
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversible crosslinking and fast stress relaxation in dynamic polymer networks via transalkylation using 1,4-diazabicyclo[2.2.2] octane†
Eveline E. L.
Maassen
 ab,
Johan P. A.
Heuts
*a and
Rint P.
Sijbesma
*a
ab,
Johan P. A.
Heuts
*a and
Rint P.
Sijbesma
*a
aSupramolecular Polymer Chemistry group, Department of Chemical Engineering and Chemistry, and Institute for Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: J.P.A.Heuts@tue.nl; R.P.Sijbesma@tue.nl
bBrightlands Materials Center (BMC), P.O. Box 18, 6160 MD Geleen, The Netherlands
First published on 2nd June 2021
Abstract
Dynamically crosslinked polymers offer the possibility to design materials that have the robustness of thermosetting materials and the reshapability of thermoplastic materials. A reversible crosslinker based on transalkylation chemistry using 1,4-diazabicyclo[2.2.2]octane (DABCO) was developed. The kinetics of the exchange reaction of benzyl bromide with DABCO was studied in detail by varying molar ratios and temperature. The system showed efficient exchange without addition of a catalyst. Reversibility in a polymer was studied by the crosslinking of a linear, benzylic bromide functionalized polyacrylate with DABCO. The resulting network was insoluble in common solvents, but it readily dissolved in the presence of DABCO. Rheological measurements showed that the crosslinks are dynamic, with a strong temperature dependent relaxation that at 140 °C is faster than for other transalkylation based dynamic networks reported in the literature.
Introduction
Thermosetting polymers are covalently crosslinked networks with excellent properties such as solvent resistance and high thermal and dimensional stability.1,2 Although these properties are highly advantageous for a wide range of applications such as in the coatings,3 adhesives,4 construction,5 and automotive6 industries, recycling or reprocessing these products is very challenging because they are unable to flow upon heating. By introducing exchangeable crosslinks, enhanced properties such as reshapability,7 recyclability,7,8 (self-)healing,9,10 or stress relaxation can be obtained while retaining the advantageous properties like enhanced thermal and chemical stability.11,12 Networks containing specially designed reversible covalent crosslinks are also known as covalent adaptable networks (CANs).8,13–19 Depending on the nature of the exchangeable bond, a stimulus such as light,20 temperature,21 or pH triggers the reversibility of the bond.15 Many dynamic motifs for bond exchange have been developed over the past years, some examples are transesterification reactions,7,22–26 boronic ester exchange,27–29 disulfide exchange,30 controllable reversible addition–fragmentation reactions,31–33 transcarbonation exchange,34 and (vinylogous) urethane exchange.35–38 Because nucleophilic substitutions are usually considered to be non-reversible, they have often been overlooked for use in CANs.15 However, the reversibility of specific nucleophilic substitutions has been known for years,39 and has recently been employed in CANs.40 Kulchat and Lehn investigated the transalkylation of quaternary ammonium salts,41 and showed their potential for the development of dynamic covalent chemistry. Drockenmuller and coworkers were the first to report a dynamic polymeric network based on transalkylation exchange of C–N bonds.42 Network polymers containing 1,2,3-triazolium salts as reversible crosslinks were synthesized and could be recycled by compression molding. After two recycling cycles, samples with an extension at break similar to the starting material were obtained, indicating efficient network reformation. Furthermore, efficient stress relaxation was observed in rheological experiments. At a temperature of 200 °C, the characteristic relaxation time τ was only a few seconds. Following this work, Konkolewicz and coworkers designed acrylic polymeric materials which were dynamically crosslinked with anilinium salts.43 These dynamic polymeric materials show excellent malleability at 60 °C and moreover could be quantitively healed by mild heating to 60 °C or 80 °C. A more detailed kinetic study elucidated the exchange mechanism and it was found that although the exchange follows a dissociative pathway, they have low degrees of de-crosslinking and therefore the networks have properties resembling those of associative CANs.44 Du Prez and coworkers developed a reversible network via C–S transalkylation of trialkylsulfonium salts.45 With kinetic studies they showed that alkylbrosylates are efficient transalkylation agents to convert thioesters into trialkylsulfonium salts. These salts showed complete relaxation at elevated temperatures in rheological stress relaxation experiments, and remolding by compression molding yielded defect free samples. Recycled samples showed no significant change in mechanical properties when measured in DMTA and tensile tests. Zhang and coworkers utilized similar C–S transalkylation reactions to recycle sulfur-cured rubber.46Inspired by these systems, we aimed at developing a transalkylation system that can be implemented in (meth)acrylate polymers. Multifunctional (meth)acrylate polymers are a typical example of thermosetting polymers and due to their fast curing behavior and compatibility with many different functionalities,47 they have many industrial applications e.g. in dentistry,48 coatings,3,49 liquid-crystal displays,50 inks and adhesives.4,51 The use of a two-step approach of linear polymerization followed by reversible crosslinking offers opportunities for the introduction of dynamic behavior into commercially available methacrylic polymers.38 A number of different tertiary amines were considered as reversible crosslinking agent. 1,4-Diazabicyclo[2.2.2]octane (DABCO) was selected as the most promising candidate for fast exchange reactions (Scheme 1). Compared to the popular nucleophilic catalyst 4-dimethylaminopyridine (DMAP), a pyridine derivate, the bicyclic aliphatic amine DABCO is a significantly better nucleophile, yet also a much better leaving group (by a factor of 103 and 106 respectively)52 will transalkylate much faster than DMAP derivatives. An additional advantage is that one DABCO molecule can form a crosslink between two polymer chains, because it contains two tertiary amine functional groups. Finally, DABCO can easily be alkylated at low temperatures, making the alkylation process compatible with (meth)acrylates.53
 | ||
| Scheme 1 Schematic representation of the topology rearrangements due to the transalkylation of DABCO by pendant benzyl bromide groups. | ||
The work reported here investigates the potential of DABCO in transalkylation reactions to create dynamically crosslinked acrylate networks. The kinetics of the exchange reaction of benzyl bromide with DABCO was studied in detail using a small molecule model system. Additionally, we demonstrate the formation of a dynamic covalent network by crosslinking a linear benzylic bromide functionalized methacrylic prepolymer in a single step with DABCO. The cured network can be reprocessed and shows efficient stress relaxation in rheological measurements.
Experimental
Materials
All commercial chemicals and solvents were used as received, unless stated otherwise. 4-(Bromomethyl)benzoic acid and 2-hydroxyethyl methacrylate were obtained from TCI EUROPE. All other chemicals for the synthesis were purchased from Sigma Aldrich. Solvents were obtained from Biosolve.Characterization methods
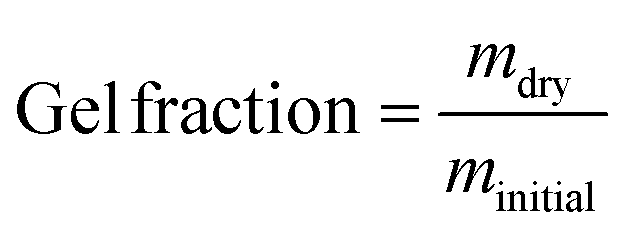 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 g mol−1. The used eluent was THF with a flow rate of 1 mL min−1 at 40 °C. The molar mass was determined relative to narrow dispersity polystyrene standards purchased from Agilent.
000000 g mol−1. The used eluent was THF with a flow rate of 1 mL min−1 at 40 °C. The molar mass was determined relative to narrow dispersity polystyrene standards purchased from Agilent.
Synthesis of small molecules
The synthesis and characterization of 1,4-dibenzyl-1,4-diazabicyclo[2.2.2]octane-1,4-diium dibromide, 1-benzyl-1,4-diazabicyclo[2.2.2]octan-1-ium bromide, 1-benzyl-4-(4-methyl- benzyl)-1,4-diazabicyclo [2.2.2]octane-1,4-diium dibromide and 1-isopropyl-1,4-diazabicyclo[2.2.2]octan-1-ium bromide are described in the ESI.†Synthesis of 2-(methacryloyloxy)ethyl 4-(bromomethyl) benzoate55
4-(Bromomethyl)benzoic acid (31.2 g, 0.14 mol) was suspended in chloroform (500 mL, 0.3 M) and 2-hydroxyethylmethacrylate (16.1 mL, 0.13 mol), N,N‘-dicyclohexylcarbodiimide (DCC, 41.p g, 0.2 mol) and 4-(dimethyl-amino)pyridinium 4-toluene-sulfonate (DPTS, 9,7 g, 0.04 mol) were added. The mixture was stirred for 3 nights at room temperature before filtration over a glass filter. The solvent was evaporated and the crude material was dissolved in methylene chloride and impregnated on silica. Column chromatography with ethyl acetate–heptane 80:20 was used to purify the mixture. The sticky material was precipitated in methanol to remove any homopolymer formed during the workup. The product (BrEMA, see Scheme 4) was obtained as a white solid, yield: 38%.
1H NMR (400 MHz, DMSO-d6): δ [ppm] = 7.94 (d, J = 8.3 Hz, 2H), 7.60 (d, J = 8.3 Hz, 2H), 6.03 (s, 1H), 5.69 (s, 1H), 4.76 (s, 2H), 4.62–4.49 (m, 2H), 4.49–4.38 (m, 2H), 1.87 (s, 3H).
13C NMR (101 MHz, DMSO-d6): δ [ppm] = 166.9, 165.6, 144.0, 136.1, 130.1, 130.0, 129.6, 126.6, 63.2, 62.8, 33.6, 18.4.
SEC analysis showed that polymeric material was not present in the final product.
Synthesis of poly(butyl methacrylate-co-2-(methacryloyloxy) ethyl 4-(bromomethyl)benzoate)
Inhibitor was removed from butyl methacrylate (BMA) monomer by filtration over basic alumina. BMA (22 mL, 138 mmol) and 2-(methacryloyloxy)ethyl 4-(bromomethyl) benzoate (BrEMA, 5 g, 15 mmol) were dissolved in THF (58 mL, 70 wt.%). 1-Decanethiol (0.1 g, 0.6 mmol) and AIBN (0.2 g, 1.2 mmol) were added to the solution. The solution was cooled to 0 °C and deoxygenated by purging with argon for 20 minutes. While under argon, the reaction mixture was heated to 50 °C to start the polymerization. After 18 hours of reaction, the conversion determined via NMR was 82% and the viscosity had strongly increased. The reaction was stopped by addition of hydroquinone. The mixture was precipitated in 7-fold v/v cold methanol, yielding a white sticky solid. The precipitate was filtered and dried in a vacuum oven at 50 °C for a period of 16 h. The final product was obtained as a white solid.
1H-NMR (400 MHz, THF-d8): δ [ppm] = 8.08 (s, Ar![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ), 7.60 (s, Ar), 4.69 (s, ArC2Br), 4.53 (s, OC2CH2O), 4.30 (s, OCH2C2O), 3.96 (s, COOC2(CH2)3), 2.14–1.79 (multiple C2 main chain peaks), 1.61 (br s, COOCH2C2CH2CH3), 1.42 (br s, COOCH2CH2C2CH3), 1.01–0.82 (multiple C3 peaks).
), 7.60 (s, Ar), 4.69 (s, ArC2Br), 4.53 (s, OC2CH2O), 4.30 (s, OCH2C2O), 3.96 (s, COOC2(CH2)3), 2.14–1.79 (multiple C2 main chain peaks), 1.61 (br s, COOCH2C2CH2CH3), 1.42 (br s, COOCH2CH2C2CH3), 1.01–0.82 (multiple C3 peaks).
The overall composition of the copolymer was determined by peak integration of the 1H NMR spectra, and was found to contain 10% BrEMA. Since copolymerization of two methacrylates are characterized by reactivity ratios close to 1, no significant composition drift is expected for this copolymerization.
SEC analysis showed a single peak with a Mn ≈ 33 × 103 g mol−1 and a PDI ≈ 1.7 with respect to polystyrene standards.
Network formation
The linear poly(BMA-co-BrEMA) (7 g) was dissolved in 70 mL tetrahydrofuran using mild sonication in an ultrasonic bath. The desired amount of DABCO (0.3 mg for a ratio of 0.6 mol DABCO per benzyl bromide group) was added and sonicated for 2 minutes, to ensure homogeneous mixing. The liquid mixture was heated for 1 hour at 60 °C resulting in the formation of an organogel. The gel was transferred to aluminum molds, and solvent was removed using a vacuum oven at 70 °C overnight.Sample preparation
The crosslinked polymer was compression molded into disk or bar shaped samples using a Collin Press 300 G with an operating temperature of 140 °C, pressure of 100 bar for 20 minutes and subsequently cooled with water. To remove any thermal history prior to rheology experiments, the samples were thermally treated by placing them into an oven overnight at 120 °C.Results and discussion
Model experiments
To evaluate the efficiency of the transalkylation reaction of DABCO a kinetic study on low molecular weight model compounds was performed. First model compound S, (Scheme 2) was prepared by the alkylation of DABCO with two molecules of benzyl bromide (see ESI† for synthetic details). For the exchange experiments, DABCO (D), was added to compound S in DMSO.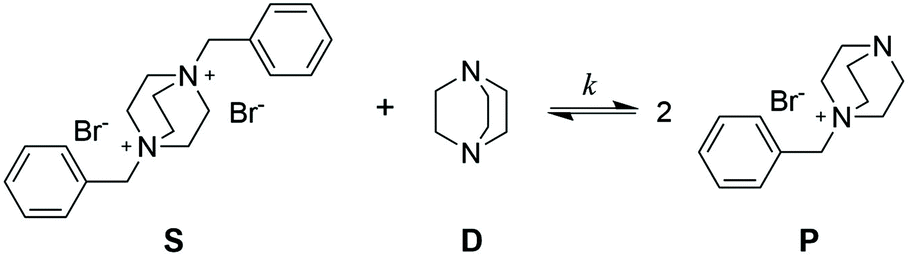 | ||
| Scheme 2 Exchange reaction of bisbenzyl-DABCO (S) and free DABCO (D) for kinetic study in DMSO. | ||
The formation of the monobenzyl-DABCO product (P) was monitored using 1H NMR. In Fig. 1a, the changes in the relevant NMR signals for the determination of the concentrations of S and P are shown (see also Fig. S1, ESI†).
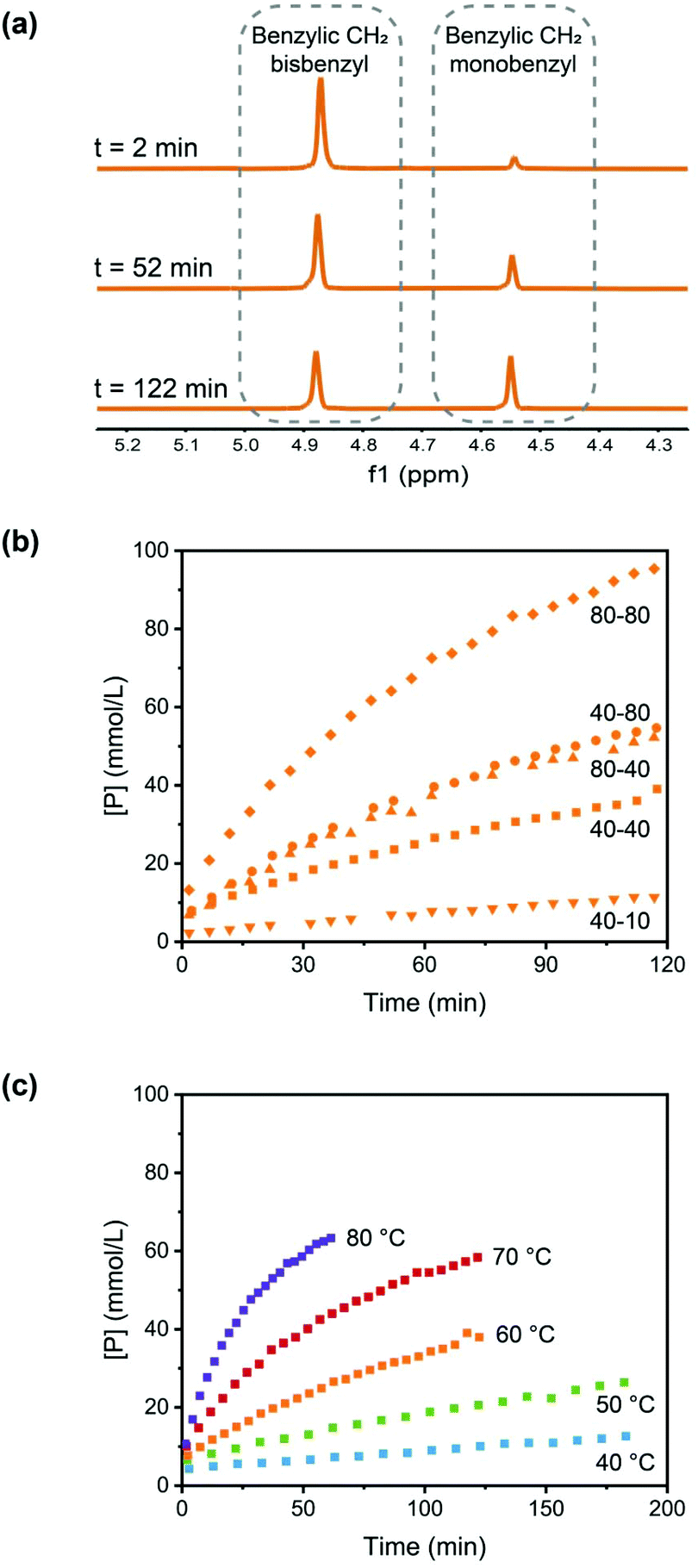 | ||
| Fig. 1 (a) NMR signals for the determination of the concentrations of monobenzyl-DABCO (P) and bisbenzyl-DABCO (S), T = 60 °C and [S]0 = [D]0 = 40 mM. (b) Formation of P from S and DABCO (D) in DMSO for a range of different initial concentrations at 60 °C, data labels in the plot denote concentrations [S]0 – [D]0 in mM. (c) Temperature dependence of the formation of P using initial concentrations [S]0 = [D]0 = 40 mM. | ||
The concentrations of the product, monobenzyl-DABCO (P) are plotted as a function of reaction time for a range of initial concentrations at a temperature of 60 °C in Fig. 1b and a range of temperatures for equimolar initial concentrations of 40 mM in Fig. 1c.
Quantitative analysis of the results shown in Fig. 1 are consistent with a reaction rate that is first-order in both reactant concentrations, and second-order rate coefficients were determined at a range of temperatures (see ESI† for more details). An Arrhenius analysis leads to an activation energy of 93 kJ mol−1 and frequency factor A ≈ 7 × 1011 L mol−1 s−1 for the second-order forward reaction rate coefficient k.
In a dynamic polymer network, the nucleophile in the exchange reaction is not DABCO, but a dangling DABCO group that is quaternized on one of the nitrogen atoms. Since the positive charge may be expected to reduce the nucleophilicity of the second nitrogen atom through electrostatic repulsion, it was relevant to investigate the reactivity of a monosubstituted DABCO with bisbenzyl-DABCO. It was found that monosubstituted DABCO is 3 to 5 times less reactive due to electrostatic effects, but still shows efficient exchange (for details, see ESI Fig. S3†).
To investigate whether exchange reactions take place in the absence of free amine, the reaction of S with excess alkylating agent 4-methylbenzyl bromide was performed (Scheme 3). Surprisingly, exchange reactions occurred and a mixed bisbenzylated product was formed, albeit at a significantly lower rate than for the reaction of S with DABCO under the same conditions (Fig. S4, ESI†).
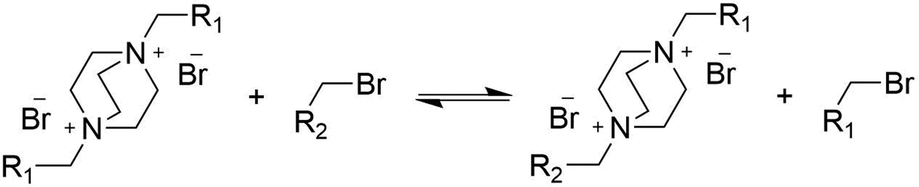 | ||
| Scheme 3 Exchange reaction of bisbenzyl-DABCO excess alkylating agent. | ||
The occurrence of trans-alkylation in the absence of free amino groups is in line with the work of Lehn and coworkers,41 who propose two pathways for transalkylation of quaternary amines, a mechanism with direct attack of the amine on the benzylic carbon atom, and an indirect mechanism in which the bromide counterion acts as a nucleophile to produce benzyl bromide and an amine (Scheme S1, ESI†). Our results strongly suggest that both the indirect and direct pathway occur with the direct one being dominant. Other dynamic networks based on trans-N-alkylation reactions like those reported by Konkolewicz44 and Drockenmuller42,56 seem to follow a dissociative mechanism but nevertheless have low degrees of de-crosslinking resulting in properties resembling those of associative CANs.57
Polymer synthesis
After the successful transalkylation reaction in the model study we aimed at using this chemistry to reversibly crosslink acrylic polymers. To this end, a monomer with a benzyl bromide functional group was synthesized via an esterification reaction of 4-(bromomethyl) benzoic acid and 2-hydroxyethyl-methacrylate. This monomer (BrEMA) was copolymerized with n-butyl methacrylate (BMA) in a free radical polymerization at 50 °C, using AIBN as an initiator and decanethiol (DT) as a chain transfer agent (Scheme 4) to yield a linear polymer with benzyl bromine side groups (SEC: Mn = 33 × 103 g mol−1 and Đ ≈ 1.7, Fig. 2a). Butyl methacrylate was selected as the comonomer to obtain a polymer with a low Tg, to facilitate chain mobility necessary for the exchange.58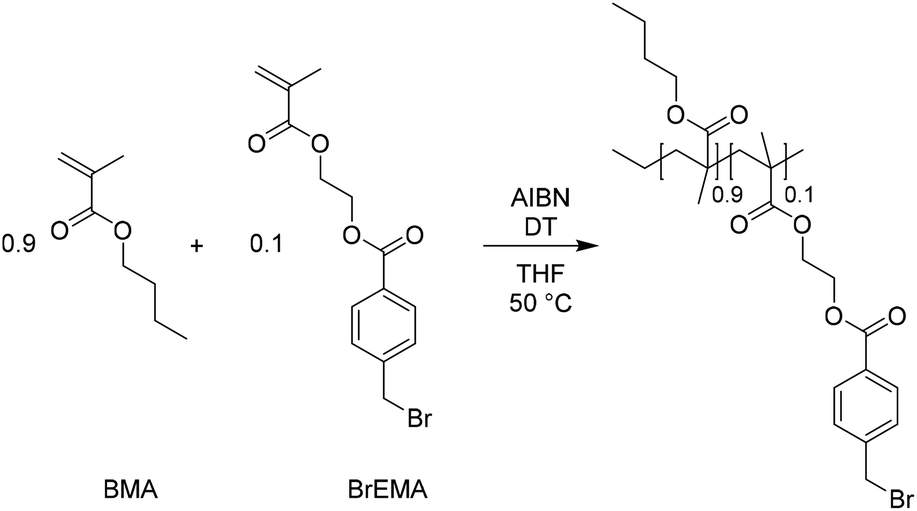 | ||
| Scheme 4 Free radical copolymerization of BMA and BrEMA. | ||
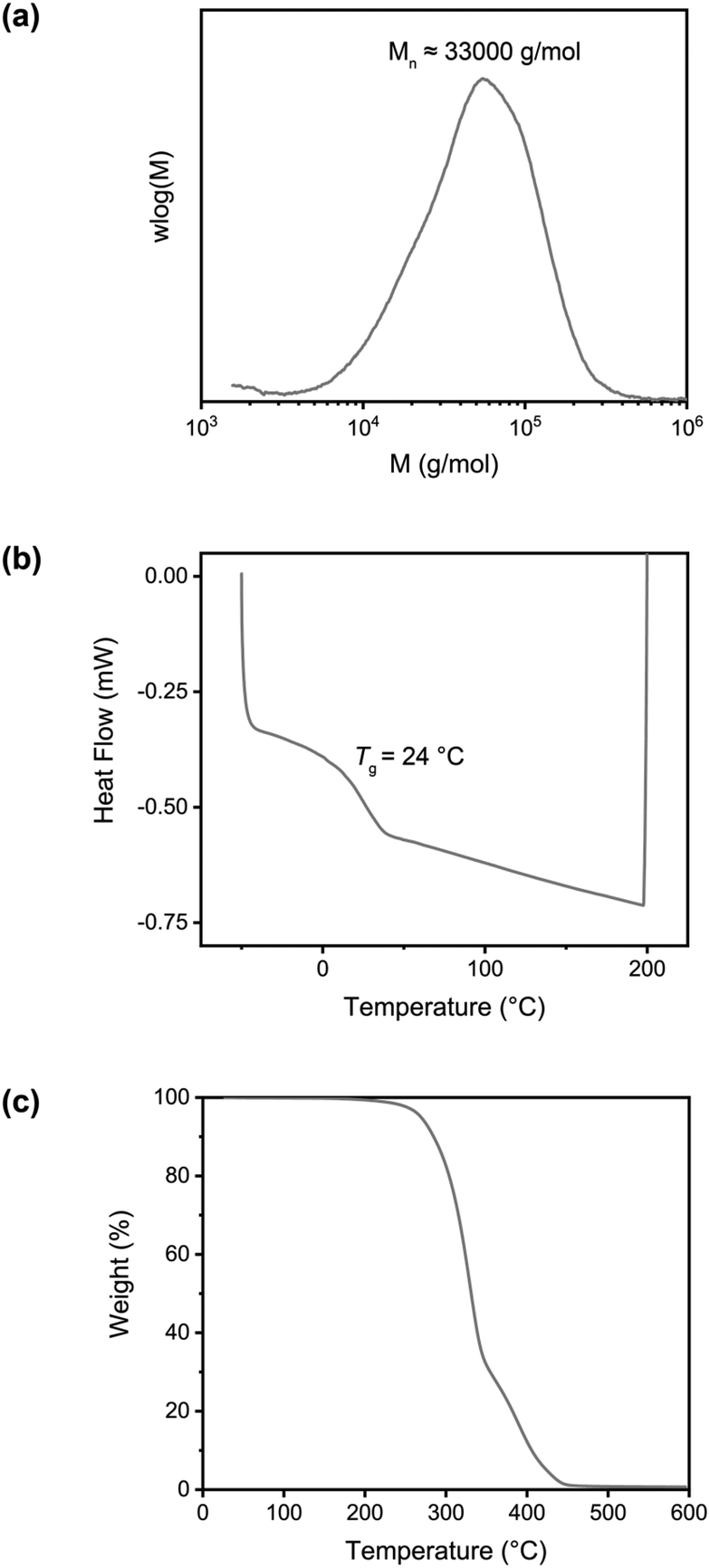 | ||
| Fig. 2 Characterization of the linear polymer with benzylbromide side groups, poly(BMA-co-BrEMA). (a) Size exclusion chromatogram (THF, PS standards). (b) DSC thermogram measured from −50 °C to 200 °C with a heating rate of 20 °C min−1. (c) TGA thermogram measured under N2 with a heating rate of 10 °C min−1. | ||
The copolymer composition was determined from 1H NMR in THF-d8 (Fig. S5, ESI†) and it was found that the synthesized polymer contained 10 mol% of BrEMA units. The Tg of the copolymer was 24 °C, as determined by DSC, Fig. 2b. The TGA thermogram, Fig. 2c, showed the typical thermal stability of a methacrylate polymer with significant thermal degradation from 260 °C onwards.
Network formation
After having established that DABCO and benzylbromide also efficiently react in THF (see ESI, Fig. S6†), crosslinking experiments with polymer were carried out in THF. The linear polymer readily dissolved in THF (700 mg in 7 mL) resulting in a clear transparent solution, Fig. 3. Upon addition of DABCO (29 mg), the mixture became slightly opaque. When left to react at room temperature the viscosity of the mixture slowly increased until after 1.5 hours a gel was obtained that was able to support its own weight upon tilting the vial. When reacted at 60 °C a gel was obtained after only 10 minutes (Fig. 3). | ||
| Fig. 3 After addition of DABCO to a solution of linear polymer in THF a gel is formed. When reacted at room temperature, top pictures, the viscosity slowly increases over time until a gel is formed. When reacted at 60 °C a gel is obtained after 10 min. | ||
An attempt was made to show the presence of DABCO adducts in the polymer by 1H and 13C NMR. A small amount of DABCO crosslinker was added to a solution of linear copolymer in THF-d8 (70 mg polymer, 0.49 mg DABCO; this corresponds to 0.0043 mmol DABCO and 0.043 mmol benzyl bromide).
After 1 hour of reaction at 60 °C an increase in viscosity was observed and the sample was analyzed by NMR.
Due to the low concentration of crosslinks, a signal of the adduct could not be observed in NMR. The DOSY NMR spectra in Fig. 4a show a clear decrease in diffusion coefficients when the copolymer was reacted with DABCO, indicating that chains are linked together by DABCO.59,60
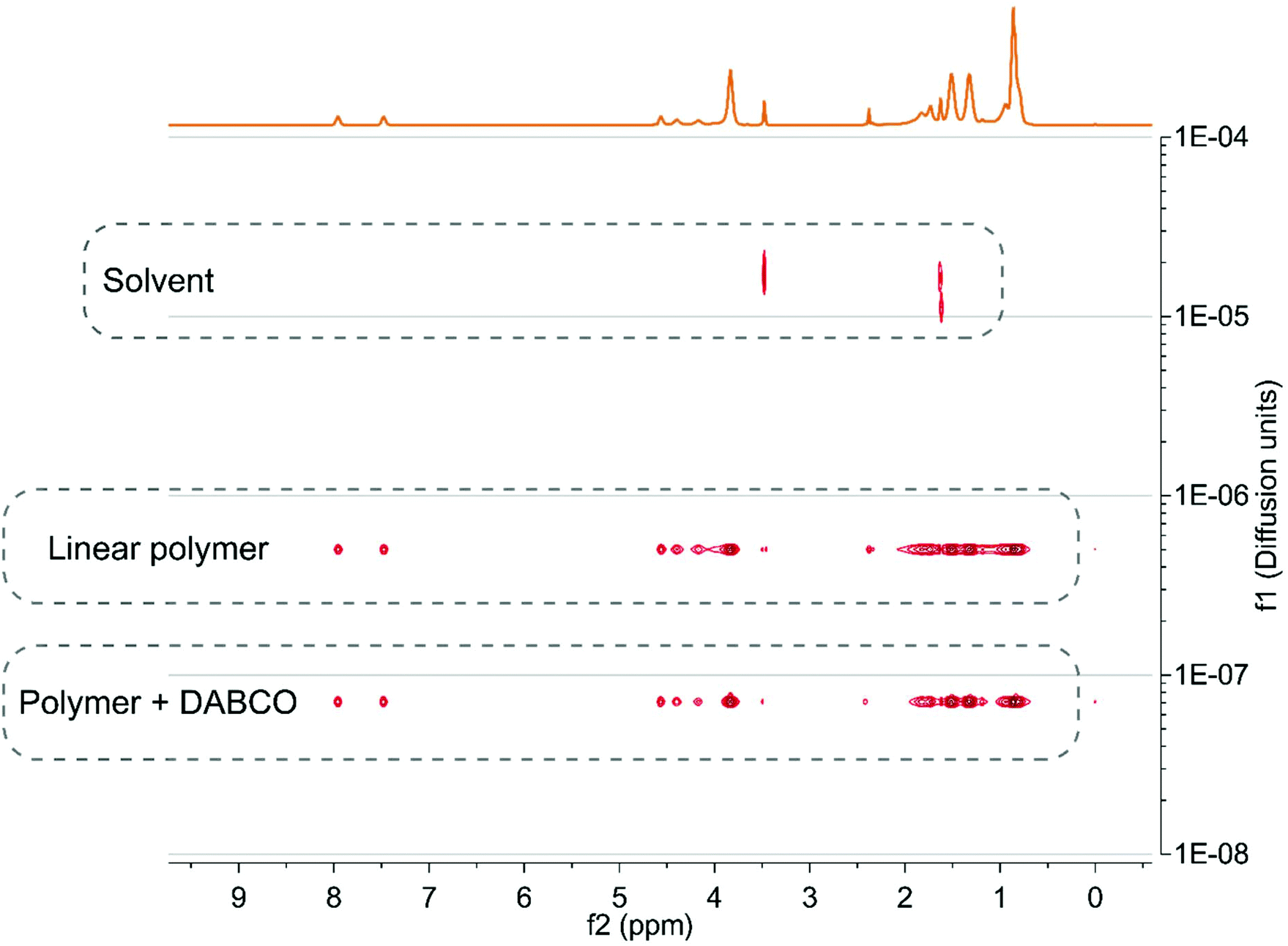 | ||
| Fig. 4 Overlay of DOSY spectra of the linear copolymer and copolymer reacted with DABCO. | ||
In order to obtain a network for mechanical testing, a batch of 7 g of linear polymer was reacted with 0.3 mg of DABCO (approximately 0.6 DABCO per benzyl bromide group) as a crosslinking agent by heating for 1 hour at 60 °C in THF. The resulting organogel was dried, yielding a solid polymer. The Tg after crosslinking was 32 °C which is slightly higher than that of the linear polymer (Fig. S7a, ESI†). The thermal stability decreased slightly as the crosslinked polymer starts degrading from 180 °C (Fig. S7b, ESI†). To quantatively check the formation of a network, the gel fraction was determined by extraction with THF at room temperature. The linear polymer readily dissolved in THF but after crosslinking, the gel fraction was ≥98%. This high gel fraction confirms the presence of a highly branched and/or network polymer.
The temperature dependence of the moduli was studied using oscillatory rheometry (Fig. 5).
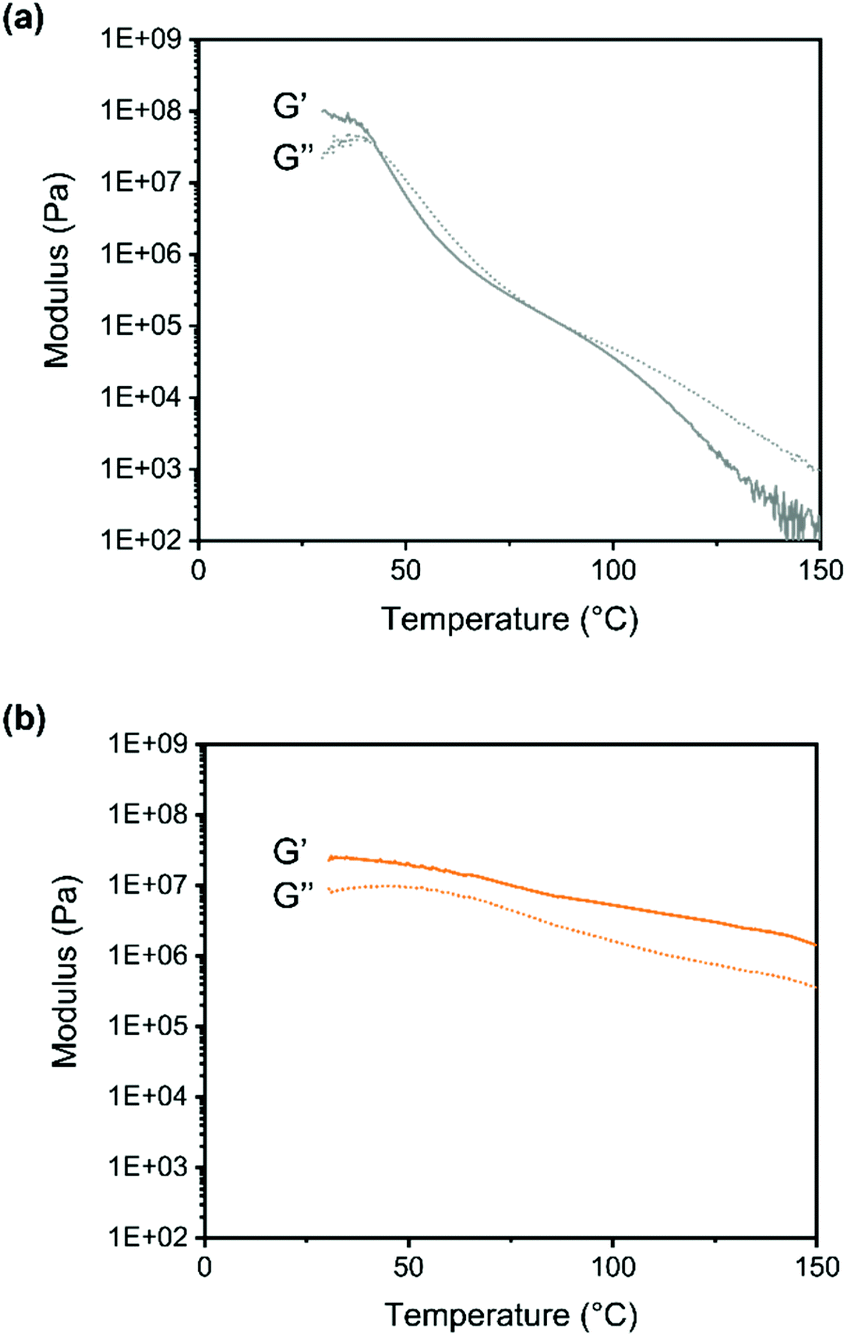 | ||
| Fig. 5 Storage (G′) and loss moduli (G′′) of (a) linear polymer (b) polymer crosslinked with DABCO, measured with temperature dependent oscillatory rheometry (0.1% strain, 1 Hz). | ||
For the linear copolymer the change from rubbery to viscous behavior is clearly seen around 95 °C. For the crosslinked system the G′ is larger than G′′ for the whole temperature range again confirming the presence of a network.
Dynamic behaviour
After crosslinking, the polymer was shaped into disks and bars by compression molding at elevated temperatures. This is already indicative of the presence of dynamic crosslinks as this type of processing is not possible for classical thermosets. When compression molded samples were heated to 100 °C, a clear difference between the linear polymer and the crosslinked polymer was observed. The non-crosslinked sample flowed at 100 °C even when no stress was applied (Fig. 6a), while the crosslinked sample maintained its shape even when heated up to 250 °C (Fig. 6b). In addition to the processability of the crosslinked sample by compression molding, the reversibility of the network was further confirmed by treating a sample with excess DABCO in THF. When this mixture was kept overnight at room temperature, the polymer bar could be taken out in one piece, but heating to 60 °C resulted in full dissolution of the bar in 120 minutes (Fig. 6c). | ||
| Fig. 6 (a) The linear polymer flows when heated to 100 °C. (b) The crosslinked polymer does not flow even when heated to 250 °C. Some yellowing occurred when heating samples above 200 °C. (c) A bar of crosslinked polymer does not dissolve when heated, whilst in the presence of excess DABCO de-crosslinking occurs, yielding a soluble polymer. | ||
For a quantitative study on the reversibility of these systems, stress relaxation experiments were performed in a rheometer. Networks with 0.6 and 0.4 mol of DABCO per mol of benzyl bromide group were tested, corresponding to excess amine and excess benzyl bromide, respectively. The experiments were performed in the linear viscoelastic regime at 1% strain. The time dependent stress relaxation moduli G(t) are shown at four different temperatures for both systems in Fig. 7. Both systems show efficient stress relaxation with a rate that strongly increases with temperature, and is faster in the system with excess amine, in accordance with the stoichiometry dependence in the small molecule kinetic studies. In contrast to stress relaxation in a number of other CANs,38,45,61 relaxation could not be adequately described by a simple exponential function (characteristic of a Maxwell model). By fitting the data to a stretched exponential, (eqn (2)) starting at a time of 1s, good fits were obtained with R2 ≥ 0.99 (see Fig. S9 ESI†).62 From the deviation from a stretched exponential below 1s, that can be seen in the plots of Fig. 7, it is evident that a fast component contributes to relaxation at short times. The best fit parameters are shown in Table 1.
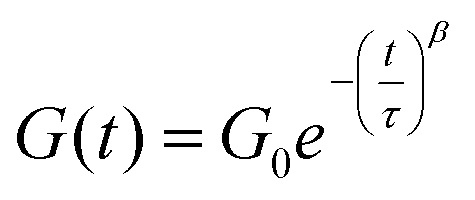 | (2) |
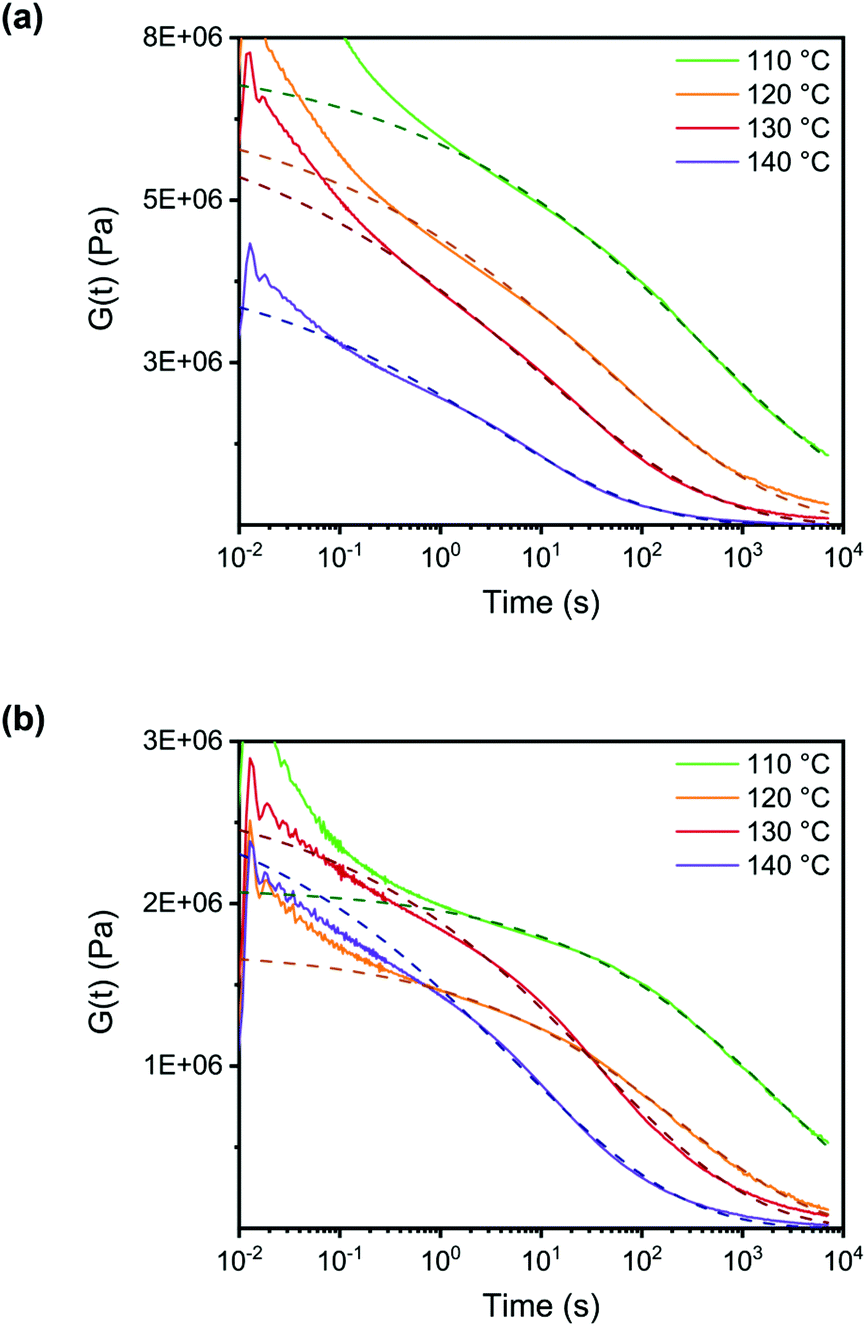 | ||
| Fig. 7 Temperature dependent stress relaxation at 1% strain. The dashed lines are fitted using a stretched exponential function for times >1 s. (a) System with 0.6 eq. of DABCO (excess amine). (b) System with 0.4 eq. of DABCO (excess benzyl bromide). | ||
| 0.6 eq. DABCO | 0.4 eq. DABCO | |||||
|---|---|---|---|---|---|---|
| Temperature [°C] | G 0 [MPa] | τ fit [s] | β | G 0 [MPa] | τ fit [s] | β |
| 110 | 7.2 | 484 | 0.25 | 2.1 | 2432 | 0.34 |
| 120 | 6.6 | 42 | 0.24 | 1.7 | 265 | 0.33 |
| 130 | 6.4 | 9 | 0.25 | 2.7 | 37 | 0.28 |
| 140 | 4.1 | 3 | 0.28 | 2.8 | 6 | 0.26 |
Relaxation times are strongly temperature dependent, and the stretching parameters changed relatively little with temperature. Therefore, we used the fitted relaxation times, τfit to calculate activation energies using the Arrhenius rate law (eqn (3)).
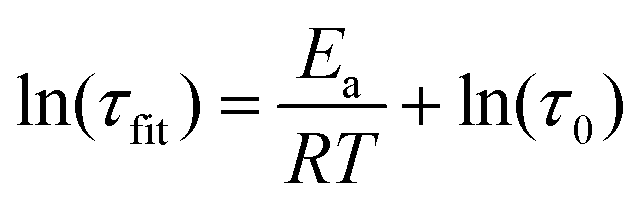 | (3) |
Values of 216 kJ mol−1 for the system with excess amine and 266 kJ mol−1 for the system with excess benzyl bromide, were obtained (Fig. S10, ESI†).
These activation energies for mechanical relaxation are significantly higher than the activation energy for the chemical reactions obtained from the small molecule kinetics. The difference is due to the influence of polymer dynamics on the overall rate of transalkylation, but also contains a contribution from differences in polarity between the polymer matrix and highly polar DMSO in the model reaction, which strongly affects the activation energy of the chemical reaction.
Mechanical relaxation in this material also has a higher activation energy and a lower characteristic relaxation time, (τ at 140 °C) than observed in other transalkylation networks (Table S2, ESI†). The triazolium based network reported by Drockenmuller and coworkers has a much lower activation energy (140 kJ mol−1) and much higher relaxation times τ (655 s) at 140 °C.42 For other systems the activation energy is even lower and relaxation times are longer. It is worth noting here that in a very recent publication63 on the triazolium system the use of benzylic N-substituents resulted in relaxation times that were about two orders of magnitude lower than those in the systems with only aliphatic N-substituents, but all of the investigated systems had activation energies between 126 and 155 kJ mol−1. These results may suggest that part of the fast relaxations that are observed in the current studies may be caused by the fact we used a benzyl bromide substituent and not an alkyl bromide substituent.
In Fig. 8, we compare the temperature dependence of our current system with a selection of previously published systems. It is clear that, at least within this selection, the current DABCO system is quite unique with its very pronounced temperature dependence. This is highly beneficial for processing purposes as exchange is fast at high temperatures but the material is stable at the use temperature.
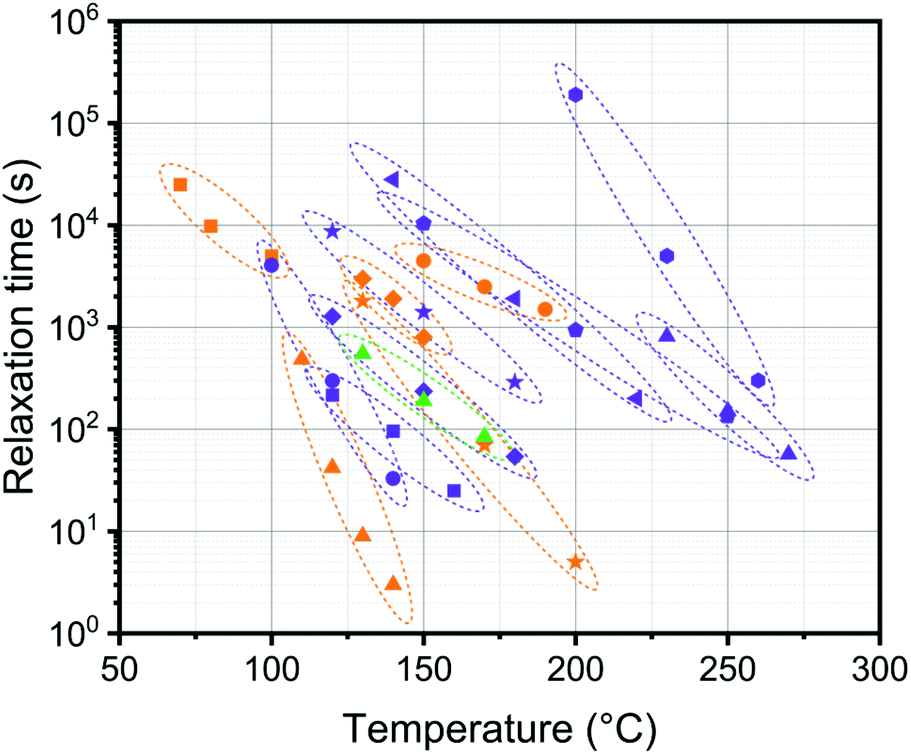 | ||
Fig. 8 Comparison of reported relaxation times at a range of temperatures for a selection of covalent dynamic networks: ( ) DABCO (0.6 eq., this work), ( ) DABCO (0.6 eq., this work), ( ) pyridinium (VSB-35),40 ( ) pyridinium (VSB-35),40 ( ) anilinium salts (PMSEA – 5% VBABr),44 ( ) anilinium salts (PMSEA – 5% VBABr),44 ( ) poly(thioether-sulfonium salts) (5%),45 ( ) poly(thioether-sulfonium salts) (5%),45 ( ) 1,2,3-triazolium,42 ( ) 1,2,3-triazolium,42 ( ) poly(butylene terephthalate)/glycerol,23 ( ) poly(butylene terephthalate)/glycerol,23 ( ) polylactide (IC:OH = 0.75:1),24 ( ) polylactide (IC:OH = 0.75:1),24 ( ) poly(methyl methacrylate)-dioxaborolanes,29 ( ) poly(methyl methacrylate)-dioxaborolanes,29 ( ) polycaprolactone/SO3H,25 ( ) polycaprolactone/SO3H,25 ( ) poly-caprolactone/COOH,25 ( ) poly-caprolactone/COOH,25 ( ) DGEBA epoxy-anhydride,7 ( ) DGEBA epoxy-anhydride,7 ( ) poly(butylene terephthalate)/pentaerythritol,61 ( ) poly(butylene terephthalate)/pentaerythritol,61 ( ) phosphate triesters,64 ( ) phosphate triesters,64 ( ) vinylogous urethanes.35 ) vinylogous urethanes.35 | ||
Conclusions
In summary, we have demonstrated that the alkylation of benzyl bromide with DABCO creates a dynamic bond with efficient exchange at elevated temperatures. A linear polymer with benzyl bromide side groups was crosslinked using DABCO as a divalent crosslinker without addition of a catalyst, resulting in a highly crosslinked polymer network. The polymer networks with excess of amine and excess of bromine both showed efficient stress relaxation in rheological step-strain stress relaxation experiments. At 140 °C, relaxation times for both stoichiometries are faster than observed in many reported systems, e.g., triazolium and trialkylsulfonium based transalkylation networks,42,45,63 and several orders of magnitude faster than observed in a range of systems based on transesterification reactions.65–68 Since the networks are made from easily accessible chemicals and the exchange reaction is efficient at relatively low temperatures, this system offers a simple route towards moldable, recyclable network polymers. We expect that changing size and nucleophilicity of the counterion will give access to networks with a wide range of relaxation temperatures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Brightlands Material Center (BMC) and TU/e impuls scheme for funding. Remon Heemskerk and Nicole Ziegenbalg are thanked for their work on the kinetic experiments. We also like to thank Jolanda Spiering and Annelore Aerts for assistance with the monomer and polymer synthesis.References
- S. H. Goodman, in Handbook of Thermoset Plastics, ed. H. Dodiuk and S. H. Goodman, Noyes Publications, Westwood, 2nd edn, 1998, pp. 0–26 Search PubMed.
- J. P. Pascault and R. J. J. Williams, in Thermosets: Structure, Properties, and Applications, ed. Q. Guo, Elsevier, Amsterdam, 2nd edn, 2017, pp. 3–34 Search PubMed.
- Radiation Curing of Polymeric Materials, ed. C. E. Hoyle and J. F. Kinstle, American Chemical Society, Washington, DC, 1990, vol. 417 Search PubMed.
- B. Goss, Int. J. Adhes. Adhes., 2002, 22, 405–408 CrossRef CAS.
- D. Song and R. K. Gupta, in Thermosets – Structure, Properties and Applications, ed. Q. B. T.-T. Guo, Woodhead Publishing, 2012, pp. 165–188 Search PubMed.
- P. K. Mallick, in Materials, Design and Manufacturing for Lightweight Vehicles, ed. P. K. Mallick, Woodhead Publishing, 2010, pp. 208–231 Search PubMed.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1703 CrossRef CAS PubMed.
- N. Van Herck and F. E. Du Prez, Macromolecules, 2018, 51, 3405–3414 CrossRef CAS.
- J. W. Wydra, C. R. Fenoli, N. B. Cramer, J. W. Stansbury and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1315–1321 CrossRef CAS.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Adv. Mater., 2017, 29, 1606100 CrossRef PubMed.
- P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed, 2019, 58, 9682–9695 Search PubMed.
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- M. Guerre, C. Taplan, J. M. Winne and F. E. Du Prez, Chem. Sci., 2020, 11, 4855–4870 RSC.
- B. Krishnakumar, R. V. S. P. Sanka, W. H. Binder, V. Parthasarthy, S. Rana and N. Karak, Chem. Eng. J., 2020, 385, 123820 CrossRef.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2015, 7, 30–38 RSC.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 784, 3–5 Search PubMed.
- J. P. Brutman, P. A. Delgado and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 607–610 CrossRef CAS.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS.
- Y. Zhou, J. G. P. Goossens, R. P. Sijbesma and J. P. A. Heuts, Macromolecules, 2017, 50, 6742–6751 CrossRef CAS.
- J. P. Brutman, P. A. Delgado and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 607–610 CrossRef CAS.
- H. Zhang, S. Majumdar, R. A. T. M. Van Benthem, R. P. Sijbesma and J. P. A. Heuts, ACS Macro Lett., 2020, 272–277 CrossRef CAS.
- T. Liu, B. Zhao and J. Zhang, Polymer, 2020, 194, 122392 CrossRef CAS.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed.
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Macromolecules, 2015, 48, 2098–2106 CrossRef CAS.
- M. Röttger, T. Domenech, R. Van Der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- A. Rekondo, R. Martin, A. Ruiz De Luzuriaga, G. Cabañero, H. J. Grande and I. Odriozola, Mater. Horiz., 2014, 1, 237–240 RSC.
- C. R. Fenoli, J. W. Wydra and C. N. Bowman, Macromolecules, 2014, 47, 907–915 CrossRef CAS.
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Angew. Chem., Int. Ed, 2011, 50, 1660–1663 Search PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- R. L. Snyder, D. J. Fortman, G. X. De Hoe, M. A. Hillmyer and W. R. Dichtel, Macromolecules, 2018, 51, 389–397 CrossRef CAS.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS PubMed.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 CrossRef PubMed.
- J. J. Lessard, L. F. Garcia, C. P. Easterling, M. B. Sims, K. C. Bentz, S. Arencibia, D. A. Savin and B. S. Sumerlin, Macromolecules, 2019, 52, 2105–2111 CrossRef CAS.
- E. D. Hughes, F. Juliusburger, S. Masterman, B. Topley and J. Weiss, J. Chem. Soc., 1935, 1525–1529 RSC.
- J. Huang, L. Zhang, Z. Tang, S. Wu and B. Guo, Compos. Sci. Technol., 2018, 168, 320–326 CrossRef CAS.
- S. Kulchat and J. M. Lehn, Chem. – Asian J., 2015, 10, 2484–2496 CrossRef CAS PubMed.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, D. Montarnal and E. Drockenmuller, J. Am. Chem. Soc., 2015, 137, 6078–6083 CrossRef CAS PubMed.
- P. Chakma, Z. A. Digby, M. P. Shulman, L. R. Kuhn, C. N. Morley, J. L. Sparks and D. Konkolewicz, ACS Macro Lett., 2019, 8, 95–100 CrossRef CAS.
- P. Chakma, C. N. Morley, J. L. Sparks and D. Konkolewicz, Macromolecules, 2020, 53, 1233–1244 CrossRef CAS.
- B. Hendriks, J. Waelkens, J. M. Winne and F. E. Du Prez, ACS Macro Lett., 2017, 6, 930–934 CrossRef CAS.
- Z. Tang, Y. Liu, Q. Huang, J. Zhao, B. Guo and L. Zhang, Green Chem., 2018, 20, 5454–5458 RSC.
- C. Decker, Polym. Int., 1998, 45, 133–141 CrossRef CAS.
- J. W. Stansbury, J. Esthet. Dent., 2000, 12, 300–308 CrossRef CAS PubMed.
- C. Decker, Macromol. Rapid Commun., 2002, 23, 1067–1093 CrossRef CAS.
- R. Penterman, S. I. Klink, H. de Koning, G. Nisato and D. J. Broer, Nature, 2002, 417, 55–58 CrossRef CAS PubMed.
- A. Hancock and L. Lin, Pigm. Resin Technol., 2004, 33, 280–286 CrossRef CAS.
- M. Baidya, S. Kobayashi, F. Brotzel, U. Schmidhammer, E. Riedle and H. Mayr, Angew. Chem., Int. Ed, 2007, 46, 6176–6179 Search PubMed.
- K. Zhang, K. J. Drummey, N. G. Moon, W. D. Chiang and T. E. Long, Polym. Chem., 2016, 7, 3370–3374 RSC.
- J. Chauvet, J. M. Asua and J. R. Leiza, Polymer, 2005, 46, 9555–9561 CrossRef CAS.
- J. S. Moore and S. I. Stupp, Macromolecules, 1990, 23, 65–70 CrossRef CAS.
- A. Jourdain, R. Asbai, O. Anaya, M. M. Chehimi, E. Drockenmuller and D. Montarnal, Macromolecules, 2020, 53, 1884–1990 CrossRef CAS.
- B. R. Elling and W. R. Dichtel, ACS Cent. Sci., 2020, 6, 1488–1496 CrossRef CAS PubMed.
- Thermal Transitions of Homopolymers: Glass Transition & Melting Point, https://www.sigmaaldrich.com/technical-documents/articles/materials-science/polymer-science/thermal-transitions-of-homopolymers.html, (accessed 27 January 2019).
- H. Barjat, G. A. Morris, S. Smart, A. G. Swanson and S. C. R. Williams, J. Magn. Reson., Ser. B, 1995, 108, 170–172 CrossRef CAS.
- H. Zhou, NMR Theory and Techniques, https://nmr.chem.ucsb.edu/education/part7.html#dosy, (accessed 20 January 2020).
- Y. Zhou, J. G. P. Goossens, S. van den Bergen, R. P. Sijbesma and J. P. A. Heuts, Macromol. Rapid Commun., 2018, 39, 1800356 CrossRef PubMed.
- L. Li, X. Chen, K. Jin and J. M. Torkelson, Macromolecules, 2018, 51, 5537–5546 CrossRef CAS.
- O. Anaya, A. Jourdain, I. Antoniuk, H. Ben Romdhane, D. Montarnal and E. Drockenmuller, Macromolecules, 2021, 54, 3281–3292 CrossRef CAS.
- S. Majumdar, H. Zhang, M. Soleimani, R. A. T. M. Van Benthem, J. P. A. Heuts and R. P. Sijbesma, ACS Macro Lett., 2020, 9, 1753–1758 CrossRef CAS.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- B. Zhang, K. Kowsari, A. Serjouei, M. L. Dunn and Q. Ge, Nat. Commun., 2018, 9, 1831 CrossRef PubMed.
- F. I. Altuna, V. Pettarin and R. J. J. Williams, Green Chem., 2013, 15, 3360–3366 RSC.
- J. Han, T. Liu, C. Hao, S. Zhang, B. Guo and J. Zhang, Macromolecules, 2018, 51, 6789–6799 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, kinetic analysis, calculation of activation energy, reaction mechanisms material characterization and supplementary figures. See DOI: 10.1039/d1py00292a |
| This journal is © The Royal Society of Chemistry 2021 |