
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of diblock copolymer spheres, worms and vesicles via RAFT aqueous emulsion polymerization of hydroxybutyl methacrylate†
Saul J.
Hunter
 a,
Joseph R.
Lovett
a,
Oleksandr O.
Mykhaylyk
a,
Elizabeth R.
Jones
b and
Steven P.
Armes
*a
a,
Joseph R.
Lovett
a,
Oleksandr O.
Mykhaylyk
a,
Elizabeth R.
Jones
b and
Steven P.
Armes
*a
aDainton Building, Department of Chemistry, The University of Sheffield, Brook Hill, Sheffield, South Yorkshire S3 7HF, UK. E-mail: s.p.armes@shefieldf.ac.uk
bDSM Applied Science Centre, P.O. Box 1066, 6160 BB Geleen, The Netherlands
First published on 9th June 2021
Abstract
There are many literature examples of reversible addition–fragmentation chain transfer (RAFT) aqueous emulsion polymerization that produce only kinetically-trapped spheres, even when targeting highly asymmetric diblock copolymer compositions. Recently, we postulated that the aqueous solubility of the vinyl monomer was likely to be a key parameter for overcoming this morphological limitation. In the present study, the RAFT aqueous emulsion polymerization of hydroxybutyl methacrylate (HBMA) has been revisited using a relatively short non-ionic poly(glycerol monomethacrylate) (PGMA) precursor as a steric stabilizer block. HBMA was selected for its relatively high aqueous solubility (∼25 g dm−3 at 50 °C). Conversions of more than 99% were achieved within 2 h at 50 °C using a low-temperature azo initiator (VA-044), as indicated by 1H NMR studies. Gel permeation chromatography analysis confirmed that high blocking efficiencies and relatively low dispersities (Mw/Mn < 1.37) could be achieved under these conditions. A pseudo-phase diagram was constructed by systematically increasing the PHBMA target DP from 10 to 120 and varying the copolymer concentration between 5 and 20% w/w. Only spheres, vesicles or mixed phases were accessible at 5% w/w copolymer concentration, with higher concentrations being required to access a pure worm phase. Transmission electron microscopy (TEM) and small-angle X-ray scattering studies indicated the formation of well-defined diblock copolymer worms and vesicles when targeting longer PHBMA blocks. The evolution in copolymer morphology when targeting PGMA41-PHBMA120 vesicles was monitored using TEM. This technique revealed intermediate morphologies that are strikingly similar to those reported during the preparation of PGMA47-PHPMA200 vesicles via RAFT aqueous dispersion polymerization (A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581). This suggests that the formation of vesicles via RAFT aqueous emulsion polymerization occurs via essentially the same mechanism. Finally, linear PGMA41-PHBMA110 vesicles were evaluated as putative Pickering emulsifiers for the stabilization of n-dodecane droplets in water. Such nano-objects survive high-shear homogenization and stabilize genuine Pickering emulsions, unlike linear PGMA45-PHPMA200 vesicles (K. L. Thompson, P. Chambon, R. Verber and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 12450).
Introduction
Over the past two decades, there has been considerable interest in using controlled living radical polymerization techniques to prepare amphiphilic diblock copolymers in the form of nanoparticles in aqueous media via polymerization-induced self-assembly (PISA).1–15 PISA occurs in situ when a water-soluble homopolymer is chain-extended with an appropriate second monomer. The growing second block gradually becomes insoluble in the reaction solution, which drives in situ self-assembly. In principle, PISA offers control over the final morphology simply by targeting appropriate diblock compositions.16 More specifically, the geometric packing parameter17 for the copolymer chains governs the preferred morphology provided that certain other criteria are met, such as a suitably short steric stabilizer block and a sufficiently high copolymer concentration.11 Typically, spheres,18–20 worms19,21 or vesicles22–28 can be obtained using this approach, but lamellae have also been observed.15,29,30 In particular, reversible addition–fragmentation chain transfer (RAFT) polymerization has enabled the controlled polymerization of many water-immiscible monomers such as styrene, n-butyl acrylate, methyl methacrylate, vinyl acetate, benzyl methacrylate, glycidyl methacrylate and 2,2,2-trifluoroethyl methacrylate.31–47In principle, such RAFT aqueous emulsion polymerization formulations provide a convenient surfactant-free route for the synthesis of sterically-stabilized nanoparticles with various morphologies.11,48,49 In practice, there are many literature examples of such syntheses that are restricted to kinetically-trapped spheres.36,40,42,43,45,50–54 Currently, this limitation is not well-understood and there is no satisfactory explanation for the relatively few exceptions that provide access to so-called higher order morphologies such as worms or vesicles.46,55–61 Recently, we proposed that the aqueous solubility of the water-immiscible monomer is a key parameter for preparing block copolymer worms or vesicles via RAFT aqueous emulsion polymerization.44,62–64 This concept was initially explored by Cockram et al.,44 who investigated the RAFT aqueous emulsion polymerization of hydroxybutyl methacrylate (HBMA). This monomer has an aqueous solubility of ∼20 g dm−3 at 70 °C, which is appreciably higher than that for more traditional vinyl monomers such as styrene or n-butyl acrylate.65 RAFT aqueous emulsion polymerization of HBMA using a partially ionized poly(methacrylic acid) stabilizer block at pH 5 produced a new non-spherical ‘monkey nut’ morphology.44 Subsequently, Foster and co-workers obtained a mixed phase of worms and vesicles by chain-extending a non-ionic poly(ethylene glycol) (PEG113) precursor with HBMA.66 Similarly, Hatton et al. explored the RAFT aqueous emulsion polymerization of GlyMA (aqueous solubility = 18–20 g dm−3 at 50 °C)67 using a non-ionic poly(glycerol monomethacrylate) (PGMA) precursor as a steric stabilizer block.45,62,64 Well-defined diblock copolymer worms and vesicles were obtained when the PGMA stabilizer block was sufficiently short, which is known to promote access to higher order morphologies.16,62–64 Furthermore, Dai and co-workers recently prepared poly(poly(ethylene glycol) methyl ether methacrylate) (PPEGMA)-PGlyMA diblock copolymer spheres, worms and vesicles via redox-initiated RAFT aqueous emulsion polymerization.49 More recently, Brotherton and co-workers reported that RAFT aqueous emulsion polymerization of 2-methoxyethyl methacrylate (MOEMA), a third methacrylic monomer with moderate aqueous solubility (19.6 g dm−3 at 70 °C).63 In this case in situ small-angle X-ray scattering (SAXS) was used to monitor the in situ evolution in copolymer morphology from spheres to worms to vesicles during the polymerization when targeting a sufficiently asymmetric diblock composition.63 Vinyl acetate is another example of a monomer with moderate aqueous solubility (26 g dm−3 at 50 °C) that allows access to higher-order morphologies during PISA syntheses.47 More specifically, Galanopoulo and co-workers reported the formation of spheres, vesicles or large compound vesicles when chain-extending a water-soluble PEG precursor with vinyl acetate via RAFT aqueous emulsion polymerization.47
In 2014 Ratcliffe et al. briefly examined the RAFT aqueous emulsion homopolymerization of HBMA as part of a broader copolymerization study.68 More specifically, a carboxylic acid functionalized PGMA60 precursor was chain-extended with HBMA at 70 °C to prepare a series of kinetically-trapped PGMA60-PHBMAx diblock copolymer spheres (x = 75–500) at around pH 5–6. In 2015 Lovett et al. reported that terminal anionic charge introduced into the steric stabilizer chains via ionization of carboxylic acid end-groups is sufficient to drive a worm-to-sphere transition.69 This explains why only kinetically-trapped spheres were observed by Ratcliffe et al.68 Herein, we revisit this PGMA-PHBMA formulation using a relatively short PGMA41 precursor to gain access to spheres, worms and vesicles. A pseudo-phase diagram is constructed to enable the reproducible targeting of pure copolymer morphologies. Furthermore, monitoring the evolution in copolymer morphology by transmission electron microscopy (TEM) when targeting vesicles for this aqueous PISA formulation reveals various transient intermediates similar to those observed during the RAFT aqueous dispersion polymerization of 2-hydroxypropyl methacrylate (HPMA) by Blanazs et al.70 Finally, the performance of linear PGMA41-PHBMA110 vesicles as a Pickering emulsifier for n-dodecane droplets is critically compared to that previously reported for the analogous linear PGMA45-PHPMA200 vesicles.71
Experimental
Materials
Glycerol monomethacrylate (GMA) was kindly donated by GEO Specialty Chemicals (Hythe, UK) and used without further purification. Hydroxybutyl methacryate (HBMA; 94% purity; comprising a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 4-hydroxybutyl methacrylate and 2-hydroxybutyl methacrylate) and 4,4′-azobis(4-cyanopentanoic acid) (ACVA; 99%) and tolylene 2,4-diisocyanate-terminated poly(propylene glycol) (PPG-TDI) were purchased from Sigma-Aldrich (UK) and were used as received. 2,2′-Azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (VA-044; ≥97%) and 2-cyano-2-propyl dithiobenzoate (CPDB) were purchased from Strem Chemicals Ltd (Cambridge, UK) and used as received. Deuterated methanol (CD3OD) was purchased from Goss Scientific Instruments Ltd (Cheshire, UK). All other solvents were purchased from Fisher Scientific (Loughborough, UK) and were used as received. Deionized water was used for all experiments.
:1 mixture of 4-hydroxybutyl methacrylate and 2-hydroxybutyl methacrylate) and 4,4′-azobis(4-cyanopentanoic acid) (ACVA; 99%) and tolylene 2,4-diisocyanate-terminated poly(propylene glycol) (PPG-TDI) were purchased from Sigma-Aldrich (UK) and were used as received. 2,2′-Azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (VA-044; ≥97%) and 2-cyano-2-propyl dithiobenzoate (CPDB) were purchased from Strem Chemicals Ltd (Cambridge, UK) and used as received. Deuterated methanol (CD3OD) was purchased from Goss Scientific Instruments Ltd (Cheshire, UK). All other solvents were purchased from Fisher Scientific (Loughborough, UK) and were used as received. Deionized water was used for all experiments.
Synthesis of the PGMA41 precursor
A round-bottomed flask was charged with GMA (20.00 g, 0.126 mol), CPDB (0.614 g, 2.22 mmol; target DP = 57), ACVA (0.124 g, 0.444 mmol; CPDB/ACVA molar ratio = 5.0), and anhydrous ethanol (30.9 g) to afford a 40% w/w solution. The resulting pink solution was purged with N2 gas for 30 min before the sealed flask was immersed into an oil bath set at 70 °C. After 140 min (70% conversion as judged by 1H NMR spectroscopy), the GMA polymerization was quenched by immersion of the flask into an ice bath and subsequently exposing the reaction mixture to air. The crude polymer was then precipitated twice into excess dichloromethane and washed three times with this solvent before being freeze-dried overnight. 1H NMR studies indicated a mean degree of polymerization of 41 via end-group analysis (by comparing the integrated aromatic RAFT end-group signals at 7.1–7.4 ppm to those assigned to the two oxymethylene protons at 3.5–4.4 ppm). Gel permeation chromatography (GPC) studies indicated an Mn of 13900 g mol−1 and an Mw/Mn of 1.18.
Synthesis of PGMA41-PHBMAx diblock copolymer nanoparticles by RAFT aqueous emulsion polymerization of HBMA
The following example is representative of the general protocol. PGMA41 precursor (0.149 g, 21.9 μmol), HBMA monomer (0.417 g, 2.63 mmol; target DP = 120), VA-044 initiator (1.77 mg, 52.3 μmol; PGMA41/VA-044 molar ratio = 4.0), and deionized water (5.11 g, 10% w/w solids) were added to a 14 mL sample vial. This reaction solution was purged using N2 gas for 30 min at 20 °C prior to immersing the flask into an oil bath set at 50 °C. After 2 h, the HBMA polymerization was quenched by exposing the contents of the flask to air, after 120 min followed by cooling to ambient temperature.Preparation of Pickering emulsions
n-Dodecane (2.0 ml) was homogenized at 20 °C with a 0.06–2.00% w/w aqueous dispersion of linear PGMA41-PHBMA110 vesicles (2.0 ml) for 2 min at 12000 rpm using a IKA Ultra-Turrax T-18 homogenizer equipped with a 10 mm dispersing tool. After appropriate dilution, the resulting oil droplets were imaged by optical microscopy and their volume-average droplet diameter was determined by laser diffraction.
Colloidosome formation
PPG-TDI (20.0 g dm−3) was weighed into a sample vial and then dissolved in n-dodecane (2.0 ml) prior to homogenization with 2.0 ml of a 0.25% w/w aqueous dispersion of linear PGMA41-PHBMA110 vesicles for 2 min at 20 °C using a IKA Ultra-Turrax T-18 homogenizer equipped with a 10 mm dispersing tool operating at 12000 rpm. The resulting stable milky-white emulsion was allowed to stand at 20 °C for several hours to allow the urethane cross-linking reaction to proceed.
1H NMR spectroscopy
Spectra were recorded in CD3OD at 20 °C using a Bruker Avance III HD 400 MHz spectrometer with 64 scans being averaged per spectrum.Dynamic light scattering (DLS)
Hydrodynamic diameters (Dh) and polydispersity indices (PDI) were determined by DLS via the cumulants method utilizing a Malvern Zetasizer NanoZS instrument. All measurements were performed on 0.1% copolymer dispersions (prepared by dilution using deionized water) using disposable plastic cuvettes. All data were averaged over three consecutive runs.Gel permeation chromatography (GPC)
Copolymer molecular weight distributions were assessed using the following GPC set-up. Two Agilent PL gel 5 μm Mixed-C columns and a guard column were connected in series to an Agilent 1260 Infinity GPC system equipped with both refractive index and UV–visible detectors (only the refractive index detector was used in the present study) operating at 60 °C. The GPC eluent was HPLC-grade DMF containing 10 mM LiBr at a flow rate of 1.0 mL min−1. Calibration was achieved using a series of ten near-monodisperse poly(methyl methacrylate) standards (Mp ranging from 625 to 618000 g mol−1). Chromatograms were analyzed using Agilent GPC/SEC software provided by the manufacturer.
Transmission electron microscopy (TEM)
Copper/palladium TEM grids (Agar Scientific, UK) were coated in-house to yield a thin film of amorphous carbon. The grids were subjected to a plasma glow discharge for 30 s. One droplet of each dilute aqueous copolymer dispersion (or aqueous emulsion) (10 μL, 0.1% w/w copolymer concentration or 0.1% oil droplets, respectively) was placed in turn on a freshly-treated grid for 1 min and then carefully blotted with filter paper to remove excess solution. To ensure sufficient electron contrast, a droplet of uranyl formate (10 μL of a 0.75% w/w solution) was placed on the sample-loaded grid for 20 s and then blotted to remove excess stain. Each grid was carefully dried using a vacuum hose. Imaging was performed using a FEI Tecnai Spirit 2 microscope operating at 80 kV and fitted with an Orius SC1000B camera.Optical microscopy (OM)
Optical microscopy images of Pickering emulsions were recorded using a Cole–Palmer compound optical microscope equipped with an LCD tablet display and a Moticam BTW digital camera.Laser diffraction
Each Pickering emulsion was sized by laser diffraction using a Malvern Mastersizer 3000 instrument equipped with a Hydro EV wet sample dispersion unit, a red HeNe laser operating at 633 nm and a LED blue light source operating at 470 nm. The stirring rate was adjusted to 1500 rpm in order to avoid creaming of the emulsion droplets during analysis. After each measurement, the cell was rinsed once with ethanol and three times with deionized water and the laser was aligned centrally to the detector prior to data acquisition.Small angle X-ray scattering (SAXS)
SAXS patterns were recorded for selected 1.0% w/w aqueous dispersions of PGMA41-PHBMAx nanoparticles at the European Synchrotron Radiation Facilities (ESRF, station ID02, Grenoble, France) using monochromatic X-ray radiation (λ = 0.0995 nm; q range = 0.002 to 0.15 Å−1, where q is the length of the scattering vector and θ is one-half of the scattering angle, such that q = 4πsinθ/λ) and a Rayonix MX-170HS CCD detector. A flow-through horizontal capillary set-up was used as the sample holder, with a 2.0 mm diameter glass capillary. Scattering data were reduced using standard routines from the beamline and were further analyzed using Irena SAS macro72 for Igor Pro. Water was used for absolute intensity calibration.
Results and discussion
RAFT emulsion polymerization of HBMA
A PGMA precursor with mean degree of polymerization (DP) of 41 was synthesized via RAFT ethanolic solution polymerization of GMA using a dithiobenzoate-based RAFT agent (CPDB) at 70 °C, as previously described.42 This PGMA precursor was then chain-extended via RAFT aqueous emulsion polymerization of HBMA at 50 °C, as shown in Scheme 1. The mean target DP of the core-forming PHBMA block was systematically varied between 10 and 120 for copolymer concentrations ranging between 5% and 20% w/w. High HBMA conversions (≥99%) were achieved for all PISA syntheses, as confirmed by 1H NMR spectroscopy studies (see Fig. S1†). | ||
| Scheme 1 RAFT aqueous emulsion polymerization of HBMA (N.B. this monomer comprises a 1:1 mixture of the 2- and 4-isomers but only the latter is depicted in this scheme) using a PGMA41 precursor provides convenient access to diblock copolymer spheres, worms or vesicles depending on the target degree of polymerization (x) for the PHBMA block. | ||
DMF GPC studies of a series of five PGMA41-PHBMAx diblock copolymers indicated relatively narrow, unimodal molecular weight distributions (dispersities ranging from 1.18 to 1.37) and high blocking efficiencies (see Fig. 1). It is worth noting that similar dispersities (Mw/Mn = 1.16 to 1.32 when targeting PHBMA DPs between 75 and 175, respectively) were reported by Ratcliffe et al. for PGMA60-PHBMAx spheres.67 In striking contrast, Cockram and co-workers obtained dispersities ranging from 1.93 to 6.13 for methylated PMAA56-PHBMAx diblock copolymers when targeting PHBMA DPs between 130 and 300, respectively.44 There are several reasons for these differences. Firstly, the batch of HBMA monomer used in the present study may contain significantly less dimethacrylate impurity than that used by Cockram and co-workers.44 Secondly, the non-ionic PGMA41-PHBMAx diblock copolymer chains prepared herein do not require any methylation prior to GPC analysis, unlike the anionic PMAA-PHBMA copolymer chains prepared by Cockram and co-workers.44 It is known that exhaustive methylation of acidic blocks can introduce GPC artefacts.44 Thirdly, rather lower PHBMA DPs are required to target spheres, worms and vesicles in the present study, which should minimize the light branching resulting from any dimethacrylate impurities.73
 | ||
| Fig. 1 Overlaid DMF GPC traces recorded for a series of five PGMA41-PHBMAx diblock copolymers prepared by RAFT aqueous emulsion polymerization of HBMA (conditions: 10% w/w, 50 °C, 2 h) for x = 30, 50, 70, 90 and 110. The GPC curve for the PGMA41 precursor is also shown as a reference. Molecular weight data are expressed relative to a series of near-monodisperse poly(methyl methacrylate) calibration standards. | ||
A pseudo-phase diagram was constructed for a series of PGMA41-PHBMAx diblock copolymer nano-objects using morphology assignments based on TEM studies, (see Fig. 2 and Fig. S2†). In general, the copolymer morphology depends on the copolymer concentration, with lower concentrations favouring the formation of spherical nanoparticles. For example, the PISA synthesis of PGMA41-PHBMA60 at 15% w/w produces a pure worm phase (Fig. 2b), whereas targeting the same composition at 5% w/w produces only spheres. The sphere phase is unusually narrow at copolymer concentrations of 10–20% w/w. Furthermore, increasing the copolymer concentration enables access to worm and vesicle phases even when targeting relatively short PHBMA blocks. In contrast, for the synthesis of PGMA28-PMOEMAx nanoparticles via RAFT aqueous emulsion polymerization, shorter hydrophobic PMOEMA blocks were required to access vesicles at lower copolymer concentrations.63 In the current study, pure vesicles can be obtained at each of the copolymer concentrations investigated, even at 5% w/w. In contrast, pure worms can only be obtained at a minimum copolymer concentration of 10% w/w. Clearly, the problem of kinetically-trapped spheres that is often encountered for RAFT aqueous emulsion polymerization formulations can be overcome by appropriate monomer selection. More specifically, the water-immiscible monomer (e.g., GlyMA, MOEMA or HBMA) should exhibit moderate aqueous solubility (∼20 g m−3). This ensures enhanced polymer chain mobility within the growing monomer-swollen nanoparticle cores within the relatively short time scale of the polymerization, thus enabling evolution of the copolymer morphology. In this context, it is perhaps also noteworthy that pure worms and vesicles can be obtained with a somewhat longer steric stabilizer block (PGMA41) when using HBMA compared to GlyMA (PGMA25) and MOEMA (PGMA29), respectively.62,63
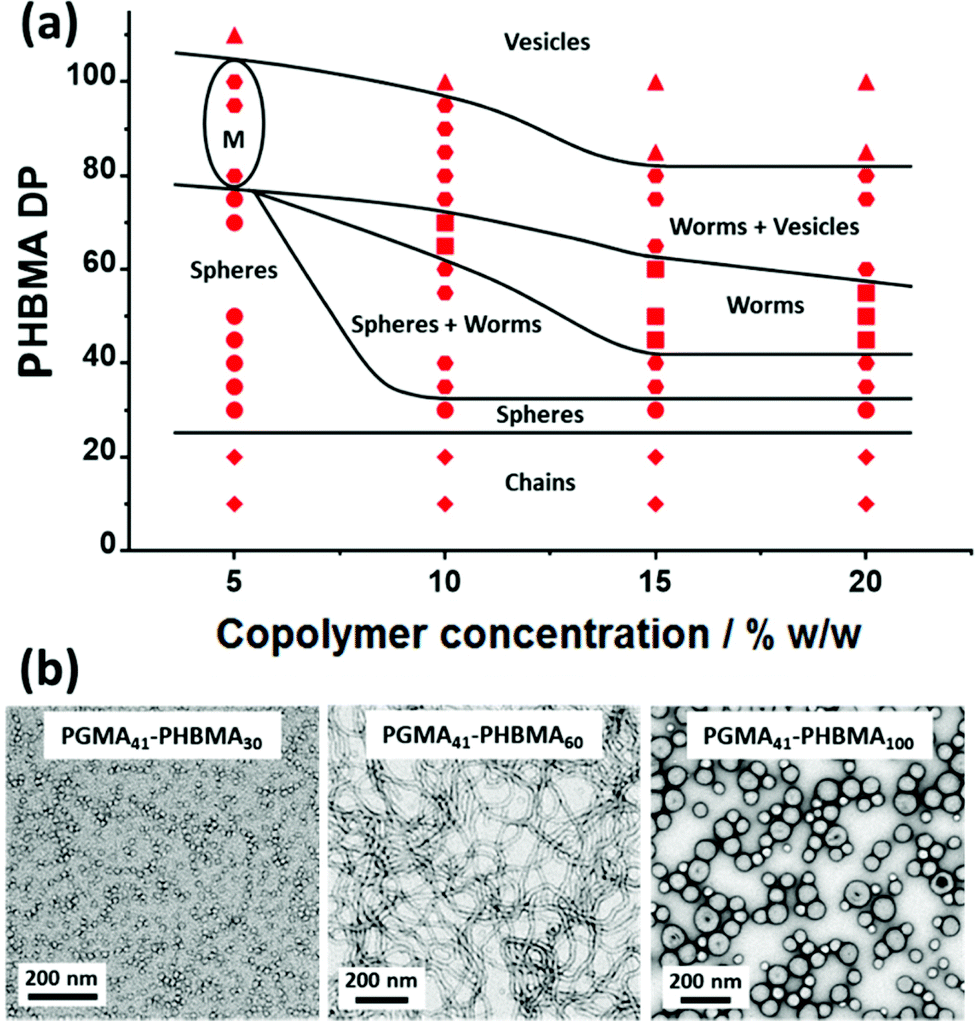 | ||
| Fig. 2 (a) Pseudo-phase diagram constructed for a series of PGMA41-PHBMAx diblock copolymer nanoparticles prepared by RAFT aqueous emulsion polymerization of HBMA using a PGMA41 precursor at a copolymer concentration of 5–20% w/w [N.B. ‘M’ denotes a mixed phase comprising spheres, worms and vesicles]. (b) Representative TEM images are shown for PGMA41-PHBMAx nano-objects prepared at 15% w/w, where x = 30 (spheres), 60 (worms) and 100 (vesicles). | ||
SAXS patterns were recorded for 1.0% w/w aqueous dispersions of PGMA41-PHBMAx spheres, worms and vesicles prepared at 10% w/w, see Fig. 3. This ten-fold dilution ensures that the structure factor can be assumed to be unity, which simplifies the data analysis.74 The SAXS pattern obtained for PGMA41-PHBMA30 can be satisfactorily fitted using a spherical micelle model75 (see Fig. 3a and Table S5† for more details). This approach enables a volume-average core diameter of 12.2 nm to be calculated. Assuming that the mean radius of gyration of the PGMA41 stabilizer chains is 2.2 nm, the overall volume-average diameter calculated from this fit is 21 nm. Bearing in mind the effect of polydispersity, this is consistent with the intensity-average diameter of 25 ± 8 nm reported by DLS. Moreover, this scattering pattern has a low q gradient of approximately zero, which is consistent with the morphology indicated by TEM studies and further indicates non-interacting spheres. The SAXS pattern recorded for PGMA41-PHBMA70 can be fitted using a worm-like micelle model,75 where the low q gradient of approximately −1 is consistent with the highly anisotropic worm morphology indicated by the corresponding TEM image (see Fig. 3b).76,77 SAXS patterns recorded for PGMA41-PHBMA100 and PGMA41-PHBMA120 could each be fitted using a vesicle model;78 a low q gradient of approximately −2 was observed in each case, which is consistent with the presence of bilayers (see Fig. 3c). These analyses indicated volume-average diameters of 108 nm and 75 nm, respectively. Given that such small vesicles are much less prone to collapse under ultrahigh vacuum conditions, this morphology could be incorrectly assigned as spheres based on TEM analysis alone. Such situations serve to highlight the value of performing SAXS studies, which readily enable discrimination between spheres and vesicles. Similar observations have been made for relatively small vesicles prepared via RAFT aqueous emulsion polymerization of MOEMA63 and GlyMA.45
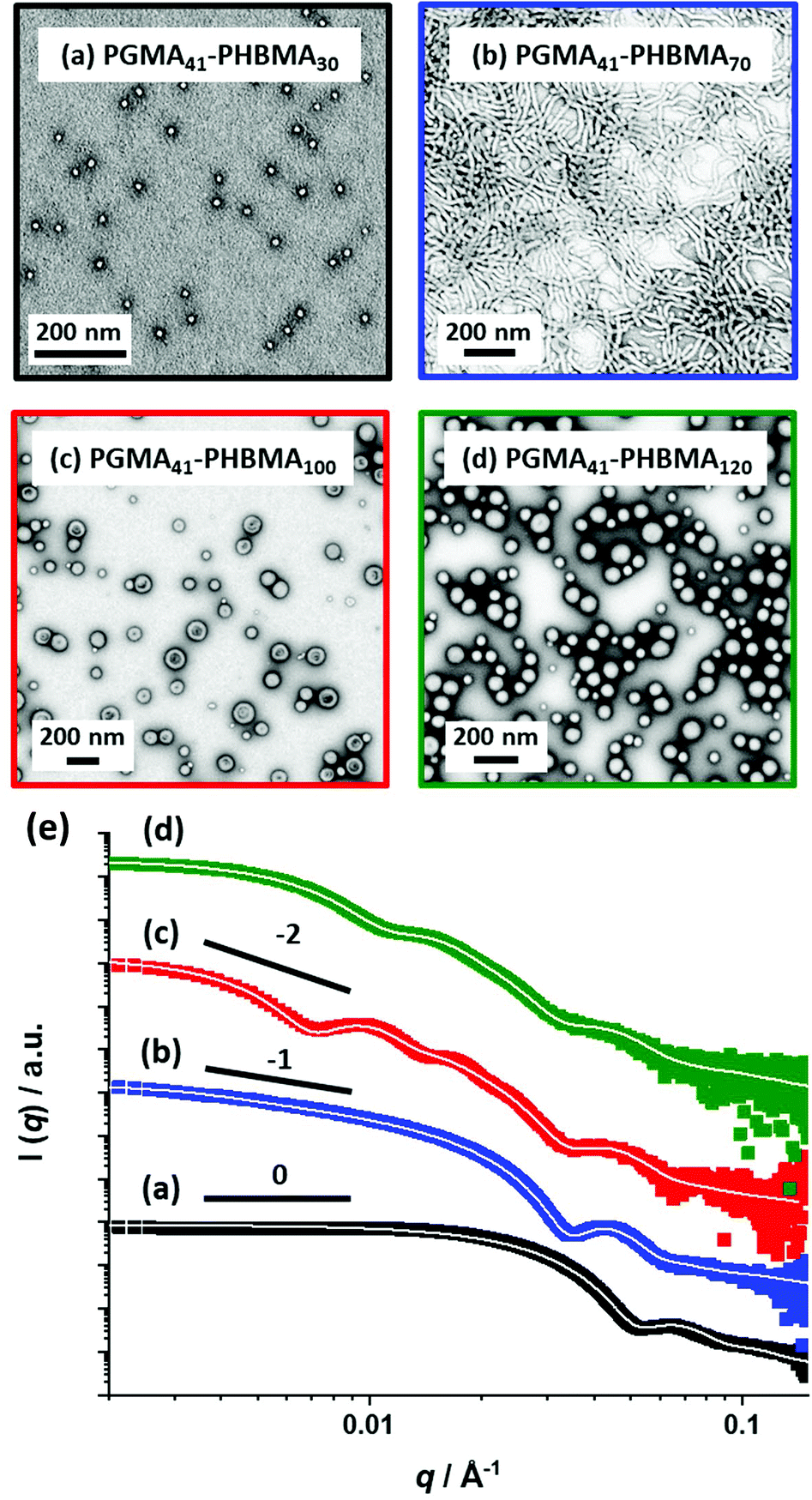 | ||
| Fig. 3 TEM images obtained for (a) PGMA41-PHBMA30 spheres, (b) PGMA41-PHBMA70 worms, (c) relatively large PGMA41-PHBMA100 vesicles and (d) relatively small PGMA41-PHBMA120 vesicles. (e) Corresponding SAXS patterns recorded at 1.0% w/w for the same four copolymer dispersions, which were each originally prepared at 10% w/w. White solid lines show the data fits obtained for each SAXS pattern using an appropriate spherical micelle, worm-like micelle or vesicle model. Each low q gradient is consistent with the corresponding TEM image. | ||
The kinetics of polymerization for the RAFT aqueous emulsion polymerization of HBMA was assessed at a copolymer concentration of 10% w/w. A PHBMA DP of 120 was targeted and more than 99% HBMA conversion was achieved within 70 min at 50 °C (see Fig. 4). Intermediate conversions were determined by periodically withdrawing small aliquots of the reaction mixture and quenching the polymerization in each case, and subsequent analysis by 1H NMR spectroscopy, DLS and TEM studies were also performed on each aliquot. A two-fold increase in the rate of HBMA polymerization was observed after around 30 min, which corresponds to approximately 15% conversion and a PHBMA DP of 18.
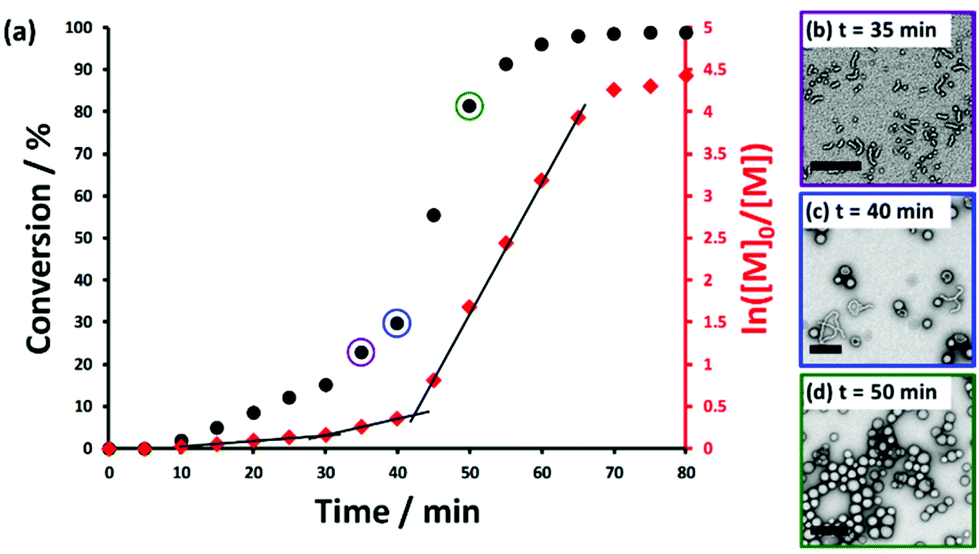 | ||
| Fig. 4 (a) Kinetic studies during the synthesis of PGMA41-PHBMA120 vesicles via RAFT aqueous emulsion polymerization of HBMA at a copolymer concentration of 10% w/w: conversion vs. time curve (black circles) and its corresponding semi-logarithmic plot (red diamonds). Inset: representative TEM images recorded for intermediate copolymer morphologies observed after (b) 35 min (23% conversion; PHBMA DP = 28), (c) 40 min (36% conversion; PHBMA DP = 43), and (d) 50 min (81% conversion; PHBMA DP = 97). Each scale bar = 200 nm. | ||
This rate enhancement is attributed to micellar nucleation, or the point at which the growing diblock copolymer chains begin to form nascent nuclei. This interpretation is supported by the substantial increase observed in the scattered light intensity (derived count rate) from 70 to 700 kcps and a DLS diameter of around 20 nm (see Fig. S3†). Similar rate acceleration effects have been reported for both RAFT dispersion polymerization70,79 and also for other RAFT aqueous emulsion polymerization formulations.46,63 TEM images recorded after 35 min indicate the presence of spheres and short worms (see Fig. 4b). A further three-fold rate enhancement was observed at 40 min (36% conversion). This corresponds to a PHBMA DP of 43, which is consistent with a sphere/worm mixed phase as indicated by the phase diagram. This suggests that this second rate acceleration is associated with the formation of worm-like nanoparticles. Such two-stage rate enhancements are not well-understood but have been recently observed for RAFT aqueous emulsion polymerization of MOEMA63 and also for RAFT dispersion polymerizations conducted in non-polar media.79 Finally, the polymerization proceeds more slowly after 55 min, presumably owing to the gradual depletion of unreacted HBMA within the monomer-swollen vesicle membranes.
TEM studies conducted during this HBMA polymerization reveal intermediate morphologies (e.g. spheres and worms) that are strikingly similar to those observed during RAFT aqueous dispersion polymerization of HPMA when targeting vesicles.70,80 These observations suggest that the mechanism for vesicle formation is likely to be the same (or at least very similar) for RAFT aqueous dispersion polymerization and RAFT aqueous emulsion polymerization.
Preparation of Pickering emulsions using PGMA-PHBMA vesicles
RAFT-mediated PISA enables the design of a wide range of surface-active block copolymer nanoparticles that can stabilize either oil42,71,81–83 or water droplets,84–87 as summarized in a recent review.88 Particular attention has been paid to vesicles since they were the initial diblock copolymer nanoparticles to be utilized as Pickering emulsifiers.71,89–91 In one such study, Thompson et al.71 attempted the preparation of oil-in-water Pickering emulsions using linear PGMA45-PHPMA200 vesicles. However, these nano-objects did not withstand the high-shear homogenization conditions required to generate the oil droplets, instead in situ dissociation occurred to form individual diblock copolymer chains.71 Such dissociation was attributed to the weakly hydrophobic nature of the core-forming PHPMA block. Since these PGMA45-PHPMA200 chains were amphiphilic, a stable emulsion was obtained but it was not a genuine Pickering emulsion.92 To ensure that the original vesicle morphology survived homogenization, a small amount of ethylene glycol dimethacrylate crosslinker could be added as a third block to form covalently-stabilized vesicles.92 Alternatively, PGMA63-PHPMA350-PBzMAx triblock copolymer framboidal vesicles can be prepared via aqueous PISA, with the third PBzMA block being sufficiently hydrophobic to prevent in situ dissociation during high shear homogenization.89The aqueous solubility of HPMA monomer is ∼100 g dm−3 at 70 °C, whereas that for HBMA is only ∼20 g dm−3 at the same temperature.44 On this basis, PHBMA homopolymer is expected to be significantly more hydrophobic than PHPMA homopolymer, which should in principle lead to stronger van de Waals interactions between such water-insoluble chains in an aqueous environment. The scientific question addressed herein is whether these stronger attractive forces are sufficient to enable linear PGMA41-PHBMA110 vesicles to serve as an emulsifier for the production of oil-in-water Pickering emulsions via high-shear homogenization. To examine this hypothesis, a PHBMA DP of 110 was targeted at a copolymer concentration of 10% w/w to produce PGMA41-PHBMA110 vesicles.
These vesicles were then evaluated as putative Pickering emulsifiers for the stabilization of n-dodecane droplets in water for copolymer concentrations ranging from 0.063% to 2.00% w/w. Thus a series of such aqueous dispersions of vesicles were homogenized with an equal volume of n-dodecane at 12000 rpm for 2 min at 20 °C to produce n-dodecane-in-water emulsions, as depicted in Scheme 2. Fig. 5 shows a digital photograph, laser diffraction data and optical microscopy images obtained for the emulsions produced at various copolymer concentrations. Lowering the copolymer concentration leads to a reduction in turbidity for the lower aqueous phase because it contains fewer excess non-adsorbed vesicles. There is a corresponding gradual increase in the volume-average droplet diameter because using fewer vesicles reduces the total surface area of the oil phase that can be coated with a monolayer of adsorbed vesicles, which in turn leads to coarser droplets. This is consistent with the formation of genuine Pickering emulsions and has been reported by various research groups.71,86,93,94 In contrast, Thompson and co-workers reported essentially no change in the mean droplet diameter with copolymer concentration when using linear PGMA45-PHPMA200 vesicles, which is consistent with the in situ break-up of these more delicate nano-objects during high-shear homogenization.71,81,92
 | ||
| Scheme 2 Schematic preparation of an oil-in-water Pickering emulsion prepared via high-shear homogenization of n-dodecane with an equal volume of an aqueous dispersion of linear PGMA41-PHBMA110 vesicles at 12000 rpm for 2 min at 20 °C. The copolymer concentration (which refers to the aqueous phase only) was systematically varied from 0.063% to 2.00% w/w during such experiments. | ||
 | ||
| Fig. 5 (a) Digital photographs obtained for the Pickering emulsions prepared using PGMA41-PHBMA110 vesicles at various copolymer concentrations (which refer to the initial aqueous phase only). (b) Relationship between volume-average droplet diameter (determined by laser diffraction) and copolymer concentration after high-shear homogenization of n-dodecane with aqueous dispersions of PGMA41-PHBMA110 diblock copolymer vesicles of varying concentration. The upturn in droplet size at low copolymer concentration is consistent with the formation of a series of n-dodecane-in-water Pickering emulsions. (c) Optical microscopy images recorded for the n-dodecane droplets prepared at various copolymer concentrations. Emulsification conditions: 12000 rpm for 2 min at 20 °C. | ||
TEM studies of the dried emulsion droplets (see Fig. 6a) confirmed that linear PGMA41-PHBMA110 vesicles do indeed remain intact after high-shear homogenization. More specifically, a densely packed monolayer of adsorbed vesicles is observed after evaporation of both the oil and aqueous phases under the ultrahigh vacuum conditions required for TEM studies. This residual superstructure provides direct evidence for the formation of genuine Pickering emulsions. It is perhaps worth emphasizing that the difference in chemical structure between the PHPMA and PHBMA core-forming blocks is rather subtle: the latter possess just one extra methylene group per monomer repeat unit. Nevertheless, this is sufficient to ensure survival of PGMA41-PHBMA110 vesicles without requiring a third block for either covalent stabilization (PEGDMA) or physical reinforcement via stronger attractive inter-chain forces (PBzMA).
 | ||
| Fig. 6 TEM images obtained for (a) a single dried n-dodecane Pickering droplet or (b) a corresponding cross-linked colloidosome prepared using a 0.25% w/w aqueous dispersion of PGMA41-PHBMA110 vesicles. Emulsification conditions: 12000 rpm for 2 min at 20 °C. | ||
PGMA58-PHPMA350-PEGDMA20 cross-linked vesicles have been previously examined by Thompson et al. for the preparation of colloidosomes.71 This protocol involved using diisocyanate-capped poly(propylene glycol) (PPG-TDI), which is an oil-soluble polymeric cross-linker that readily reacts with the hydroxyl groups on the PGMA stabilizer chains (and perhaps also the PHBMA core-forming chains) to convert the precursor Pickering emulsions into covalently cross-linked colloidosomes. In the current study, the preparation of colloidosomes involved homogenization of a 0.25% w/w aqueous dispersion of linear PGMA41-PHBMA110 vesicles with an equal volume of n-dodecane containing 20.0 g dm−3 PPG-TDI cross-linker at 12000 rpm for 2 min at 20 °C. The initial Pickering emulsion was allowed to stand at 20 °C for several hours to allow the urethane cross-linking reaction to proceed (see Fig. S4†). The resulting colloidosomes were imaged using optical microscopy (see Fig. S5a†) and TEM (see Fig. 6b). To evaluate their structural integrity, these colloidosomes were subjected to an alcohol challenge whereby excess methanol was added to remove the oil phase prior to TEM studies. As shown in Fig. S5b,† such colloidosomes were sufficiently cross-linked to withstand this alcohol challenge. However, TEM studies (see Fig. S5c†) suggest that the vesicles break up to form ill-defined aggregates in the presence of methanol.
Conclusions
In summary, a rather subtle change in monomer structure leads to the aqueous solubility of HBMA being approximately four times lower than that of HPMA, which represents the difference between aqueous emulsion polymerization and aqueous dispersion polymerization, respectively. Interestingly, chain-extending a non-ionic PGMA41 steric stabilizer via RAFT aqueous emulsion polymerization of HBMA provides convenient access to diblock copolymer spheres, worms or vesicles. Thus HBMA exhibits sufficient aqueous solubility to avoid the problem of kinetically-trapped spheres that is observed for so many RAFT aqueous emulsion polymerization formulations. More than 99% HBMA conversion can be achieved within 2 h at 50 °C using a low-temperature azo initiator, with GPC analysis indicating relatively low dispersities (Mw/Mn < 1.37) when targeting PHBMA DPs up to 110. A pseudo-phase diagram was constructed for the synthesis of PGMA41-PHBMAx nano-objects at copolymer concentrations ranging from 5% to 20% w/w when targeting x = 10 to 120. Given the observed concentration-dependent copolymer morphologies and the presence of mixed phases, this systematic approach is essential to ensure reproducible targeting of pure spheres, worms or vesicles. Pure vesicles could be obtained at just 5% w/w, whereas pure worms required higher copolymer concentrations. 1H NMR spectroscopy was used to perform kinetic studies when targeting PGMA41-PHBMA120 vesicles. A significant rate acceleration was observed at two separate stages, with TEM and DLS studies suggesting that the first stage heralds the onset of micellar nucleation while the second stage corresponds to the sphere-to-worm transition. This study provides useful insights regarding the mechanism of vesicle formation via RAFT aqueous emulsion polymerization, which appears to be similar to that reported for RAFT aqueous dispersion polymerization of HPMA when using essentially the same non-ionic PGMA stabilizer block. Finally, the Pickering emulsifier performance of linear PGMA41-PHBMA110 vesicles was assessed. Laser diffraction and TEM studies suggest that such vesicles survive intact when exposed to the high-shear conditions required for homogenization, unlike the linear PGMA45-PHPMA200 vesicles previously reported by Thompson and co-workers.71 This is attributed to the greater hydrophobic character of the PHBMA chains, which leads to stronger inter-chain attractive forces and hence enables retention of the original vesicle morphology after emulsification.Conflicts of interest
There are no conflicts to declare.Acknowledgements
EPSRC is thanked for a CDT PhD studentship to support S. J. H. (EP/L016281) and also an Established Career Particle Technology Fellowship (EP/R003009) for S. P. A. DSM (Geleen, The Netherlands) is acknowledged for partial support of this PhD project and for permission to publish this work. The authors thank Christopher Hill and Dr Svetomir Tzokov at the University of Sheffield Biomedical Science Electron Microscopy suite. The authors also thank the European Synchrotron Radiation Facility for providing beam time at the ID02 beamline (SC-4865).References
- F. Lorandi, Y. Wang, M. Fantin and K. Matyjaszewski, Angew. Chem., Int. Ed., 2018, 57, 8270–8274 CrossRef CAS PubMed.
- P. B. Zetterlund, S. C. Thickett, S. Perrier, E. Bourgeat-Lami and M. Lansalot, Chem. Rev., 2015, 115, 9745–9800 CrossRef CAS PubMed.
- C. Cheng, J. Shu, S. Gong, L. Shen, Y. Qiao and C. Fu, New J. Chem., 2010, 34, 163–170 RSC.
- Z. An, Q. Shi, W. Tang, C.-K. Tsung, C. J. Hawker and G. D. Stucky, J. Am. Chem. Soc., 2007, 129, 14493–14499 CrossRef CAS PubMed.
- G. Delaittre, M. Save and B. Charleux, Macromol. Rapid Commun., 2007, 28, 1528–1533 CrossRef CAS.
- G. Liu, Q. Qiu and Z. An, Polym. Chem., 2012, 3, 504–513 RSC.
- C. Grazon, J. Rieger, N. Sanson and B. Charleux, Soft Matter, 2011, 7, 3482–3490 RSC.
- W. Shen, Y. Chang, G. Liu, H. Wang, A. Cao and Z. An, Macromolecules, 2011, 44, 2524–2530 CrossRef CAS.
- G. Liu, Q. Qiu, W. Shen and Z. An, Macromolecules, 2011, 44, 5237–5245 CrossRef CAS.
- S. Sugihara, S. P. Armes, A. Blanazs and A. L. Lewis, Soft Matter, 2011, 7, 10787–10793 RSC.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS.
- S. Sugihara, K. Sugihara, S. P. Armes, H. Ahmad and A. L. Lewis, Macromolecules, 2010, 43, 6321–6329 CrossRef CAS.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., 2020, 59, 8368–8392 CrossRef PubMed.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS PubMed.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Trans., 1976, 72, 1525–1568 CAS.
- M. J. Derry, L. A. Fielding and S. P. Armes, Polym. Chem., 2015, 6, 3054–3062 RSC.
- S. J. Byard, M. Williams, B. E. McKenzie, A. Blanazs and S. P. Armes, Macromolecules, 2017, 50, 1482–1493 CrossRef CAS PubMed.
- S. Parkinson, N. S. Hondow, J. S. Conteh, R. A. Bourne and N. J. Warren, React. Chem. Eng., 2019, 4, 852–861 RSC.
- M. J. Rymaruk, S. J. Hunter, C. T. O'Brien, S. L. Brown, C. N. Williams and S. P. Armes, Macromolecules, 2019, 52, 2822–2832 CrossRef CAS.
- L. D. Blackman, K. E. B. Doncom, M. I. Gibson and R. K. O'Reilly, Polym. Chem., 2017, 8, 2860–2871 RSC.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 1023–1033 CrossRef CAS PubMed.
- C. Gonzato, M. Semsarilar, E. R. Jones, F. Li, G. J. P. Krooshof, P. Wyman, O. O. Mykhaylyk, R. Tuinier and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 11100–11106 CrossRef CAS PubMed.
- A. Czajka and S. P. Armes, Chem. Sci., 2020, 11, 11443–11454 RSC.
- Q. Zhang, R. Zeng, Y. Zhang, Y. Chen, L. Zhang and J. Tan, Macromolecules, 2020, 53, 8982–8991 CrossRef CAS.
- S. Varlas, J. C. Foster, P. G. Georgiou, R. Keogh, J. T. Husband, D. S. Williams and R. K. O'Reilly, Nanoscale, 2019, 11, 12643–12654 RSC.
- D. Li, M. Huo, L. Liu, M. Zeng, X. Chen, X. S. Wang and J. Y. Yuan, Macromol. Rapid Commun., 2019, 40, 5 Search PubMed.
- X. Wang, J. Zhou, X. Lv, B. Zhang and Z. An, Macromolecules, 2017, 50, 7222–7232 CrossRef CAS.
- P. Yang, O. O. Mykhaylyk, E. R. Jones and S. P. Armes, Macromolecules, 2016, 49, 6731–6742 CrossRef CAS PubMed.
- S. W. Prescott, M. J. Ballard, E. Rizzardo and R. G. Gilbert, Aust. J. Chem., 2002, 55, 415–424 CrossRef CAS.
- S. W. Prescott, M. J. Ballard, E. Rizzardo and R. G. Gilbert, Macromol. Theory Simul., 2006, 15, 70–86 CrossRef CAS.
- C. J. Ferguson, R. J. Hughes, B. T. T. Pham, B. S. Hawkett, R. G. Gilbert, A. K. Serelis and C. H. Such, Macromolecules, 2002, 35, 9243–9245 CrossRef CAS.
- C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such and B. S. Hawkett, Macromolecules, 2005, 38, 2191–2204 CrossRef CAS.
- E. Groison, S. Brusseau, F. D'Agosto, S. Magnet, R. Inoubli, L. Couvreur and B. Charleux, ACS Macro Lett., 2012, 1, 47–51 CrossRef CAS.
- N. P. Truong, M. V. Dussert, M. R. Whittaker, J. F. Quinn and T. P. Davis, Polym. Chem., 2015, 6, 3865–3874 RSC.
- D. E. Ganeva, E. Sprong, H. de Bruyn, G. G. Warr, C. H. Such and B. S. Hawkett, Macromolecules, 2007, 40, 6181–6189 CrossRef CAS.
- I. Chaduc, A. Crepet, O. Boyron, B. Charleux, F. D'Agosto and M. Lansalot, Macromolecules, 2013, 46, 6013–6023 CrossRef CAS.
- I. Chaduc, W. Zhang, J. Rieger, M. Lansalot, F. D'Agosto and B. Charleux, Macromol. Rapid Commun., 2011, 32, 1270–1276 CrossRef CAS PubMed.
- I. Chaduc, M. Girod, R. Antoine, B. Charleux, F. D'Agosto and M. Lansalot, Macromolecules, 2012, 45, 5881–5893 CrossRef CAS.
- C. K. Poon, O. Tang, X.-M. Chen, B. T. T. Pham, G. Gody, C. A. Pollock, B. S. Hawkett and S. Perrier, Biomacromolecules, 2016, 17, 965–973 CrossRef CAS PubMed.
- V. J. Cunningham, A. M. Alswieleh, K. L. Thompson, M. Williams, G. J. Leggett, S. P. Armes and O. M. Musa, Macromolecules, 2014, 47, 5613–5623 CrossRef CAS.
- B. Akpinar, L. A. Fielding, V. J. Cunningham, Y. Ning, O. O. Mykhaylyk, P. W. Fowler and S. P. Armes, Macromolecules, 2016, 49, 5160–5171 CrossRef CAS PubMed.
- A. A. Cockram, T. J. Neal, M. J. Derry, O. O. Mykhaylyk, N. S. J. Williams, M. W. Murray, S. N. Emmett and S. P. Armes, Macromolecules, 2017, 50, 796–802 CrossRef CAS PubMed.
- F. L. Hatton, J. R. Lovett and S. P. Armes, Polym. Chem., 2017, 8, 4856–4868 RSC.
- W. Zhang, F. D'Agosto, P.-Y. Dugas, J. Rieger and B. Charleux, Polymer, 2013, 54, 2011–2019 CrossRef CAS.
- P. Galanopoulo, P.-Y. Dugas, M. Lansalot and F. D'Agosto, Polym. Chem., 2020, 11, 3922–3930 RSC.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- X. Dai, L. Yu, Y. Zhang, L. Zhang and J. Tan, Macromolecules, 2019, 52, 7468–7476 CrossRef CAS.
- J. Rieger, F. Stoffelbach, C. Bui, D. Alaimo, C. Jérôme and B. Charleux, Macromolecules, 2008, 41, 4065–4068 CrossRef CAS.
- J. Rieger, G. Osterwinter, C. Bui, F. Stoffelbach and B. Charleux, Macromolecules, 2009, 42, 5518–5525 CrossRef CAS.
- J. Rieger, W. Zhang, F. Stoffelbach and B. Charleux, Macromolecules, 2010, 43, 6302–6310 CrossRef CAS.
- W. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 7584–7593 CrossRef CAS.
- O. J. Deane, O. M. Musa, A. Fernyhough and S. P. Armes, Macromolecules, 2020, 53, 1422–1434 CrossRef CAS.
- S. Boissé, J. Rieger, K. Belal, A. Di-Cicco, P. Beaunier, M.-H. Li and B. Charleux, Chem. Commun., 2010, 46, 1950–1952 RSC.
- S. Boissé, J. Rieger, G. Pembouong, P. Beaunier and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3346–3354 CrossRef.
- W. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2012, 45, 4075–4084 CrossRef CAS.
- J. Lesage de la Haye, X. Zhang, I. Chaduc, F. Brunel, M. Lansalot and F. D'Agosto, Angew. Chem., Int. Ed., 2016, 55, 3739–3743 CrossRef CAS PubMed.
- B. T. T. Pham, D. Nguyen, V. T. Huynh, E. H. Pan, B. Shirodkar-Robinson, M. Carey, A. K. Serelis, G. G. Warr, T. Davey, C. H. Such and B. S. Hawkett, Langmuir, 2018, 34, 4255–4263 CrossRef CAS PubMed.
- D. Nguyen, V. Huynh, N. Pham, B. Pham, A. Serelis, T. Davey, C. Such and B. Hawkett, Macromol. Rapid Commun., 2019, 40, 1800402 CrossRef PubMed.
- J. Tan, X. Dai, Y. Zhang, L. Yu, H. Sun and L. Zhang, ACS Macro Lett., 2019, 8, 205–212 CrossRef CAS.
- F. L. Hatton, A. M. Park, Y. Zhang, G. D. Fuchs, C. K. Ober and S. P. Armes, Polym. Chem., 2019, 10, 194–200 RSC.
- E. E. Brotherton, F. L. Hatton, A. A. Cockram, M. J. Derry, A. Czajka, E. J. Cornel, P. D. Topham, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2019, 141, 13664–13675 CrossRef CAS PubMed.
- F. L. Hatton, M. J. Derry and S. P. Armes, Polym. Chem., 2020, 11, 6343–6355 RSC.
- W. H. Lane, Ind. Eng. Chem., Anal. Ed., 1946, 18, 295–296 CrossRef CAS.
- J. C. Foster, S. Varlas, B. Couturaud, J. R. Jones, R. Keogh, R. T. Mathers and R. K. O'Reilly, Angew. Chem., Int. Ed., 2018, 57, 15733–15737 CrossRef CAS PubMed.
- L. P. D. Ratcliffe, A. J. Ryan and S. P. Armes, Macromolecules, 2013, 46, 769–777 CrossRef CAS.
- L. P. D. Ratcliffe, A. Blanazs, C. N. Williams, S. L. Brown and S. P. Armes, Polym. Chem., 2014, 5, 3643–3655 RSC.
- J. R. Lovett, N. J. Warren, L. P. D. Ratcliffe, M. K. Kocik and S. P. Armes, Angew. Chem., Int. Ed., 2015, 54, 1279–1283 CrossRef CAS PubMed.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- K. L. Thompson, P. Chambon, R. Verber and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 12450–12453 CrossRef CAS PubMed.
- J. Ilavsky and P. R. Jemian, J. Appl. Crystallogr., 2009, 42, 347–353 CrossRef CAS.
- I. Bannister, N. C. Billingham, S. P. Armes, S. P. Rannard and P. Findlay, Macromolecules, 2006, 39, 7483–7492 CrossRef CAS.
- J. S. Pedersen and C. Svaneborg, Curr. Opin. Colloid Interface Sci., 2002, 7, 158–166 CrossRef CAS.
- J. Pedersen, J. Appl. Crystallogr., 2000, 33, 637–640 CrossRef CAS.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
- V. J. Cunningham, L. P. D. Ratcliffe, A. Blanazs, N. J. Warren, A. J. Smith, O. O. Mykhaylyk and S. P. Armes, Polym. Chem., 2014, 5, 6307–6317 RSC.
- J. Bang, S. Jain, Z. Li, T. P. Lodge, J. S. Pedersen, E. Kesselman and Y. Talmon, Macromolecules, 2006, 39, 1199–1208 CrossRef CAS.
- E. J. Cornel, S. van Meurs, T. Smith, P. S. O'Hora and S. P. Armes, J. Am. Chem. Soc., 2018, 140, 12980–12988 CrossRef CAS.
- M. J. Derry, L. A. Fielding, N. J. Warren, C. J. Mable, A. J. Smith, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2016, 7, 5078–5090 RSC.
- S. J. Hunter, K. L. Thompson, J. R. Lovett, F. L. Hatton, M. J. Derry, C. Lindsay, P. Taylor and S. P. Armes, Langmuir, 2019, 35, 254–265 CrossRef CAS PubMed.
- C. J. Mable, K. L. Thompson, M. J. Derry, O. O. Mykhaylyk, B. P. Binks and S. P. Armes, Macromolecules, 2016, 49, 7897–7907 CrossRef CAS PubMed.
- Y. Zhang, L. Yu, X. Dai, L. Zhang and J. Tan, ACS Macro Lett., 2019, 8, 1102–1109 CrossRef CAS.
- K. L. Thompson, L. A. Fielding, O. O. Mykhaylyk, J. A. Lane, M. J. Derry and S. P. Armes, Chem. Sci., 2015, 6, 4207–4214 RSC.
- K. L. Thompson, C. J. Mable, J. A. Lane, M. J. Derry, L. A. Fielding and S. P. Armes, Langmuir, 2015, 31, 4137–4144 CrossRef CAS PubMed.
- C. György, S. J. Hunter, C. Girou, M. J. Derry and S. P. Armes, Polym. Chem., 2020, 11, 4579–4590 RSC.
- M. J. Rymaruk, K. L. Thompson, M. J. Derry, N. J. Warren, L. P. D. Ratcliffe, C. N. Williams, S. L. Brown and S. P. Armes, Nanoscale, 2016, 8, 14497–14506 RSC.
- S. J. Hunter and S. P. Armes, Langmuir, 2020, 36, 15463–15484 CrossRef CAS PubMed.
- C. J. Mable, N. J. Warren, K. L. Thompson, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2015, 6, 6179–6188 RSC.
- Q. Xu, Y. Zhang, X. Li, J. He, J. Tan and L. Zhang, Polym. Chem., 2018, 9, 4908–4916 RSC.
- Z. P. Wang, M. C. M. van Oers, F. Rutjes and J. C. M. van Hest, Angew. Chem., Int. Ed., 2012, 51, 10746–10750 CrossRef CAS PubMed.
- K. L. Thompson, C. J. Mable, A. Cockram, N. J. Warren, V. J. Cunningham, E. R. Jones, R. Verber and S. P. Armes, Soft Matter, 2014, 10, 8615–8626 RSC.
- R. Aveyard, B. P. Binks and J. H. Clint, Adv. Colloid Interface Sci., 2003, 100, 503–546 CrossRef.
- B. P. Binks and C. P. Whitby, Langmuir, 2004, 20, 1130–1137 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectrum recorded for PGMA41-PHBMA120; additional TEM images for PGMA41-PHBMAx prepared at 5% and 20% w/w; monomer conversions and molecular weight data for PGMA41-PHBMAx nanoparticles; structural parameters obtained from fitting SAXS patterns recorded for PGMA41-PHBMAx nanoparticles; further kinetic data for PGMA41-PHBMA120 vesicles; further details for the scattering models and general analytical approach; TEM and optical microscopy images of colloidosomes diluted using either water or methanol. See DOI: 10.1039/d1py00517k |
| This journal is © The Royal Society of Chemistry 2021 |