
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effects of cononsolvents on the synthesis of responsive particles via polymerisation-induced thermal self-assembly†
Marissa D.
Morales-Moctezuma
and
Sebastian G.
Spain
 *
*
Polymer and Biomaterials Chemistry Laboratories, Department of Chemistry, University of Sheffield, Sheffield S3 7HF, UK. E-mail: s.g.spain@sheffield.ac.uk
First published on 20th July 2021
Abstract
Nanogels have emerged as innovative platforms for numerous biomedical applications including gene and drug delivery, biosensors, imaging, and tissue engineering. Polymerisation-induced thermal self-assembly (PITSA) has been shown to be a suitable route for the synthesis of temperature-responsive nanogels, and cononsolvents have been used to improve the solubility of comonomers. Here, we show that use of cononsolvents during synthesis also has significant effects on the properties of the final nanogels. Responsive nanogels consisting of a poly(acrylic acid) corona and a crosslinked poly(N-isopropylacrylamide) core were synthesised via RAFT-mediated PITSA in water and water/EtOH mixtures. Nanogels synthesised in water were found to increase in size as the degree of polymerisation (DPn) of the pNIPAM block was increased. For a fixed pNIPAM DPn, particle sizes were seen to decrease with increasing EtOH content in the synthesis solvent. This is rationalised by the decreasing solubility of the growing pNIPAM in the reaction medium resulting in earlier particle nucleation. The temperature response of the particles was also found to be dependent on the synthesis conditions with increasing EtOH content in the synthesis solvent increasing the volume phase transition temperature. Model poly(acrylic acid)-block-poly(N-isopropylacrylamide) copolymers synthesised under similar conditions were seen to have differing tacticities dependent on the synthesis solvent with increasing EtOH content resulting in increased syndiotacticity and increased cloud point temperatures. We propose that the tacticity changes are due to the PITSA process, with the constrained environment of the particle resulting in increased syndiotacticity.
Introduction
Nanogels/microgels are colloidal particles with a diameter in the sub-micron region and have emerged as innovative materials for numerous biomedical applications including gene and drug delivery,1,2 biosensors,3 imaging,4 and tissue engineering.5 Various methodologies have been reported for the preparation of nanogels. They include the crosslinking of prepolymers by non-covalent interactions (e.g. hydrophobic or ionic interactions)6–11 or the formation of covalent crosslinks between reactive functional groups of polymer chains using chemistries such as thiol-disulfide exchange,12–15 click reactions,16,17 amidation of activated esters,18–20 and photoreactions.21–25 Moreover, covalently crosslinked nanogels are also prepared by the heterogeneous radical (co)polymerisation of vinyl monomers and a crosslinker in a one-pot synthesis. Inverse precipitation,26–29 (inverse) mini- or microemulsion,30–38 and dispersion39,40 processes are relevant techniques used in the synthesis of nanogels by either free radical or controlled radical polymerisation techniques. Although well-defined crosslinked nanoparticles can be prepared by these heterogeneous techniques, their main drawbacks are the use of molecular surfactants or potentially toxic solvents.41 In order to avoid these issues, nanogel formation using surfactant-free aqueous dispersion polymerisation has emerged as a versatile way to prepare colloidally stable nanoparticles from solvophilic precursors.42,43 For example, An et al. used poly(N,N-dimethylacrylamide) (pDMA) to both stabilise the in situ formation of nanoparticles and to mediate the reversible addition–fragmentation chain transfer (RAFT) polymerisation of N-isopropylacrylamide (NIPAM) in the presence of N,N′-methylenebisacrylamide (BIS) crosslinker.43 This aqueous RAFT dispersion approach is based on a polymerisation-induced self-assembly (PISA) process. PISA is a polymerisation technique whereby a soluble homopolymer (stabiliser) is chain extended with a second monomer that forms an insoluble polymer in the polymerisation solvent. As the second block grows and becomes insoluble, polymer chains assemble into nanoparticles stabilised with the soluble block and the insoluble block as the core (Scheme 1a). Further extension of the insoluble block allows control of particle size as well as access to higher morphologies such as cylindrical micelles (or worms) and vesicles (polymersomes).44–46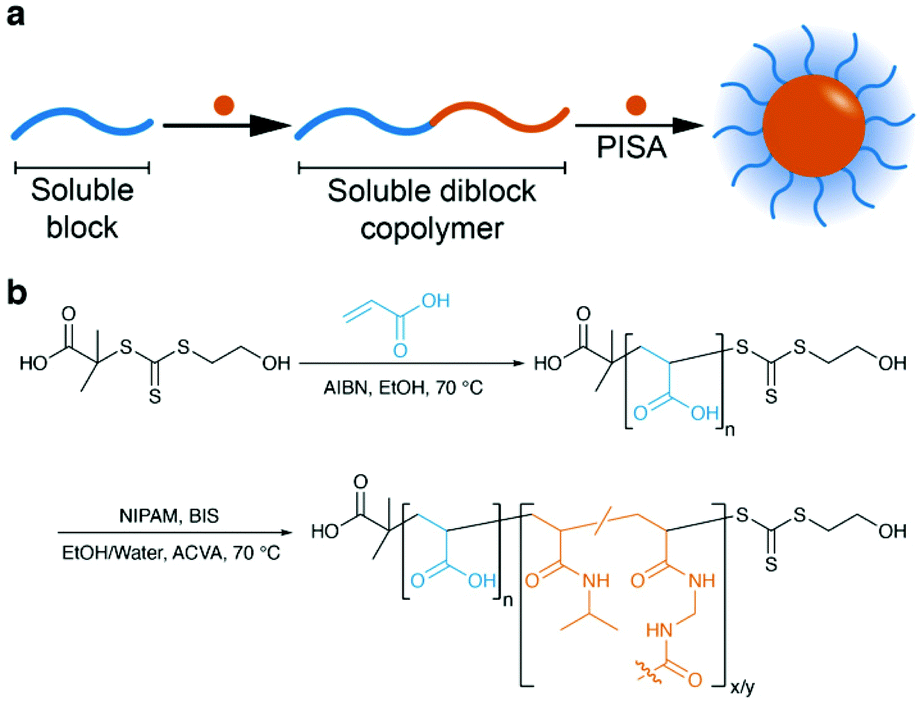 | ||
| Scheme 1 (a) Schematic of the polymerisation-induced self-assembly process. (b) Synthesis of pAAn-b-pNIPAMx/BISy nanogels. | ||
Polymerisation-induced thermal self-assembly (PITSA) is a derivative of PISA whereby a temperature-responsive polymer, e.g. pNIPAM, is grown from the stabiliser block above its lower critical solution temperature (LCST) resulting in thermally-induced self-assembly during the polymerisation.43,47 Addition of a crosslinker during the PITSA process allows the synthesis of temperature-responsive pNIPAM nanogels that do not dissociate on cooling, instead they shrink and swell above and below a volume phase transition temperature (VPTT), respectively.43
So far, PITSA has been used to synthesise nanogels under aqueous dispersion conditions from temperature-responsive monomers such as NIPAM,43,48–50N,N-diethylacrylamide (DEAM),51,52 oligo(ethylene glycol)methacrylates (OEGMAs),53 oligo(ethylene glycol)acrylates (OEGAs),49,54 and 2-(dimethylamino)ethyl methacrylate (DMAEMA).55 It is noteworthy that while this technique is environmentally friendly since it is performed in water and high-yielding, its use is limited to water-soluble monomers whose polymer becomes less soluble during polymerisation. To overcome this limitation, some researchers have explored the addition of small quantities of a solvent with low toxicity such as alcohols to water as a route to include hydrophobic components (e.g. additional monomers) into the nanogel structure56,57 or to promote the immiscibility of growing chains during polymerisation.57–59 Evidently, water is chosen as the major solvent and an alcohol (e.g. ethanol,56–58tert-butanol,59 or 2-propanol60–62) is selected as the minor solvent. For example, Xu et al. used a mixture of water/ethanol (75/25, v/v) for the RAFT dispersion polymerisation of NIPAM and functional comonomers (e.g. hydrophobic monomers, fluorophores) with ethanol allowing solubilisation of the additional monomers.56 This binary solvent strategy was later used for the synthesis of doubly thermoresponsive triblock nanogels that consisted of a poly(diethylene glycol ethyl acrylate) (pDEGA) outer block, a pDMA midblock, and a crosslinked pNIPAM core block using a mixture of water/ethanol (65/35, v/v).58 In this case, ethanol allowed better solubilisation of pDEGA thus promoting the location of pDEGA in the corona and the pNIPAM block in the core of the nanogel. In a traditional aqueous PISA, the block with the lowest LCST (i.e. pDEGA) would otherwise be located in the core of the structure and the block with the highest LCST (i.e. pNIPAM) in the outer shell.58 By using this approach, the library of nanogels synthesised by RAFT dispersion polymerisation was expanded to different compositions and functionalities that were inaccessible by using only water during synthesis. Nonetheless, it is well-established that mixtures of water and ethanol show cononsolvency for pNIPAM at certain mole fractions despite being good solvents for pNIPAM individually.63 For instance, the lower critical solution temperature (LCST) of pNIPAM is around 31–35 °C in pure water,64,65 but in mixtures of cononsolvents (e.g. ethanol,66,67 methanol,68–70 THF,71,72 DMSO,73,74 and acetone74,75) its LCST is lowered when small amounts of the organic solvent are added. Hence, the cononsolvent composition is likely to influence the in situ self-assembly process when conducting the RAFT dispersion polymerisation of NIPAM.
Herein, we aimed to investigate the effects of the synthesis cononsolvent composition on the properties of pNIPAM nanogels prepared by RAFT-mediated PITSA in water and water/ethanol mixtures. To achieve this, the RAFT dispersion polymerisation of NIPAM in the presence of BIS was conducted using poly(acrylic acid) (pAA) as the macromolecular chain transfer agent (macroCTA) to synthesise multi-responsive crosslinked particles (pAA-b-pNIPAM/BIS) (Scheme 1b). The nanogel sizes were found to be strongly dependent on the solvent mixture used during their synthesis. The thermal properties of the nanogels were also found to be dependent on the solvent used during the synthesis and this is demonstrated to be due to the different tacticity of the pNIPAM core.
Results and discussion
Synthesis of poly(acrylic acid) macro-chain transfer agents (pAA macroCTAs)
The synthesis of pAA-b-pNIPAM/BIS is outlined in Scheme 1b. Poly(acrylic acid) was chosen as the stabiliser block as it is soluble in all water/EtOH compositions and highly charged stabiliser blocks are known to prevent the formation of higher order morphologies (e.g. worm-like micelles and vesicles) during PISA,76–78 thereby simplifying data analysis by minimising the probability of additional phase transitions that may complicate data interpretation. A poly(acrylic acid) (pAA) macroCTA was synthesised by RAFT polymerisation of acrylic acid in ethanol with 2-(hydroxyethylthiocarbonothioylthio)-2-methylpropanoic acid (HEMP) as the chain transfer agent. An initial kinetic experiment determined that high acrylic acid conversions (∼90%) were obtained after 5 h of polymerisation. Subsequent polymerisations were stopped at approximately 70% conversion to ensure high end-group fidelity in the subsequent block copolymer and nanogel syntheses.The number-average degree of polymerisation (DPn) for the pAA macroCTA was 45 as determined by end-group analysis by 1H NMR spectroscopy. The pAA macroCTA was characterised by size exclusion chromatography (SEC) in THF/AcOH against pMMA standards after methylation with trimethylsilyl diazomethane and found to be mono-modal and of low dispersity (Mn = 4.6 kg mol−1, Mw/Mn = 1.19). Full characterisation (1H NMR spectra, SEC chromatograms and molar masses) of the pAA macroCTA may be found in the ESI (Fig. S1–5 and Table S1†).
Synthesis and characterisation of pAA-b-pNIPAM/BIS nanogels in water
Prior to investigating the effect of cononsolvency on nanogel synthesis a series of pAA45-b-pNIPAMx/BIS3 (x = 87, 175 and 262) nanogels were synthesised in water. The pAA45 macroCTA was chain extended with N-isopropylacrylamide (NIPAM) and N,N′-methylene bisacrylamide (BIS) via RAFT-mediated aqueous dispersion polymerisation at pH 6.7. This pH was chosen to ensure that the pAA was in a highly charged state (pKa 5.8) and was reached by addition of a minimal volume of NaOH solution to minimise any changes to ionic strength that may affect the self-assembly. The resulting nanogel dispersions were purified by dialysis against deionised water and characterised with dynamic light-scattering (DLS), transmission electron microscopy (TEM) and ζ-potential measurements (Table 1, entries 1–3).| Entry | Target composition [pAA45]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[NIPAM]:[BIS]a :[NIPAM]:[BIS]a |
X EtOH | Conversion c/% | DPn,theod |
M
n,theoe/kg mol−1 |
D h ± S.D./nm (PDI)f | ζ-Potential ± S.D.g/mV |
|---|---|---|---|---|---|---|---|
| a General reaction conditions: Targeted [pAA45]/[ACVA] = 5, solids content 10% w/w, 70 °C for 18 h. b Mole fraction of ethanol used during nanogel synthesis. c Gravimetric determination by moisture analysis of solids content against predicted solids content. d For an equivalent diblock synthesised without crosslinker, estimated from [NIPAM/[pAA45] × conversion. e Calculated using Mn, theo = Me + nMAA + xMNIPAM + yMBIS, where Me is the molecular weight of the CTA end groups, n, x and y are the DPn of pAA, pNIPAM, and BIS respectively. f DLS data obtained from 0.1% w/v nanogels dispersions in DI water at pH 7 and 25 °C. g Average zeta-potential and the standard deviation of five run measurements for 0.1% w/v nanogels dispersions in DI water with 1 mM KCl at pH 7 and 25 °C. | |||||||
| 1 | [1]:[87]:[3] |
0 | 74 | 65 | 11.3 | 80 ± 26 (0.106) | −40.9± 0.7 |
| 2 | [1]:[175]:[3] |
0 | 90 | 158 | 21.8 | 231 ± 64 (0.076) | −34.2 ± 2.0 |
| 3 | [1]:[262]:[3] |
0 | 84 | 220 | 28.8 | 483 ± 121 (0.063) | −29.9 ± 0.7 |
| 4 | [1]:[175]:[3] |
0.06 | 98 | 172 | 23.4 | 159 ± 37 (0.053) | −31.1 ± 1.9 |
| 5 | [1]:[175]:[3] |
0.12 | 100 | 175 | 23.7 | 97 ± 41 (0.175) | −34.0 ± 2.7 |
| 6 | [1]:[175]:[3] |
0.14 | 92 | 161 | 22.2 | 90 ± 42 (0.222) | −22.8 ± 3.6 |
During the PITSA process, the growing pNIPAM chains will reach a critical degree of polymerisation at which they will undergo a coil-to-globule transition and aggregate into nascent particles with a pNIPAM core and a pAA shell. As pAA is highly negatively charged at neutral pH, it was expected that the synthesised nanogels would be highly negatively charged and this is confirmed by their measured ζ-potentials (−41 mV to −29 mV). As the length of the pNIPAM block is increased, it is expected that the size of the core will increase with the increasing volume fraction of pNIPAM. From DLS measurements (Fig. 1a, Fig. S6† and Table 1, entries 1–3), it is clear that the hydrodynamic diameter, Dh, increases from 80 nm for pAA45-b-pNIPAM87/BIS3 nanogels to 483 nm for pAA45-b-pNIPAM262/BIS3 nanogels. As the volume fraction of pNIPAM is increased, it is also expected that higher order morphologies may be formed. For example, Figg et al. found that nano-objects formed during PITSA of NIPAM from a pDMA34-b-p(DMA-st-AA)14/6 macroCTA were spherical when the pNIPAM DPn was 11 and 52 but worms and vesicles were observed with DPn of 101 and 137, respectively but required post-polymerisation crosslinking as a way to prevent dissolution of the chains during cooling.47
 | ||
| Fig. 1 Characterisation of pAA45-b-pNIPAMx/BIS3 (x = 87, 175 and 262) nanogels synthesised in water. (a) Intensity average log normal size distributions from dynamic light-scattering. (b) Transmission electron micrographs. DPn indicates the targeted DP of pNIPAM. DLS and TEM samples were prepared from 0.1% w/v nanogel dispersions in water at pH 7 at 25 °C. TEM samples were stained with phosphotungstic acid (0.75% w/v, pH 7). | ||
Here, when a pNIPAM DPn of 87 was targeted only spheres were evident in the TEM micrographs (Fig. 1b). For higher pNIPAM DPn (175 and 262) particle morphologies were irregular and appear to be aggregates of smaller spherical particles. The formation of spheres rather than higher morphologies has been seen in PISA systems where a highly-charged stabiliser block is used76,78 and is thought to be due to charge repulsion preventing particle fusion to form worms.77 Additionally, the presence of a crosslinker in the system may result in particles being trapped in a spherical morphology as the chains cannot reorganise. This may also explain the aggregate morphology seen in the pNIPAM samples with DPn 175 and 262 as particle growth also occurs through coalescence of smaller particles which will be inhibited by partial crosslinking during the polymerisation and this is supported by asymmetric flow field flow fractionation studies of related nanogels.79
Cononsolvents used during synthesis affect the size of pAA-b-pNIPAM/BIS nanogels
Despite individually being good solvents for pNIPAM, water and ethanol are known to display cononsolvency for pNIPAM at certain mole fractions.66,67 Xu et al. utilised this cononsolvency to synthesise poly(N,N-dimethylacrylamide)-b-pNIPAM/BIS nanogels containing a range of functional monomers with the addition of ethanol allowing solvation of hydrophobic monomers.56 The composition of the cononsolvent is known to have a significant effect on the lower critical solution temperature (LCST) of pNIPAM80 and can also cause an upper critical solution temperature at high mole fractions of the organic solvent.80 Additionally, Wen et al. have recently demonstrated that solvent quality for the core-forming block can affect self-assembly during PISA.81 Consequently, it was expected that it would also have an effect on the nucleation and self-assembly processes during PITSA.pAA45-b-pNIPAM175/BIS3 nanogels were synthesised as before in water/EtOH mixtures at pH 6.7. The resulting nanogel dispersions were purified by dialysis against deionised water and characterised with dynamic light-scattering (DLS), transmission electron microscopy (TEM) and ζ-potential measurements (Fig. 2, Fig. S7† and Table 1, entries 4–6). For all nanogels, the ζ-potentials were strongly negative (−34 to −23 mV) as expected. Nanogel sizes measured by DLS in water were found to be strongly dependent on the composition of the solvent used during the synthesis. When synthesised in water alone, pAA45-b-pNIPAM175/BIS3 nanogels had a hydrodynamic diameter of 231 nm. For nanogels synthesised in 0.06 mol% EtOH (XEtOH = 0.06), Dh was drastically reduced to 159 nm with no increase in the polydispersity of the particles. Further increases in EtOH concentration resulted in further reduction in nanogel hydrodynamic diameter (Dh = 97 nm and 90 nm for XEtOH = 0.12 and XEtOH = 0.14, respectively). TEM micrographs of the nanogels showed a similar reduction in size as well as a shift from the “aggregated spherical particle” morphology to individual spherical particles as the ethanol content was increased (Fig. 2 and S8†). Similar behaviour was seen for nanogels synthesised with targeted pNIPAM DPn of 87 and 262 in cononsolvents (Fig. S9 and S10, Table S2†).
 | ||
| Fig. 2 Characterisation of pAA45-b-pNIPAM175/BIS3 nanogels synthesised in water/EtOH mixtures. (a) Intensity average log normal size distributions distributions from dynamic light-scattering. (b) Transmission electron micrographs. DLS and TEM samples were prepared from 0.1% w/v nanogel dispersions in water at pH 7 at 25 °C. TEM samples were stained with phosphotungstic acid (0.75% w/v, pH 7). | ||
Wen et al. recently reported a similar effect in the PISA synthesis of poly(potassium 3-sulfopropyl methacrylate)-b-poly(benzyl methacrylate) nanoparticles where synthesis in alcohol/water mixtures resulted in increased particle size compared to the synthesis in water alone.81 This increase was rationalised by the increased solubility of the benzyl methacrylate block with increasing alcohol content allowing greater chain mobility and solubility and therefore the greater exchange of chains being growing particles. Here, the decrease in nanogel diameter with increasing ethanol content during the PITSA process is not unexpected and can be rationalised when the effect of the cononsolvent on the self-assembly process is considered. During PISA nascent particles are nucleated when the core-forming block reaches a critical DPn where it becomes insoluble. In PITSA, this critical DPn should be related to the LCST of the core-forming polymer in the synthesis solvent. pNIPAM is generally considered to have an LCST of 32 °C in water but cloud point measurements indicate this is reduced to below 25 °C when XEtOH = 0.05 and below 10 °C when the XEtOH = 0.1 (Fig. S11†), indicating that pNIPAM is significantly less soluble in the cononsolvent mixtures than in water alone. Consequently, the critical DPn at which particle nucleation occurs should be lower with increasing XEtOH within the range of cononsolvent compositions investigated here. To confirm this, pNIPAM homopolymers were synthesised via precipitation polymerisation in water and water/EtOH mixtures and it was found that the Mn of the resulting polymers was strongly dependent on the synthesis solvent (Fig. S12†) with increasing EtOH content during the synthesis reducing the Mn of the pNIPAM (e.g. XEtOH = 0, Mn = 280 kg mol−1versus XEtOH = 0.15, Mn = 35 kg mol−1). These data indicate that pNIPAM becomes insoluble at a lower DPn as the EtOH content increases for the molar fractions tested here, consequently the critical DPn should be lower. As the critical DPn is lower, then particle nucleation should occur earlier in the polymerisation. As crosslinking is more probable within particles, where the polymer (and possibly monomer) concentration is higher, than in between dissolved chains then earlier nucleation of particles should lead to smaller particles due to the spherical morphology being “trapped” earlier in the polymerisation. It should be noted that variations in pH or ionic strength could also affect the PITSA process by changing the conformation/volume of the pAA stabiliser. However, addition of ethanol changes the dielectric constant of the solvent82 which will also effect the pKa of the pAA stabiliser and the apparent ionic strength and will likely have a larger effect than any small differences the quantities of reagents added.
Cononsolvents used during synthesis affect the thermal properties of nanogels
As the nanogels contain a pNIPAM core, they are expected to display temperature-responsive properties. These nanogels are already a dispersion and therefore do not display a cloud point or lower critical solution temperature in the same sense as a linear pNIPAM sample. Instead, these nanogels display a volume phase transition temperature (VPTT) at which the crosslinked pNIPAM core collapses, resulting in a decrease in the particle diameter at this temperature. Particle size changes with temperature were measured using variable temperature DLS studies at an aqueous pH of 7. As expected, all of the nanogels were temperature-responsive (Fig. 3a). The nanogels synthesised in XEtOH 0, 0.06 and 0.12 clearly decrease in volume upon heating but this decrease is less pronounced for smaller nanogels where the core will have a smaller contribution to the overall particle volume. VPTTs of these nanogels were determined as the halfway point between the swollen and collapsed states (eqn (S1)†). Interestingly, the VPTT increased with the XEtOH used during the synthesis. When synthesised in water alone the pAA45-b-pNIPAM175/BIS3 nanogel had a VPTT of 37 °C, but nanogels synthesised in XEtOH = 0.06 and 0.12 had VPTTs of 39 °C and 42 °C, respectively. VPTTs were higher than the VPTT for a pNIPAM gel (∼32 °C)83 due the hydrophilic characteristics of pAA at pH 7.84 Note that, although there may be small variations in ionic strength between samples, the LCST of pNIPAM is relatively insensitive to salts at the low concentrations that may occur due to experimental variation with the LCST of pNIPAM-coated silica particles only decreasing by 1 °C in the presence of 50 mM KCl.85 Since the nanogels had similar monomer compositions, the shift in the VPTT with increasing XEtOH used during synthesis should be related to differences caused by the particle nucleation. The VPTT shift is more clear when the sizes are normalised (Fig. 3b). Notably, the onset of shrinking is also affected by the cononsolvent composition used during the synthesis. The nanogels synthesised in water alone had an onset of shrinking at 21 °C, whereas the ones synthesised in XEtOH 0.06 and 0.12 were at 29 °C and 34 °C, respectively. For nanogels synthesised in XEtOH 0.14, there is a second transition with an increase in Dh at approximately 38 °C indicating that flocculation occurs above this temperature, which is supported by an increase in the PDI (Fig. S13†). | ||
| Fig. 3 (a) Variable temperature DLS data for pAA45-b-(pNIPAM175/BIS3) nanogels synthesised in water and water/EtOH mixtures. (b) Normalised sizes. DLS measurements were obtained from 0.1% w/v nanogel aqueous dispersions at pH 7. | ||
Synthesis and properties of pAA-b-pNIPAM block copolymers
The LCST of pNIPAM (and VPTT of pNIPAM gels) is known to be sensitive to end groups or, in the case of block copolymers, the other blocks.86 For example, Xia et al. demonstrated that that an N-phenyl amide end group lowered the cloud point of pNIPAM by as much as 9 °C compared to the unsubstituted amide.87 Additionally, the tacticity of pNIPAM is also known to affect its LCST and the tacticity can be affected by the synthesis conditions, e.g. the solvent.88,89 For example, Biswas et al. demonstrated that the isotacticity of pNIPAM synthesised in presence of 0.1 M Y(OTf)3 increased from 55% to 86% as the methanol mole fraction in water was increased from 31 to 100 mol%.89 Additionally, the cloud point of nanoparticles coated with responsive polymers is known to be dependent on the particle size.90To understand how these factors affect the VPTT of pAA-b-pNIPAM/BIS nanogels, pAA-b-pNIPAM diblock copolymers were synthesised as soluble analogues that are more amenable to detailed characterisation.
A pAA34 macroCTA (Mn = 3.2 kg mol−1, Mw/Mn = 1.20) was used to prepare a series of pAA34-b-pNIPAMx (x = 92, 184 and 280) diblock copolymers by RAFT dispersion polymerisation of NIPAM in either water or water/ethanol mixtures targeting nominal pNIPAM DPn between 92 and 280 (Table S3†). The shorter pAA macroCTA was chosen to reduce the pAA contribution during later characterisation (e.g. in the 1H NMR spectra). These conditions were chosen to mimic those used in the nanogel synthesis but in the absence of crosslinker. In all cases, the NIPAM conversions were at least 99% as determined by 1H NMR spectroscopy. pAA34-b-pNIPAMx diblock copolymers were purified by exhaustive dialysis against deionised water for 48 h with repeated water changes before characterisation (Table 2 and Table S3†). 1H NMR spectra of the purified copolymers (Fig. 4) were all very similar with only minor variations in the resonances for the backbone protons (δ 1.4–2.1 ppm) and the DPn measured to be as expected. As these nano-objects scatter light, it is possible to measure the cloud point of the diblock copolymers using turbidimetry. A clear increase in the cloud point temperature, Tcp, from 34 °C to 39 °C was observed as XEtOH was increased from 0 to 0.15 (Table 2, Table S3 and Fig. S14†). The diblock copolymers were also found to self-assemble above their Tcp to form defined nano-objects. For the pAA34-b-pNIPAM184 diblock copolymers there is a clear difference in the size of the resulting nano-objects depending on the XEtOH used during the synthesis with the copolymer synthesized in water alone forming smaller dispersions (Dh = 49 nm) than those synthesis in water/EtOH mixtures (Dh = 67–140 nm). It should be noted that this size dependence is the reverse of that seen for the sizes of nanogels synthesised in different cononsolvent compositions. However, the diblocks were assembled in water alone and from solution which will result in different structures to those self-assembled in situ in varying solvent mixtures and the presence of crosslinker. As noted above, NMR spectroscopy showed very little difference between copolymers so such large differences in the self-assembly were unexpected. However, no suitable conditions for SEC analysis could be found for these diblock copolymers, therefore differences in architecture, Mw/Mn or homopolymer contamination cannot be completely eliminated. Similar trends in both nano-object size and were seen for pAA34-b-pNIPAM92 and pAA34-b-pNIPAM280 diblock copolymers series (Table S3†).
 | ||
| Fig. 4 1H NMR spectra of purified pAA34-b-pNIPAM184 diblock copolymers synthesised in water and water/EtOH mixtures. The r and m labels are used to denote the resonances attributed to the racemic and meso diads, respectively. Spectra were recorded in D2O. | ||
| Targeted pNIPAM DPna |
X
EtOHb |
Conversionc/% | DPn,theod |
Cloud point (Tc)e/°C | D h ± S.D.f/nm (PDI) |
|---|---|---|---|---|---|
| a General reaction conditions: Targeted [pAA34]/[ACVA] = 5, solids content 10% w/w,70 °C for 18 h. b Mole fraction of ethanol used during nanogel synthesis. c Determined by 1H NMR spectroscopy using the ratio of the sum of vinyl proton integrals of NIPAM and the polymer backbone. d Estimated from [NIPAM/[pAA34] × conversion. e Cloud points were estimated from the inflection point of the normalised absorbance curve of 1% w/v dispersions in water at 550 nm at a heating rate of 0.12 °C min−1. f DLS data obtained from 0.1% w/v dispersions in DI water at pH 7 and 50 °C. | |||||
| 184 | 0 | >99 | 184 | 34 | 49 ± 12 (0.06) |
| 184 | 0.06 | >99 | 184 | 36 | 67 ± 31 (0.21) |
| 184 | 0.12 | >99 | 184 | 39 | 93 ± 44 (0.22) |
| 184 | 0.15 | 99 | 182 | 39 | 140 ± 56 (0.16) |
It is well known that solvents and additives (e.g. Lewis acids) used during synthesis can affect the microstructure (tacticity) of polymers.91 Biswas et al. reported that pNIPAM synthesised in different water/MeOH mixtures with Yb(OTf)3 have different tacticities and that the cloud point was dependent on the tacticity of the pNIPAM, with pNIPAM with higher isotactic content having a lower cloud point.89 Further inspection of the 1H NMR spectra (Fig. 4) in the 1.2–1.8 ppm region resulting from the pNIPAM backbone methylene group showed small but distinct differences in the tacticity of the pNIPAM block with an increase in syndiotactic (racemic diad) from approximately 53% to 59% as XEtOH is increased from 0 to 0.15.
The shift in the Tcp was previously attributed to the different content of meso diads, where diblock copolymers synthesised in water were more meso-rich (i.e. less hydrophilic) than those synthesised in cononsolvents, hence these have a lower Tcp.92 An increase in racemic diads generates an alternating chirality where geometrical constraints make the formation of local hydrogen bonds between amide groups difficult and hence water molecules interact more strongly with the polymer, thus explaining the enhanced hydrophilicity and higher Tcp.
If the polymerisation solvent is causing the change in tacticity of pNIPAM, as has been reported in literature, then the homopolymers synthesised in different water/EtOH mixtures would also be expected to have differences in the tacticity. However, 1H NMR spectroscopy showed no change in the 1.2–1.8 ppm region of these polymers (Fig. S15†). Changes in tacticity of poly(methyl methacrylate) have been reported in heterogeneous polymerisations compared to bulk polymerisation.93,94 In these cases, the volume of the polymer chain is comparable to the volume of the polymerisation loci so the increased syndiotacticity has been attributed to the confinement of the chains requiring more gauche conformations along the polymer backbone to enable it to fold back into the particle/loci. This restriction on the chain conformation may lead to a preferred conformation at the propagating centre and therefore the formation of different triads.
Although the particles here are unlikely to be single chain, their small volume is still likely to lead to the chains being conformationally restricted. Therefore, we suggest that the change in tacticity is due to the PITSA process with the constraints placed upon the propagating centre and accessible monomer resulting in a preference for syndiotacticity. The differences between polymers synthesised in different water/EtOH mixtures can be explained by the critical degrees of polymerisation required for nucleation. As XEtOH is increased, the critical degree of polymerisation is reduced, resulting in earlier nucleation and the polymer chain growing under constrained conditions for longer, increasing the length of chain with higher syndiotactity and the overall average syndiotacticity of the sample.
Materials and methods
Materials
2-Mercaptoethanol (≥99%), carbon disulfide (≥99%), (trimethylsilyl)diazomethane (2 M solution in diethyl ether) and 4,4′-azobis(4-cyanovaleric acid) (ACVA, 98%) were purchased from Sigma-Aldrich. Potassium phosphate tribasic (97%) was purchased from Alfa Aesar. 2-Bromoisobutyric acid (98%) was purchased from Acros Organics. Acrylic acid (AA, Merck, ≥99%) was passed through a column of basic Al2O3 prior to use. N-Isopropyl acrylamide (NIPAM, Fluorochem) and 2,2′-azobis(2-methylpropionitrile) (AIBN) (Sigma-Aldrich) were recrystallised from n-hexane and dried before use. N,N′-Methylene bisacrylamide (BIS) (Sigma-Aldrich) was recrystallised from hot methanol and dried before use. 2-(Hydroxyethylthiocarbonothioylthio)-2-methylpropanoic acid (HEMP) was synthesised as previous reported.79 All other reagents were used as received. SnakeSkin™ dialysis tubing (ThermoFisher scientific) of 3.5 kDa molecular weight cut-off (MWCO) was used to purify the macro-CTAs and nanogel solutions. For nuclear magnetic resonance (NMR) experiments, samples were prepared in either D2O or CDCl3 (Sigma Aldrich).Synthesis of poly(acrylic acid) macroCTA via ethanolic RAFT solution polymerisation
Poly(acrylic acid) macroCTAs with different DPn were synthesised similarly but targeting different molar ratios of [AA]:[HEMP]. Using the pAA45 synthesis as an example: A mixture of HEMP (0.56 g, 2.31 mmol), AA (10 g, 138.78 mmol), AIBN (0.076 g, 0.46 mmol) and ethanol to give a 25% w/w solids solution was purged thoroughly with N2 for 30 min. The flask was then placed onto a DrySyn® heating block preheated to 70 °C and left to react for 130 min. The reaction was quenched by removing the flask from the heat source and opening to air. AA conversion (78%) was calculated by 1H NMR spectroscopy. The product was recovered by precipitation from diethyl ether (400 mL). Further product purification was performed by dialysis against water and freeze-drying to give a pale yellow solid. δH (400 MHz; D2O, 25 °C) (ppm): 3.83 (2H, t, CH2OH), 3.60 (2H, t, CH2SC), 2.39 (1H, br. s, –CH–), 1.93–1.60 (2H, br. t, –CH2–); δC (100 MHz; D2O, 25 °C): 34.3 (–CH2–), 41.5 (–CH–), 178.9 (C(O));  (ATR) cm−1: 2935 (br. s, R-COOH), 1701 (vs, C
(ATR) cm−1: 2935 (br. s, R-COOH), 1701 (vs, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1449 (w, –CH2–), 1411 (w, R-CH2-S), 1214 (m, –COOH), 1162 (s, CS), 794 (m, –C–(CH3)2). Mn, theor = 3987 g mol−1; Mn,SEC = 4.7 kg mol−1, Mw/Mn = 1.19.
O), 1449 (w, –CH2–), 1411 (w, R-CH2-S), 1214 (m, –COOH), 1162 (s, CS), 794 (m, –C–(CH3)2). Mn, theor = 3987 g mol−1; Mn,SEC = 4.7 kg mol−1, Mw/Mn = 1.19.
Synthesis of poly(acrylic acid)-b-poly(N-isopropylacrylamide-st-N,N′-methylene bisacrylamide) nanogels via RAFT dispersion polymerisation in cononsolvents
RAFT dispersion polymerisation of NIPAM and BIS was performed in either water or different water–ethanol (H2O:EtOH) mole fractions at 10% w/w solids content. The mole ratio of [NIPAM]/[pAA45] was adjusted between 87 to 262 to give a range of nanogels with different sizes. An example of nanogel synthesis targeting a pAA45-b-pNIPAM175/BIS3 in XEtOH = 0.06 was as follows: pAA45 (0.0352 g, 0.010 mmol), NIPAM (0.2003 g, 1.770 mmol), BIS (0.0041 g, 0.027 mmol), were dissolved in a mixture of H2O:EtOH (1.8724 g of H2O and 0.2112 g of ethanol). The pH of the solution was adjusted to pH 6.68 using an aqueous solution of NaOH (0.07 mL, 0.35 mmol). The mixture was purged with N2 for 30 minutes, followed by the addition of the degassed ACVA solution in ethanol (0.124 mL, 14.27 mM) via microsyringe. All the quantities of solids and volume of solutions added were calculated in advance to give a final concentration of 10% w/w solids before reaction. The mixture was then placed into an oil bath previously set at 70 °C and left to react for 18 h. Total monomer conversion (98%) was estimated by moisture analysis. The product dispersion was purified by dialysis against DI water and was later stored at 4 °C.  (ATR-FTIR) cm−1: 3290 (br. m, –CONH), 2973 (m, R-COOH), 2934 and 2873 (m, –CH2), 1640 and 1539 (s, –CONR2), 1457 (m, –CH2–), 1409 (w, –COOH), 1386 and 1367 (m, –C(CH3)2), 1172 (m, CS), 1130 (m, –C(CH3)2), 879 (w, SC(S)S).
(ATR-FTIR) cm−1: 3290 (br. m, –CONH), 2973 (m, R-COOH), 2934 and 2873 (m, –CH2), 1640 and 1539 (s, –CONR2), 1457 (m, –CH2–), 1409 (w, –COOH), 1386 and 1367 (m, –C(CH3)2), 1172 (m, CS), 1130 (m, –C(CH3)2), 879 (w, SC(S)S).
Synthesis of poly(acrylic acid)-b-poly(N-isopropylacrylamide) copolymers in cononsolvents
A pAA34 macroCTA was chain extended with NIPAM at different [NIPAM]/[pAA34] mole ratios in either water or water–ethanol (H2O:EtOH) cononsolvents mixes at a 10% w/w solids content. An example of a diblock copolymer synthesis targeting a mole ratio of [NIPAM]:[pAA34] of 184 in XEtOH = 0.06 was as follows: the pAA34 macroCTA (0.0258 g, 0.0096 mmol), and NIPAM monomer (0.2001 g, 1.77 mmol) were dissolved in a mixture of H2O:EtOH (1.7542 g of H2O and 0.1983 g of ethanol). The pH of the solution was then adjusted to pH 6.73 using an aqueous solution of NaOH (0.05 mL, 0.25 mmol). All the quantities of solids and volume of solutions added were calculated in advance to give a final concentration of 10% w/w solids before reaction. The solution was then degassed under nitrogen for 30 minutes, followed by the addition of the degassed ACVA solution in ethanol (0.14 mL, 14.27 mM) via microsyringe. The solution was immersed into an oil bath preheated to 70 °C and left to react for 18 hours. NIPAM monomer conversion (99%) was estimated by 1H NMR spectroscopy. The crude product was purified by dialysis against DI water and freeze-dried to yield a white solid. Mn,theo = 28644 g mol−1; δH (400 MHz, D2O, 25 °C): 3.85 (br. d, H, –NCH), 2.25–1.96 (br. d, 1H, –CH–), 1.53 (br. t, 2H, –CH2–), 1.09 (br. s, 6H, –CH(CH3)2); δC (100 MHz; D2O, 25 °C): 21.5 (–CH2–), 41.7 (–CH–), 175.2 (C(O));  (ATR) cm−1: 3283 (br. m, –CONH), 2973 (m, R-COOH), 2929 and 2876 (w, –CH2–), 1634 and 1538 (s, –CON-R2), 1459 (m, –CH2–), 1387 and 1367 (m, –C(CH3)2), 1172 (w, CS).
(ATR) cm−1: 3283 (br. m, –CONH), 2973 (m, R-COOH), 2929 and 2876 (w, –CH2–), 1634 and 1538 (s, –CON-R2), 1459 (m, –CH2–), 1387 and 1367 (m, –C(CH3)2), 1172 (w, CS).
NMR spectroscopy
Nuclear Magnetic Resonance (NMR) spectra were recorded at 25 °C on a 400 MHz Bruker Avance III HD spectrometer. Each collected spectrum was calibrated using the residual solvent peak as reference. 1H NMR spectra were collected over 64 scans averaged per spectrum. Spectra were analysed with Bruker Topspin 3.0.Size exclusion chromatography (SEC)
Molar masses and dispersities of pAA were determined by SEC was determined after esterification of the carboxylic groups. pAA samples were dissolved in THF/methanol followed by the dropwise addition of trimethylsilyldiazomethane. Addition of the methylation agent ended when the production of N2 stopped, and the yellow colour remain unchanged. The solutions were left to stir allowing the solvents to evaporate overnight. Esterified samples were dissolved in the SEC eluent. Samples were analysed on an Agilent PL-GPC 50 system fitted with 2 × PLgel Mixed-C 5 μm (300 × 7.5 mm) columns. THF containing 4%v/v acetic acid and 0.025%w/v BHT was used as eluent at a flow rate of 1 mL min−1 at 25 °C. The equipment was calibrated with near-monodisperse poly(methyl methacrylate) standards (molecular weight range between 5.45 × 102–2.00 × 106 g mol−1). Analyte samples were prepared at a concentration of 2 mg mL−1 in THF (HPLC, 4%v/v acetic acid, 0.025%w/v BHT) and filtered through a 0.45 μm polytetrafluoroethylene (PTFE) filter before injection (100 μL). Molar masses and dispersities were calculated with Agilent GPC Software.Fourier transform infrared spectroscopy (ATR-FTIR)
ATR-FTIR was used to assess the chemical composition of materials. IR spectra were recorded on a PerkinElmer Spectrum™ 100 FT-IR spectrometer using a universal diamond ATR (UATR) accessory. Data was collected over 5 scans in the 450 to 4000 cm−1 region. The IR data was recorded and extracted from a Bruker software to further processing in Excel.Gravimetric analysis
The monomer conversion for the synthesis of nanogels was estimated gravimetrically using a KERN DAB 100-3 electronic moisture analyser. Samples were weighed and heated up to 190 °C to determine their solid content.Dynamic light scattering (DLS)
Determination of the hydrodynamic diameter of the purified nanogels (0.1% w/w) was obtained using a NanoBrook Omni particle analyser at a scattering angle of 173° using a 35 mW diode laser. Each DLS measurement consisted on 5 runs of 3 minutes at count rates between 440–550 kcps. Temperature dependent DLS experiments were performed from low to high temperatures with increments of 2.5 °C with 8 minutes in-between intervals to allow thermal stabilization. The pH of the solutions was manually adjusted by addition of NaOH or HCl solutions using a Hach H160 ISFET probe. The method of the constrained regularization method for inverting data (CONTIN) was used to obtain the size distribution from the autocorrelation function.Cloud point measurements by UV-vis spectrometry
The absorbance of the copolymer aqueous solutions (1% w/w) at 550 nm as a function of temperature was recorded on a Cary 300 Bio UV-vis spectrometer fitted with a Peltier temperature controlled multi-cell block. The temperature was increased from 10 to 60 °C at a heating rate of 0.12 °C min−1. The cloud points were calculated from the inflection point of each absorbance curve.Aqueous electrophoresis
ζ-Potential measurements for the aqueous nanogel dispersions (0.1% w/w) containing background KCl (1 mM) were determined on a Malvern Zetasizer Nano-ZS instrument. The Smoluchowski relationship was used to determine the zeta potential from the electrophoretic mobility.Transmission electron microscopy (TEM)
TEM images were collected using a Phillips CM100 microscope adapted with a Gatan CCD camera. Nanogel solutions (0.1% w/w, 10 μL) were placed onto freshly glow discharged carbon-coated grids for 1 min and then blotted with filter paper. Phosphotungstic acid (0.75% w/w, 5 μL) stain was then placed onto the sample and left for 20 s to finally blot it again with filter paper to remove the excess of stain. The grid was then dried with an adapted vacuum hose.Conclusions
A series of responsive nanogels were prepared by RAFT-mediated PITSA of NIPAM and BIS from a PAA macroCTA in water and water/EtOH mixtures to investigate the effects of the synthesis cononsolvent composition on the properties of pNIPAM nanogels. Characterisation of the crosslinked particles via DLS and TEM showed that the size of the nanogels synthesised in water increased as the DPn of the pNIPAM block was increased whereas for a fixed pNIPAM DPn particle size was seen to decrease with increasing EtOH content. The latter was attributed to the fact that solubility of the growing pNIPAM is reduced in the reaction medium resulting in earlier particle nucleation. Furthermore, the synthesis cononsolvent composition was found to affect the thermal properties of the final nanogels. For nanogels with a fixed pNIPAM DPn, the VPTT was found to increase to higher temperatures with increasing EtOH mole fraction in water used during synthesis. Detailed characterisation of analogue pAA-b-pNIPAM diblock copolymers with a fixed pNIPAM DPn showed that the copolymers had differences in the tacticity according to the synthesis solvent composition. The copolymers were found to have higher syndiotacticity and increased cloud point temperatures in accordance with the increasing EtOH mole fraction used in their synthesis. These differences were rationalised to the PITSA process, where an earlier nucleation caused by changes in the solubility of growing pNIPAM can lead to constrained conditions resulting in increasing the average syndiotacticity of the copolymer. Our study provides new insights into the effects of using mixtures of solvents on the properties of pNIPAM nanogels prepared by a RAFT-mediated PITSA approach. This is relevant for researchers that use a cononsolvency strategy to incorporate functionalities to pNIPAM nanogels using a RAFT dispersion polymerisation. It is noteworthy that the properties of the nanogels prepared in this study were evaluated at a fixed dispersion pH and since these are composed of PAA their conformation may be sensitive to changes in pH and ionic strength. However, this will be addressed in a future publication.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank the Consejo Nacional de Ciencia y Tecnología (CONACYT scholarship 461300, PhD studentship for MMM), the University of Sheffield and the UK Engineering and Physical Sciences Research Council [EPSRC grant EP/P027989/1] for funding. Dr Svet Tzokov is thanked for his assistance with TEM.Notes and references
- R. T. Chacko, J. Ventura, J. Zhuang and S. Thayumanavan, Adv. Drug Delivery Rev., 2012, 64, 836–851 CrossRef CAS.
- D. Li, C. F. van Nostrum, E. Mastrobattista, T. Vermonden and W. E. Hennink, J. Controlled Release, 2017, 259, 16–28 CrossRef CAS.
- P. Manickam, M. Pierre, R. D. Jayant, M. Nair and S. Bhansali, in Nanogels for Biomedical Applications, The Royal Society of Chemistry, 2018, vol. 30, ch. 13, pp. 261–282 Search PubMed.
- S. Maya, B. Sarmento, A. Nair, N. S. Rejinold, S. V. Nair and R. Jayakumar, Curr. Pharm. Des., 2013, 19, 7203–7218 CrossRef CAS PubMed.
- A. Sivashanmugam, R. A. Kumar, M. V. Priya, S. V. Nair and R. Jayakumar, Eur. Polym. J., 2015, 72, 543–565 CrossRef CAS.
- K. Akiyoshi, S. Deguchi, N. Moriguchi, S. Yamaguchi and J. Sunamoto, Macromolecules, 1993, 26, 3062–3068 CrossRef CAS.
- E. Akiyama, N. Morimoto, P. Kujawa, Y. Ozawa, F. M. Winnik and K. Akiyoshi, Biomacromolecules, 2007, 8, 2366–2373 CrossRef CAS PubMed.
- S. A. Ferreira, P. J. G. Coutinho and F. M. Gama, Langmuir, 2010, 26, 11413–11420 CrossRef CAS PubMed.
- M. Takeo, T. Mori, T. Niidome, S. Sawada, K. Akiyoshi and Y. Katayama, J. Colloid Interface Sci., 2013, 390, 78–84 CrossRef CAS PubMed.
- D. Y. Ko, H. J. Moon and B. Jeong, J. Mater. Chem. B, 2015, 3, 3525–3530 RSC.
- J. P. Quiñones, H. Peniche and C. Peniche, Polymers, 2018, 10, 235 CrossRef.
- J.-H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 17227–17235 CrossRef CAS.
- D. C. González-Toro, J.-H. Ryu, R. T. Chacko, J. Zhuang and S. Thayumanavan, J. Am. Chem. Soc., 2012, 134, 6964–6967 CrossRef PubMed.
- L. Li, K. Raghupathi, C. Yuan and S. Thayumanavan, Chem. Sci., 2013, 4, 3654–3660 RSC.
- Z. Jiang, W. Cui, J. Mager and S. Thayumanavan, Ind. Eng. Chem. Res., 2019, 58, 6982–6991 CrossRef CAS.
- X. T. Cao, A. M. Showkat and K. T. Lim, Eur. Polym. J., 2015, 68, 267–277 CrossRef CAS.
- B. Aktan, L. Chambre, R. Sanyal and A. Sanyal, Biomacromolecules, 2017, 18, 490–497 CrossRef CAS PubMed.
- J. Zhuang, S. Jiwpanich, V. D. Deepak and S. Thayumanavan, ACS Macro Lett., 2012, 1, 175–179 CrossRef CAS.
- C. W. Park, H.-M. Yang, H. J. Lee and J.-D. Kim, Soft Matter, 2013, 9, 1781–1788 RSC.
- B. Liu and S. Thayumanavan, J. Am. Chem. Soc., 2017, 139, 2306–2317 CrossRef CAS PubMed.
- W. I. Choi, G. Tae and Y. H. Kim, J. Mater. Chem., 2008, 18, 2769–2774 RSC.
- J. He, X. Tong and Y. Zhao, Macromolecules, 2009, 42, 4845–4852 CrossRef CAS.
- Y.-C. Wang, J. Wu, Y. Li, J.-Z. Du, Y.-Y. Yuan and J. Wang, Chem. Commun., 2010, 46, 3520–3522 RSC.
- W. Chen, Y. Hou, Z. Tu, L. Gao and R. Haag, J. Controlled Release, 2017, 259, 160–167 CrossRef CAS PubMed.
- D. Lu, M. Zhu, S. Wu, W. Wang, Q. Lian and B. R. Saunders, Polym. Chem., 2019, 10, 2516–2526 RSC.
- D. Steinhilber, M. Witting, X. Zhang, M. Staegemann, F. Paulus, W. Friess, S. Küchler and R. Haag, J. Controlled Release, 2013, 169, 289–295 CrossRef CAS PubMed.
- M. Giulbudagian, M. Asadian-Birjand, D. Steinhilber, K. Achazi, M. Molina and M. Calderón, Polym. Chem., 2014, 5, 6909–6913 RSC.
- L. I. Vossen, S. Wedepohl and M. Calderón, Polymers, 2018, 10, 398 CrossRef PubMed.
- A. Oehrl, S. Schötz and R. Haag, Colloid Polym. Sci., 2020, 298, 719–733 CrossRef CAS.
- K. McAllister, P. Sazani, M. Adam, M. J. Cho, M. Rubinstein, R. J. Samulski and J. M. DeSimone, J. Am. Chem. Soc., 2002, 124, 15198–15207 CrossRef CAS PubMed.
- D. J. Bharali, S. K. Sahoo, S. Mozumdar and A. Maitra, J. Colloid Interface Sci., 2003, 258, 415–423 CrossRef CAS PubMed.
- J. K. Oh, D. J. Siegwart, H.-i. Lee, G. Sherwood, L. Peteanu, J. O. Hollinger, K. Kataoka and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 5939–5945 CrossRef CAS PubMed.
- P. B. Zetterlund, N. Alam and M. Okubo, Polymer, 2009, 50, 5661–5667 CrossRef CAS.
- P. Bhardwaj, V. Singh, S. Aggarwal and U. K. Mandal, J. Macromol. Sci., Part A: Pure Appl.Chem., 2009, 46, 1083–1094 CrossRef CAS.
- W. Li, K. Matyjaszewski, K. Albrecht and M. Möller, Macromolecules, 2009, 42, 8228–8233 CrossRef CAS.
- S. F. Medeiros, A. M. Santos, H. Fessi and A. Elaissari, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3932–3941 CrossRef CAS.
- R. W. Graff, Y. Shi, X. Wang and H. Gao, Macromol. Rapid Commun., 2015, 36, 2076–2082 CrossRef CAS PubMed.
- D. S. Spencer, B. C. Luu, D. W. Beckman and N. A. Peppas, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1536–1544 CrossRef CAS PubMed.
- X. Li, J. Zuo, Y. Guo and X. Yuan, Macromolecules, 2004, 37, 10042–10046 CrossRef CAS.
- J. Moselhy, T. Vira, F. F. Liu and X. Y. Wu, Int. J. Nanomed., 2009, 4, 153–164 CrossRef CAS.
- D. Pamfil and C. Vasile, in Polymer Gels: Perspectives and Applications, ed. V. K. Thakur, M. K. Thakur and S. I. Voicu, Springer Singapore, Singapore, 2018, pp. 71–110, DOI:10.1007/978-981-10-6080-9_4.
- G. Delaittre, M. Save and B. Charleux, Macromol. Rapid Commun., 2007, 28, 1528–1533 CrossRef CAS.
- Z. An, Q. Shi, W. Tang, C.-K. Tsung, C. J. Hawker and G. D. Stucky, J. Am. Chem. Soc., 2007, 129, 14493–14499 CrossRef CAS.
- W.-M. Wan, C.-Y. Hong and C.-Y. Pan, Chem. Commun., 2009, 5883–5885 RSC.
- G. Delaittre, C. Dire, J. Rieger, J.-L. Putaux and B. Charleux, Chem. Commun., 2009, 2887–2889 RSC.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- C. A. Figg, A. Simula, K. A. Gebre, B. S. Tucker, D. M. Haddleton and B. S. Sumerlin, Chem. Sci., 2015, 6, 1230–1236 RSC.
- X.-b. Liu, J.-f. Zhou and X.-d. Ye, Chin. J. Chem. Phys., 2012, 25, 463–468 CrossRef CAS.
- L. Hou, K. Ma, Z. An and P. Wu, Macromolecules, 2014, 47, 1144–1154 CrossRef CAS.
- R. Rajan and K. Matsumura, Macromol. Rapid Commun., 2017, 38, 1700478 CrossRef.
- J. Rieger, C. Grazon, B. Charleux, D. Alaimo and C. Jérôme, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2373–2390 CrossRef CAS.
- C. Grazon, J. Rieger, N. Sanson and B. Charleux, Soft Matter, 2011, 7, 3482–3490 RSC.
- W. Shen, Y. Chang, G. Liu, H. Wang, A. Cao and Z. An, Macromolecules, 2011, 44, 2524–2530 CrossRef CAS.
- G. Liu, Q. Qiu, W. Shen and Z. An, Macromolecules, 2011, 44, 5237–5245 CrossRef CAS.
- L. Yan and W. Tao, Polymer, 2010, 51, 2161–2167 CrossRef CAS.
- Y. Xu, Y. Li, X. Cao, Q. Chen and Z. An, Polym. Chem., 2014, 5, 6244–6255 RSC.
- W. Fu, C. Luo, E. A. Morin, W. He, Z. Li and B. Zhao, ACS Macro Lett., 2017, 6, 127–133 CrossRef CAS.
- Y. Li, Z. Ye, L. Shen, Y. Xu, A. Zhu, P. Wu and Z. An, Macromolecules, 2016, 49, 3038–3048 CrossRef CAS.
- K. Ma, Y. Xu and Z. An, Macromol. Rapid Commun., 2015, 36, 566–570 CrossRef CAS PubMed.
- N. Bhuchar, R. Sunasee, K. Ishihara, T. Thundat and R. Narain, Bioconjugate Chem., 2012, 23, 75–83 CrossRef CAS.
- Y. Kotsuchibashi and R. Narain, Polym. Chem., 2014, 5, 3061–3070 RSC.
- R. Narain, Y. Wang, M. Ahmed, B. F. L. Lai and J. N. Kizhakkedathu, Biomacromolecules, 2015, 16, 2990–2997 CrossRef CAS PubMed.
- H. M. Crowther and B. Vincent, Colloid Polym. Sci., 1998, 276, 46–51 CrossRef CAS.
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Part A: Pure Appl.Chem., 1968, 2, 1441–1455 CrossRef CAS.
- H. G. Schild and D. A. Tirrell, J. Phys. Chem., 1990, 94, 4352–4356 CrossRef CAS.
- T. López-León, D. Bastos-González, J. L. Ortega-Vinuesa and A. Elaïssari, ChemPhysChem, 2010, 11, 188–194 CrossRef.
- M. J. A. Hore, B. Hammouda, Y. Li and H. Cheng, Macromolecules, 2013, 46, 7894–7901 CrossRef CAS.
- H. G. Schild, M. Muthukumar and D. A. Tirrell, Macromolecules, 1991, 24, 948–952 CrossRef CAS.
- G. Zhang and C. Wu, J. Am. Chem. Soc., 2001, 123, 1376–1380 CrossRef CAS.
- C. Scherzinger, P. Lindner, M. Keerl and W. Richtering, Macromolecules, 2010, 43, 6829–6833 CrossRef CAS.
- S. Zhou, S. Fan, S. C. F. Au-yeung and C. Wu, Polymer, 1995, 36, 1341–1346 CrossRef CAS.
- J. Hao, H. Cheng, P. Butler, L. Zhang and C. C. Han, J. Chem. Phys., 2010, 132, 154902 CrossRef PubMed.
- H. Yamauchi and Y. Maeda, J. Phys. Chem. B, 2007, 111, 12964–12968 CrossRef CAS PubMed.
- J. Wang, N. Wang, B. Liu, J. Bai, P. Gong, G. Ru and J. Feng, Phys. Chem. Chem. Phys., 2017, 19, 30097–30106 RSC.
- H. A. Pérez-Ramírez, C. Haro-Pérez, E. Vázquez-Contreras, J. Klapp, G. Bautista-Carbajal and G. Odriozola, Phys. Chem. Chem. Phys., 2019, 21, 5106–5116 RSC.
- E. R. Jones, M. Semsarilar, P. Wyman, M. Boerakker and S. P. Armes, Polym. Chem., 2016, 7, 851–859 RSC.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS.
- M. Williams, N. J. W. Penfold, J. R. Lovett, N. J. Warren, C. W. I. Douglas, N. Doroshenko, P. Verstraete, J. Smets and S. P. Armes, Polym. Chem., 2016, 7, 3864–3873 RSC.
- E. Niezabitowska, A. R. Town, B. Sabagh, M. D. Morales-Moctezuma, V. R. Kearns, S. G. Spain, S. P. Rannard and T. O. McDonald, Nanoscale Adv., 2020, 2, 4713–4721 RSC.
- R. O. R. Costa and R. F. S. Freitas, Polymer, 2002, 43, 5879–5885 CrossRef CAS.
- S.-P. Wen, J. G. Saunders and L. A. Fielding, Polym. Chem., 2020, 11, 3416–3426 RSC.
- J. Wyman, J. Am. Chem. Soc., 1931, 53, 3292–3301 CrossRef CAS.
- J. Brijitta, B. V. R. Tata and T. Kaliyappan, J. Nanosci. Nanotechnol., 2009, 9, 5323–5328 CrossRef CAS PubMed.
- M. B. Huglin, Y. Liu and J. Velada, Polymer, 1997, 38, 5785–5791 CrossRef CAS.
- B. A. Humphreys, E. J. Wanless and G. B. Webber, J. Colloid Interface Sci., 2018, 516, 153–161 CrossRef CAS PubMed.
- A. Halperin, M. Kröger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342–15367 CrossRef CAS.
- Y. Xia, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2006, 39, 2275–2283 CrossRef CAS.
- Y. Katsumoto and N. Kubosaki, Macromolecules, 2008, 41, 5955–5956 CrossRef CAS.
- C. S. Biswas, K. Mitra, S. Singh and B. Ray, J. Chem. Sci., 2016, 128, 415–420 CrossRef CAS.
- M. I. Gibson, D. Paripovic and H.-A. Klok, Adv. Mater., 2010, 22, 4721–4725 CrossRef CAS PubMed.
- Y. Isobe, D. Fujioka, S. Habaue and Y. Okamoto, J. Am. Chem. Soc., 2001, 123, 7180–7181 CrossRef CAS PubMed.
- T. Hirano, A. Ono, H. Yamamoto, T. Mori, Y. Maeda, M. Oshimura and K. Ute, Polymer, 2013, 54, 5601–5608 CrossRef CAS.
- W. Jiang, W. Yang, X. Zeng and S. Fu, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 733–741 CrossRef CAS.
- S. C. Pilcher and W. T. Ford, Macromolecules, 1998, 31, 3454–3460 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, SEC chromatograms and summary data for pAA macroCTAs; synthesis and cloud point measurements of pNIPAM in different solvents; DLS (including correlation functions), TEM and additional summary data for nanogels; determination of nanogel VPTT, NMR spectra and SEC chromatograms for pNIPAM synthesised by precipitation polymerisation in different solvents; cloud points and summary data for diblock copolymers. See DOI: 10.1039/d1py00396h |
| This journal is © The Royal Society of Chemistry 2021 |