factors and possibly decomposition of 2-methylbut-3-yn-2-ol under basic conditions.48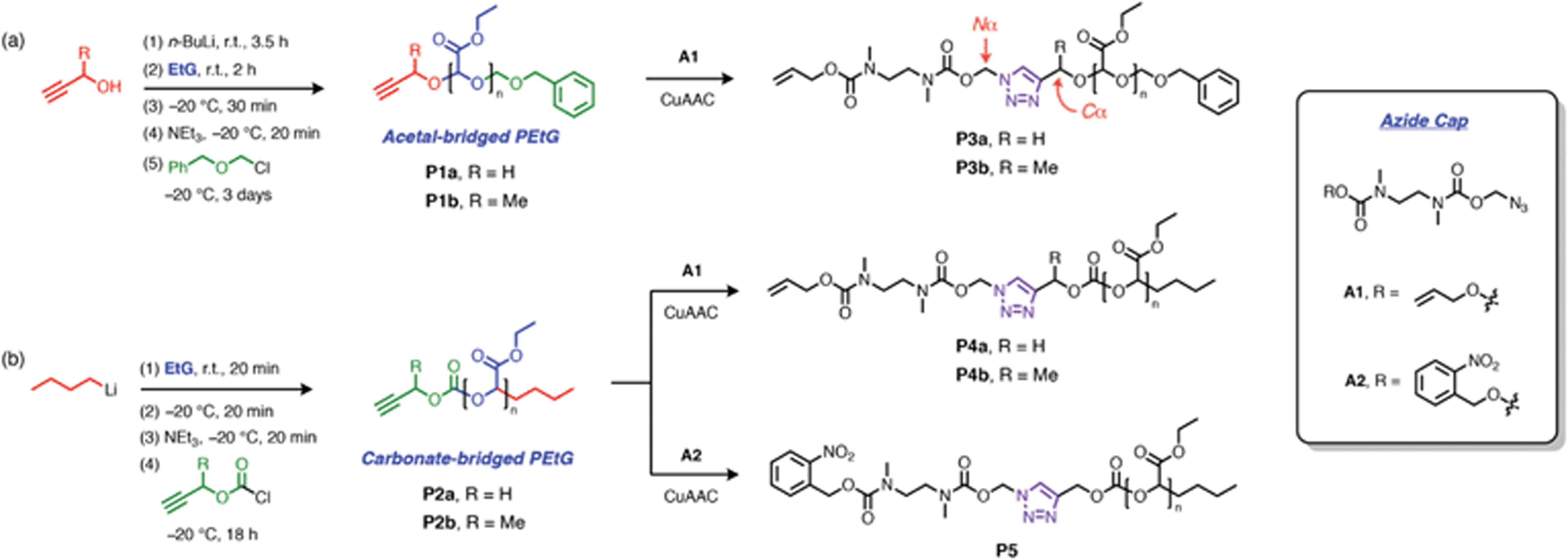 | ||
| Scheme 3 Synthesis and post-synthetic capping of alkyne-terminated PEtGs. (a) Synthesis of acetal-bridged PEtG derivatives. The Nα and Cα positions adjacent to the triazole ring are annotated on P3a–b. (b) Synthesis of carbonate-bridged PEtG derivatives. CuAAC Conditions: CuSO4·5H2O (0.3 equiv.), sodium ascorbate (0.9 equiv.), DMF, 50 °C, 16–24 h. | ||
| Entry | M n,NMR | DPNMR | M n,SEC | Đ |
|---|---|---|---|---|
| P1a | 5.4 | 51 | 4.0 | 1.32 |
| P1b | 11.1 | 107 | 10.3 | 1.17 |
| P2a | 4.3 | 41 | 5.3 | 1.31 |
| P2b | 3.8 | 36 | 4.7 | 1.27 |
| P3a | 5.1 | 46 | 3.9 | 1.37 |
| P3b | 12.3 | 116 | 10.4 | 1.12 |
| P4a | 4.2 | 37 | 5.7 | 1.27 |
| P4b | 3.0 | 25 | 4.0 | 1.36 |
| P5 | 5.9 | 53 | 6.9 | 1.21 |
Post-synthetic ‘click’ activation of PEtGs P1a–b and P2a–b was initially investigated using allyl-protected cap A1, which has served as a reliable and conveniently studied model cap in previous work.44,45 Alkyne-terminated PEtGs were treated with azide A1 (1.5–2.0 equiv.) in DMF at 50 °C under CuAAC conditions using a copper(II) sulfate and sodium ascorbate catalyst system. Reaction mixtures were heated for 16–24 h, then dissolved in EtOAc and washed with aqueous Na2EDTA to remove copper salts. Excess A1 was removed by dissolving the polymers in CH2Cl2 followed by dropwise addition of Et2O, resulting in precipitation of the polymers. Upon vacuum drying, alloc-capped PEtGs P3a–b and P4a–b were obtained as tacky solids. Successful introduction of the SIT caps was confirmed by 1H NMR spectroscopic analyses, which showed peaks characteristic of the attached SIT linker in all cases (ESI, section S4†). DOSY NMR spectroscopy also showed that the end-group and polymer backbone resonances shared similar diffusion coefficients, further supporting successful post-synthetic capping of the PEtGs. SEC traces of polymers P3a–b and P4a–b did not change appreciably following CuAAC modification and purification (Table 1).
Kinetics I: acetal-bridged PEtGs
Palladium(0)-triggered degradation of P3a–b was studied by 1H NMR spectroscopy in DMSO-d6/D2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 65 °C in the presence of morpholine (10 equiv.) to regenerate the catalyst and assist with base-mediated fragmentation of the SIT linker. This solvent mixture was chosen to demonstrate the compatibility of the linker with the presence of water; however, solubility limitations precluded higher aqueous fractions. A control experiment showed that the polymers were stable towards morpholine in the absence of catalyst (ESI, section S9.2†). Self-immolation was initiated by addition of Pd(PPh3)4 (0.2 equiv. per trigger) to an NMR tube containing the polymer solution. Broad 1H resonances of the PEtG obscured peaks of the allyl end-cap and Cα-environment, preventing direct observation of trigger cleavage and 1,4-elimination for both polymers. However, the progress of subsequent diamine cyclization could be tracked by monitoring loss of the Nα-methylene group adjacent to the triazole ring and the concomitant appearance of the cyclization product 1,3-dimethyl-2-imidazolidinone (DMI) (Scheme 4b). For both P3a and P3b, complete cyclization of the spacer occurred within 25 min (Scheme 4b). However, in both cases the integral of the PEtG backbone did not decrease appreciably even after 3 h at 65 °C, and peaks corresponding to the hydrated PEtG degradation product, ethyl glyoxylate hydrate (EtGH), were not observed. These results suggest that degradation of the SIT linker was interrupted, presumably due to unsuccessful 1,4-elimination of the triazole moiety, thereby preventing degradation of the polymer backbone.
:1) at 65 °C in the presence of morpholine (10 equiv.) to regenerate the catalyst and assist with base-mediated fragmentation of the SIT linker. This solvent mixture was chosen to demonstrate the compatibility of the linker with the presence of water; however, solubility limitations precluded higher aqueous fractions. A control experiment showed that the polymers were stable towards morpholine in the absence of catalyst (ESI, section S9.2†). Self-immolation was initiated by addition of Pd(PPh3)4 (0.2 equiv. per trigger) to an NMR tube containing the polymer solution. Broad 1H resonances of the PEtG obscured peaks of the allyl end-cap and Cα-environment, preventing direct observation of trigger cleavage and 1,4-elimination for both polymers. However, the progress of subsequent diamine cyclization could be tracked by monitoring loss of the Nα-methylene group adjacent to the triazole ring and the concomitant appearance of the cyclization product 1,3-dimethyl-2-imidazolidinone (DMI) (Scheme 4b). For both P3a and P3b, complete cyclization of the spacer occurred within 25 min (Scheme 4b). However, in both cases the integral of the PEtG backbone did not decrease appreciably even after 3 h at 65 °C, and peaks corresponding to the hydrated PEtG degradation product, ethyl glyoxylate hydrate (EtGH), were not observed. These results suggest that degradation of the SIT linker was interrupted, presumably due to unsuccessful 1,4-elimination of the triazole moiety, thereby preventing degradation of the polymer backbone.
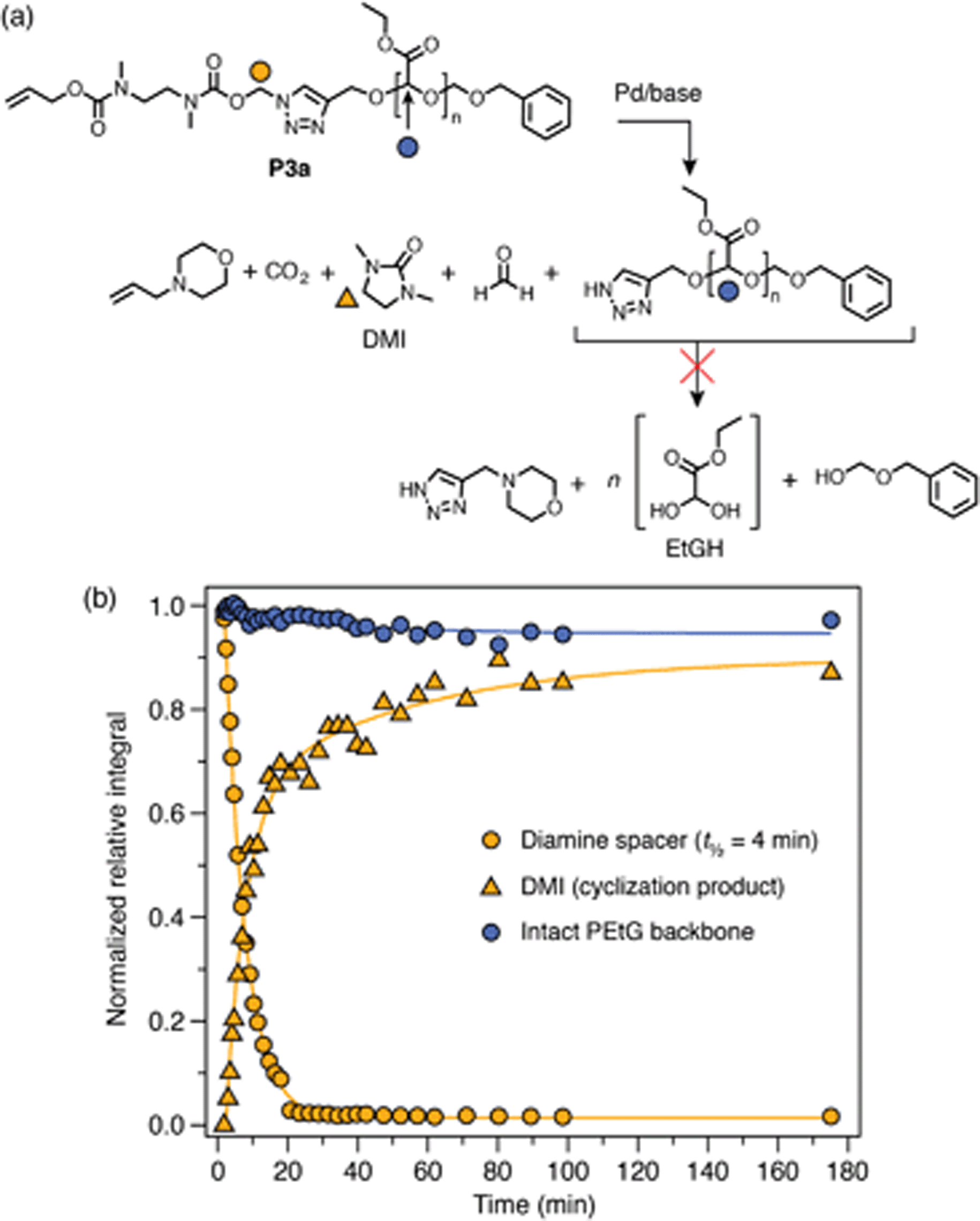 | ||
| Scheme 4 Representative degradation profile of polymer P3a. Full data included in the ESI (section S9.2†). (a) Scheme depicting the proposed degradation pathway. Markers denote 1H environments monitored for kinetics. (b) Degradation plot, measured by 1H NMR (∼10 mmol in DMSO-d6/D2O 9:1, 65 °C; 10 equiv. morpholine) showing complete cyclization of the diamine spacer, but no appreciable decrease in the integral of peaks representing the PEtG backbone. Diamine cyclization was modelled as a first-order process to estimate its half-life. | ||
We prepared acetal-bridged model compounds 1a–c (Scheme 5a) to better understand why polymers P3a–b did not undergo complete self-immolation. These model compounds have much simpler NMR spectra than the corresponding polymers, which enabled more detailed analysis of their degradation profiles. Additionally, geminal dimethyl-substituted 1c was successfully synthesized, providing an additional congener for studying the potential influence of Cα-substitution on the self-immolation kinetics.
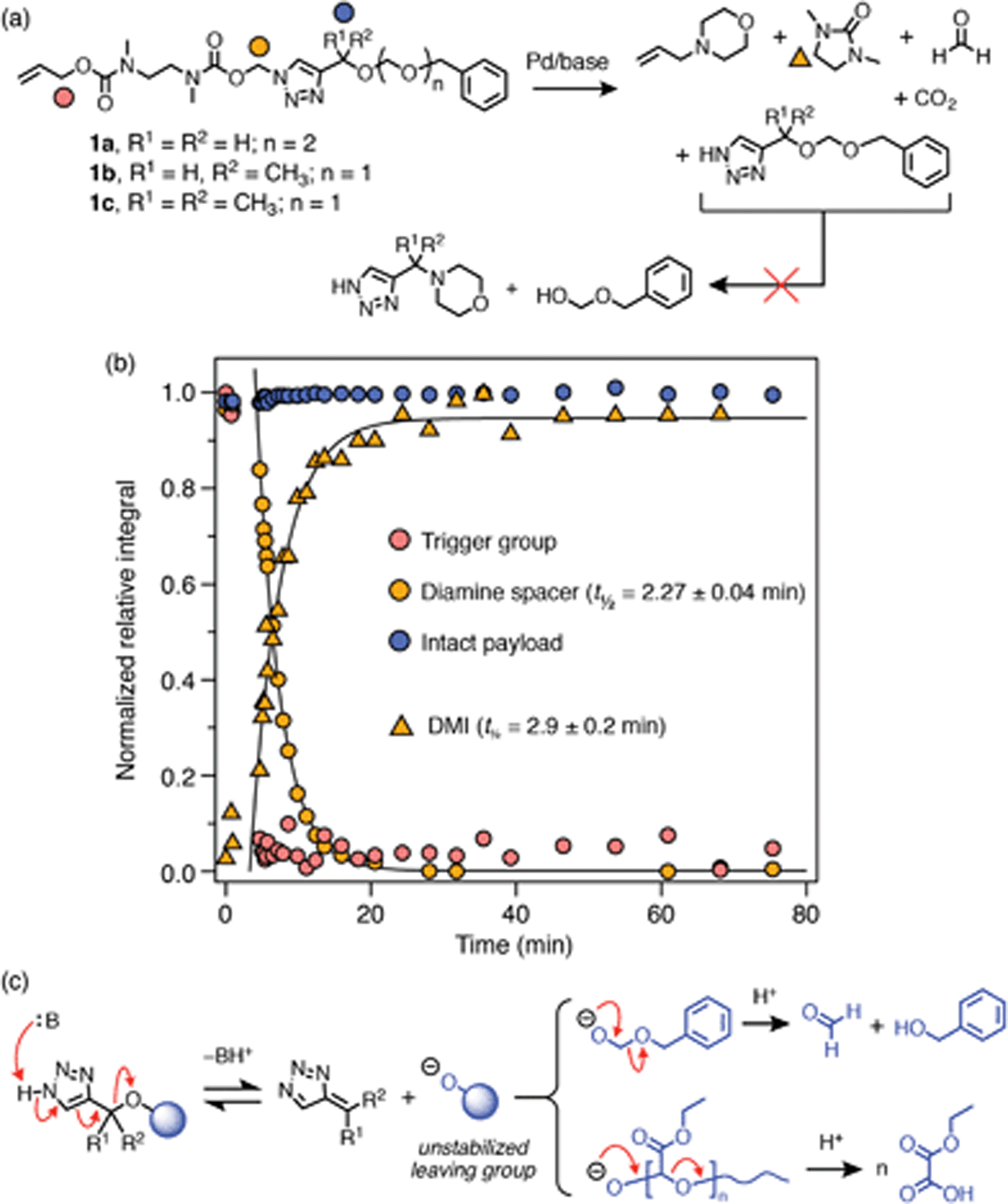 | ||
| Scheme 5 Self-immolation of acetal-bridged model compounds. (a) Degradation reaction scheme for 1a–c. Markers denote 1H environments monitored for kinetics. (b) Representative degradation profile of 1c, measured by 1H NMR (25 mM in DMSO-d6/D2O 8:2, 65 °C; 50 equiv. morpholine). Half-lives were estimated by approximating the degradation profiles as first-order processes. (c) Scheme illustrating the putative 1,4-elimination products of acetal-bridged SIT derivatives, which would result in the formation of an unstable deprotonated hemiacetal anion before further stepwise fragmentation. | ||
Self-immolation reactions of 1a–c were performed under slightly more forcing conditions than P3a–b (50 equiv. morpholine in DMSO-d6/D2O 8:2, 65 °C) to ascertain if 1,4-elimination was possible with the acetal-bridged linker design. Following Pd(0) addition, trigger cleavage and diamine cyclization were complete within 25 min for all three compounds (Scheme 5b; ESI, section S9.2†). However, in all cases 1,4-elimination of the triazole moiety was not observed, which agrees with the behavior of P3a–b. Failure of the triazole groups to undergo 1,4-elimination, even under ostensibly more favorable conditions, suggests that the acetal bridge has a high barrier to elimination irrespective of the degree of substitution at the Cα-position. We attribute this barrier to poor leaving group ability of the deprotonated hemiacetal elimination product that would result from scission of the triazolylmethoxy C–O bond (Scheme 5c). For the PEtGs, the subsequent depolymerization cascade was expected to provide a thermodynamic driving force that would encourage 1,4-elimination across the triazole ring. However, this hypothesis was not supported by our observations.
Kinetics II: carbonate-bridged PEtGs
Next, carbonate-bridged polymers P4a–b were investigated. These polymers were expected to be more amenable to degradation due to the enhanced leaving group ability of the carbonate, as well as the irreversible expulsion of CO2 upon 1,4-elimination. To test this hypothesis, P4a–b were subjected to the same degradation conditions as the previous polymers, undergoing rapid trigger cleavage upon addition of Pd(PPh3)4, followed by cyclization of the diamine spacer (Scheme 6b; ESI, section S9.3†). In both cases, signals of the PEtG backbone were also observed to decrease, indicating successful depolymerization that reached ∼80% degradation after 2 h. Interestingly, cyclization of the diamine spacer in P4a–b was significantly slower than for the acetal-bridged polymers and model compounds, which we attribute to a reversible side-reaction between the deprotected diamine spacer and ethyl glyoxylate to form a transient hemiaminal species (see ESI, section S10,† for auxiliary discussion). A catalyst-free control experiment for P4a revealed that the PEtG backbone undergoes a small degree of background degradation (Scheme 6c) via base hydrolysis of the carbonate bridge. However, background degradation is considerably slower than triggered self-immolation, indicating that they are distinct processes. Thus, successful degradation of P4a–b demonstrates that the carbonate bridge functions as a better leaving group for triazole 1,4-elimination than the acetal analogue.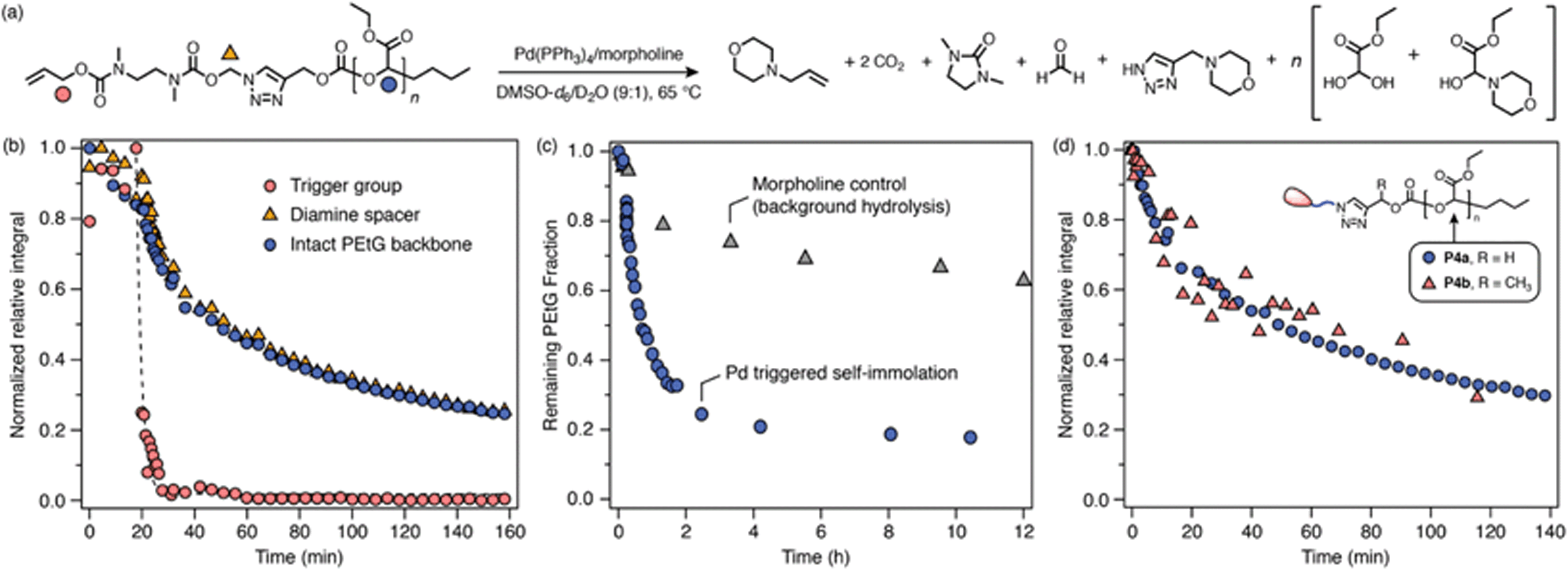 | ||
| Scheme 6 Representative degradation kinetics of P4a. Degradation profiles were measured by 1H NMR (∼10 mmol in DMSO-d6/D2O 9:1, 65 °C; 10 equiv. morpholine) (full data included in ESI, section S9.3†). (a) Proposed degradation pathway. Markers denote the proton environments monitored in (b). (b) Degradation profile showing rapid trigger cleavage, then simultaneous cyclization and depolymerization steps. (c) Comparison of background hydrolysis and depolymerization profiles of P4a over 10 h. The integral of the PEtG acetal CH environment was monitored in both cases. (d) Comparison of depolymerization profiles of P4a and P4b, showing that the depolymerization is not sensitive to the Cα-substituent. | ||
An interesting feature in the degradation profiles of P4a–b is the simultaneous rates of diamine cyclization and depolymerization (Scheme 6b; ESI Fig. S65†), which indicates that cyclization of the SIT linker is the rate-determining step of the overall cascade. Moreover, the normalized depolymerization profiles of P4a and P4b are nearly identical despite having different Cα-substituents (Scheme 6d), which also suggests that triazole 1,4-elimination is not rate-determining. This contrasts with the behavior of previously reported carbamate-bridged SIT linkers,44 for which 1,4-elimination is generally the slowest self-immolation step.
The high rate of 1,4-elimination relative to cyclization for P4a–b is consistent with carbonate being a better leaving group than a carbamate or a deprotonated hemiacetal. To verify this observation, we prepared model compounds 2a–b and examined their degradation profiles under the same conditions as P4a–b (DMSO-d6/D2O 9:1, 65 °C, 10 equiv. morpholine). Both compounds exhibited identical cyclization rates (Scheme 7) that agreed closely with cyclization rates for the acetal-bridged compounds (t½ ∼ 3 min). The rates of 1,4-elimination were either concomitant with (2b) or slightly slower (2a) than cyclization, demonstrating that the carbonate bridge is an excellent leaving group. Interestingly, the control experiment for 2a also revealed that the carbonate bridge was completely stable for at least 1 h both in DMSO-d6/D2O (9:1) with 10 equiv. morpholine, and DMSO-d6/D2O (8:2) with 50 equiv. morpholine (ESI, section S9.3†). This result suggests that background hydrolysis observed for P4a–b may be partly driven by the large increase in entropy upon depolymerization.
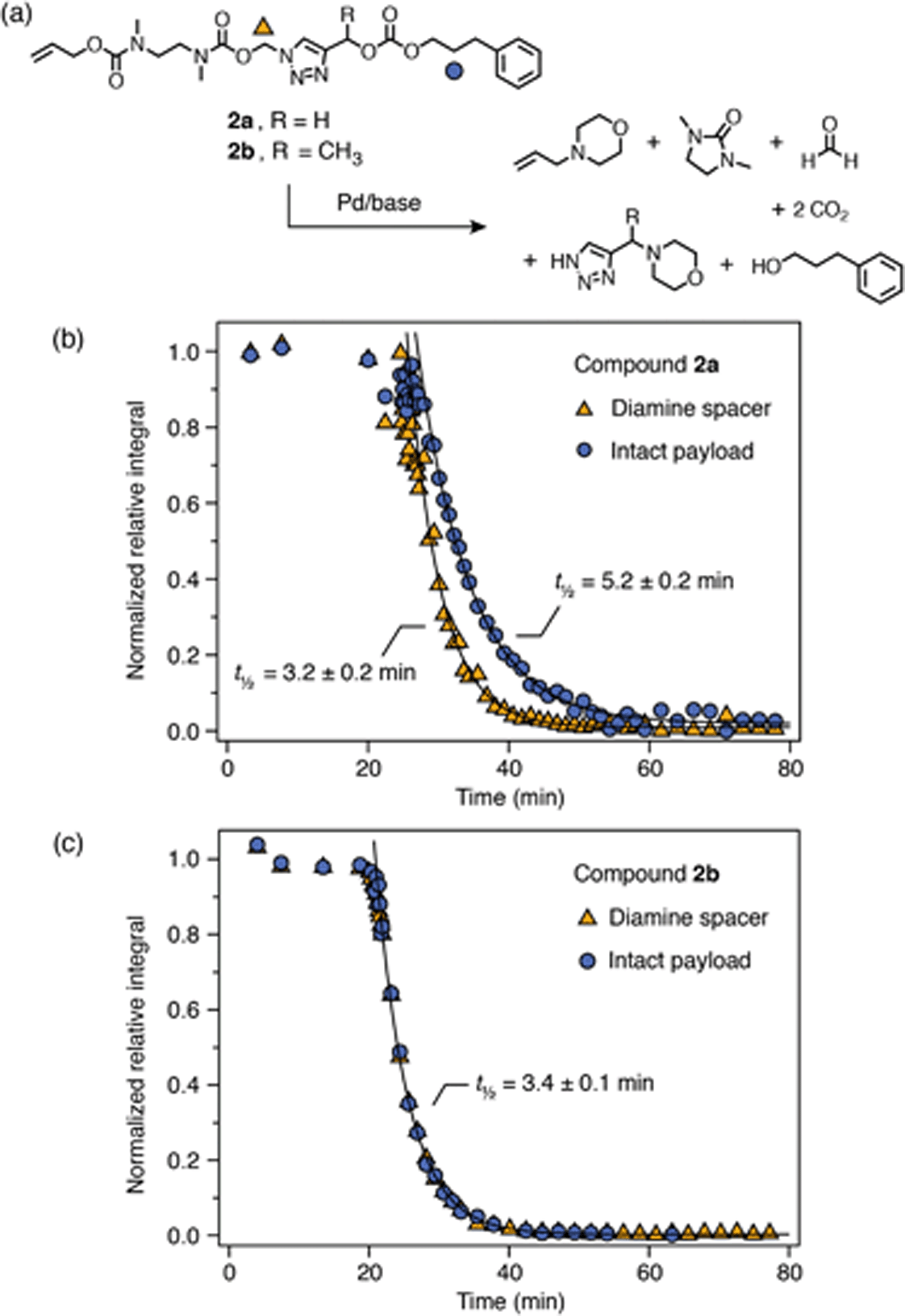 | ||
| Scheme 7 Self-immolation of carbonate-bridged model compounds measured by 1H NMR spectroscopy (25 mM in DMSO-d6/D2O 9:1, 65 °C; 10 equiv. morpholine). Half-lives were estimated by modelling the degradation profiles as first-order processes. (a) Proposed degradation reaction for 2a–b. Markers denote 1H environments monitored for kinetics (circles for 2a and diamonds for 2b). (b) Degradation profile of 2a. (c) Degradation profile of 2b. | ||
Kinetics III: light-cleavable SIT-cap
Kinetics results for the carbonate-bridged compounds demonstrated that their degradation is largely insensitive to substitution at the Cα-position. A fortunate consequence of this insensitivity is that the least hindered alkyne-PEtG precursor (P2a) can be used as a parent compound for other post-synthetic activation reactions. To demonstrate the versatility of this method, we synthesized novel azide linker A2, which carries an o-nitrobenzyl carbamate trigger group that can be cleaved by UV photoirradiation (Scheme 8). Treatment of P2a with A2 under CuAAC conditions afforded ‘click’-activated derivative P5. As for the other ‘click’-activated polymers, 1H NMR and DOSY spectroscopy confirmed successful attachment of the SIT linker and SEC analysis showed that there were no substantial changes in the molecular weight distribution of the polymer (ESI, section S8†).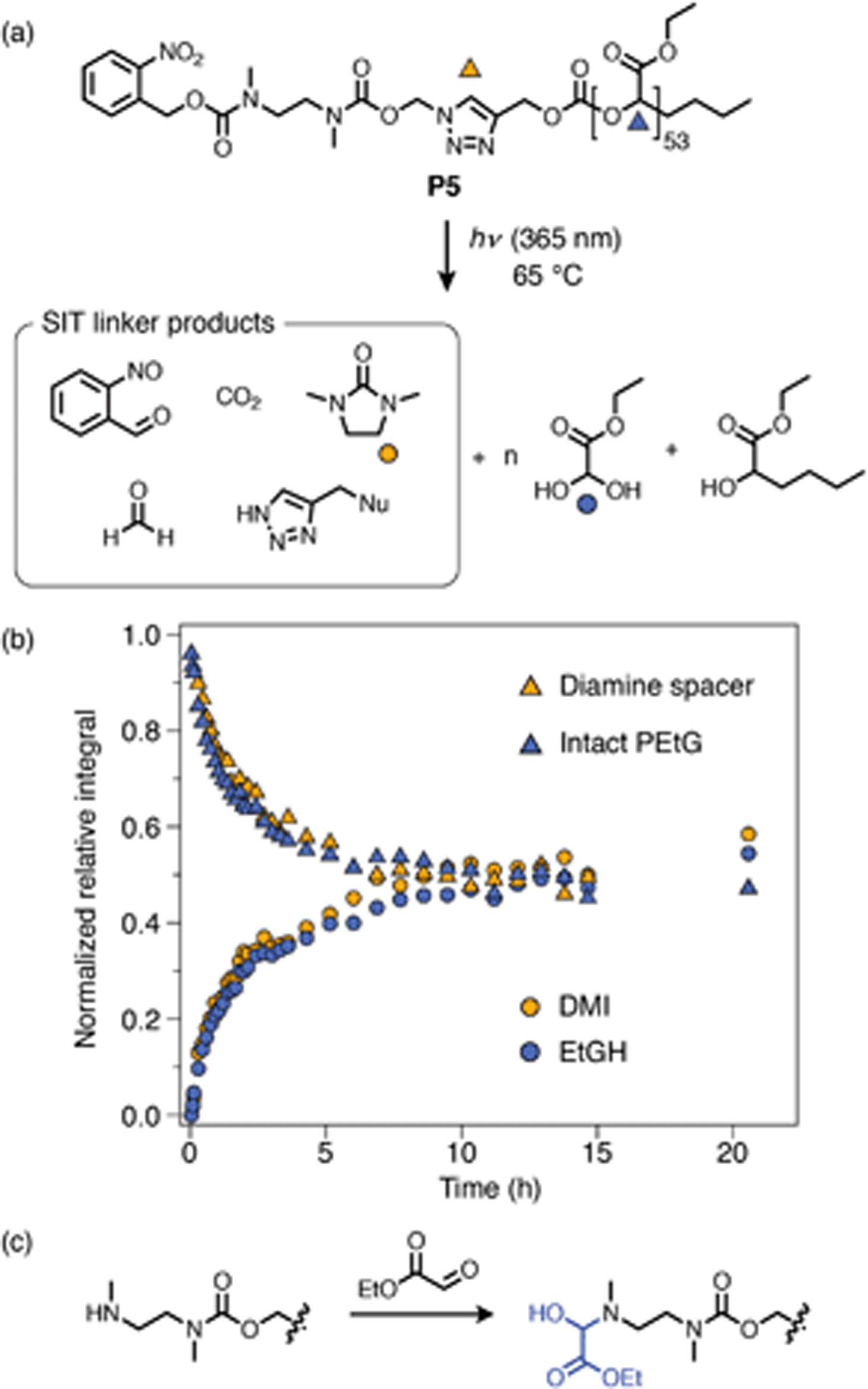 | ||
| Scheme 8 Degradation analysis of o-nitrobenzyl capped P5. (a) Proposed degradation pathway. Markers denote the proton environments monitored in (b). ‘Nu’ denotes a generic nucleophilic residue, most likely –OH under these conditions. (b) Degradation kinetics profile following irradiation at 365 nm, measured by 1H NMR analysis (∼10 mmol in DMSO-d6/D2O 9:1, 65 °C) (full data shown in ESI, section S9.4†). (c) Proposed amine passivation mechanism, whereby liberated EtGH reacts with the deprotected cyclization spacer to form a hemiaminal when no other sacrificial nucleophiles are present (e.g., morpholine, which was present for self-immolation experiments using P4a–b). | ||
Self-immolation of P5 was initiated by irradiating a solution of the polymer in DMSO-d6/D2O (9:1) at 365 nm for 30 min, followed by incubation at 65 °C. The o-nitrobenzyl group was completely cleaved within this 30 min window as confirmed by 1H NMR spectroscopy (ESI, Fig. S73†). Subsequent in situ1H NMR monitoring showed successful cyclization of the diamine linker and depolymerization of the PEtG backbone, with cyclization being the rate-determining step, as was observed for P4a–b. A negative control experiment, whereby P5 was incubated at 65 °C in DMSO-d6/D2O (9:1) without irradiation, showed no degradation over at least 1 h (ESI, Fig. S72†), confirming that degradation of the PEtG backbone was initiated by UV-induced cleavage of the SIT end-group. Degradation of P5 proceeded much more slowly than P4a–b, reaching a limiting degree of depolymerization of 50–60% after ∼10 h. The slower rate and incomplete degradation of P5 is consistent with putative hemiaminal formation, which is irreversible in the absence of a competing base such as morpholine (see ESI section S10.2† for auxiliary discussion). Nonetheless, degradation of P5 demonstrates successful introduction of a cleavable o-nitrobenzyl cap via PSM and demonstrates that the SIT linker can undergo self-immolation without additional base, which has not previously been shown with this linker design.
Conclusions
In conclusion, we have demonstrated a new method for the post-polymerization attachment of SIT end-groups to PEtG SIPs using the CuAAC reaction. Our approach involves the reaction of alkyne-terminated PEtGs with a dual-protected diamine precursor (A) carrying a cleavable carbamate ‘trigger’ group and an azidomethyl carbamate ‘click’ handle. Degradation profiles of the SIT-capped PEtGs and related small molecule model compounds were analyzed by in situ NMR spectroscopy experiments. Our results demonstrate that the ability of the SIT linker to undergo complete degradation depended strongly on the moiety connecting the SIT linker to the PEtG backbone. When this bridging group was an acetal, the SIT linker could not undergo 1,4-elimination, preventing depolymerization of the PEtG. However, when the bridging group was a carbonate, 1,4-elimination across the triazole ring proceeded rapidly, leading to successful degradation of the PEtG. Interestingly, unlike in previous work, kinetic profiles of the carbonate-bridged compounds showed that degradation of the SIT linker was not strongly influenced by substitution at the triazole Cα-position, as the diamine cyclization was the rate limiting step in the reaction sequence. We anticipate that, instead of tuning the end-cap cleavage rate using the triazole Cα-position, rate tuning can likely still be achieved by tuning the rate-limiting cyclization step through the incorporation of different cyclization spacers. Overall, the approach described herein can be used to introduce a variety of stimuli-responsive trigger groups to pre-synthesized SIPs via post-polymerization ‘click’ modification and may offer a new route for introducing self-immolative triazole junctions into polymer architectures.Author contributions
PGM, XL, AH, OA, ALK, YCL, TNF and HZ performed synthesis, characterization, and data analysis. DAR performed self-immolation kinetics experiments. ERG and DAR designed and supervised the project. All authors assisted in preparation of the manuscript and ESI.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Australian Research Council (DE190100797), the Australian Academy of Science Regional Collaborations Programme, and the Natural Sciences and Engineering Research Council of Canada (Discovery Grant 2016-04636). The authors also acknowledge the facilities and the scientific and technical assistance of Sydney Analytical, a core research facility at the University of Sydney. We thank Dr Ian Luck (NMR) and Dr Nicholas Proschogo (MS) for technical assistance, and Dr Karen Hakobyan for maintenance of the analytical SEC systems.Notes and references
- R. E. Yardley, A. R. Kenaree and E. R. Gillies, Macromolecules, 2019, 52, 6342–6360 CrossRef CAS.
- M. E. Roth, O. Green, S. Gnaim and D. Shabat, Chem. Rev., 2015, 116, 1309–1352 CrossRef PubMed.
- G. I. Peterson, M. B. Larsen and A. J. Boydston, Macromolecules, 2012, 45, 7317–7328 CrossRef CAS.
- S. Tang, L. Tang, X. Lu, H. Liu and J. S. Moore, J. Am. Chem. Soc., 2018, 140, 94–97 CrossRef CAS PubMed.
- Z. Deng, Y. Qian, Y. Yu, G. Liu, J. Hu, G. Zhang and S. Liu, J. Am. Chem. Soc., 2016, 138, 10452–10466 CrossRef CAS PubMed.
- J. Tan, Z. Deng, G. Liu, J. Hu and S. Liu, Biomaterials, 2018, 178, 608–619 CrossRef CAS PubMed.
- X. Wang, C. Yao, G. Zhang and S. Liu, Nat. Commun., 2020, 11, 1524 CrossRef CAS PubMed.
- Y. Xiao, X. Tan, Z. Li and K. Zhang, J. Mater. Chem. B, 2020, 8, 6697–6709 RSC.
- M. T. Gambles, B. Fan, A. Borecki and E. R. Gillies, ACS Omega, 2018, 3, 5002–5011 CrossRef CAS PubMed.
- M. Gisbert-Garzaran, D. Lozano, M. Vallet-Regi and M. Manzano, RSC Adv., 2017, 7, 132–136 RSC.
- S. M. Heuchan, B. Fan, J. J. Kowalski, E. R. Gillies and H. A. L. Henry, J. Agric. Food Chem., 2019, 67, 12720–12729 CrossRef CAS PubMed.
- S. M. Heuchan, J. P. MacDonald, L. A. Bauman, B. Fan, H. A. L. Henry and E. R. Gillies, ACS Omega, 2018, 3, 18603–18612 CrossRef CAS PubMed.
- M. S. Baker and S. T. Phillips, Org. Biomol. Chem., 2012, 10, 3595–3599 RSC.
- S. Gnaim and D. Shabat, Acc. Chem. Res., 2019, 52, 2806–2817 CrossRef CAS PubMed.
- R. Weinstain, P. S. Baran and D. Shabat, Bioconjugate Chem., 2009, 20, 1783–1791 CrossRef CAS PubMed.
- K. Yeung, H. Kim, H. Mohapatra and S. T. Phillips, J. Am. Chem. Soc., 2015, 137, 5324–5327 CrossRef CAS PubMed.
- E. M. Lloyd, H. Lopez Hernandez, E. C. Feinberg, M. Yourdkhani, E. K. Zen, E. B. Mejia, N. R. Sottos, J. S. Moore and S. R. White, Chem. Mater., 2018, 31, 398–406 CrossRef.
- M. S. Baker, H. Kim, M. G. Olah, G. G. Lewis and S. T. Phillips, Green Chem., 2015, 17, 4541–4545 RSC.
- A. Sagi, R. Weinstain, N. Karton and D. Shabat, J. Am. Chem. Soc., 2008, 130, 5434–5435 CrossRef CAS PubMed.
- R. Erez and D. Shabat, Org. Biomol. Chem., 2008, 6, 2669–2672 RSC.
- S. Gnaim and D. Shabat, Acc. Chem. Res., 2014, 47, 2970–2984 CrossRef CAS PubMed.
- M. G. Olah, J. S. Robbins, M. S. Baker and S. T. Phillips, Macromolecules, 2013, 46, 5924–5928 CrossRef CAS.
- X.-W. Kan, L.-J. Zhang, Z.-Y. Li, F.-S. Du and Z.-C. Li, Macromol. Rapid Commun., 2021, 2100169 CrossRef CAS PubMed.
- E. K. Y. Chen, R. A. McBride and E. R. Gillies, Macromolecules, 2012, 45, 7364–7374 CrossRef CAS.
- K. Kumar and A. P. Goodwin, ACS Macro Lett., 2015, 4, 907–911 CrossRef CAS PubMed.
- J. M. Lobez and T. M. Swager, Angew. Chem., Int. Ed., 2010, 49, 95–98 CrossRef CAS PubMed.
- O. P. Lee, H. Lopez Hernandez and J. S. Moore, ACS Macro Lett., 2015, 4, 665–668 CrossRef CAS.
- B. Fan, J. F. Trant, A. D. Wong and E. R. Gillies, J. Am. Chem. Soc., 2014, 136, 10116–10123 CrossRef CAS PubMed.
- C. E. Diesendruck, G. I. Peterson, H. J. Kulik, J. A. Kaitz, B. D. Mar, P. A. May, S. R. White, T. J. Martínez, A. J. Boydston and J. S. Moore, Nat. Chem., 2014, 6, 623–628 CrossRef CAS PubMed.
- R. Weinstain, A. Sagi, N. Karton and D. Shabat, Chem. – Eur. J., 2008, 14, 6857–6861 CrossRef CAS PubMed.
- Y. Xiao, Y. Li, B. Zhang, H. Li, Z. Cheng, J. Shi, J. Xiong, Y. Bai and K. Zhang, ACS Macro Lett., 2019, 8, 399–402 CrossRef CAS.
- Y. Xiao, H. Li, B. Zhang, Z. Cheng, Y. Li, X. Tan and K. Zhang, Macromolecules, 2018, 51, 2899–2905 CrossRef CAS PubMed.
- R. E. Yardley, A. Rabiee Kenaree, X. Liang and E. R. Gillies, Macromolecules, 2020, 53, 8600–8609 CrossRef CAS.
- Q. E. A. Sirianni, A. Rabiee Kenaree and E. R. Gillies, Macromolecules, 2018, 52, 262–270 CrossRef.
- B. Fan and E. R. Gillies, Mol. Pharm., 2017, 14, 2548–2559 CrossRef CAS PubMed.
- G. Liu, X. Wang, J. Hu, G. Zhang and S. Liu, J. Am. Chem. Soc., 2014, 136, 7492–7497 CrossRef CAS PubMed.
- G. Liu, G. Zhang, J. Hu, X. Wang, M. Zhu and S. Liu, J. Am. Chem. Soc., 2015, 137, 11645–11655 CrossRef CAS PubMed.
- R. M. Arnold, N. E. Huddleston and J. Locklin, J. Mater. Chem., 2012, 22, 19357–19365 RSC.
- K. A. Günay, P. Theato and H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1–28 CrossRef.
- Y. Zhong, B. J. Zeberl, X. Wang and J. Luo, Acta Biomater., 2018, 73, 21–37 CrossRef CAS PubMed.
- S. Tempelaar, L. Mespouille, O. Coulembier, P. Dubois and A. P. Dove, Chem. Soc. Rev., 2013, 42, 1312–1336 RSC.
- D. A. Roberts, T. W. Schmidt, M. J. Crossley and S. Perrier, Chem. – Eur. J., 2013, 19, 12759–12770 CrossRef CAS PubMed.
- G. Gody, D. A. Roberts, T. Maschmeyer and S. Perrier, J. Am. Chem. Soc., 2016, 138, 4061–4068 CrossRef CAS PubMed.
- D. A. Roberts, B. S. Pilgrim, T. N. Dell and M. M. Stevens, Chem. Sci., 2020, 11, 3713–3718 RSC.
- T. N. Forder, P. G. Maschmeyer, H. Zeng and D. A. Roberts, Chem. – Asian J., 2021, 16, 287–291 CrossRef CAS PubMed.
- A. Rabiee Kenaree and E. R. Gillies, Macromolecules, 2018, 51, 5501–5510 CrossRef CAS.
- C. A. Blencowe, D. W. Thornthwaite, W. Hayes and A. T. Russell, Org. Biomol. Chem., 2015, 13, 8703–8707 RSC.
- J. Gordon, 2-Methylbut-3-yn-2-ol in Encyclopedia of Reagents for Organic Synthesis, 2001. DOI:10.1002/047084289X.rm157.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py01169c |
| This journal is © The Royal Society of Chemistry 2021 |