
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of ethylene–norbornene–1-octene terpolymers with high 1-octene contents, molar masses, and tunable Tg values, in high yields using half-titanocene catalysts†
Laura
Boggioni
 a,
Hitoshi
Harakawa
b,
Simona
Losio
a,
Kotohiro
Nomura
*b and
Incoronata
Tritto
*a
a,
Hitoshi
Harakawa
b,
Simona
Losio
a,
Kotohiro
Nomura
*b and
Incoronata
Tritto
*a
aIstituto di Scienze e Tecnologie Chimiche “G. Natta”, Consiglio Nazionale delle Ricerche, Via A. Corti, 12, 20133 Milano, Italy. E-mail: incoronata.tritto@scitec.cnr.it
bDepartment of Chemistry, Tokyo Metropolitan University, 1-1 Minami Osawa, Hachioiji, Tokyo 192-0397, Japan. E-mail: ktnomura@tmu.ac.jp
First published on 2nd July 2021
Abstract
Terpolymerizations of ethylene (E) and norbornene (N) with 1-octene (O) using different aryloxo- or ketimide-modified half-titanocene catalysts, Cp′TiCl2(O-2,6-iPr2C6H3) [Cp′ = indenyl (1), C5Me5 (Cp*, 2), tBuC5H4 (3)] and Cp′TiCl2(N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CtBu2) [Cp′ = tBuC5H4 (4), Cp (5)], in the presence of MAO cocatalysts were investigated. The microstructures, termonomer contents, and molecular weights of the resultant terpolymers, and their thermal properties were determined. The ketimide-modified Cp′TiCl2(NCtBu2) (4 and 5) exhibited higher catalytic activities and yielded higher molecular weight terpolymers, poly(E-ter–N-ter–O)s, than the aryloxo-modified Cp′TiCl2(O-2,6-iPr2C6H3) (1–3); the 1-octene incorporation is often higher than that prepared using ansa-metallocenes, noticeably reaching 13.5 mol%. Terpolymers prepared using ketimide-modified Cp′TiCl2(NCtBu2) possessed significantly higher Mn values than the E/N copolymers; the same trend is observed in the terpolymers prepared using the Cp*-aryloxo analogue (2). This has been demonstrated to be correlated with the lower propensity of catalysts 4 and 5 to provide β-H elimination, which occurred at the last-inserted E unit, as suggested through the analysis of the vinyl end groups observed in the 1H NMR spectra. This unique behaviour is quite different from that of well-developed ansa-metallocenes. Thus, poly(E-ter–N-ter–O)s with high O contents, molar masses, and Tg values from 23 °C to 153 °C were obtained in high yields using 2, 4, and 5 half-titanocene catalysts.
CtBu2) [Cp′ = tBuC5H4 (4), Cp (5)], in the presence of MAO cocatalysts were investigated. The microstructures, termonomer contents, and molecular weights of the resultant terpolymers, and their thermal properties were determined. The ketimide-modified Cp′TiCl2(NCtBu2) (4 and 5) exhibited higher catalytic activities and yielded higher molecular weight terpolymers, poly(E-ter–N-ter–O)s, than the aryloxo-modified Cp′TiCl2(O-2,6-iPr2C6H3) (1–3); the 1-octene incorporation is often higher than that prepared using ansa-metallocenes, noticeably reaching 13.5 mol%. Terpolymers prepared using ketimide-modified Cp′TiCl2(NCtBu2) possessed significantly higher Mn values than the E/N copolymers; the same trend is observed in the terpolymers prepared using the Cp*-aryloxo analogue (2). This has been demonstrated to be correlated with the lower propensity of catalysts 4 and 5 to provide β-H elimination, which occurred at the last-inserted E unit, as suggested through the analysis of the vinyl end groups observed in the 1H NMR spectra. This unique behaviour is quite different from that of well-developed ansa-metallocenes. Thus, poly(E-ter–N-ter–O)s with high O contents, molar masses, and Tg values from 23 °C to 153 °C were obtained in high yields using 2, 4, and 5 half-titanocene catalysts.
Introduction
Transition metal catalyzed olefin polymerization plays a key role in the synthesis of polyolefins and accounts for almost 50% of the global production of commercialized synthetic polymers. In this research field, great attention has been paid to the synthesis of new polymers with specific functions that are very difficult to synthesize by ordinary catalysts; many studies in the development of metallocenes,1,2 constrained geometry catalysts,3,4 nonbridged half-metallocenes,5–7 and so called non-metallocene type8–13 catalysts have thus been reported for the purpose. Copolymerization is an important tool that allows the modification of the physical and mechanical properties of the resultant polymers by varying the comonomer ratio and microstructure, which is possible by advances in catalyst design.1–23Cyclic olefin copolymers (COCs), prepared by metal catalyzed coordination polymerization with cyclic olefins, have promising commercial applications exemplified in optics, capacitor films, packaging, and medical containers. In particular ethylene (E)/norbornene (N) copolymers with more than 20 mol% of N content prepared using ansa-metallocene-based catalysts24,25 are amorphous thermoplastic materials, which exhibit interesting properties like excellent humidity barrier, variable and high glass transition temperatures, stiffness, thermoformability, notable biocompatibility, and chemical inertness. These properties are affected by the comonomer content and microstructure which are governed by the structure of the catalyst employed.26–39 Trade names of commercial grades of these copolymers are TOPAS40 and APEL.41
The introduction of a third monomer into the poly(E-co-N) chain may result in polymers with enhanced mechanical or other physical properties. However, only several reports for terpolymerization with ethylene and norbornene have been known.42–49 α-Olefins of linear 1-octene and alicyclic vinylcyclohexane were considered in the terpolymerization with E and N by two ansa-metallocenes.47 The presence of a long linear α-olefin within the copolymer backbone resulted in obtaining promising terpolymers with variable Tg and molar masses.
Nonbridged half-titanocenes containing anionic donor ligands of type, Cp′TiX2(Y) (Cp′ = cyclopentadienyl group and Y = aryloxo, ketimide, phosphinimide, etc.), are reported to be active for syntheses of new ethylene copolymers.50 The aryloxo or the ketimide analogues, Cp′TiCl2(O-2,6-iPr2C6H3) or Cp′TiCl2(NCtBu2), are, in particular, efficient for the copolymerization of ethylene with sterically encumbered olefins or cyclic olefins. Recently, ketimide-modified half-titanocenes, CpTiCl2(NCtBu2), known as active catalysts for E-co-N copolymerization,50 showed high catalytic activity with efficient N incorporation in the copolymerization of N with α-olefins affording high molecular weight copolymers with a high N content, unimodal molecular weight distribution and composition.51 However, as far as we know, examples of terpolymerization of ethylene, norbornene and 1-octene with aryloxo or the ketimide modified analogues are not found in the literature. Thus, we herein explored the synthesis of terpolymers of ethylene and norbornene with 1-octene, using a series of selected half-titanocenes (1–5, Scheme 1) with the aim of expanding the copolymer properties.
 | ||
| Scheme 1 Studied half-titanocene-based catalyst precursors used in the terpolymerization of ethylene (E), norbornene (N) and 1-octene (O). | ||
Terpolymerizations were investigated by employing half-titanocene based precursors shown in Scheme 1 activated with methylaluminoxane (MAO). In particular, the effect of termonomer (1-octene) concentration (conducted at high polymerization pressures and temperatures) on the polymerization behaviour was explored. Polymer analysis, including microstructure and end group analyses and determination of molar mass and thermal properties, shed light on the factors that determine termonomer insertion and chain termination. We herein demonstrate that these half-titanocene catalysts, unlike ansa-metallocenes, allow one to obtain poly(E-ter–N-ter–O)s with both high molar masses and O content, to modulate O and N incorporation in terpolymers and hence their Tg values in a range from 23 °C to 153 °C.
Experimental
Materials and general
All experiments with air-sensitive compounds were carried out under a nitrogen atmosphere using the standard Schlenk technique or using a drybox. Ethylene and nitrogen gases were purified by passing through a column of BTS-catalysts and molecular sieves. Freshly distilled (anhydrous, oxygen free) toluene over Na was used. Norbornene (Sigma-Aldrich Co. LLC) was also distilled from Na under a nitrogen atmosphere and used as a stock solution dissolved in toluene. 1-Octene (anhydrous, 98%, Sigma-Aldrich Co. LLC) distilled over CaH2 was stored under a nitrogen atmosphere in the presence of molecular sieves. Methylaluminoxane (MAO, 10 wt% solution in toluene, Crompton) was dried prior to use (50 °C, 3 h, 0.1 mmHg) by removing toluene and unreacted AlMe3 (TMA). (Indenyl)TiCl2(O-2,6-iPr2C6H3)16 (1), Cp*TiCl2(O-2,6-iPr2C6H3) (2),16 (tBuC5H4)TiCl2(O-2,6-iPr2C6H3) (3),16 (tBuC5H4)TiCl2(NCtBu2) (4),15 and CpTiCl2(NCtBu2) (5)15 were prepared according to the reported procedure. C2D2Cl4 was purchased from Merck KGaA and used as received.
Analytical measurements
NMR spectra for analysis of the resultant copolymers were recorded on a Bruker NMRAVANCE400 spectrometer (400 MHz, 1H; 100.58 MHz, 13C) operating at 100.58 MHz (13C) in the PFT mode working at 103 °C. The applied conditions were as follows: 10 mm probe, 90° pulse angle 13.5 μs power, 64K data points; acquisition time 4.52 s; relaxation delay 16 s; and 3K–4K transient. Proton broad-band decoupling was achieved with a 1D sequence using bi-waltz-16-32-power gate decoupling. The terpolymer sample (ca. 100 mg) was dissolved in tetrachloroethane-d2 in a 10 mm NMR tube. Chemical shifts for 1H NMR spectra were referred to tetrachloroethane-d2 (5.86 ppm) and hexamethyldisiloxane (HMDS) was used as the reference in the 13C spectra. Size exclusion chromatography (SEC, Waters, GPCV2000 high system) in o-dichlorobenzene (containing 0.05 wt/v% 2,6-di-tert-butyl-p-cresol) was conducted at 145 °C for the measurement of molecular weights and their distributions in the copolymers. The molecular weight was estimated on the basis of a calibration curve using standard polystyrene samples. The thermal properties of the resultant polymers were analysed by differential scanning calorimetry (DSC, PerkinElmer Pyris 1). The polymer samples were heated from −30 °C to 200 °C and then cooled (20 °C min−1) under a nitrogen atmosphere; these heating/cooling cycles were repeated twice; and the glass transition temperature (Tg) was thus determined in the second heating scan (middle of the transition curve).Polymerization procedure
Terpolymerization reactions were performed at 60 °C according to the following procedure. Into a 1-L Büchi BEP2000 laboratory autoclave (a type-I glass pressure vessel equipped with a heat jacket) pre-evacuated at 80 °C (for 60 min), a prescribed amount of toluene, norbornene, MAO were added under a nitrogen atmosphere. 1-Octene (prescribed amount) was then added to the solution. The reactor was then heated to 60 °C (with thermal equilibration), and ethylene was then filled at the prescribed pressure. Polymerization was initiated by injection of a toluene solution containing the precatalyst. During the polymerization, the ethylene pressure was kept constant (4 bar). The polymerization was terminated by injection of ethanol (2 mL) and HCl, and the resultant polymer was precipitated in acetone. The polymer slurry was stirred overnight, and the precipitates were collected by filtration, and the resultant polymers were suspended again in acetone with stirring. The polymers were then collected and dried in vacuo at 70 °C until their weights are constant.Results and discussion
Synthesis and characterization of poly(ethylene-ter–norbornene-ter–1-octene)s
Selected results of terpolymerizations of ethylene (E) and norbornene (N) with 1-octene (O) by using half-titanocenes, [Cp′TiCl2(O-2,6-iPr2C6H3), Cp′ = indenyl (1), Cp* (2), tBuC5H4 (3), Cp′TiCl2(NCtBu2), Cp′ = tBuC5H4 (4), and Cp (5)], activated with MAO, are presented in Table 1. The results of E/N copolymerization conducted under the same conditions are also presented in the table for comparison. The catalyst precursors were chosen as representative half-titanocenes concerning the synthesis of high molecular weight poly(E-co-N)s (4),50 remarkable catalytic activity (5),50 and high N incorporation (1).7,34 The [N]/[E] molar ratio was kept constant at 4.0 to prepare terpolymers with N contents higher than 30 mol%, and thus possess high glass transition temperatures (Tgs). The termonomer (1-octene, O) concentration was varied as much as possible in order to obtain the high molecular weight (Mw) terpolymers with various (high) termonomer contents. The effect of feed composition was investigated at 60 °C under an ethylene pressure of 4.0 bar. Their microstructures and contents in each monomer were determined by means of 13C NMR spectroscopy. The molar masses (molecular weights) and the Tg values in the resultant polymers were determined by SEC and DSC measurements, respectively.
CtBu2) [Cp′ = tBuC5H4 (4) and Cp (5)]-MAO catalyst systemsa
| Entry | Catalyst | N/E/O | Time (h) | Activityb | Ec (mol %) | Nc (mol%) | Oc (mol%) | M n ×10−3 | M w/Mn (PDI)d | T g (Tm)e (°C) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: catalyst = 5 μmol, [MAO]/[Ti] = 10.000, ethylene pressure = 4.0 bar, solvent = toluene, total volume of solution = 200 mL, and temperature = 60 °C. b Activity = (kg per mol Ti h). c Determined from the 13C NMR spectra in C2D2Cl4 at 103 °C with HMDS as the reference. d Number average molecular weight (Mn, g mol−1) determined by SEC analysis. e Determined from the DSC thermogram. f Entry 4 ΔH = 1.36 J g−1; entry 5 ΔH = 0.77 J g−1; entry 6 ΔH = 0.25 J g−1. g n.d. = not determined. | ||||||||||
| 1 | 1 | 4/1/0 | 0.75 | 405 | 32.8 | 67.2 | — | 8.6 | Bimodal | 131 |
| 2 | 1 | 4/1/0.5 | 0.50 | 413 | 34.4 | 58.0 | 7.6 | 8.4 | 2.99 | 94 |
| 3 | 1 | 4/1/1 | 0.25 | 879 | 31.5 | 59.1 | 9.4 | 8.0 | 2.11 | 81 |
| 4 | 2 | 4/1/0 | 0.50 | 109 | 69.8 | 30.2 | — | 59 | 3.01 | 65 (124)f |
| 5 | 2 | 4/1/0.5 | 0.50 | 456 | 58.3 | 34.2 | 7.5 | 60 | 1.78 | 35 (125)f |
| 6 | 2 | 4/1/1 | 0.17 | 1230 | 52.1 | 34.4 | 13.5 | 81 | 1.53 | 23 (127)f |
| 7 | 3 | 4/1/0 | 0.50 | n.d.g | 53.2 | 46.8 | — | 14.6 | Bimodal | — |
| 8 | 3 | 4/1/0.5 | 0.50 | 99 | 38.2 | 56.1 | 5.7 | 11 | 2.52 | 123 |
| 9 | 3 | 4/1/1 | 0.50 | 43 | 38.1 | 52.0 | 9.9 | 9.0 | 2.59 | — |
| 10 | 4 | 4/1/0 | 0.50 | 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 900 |
47.1 | 52.9 | — | 222 | 2.52 | n.d. |
| 11 | 4 | 4/1/0.5 | 0.17 | 4640 | 49.6 | 46.9 | 3.5 | 420 | 2.12 | 104 |
| 12 | 4 | 4/1/1 | 0.08 | 10400 |
50.7 | 44.8 | 4.5 | 442 | 1.86 | 103 |
| 13 | 5 | 4/1/0 | 0.17 | 21300 |
40.9 | 59.1 | — | 203 | 3.96 | n.d. |
| 14 | 5 | 4/1/0.5 | 0.08 | 34700 |
39.6 | 57.6 | 2.8 | 278 | 2.91 | 153 |
| 15 | 5 | 4/1/1 | 0.05 | 97700 |
42.2 | 52.2 | 5.6 | 361 | 1.95 | 132 |
It is clear that the activities of E/N copolymerization and E/N/O terpolymerization by the ketimide modified, Cp′TiCl2(NCtBu2) (4, 5), catalyst systems were much higher than those by the aryloxo modified, Cp′TiCl2(O-2,6-iPr2C6H3) (1–3), catalyst systems. This is especially evident from the results in the presence of the tBuC5H4 analogues (3 and 4). As reported in the E/N copolymerization,6,7,34,50 the activity was dependent upon the substituent on the cyclopentadienyl ligand employed; the tert-butyl substituted 3 is the least active among the aryloxo catalysts, whereas the unsubstituted CpTiCl2(NCtBu2) (5) is the most active among the ketimide modified catalysts. Moreover, it should be noted that the catalytic activities of 1, 2, and 5 increased by increasing the initial 1-octene concentration in the polymerizations. To our knowledge, the observed activities (by 5: 34700–97700 kg polymer per mol Ti h) are the highest among those reported for E/N/O terpolymerization by transition metal catalysts.
It was revealed that norbornene incorporation in the resultant copolymers prepared using the aryloxo catalyst systems, Cp′TiCl2(O-2,6-iPr2C6H3) (conducted under the same conditions), follows the order 1 ≥ 3 ≫ 2, suggesting that the steric hindrance in Cp′ leads to the lower N incorporation. The same trend was seen with the ketimide catalyst systems, Cp′TiCl2(NCtBu2), and N incorporation with 5 is higher than that with 4.
Interestingly, the addition of 1-octene resulted in unexpected features in comonomer incorporation. First, it is worth noting that 1-octene incorporation is often higher than that obtained under similar conditions with ansa-metallocenes,47 noticeably reaching 13.5 mol%. Instructive is that 1-octene generally competes more with ethylene than with norbornene in the copolymerization using a series of the aryloxo catalysts, Cp′TiCl2(O-2,6-iPr2C6H3). For example, the resultant copolymer in the E/N copolymerization by the Cp*-aryloxo analogue (2) contains 69.8 mol% of E and 30.2 mol% of N (entry 4), while the resultant terpolymer prepared at the same initial N/E molar ratio (with the addition of O) contains 52.1 mol% of E, 34.4 mol% of N and 13.5 mol% of O (entry 6). The same trend was also demonstrated by the tert-BuCp-aryloxo analogue (3, entry 7 vs. 8, 9). This trend, however, does not apply to that synthesized with the ketimide catalysts, Cp′TiCl2(NCtBu2), in which 1-octene competes with norbornene. For example, the resultant copolymer in the E/N copolymerization prepared using the Cp-ketimide analogue (5) contains 40.9 mol% of E and 59.1 mol% of N (entry 13), while the resultant terpolymer prepared at the same initial N/E molar ratio (with the addition of O) contains 42.2 mol% of E, 52.2 mol% of N and 5.6 mol% of O (entry 15). Similarly, the resultant copolymer in the E/N copolymerization prepared using the tert-BuCp-ketimide analogue (4) contains 47.1 mol% of E and 52.9 mol% of N (entry 10), while the terpolymer contains 50.7 mol% of E, 44.8 mol% of N, and 4.5 mol% of O (entry 12). It seems that 1-octene substitutes the comonomer present in the copolymer in a greater amount.
It was revealed that the Mn values (determined by SEC) of the terpolymers obtained using the ketimide modified catalysts, Cp′TiCl2(NCtBu2), are significantly higher than those obtained using the aryloxo catalysts, Cp′TiCl2(O-2,6-iPr2C6H3). Interestingly, the Mn values of terpolymers obtained using the tert-BuCp-ketimide analogue (4) are higher than those obtained using the Cp-ketimide analogue (5), and the Mn values increased upon increasing the initial O concentration in the feed. This is unexpected and in contrast to the results obtained for the terpolymer prepared using ansa-metallocenes.47 This unexpected behaviour was also observed in the terpolymerization using the Cp*-aryloxo analogue (2), Cp*TiCl2(O-2,6-iPr2C6H3), which exhibited the highest activities affording the copolymers with the highest Mn values among the aryloxo catalyst systems. The Mn values in the terpolymers prepared using 1 and 3 were relatively low, and the values decreased upon increasing the O content. It thus seems that higher molar masses were obtained with an increase in the 1-octene content in the copolymer parallel to the increase in activities.
Microstructure analysis of poly(ethylene-ter–norbornene-ter–1-octene)s
The resultant terpolymers were analysed by 13C NMR spectroscopy to estimate the monomer content and their microstructure. In the past, many efforts were dedicated to elucidating the 13C NMR spectra of poly(E-co-N) synthesized by metallocene based catalysts.31–38 It is possible to clarify the polymer microstructure at tetrad and pentad levels, which is dictated by the symmetry and ligand framework in the metallocene catalysts. The 13C NMR spectra of E–N terpolymers are of course more complex and general assignments have been made by comparison with the 13C NMR spectra of poly(E-co-N) and poly(E-ter–N-ter–O)47 prepared with different catalytic systems in the literature.
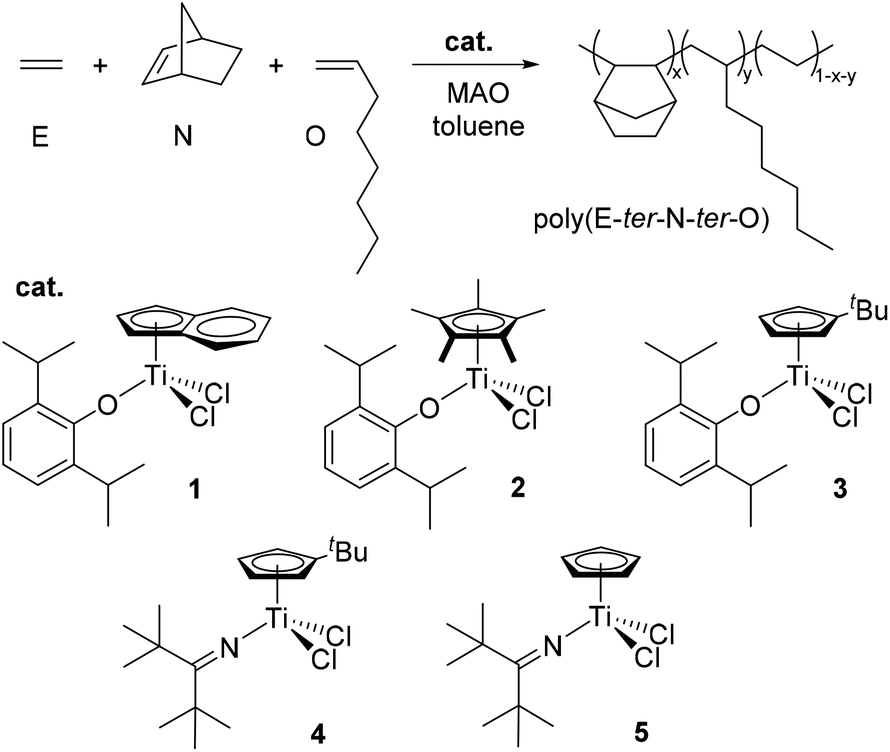
Fig. 1 shows the 13C NMR spectra of poly(E-co-N)s synthesized with ketimide catalysts Cp′TiCl2(NCtBu2). It is clear that these catalysts produce copolymers containing mostly racemic ENNE tetrads52 and a small number of meso tetrads in a ratio of 8/1 as well as racemic–racemic (r) and racemic–meso (rm) ENNN tetrads.36,37,52 Also m and r alternating NENE sequences are visible; in copolymers prepared using 4, the ratio between NENE/ENNE is 1.1 and in those prepared using 5, it is 0.73.
 | ||
| Fig. 1
13C NMR spectra of poly(E-co-N)s obtained using (a) (tBuC5H4)TiCl2(NCtBu2) (4, entry 10) and (b) CpTiCl2(NCtBu2) (5, entry 13). | ||
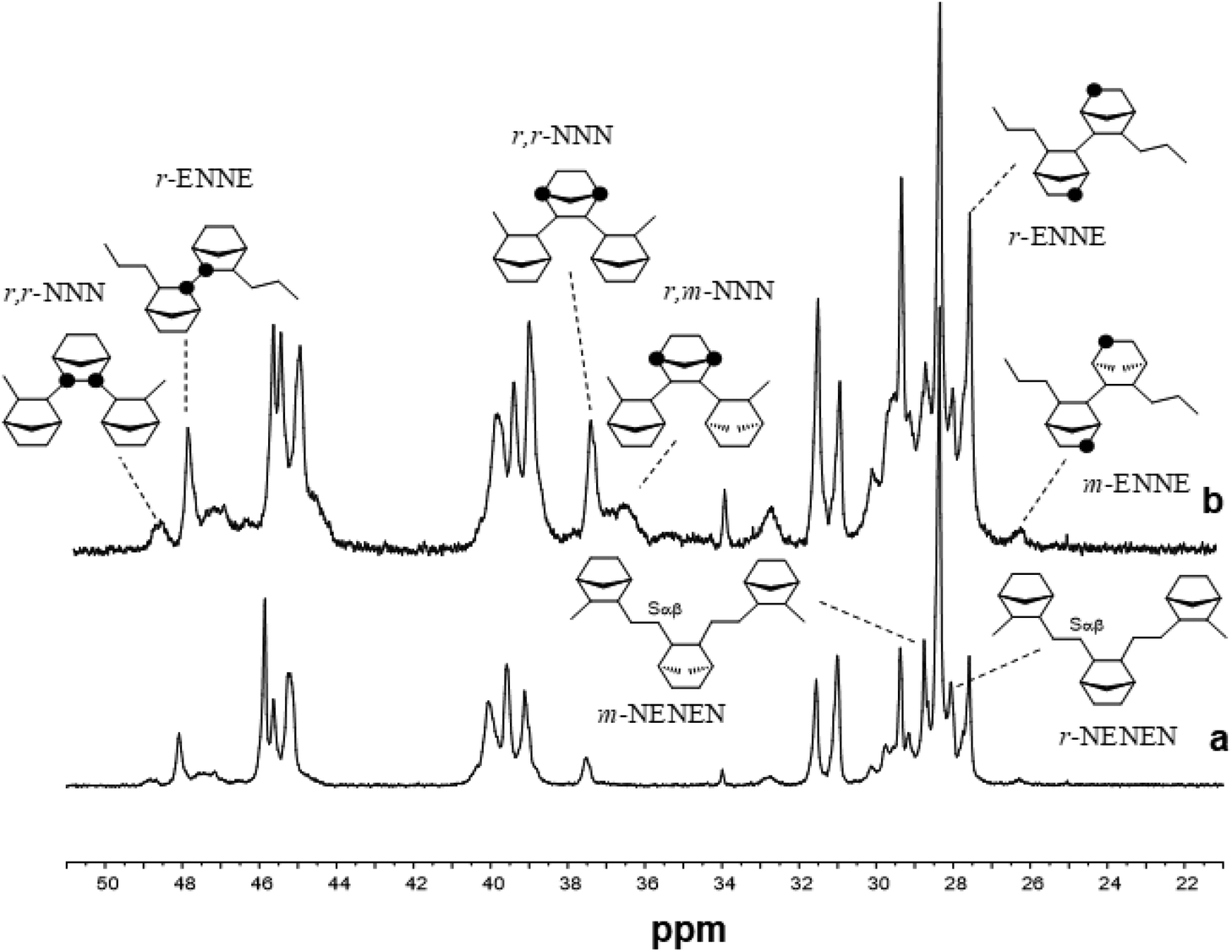
Fig. 2 shows the 13C NMR spectra of poly(E-co-N)s synthesized with the aryloxo Cp′TiCl2(O-2,6-iPr2C6H3) catalysts. The microstructure analysis of poly(E-co-N)s prepared using the Cp*-aryloxo catalyst (2), which afforded the E/N copolymer with the lowest norbornene content possessed a significant degree of NEEN (16.2 mol%) and ENEE (16.4 mol%) tetrads along with a lower amount of alternating m NENE tetrads (8.4 mol%) and r NENE (6.3 mol%) tetrads. Higher selectivity for producing r ENNE tetrads has been demonstrated with respect to the meso ones, while less than 0.5 mol% of NNNE tetrads have been calculated.
 | ||
| Fig. 2 13C NMR spectra of poly(E-co-N)s obtained using (a) Cp*TiCl2(O-2,6-iPr2C6H3) (2, entry 4), (b) (Ind)TiCl2(O-2,6-iPr2C6H3) (1, entry 1) and (c) (tBuC5H4)TiCl2(O-2,6-iPr2C6H3) (3, entry 7). | ||
The analysis of various stereosequences of poly(E-co-N) obtained by the indenyl catalyst (1) shows a high tendency of this catalyst to form ENNE tetrads (16.1 mol%) in the amount of r (9.0 mol%) and m (7.1 mol%) as well as mm NNN triads (1.2 mol%). Similar behaviour is shown by catalyst 3 with a marked tendency to form NNNE tetrads and NN dyads both r and m in a 2/1 ratio.
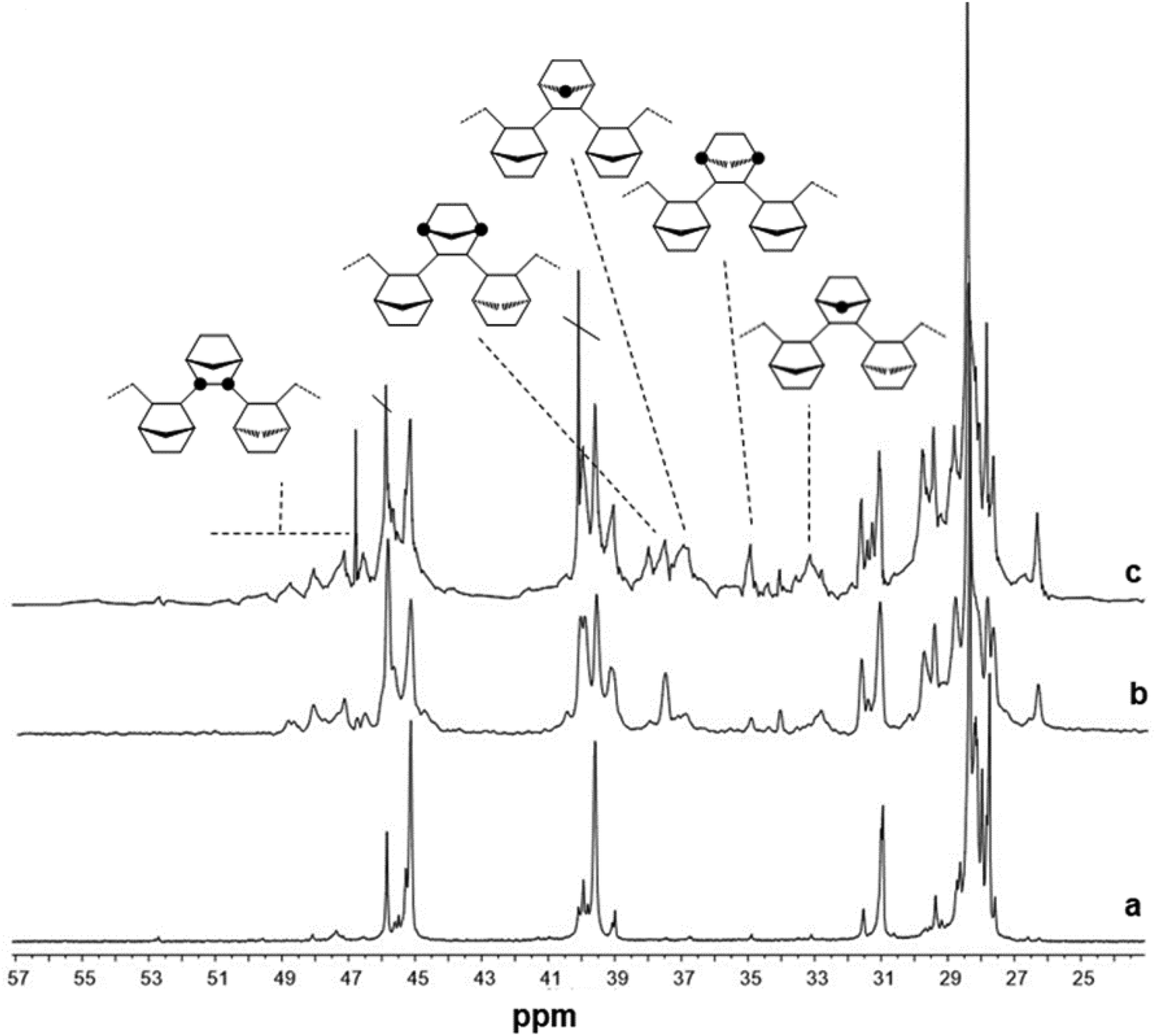
The 13C NMR spectra of poly(E-ter–N-ter–O) with the N/E/O molar ratio in the feed of 4/1/1 prepared using catalysts (tBuC5H4)TiCl2(NCtBu2) (4), CpTiCl2(NCtBu2) (5) and Cp*TiCl2(O-2,6-iPr2C6H3 (2) are shown for comparison in Fig. 3. A general structure of terpolymers with the carbon atom labelling based on accepted nomenclature47 is also included in the figure. The analysis of various stereosequences of the terpolymers with a higher amount of 1-octene in the feed (entries 6, 12, 15, 3, and 9) shows the presence of r NN dyads; entries 15, 3 and 9 present m NN dyads in a small amount.
 | ||
| Fig. 3
13CNMR spectra of poly(E-ter–N-ter–O) obtained using (a) (tBuC5H4)TiCl2(NCtBu2) (4, Table 1, entry 12), (b) CpTiCl2(NCtBu2) (5, Table 1, entry 15) and (c) Cp*TiCl2(O-2,6-iPr2C6H3 (2, Table 1, entry 6) catalysts with an N/E/O molar ratio in the feed of 4/1/1. | ||
The complete analysis of the spectra at the tetrad level38 showed that as in copolymers, catalyst 2 produced terpolymers with the lowest norbornene incorporation, but the highest incorporation of 1-octene compared to the other catalysts. The microstructure analysis revealed that the polymer possessed a high percentage of isolated norbornene units [NEEE (17.7 mol%) and ENEE (19.9 mol%)], a significant amount of NENE (12.5 mol%), and r ENNE sequences (4.2 mol%) in a moderate percentage. Instead, the microstructure analysis of the terpolymer prepared using catalyst 1 proved that the resultant terpolymers possess marked selectivity with a high degree of alternating NENE (15.2 mol%) and ENNE tetrads (13.8 mol%), 8.7 mol% racemic and 5.1 mol% meso dyads, and gave a small amount of NNEN stereosequences (3.8 mol%). Catalyst 3, as catalyst 1, has a noticeable selectivity for producing ENNE sequences (15.7 mol%) mainly racemic (10.0 mol%) and alternating NENE sequences (14.9 mol%).
As to terpolymers obtained using ketimide Cp′TiCl2(NCtBu2), it is clear from the spectra that 1-octene competes mainly with norbornene since the signals of ENNE and ENNN sequences are smaller than those of the corresponding copolymers. Terpolymers produced using catalyst 4 show a more alternating microstructure than the corresponding copolymers, with 18.0 mol% of NENE, 6.7 mol% of r ENNE and no m tetrads. Finally, catalyst 5 with the highest amount of r ENNE tetrads (15.4 mol%) and a small percentage of meso ones (1.7 mol%).
Chain end analysis and molar masses
As mentioned above, poly(E-ter–N-ter–O)s synthesized using 2, 4, and 5 catalyst systems have higher molar masses than the corresponding poly(E-co-N) copolymers, as evident from Fig. 4. This is a remarkable contrast, since the molecular weight decreased upon increasing the termonomer feed concentration in the terpolymerization with ansa-metallocene catalysts, clearly suggesting that 1-octene plays a role as chain termination and/or transfer agents, which means that 1-octene plays a role in terminating/reinitiating a growing polymer chain.47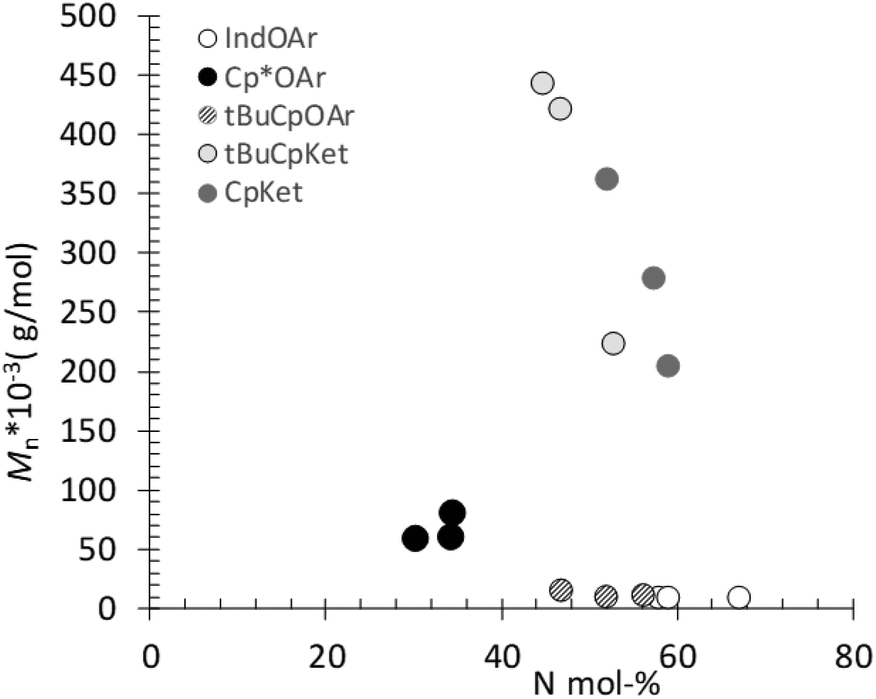 | ||
| Fig. 4 Molar masses (Mn) versus N content in poly(E-ter–N-ter–O), evidencing the increase of Mn with the 1-octene content. | ||
Polymer chain end analysis can provide evidence for the reactions involved in chain growth termination in a catalytic system. The relative areas of signals characteristic of unsaturated end-groups, deriving from β-H transfer to the metal or to the monomer, designate the possible chain transfer pathways.
Fig. 5 and 6 show the olefinic region of the 1H NMR spectra of poly(E-ter–N-ter–O) and poly(E-co-N) prepared using catalysts 2 and 5, respectively, recorded under the same conditions. The spectra revealed the existence of various olefinic double bonds, which are associated with terminal and internal unsaturations. Signal assignments made for poly(E-ter–N-ter–O)s are presented in Table 1S.†
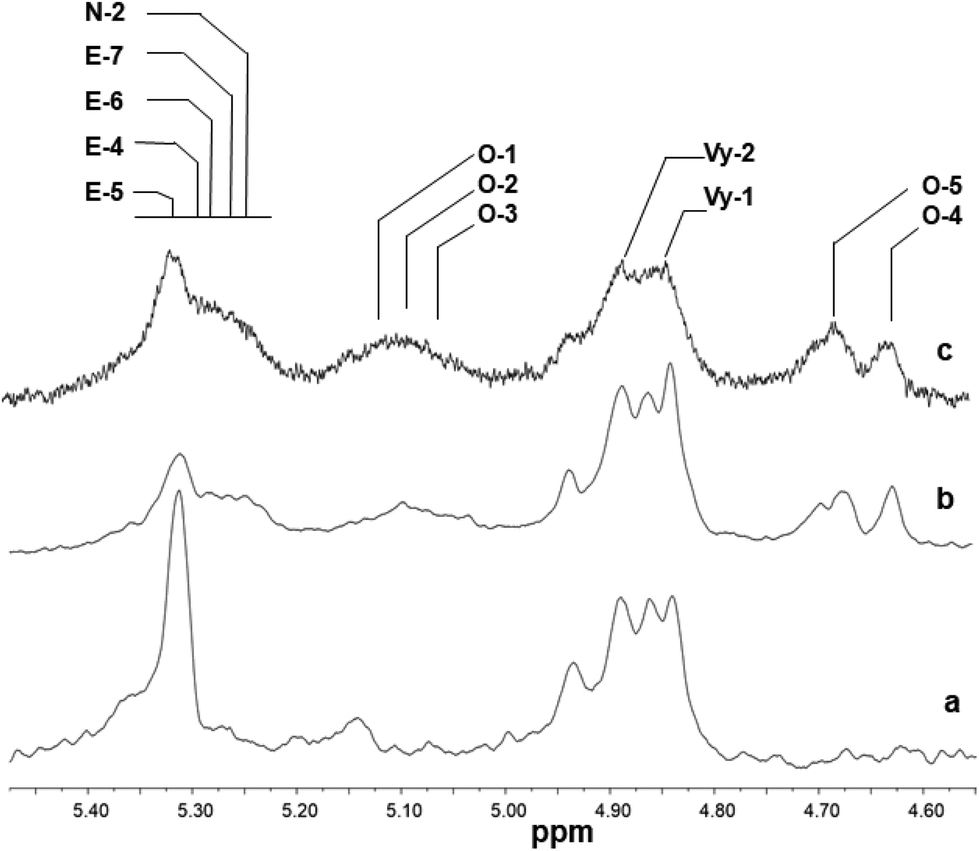 | ||
| Fig. 5 1H NMR spectra (expanded in the olefinic region, in tetrachloroethane-d2 at 103 °C) of polymers prepared using Cp*TiCl2(O-2,6-iPr2C6H3) (2): (a) poly(E-co-N) (Table 1, entry 4); (b) poly(E-ter–N-ter–O) (Table 1, entry 5); and (c) poly(E-ter–N-ter–O) (Table 1, entry 6). | ||
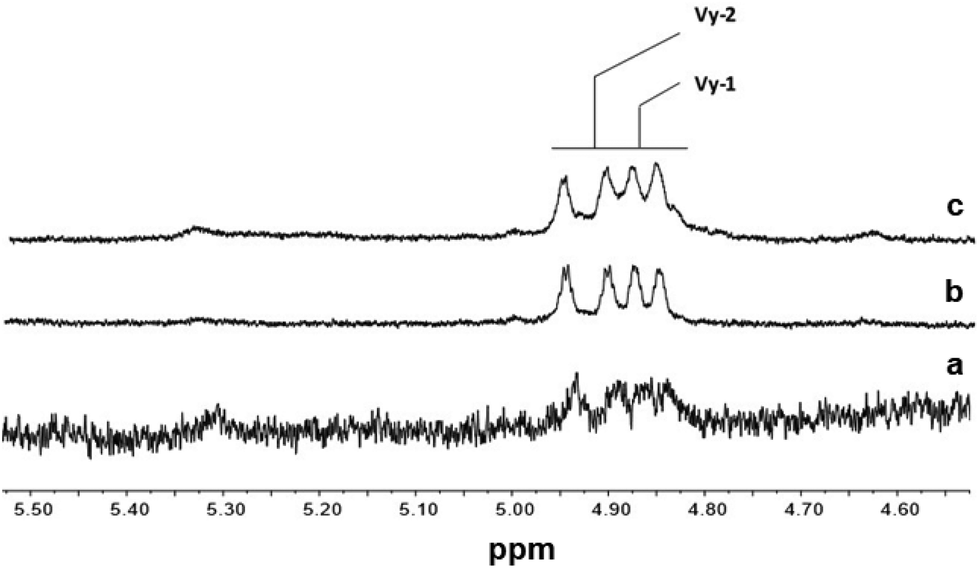 | ||
| Fig. 6
1H NMR spectra (expanded in the olefinic region, in tetrachloroethane-d2 at 103 °C) of polymers prepared using CpTiCl2(NCtBu2) (5): (a) poly(E-co-N) (Table 1, entry 13); (b) poly(E-ter–N-ter–O) (Table 1, entry 14); and (c) poly(E-ter–N-ter–O) (Table 1, entry 15). | ||
Possible pathways accounting for the formation of terpolymer chain end unsaturations that occurred at the last inserted E, O, or N unit are shown in Scheme 2.
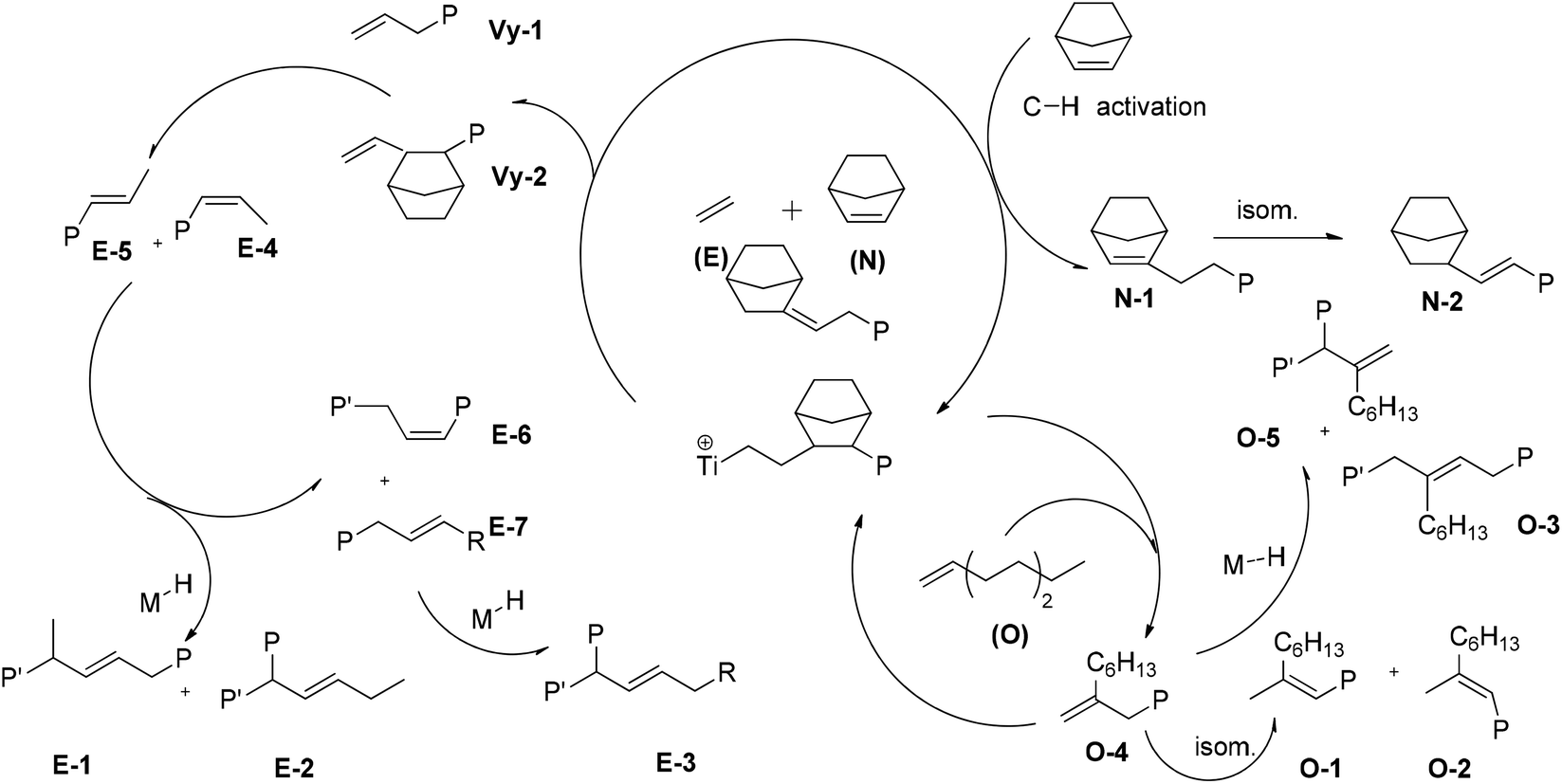 | ||
| Scheme 2 Possible pathways accounting for the observed terpolymer chain unsaturations. | ||
The 1H NMR spectra of polymers synthesized using 2, shown in Fig. 5, are different from those of poly(E-co-N) obtained using 5 (Fig. 6). It is evident that in the 1H NMR spectra of the polymers prepared using the ketimide modified CpTiCl2(NCtBu2) (5) catalyst, which gives co- and terpolymers with low 1-octene and very high molar masses, only Vy-1 and Vy-2 chain ends are observed, while in the spectra of co- and terpolymers prepared using 2 we observe a more complex pattern; signals of the groups N-2 + E-7 + E-4 + E-5 + E-6 and of E-1, E-2, and E-3 are visible. We recall that E-4 + E-5 derives from the secondary insertion of Vy-1 followed by β-H elimination and N-2 derives from CH activation followed by isomerisation. Fig. 7 shows a comparison of the 1H NMR spectra of poly(E-co-N)s prepared using 2 and 3. The difference between the two is in the pattern of Vy groups, which is more complex in the spectra of copolymers prepared using 3 and indicates that these copolymers, with high N contents and low molar masses, terminate more easily using β-H elimination occurred at the last E unit inserted after 2 or 3 N units. Thus, the factors that allow this catalyst to insert two or three consecutive N units are also those that induce low activity and low copolymer molar masses. Moreover, O-4 chain ends are well visible in the spectra of terpolymers as well as the signals of O-1, O-2, and O-3 end groups, which also arise from β-H elimination and 2-1 reinsertion.
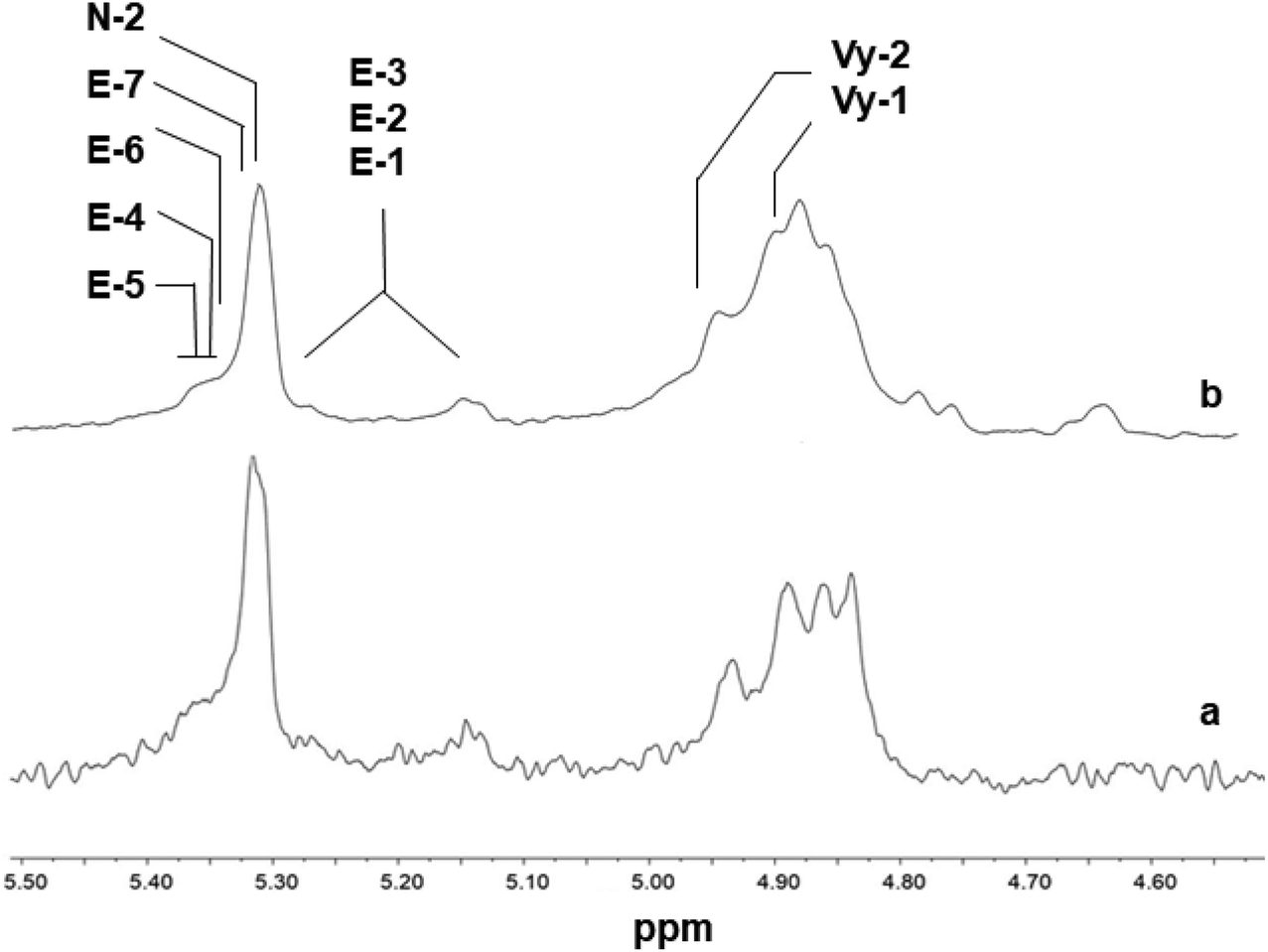 | ||
| Fig. 7 1H NMR spectra (expanded in the olefinic region, in tetrachloroethane-d2 at 103 °C) of poly(E-co-N)s synthesized using (a) Cp*TiCl2(O-2,6-iPr2C6H3) (2, Table 1, entry 4, % N = 30.2) and (b) (tBuC5H4)TiCl2(O-2,6-iPr2C6H3) (3, Table 1, entry 7, % N = 46.8). | ||
The 1H NMR spectra of terpolymers prepared using catalysts 1 and 3 (Fig. 8) are quite similar to those of 2. Some changes seem to arise from the vicinity at the chain ends of NN dyads or NNN triads with different microstructures.
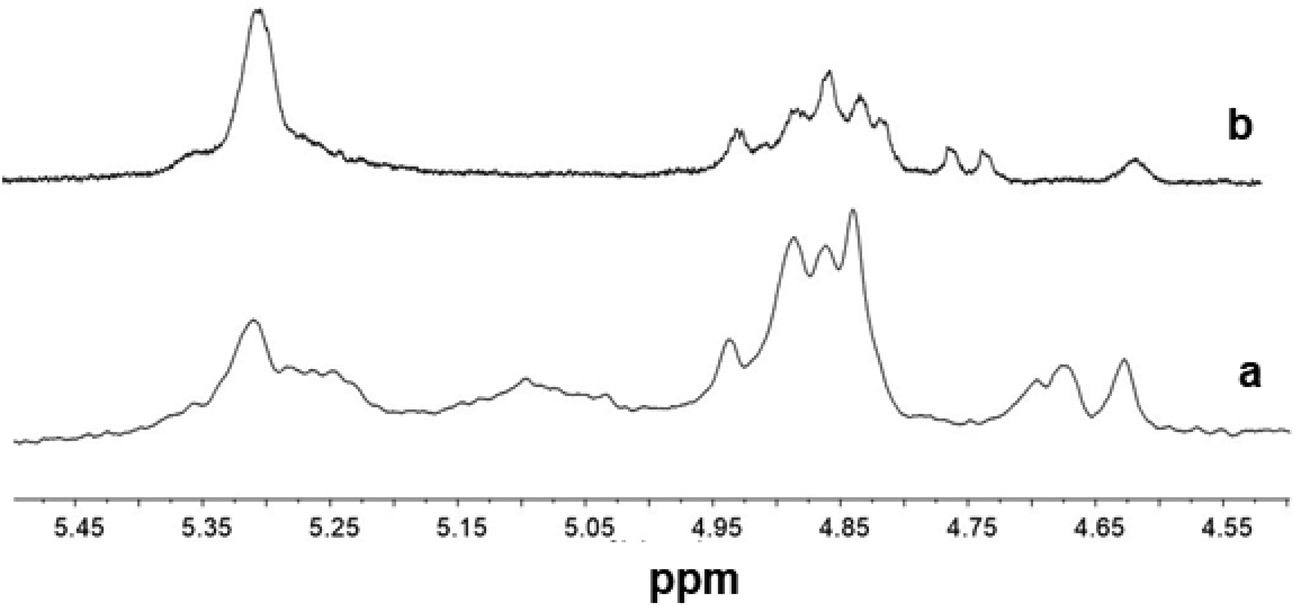 | ||
| Fig. 8 1H NMR spectra (expanded in the olefinic region, in tetrachloroethane-d2 at 103 °C) of poly(E-ter–N-ter–O)s synthesized using (a) Cp*TiCl2(O-2,6-iPr2C6H3) (2, Table 1, entry 5) and (b) (Ind)TiCl2(O-2,6-iPr2C6H3) (1, Table 1, entry 2). | ||
Thermal analysis
The thermal properties of terpolymers synthesized with catalysts 1, 3, 4, and 5, by DSC thermal analysis, showed obvious single glass transition temperatures (Tg). This observation indicates that these terpolymers have a completely amorphous morphology. The terpolymers prepared using 2 with a higher E content and an atactic alternating microstructure also showed a small endothermic peak corresponding to the melting of the crystalline phase. The already small copolymer crystallinity ΔH = 1.36 J g−1 decreases with an increase in the O content in the terpolymers ΔH = 0.77 and 0.25 J g−1 for entries 5 and 6, respectively (see Fig. 1S†). Glass transition temperatures spanned from 23 °C to 153 °C.In Fig. 9, the Tg values of all the synthesized polymers are shown versus the norbornene (Fig. 9a) and 1-octene (Fig. 9b) contents calculated from the 13C NMR spectra. It turned out that the Tg values of the polymers from 2 are the lowest, and since they have quite similar N content, their Tg values are strongly affected by the termonomer (O) content, as shown in Fig. 9b. A similar trend is visible in the Tg values of the polymers prepared using the other two aryloxo catalysts 1 and 3, and these Tg values are higher than those obtained using 2 due to the high N content of the terpolymers despite their low molar masses. The Tg values of the polymers with high molar masses in the series prepared using ketimide catalysts 4 and 5 clearly depend on the N content in the terpolymer and increase with an increase in the cyclic monomer content as expected. Fig. 9b clearly shows the strong dependence of Tg values on the O content of polymers synthesized using each catalyst.
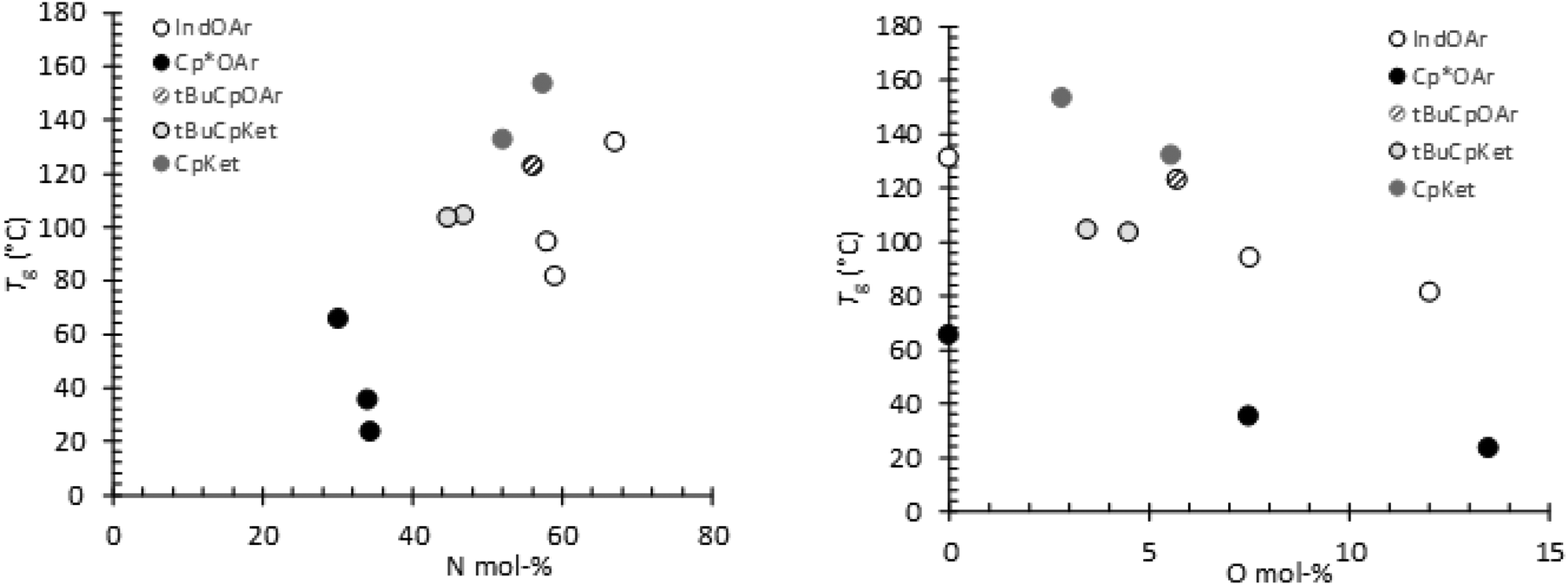 | ||
| Fig. 9 T g versus N content (left – a) and O content (right – b) in poly(E-ter–N-ter–O)s. | ||
Mechanistic considerations
From the results reported above, it is clear that aryloxo Cp′TiCl2(O-2,6-iPr2C6H3) (1–3) and ketimide Cp′TiCl2(NCtBu2) (4 and 5) precatalysts have a unique behaviour in E–N terpolymerization with 1-octene regarding 1-octene incorporation, 4 and 5 also regarding terpolymer molar masses, especially when compared to similar terpolymerization with ansa-metallocenes.47
The analysis of the copolymer microstructure reveals that CpTiCl2(NCtBu2) (5) behaves somehow similar to CsiPr(Cp)(Flu)ZrCl2 or Ph2C(Cp)(Flu)ZrCl2, which gives copolymers with only r ENNE sequences, while CpTiCl2(NCtBu2) produces copolymers with a higher N content, which contain besides r ENNE also a smaller amount of m ENNE and some rr and mr ENNN sequences. The same catalyst 5 allows higher O incorporation than ansa-metallocenes; in terpolymers ENNE and ENNN sequences decrease. This along with the great increase in the molar masses of terpolymers by the ketimide catalysts indicate that olefin insertion after NN dyads and NNN triads is relatively slow, though these catalysts allow high N incorporation. Thus, after two or three N units a kind of “semidormant state” is formed, which is reactivated by 1-octene insertion and this may explain why 1-octene competes with norbornene. The exceptional molar masses of these terpolymers, compared with those by ansa-metallocenes, arise from less tendency to give β-H elimination.
As to tert-butyl substitution, the copolymers obtained using (tBuC5H4)TiCl2(NCtBu2) (4) are characterized by a lower N content with respect to the copolymers prepared using the Cp analogue (5), still containing mainly r ENNE sequences. In ansa-metallocenes, iPr(3-iPr-Cp)(Flu)ZrCl235 or iPr(3-tBu-Cp)(Flu)ZrCl2 at a high N/E ratio produces mainly isotactic alternating poly(E-co-N) in contrast to the unsubstituted iPr(Cp)(Flu)ZrCl2 which gives copolymers with only r ENNE sequences.
Probably, norbornene and ethylene insertions occur at the same site by a Cossee's migratory mechanism as in ansa-metallocenes. However, the copolymer chain, owing to a more open structure of ketimide catalysts does not need to always back-skip to the original position to proceed with the next insertion, allowing 5 to form some m ENNE sequences. tert-Bu catalyst 4 makes a second N insertion possible without the need for the copolymer chain to back-skip to the original position which results in the isotactic alternating microstructure of poly(E-co-N)s by iPr or tBu(Cp)(Flu)ZrCl2.
For understanding the catalytic behavior of aryloxo Cp′TiCl2(O-2,6-iPr2C6H3) catalytic systems, it can be useful to compare the copolymers prepared using Cp*TiCl2(O-2,6-iPr2C6H3) (2) with those prepared using Me2Si(Me4Cp)(NtBu)TiCl2, the so-called constrained geometry catalysts (CGCs) with an open structure. The metal center of CGCs when activated with MAO gives copolymers containing both m and r alternating NEN-sequences and isolated norbornene units. It has been shown that this catalyst allows for poly(E-co-N) containing long-chain branches formed by the insertion of a macromonomer obtained after β-H elimination.33 Thus, it is reasonable that 2 incorporates a high amount of 1-octene.
It is worth recalling that the 1H NMR spectra of all poly(E-co-N)s and poly(E-ter–N-ter–O), obtained using Cp*TiCl2(O-2,6-iPr2C6H3) and from the other aryloxo Cp′TiCl2(O-2,6-iPr2C6H3) catalyst systems, show signals of E-1, E-2 and E-3 trans vinyl chain ends which derive from the isomerization of Vy-1 and Vy-2 chain ends. Moreover, in the spectra of terpolymers, terminal groups at the last 1-octene unit arising from β-H elimination and 2-1 reinsertion are visible. The spectra of copolymers prepared using 1 indicate that these copolymers, with high N content and low molar masses, terminate more easily by β-H elimination at the last E unit inserted after 2 or 3 N units. Thus, the factors that allow this catalyst to insert two or three consecutive N units are also those that induce low activity and low copolymer molar masses.
This comparison indicates that ketimide catalysts have a more rigid structure than aryloxo catalysts. The higher mobility of the framework of the aryloxo catalysts is responsible for the lower molar masses and for the high O and N incorporation. This explains the high incorporation of comonomers with highly steric bulk such as 2-methyl-1-pentene, cyclohexene, and tert-butylethylene in ethylene copolymerization using the aryloxide analogues.6,7
Finally, an increase in molar masses along with the O content, which is the novelty in these terpolymerizations, arises also from the lower tendency of these catalysts to give β-H elimination.
Conclusions
Investigation of E–N terpolymerization with 1-octene as the α olefin using aryloxo Cp′TiCl2(O-2,6-iPr2C6H3) (1–3) and ketimide Cp′TiCl2(NCtBu2) (4–5) precatalysts sheds light on the factors that influence the growth or the termination of terpolymer chains with these half-titanocene precatalysts.
The activities of copolymerization and terpolymerization by the ketimide modified Cp′TiCl2(NCtBu2) are higher than those using the aryloxo Cp′TiCl2(O-2,6-iPr2C6H3) catalyst systems. The steric hindrance of ligand substitution of the catalyst also affects activities; tert-butyl substituted 3 and 4 are the least active, among the aryloxo and ketimide modified catalysts, respectively.
Regarding 1-octene incorporation, it is very interesting that O incorporation is often higher than that obtained under similar conditions with ansa-metallocenes, noticeably reaching 13.5 mol%. Moreover, in the series with aryloxo Cp′TiCl2(O-2,6-iPr2C6H3) catalysts, in general, 1-octene competes more with ethylene than with norbornene, while in the series synthesized with the ketimide Cp′TiCl2(NCtBu2) catalysts 1-octene competes with norbornene.
Microstructural analysis of co- and terpolymers reveals that those prepared using aryloxo Cp′TiCl2(O-2,6-iPr2C6H3) catalysts are mainly atactic, while those prepared using the ketimide Cp′TiCl2(NCtBu2) catalysts are more syndiotactic.
As far as polymer molar masses are concerned, the molar masses of co- and terpolymers synthesized using the ketimide precatalysts are much higher than those synthesized using the aryloxo precatalysts. Interestingly, terpolymers prepared using the ketimide modified Cp′TiCl2(NCtBu2) catalysts have significantly higher molar masses than copolymers, and the same trend is observed in terpolymers obtained using the aryloxo Cp*TiCl2(O-2,6-iPr2C6H3) catalyst system 2. This behaviour is unique and is very different from that of well-developed ansa-metallocenes, important catalysts for α-olefin and norbornene copolymerization, where 1-octene behaves as a chain termination/transfer agent.
Chain end group analysis gave evidence for the low tendency of catalysts 4 and 5 to give β-H elimination at the last inserted E or O unit, while in the 1H NMR spectra of poly(E-co-N) polymers prepared using 1–3 catalysts, signals of the groups arising from β-H elimination and reinsertion and subsequent isomerisation are visible. In the 1H NMR spectra of poly(E-ter–N-ter–O) with high O contents and low molar masses using the same catalysts, signals of chain end groups formed after the last O insertion are also well visible.
Finally, DSC thermal analysis showed clearly the strong dependence of Tg values on the O content of polymers synthesized using each catalyst. In particular, 2, 4, and 5 half-titanocene catalysts allow one to obtain poly(E-ter–N-ter–O) with high molar masses and to modulate O and N incorporation and thus Tg values from 23 °C to 153 °C. These amorphous and more flexible polymers, due to their expected excellent optical properties, will allow the broadening of COC applications, for example, in electronic flexible devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Fabio Bertini, Fulvia Greco and Daniele Piovani for their help with the DSC, NMR and SEC characterization, respectively. This project by KN was partly supported by the Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS, no. 18H01982 and 21H01942).Notes and references
- H. H. Brintzinger, D. Fischer, R. Mülhaupt, B. Rieger and R. M. Waymouth, Angew. Chem., Int. Ed. Engl., 1995, 34, 1143–1170 CrossRef CAS.
- W. Kaminsky and M. Arndt, Adv. Polym. Sci., 1997, 127, 143–187 CrossRef CAS.
- A. L. McKnight and R. M. Waymouth, Chem. Rev., 1998, 98, 2587–2598 CrossRef CAS PubMed.
- H. Braunschweig and F. M. Breitling, Coord. Chem. Rev., 2006, 250, 2691–2720 CrossRef CAS.
- K. Nomura, J. Liu, S. Padmanabhan and B. Kitiyanan, J. Mol. Catal. A: Chem., 2007, 267, 1–29 CrossRef CAS.
- K. Nomura, Dalton Trans., 2009, 41, 8811–8823 RSC.
- K. Nomura and J. Liu, Dalton Trans., 2011, 40, 7666–7682 RSC . References including a list of reported complexes are cited therein.
- G. J. P. Britovsek, V. C. Gibson and D. F. T. Wass, Angew. Chem., Int. Ed., 1999, 38, 428–447 CrossRef CAS PubMed.
- P. D. Bolton and P. Mountford, Adv. Synth. Catal., 2005, 347, 355–366 CrossRef CAS.
- K. Nomura and S. Zhang, Chem. Rev., 2011, 111, 2342–2362 CrossRef CAS PubMed . References (concerning vanadium complex catalyzed olefin polymerization) are cited therein.
- H. Makio, H. Terao, A. Iwashita and T. Fujita, Chem. Rev., 2011, 111, 2363–2449 CrossRef CAS.
- C. Redshaw and Y. Tang, Chem. Soc. Rev., 2012, 41, 4484–4510 RSC.
- J. A. Gladysz, Chem. Rev., 2000, 100, 1167–1168 CrossRef CAS PubMed.
- H. G. Alt, Coord. Chem. Rev., 2006, 250, 1–272 CrossRef CAS.
- K. Nomura, N. Naga, M. Miki and K. Yanagi, Macromolecules, 1998, 31, 7588–7597 CrossRef CAS.
- K. Nomura, K. Fujita and M. Fujiki, Organometallics, 1998, 17, 2152–2154 CrossRef CAS.
- Metal-catalysed Polymerisation, ed. B. Milani and C. Claver, Dalton Trans., 2009, vol. 41, pp. 8769–9076 Search PubMed.
- Metal-catalyzed polymerization of olefins, ed. B. Liu, M. Terano, V. Busico, W.-Y. Wong and Y. Tang, J. Organomet. Chem., 2015, vol. 798, pp. 291–436 Search PubMed.
- G. J. Domski, J. M. Rose, G. W. Coates, A. D. Bolig and M. Brookhart, Prog. Polym. Sci., 2007, 32, 30–92 CrossRef CAS.
- J. P. McInnis, M. Delferro and T. Marks, Chem. Rev., 2011, 111, 2450–2485 CrossRef PubMed.
- J. P. McInnis, M. Delferro and T. Marks, Acc. Chem. Res., 2014, 47, 2545–2557 CrossRef CAS PubMed.
- A. Valente, A. Mortreux, M. Visseaux and P. Zinck, Chem. Rev., 2013, 113, 3836–3857 CrossRef CAS PubMed.
- H. Harakawa, S. Patamma, A. C. Boccia, L. Boggioni, D. R. Ferro, S. Losio, K. Nomura and I. Tritto, Macromolecules, 2018, 51, 853–863 CrossRef CAS.
- W. Kaminsky, A. Bark and R. Steiger, J. Mol. Catal., 1992, 72, 109–119 CrossRef.
- W. Kaminsky and M. Arndt-Rosenau, in Metallocene-based Polyolefins, ed. J. Scheirs and W. Kaminsky, Wiley, Chichester, 2000, p. 89 and references therein Search PubMed.
- D. Ruchatz and G. Fink, Macromolecules, 1998, 31, 4674–4680 CrossRef CAS PubMed.
- N. Herfert, P. Montag and G. Fink, Makromol. Chem., 1993, 194, 3167–3182 CrossRef CAS.
- M. Arndt-Rosenau and I. Beulich, Macromolecules, 1999, 32, 7335–7343 CrossRef CAS.
- I. Tritto, L. Boggioni, M. C. Sacchi and P. Locatelli, J. Mol. Catal. A: Chem., 1998, 133, 139–150 CrossRef CAS.
- A. Provasoli, D. R. Ferro, I. Tritto and L. Boggioni, Macromolecules, 1999, 32, 6697–6706 CrossRef CAS.
- I. Tritto, C. Marestin, L. Boggioni, L. Zetta, A. Provasoli and D. R. Ferro, Macromolecules, 2000, 33, 8931–8944 CrossRef CAS.
- I. Tritto, C. Marestin, L. Boggioni, L. Zetta, A. Provasoli, H. H. Brintzinger and D. R. Ferro, Macromolecules, 2001, 34, 5770–5777 CrossRef CAS.
- K. Thorshaug, R. Mendichi, I. Tritto, S. Trinkle, C. Friedrich and R. Mülhaupt, Macromolecules, 2002, 35, 2903–2911 CrossRef CAS.
- K. Nomura, M. Tsubota and M. Fujiki, Macromolecules, 2003, 36, 3797–3799 CrossRef CAS.
- I. Tritto, L. Boggioni and D. R. Ferro, Macromolecules, 2004, 37, 9681–9693 CrossRef CAS.
- T. Hasan, T. Shiono and T. Ikeda, Makromol. Symp., 2004, 213, 123–129 CrossRef CAS.
- T. Hasan, T. Shiono and T. Ikeda, Macromolecules, 2004, 23, 8503–8509 CrossRef.
- I. Tritto, L. Boggioni and D. R. Ferro, Coord. Chem. Rev., 2006, 250, 212–241 CrossRef CAS.
- I. Tritto, A. Ravasio, L. Boggioni, F. Bertini, J. Hitzbleck and J. Okuda, Macromol. Chem. Phys., 2010, 211, 897–904 CrossRef CAS.
- see http//www.topas.com and M.-J. Brekner, F. Osan, J. Rohrmann and M. Antberg, Process for the preparation of chemically homogeneous cycloolefin copolymers, U.S. Patent5324801, 1994 Search PubMed.
- See https://jp.mitsuichemicals.com/.
- H.-T. Ban, N. Nishii, Y. Tsunogae and T. Shiono, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2765–2773 CrossRef CAS.
- S. Liu, Z. Yao, K. Cao, B. Li and S. Zhu, Macromol. Rapid. Commun., 2009, 30, 548–553 CrossRef CAS PubMed.
- H. Lasarov and T. T. Pakkanen, Macromol. Rapid. Commun., 2001, 22, 434–438 CrossRef CAS.
- R. A. Wendt and G. Fink, Macromol. Chem. Phys., 2000, 201, 1365–1373 CrossRef CAS.
- F. G. Sernetz and R. Mülhaupt, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 2549–2560 CrossRef CAS.
- R. Marconi, A. Ravasio, L. Boggioni, F. Di Colo, I. Tritto and U. M. Stehling, Macromolecules, 2011, 44, 795–804 CrossRef CAS.
- R. Tanaka, M. Goda, Z. Cai, Y. Nakayama and T. Shiono, Polymer, 2015, 70, 252–256 CrossRef CAS.
- L. Boggioni, N. G. Galotto, F. Bertini and I. Tritto, Polymers, 2016, 8, 60–78 CrossRef.
- K. Nomura, W. Wang, M. Fujiki and J. Liu, Chem. Commun., 2006, 25, 2659–2661 RSC.
- W. Zhao and K. Nomura, Macromolecules, 2016, 49, 59–70 CrossRef CAS.
- C2/C3 of norbornene can be either S/R or R/S, so the relationship between two subsequent norbornene units (NN) or two norbornenes in alternating NEN sequences can be either erythro di-isotactic (meso, m) or erythro di-syndiotactic (racemic, r); for more details see ref. 38.
Footnote |
| † Electronic supplementary information (ESI) available: Determination of the termonomer content; 1H NMR data of the olefinic chain ends in the investigated terpolymers (Table 1S). Fig. 1S: DSC curves of entries 4–6 obtained using catalyst 2; Fig. 2S: DSC; and Fig. 3S: GPC. See DOI: 10.1039/d1py00647a |
| This journal is © The Royal Society of Chemistry 2021 |