
Effect of polycyclosilane microstructure on thermal properties†
Qifeng
Jiang
 ,
Sydnee
Wong
and
Rebekka S.
Klausen
*
,
Sydnee
Wong
and
Rebekka S.
Klausen
*
Department of Chemistry, Johns Hopkins University, 3400 N. Charles St, Baltimore, MD 21218, USA. E-mail: Klausen@jhu.edu
First published on 30th April 2021
Abstract
Thermal characterization of polysilanes has focused on the influence of organic side chains, whereas little is understood about the influence of silane backbone microstructure on thermal stability, phase properties, and pyrolysis. To address this knowledge gap, we prepared three distinct polycyclosilanes: linear polymers synthesized from the cyclosilane building blocks 1,4Si6 and 1,3Si6, as well as a cyclic polymer of 1,3Si6. Thermal properties across the temperature range 25 to 600 °C were investigated using differential scanning calromietry (DSC) and thermogravimetric analysis (TGA). We found differences between linear and cyclic materials, including a phase transition unique to the cyclic polymer and lower rates of mass loss during pyrolysis. Density functional theory (DFT) calculations provided insight into microstructure-dependent pyrolysis.
Introduction
Herein we report a systematic investigation of the thermal properties of polycyclosilanes, a novel class of organometallic polymers. Hydrocarbon-based elastomers, thermoplastics, and thermosets are distinguished by their thermal properties, such as the operating temperature (above or below the glass transition temperature, Tg) and physical behaviour above the phase transition (e.g. flow).1 Models can quantitatively relate polymer Tg to structural phenomena (e.g. the Flory–Fox equation1,2 and the number-average molecular weight). In addition to reversible phenomena that occur at relatively low temperature like the phase transitions Tg or Tm, there is interest in the chemical reactivity of polymers at high temperature (pyrolysis).3 In contrast to hydrocarbon polymers, structure-based understanding of polysilane thermal property relationships4,5 is more limited and has focused on the influence of organic side chains.6,7 Two possible reasons for this issue are (1) the synthetic challenge of creating a structurally well-defined homologous series of polysilanes and (2) the propensity of some polysilanes to undergo skeletal rearrangement to polycarbosilanes at elevated temperatures.8,9 The polysilane to polycarbosilane rearrangement precedes the ultimate formation of silicon carbide (SiC) fibers.10–12We recently described the synthesis and Cp2ZrCl2/n-BuLi-mediated13,14 dehydrocoupling polymerization of the bifunctional cyclosilane monomers 1,4Si6 and 1,3Si6 (Fig. 1a).15–20 These directional building blocks template distinct linear or cyclic polymeric architectures: polymers lin-poly(1,4Si6) and cyc-poly(1,3Si6) were obtained in comparable molecular weights (Mnca. 3000 g mol−1), but cyc-poly(1,3Si6) lacked spectroscopic signatures consistent with end groups. We considered these materials an opportunity to investigate the effect of polysilane backbone on thermal properties without variation of the side chain, which in all cases were methyl or hydro groups.
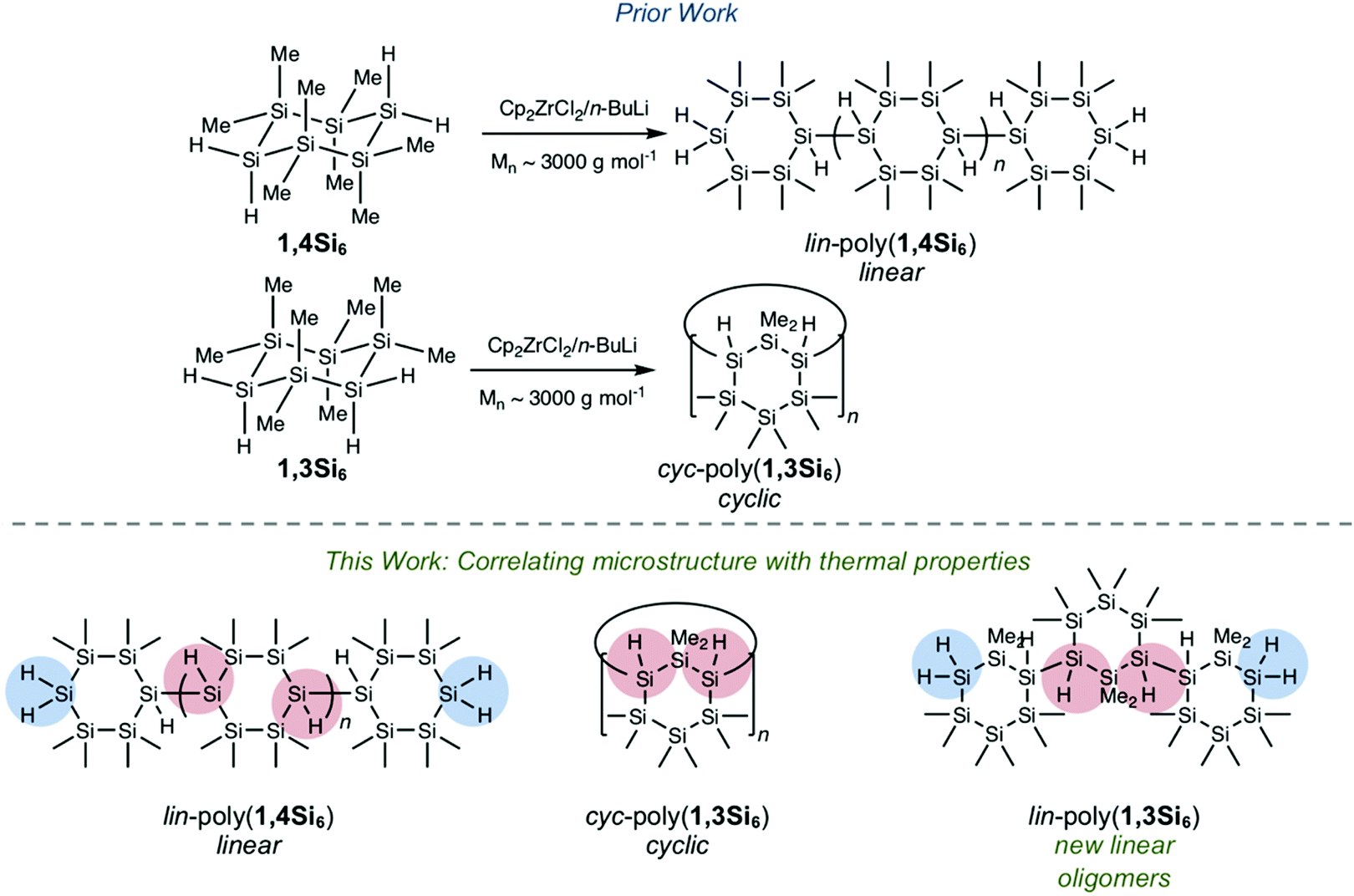 | ||
| Fig. 1 (a) Prior work: Cp2ZrCl2/n-BuLi-mediated dehydrocoupling polymerization of directional building blocks 1,4Si6 and 1,3Si6. (b) This work: Synthesis of new linear lin-poly(1,3Si6) and analysis of relationships between microstructure and thermal decomposition. | ||
We hypothesized that lin-poly(1,4Si6) and cyc-poly(1,3Si6) would exhibit distinct thermal behaviour and reactivity. Cyclic polymers, both experimentally and computationally, show markedly different properties from linear variants despite similar chemical compositions.21–28 For example, Roovers et al. reported that low molecular weight cyclic polystyrene (Mw = 4700 g mol−1) has a glass transition temperature 21 °C higher than linear polystyrene of the same molecular weight.27 In addition, the SiH2 end groups present in lin-poly(1,4Si6) are significantly stronger than SiH internal groups (Fig. 1b) due to the weakening effect of silyl substitution on Si–H bonds,29 which may perturb the sequence of chemical steps during thermolysis. To probe these differences, we also sought the synthesis of a linear polymer of 1,3Si6 (lin-poly(1,3Si6)) which would have the same relative connectivity of cyc-poly(1,3Si6) but would possess SiH2 end groups.
Herein, we report that replacement of the standard Cp2ZrCl2/nBuLi catalyst with Cp2ZrMe2 provided lin-poly(1,3Si6), the desired linear polymer of 1,3Si6, a structural assignment supported by 1H and 29Si NMR spectroscopy. We investigate the influence of polycyclosilane microstructure on thermal behavior and decomposition using a combined theoretical and experimental study. Thermal stability was investigated by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) complemented by density functional theory (DFT) calculations. All three samples were glassy solids at room temperature. A glass transition was observed at 108 °C in cyc-poly(1,3Si6), but no reversible phase transitions were found below 200 °C in lin-poly(1,4Si6) and lin-poly(1,3Si6). Above 250 °C, all three polycyclosilanes showed significant thermal decomposition that proceeded in two phases. A generic thermolysis process of polycyclosilanes is proposed based on this combined theoretical and experimental study. As a polymeric material combining cyclic subunits and mixed H- and Me-termination, polycyclosilanes may find utility as ceramic precursors.
Experimental
Methods and materials
Unless otherwise specified, all chemicals were used as purchased without further purification. Solvents THF (Fisher, HPLC grade), pentane (Fisher, certified ACS), and toluene (Fisher, certified ACS) were dried on a J. C. Meyer Solvent Dispensing System (SDS) using stainless steel columns packed with neutral alumina (except for toluene which is dried with neutral alumina and Q5 reactant, a copper(II) oxide oxygen scavenger), following the manufacturer's recommendations for solvent preparation and dispensation unless otherwise noted. Bis(cyclopentadienyl)zirconium(IV) dichloride (zirconocene dichloride, Cp2ZrCl2), n-butyllithium (2.5 M in hexanes) and Celite were purchased from Sigma Aldrich. The alkyllithium reagent was diluted in toluene and titrated with diphenylacetic acid before use.30 Bis(cyclopentadienyl)dimethylzirconium(IV) (Cp2ZrMe2) was purchased from Strem Chemicals. Polycyclosilanes lin-poly(1,4Si6) and cyc-poly(1,3Si6), were synthesized via dehydro-coupling polymerization of bifunctional monomers (1,4Si6 and 1,3Si6) according to the literature procedure.16,171H NMR and 29Si {1H} NMR were recorded on either a Bruker Avance 300, 400 or III HD 400 MHz Spectrometer and chemical shifts are reported in parts per million (ppm). Spectra were recorded in benzene-d6 with the residual solvent peak as the internal standard (1H NMR: C6H6δ = 7.16). Molecular weights were measured by gel permeation chromatography (GPC) on a Tosoh Bioscience EcoSEC GPC workstation with UV detection at 254 nm using butylated hydroxytoluene stabilized tetrahydrofuran (THF) as the eluent (0.35 mL min−1, 40 °C) through TSKgel SuperMultipore HZ-M guard column (4.6 mm ID × 2.0 cm, 4 μm, Tosoh Bioscience) and a TSKgel SuperMultipore HZ-M column (4.6 mm ID × 15 cm, 4 μm, Tosoh Bioscience). Polystyrene standards (EasiVial PS-M, Agilent) were used to build a calibration curve. Processing was performed using EcoSEC Data Analysis software (Version 1.14, Tosoh Bioscience). The samples were dissolved in THF (1 mg mL−1), filtered through syringe filters (Millex-FG Syringe Filter Unit, 0.20 μm, PTFE, EMD Millipore), and injected by an auto-sampler (10 μL). Differential scanning calorimetry (DSC) was conducted using a TA Instruments DSC Q20 V24.11 Build 124 and processing was performed using Universal V4.5A (TA Instruments). Samples were sealed in hermetic aluminum pans, heated from 35 to 200 °C (3 °C min−1 for lin-poly(1,4Si6) and 20 °C min−1 for cyc-poly(1,3Si6) and lin-poly(1,3Si6), and cooled from 200 to 35 °C, for two cycles under a purge gas of nitrogen (25 mL min−1). Glass transition temperatures (Tg) were calculated from the second heating cycle. Thermogravimetric analysis (TGA) was conducted using a TA Instruments SDT Q600 under flowing Ar at a heating rate of 5.0 °C min−1 from 40 to 600 °C.
Cp2ZrMe2 synthesis of lin-poly(1,3Si6)
In a nitrogen atmosphere glove box, Cp2ZrMe2 (0.056 equiv., 0.019 mmol, 4.8 mg) was added to an oven-dried 2-dram vial equipped with a stir bar. 1,3Si6 (1.0 equiv., 0.34 mmol, 0.100 g) was added by syringe. The vial was sealed with a septum cap equipped with a vent needle. The mixture was stirred at room temperature in the glove box. After 24 hours, the stir bar was trapped in a dark red glass in the vial. The solid was dissolved in 10 mL of pentane and 0.5 grams of Celite was added. The solution was stirred for 2 hours. The Celite was filtered through a fritted funnel and washed with 5 mL of pentane. The filtrate was then concentrated to a yellow solid under vacuum. The procedure was repeated until a white solid was yielded, which indicated that the catalyst had been fully removed. The solid was further dried under vacuum at 60 °C for 3 hours (85.8 mg, 87%).Cp2ZrMe2 synthesis of cyc-poly(1,3Si6)
In a nitrogen atmosphere glove box, Cp2ZrMe2 (0.056 equiv., 0.019 mmol, 4.8 mg) was added to an oven-dried 2-dram vial equipped with a stir bar and dissolved by 0.5 mL of toluene. 1,3Si6 (1.0 equiv., 0.34 mmol, 0.100 g) was weighed in a 1-dram vial and diluted by 0.5 mL of toluene. The solution of 1,3Si6 was added dropwise to the 2-dram vial by syringe. 0.25 mL of toluene was used to rinse the vial and syringe, then added to the 2-dram vial as well. The 2-dram vial was sealed with a septum cap equipped with a vent needle. The mixture was stirred at room temperature in the glove box. After 24 hours, the reaction mixture turned yellow, and deep orange after 4 days. 7 days later, volatile materials were removed under vacuum and the residual solid was dissolved in 10 mL of pentane. 0.5 grams of Celite was added and the solution was stirred for 2 hours. The Celite was filtered through a fritted funnel and washed with 5 mL of pentane. The filtrate was then concentrated to a yellow solid under vacuum. The procedure was repeated until a white solid was yielded, which indicated that the catalyst had been fully removed. The solid was further dried under vacuum at 60 °C for 3 hours (83.4 mg, 85%).Results and discussion
Synthesis of lin-poly(1,3Si6)
During our initial work on 1,3Si6 polymerization with Cp2ZrCl2/n-BuLi, we identified two fractions by gel permeation chromatography (GPC): a dominant higher molecular weight fraction corresponding to cyc-poly(1,3Si6) and a minor low molecular weight fraction attributed to linear oligomers too short to cyclize (Fig. 2, dashed line).17 Spectroscopic support for this hypothesis arose from observation of minor signals consistent with SiH2 end groups in 1H NMR and 29Si DEPT spectra.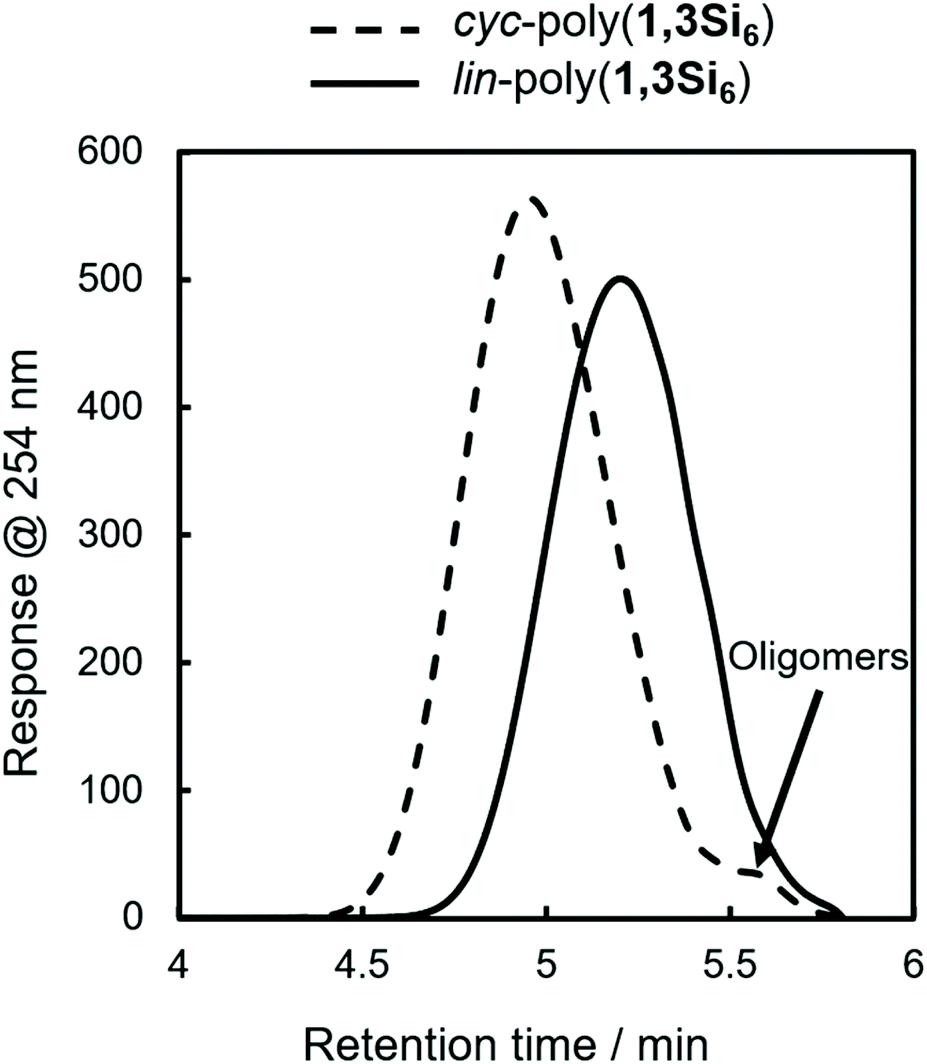 | ||
| Fig. 2 GPC of cyc-poly(1,3Si6) (dotted line) and lin-poly(1,3Si6) (solid line). Determined relative to polystyrene standards at 254 nm (THF, [lin-poly(1,3Si6)] = 1 mg mL−1, 40 °C, 0.35 mL min−1, 10 μL injection). | ||
Hypothesizing that at a lower average degree of polymerization, poly(1,3Si6) might exist exclusively of linear oligomers we reinvestigated catalysis of 1,3Si6 polymerization. We found that Cp2ZrMe2-polymerization could provide either linear or cyclic samples depending on solvent. Bulk polymerization of 1,3Si6 with Cp2ZrMe2 (Scheme 1) provided lin-poly(1,3Si6) in 87% yield that subsequent NMR analysis indicated was consistent with structural assignment to a predominantly linear structure (vide infra). GPC analysis indicated lin-poly(1,3Si6) consisted of a single fraction (Fig. 2). Dilution of the 1,3Si6/Cp2ZrMe2 polymerization with toluene resulted in cyc-poly(1,3Si6), which NMR characterization suggested was consistent with a predominantly cyclic architecture (Table 1). A 7-day reaction time was required for Cp2ZrMe2 catalysis in toluene, as this catalyst requires a 4-day induction period.31,32 Heating the reaction at 65 °C shortened the induction period to 24 hours.
 | ||
| Scheme 1 Synthesis of lin-poly(1,3Si6), a linear polymer of 1,3Si6. | ||
| Polymer | Monomer | Catalyst | Solvent | Reaction time | Structure |
M
n,GPC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (kg mol−1) a (kg mol−1) |
M
w,GPCa (kg mol−1) |
Đ |
c |
|---|---|---|---|---|---|---|---|---|---|
a Determined by GPC at 254 nm relative to a polystyrene standard.
b
Đ = Mw/Mn.
c 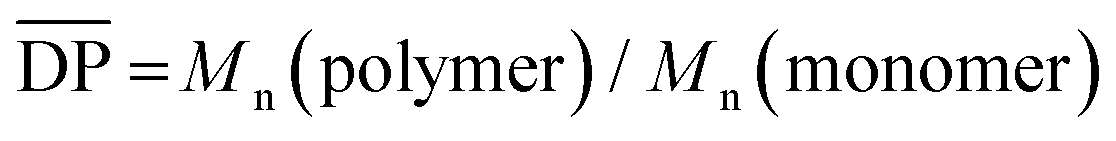 , as determined by GPC analysis. , as determined by GPC analysis.
|
|||||||||
| lin-poly(1,4Si6) | 1,4Si6 | Cp2ZrCl2/n-BuLi | Toluene | 24 h | Linear | 2.59 | 3.29 | 1.27 | 9 |
| cyc-poly(1,3Si6) | 1,3Si6 | Cp2ZrCl2/n-BuLi | Toluene | 24 h | Cyclic | 3.07 | 4.52 | 1.48 | 10 |
| lin-poly(1,3Si6) | 1,3Si6 | Cp2ZrMe2 | None | 24 h | Linear | 1.77 | 2.39 | 1.35 | 6 |
| cyc-poly(1,3Si6) | 1,3Si6 | Cp2ZrMe2 | Toluene | 7 d | Cyclic | 3.84 | 7.27 | 1.90 | 13 |

GPC analysis also indicated that cyc-poly(1,3Si6) was obtained in higher molecular weight (ca. 3000 g mol−1) than lin-poly(1,3Si6) (ca. 1700 g mol−1) which supports the hypothesis that cyclization is inhibited at lower degrees of polymerization presumably due to strain in forming smaller cycles. However, the known tendency of cyclic polymers to adopt more compact conformations that can lead to anomalous elution on GPC affects confidence in the GPC-estimated molecular weight characteristics of cyc-poly(1,3Si6) and lin-poly(1,3Si6). For example, Veige et al. showed that GPC overestimated the molecular weight of cyclic polystyrene by 10000 g mol−1, or approximately 100 degrees of polymerization.26 For the cyclosilane system, however, GPC is unlikely to significantly misestimate polycyclosilane molecular weight as silane dehydropolymerization rarely exceeds 20 degrees of polymerization.33
We suggest that linear oligomers were formed during bulk polymerization due to a change in physical properties. In the absence of solvent, the mixture of monomer and Cp2ZrMe2 gradually changes from a liquid to a glass. This may sequester oligomers from further reaction, providing a single fraction of low molecular weight oligomers too short to cyclize. This hypothesis is consistent with the observation that dilution with toluene actually increased molecular weight and lead to cyclized polymer. Dilution typically inhibits intermolecular reactions, such as chain extension.
Mechanistic differences between the Cp2ZrMe2 and Cp2ZrCl2/n-BuLi catalysts may also play a role in the results. Group 4 metallocene derivatives are among the best-studied catalysts for hydrosilane dehydropolymerization and have long served an important role in understanding novel inorganic mechanisms.33,34 Corey et al. first reported that in situ activation of Cp2ZrCl2 in toluene with two equivalents of n-butyllithium yielded an effective catalyst for secondary silane polymerization.13,14 Chloride substitution by the alkyllithium yields an intermediate dialkyl zirconocene, which further reacts via β-hydrogen abstraction to yield the active, undercoordinated catalyst, although the exact catalyst structure remains ambiguous.35 In contrast, dimethyl zirconocene (Cp2ZrMe2), first reported by Harrod et al., does not require activation by an exogenous agent and can therefore be used either neat or with added solvent.31,36,37 However, reactions with Cp2ZrMe2 can show an induction period,36 presumably due to slow formation of the active undercoordinated catalyst in the absence of β-hydrogens.
Polycyclosilane microstructure
Polycyclosilanes feature a well-defined, periodic alternation of methylated and hydrogenated silicon atoms (Fig. 1b). This structural pattern provides diagnostic features for spectroscopic characterization based on the unique signatures of tertiary (Si3SiH), secondary (Si2SiH2), and primary (SiSiH3) bonds. Variable silyl- and hydro-substitution strongly influences chemical shift in 29Si NMR spectroscopy.38,39 The degree of silyl substitution has a marked weakening effect on Si–H bond strength,29 which is reflected in a significant shift to lower frequency FTIR resonances for the weaker Si3SiH internal groups relative to stronger Si2SiH2 end groups.171H NMR spectra are shown in Fig. 3a. The spectrum of cyc-poly(1,3Si6) is dominated by a single broad resonance at δ 3.25, consistent with its high symmetry macrocyclic structure.17lin-Poly(1,4Si6) has a series of resonances in the SiHx region (δ 3.5–3.0), which were assigned to SiH2 end groups or SiH internal sites by 1H–29Si HSQC.17 The 1H NMR spectrum of lin-poly(1,3Si6) is strikingly similar to lin-poly(1,4Si6) with several broad resonances from δ 3.45 to 3.10, even though each is a polymer of an isomeric cyclosilane. These spectral data are consistent with assignment of lin-poly(1,3Si6) to a lower-symmetry linear structure rather than a macrocycle.
 | ||
| Fig. 3 (a) Cropped 1H NMR spectra of lin-poly(1,4Si6), cyc-poly(1,3Si6) and lin-poly(1,3Si6). Only the SiH region is shown. (b) 1H–29Si HSQC NMR spectrum of lin-poly(1,3Si6). Crosspeaks are labelled. (c) Cropped 29Si{1H} DEPT spectra of lin-poly(1,4Si6), cyc-poly(1,3Si6) and lin-poly(1,3Si6). Only the SiH region is shown. (d) Cropped ATR-FTIR spectra of cyc-poly(1,3Si6) and lin-poly(1,3Si6) highlighting the Si–H and Si–Me regions. HSQC = heteronuclear single quantum coherence; DEPT = distortionless enhancement by polarization transfer. | ||
Further support for assignment of lin-poly(1,3Si6) to a predominantly linear structure comes from the 1H–29Si HSQC (Fig. 3b) and 29Si {1H} DEPT (Fig. 3c) spectra. Like lin-poly(1,4Si6), the 29Si {1H} DEPT spectrum of lin-poly(1,3Si6) possesses two sets of strong resonances consistent with Si3SiH tertiary silanes (internal sites, δ −115.3) and Si2SiH2 secondary silanes (end groups, δ −97.46). A proton-coupled 29Si INEPT+ spectrum showed the expected multiplicities for the number of attached protons, confirming these assignments (Fig. S1†). In the 1H–29Si HSQC spectrum of lin-poly(1,3Si6), crosspeaks were observed between the 1H resonances at δ 3.25 to 3.10 and 29Si NMR resonances centered at δ −112 assigned to Si3SiH sites (Fig. 3b). Meanwhile, 1H resonances at δ 3.44 to 3.28 correlated with the 29Si NMR resonances centered at δ −97 assigned to Si2SiH2 sites. The 2-D spectral correlations confirm the assignment of the lin-poly(1,3Si6) structure to a linear structure containing both tertiary silane internal sites and secondary silane end groups.
Finally, lin-poly(1,3Si6)'s Fourier transform infrared (FTIR) spectrum showed two resonances in the SiHx region consistent with stronger Si2SiH2 and weaker Si3SiH bonds (Fig. 3d). cyc-Poly(1,3Si6) lacking Si2SiH2 end groups has a more symmetric band in this region centered at lower frequency. The spectroscopic studies described here support assignment of lin-poly(1,3Si6) to a largely linear structure.
Differential scanning calorimetry
While the thermal properties of Si–O polymers are well-characterized, and cyclic and linear polydimethylsiloxane (PDMS) were foundational models for understanding the effect of architecture on the Tg,23 systematic investigation of polysilane thermal properties has been largely limited to understanding the role of organic side chains in influencing backbone conformation and optical properties. Polysilanes with longer side chains (hexyl, heptyl, octyl) exhibit solid-state thermochromism,40,41 while polysilanes with shorter side chains (butyl, pentyl) do not.42 Significant experimentation supports the following model:(1) Polysilanes with short side chains adopt a helical Si–Si conformation at room temperature that absorbs higher energy light (λ = 317 nm).
(2) In contrast, polysilanes with longer side chains adopt an all-anti conformation at room temperature that maximizes σ-conjugation and results in a bathochromically shifted absorption band (λ = 375 nm). This is attributed to side chain crystallization that locks the Si–Si backbone into the anti-conformation. At elevated temperatures, side chain melting results in a backbone disordering that shifts the absorption band back to higher energies (λ = 317 nm).
Rabolt et al. found by differential scanning calorimetry (DSC) a reversible first-order transition at ca. 40 °C in poly(di-n-hexylsilane) that was assigned to the side-chain melting.41 Schilling et al. reported that DSC analysis of poly(di-n-butylsilane) and poly(di-n-pentylsilane) yielded significantly different results, including the lack of a transition comparable to side-chain melting (a weaker transition to a more disordered phase was found above ca. 80 °C).42 In addition, Schilling et al. reported a very weak second-order transition at 36 °C (only apparent in poly(di-n-butylsilane) during slow DSC runs) and an intense second-order transition at −40 °C. On the basis of solid-state NMR experiments (13C CP-MAS), all phase transitions were attributed to side chains.
The poly(di-n-alkylsilanes) studied by Miller,41 Schilling,42 and others were prepared by Wurtz polymerization of the corresponding dichlorodialkylsilane. Wurtz polymerization typically results in higher molecular weight materials than those prepared by dehydropolymerization and the products also differ in possessing two alkyl side chains rather than one (e.g. poly(SiR2) vs. poly(SiHR)). While monoalkyl polysilanes surely possess different thermal properties than dialkyl, we have identified only one report of thermal analysis of a polysilane prepared by dehydropolymerization. Corey et al. reported dehydropolymerization of p-tolylsilane and that DSC analysis of poly(SiH(p-tol)) (degree of polymerization ca. 20) revealed no phase transitions over the temperature range 25 to 200 °C.43 Some insight into the influence of hydro side chains arises from comparison of the copolymer poly(SiPh2-co-SiMeH) to poly(SiMePh).44 Both poly(SiMePh) and poly(SiPh2-co-SiMeH) exhibited a single glass transition, but the poly(SiPh2-co-SiMeH) Tg decreased with increasing methylhydro composition (Tg = 64 to 85 °C vs. 88 °C for poly(SiMePh)). These data were interpreted as showing greater flexibility and mobility in hydro-substituted polysilanes.
While prior work has provided insight into side chain effects, little is understood about how the silane architecture might influence thermal properties. We therefore performed DSC analysis of the three polycyclosilanes in the temperature range 35 to 200 °C as thermogravimetric analysis (TGA, vide infra) indicated polymer decomposition above 200 °C. Heating rates between 3 and 20 °C min−1 were tested.
For the two linear systems lin-poly(1,4Si6) and lin-poly(1,3Si6), no reversible phase transition was observed below 200 °C at any heating rate (Fig. 4a and c). In contrast, for cyc-poly(1,3Si6) we observed an apparent second-order phase transition at 108 °C (Fig. 4b). The transition was only observed with a heating rate of 20 °C min−1 and a cooling rate of 3 °C min−1; no transition was apparent at slower heating rates. The reversibility of the 108 °C transition is unclear: no transition is seen on cooling (Fig. S2†) but the transition observed on heating is reproducible (second and third heating cycles overlay, Fig. S3†), which is inconsistent with a change in chemical structure. The 108 °C transition was apparent in all samples of cyc-poly(1,3Si6) evaluated, regardless of preparative method.
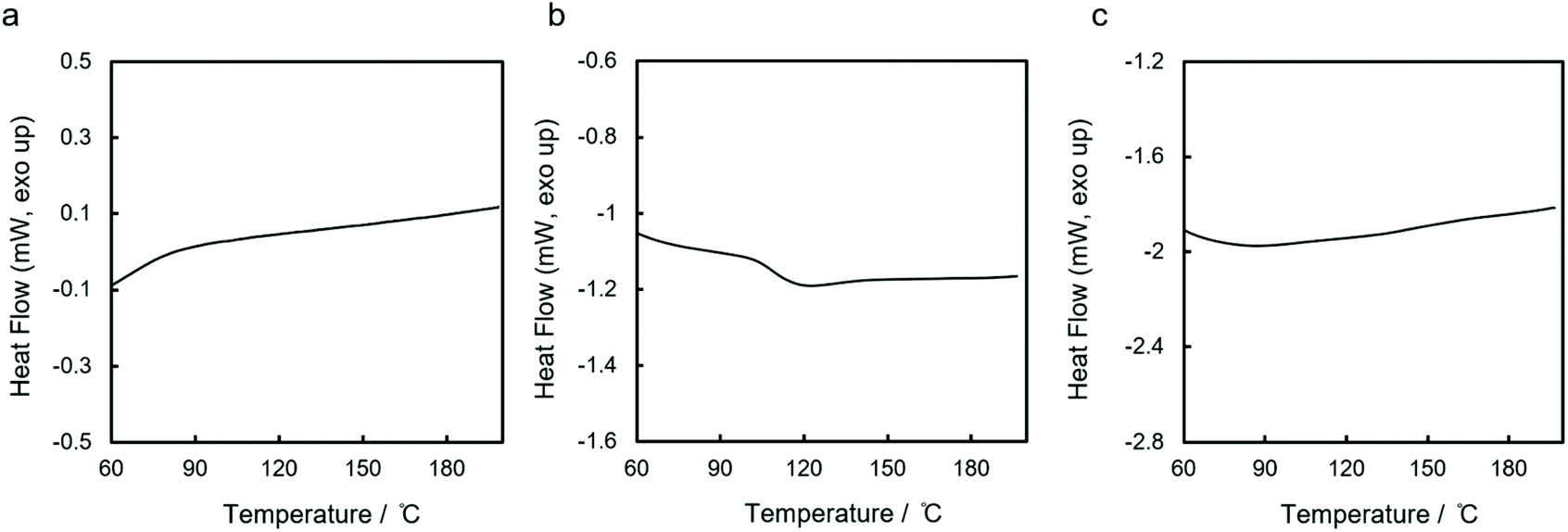 | ||
| Fig. 4 DSC curves of (a) lin-poly(1,4Si6), (b) cyc-poly(1,3Si6), and (c) and lin-poly(1,3Si6). The second cycle is shown. Heating rate: 3 °C min−1 for lin-poly(1,4Si6), 20 °C min−1 for cyc-poly(1,3Si6) and lin-poly(1,3Si6). | ||
The DSC analysis of lin-poly(1,4Si6) and lin-poly(1,3Si6) is similar to that reported by Corey et al. for linear poly(SiH(p-tol)).43 The comparatively high mobility of monoalkyl segments in linear polysilanes may complicate observation of the Tg in polymers with the general formula poly(SiHR). The observation of a 108 °C phase transition is unprecedented. We suggest assignment to a glass transition. While the Tg is not apparent on cooling, this may arise from crystallization during the heating phase. Assignment to a glass transition is also consistent with the known tendency of cyclic polymers to exhibit anomalous glass transitions relative to linear polymers.
Thermal stability of polycyclosilanes
Polymers based on main group elements find application as solution-processable precursors to ceramics (polymer-derived ceramics, PDCs).11,45 Yajima et al. discovered pyrolysis of polydimethylsilane ([SiMe2]n) yielding silicon carbide (SiC) fibers.46,47 Polysilazane pyrolysis yields silicon nitride (Si3N4) or Si/C/N ceramics depending on the precursor and thermolysis conditions.45,48,49 Several studies have demonstrated the influence of precursor structure on PDC structure and properties. Schilling et al. demonstrated the positive influence of backbone branching on SiC ceramic yield using polycarbosilanes prepared by potassium reduction of vinylmethyldichlorosilane.50 Polysilanes bearing both hydrogen and alkyl substituents51 are particularly attractive to circumvent carbon formation during pyrolysis, as shown by Laine et al. in the decomposition of polymethylsilane (–[MeSiH]x–).52The thermal decomposition of polycyclosilanes was studied by thermogravimetric analysis (Fig. 5). The three polymers showed overall similar thermal decomposition behavior. A minor mass loss at 65 °C in lin-poly(1,4Si6) was attributed to residual catalyst (Fig. 5a), which was easier to remove from the more soluble cyc-poly(1,3Si6) and lin-poly(1,3Si6) samples. TGA curves of derivative weight change reveal two main phases of weight loss when samples are heated from 40 to 600 °C in argon flow. When the temperature increased to 500 °C, about 50% weight loss is observed in all three samples. Then, the weight became constant above 550 °C. A black solid residue remained in the sample pan after the TGA measurement. These observations are consistent with ceramization,8 although the product was not characterized in this study.
 | ||
| Fig. 5 TGA curves of (a) lin-poly(1,4Si6), (b) cyc-poly(1,3Si6) and (c) lin-poly(1,3Si6). Solid lines: derivative weight change; dotted lines: percentage weight change. | ||
Some differences between the polymer systems were also apparent between 200 to 400 °C. The linear polymers both showed similar derivative weight change in this region, with a peak value greater than 0.6, while for cyc-poly(1,3Si6) the peak value was lower than 0.5 (Fig. 5b). Both linear polymers lost approximately 45% of their weight in this phase, while only 33% for cyc-poly(1,3Si6). This indicates that linear polycyclosilanes decomposed more rapidly than cyc-poly(1,3Si6) in this temperature range. In addition, multiple features (e.g. peaks and shoulders) appeared in the derivative weight change curve of lin-poly(1,4Si6), while smoother curves were obtained in the measurements of cyc-poly(1,3Si6) and lin-poly(1,3Si6).
These observations point to microstructure-dependent differences in early stage polycyclosilane pyrolysis. The first phase of polysilane pyrolysis to SiC is skeletal rearrangement from a polysilane to a polycarbosilane;53 in poly(SiMeH) pyrolysis, this rearrangement occurs at 400 °C.52 The pyrolysis of hexamethyldisilane is a model system for polysilane thermolysis and several reactive intermediates have been proposed, including radical,53,54 silylene (R2Si),55 and silene (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si) species.54 In all cases, homolysis of a weak bond (e.g. Me3Si–SiMe3 to 2Me3Si˙) initiates skeletal rearrangement. The different polycyclosilane microstructures, especially with respect to Si–H bond strengths, may influence the relative rates of processes implicated in polycyclosilane thermolysis.
Si) species.54 In all cases, homolysis of a weak bond (e.g. Me3Si–SiMe3 to 2Me3Si˙) initiates skeletal rearrangement. The different polycyclosilane microstructures, especially with respect to Si–H bond strengths, may influence the relative rates of processes implicated in polycyclosilane thermolysis.
Bond dissociation energy calculations
To understand the influence of polycyclosilane microstructure on thermolysis, we carried out density functional theory (DFT) calculations of bond dissociation energy on monomers and model molecules. As a compromise between computational ease and experimental relevance, the monomer 1,4Si6 and its linear trimer were selected for study. Geometries were fully optimized without symmetry restrictions using B3LYP hybrid exchange–correlation functional with the 6-31G(d) basis set. The basis set was utilized to investigate structures and expected characteristics of linear and cyclic forms of poly(1,3Si6).17 To ensure optimized geometries were local minima on their potential energy surfaces, the same level of theory was utilized to perform frequency calculations and showed no imaginary frequencies. The optimized geometry of 1,4Si6 is a chair conformation (consistent with a prior X-ray crystallographic study),16 whereas in the linear trimer rings were slightly twisted. Table 2 summarizes calculated homolytic bond dissociation energies (BDEs) of 1,4Si6 and its trimer.|
|
|||
|---|---|---|---|
| 1,4Si6 | lin-(1,4Si6)3 | ||
| Bond | BDE (kcal mol−1) | Bond | BDE (kcal mol−1) |
| Si(1)–Ha | 88.0 | Si(1)–Ha | 88.4 |
| Si(1)–He | 86.5 | Si(1)–He | 86.7 |
| Si(2)–Mea | 80.0 | Si(4)–H | 85.6 |
| Si(2)–Mee | 78.9 | Si(6)–Mea | 73.4 |
| Si(1)–Si(2) | 68.1 | Si(6)–Mee | 73.3 |
| Si(2)–Si(2) | 68.4 | Si(2)–Si(3) | 63.0 |
| Si(3)–Si(4) | 59.6 | ||
| Si(4)–Si(5) | 54.9 | ||
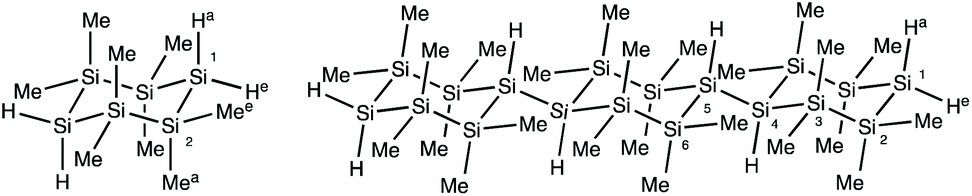
In 1,4Si6, Si–Si bonds were significantly weaker than Si–Me and Si–H bonds by 12–18 kcal mol−1. A similar trend in relative bond strengths (from high to low: Si–H, Si–Me, and Si–Si) was observed in lin-(1,4Si6)3. The Si–Si bond between repeat units was notably weaker than endocyclic Si–Si bonds by 5 to 8 kcal mol−1. These data suggest that the Si–Si bonds between monomers are most likely to homolyse first in polycyclosilane thermolysis. Small, volatile silanes generated from homolysis near an end group in linear polycyclosilanes would result in a significant weight decrease, which is consistent with the major weight loss observed between 200 to 400 °C in TGA measurements. In cyc-poly(1,3Si6), fewer volatile products may form as at least two Si–Si bond rupture events would be required to form a volatile byproduct. This may account for the slower mass loss observed in cyc-poly(1,3Si6) relative to linear polycyclosilanes.
At higher temperatures, organic side groups and hydrogen atoms start to detach from silicon leading to the second phase of weight loss. Combining experimental measurements and BDE calculations, the thermolysis process of polycyclosilanes is proposed as shown in Fig. 6.
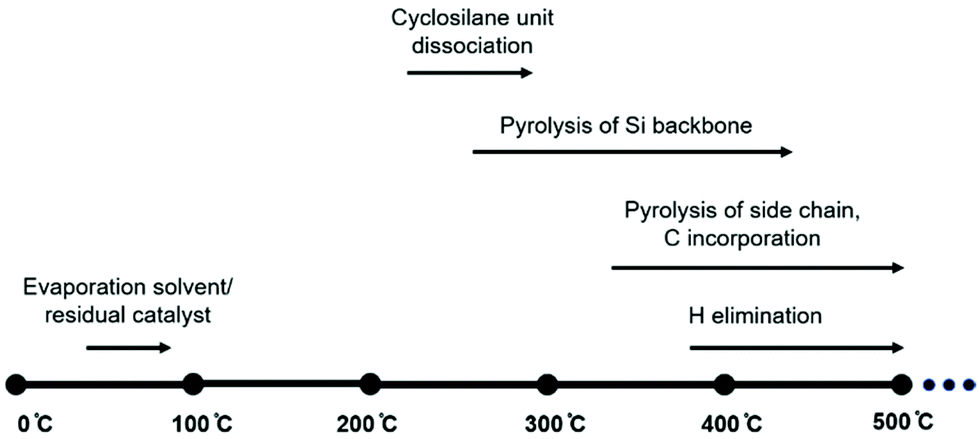 | ||
| Fig. 6 Proposed polycyclosilane pyrolysis. | ||
Conclusions
Herein, we investigated the microstructure-dependent thermal properties of polycyclosilanes. We prepared a known linear polymer of 1,4Si6 (lin-poly(1,4Si6)) and a predominantly cyclic polymer of 1,3Si6 (cyc-poly(1,3Si6)). In the course of these efforts, we also synthesized a new, linear oligomer of cyclosilane 1,3Si6 (lin-poly(1,3Si6)). Thermal analysis by DSC found no phase transitions for linear polycyclosilanes below the decomposition temperature, but a phase transition at 108 °C was found for cyc-poly(1,3Si6). We suggested assignment to a glass transition temperature. Polycyclosilane thermolysis was investigated by TGA and investigated by density functional theory calculations. Lower rates of mass loss were found for cyc-poly(1,3Si6), which was attributed to the need for two bond homolysis events to form volatile byproducts. These studies provide insights into how synthetic control of polysilane microstructure can predictably influence thermal properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was primarily supported by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences, under Award No. DE-SC0020681 (building block synthesis, structural characterization). DFT calculations were conducted using scientific computing services at the Maryland Advanced Research Computing Center (MARCC).Notes and references
- G. Odian, Principles of Polymerization, John Wiley & Sons, Inc., 4th edn, 2004 Search PubMed.
- T. G. Fox and P. J. Flory, Second–Order Transition Temperatures and Related Properties of Polystyrene. I. Influence of Molecular Weight, J. Appl. Phys., 1950, 21(6), 581–591, DOI:10.1063/1.1699711.
- S. D. Anuar Sharuddin, F. Abnisa, W. M. A. Wan Daud and M. K. Aroua, A Review on Pyrolysis of Plastic Wastes, Energy Convers. Manage., 2016, 308–326, DOI:10.1016/j.enconman.2016.02.037.
- J. M. Zeigler, Polysilanes - Prototypical σ-Conjugated Polymers, Synth. Met., 1989, 28(1–2), 581–591, DOI:10.1016/0379-6779(89)90577-8.
- R. D. Miller and J. Michl, Polysilane High Polymers, Chem. Rev., 1989, 89(6), 1359–1410, DOI:10.1021/cr00096a006.
- S. Demoustier-Champagne, S. Cordier and J. Devaux, Polymer Blends of Polymethylphenylsilane with Polystyrene and Thermal Properties of Polymethylphenylsilane, Polymer, 1995, 36(5), 1003–1007, DOI:10.1016/0032-3861(95)93600-Q.
- T. Asuke and R. West, Poly(n-Butyl-n-Hexylsilylene), a Liquid-Crystalline Polysilane, Macromolecules, 1991, 24(1), 343–344, DOI:10.1021/ma00001a058.
- M. Birot, J. P. Pillot and J. Dunoguès, Comprehensive Chemistry of Polycarbosilanes, Polysilazanes, and Polycarbosilazanes as Precursors of Ceramics, Chem. Rev., 1995, 95(5), 1443–1477, DOI:10.1021/cr00037a014.
- K. Shiina and M. Kumada, Thermal Rearrangement of Hexamethyldisilane to Trimethyl(Dimethylsilylmethyl)-Silane, J. Org. Chem., 1958, 23(1), 139, DOI:10.1021/jo01095a635.
- S. Yajima, Y. Hasegawa, K. Okamura and T. Matsuzawa, Development of High Tensile Strength Silicon Carbide Fibre Using an Organosilicon Polymer Precursor, Nature, 1978, 273(5663), 525–527, DOI:10.1038/273525a0.
- P. Colombo, G. Mera, R. Riedel and G. D. Sorarù, Polymer-Derived Ceramics: 40 Years of Research and Innovation in Advanced Ceramics, J. Am. Ceram. Soc., 2010, 93(7), 1805–1837, DOI:10.1111/j.1551-2916.2010.03876.x.
- G. Barroso, Q. Li, R. K. Bordia and G. Motz, Polymeric and Ceramic Silicon-Based Coatings-a Review, J. Mater. Chem. A, 2019, 1936–1963, 10.1039/c8ta09054h.
- J. Y. Corey, X. H. Zhu, T. C. Bedard and L. D. Lange, Catalytic Dehydrogenative Coupling of Secondary Silanes with Cp2MCl2-BuLi, Organometallics, 1991, 10(4), 924–930, DOI:10.1021/om00050a024.
- L. S. Chang and J. Y. Corey, Dehydrogenative Coupling of Diarylsilanes, Organometallics, 1989, 8(8), 1885–1893, DOI:10.1021/om00110a010.
- E. A. Marro, C. P. Folster, E. M. Press, H. Im, J. T. Ferguson, M. A. Siegler and R. S. Klausen, Stereocontrolled Syntheses of Functionalized Cis- And Trans-Siladecalins, J. Am. Chem. Soc., 2019, 141(44), 17926–17936, DOI:10.1021/jacs.9b09902.
- E. M. Press, E. A. Marro, S. K. Surampudi, M. A. Siegler, J. A. Tang and R. S. Klausen, Synthesis of a Fragment of Crystalline Silicon: Poly(Cyclosilane), Angew. Chem., Int. Ed., 2017, 56(2), 568–572, DOI:10.1002/anie.201610208.
- E. A. Marro, E. M. Press, M. A. Siegler and R. S. Klausen, Directional Building Blocks Determine Linear and Cyclic Silicon Architectures, J. Am. Chem. Soc., 2018, 140(8), 5976–5986, DOI:10.1021/jacs.8b02541.
- C. P. Folster and R. S. Klausen, Metallocene Influence on Poly(Cyclosilane) Structure and Properties, Polym. Chem., 2018, 9, 1938–1941, 10.1039/C8PY00312B.
- E. A. Marro and R. S. Klausen, Conjugated Polymers Inspired by Crystalline Silicon, Chem. Mater., 2019, 31(7), 2202–2211, DOI:10.1021/acs.chemmater.9b00131.
- R. W. Dorn, E. A. Marro, M. P. Hanrahan, R. S. Klausen and A. J. Rossini, Investigating the Microstructure of Poly(Cyclosilane) by 29Si Solid-State NMR Spectroscopy and DFT Calculations, Chem. Mater., 2019, 31(21), 9168–9178, DOI:10.1021/acs.chemmater.9b03606.
- Z. Jia and M. J. Monteiro, Cyclic Polymers: Methods and Strategies, J. Polym. Sci., Part A: Polym. Chem., 2012, 50(11), 2085–2097, DOI:10.1002/pola.25999.
- D. J. Bannister and J. A. Semlyen, Studies of Cyclic and Linear Poly(Dimethyl Siloxanes): 6. Effect of Heat, Polymer, 1981, 22(3), 377–381, DOI:10.1016/0032-3861(81)90050-1.
- S. J. Clarson, K. Dodgson and J. A. Semlyen, Studies of Cyclic and Linear Poly(Dimethylsiloxanes): 19. Glass Transition Temperatures and Crystallization Behaviour, Polymer, 1985, 26(6), 930–934, DOI:10.1016/0032-3861(85)90140-5.
- D. J. Orrah, J. A. Semlyen and S. B. Ross-Murphy, Studies of Cyclic and Linear Poly(Dimethylsiloxanes): 27. Bulk Viscosities above the Critical Molar Mass for Entanglement, Polymer, 1988, 29(8), 1452–1454, DOI:10.1016/0032-3861(88)90310-2.
- J. Roovers and P. M. Toporowski, Synthesis of High Molecular Weight Ring Polystyrenes, Macromolecules, 1983, 16(6), 843–849, DOI:10.1021/ma00240a002.
- C. D. Roland, H. Li, K. A. Abboud, K. B. Wagener and A. S. Veige, Cyclic Polymers from Alkynes, Nat. Chem., 2016, 8(8), 791–796, DOI:10.1038/nchem.2516.
- P. G. Santangelo, C. M. Roland, T. Chang, D. Cho and J. Roovers, Dynamics near the Glass Temperature of Low Molecular Weight Cyclic Polystyrene, Macromolecules, 2001, 34(26), 9002–9005, DOI:10.1021/ma011069+.
- L. Gao, J. Oh, Y. Tu, T. Chang and C. Y. Li, Glass Transition Temperature of Cyclic Polystyrene and the Linear Counterpart Contamination Effect, Polymer, 2019, 170, 198–203, DOI:10.1016/j.polymer.2019.03.018.
- J. M. Kanabus-Kaminska, J. A. Hawari, D. Griller and C. Chatgilialoglu, Reduction of Silicon-Hydrogen Bond Strengths, J. Am. Chem. Soc., 1987, 109(17), 5267–5268, DOI:10.1021/ja00251a035.
- W. G. Kofron and L. M. Baclawski, A Convenient Method for Estimation of Alkyllithium Concentrations, J. Org. Chem., 1976, 1879–1880, DOI:10.1021/jo00872a047.
- C. Aitken, J. F. Harrod and U. S. Gill, Structural Studies of Oligosilanes Produced by Catalytic Dehydrogenative Coupling of Primary Organosilanes, Can. J. Chem., 1987, 65(8), 1804–1809, DOI:10.1139/v87-303.
- Y. Mu, C. Aitken, B. Cote, J. F. Harrod and E. Samuel, Reactions of Silanes with Bis(Cyclopentadienyl)Dialkylzirconium Complexes, Can. J. Chem., 1991, 69(2), 264–276, DOI:10.1139/v91-042.
- T. D. Tilley, The Coordination Polymerization of Silanes to Polysilanes by a “σ-Bond Metathesis” Mechanism. Implications for Linear Chain Growth, Acc. Chem. Res., 1993, 26(1), 22–29, DOI:10.1021/ar00025a004.
- R. Waterman, Mechanisms of Metal-Catalyzed Dehydrocoupling Reactions, Chem. Soc. Rev., 2013, 42, 5621–5980, 10.1039/c3cs60082c.
- V. K. Dioumaev and J. F. Harrod, Nature of the Species Present in the Zirconocene Dichloride-Butyllithium Reaction Mixture, Organometallics, 1997, 16(7), 1452–1464, DOI:10.1021/om960543a.
- C. Aitken, J. F. Harrod and E. Samuel, Synthesis and Structural Characterization of an Unusual Silylzirconium Hydride Complex, Can. J. Chem., 1986, 64(8), 1677–1679, DOI:10.1139/v86-276.
- W. H. Campbell, T. K. Hilty and L. Yurga, Dimethylzirconocene-Catalyzed Polymerization of Alkylsilanes, Organometallics, 1989, 8(11), 2615–2618, DOI:10.1021/om00113a016.
- T. A. Blinka, B. J. Helmer and R. West, Polarization Transfer NMR Spectroscopy for Silicon-29: The INEPT and DEPT Techniques, Adv. Organomet. Chem., 1984, 23(C), 193–218, DOI:10.1016/S0065-3055(08)60611-5.
- M. P. Hanrahan, E. L. Fought, T. L. Windus, L. M. Wheeler, N. C. Anderson, N. R. Neale and A. J. Rossini, Characterization of Silicon Nanocrystal Surfaces by Multidimensional Solid-State NMR Spectroscopy, Chem. Mater., 2017, 29(24), 10339–10351, DOI:10.1021/acs.chemmater.7b03306.
- R. D. Miller, D. Hofer, J. Rabolt and G. N. Fickes, Anomalously Long-Wavelength Electronic Transition in Conformationally Locked Organosilane High Polymers, J. Am. Chem. Soc., 1985, 107(7), 2172–2174, DOI:10.1021/ja00293a058.
- J. F. Rabolt, D. Hofer, R. D. Miller and G. N. Fickes, Studies of Chain Conformational Kinetics in Poly(Di-n-Alkylsilanes) by Spectroscopic Methods. 1. Poly(Di-n-Hexylsilane), Poly(Di-n-Heptylsilane), and Poly(Di-n-Octylsilane), Macromolecules, 1986, 19(3), 611–616, DOI:10.1021/ma00157a021.
- F. C. Schilling, A. J. Lovinger, J. M. Zeigler, D. D. Davis and F. A. Bovey, Solid-State Structures and Thermochromism of Poly(Di-n-Butylsilylene) and Poly(Di-n-Pentylsilylene), Macromolecules, 1989, 22(7), 3055–3063, DOI:10.1021/ma00197a028.
- B. J. Grimmond and J. Y. Corey, Synthesis and Characterization of Atactic Poly(p-Tolylsilane) via the Catalytic Dehydrocoupling of p-Tolylsilane, Organometallics, 2000, 19(19), 3776–3783, DOI:10.1021/OM000441R.
- L. Sacarescu, A. Siokou, G. Sacarescu, M. Simionescu and I. Mangalagiu, Methylhydrosilyl Chemostructural Effects in Polyhydrosilanes, Macromolecules, 2008, 41(3), 1019–1024, DOI:10.1021/ma071853f.
- M. Birot, J. P. Pillot and J. Dunoguès, Comprehensive Chemistry of Polycarbosilanes, Polysilazanes, and Polycarbosilazanes as Precursors of Ceramics, Chem. Rev., 1995, 95, 1443–1477, DOI:10.1021/cr00037a014.
- S. Yajima, J. Hayashi and M. Omori, Continuous Silicon Carbide Fiber of High Tensile Strength, Chem. Lett., 1975, 4(9), 931–934, DOI:10.1246/cl.1975.931.
- S. Yajima, Y. Hasegawa, K. Okamura and T. Matsuzawa, Development of High Tensile Strength Silicon Carbide Fibre Using an Organosilicon Polymer Precursor, Nature, 1978, 273(5663), 525–527, DOI:10.1038/273525a0.
- H. N. Han, D. A. Lindquist, J. S. Haggerty and D. Seyferth, Pyrolysis Chemistry of Poly(Organosilazanes) to Silicon Ceramics, Chem. Mater., 1992, 4(11), 705–711, DOI:10.1021/cm00021a038.
- D. Seyferth and G. H. Wiseman, High–Yield Synthesis of Si3N4/SiC Ceramic Materials by Pyrolysis of a Novel Polyorganosilazane, J. Am. Ceram. Soc., 1984, 67, C132–C133, DOI:10.1111/j.1151-2916.1984.tb19620.x.
- C. L. Schilling, J. P. Wesson and T. C. Williams, Polycarbosilane Precursors for Silicon Carbide, J. Polym. Sci., Polym. Symp., 1983, 70(1), 121–128, DOI:10.1002/polc.5070700110.
- D. Seyferth, T. G. Wood, H. J. Tracy and J. L. Robison, Near-Stoichiometric Silicon Carbide from an Economical Polysilane Precursor, J. Am. Ceram. Soc., 1992, 75(5), 1300–1302, DOI:10.1111/j.1151-2916.1992.tb05578.x.
- Z.-F. Zhang, F. Babonneau, R. M. Laine, Y. Mu, J. F. Harrod and J. A. Rahn, Poly(Methylsilane)-A High Ceramic Yield Precursor to Silicon Carbide, J. Am. Ceram. Soc., 1991, 74(3), 670–673, DOI:10.1111/j.1151-2916.1991.tb04080.x.
- S. Yajima, Y. Hasegawa, J. Hayashi and M. Iimura, Synthesis of Continuous Silicon Carbide Fibre with High Tensile Strength and High Young's Modulus, J. Mater. Sci., 1978, 13(12), 2569–2576, DOI:10.1007/BF02402743.
- I. M. T. Davidson and A. V. Howard, Mechanism of Thermolysis of Hexamethyldisilane and the Silicon-Silicon Bond Dissociation Energy, J. Chem. Soc., Faraday Trans. 1, 1975, 71, 69–77, 10.1039/F19757100069.
- I. M. T. Davidson, Some Examples of the Contribution of Gas-Kinetic Studies to the Understanding of Organosilicon Reaction Mechanisms, J. Organomet. Chem., 1988, 255–265, DOI:10.1016/0022-328X(88)89081-8.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00383f |
| This journal is © The Royal Society of Chemistry 2021 |