
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect synthesis via RAFT of amphiphilic diblock polyelectrolytes facilitated by the use of a polymerizable ionic liquid as a monomer†
Aleksander
Guzik
 ab and
Patrizio
Raffa
*a
ab and
Patrizio
Raffa
*a
aDepartment of Chemical Engineering – Product Technology, ENTEG Institute, Faculty of Science and Engineering, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: p.raffa@rug.nl
bDPI, P.O. Box 902, 5600 AX Eindhoven, The Netherlands
First published on 8th September 2021
Abstract
A new method for obtaining amphiphilic block polyelectrolytes with a strongly hydrophobic block under homogeneous conditions is described and demonstrated by synthesizing poly(2-acrylamido-2-methanepropanesulfonate-b-styrene) block copolymers using RAFT polymerization. The technique involves the transformation of the 2-acrylamido-2-methanepropanesulfonate (AMPS) monomer into a polymerizable ionic liquid (PIL) by neutralization with triethylamine, which renders the resulting P(Et3N-AMPS) macroCTA soluble in DMF, DMSO, and ethanol. Consequently, straightforward homogeneous chain extension with a strongly hydrophobic monomer such as styrene is possible under homogeneous conditions. Homopolymerization of the PIL monomer is studied, revealing pseudo-1st-order kinetics and polydispersities slightly higher than those for the Na-AMPS monomer. High end group fidelity is demonstrated by chain extension experiments. Amphiphilic diblock copolymers are synthesized in a range of targeted chain compositions, and their formation is confirmed by SEC and 1H-DOSY, and by investigating self-assembled structures with DLS. The reverse approach based on styrene-first polymerization also proved successful, further expanding the scope of this work. We believe that this provides an interesting alternative to protection–deprotection methods and emulsion polymerization methods, when strongly anionic block copolymers are desired.
Introduction
Strong polyelectrolytes are well-established polymeric materials. Compared to weak ones, they may present some advantages, depending on the intended application, such as pH-independent ionization, solution stability in the presence of divalent cations, and strong interactions between chains. 2-Acrylamido-2-methylpropanesulfonic acid (AMPS) is a sulfonate-bearing monomer that can be used as an alternative for styrene sulfonate to introduce strong ionic groups into a copolymer, affording strong polyelectrolytes. AMPS has a fast rate of propagation and is hydrolytically stable. For these reasons, AMPS has been applied in biomaterials,1 hydrogels,2 colloid stabilizers,3,4 solid electrolytes for fuel cells,5 and polymers for enhanced oil recovery.6–8These applications can be potentially greatly expanded by producing AMPS-containing well-defined block copolymers. Such materials are generally easily made using a range of controlled radical polymerization methods (CRP),9 one of which is reversible addition–fragmentation transfer polymerization (RAFT).10 RAFT is based on the presence of a chain transfer agent (CTA), typically in the form of a dithiobenzoate, trithiocarbonate or a xanthate, in a radical polymerization mixture. It is easy to conduct and set up, and it allows tailoring both the polymer composition and, in a recent development, also its polydispersity.11
AMPS (as well as other similar strongly anionic sulfonate monomers) has been polymerized using controlled radical polymerization methods, either on its own to form the corresponding homopolymer12 or co-polymerized with other monomers such as: N,N-dimethylacrylamide,13 2-hydroxyethyl methacrylate,14N-hydroxyethyl acrylamide and 4-acryloylmorpholine,15N-isopropylacrylamide,16–18 3-acrylamido-3-methylbutanoate12 and methyl methacrylate,19 yielding either block or statistical copolymers. However, the co-polymerization of AMPS with strongly hydrophobic monomers to produce amphiphilic block copolymers is more challenging. Choosing a solvent poses a serious problem, as the commonly applied sodium salt of AMPS produces a polymer that tends to be insoluble in non-aqueous solvents. P(Na-AMPS-block-n-butyl acrylate) was synthesized using homogeneous RAFT, by dissolving the acid form of AMPS in NMP.20 However, NMP is known to be toxic and difficult to remove from the product and its use should be limited. Moreover, the attained conversions were low (17–53%), which is undesirable, especially when large molecular weights are required. Analogous block copolymers were synthesized in DMSO, but the targeted molecular weight was low, leading to less than 10 units of AMPS in the product.21 Moreover, the use of the acid form of AMPS results in extremely low pH values which, even though acidic RAFT can be carried out successfully,22 may cause solubility problems23 and may not be tolerated by other components of the reaction mixture. Additionally, many SEC columns cannot operate in strongly acidic environments, which imposes a requirement of chemical modification of every aliquot, if the evolution of Mn is to be tracked with SEC.
One common workaround is the use of protected, less polar, versions of the hydrophilic monomer. For example, polyelectrolyte block copolymers based on acrylic acid are often obtained by the polymerization of a corresponding ester, followed by a post-polymerization process of ester hydrolysis.24–26 Strongly ionic amphiphilic block copolymers can be synthesized in a similar manner, by following protection–deprotection chemistry, an example being the polymerization of vinyl sulfonates.27 Notably, such a method has been recently developed for 3-sulfopropyl methacrylate, to make P(Na-SPMA-block-styrene) copolymers.28 Even though this process gives good yield and is suitable for producing multiple grams of products, it is time-consuming and calls for dangerous chemicals, such as thionyl chloride or oxalyl chloride.
Another approach can be based on heterogeneous methods. P(Na-AMPS-b-styrene) has been produced by RAFT-mediated Polymerization-Induced Self-Assembly (PISA), which is a variant of emulsion polymerization, whereby the macroRAFT agent is also the surfactant.29 This approach, however, is not as straightforward as bulk polymerization, as the design of the composition has to take into account mass transfer between the two phases (e.g. radical entry). It is likely that higher molecular weight macroCTAs cannot be used for chain extensions because of increased viscosity. Finally, the order of monomer addition is fixed by the requirement of water solubility of the first block, serving as the surfactant. In our opinion, the PISA method is best suited for in situ synthesis of diblock copolymer nano-objects, which are never meant to be isolated from the latex.30–32 We propose here another approach based on polymerizable ionic liquids, which allows us to overcome the mentioned limitations.
Polymerizable ionic liquids (PILs) are a subset of organic ionic liquids that contain vinyl functionalities, allowing them to undergo free radical polymerization.33 They are generally constituted by ionic groups neutralized by some bulky organic counterions that favor them to remain dissociated and in liquid form at room temperature by hindering the formation of a strong ordered lattice. AMPS-based PILs are reported in the literature as precursors for functional materials obtained by free radical polymerization. The AMPS monomer has been previously transformed into a polymerizable ionic liquid by neutralization with triethylamine. The resultant monomer has been copolymerized with styrene34 and with acrylamide to yield superabsorbent gels.35 Copolymer gels were also attained by polymerization of AMPS neutralized with butylamine or 2-(N,N-dimethylamino)ethyl methacrylate.36 The 1-methylimidazole-AMPS monomer has been co-polymerized with styrene, yielding a material for proton exchange membranes.37 Other imidazole derivatives have also been used to produce protein-adsorbing polymers from AMPS.38 Surface-active polymers were made by using a surfactant amine (cetyltrimethylammonium hydroxide).39 AMPS has been polymerized and then the chains have been neutralized by tetrabutylammonium hydroxide to allow the polymer chains to be studied in a range of organic solvents.40,41 Although PILs have been polymerized with CRP, also to make block copolymers,42–44 we are not aware of any attempts to attain control of the polymerization of AMPS-based PIL by means of CRP, nor to utilize this approach to access strongly ionic non-PIL polyelectrolytes, which could be used as, e.g., water viscosifiers.
In this work we investigated the possibility of the synthesis of strongly charged amphiphilic Na-AMPS-block-styrene block copolymers according to the reaction sequence shown in Scheme 1. Neutralization of AMPS with triethylamine leads to an organic-soluble (Et3N-AMPS)-RAFT macroCTA, which can be chain extended with the hydrophobic monomer styrene in a homogenous mixture.
 | ||
| Scheme 1 Reaction sequence leading to the amphiphilic diblock polyelectrolytes studied in this work. | ||
Experimental
Materials
Styrene (S, Sigma-Aldrich, ≥99%) was passed through a basic alumina column to remove the inhibitor and was purged with argon before use. 4,4′-Azobis(4-cyanovaleric acid) (ACVA, Sigma-Aldrich, ≥98%) and 2,2′-azobis(2-methylpropionitrile) (AIBN, Sigma-Aldrich, 98%) were recrystallized from methanol prior to use. 2-Acrylamido-2 methylpropanesulfonic acid (AMPS, Sigma-Aldrich, 99%), triethylamine (Et3N, Sigma-Aldrich, ≥99%) and 4-((((2-carboxyethyl)thio)carbonothioyl)thio)-4 cyanopentanoic acid (CVPTTC, Boron Molecular, 98%) were used as-received, without further purification. Water used for the reactions was obtained from a Merck Milli-Q filtration system. Synthesis of BDMAT29 and MCEBTTC45 RAFT agents was performed according to the literature reports. Synthesis of polystyrene-RAFT macroCTA is also described elsewhere.46 Structures of RAFT agents and RAFT macroCTAs are reported in Scheme 2.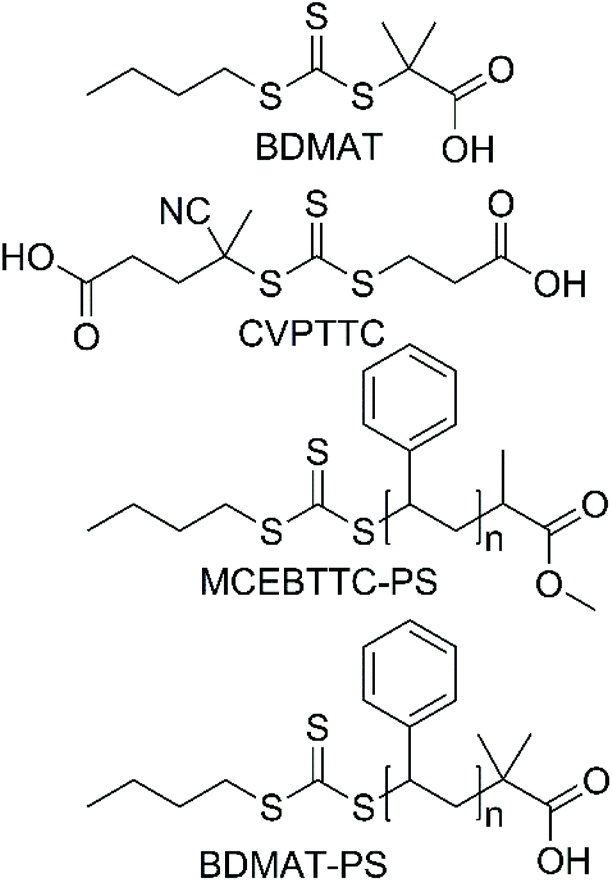 | ||
| Scheme 2 Structures of RAFT agents used in this research. | ||
Nuclear magnetic resonance
1H NMR data for the kinetic study in Fig. 1A were recorded in D2O on a 500 MHz Varian Inova. NMR spectra in Fig. 3C were recorded in d6-DMSO on a 600 MHz Bruker Avance NEO. All remaining NMR measurements were performed on a 400 MHz Varian Mercury Plus, using d6-DMSO. 1H DOSY experiments were carried out on a 600 MHz Bruker Avance NEO, using the bPGSTE pulse sequence. The raw data were processed using options available in the Mnova package. The PAMPS degree of polymerization was calculated based on the ratio of integrals of σ 1.41 (6H) singlet (AMPS methyl groups) to σ 2.45 (2H) and σ 2.73 (2H) multiplets (–CH2–CH2– from polymer end groups). Monomer conversions for the kinetic studies were determined by relating the integral of vinyl protons to the integral of the σ 1.41 signal.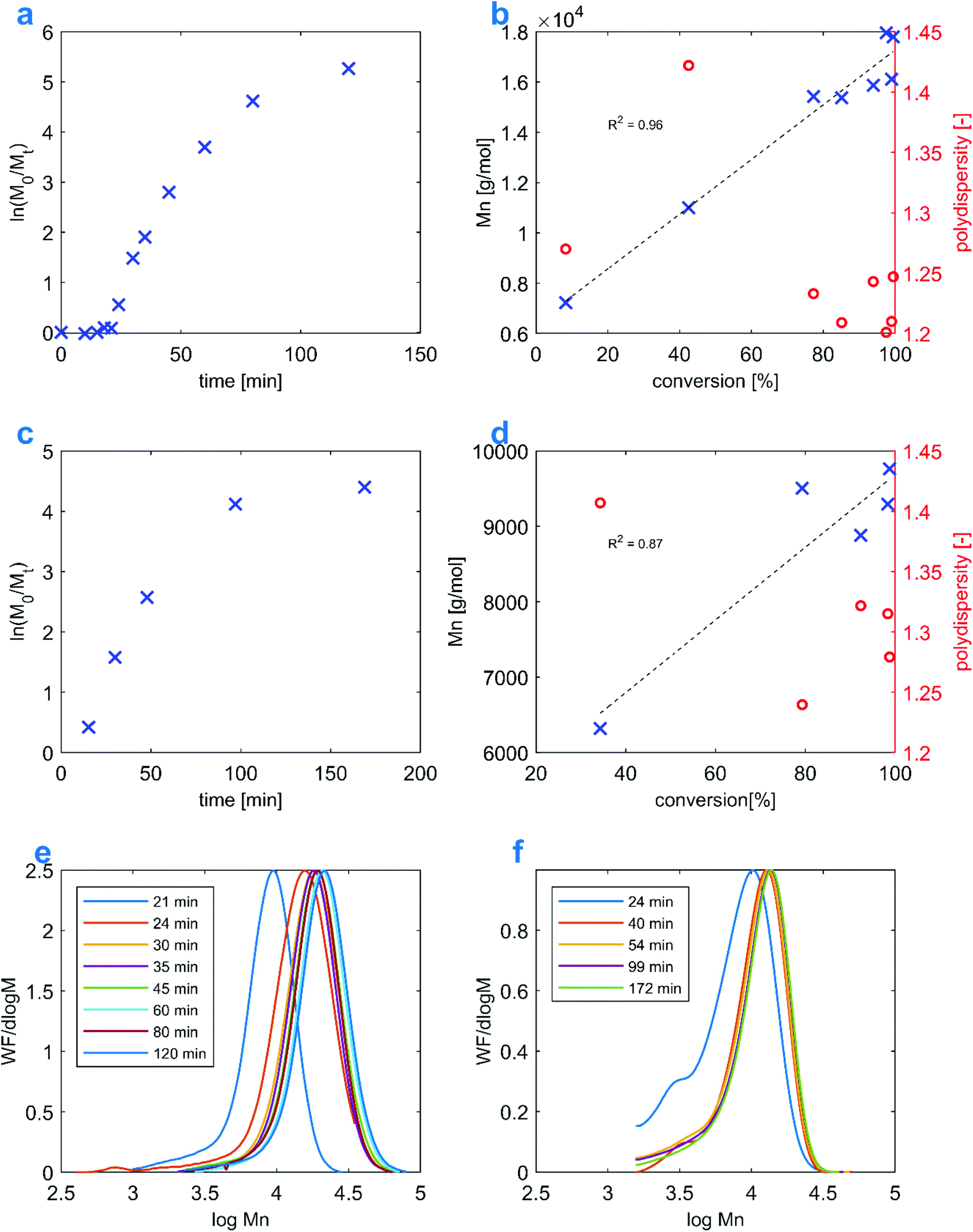 | ||
| Fig. 1 Comparison between the polymerization kinetics of the Et3N-AMPS monomer involving two different RAFT agents: BDMAT and CVPTTC. Reactions correspond to Table 1 entries: 6 (a, b, and e) and 12 (c, d, and f). | ||
UV-VIS
A Thermo Spectronic Aquamate UV-VIS spectrophotometer was used for all measurements. The samples were measured in glass cuvettes, immediately after preparation. The concentration of the samples was adjusted so that the signal was within the limits of the instrument.Gel permeation chromatography
The GPC traces of DMF solutions reported in Fig. 1, Fig. S11 and S12† were measured on a Viscotek GPCmax equipped with model 302 TDA detectors, with two columns (Agilent Technologies-PolarGel-L and M, 8 μm, 30 cm) at a flow rate of 1.0 ml min−1 and at a temperature of 50 °C. The eluent was a 10 mM solution of LiBr in DMF. The system was calibrated using PMMA standards.The chromatograms in Fig. S12† were deconvoluted by fitting a lognormal distribution using Fityk software.47
The GPC traces and values for DMF solutions reported in Table 2 and Fig. 3 were measured on an Agilent 1200 Series system, equipped with an Agilent 1200 RI detector, and a 3× PSS Gram Analytical Linear column (particle size: 10 μm; 8 × 300 mm). The system used 10 mM solution of LiBr in DMF at 50 °C and a flow rate of 1 mL min−1. The system was calibrated with polystyrene standards and used a toluene flow marker.
All aqueous solutions were recorded with the Agilent 1200 Series system using a PSS SUPREMA column and a PSS SECurity RI detector. The system was calibrated with polyacrylamide standards and an ethylene glycol flow marker. The eluent was 0.05 M NaNO3 with a 1.0 mL min−1 flow rate.
Examples of PIL homopolymerization procedures
Examples of chain extension procedures
Results and discussion
Et3N-AMPS homopolymerization
In order to estimate the reactivity and degree of control over the RAFT process, the Et3N-AMPS monomer has been compared with Na-AMPS with respect to its behavior in a polymerization reaction. Table 1 summarizes the homopolymerization reactions. Several observations can be made on the polydispersity of the P(Et3N-AMPS) polymers as a function of the solvent and RAFT agent employed. When BDMAT is used with water as solvent, the PDI values are approximately in the range 1.3–1.4, which is significantly less than the value for a product of free radical polymerization, with PDI = 3.16; this confirms that the RAFT agent is effective. A clear trend of PDI increasing with the targeted degree of polymerization (DP) can be identified, with the maximal values of 1.6–1.7 occurring for a reaction involving CVPTTC and targeting DP = 200. The discrepancy between the effects of BDMAT and CVPTTC on the final PDI value could be due to the BDMAT RAFT agent releasing a more reactive radical in the course of the RAFT mechanism, but it could also be explained by a known effect of improved control over acrylate polymerization in water over other solvents.48 The differences between both RAFT agents are discussed in the following section. Interestingly, the results when using CVPTTC seem to be nearly independent of the solvent, with water yielding only slightly lower PDIs than DMSO, at a given target DP. Conducting the aqueous process at a temperature of 50 °C instead of 80 °C is another way to reduce the PDI, albeit at a cost of a significantly slower rate of conversion. With the Na-AMPS monomer, a similar PDI vs. DP trend is observed; however, the PDI values are clearly lower, even for DP = 200. Unlike Et3N-AMPS, Na-AMPS seems to produce consistent PDI values irrespective of the choice of the RAFT agent. The conversion rates are similar in both monomers, and near-quantitative conversion is reached in 2–3 hours at 80 °C. The homopolymerization of Et3N-AMPS is also possible in ethanol (Table 1 entry 13), as both the monomer and the polymer are soluble in this solvent. This reaction has not been studied by us in detail, as its purpose is demonstration of the feasibility of using yet another solvent for the described process and chain extension with styrene could give solubility problems; however, a kinetics plot and a SEC trace of the product are available in the ESI (Fig. S5†). Similarly, Table 1 entry 14 refers to an attempt to polymerize the AMPS form in DMSO. We found that the product of this reaction is unstable, presumably owing to the presence of strongly acidic non-neutralized sulfonate moieties. See the ESI (Fig. S6†) for a photograph of the decomposed sample and an 1H NMR spectrum.| Entry | Monomer | Target Mn [kg mol−1] (DP) | [CTA]0/[I]0 | [M]0 | RAFT agent | Solvent | Temp. [°C] | M n NMR [kg mol−1] | M n SECa [kg mol−1] | PDI, SEC | Conversionb [%] |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Measured in 0.05 M NaNO3 eluent. b Measured by NMR; 100% means no integrable vinyl signals. | |||||||||||
| 1 | Et3N-AMPS | 15.2 (48) | 10.0 | 1.13 | CVPTTC | DMSO | 80 | 13.8 | 15.4 | 1.24 | 85 |
| 2 | 12.9 (41) | 9.1 | 2.9 | CVPTTC | 80 | 13.0 | 17.3 | 1.24 | 99 | ||
| 3 | 62.0 (200) | 5.0 | 3.0 | CVPTTC | 80 | 30.3 | 35.5 | 1.70 | 96 | ||
| 4 | 62.0 (200) | 10.0 | 2.0 | CVPTTC | 80 | 26.1 | 29.3 | 1.60 | 95 | ||
| 5 | FRP | FRP | 2.0 | CVPTTC | 80 | — | 45.2 | 3.16 | 98 | ||
| 6 | 15.7 (50) | 10.0 | 2.0 | CVPTTC | Water | 80 | — | See Fig. 1B | See Fig. 1B | See Fig. 1B | |
| 7 | 31.2 (100) | 10.0 | 2.0 | CVPTTC | 70 | — | 30.7 | 1.31 | 100 | ||
| 8 | 62.0 (200) | 10.0 | 2.0 | CVPTTC | 80 | 27.8 | 59.1 | 1.64 | 99 | ||
| 9 | 62.0 (200) | 10.0 | 2.0 | CVPTTC | 50 | — | 46.2 | 1.55 | 98 | ||
| 10 | 62.0 (200) | 10.0 | 2.0 | BDMAT | 80 | — | 48.0 | 1.34 | 100 | ||
| 11 | 62.0 (200) | 10.0 | 2.0 | BDMAT | 60 | — | 61.7 | 1.39 | 100 | ||
| 12 | 15.7 (50) | 10 | 2.0 | BDMAT | 70 | — | See Fig. 1D | See Fig. 1D | See Fig. 1D | ||
| 13 | 6.4 (20) | 10 | 1.5 | BDMAT | Ethanol | Reflux | — | 6.5 | 1.26 | 95 | |
| 14 | AMPS acid | 37.1 (178) | 9 | 2.2 | BDMAT | DMSO | 80 | — | — | — | 95 |
| 15 | Na-AMPS | 1.1 (5) | 9.12 | 1.73 | CVPTTC | Water | 70 | — | 4.0 | 1.25 | 100 |
| 16 | 4.6 (20) | 5.09 | 2.0 | CVPTTC | 70 | — | 8.4 | 1.17 | 100 | ||
| 17 | 11.5 (50) | 12 | 2.0 | BDMAT | 80 | — | 21.0 | 1.23 | 100 | ||
| 18 | 19.5 (85) | 4.85 | 2.0 | CVPTTC | 70 | — | 20.4 | 1.20 | 100 | ||
| 19 | 45.8 (200) | 10 | 2.0 | CVPTTC | 80 | — | 41.8 | 1.30 | 99 | ||
The kinetics of Et3N-AMPS homopolymerization was investigated in detail. To this end, two RAFT polymerization reactions targeting DP = 50 were conducted in water, each using a different RAFT agent. Both processes were studied by tracking the conversion, Mn and PDI with time. Fig. 1B and D show that the evolution of molecular weight with conversion is approximately linear for both reactions, as expected for a controlled process. The PDIs also decrease over time, which is typical of RAFT.49 Comparing Fig. 1A with C, it can be seen that in both cases the kinetics conforms to the pseudo-first-order model; what stands out, however, is the presence of a significant induction period (taking up approximately 25% of the total reaction time) for the reaction using the CVPTTC RAFT agent, which is virtually absent for the process using BDMAT. In general, such an induction period indicates either a slow propagation constant of the monomer or insufficient re-initiating capability of the R˙ radical.50 In this case, the latter is speculated to be more probable, as the cyano-stabilized tertiary radical from CVPTTC is far more stable than a radical formed from BDMAT.
Chain extension experiments
Before proceeding to the synthesis of amphiphilic block copolymers, to investigate chain end retention, we conducted chain extension experiments with Et3N-AMPS on a P(Et3N-AMPS)20 macroRAFT agent. In this way, the polymer could be characterized by aqueous SEC without the need to switch to a different solvent and column. Fig. 2 shows the pseudo-1st order kinetics and the evolution of the molecular weight and PDI. The SEC traces were monomodal and shifted to higher molecular weights as the reaction progressed, which indicates that the process was controlled and that the macroCTA had a high proportion of living chains.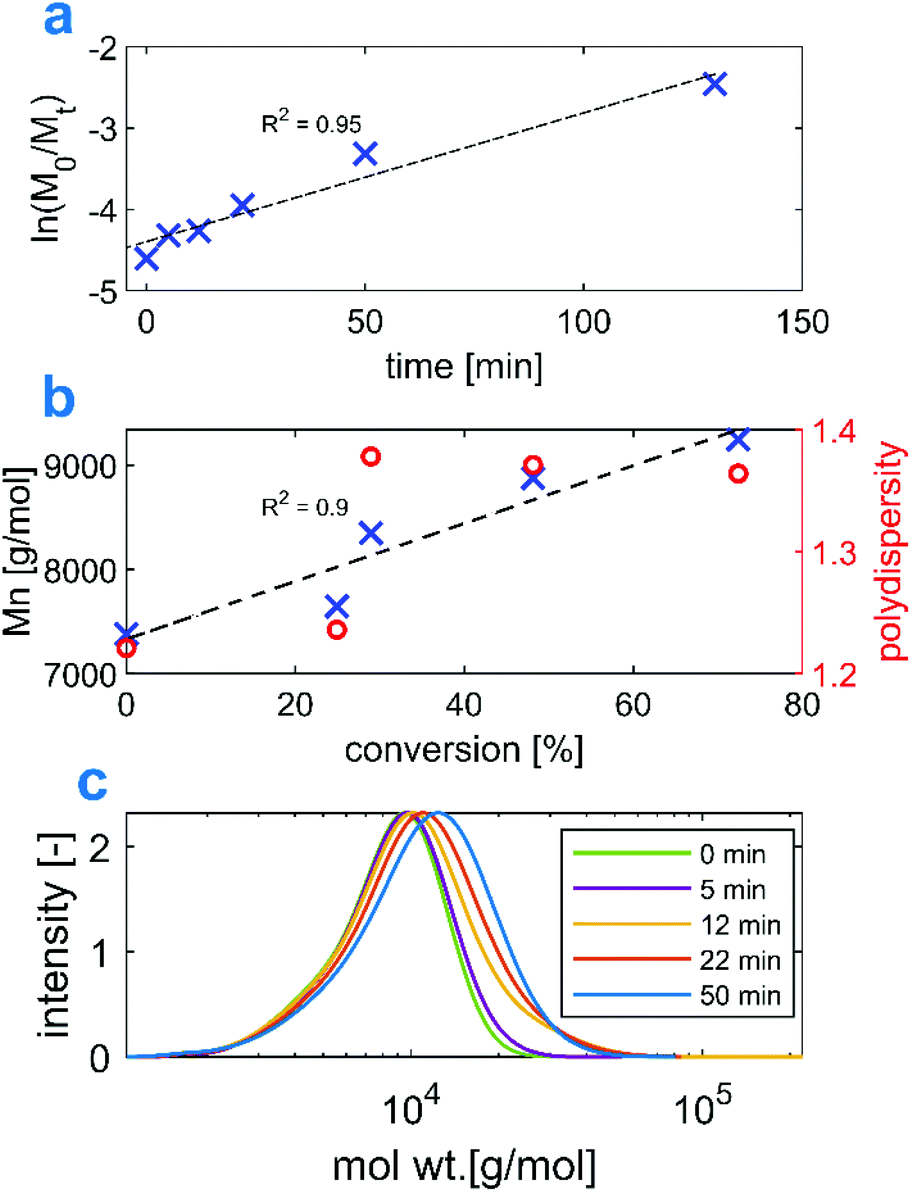 | ||
| Fig. 2 Kinetic study of a polymerization of Et3N-AMPS using a P(Et3N-AMPS)-BDMAT RAFT agent. | ||
Synthesis of amphiphilic diblocks
Amphiphilic diblock copolymers were synthesized by following either the “PAMPS-first” or “PS-first” approach. In the first type of reaction, the hydrophobic monomer styrene is polymerized in the presence of P(Et3N-AMPS)-RAFT macroCTA, while in the other case the Et3N-AMPS monomer is polymerized in the presence of PS-RAFT macroCTA. All the reaction conditions, along with the attained conversions, SEC results and DOSY diffusion coefficients are summarized in Table 2. Even though the homopolymerization of Et3N-AMPS using CVPTTC or BDMAT RAFT agents is rapid (approx. 2 hours of reaction time) and results in near-quantitative conversion, it is not the case for polymerization using polystyrene macroCTA. The attained conversions for all “PS-first” chain extensions (entries 1–4 in Table 2) were never full, despite over 20 hours of polymerization time (also see Fig. S7†). This can be attributed to the fact that in the “PS-first” scheme, the less-activated monomer (styrene) is polymerized first, while from the point of view of the RAFT mechanism it is advantageous to first polymerize the more-activated monomer. The dispersity values reported for all “PS-first” reactions (entries 1–4 in Table 2) are between 1.08 and 1.21. While these values are low and therefore suggest very good control over the polymerization, they may have been affected by the purification by acetone reprecipitation, which can selectively separate dead PS chains and shorter diblocks. This may also explain the overshot PAMPS/PS ratios and Mn values, measured by NMR and SEC, respectively.| Entry | Target DP (AMPS + styrene) | [CTA]0/[I]0 | [M]0 | [M]0/[I]0 | MacroCTA | Solvent | Temp [°C] | Conversion, NMR [%] | M n SEC [kg mol−1] | PDI, SEC | [St]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[AMPS] :[AMPS] |
Diffusion coefficient@1.41 ppm [107 cm2 s−1] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 50 + 11 | 5 | 2 | 250 | (S)11-MCEBTTC | DMF | 80 | 77 | 15.1 | 1.17 | 4.4 | 3.91 |
| 2 | 50 + 30 | 10 | 2 | 500 | S28-BDMAT | DMF | 80 | 56 | 22.7 | 1.08 | 3.9 | 3.83 |
| 3 | 100 + 30 | 10 | 2 | 1000 | S28-BDMAT | DMF | 80 | 90 | 31.6 | 1.13 | 5.5 | 3.33 |
| 4 | 200 + 30 | 10 | 2 | 2000 | S28-BDMAT | DMF | 80 | 85 | 48.7 | 1.21 | 10.1 | 2.3 |
| 5 | 41 + 48 | 10 | 1.59 | 480 | (Et3N-AMPS)41-CVPTTC | DMSO | 80 | 48 | 10.7 | 1.81 | 7.8 | 4.06 |
| 6 | 32 + 22 | 5 | 2 | 110 | (Et3N-AMPS)32-CVPTTC | DMF | 80 | 79 | 15.9 | 1.28 | 9.0 | 3.51 |
| 7 | 100 + 15 | 10 | 0.38 | 150 | (Et3N-AMPS)100-CVPTTC | DMSO | 80 | 78 | 38.3 | 1.18 | 15.2 | 2.16 |
| 8 | 200 + 15 | 5 | 0.31 | 75 | (Et3N-AMPS)200-CVPTTC | DMSO | 80 | 83 | 67.9 | 1.29 | 11.2 | 1.46 |
| 9 | 200 + 37 | 10 | 0.40 | 370 | (Et3N-AMPS)200-BDMAT | DMSO | 80 | 48 | 75.9 | 1.25 | 16.8 | 1.38 |
After purification by precipitation in acetone, the polymers were characterized by 1H-NMR, UV-VIS, elemental analysis, 1H DOSY, and DLS. Results for the sample from entry 9 can be seen in Fig. 3. The UV-VIS spectrum of the 10 mg mL−1 aqueous solution of the diblock product features several weak bands in the range 250–280 nm, which confirm the presence of aromatic compounds in the product. The spectra were recorded in aqueous solutions, as both DMSO and DMF absorb UV light in the region of interest. Therefore, the signals may have been attenuated by the formation of PS-core self-assembled structures in the solution. A qualitative presence, however, is clearly visible when compared with a homopolymer macroCTA's spectrum (Fig. 3A). Similar observation can be made for the remaining samples in Table 2 (see Fig. S9†).
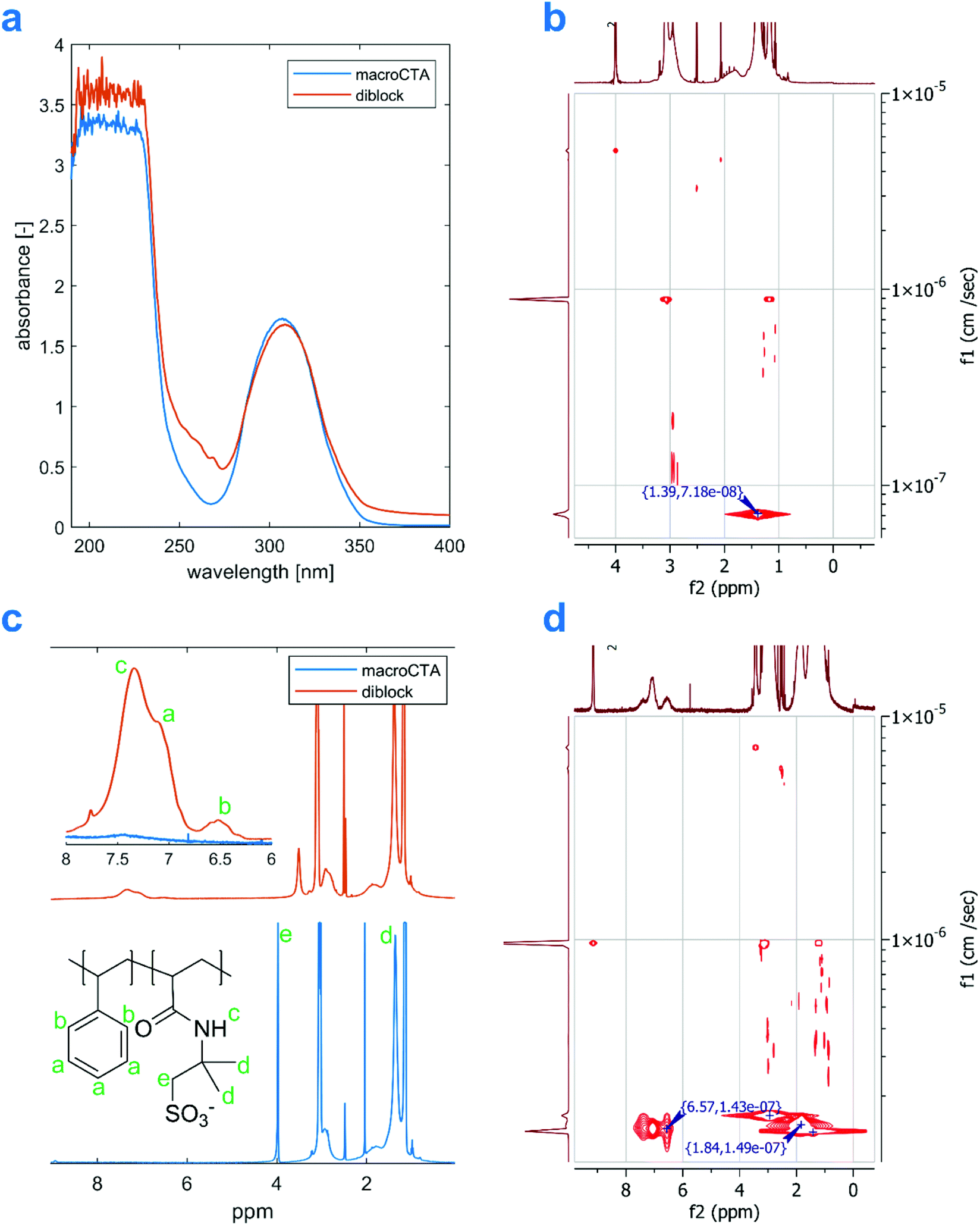 | ||
| Fig. 3 Comparison between a (Et3N-AMPS)200 macroCTA and an amphiphilic diblock produced from it (Table 2 entry 9): A – UV-VIS spectra of a 10 mg mL−1 aqueous solutions, B – 1H DOSY projection of the macroCTA, C – 1H NMR in d6-DMSO (amide proton signal in the macroCTA's spectrum has been cleared by the addition of D2O), and D – 1H DOSY projection of the diblock. | ||
The presence of styrene units in the copolymer is also evident from the NMR spectra in Fig. 3C, showing two broad peaks around 6.5 ppm and 7.1 ppm coming from the polystyrene block, which are not present in the spectrum of the homopolymer (for more 1H-NMR spectra see Fig. S3†). As the aromatic signals might be subject to significant intensity changes following chain self-assembly,20,51 we calculated the diblocks’ compositions only based on NMR spectra recorded with a high recycle delay of d1 = 30 s. The aromatic peaks are overlapped with the amide proton peak from AMPS, which can be deconvoluted in the software before integration. Finally, to further demonstrate that the polystyrene blocks are covalently joined with the P(Et3N-AMPS) blocks, 1H-DOSY experiments were performed. Briefly, this method allows us to assign a diffusion coefficient to every peak in an 1H-NMR spectrum. If two peaks are assigned similar diffusion coefficients, it can be inferred they come from the same molecule. In the 1H-DOSY plot in Fig. 3D, both the polystyrene signal at around 6.5 ppm and the –CH3 AMPS signal at 1.39 ppm have a diffusion coefficient close to 10−7 cm2 s−1, characteristic of a very large molecule. Thus, we conclude that the architecture of the products is indeed that of a diblock copolymer. A similar conclusion can be drawn for all the samples listed in Table 2 (see Fig. S10†). The small variations in the measured diffusion coefficients for AMPS and aromatic peaks are a result of a single-exponential fitting and are expected. Elemental analysis results for entry 9, which confirm a smaller proportion of nitrogen in the diblock product, compared with the macroCTA, are available in Table S1 (ESI†).
SEC analysis of the amphiphilic diblock copolymers
All the diblock samples outlined in Table 2 along with their corresponding macroCTAs have been analysed using a an SEC system using DMF (Fig. 4). The diblocks’ peaks are monomodal, indicating a high end-group retention in the macroCTAs. The shifting of the SEC traces towards lower elution volumes in the course of the chain extension process was not reliable. This unusual behavior is likely a consequence of the diblock structure. Problematic SEC characterization of block copolymers has been recently outlined,52 stressing that aspects such as a rod–coil architecture and strong block–block and block–column interactions exacerbate this issue. In the case of Et3N-AMPS-block-St copolymers, the extreme difference in polarities of the blocks and the difference in block rigidities are factors that could cause such behavior. Further evidence that this effect is related to non-ideal chain behavior comes from the fact that it seems to depend on the type of column used (compare Fig. 4 to Fig. S11,† which contains SEC traces for the same samples, measured using a different SEC apparatus with the DMF eluent).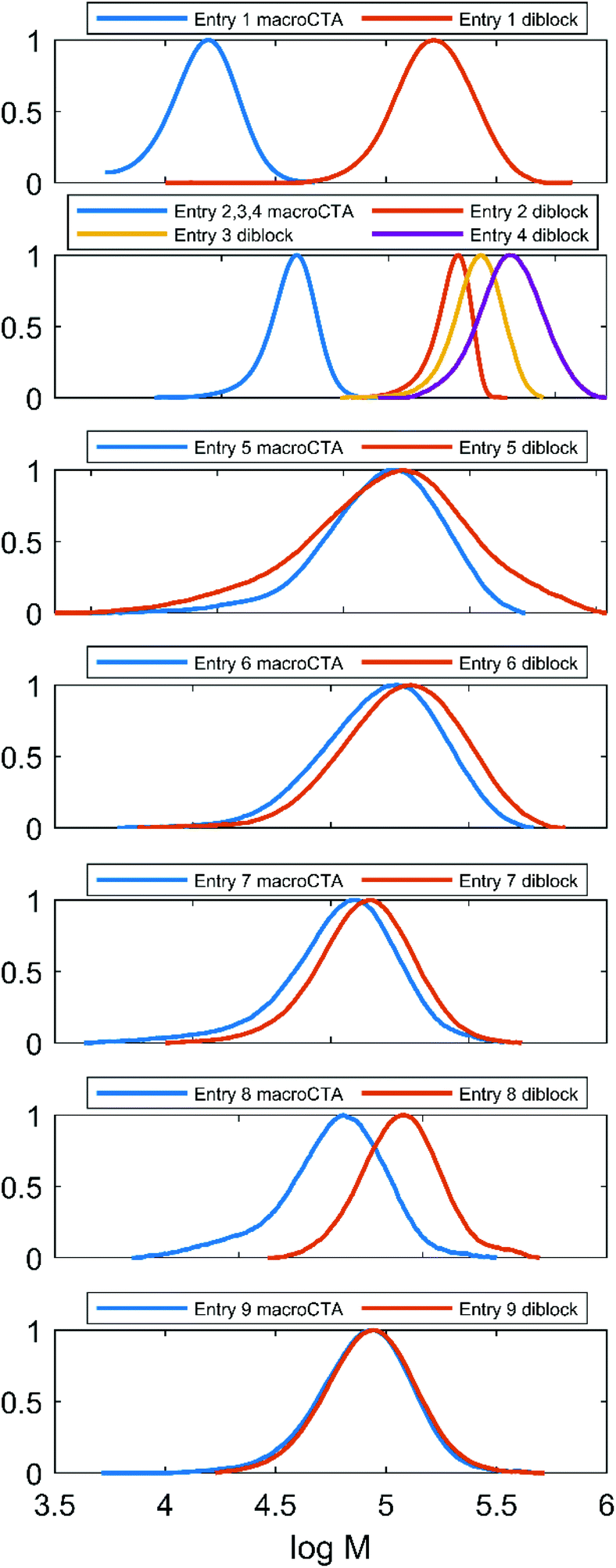 | ||
| Fig. 4 SEC traces in DMF/LiBr of all amphiphilic diblock copolymers enumerated in Table 2, together with the corresponding macroCTAs. The anomalous peak “shuffling” is discussed in text. | ||
This phenomenon is observed only for some of the “PAMPS-first” samples (entries 5–9, Fig. 4), while the “PS-first” SEC traces behave as expected (entries 1–4, Fig. 4). In order to establish whether the “PAMPS-first” is a valid method of producing the amphiphilic diblock copolymers, we examined this effect in more detail, as follows. A “PAMPS-first” reaction has been performed, targeting a large (DP = 200) molecular weight of the polystyrene block, in order to maximize the chances that the SEC peak shifting would be visible. DMF SEC samples were drawn periodically from the reaction (Fig. S12†). The trace at t = 0 min corresponds to the P(Et3N-AMPS) homopolymer macroCTA. Interestingly, the samples taken at short times (low styrene conversion) produced SEC traces that were shifted to lower apparent molecular weights than that of the macroCTA. However, within the series of traces corresponding to the growing diblock, the shifting is monotonic towards higher molecular weights, as expected. Therefore, there seems to be a discontinuity in the behavior of the polymer chains in the SEC system as the type of polymer changes from a homopolymer to an amphiphilic diblock. This initial shift towards lower apparent molecular weights has to be overcome by growing the polystyrene chain by a given amount of units before the measured “net” trace is shifted in the expected direction with respect to the original macroCTA. Indeed, an analogous situation is described for highly incompatible block copolymer systems in the previously cited work,52 where the elution time initially increases as the block copolymer is formed.
The discussed effect could be sufficient to explain the unreliable SEC trace shifting of some entries in Table 2. In “PAMPS-first” reactions, the targeted low DP of the polystyrene block along with non-quantitative conversions of the styrene monomer means that the expected increment in molecular weight is always low (ranging from approximately 2% for the sample in entry 8 to 19% for entry 5). As discussed above, this increment might not have always been enough to compensate for the initial lowering of the apparent molecular weight. On the other hand, for “PS-first” reactions, the expected molecular weight increment was always large, ranging from 300% for entry 2 to 1800% for entry 4. Given this explanation and the fact that other presented results do not differentiate between the “PS-first” and “PAMPS-first” methods, we conclude that both are valid approaches towards the synthesis of PS-block-PAMPS amphiphilic block polyelectrolytes.
Formation of an amphiphilic product was further confirmed by studying the properties of its aqueous solution. Fig. 5A shows the comparison between hydrodynamic diameters, measured by DLS, of 0.5% solutions of P(Et3N-AMPS) macroCTAs and 0.5% solutions of the corresponding diblock products. In every case, the diameters measured in the solutions of the copolymers were close to or above 100 nm, while the macroCTAs were close to 1 nm. This indicates that, in fact, the copolymers form self-assembled structures in solution, while the macroCTAs (being fully hydrophilic) do not. Furthermore, upon addition of salt solution to the diblock DLS samples, their hydrodynamic diameters decreased in all-but-one case. This could be attributed to the collapse of the negatively charged coronas as a result of increased ionic strength, although this cannot be stated conclusively as the effects related to the thickness of the electrical double layer could also play a role. Note that the DLS results shown here have not been corrected for viscosity, which is bound to introduce a small positive error in the absolute values of the hydrodynamic diameters, but it is unlikely to affect the overall discussed trend. DLS number-average diameter distributions and the values of number-average mean diameters, Z-average mean diameters and polydispersity indices are shown in Fig. S8 and Table S2.† As a final piece of evidence towards the amphiphilic nature of the copolymers, the photograph in Fig. 5B shows the formation of persistent foam after agitation of a 0.5% diblock solution, while the foam does not persist in the macroCTA solution. Observation of foaming has been used before as a qualitative test of interfacial adsorption of amphiphilic species.53–56
 | ||
| Fig. 5 Solution properties of the amphiphilic diblocks. A: DLS comparison between hydrophilic macroCTAs and amphiphilic diblock copolymers. Entries correspond to those in Table 2 B: appearance of 0.5 wt% aqueous solutions of Table 2 entry 8 macroCTA (left) and diblock (right), after being shaken by hand for 30 s and left steady for 15 min. | ||
Conclusions
There are several approaches available to synthesize strongly ionic amphiphilic block copolymers. The main practical difficulty with many of these systems is the high incompatibility of a polyelectrolytic block and a strongly hydrophobic one. Heterogeneous methods, such as PISA,29 can be applied, as well as methods based on protection–deprotection chemistry.26,28 Some anionic monomers, when in acid form, may also be polymerizable in universal solvents such as DMSO21 or NMP.20 In this work, we have shown that it might be advantageous to proceed via RAFT polymerization of a poly(ionic liquid), obtained by neutralizing the monomer (here AMPS) with an organic amine such as triethylamine, in order to ensure the solubility of the resulting polymer macroCTA in organic solvents. In particular, we showed that AMPS-block-styrene copolymers can be prepared by homogeneous RAFT using either a “PAMPS-first” or a “PS-first” approach, with the latter being slower, but providing lower dispersities, in various organic solvents, such as DMF or DMSO. The polymerization kinetics has been followed, and the formation of block copolymers with good control and high end-group fidelity has been demonstrated with the support of chain extension experiments and several characterization methods such as 1H-NMR, UV-vis, SEC, 1H-NMR-DOSY, and DLS.The design of the homogeneous RAFT process is easier than that for emulsion polymerization, and additionally the acidic monomer is neutralized, which prevents polymer degradation and allows for facile SEC characterization, both in organic and aqueous eluents. The improvement over protection–deprotection methods is the reduced number of steps and the lack of chemical alterations to the monomer, which tend to reduce the overall yield. The last step of cation exchange leads to the final sodium-salt product (see Fig. S4 in the ESI†), suitable for areas of application where the presence of common inorganic ions is more desirable, such as industrial water viscosifiers and polymeric surfactants.
Since the process is homogeneous, we have demonstrated here that it is possible to arrive at the block copolymer either by hydrophilic-first or hydrophobic-first sequences, which would not work with PISA without changing the emulsion type. Even though there might be other requirements of the order of monomer addition stemming from the RAFT mechanism, this is still an advantage given the difficulties in studying the diblock product with SEC: as the macroCTA homopolymer can usually be fully characterized, it is possible to design the synthetic procedure according to research requirements, for instance for structure–property relationship studies, when the characterization of one of the blocks could be more important or difficult than the other.
We believe that this work provides an interesting alternative to existing methods of synthesis of amphiphilic block polyelectrolytes characterized by a strongly hydrophobic block. This approach could be expanded to other anionic monomers. Further studies in this matter could also involve a thorough comparison of all available approaches, with respect to the reactivities of the monomer, attainable PDIs, stability of the products and reaction flexibility. Such a database would enable an informed choice for researchers wishing to access strongly anionic block copolymers. Finally, the characterization of solution behavior and aggregation of the obtained block copolymers is certainly within the scope of possible future research.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is part of the research programme of DPI, project #821, which is kindly acknowledged for funding.References
- A. K. Saikia, S. Aggarwal and U. K. Mandal, Preparation and Controlled Drug Release Characteristics of Thermoresponsive PEG/Poly (NIPAM-Co-AMPS) Hydrogels, Int. J. Polym. Mater., 2013, 62(1), 39–44, DOI:10.1080/00914037.2012.664208.
- S. Durmaz and O. Okay, Acrylamide/2-Acrylamido-2-Methylpropane Sulfonic Acid Sodium Salt-Based Hydrogels: Synthesis and Characterization, Polymer, 2000, 41(10), 3693–3704, DOI:10.1016/S0032-3861(99)00558-3.
- H. Bouhamed, S. Boufi and A. Magnin, Dispersion of Alumina Suspension Using Comb-like and Diblock Copolymers Produced by RAFT Polymerization of AMPS and MPEG, J. Colloid Interface Sci., 2007, 312(2), 279–291, DOI:10.1016/j.jcis.2007.03.060.
- H. Müller, W. Leube, K. Tauer, S. Förster and M. Antonietti, Polyelectrolyte Block Copolymers as Effective Stabilizers in Emulsion Polymerization, Macromolecules, 1997, 30(8), 2288–2293, DOI:10.1021/ma9615516.
- S. Zhong, X. Cui, H. Cai, T. Fu, K. Shao and H. Na, Crosslinked SPEEK/AMPS Blend Membranes with High Proton Conductivity and Low Methanol Diffusion Coefficient for DMFC Applications, J. Power Sources, 2007, 168(1), 154–161, DOI:10.1016/j.jpowsour.2007.03.028.
- A. Scott, M. Riahinezhad and A. Penlidis, Optimal Design for Reactivity Ratio Estimation: A Comparison of Techniques for AMPS/Acrylamide and AMPS/Acrylic Acid Copolymerizations, Processes, 2015, 3(4), 749–768, DOI:10.3390/pr3040749.
- F. Li, W. Zhu, D. Yu, H. Song and K. Wang, Rheological Properties and Enhanced Oil Recovery Performance of a Novel Sulfonate Polyacrylamide, J. Macromol. Sci., Part A: Pure Appl.Chem., 2018, 55(6), 449–454, DOI:10.1080/10601325.2018.1470462.
- X. Yu, W. Pu, D. Chen, J. Zhang, F. Zhou, R. Zhang and S. Gu, Degradable Cross-Linked Polymeric Microsphere for Enhanced Oil Recovery Applications, RSC Adv., 2015, 5(77), 62752–62762, 10.1039/C5RA05366H.
- K. Parkatzidis, H. S. Wang, N. P. Truong and A. Anastasaki, Recent Developments and Future Challenges in Controlled Radical Polymerization: A 2020 Update, Chem, 2020, 6(7), 1575–1588, DOI:10.1016/j.chempr.2020.06.014.
- S. Perrier, 50th Anniversary Perspective: RAFT Polymerization—A User Guide, Macromolecules, 2017, 50(19), 7433–7447, DOI:10.1021/acs.macromol.7b00767.
- R. Whitfield, K. Parkatzidis, N. P. Truong, T. Junkers and A. Anastasaki, Tailoring Polymer Dispersity by RAFT Polymerization: A Versatile Approach, Chem, 2020, 6(6), 1340–1352, DOI:10.1016/j.chempr.2020.04.020.
- B. S. Sumerlin, M. S. Donovan, Y. Mitsukami, A. B. Lowe and C. L. McCormick, Water-Soluble Polymers. 84. Controlled Polymerization in Aqueous Media of Anionic Acrylamido Monomers via RAFT, Macromolecules, 2001, 34(19), 6561–6564, DOI:10.1021/ma011288v.
- E. Read, A. Guinaudeau, D. James Wilson, A. Cadix, F. Violleau and M. Destarac, Low Temperature RAFT/MADIX Gel Polymerisation: Access to Controlled Ultra-High Molar Mass Polyacrylamides, Polym. Chem., 2014, 5(7), 2202–2207, 10.1039/c3py01750h.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Anionic Polyelectrolyte-Stabilized Nanoparticles via RAFT Aqueous Dispersion Polymerization, Langmuir, 2012, 28(1), 914–922, DOI:10.1021/la203991y.
- C. Bray, R. Peltier, H. Kim, A. Mastrangelo and S. Perrier, Anionic Multiblock Core Cross-Linked Star Copolymers via RAFT Polymerization, Polym. Chem., 2017, 8(36), 5513–5524, 10.1039/C7PY01062A.
- S. Bekhradnia, J. S. Diget, T. Zinn, K. Zhu, S. A. Sande, B. Nyström and R. Lund, Charged Star Diblock Copolymers in Dilute Solutions: Synthesis, Structure, and Chain Conformations, Macromolecules, 2015, 48(8), 2637–2646, DOI:10.1021/ma502488u.
- S. Yusa, Y. Shimada, Y. Mitsukami, T. Yamamoto and Y. Morishima, Heat-Induced Association and Dissociation Behavior of Amphiphilic Diblock Copolymers Synthesized via Reversible Addition–Fragmentation Chain Transfer Radical Polymerization, Macromolecules, 2004, 37(20), 7507–7513, DOI:10.1021/ma0492519.
- Y. Kotsuchibashi, M. Ebara, K. Yamamoto and T. Aoyagi, Tunable Stimuli-Responsive Self-Assembly System That Forms and Stabilizes Nanoparticles by Simple Mixing and Heating/Cooling of Selected Block Copolymers, Polym. Chem., 2011, 2(6), 1362–1367, 10.1039/c1py00004g.
- G. Masci, D. Bontempo, N. Tiso, M. Diociaiuti, L. Mannina, D. Capitani and V. Crescenzi, Atom Transfer Radical Polymerization of Potassium 3-Sulfopropyl Methacrylate: Direct Synthesis of Amphiphilic Block Copolymers with Methyl Methacrylate, Macromolecules, 2004, 37(12), 4464–4473, DOI:10.1021/ma0497254.
- S. Garnier and A. Laschewsky, Synthesis of New Amphiphilic Diblock Copolymers and Their Self-Assembly in Aqueous Solution, Macromolecules, 2005, 38(18), 7580–7592, DOI:10.1021/ma0506785.
- J. Garnier, J. Warnant, P. Lacroix-Desmazes, P.-E. Dufils, J. Vinas and A. van Herk, Sulfonated Macro-RAFT Agents for the Surfactant-Free Synthesis of Cerium Oxide-Based Hybrid Latexes, J. Colloid Interface Sci., 2013, 407, 273–281, DOI:10.1016/j.jcis.2013.06.037.
- M. H. Allen, S. T. Hemp, A. E. Smith and T. E. Long, Controlled Radical Polymerization of 4-Vinylimidazole, Macromolecules, 2012, 45(9), 3669–3676, DOI:10.1021/ma300543h.
- E. A. Hoff, B. A. Abel, C. A. Tretbar, C. L. McCormick and D. L. Patton, Aqueous RAFT at PH Zero: Enabling Controlled Polymerization of Unprotected Acyl Hydrazide Methacrylamides, Polym. Chem., 2017, 8(34), 4978–4982, 10.1039/C6PY01563H.
- C. Charbonneau, C. Chassenieux, O. Colombani and T. Nicolai, Controlling the Dynamics of Self-Assembled Triblock Copolymer Networks via the PH, Macromolecules, 2011, 44(11), 4487–4495, DOI:10.1021/ma2002382.
- E. Lejeune, M. Drechsler, J. Jestin, A. H. E. Müller, C. Chassenieux and O. Colombani, Amphiphilic Diblock Copolymers with a Moderately Hydrophobic Block: Toward Dynamic Micelles, Macromolecules, 2010, 43(6), 2667–2671, DOI:10.1021/ma902822g.
- P. Raffa, P. Brandenburg, D. A. Z. Wever, A. A. Broekhuis and F. Picchioni, Polystyrene–Poly(Sodium Methacrylate) Amphiphilic Block Copolymers by ATRP: Effect of Structure, PH, and Ionic Strength on Rheology of Aqueous Solutions, Macromolecules, 2013, 46(17), 7106–7111, DOI:10.1021/ma401453j.
- H. Mori, E. Kudo, Y. Saito, A. Onuma and M. Morishima, RAFT Polymerization of Vinyl Sulfonate Esters for the Controlled Synthesis of Poly(Lithium Vinyl Sulfonate) and Sulfonated Block Copolymers, Macromolecules, 2010, 43(17), 7021–7032, DOI:10.1021/ma100905w.
- A. H. Hofman, R. Fokkink and M. Kamperman, A Mild and Quantitative Route towards Well-Defined Strong Anionic/Hydrophobic Diblock Copolymers: Synthesis and Aqueous Self-Assembly, Polym. Chem., 2019, 10(45), 6109–6115, 10.1039/C9PY01227C.
- P. Gurnani, C. P. Bray, R. A. E. Richardson, R. Peltier and S. Perrier, Heparin-Mimicking Sulfonated Polymer Nanoparticles via RAFT Polymerization-Induced Self-Assembly, Macromol. Rapid Commun., 2019, 19 Search PubMed.
- S. L. Canning, G. N. Smith and S. P. Armes, A Critical Appraisal of RAFT-Mediated Polymerization-Induced Self-Assembly, Macromolecules, 2016, 49(6), 1985–2001, DOI:10.1021/acs.macromol.5b02602.
- X. Wang and Z. An, New Insights into RAFT Dispersion Polymerization-Induced Self-Assembly: From Monomer Library, Morphological Control, and Stability to Driving Forces, Macromol. Rapid Commun., 2019, 40(2), 1800325, DOI:10.1002/marc.201800325.
- C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such and B. S. Hawkett, Ab Initio Emulsion Polymerization by RAFT-Controlled Self-Assembly, Macromolecules, 2005, 38(6), 2191–2204, DOI:10.1021/ma048787r.
- J. Yuan and M. Antonietti, Poly(Ionic Liquid)s: Polymers Expanding Classical Property Profiles, Polymer, 2011, 52(7), 1469–1482, DOI:10.1016/j.polymer.2011.01.043.
- J. C. Salamone, C. C. Tsai, A. P. Olson and A. C. Watterson, Ampholytic Polystyrene Ionomers from Cationic–Anionic Monomer Pairs, J. Polym. Sci., Polym. Chem. Ed., 1980, 18(10), 2983–2992, DOI:10.1002/pol.1980.170181008.
- T. Weng, J. Guo, X. Li, Y. Cui, X. Yang, K. Zhang, B. Zhang, G. Yin, S. V. Mikhalovsky, L. I. Mikhalovska, I. N. Savina, C. A. Howel and S. R. Sandeman, Synthesis of the Polymerizable Room Temperature Ionic Liquid AMPS – TEA and Superabsorbency for Organic Liquids of Its Copolymeric Gels with Acrylamide, Des. Monomers Polym., 2014, 17(2), 140–146, DOI:10.1080/15685551.2013.840480.
- X. Yang, Y. Fang, X. Li, K. Zhang, Y. Cui, B. Zhang and G. Yin, Synthesis of Two AMPS-Based Polymerizable Room Temperature Ionic Liquids and Swelling Difference between Their Co-Polymeric Gels with HEMA, e-Polym., 2014, 14(5), 335–343, DOI:10.1515/epoly-2014-0070.
- H. Diao, F. Yan, L. Qiu, J. Lu, X. Lu, B. Lin, Q. Li, S. Shang, W. Liu and J. Liu, High Performance Cross-Linked Poly(2-Acrylamido-2-Methylpropanesulfonic Acid)-Based Proton Exchange Membranes for Fuel Cells, Macromolecules, 2010, 43(15), 6398–6405, DOI:10.1021/ma1010099.
- M. Dang, Q. Deng, G. Fang, D. Zhang, J. Liu and S. Wang, Preparation of Novel Anionic Polymeric Ionic Liquid Materials and Their Potential Application to Protein Adsorption, J. Mater. Chem. B, 2017, 5(31), 6339–6347, 10.1039/C7TB01234A.
- A. Y. Bilibin, T. M. Shcherbinina, N. V. Girbasova, V. T. Lebedev, Y. V. Kulvelis, V. S. Molchanov and I. M. Zorin, Colloidal Properties of Polymerizable Counterion Surfmers Solutions Based on Alkylamino 2-Acrylamido-2-Methylpropanesulfonates in Different Solvents, Des. Monomers Polym., 2016, 19(5), 369–380, DOI:10.1080/15685551.2016.1169371.
- L. W. Fisher, A. R. Sochor and J. S. Tan, Chain Characteristics of Poly(2-Acrylamido-2-Methylpropanesulfonate) Polymers. 1. Light-Scattering and Intrinsic-Viscosity Studies, Macromolecules, 1977, 10(5), 949–954, DOI:10.1021/ma60059a012.
- L. W. Fisher, A. R. Sochor and J. S. Tan, Chain Characteristics of Poly(2-Acrylamido-2-Methylpropanesulfonate) Polymers. 2. Comparison of Unperturbed Dimensions and Persistence Lengths, Macromolecules, 1977, 10(5), 955–959, DOI:10.1021/ma60059a013.
- R. L. Weber, Y. Ye, A. L. Schmitt, S. M. Banik, Y. A. Elabd and M. K. Mahanthappa, Effect of Nanoscale Morphology on the Conductivity of Polymerized Ionic Liquid Block Copolymers, Macromolecules, 2011, 44(14), 5727–5735, DOI:10.1021/ma201067h.
- Y. Gu and T. P. Lodge, Synthesis and Gas Separation Performance of Triblock Copolymer Ion Gels with a Polymerized Ionic Liquid Mid-Block, Macromolecules, 2011, 44(7), 1732–1736, DOI:10.1021/ma2001838.
- P. Coupillaud, M. Fèvre, A.-L. Wirotius, K. Aissou, G. Fleury, A. Debuigne, C. Detrembleur, D. Mecerreyes, J. Vignolle and D. Taton, Precision Synthesis of Poly(Ionic Liquid)-Based Block Copolymers by Cobalt-Mediated Radical Polymerization and Preliminary Study of Their Self-Assembling Properties, Macromol. Rapid Commun., 2014, 35(4), 422–430, DOI:10.1002/marc.201300776.
- K. O. Sebakhy, S. Kessel and M. J. Monteiro, Nanoreactors to Synthesize Well-Defined Polymer Nanoparticles: Decoupling Particle Size from Molecular Weight, Macromolecules, 2010, 43(23), 9598–9600, DOI:10.1021/ma1019889.
- M. Siauw, B. S. Hawkett and S. Perrier, RAFT Polymerization: A Powerful Tool for the Synthesis and Study of Oligomers, in Progress in Controlled Radical Polymerization: Materials and Applications, ACS Symposium Series, American Chemical Society, 2012, vol. 1101, pp. 13–25. DOI:10.1021/bk-2012-1101.ch002.
- M. Wojdyr, Fityk: A General-Purpose Peak Fitting Program, J. Appl. Crystallogr., 2010, 43(5–1), 1126–1128, DOI:10.1107/S0021889810030499.
- X. Pan, F. Zhang, B. Choi, Y. Luo, X. Guo, A. Feng and S. H. Thang, Effect of Solvents on the RAFT Polymerization of N-(2-Hydroxypropyl) Methacrylamide, Eur. Polym. J., 2019, 115, 166–172, DOI:10.1016/j.eurpolymj.2019.03.016.
- A. Goto and T. Fukuda, Kinetics of Living Radical Polymerization, Prog. Polym. Sci., 2004, 29(4), 329–385, DOI:10.1016/j.progpolymsci.2004.01.002.
- P. Vana, T. P. Davis and C. Barner-Kowollik, Kinetic Analysis of Reversible Addition Fragmentation Chain Transfer (RAFT) Polymerizations: Conditions for Inhibition, Retardation, and Optimum Living Polymerization, Macromol. Theory Simul., 2002, 11(8), 823–835, DOI:10.1002/1521-3919(20021101)11:8<823::AID-MATS823>3.0.CO;2-R.
- J. Spěváček, 1H NMR Study of Styrene-Butadiene Block Copolymer Micelles in Selective Solvents, Makromol. Chem., Rapid Commun., 1982, 3(10), 697–703, DOI:10.1002/marc.1982.030031009.
- K. Philipps, T. Junkers and J. Michels, The Block Copolymer Shuffle in Size Exclusion Chromatography: The Intrinsic Problem with Using Elugrams to Determine Chain Extension Success, Polym. Chem., 2021, 12, 2522–2531, 10.1039/D1PY00210D.
- H. Matsuoka, M. Matsutani, E. Mouri and K. Matsumoto, Polymer Micelle Formation without Gibbs Monolayer Formation: Synthesis and Characteristic Behavior of an Amphiphilic Diblock Copolymer Having Strong Acid Groups, Macromolecules, 2003, 36(14), 5321–5330, DOI:10.1021/ma0215161.
- H. Matsuoka, S. Maeda, P. Kaewsaiha and K. Matsumoto, Micellization of Non-Surface-Active Diblock Copolymers in Water. Special Characteristics of Poly(Styrene)- Block -Poly(Styrenesulfonate), Langmuir, 2004, 20(18), 7412–7421, DOI:10.1021/la0492153.
- S. Garnier and A. Laschewsky, New Amphiphilic Diblock Copolymers: Surfactant Properties and Solubilization in Their Micelles, Langmuir, 2006, 22(9), 4044–4053, DOI:10.1021/la0600595.
- H. Matsuoka, H. Chen and K. Matsumoto, Molecular Weight Dependence of Non-Surface Activity for Ionic Amphiphilic Diblock Copolymers, Soft Matter, 2012, 8(35), 9140–9146, 10.1039/c2sm25710f.
Footnote |
| † Electronic supplementary information (ESI) available: Additional 1H-NMR spectra, UV-VIS, elemental analysis results, SEC traces, DOSY results and additional DLS information. See DOI: 10.1039/d1py00801c |
| This journal is © The Royal Society of Chemistry 2021 |