
Reprocessable covalent adaptable networks with excellent elevated-temperature creep resistance: facilitation by dynamic, dissociative bis(hindered amino) disulfide bonds†
Mohammed A.
Bin Rusayyis
 a and
John M.
Torkelson
*ab
a and
John M.
Torkelson
*ab
aDepartment of Materials Science and Engineering, Northwestern University, Evanston, IL 60208, USA. E-mail: j-torkelson@northwestern.edu
bDepartment of Chemical and Biological Engineering, Northwestern University, Evanston, IL 60208, USA
First published on 14th April 2021
Abstract
Conventionally cross-linked polymer networks known as thermosets contain permanent cross-links which prevent their recyclability, leading to major sustainability and environmental challenges. To overcome this problem, covalent adaptable networks (CANs) containing dynamic covalent bonds have been studied over the past two decades. Because of their dynamic nature, CANs are capable of undergoing reversible or exchange reactions rendering them reprocessable, offering a sustainable alternative to thermosets. However, unlike thermosets with static cross-links, CANs are considered to be highly susceptible to creep especially at elevated temperature, which limits their utility in many high-value applications. Here, we use the dynamic cross-linker bis(2,2,6,6-tetramethylpiperidin-1-yl) disulfide methacrylate (BiTEMPS methacrylate) in the free radical polymerization of reprocessable poly(hexyl methacrylate) networks with different degrees of cross-linking. Full recovery of cross-link density was achieved after multiple recycling steps. We show that BiTEMPS chemistry is capable of arresting creep at elevated temperature up to 90 °C. Poly(hexyl methacrylate) networks containing 5 mol% BiTEMPS exhibited almost no creep with strain values of 0.07% and 0.38% at 70 °C and 90 °C, respectively, after 13.9 h of continuous, 3 kPa shear stress. This excellent creep resistance is comparable to the creep response of static networks. The temperature-dependent viscosity of a BiTEMPS-cross-linked dissociative network calculated from creep data followed an Arrhenius relationship. The viscous flow activation energy from creep and the stress relaxation activation energy were very similar to the bond dissociation energy of disulfide bonds in BiTEMPS, indicating that the creep and stress relaxation mechanisms are both dominated by the dynamic chemistry in the network. This work indicates that BiTEMPS chemistry offers a simple method to synthesize CANs with excellent elevated-temperature creep resistance while achieving full recovery of cross-link density after recycling.
1. Introduction
Thermoset materials are highly cross-linked polymers having a network structure. They are used in an extensive array of applications ranging from kettle handles to aerospace materials. Because of their covalently bonded chains that are interconnected by permanent cross-links, thermosets can offer outstanding mechanical properties, chemical and heat resistance, and dimensional stability. However, conventional thermosets cannot be reshaped or reprocessed into high-value products because of the permanent and irreversible cross-links, rendering these materials generally non-recyclable.1,2 To address this issue, several strategies have been developed to produce reprocessable and, hence recyclable, polymer networks. An increasingly commonly researched strategy to make recyclable polymer networks is to introduce dynamic covalent bonds into these materials.1–10 Network materials containing dynamic covalent bonds are often termed dynamic covalent polymer networks (DCPNs)4 or covalent adaptable networks (CANs).7,8 Most dynamic covalent chemistries employed in CANs have been characterized as either associative or dissociative.3 Associative dynamic chemistries, e.g., transesterification11–13 and transamination,14–16 involve exchange reactions in which bonds break and new bonds are formed simultaneously such that the total number of bonds remains constant. Dissociative dynamic chemistries, e.g., the Diels–Alder reaction5,17–20 and alkoxyamine chemistry,21–27 involve reversible reactions in which dynamic bonds break under applied stimulus and reform when the stimulus is removed. Some dynamic chemistries, e.g., urethane,29 hydroxyurethane,29–31 and thiourethane32 chemistries, involve both associative and dissociative mechanisms.Despite significant advancements in the development of CANs, their susceptibility to creep at elevated temperature (T) has been considered a major technological stumbling block. Creep is the continuous, time-dependent deformation of a material under constant stress and T.33 Conventional thermosets with permanent cross-links are highly creep-resistant as they show near zero or negligible creep at elevated T.34–36 They also do not exhibit significant stress relaxation even at high T below their degradation T.37–39 While stress relaxation of CANs at high T allows them to be malleable and, hence, is favorable for effective reprocessing, creep at elevated T is highly undesirable as it prevents these reprocessable networks from being used in many high-value applications.
A number of strategies have been studied to enhance the creep resistance of CANs.13,38,40–67 One effective but limited strategy was reported in 2018 by Torkelson and coworkers13 and Sumerlin and coworkers40 in which they incorporated some permanent cross-links into associative CANs. Similar approaches were reported later by other groups.41,42 With careful design and judicious control of the ratio of static and dynamic cross-links contained within the network, high-T creep can be reduced significantly while maintaining full reprocessability.13 However, given that the fraction of added static cross-links cannot exceed certain levels to avoid the formation of a percolated network, this strategy cannot be used to completely arrest creep without losing reprocessability.9,13 Other studies have also reported CANs with improved creep resistance.43–46,65–67 All of these studies were focused on associative networks, most of which were prepared via step-growth processes. More importantly, three of these studies reported CANs with arrested creep at elevated T,44,45,66 but all three studies only showed short-term creep measurements with networks tested for 10–30 min, which may not reflect their creep performance under continuous long-term stress, as we demonstrate here.
Another strategy with the potential to arrest elevated-T creep of CANs is to employ a dynamic chemistry with high activation energy (Ea),46–48 which should allow for excellent creep resistance some tens of degrees below the reprocessing temperature. In a recent study,47,48 our group incorporated dynamic cross-links based on dissociative alkoxyamine chemistry into networks and network composites similar to those used in the tire industry that were reprocessable at 140–160 °C. Because of the high activation energy (∼120 kJ mol−1) associated with the alkoxyamine dynamic chemistry, creep was effectively arrested at 80 °C with extremely small creep values comparable to permanent networks, even when the networks are subjected to 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s (13.9 h) of continuous, 3 kPa stress. However, alkoxyamine-based networks made from only monomers tend to exhibit reduced cross-link density after recycling in the absence of excess carbon–carbon double bonds that can compensate for the termination of the generated carbon-centered radicals under processing conditions.26 Thus, there remains a need to develop CANs that show little long-term reactivity at elevated T to avoid creep at those conditions but that can be processed at higher, but reasonable T over an acceptable time scale while maintaining full cross-link density recovery after recycling.
000 s (13.9 h) of continuous, 3 kPa stress. However, alkoxyamine-based networks made from only monomers tend to exhibit reduced cross-link density after recycling in the absence of excess carbon–carbon double bonds that can compensate for the termination of the generated carbon-centered radicals under processing conditions.26 Thus, there remains a need to develop CANs that show little long-term reactivity at elevated T to avoid creep at those conditions but that can be processed at higher, but reasonable T over an acceptable time scale while maintaining full cross-link density recovery after recycling.
Disulfide chemistry is among the most important and widely studied dynamic covalent chemistries.68–82 Although much stronger than O–O single bonds, S–S single bonds in disulfides are known to be relatively weak covalent bonds rendering disulfide chemistries dynamic.83–87 While often viewed as an associative exchange chemistry, the dynamic mechanism in disulfides is rather complex and involves several mechanisms that depend on both the use conditions and substituents on the disulfides.3,39 Several reports have indicated that the dynamic mechanism in disulfides is based on radical-mediated exchange reactions69,87–90 as shown in Scheme 1. This mechanism was confirmed by Asua and coworkers,91 who also noted that disulfide bonds generally undergo homolytic dissociation into radicals prior to exchange. (We do not discount the possibility that steric effects in bis(hindered amino) disulfide bonds could disallow exchange reactions, further enforcing the dissociative nature of the associated dynamic chemistry.) Although research has demonstrated some utility of disulfide linkages (RS-SR) in CANs, the very high bond dissociation energy of disulfides (BDE = 250–300 kJ mol−1)83 is too large to be effectively used in reprocessable networks on a large scale and often requires the use of external catalysts.39,46,86 Because of their high BDE, disulfide bonds in dialkyl disulfides can be inert even at high T, limiting their utility in the development of CANs. For example, poly(hexyl methacrylate) networks containing dialkyl disulfide linkages show almost no stress relaxation at 140 °C.37
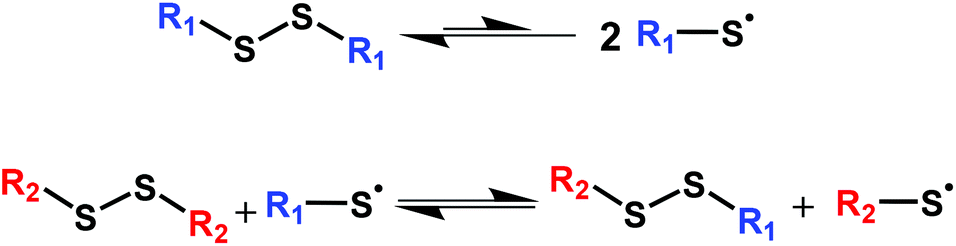 | ||
| Scheme 1 Radical-mediated dynamic exchange mechanism in disulfides. | ||
In contrast, disulfide linkages in bis(dialkylamino) disulfides (R2NS-SNR2) have a high yet substantially lower BDE (∼110–130 kJ mol−1),92,93 making them better candidates for dynamic covalent chemistry in large-scale applications. The reduction in BDE of the disulfides is attributed to the additional stabilization of the generated thionitroxide radicals contributed by the lone pair of electrons on the nitrogen atom.92 The first study to employ dialkylamino disulfide linkages in CANs was reported in 2017 by Otsuka and coworkers.94 They described a multi-step synthesis of a dialkylamino disulfide cross-linker based on bis(2,2,6,6-tetramethylpiperidin-1-yl) disulfide (BiTEMPS) and studied both the thermally reversible dissociation of the disulfide bond in BiTEMPS and the utility of BiTEMPS in synthesizing dynamic polyurethane networks. Later, they reported a study of poly(hexyl methacrylate) networks containing dynamic BiTEMPS functionalities in which they demonstrated stress relaxation in the 120–140 °C T range.37 In 2020, we reported a simple one-step method to synthesize a BiTEMPS-based dynamic cross-linker which we exploited in the free radical polymerization of CANs, and we obtained full property recovery associated with cross-link density after multiple processing steps with the CANs.95
Here, using the same approach we reported in ref. 95, we have prepared catalyst-free, reprocessable poly(hexyl methacrylate) networks containing different levels of BiTEMPS functionalities. The processability and recyclability as well as the complete recovery of cross-link density after recycling are re-demonstrated. Using the T-dependent storage modulus (E′) of these reprocessable networks, we demonstrate the dissociative nature of BiTEMPS dynamic chemistry. We further demonstrate that the prepared dissociative CANs with low but sufficient levels of dynamic bis(hindered amino) disulfide bonds exhibit excellent creep resistance when subjected to 50000 s (13.9 h) of continuous stress at elevated T up to 90 °C, only 40 °C below the processing T. To the best of our knowledge, this is the first report of dissociative CANs made from only monomers that exhibit both elevated-T creep resistance comparable to permanently cross-linked networks and full recovery of cross-link density after recycling. We also address stress relaxation in these networks and show that the viscoelastic responses of both creep and stress relaxation are dominated by the dynamic chemistry in these materials.
2. Experimental section
2.1. Materials
All chemicals are commercially available and used as received unless otherwise noted. 2,2,6,6-Tetramethyl-4-piperidyl methacrylate (TMPM) was from TCI America. Dichloromethane (Certified ACS) and methanol (99.9%) were from Fisher. Low-T azo initiator V-70 was supplied by FUJIFILM Wako Chemicals. Petroleum ether (anhydrous), sulfur monochloride (98%), hexyl methacrylate (HMA, 98%), N,N-dimethylacetamide (DMAc, anhydrous, 99.8%), toluene (99.9%) and chloroform-d (99.8 atom% D) were from Sigma-Aldrich. Hexyl methacrylate (HMA) monomer was de-inhibited using inhibitor remover (Sigma Aldrich, 311340) in the presence of calcium hydride (Sigma Aldrich, 90%). Petroleum ether and DMAc were dried over 4 Å molecular sieves for at least 48 hours before use.2.2. Cross-linker synthesis
Bis(2,2,6,6-tetramethyl-4-piperidyl methacrylate) disulfide (BiTEMPS methacrylate) was synthesized using the procedure reported in ref. 95. The synthesis starts by dissolving 2,2,6,6-tetramethyl-4-piperidyl methacrylate (8.81 g, 39.09 mmol) in pre-dried petroleum ether (∼90 ml). The solution was cooled to −70 °C and then sulfur monochloride (1.31 g, 9.72 mmol) mixed with pre-dried petroleum ether (1.30 ml) was added to the solution dropwise with continuous stirring. The solution was allowed to stir at −70 °C for 15 min followed by stirring at room T for additional 30 min. The mixture was then poured in distilled water and stirred at room T overnight. The formed creamy precipitates were filtered off and dried in a vacuum oven at 40 °C for 48 h to give BiTEMPS methacrylate (2.28 g, 46%). Anal. Calcd for C26H44N2O4S2: N, 5.46; S, 12.51. Found: N, 5.40; S, 12.59.2.3. Network synthesis
In a typical synthesis of poly(hexyl methacrylate) networks, HMA, BiTEMPS methacrylate, and pre-dried DMAc (4.8 ml) were mixed in a 20 mL scintillation vial. The cross-linker, BiTEMPS methacrylate, was added in either 2 mol% (for XLPHMA-2 sample) or 5 mol% (for XLPHMA-5 sample) level with respect to the total amount of monomer and cross-linker. DMAc was added (1.2 ml DMAc per g HMA) to facilitate the dissolution of cross-linker in the monomer. The mixture was stirred at room T until a homogeneous solution was obtained. The solution was then bubbled with N2 gas for 20 min at room T followed by the addition of V-70 initiator (1 mol% w.r.t. total amount of monomer and cross-linker). N2 gas was bubbled again into the solution for an additional 50 min at 25 °C. After 50 min, N2 bubbling was stopped, but N2 gas was allowed to flow continuously into the vial and the reaction was allowed to proceed overnight. After 24 h, the reaction was quenched by exposing it to air. The obtained solid was cut into pieces and washed with DCM/methanol mixtures (3/1, 1/1, 1/3 ratios) three times to remove any unreacted reagents. The pieces were then dried in a vacuum oven at 80 °C for 24 h.2.4. Fourier transform infrared (FTIR) spectroscopy
Attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy employed a Bruker Tensor 37 FTIR spectrophotometer with a diamond/ZnSe attachment. Sixteen scans were collected at room T over the 4000 to 600 cm−1 range at 4 cm−1 resolution. FTIR spectroscopy was done to confirm the conversion of the amine groups in TMPM, which was determined by the disappearance of the N–H stretch peak at 3312 cm−1.2.5. 1H NMR spectroscopy
1H NMR spectroscopy was performed at room T using a Bruker Avance III 500 MHz NMR spectrometer. Deuterated chloroform (CDCl3) was used as solvent, and the spectrum was reported relative to tetramethylsilane.2.6. Molding and reprocessing of networks
Dried networks were cut into millimeter-sized pieces and processed using a PHI press (Model 0230C-X1). To mold the samples used in swelling, dynamic mechanical analysis, and stress relaxation experiments, network pieces were hot pressed into ∼1 mm-thick films at 130 °C with a 10-ton ram force for 1 h. After molding, samples were cooled to room T in a cold compression mold with a 4-ton ram force for 5 min. Such films are considered to be the 1st molded sample. 2nd molded samples were prepared in a similar way where a 1st molded sample was cut into small pieces and pressed again to obtain 2nd molded sample. 2nd molded samples were cut into pieces and reprocessed using the same processing conditions to give 3rd molded samples. Disk samples for creep characterization were molded using the same press at 130 °C and 10-ton ram force for 1 h.2.7. Swelling
Small network pieces were placed in ∼20 ml of toluene in a glass vial at room T. The networks were swollen for 72 h, after which the solvent was decanted. The swollen networks were immediately weighed after removing the remaining solvent on the network surface. The networks were then dried in a vacuum oven for 3 days and weighed afterwards. Swelling tests were performed to determine the gel fractions and the swelling ratios of the as-synthesized and the molded networks. The gel fraction was calculated as md/m0, and the swelling ratio was calculated as (ms − md)/md, where m0 is the original mass of the sample before the swelling test, ms is the mass of the swollen sample, and md is the mass of dried sample after swelling.2.8. Differential scanning calorimetry (DSC)
The glass transition temperatures (Tgs) of as-synthesized and molded networks were obtained by DSC using a Mettler Toledo DSC822e. As the network samples were rubbery at room T and thus had not undergone physical aging, they were cooled to −30 °C at a rate of −20 °C min−1 followed by heating to 80 °C at 10 °C min−1. The Tg values were obtained from the heating ramp using the 1/2 ΔCp method.2.9. Dynamic mechanical analysis (DMA)
DMA was performed using a TA Instruments RSA-G2 Solids Analyzer where the tensile storage modulus (E′), the tensile loss modulus (E′′), and the damping ratio (tanδ = E′′/E′) of the networks were measured as functions of T under nitrogen atmosphere. The network rectangular specimens were mounted on the fixture and underwent a heating ramp from −55 °C to 150 °C (or 250 °C) at a heating rate of 3 °C min−1. The tension-mode measurements were collected at a frequency of 1 Hz and 0.03% oscillatory strain.
2.10. Stress relaxation
Tensile stress relaxation was characterized using a TA Instruments RSA-G2 Solids Analyzer. Rectangular specimens were mounted on the fixture and allowed to equilibrate at the test T for 10 min before starting the test. Once thermal equilibrium was reached, samples were subjected to an instantaneous 5% strain, which was maintained throughout the test. The stress relaxation modulus was recorded until at it had relaxed to 20% of its initial value.2.11. Creep
Shear creep and creep-recovery experiments were performed on ∼2 mm-thick disk samples using an Anton-Paar MCR 302 rheometer with 25 mm parallel-plate fixtures. The samples were equilibrated at the test T for at least 5 min before starting the experiment. In creep-recovery tests, the stress (3 kPa unless otherwise noted) was applied for 50000 s followed by 7200 s of recovery (zero stress). Creep strain values (Δε) reported in Table 2 were calculated as the difference in strain at t = 50000 s and t = 1800 s (Δε = ε50000 − ε1800) in order to consider only pure creep, not the initial delayed elastic deformation.36 The network viscosity (η) was calculated as a function of T as follows:η = σ/![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a2.gif) | (1) |
is the shear strain rate calculated from the fitted slope of the linear part of the creep curves (employing creep data between t = 30000 s to t = 50000 s).
| Sample | E′ (MPa) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 70 °C | 80 °C | 90 °C | 100 °C | 110 °C | 120 °C | 130 °C | 140 °C | 150 °C | |
| 1st mold | 2.72 ± 0.05 | 2.25 ± 0.04 | 2.10 ± 0.06 | 1.94 ± 0.04 | 1.78 ± 0.02 | 1.70 ± 0.06 | 1.64 ± 0.07 | 1.59 ± 0.08 | 1.56 ± 0.10 |
| 2nd mold | 2.65 ± 0.21 | 2.25 ± 0.15 | 2.03 ± 0.11 | 1.88 ± 0.12 | 1.76 ± 0.11 | 1.68 ± 0.10 | 1.61 ± 0.11 | 1.58 ± 0.08 | 1.56 ± 0.11 |
| 3rd mold | 2.82 ± 0.04 | 2.34 ± 0.17 | 2.12 ± 0.14 | 1.95 ± 0.07 | 1.85 ± 0.11 | 1.73 ± 0.04 | 1.65 ± 0.03 | 1.61 ± 0.02 | 1.59 ± 0.04 |
| T (°C) | η (Pa s) | Δεb | Instant strain recoveryc (%) | Total strain recoveryd (%) | Residual straind |
|---|---|---|---|---|---|
|
a Viscosity (η) was calculated using eqn (1) with the strain rate calculated from the slope of the creep curves between t = 30000 s and t = 50000 s.
b Creep strain (Δε) was calculated as the difference in strain between t = 50000 s and t = 1800 s.
c Measured 2 s after the stress was removed.
d Measured 1.4 h after the creep was removed for 70 °C and 2.0 h after the creep was removed for 90 °C and 100 °C.
|
|||||
| 70 | 3.85 × 1011 | 0.0007 | 78.2 | 99.98 | 0.00000355 |
| 90 | 4.48 × 1010 | 0.0038 | 73.3 | 81.85 | 0.00541 |
| 100 | 1.71 × 1010 | 0.019 | 45.9 | 51.29 | 0.0246 |
| 130 | 1.45 × 109 | 0.125 | — | — | — |
3. Results and discussion
Bis(2,2,6,6-tetramethyl-4-piperidyl methacrylate) disulfide (BiTEMPS methacrylate) was synthesized by reacting 2,2,6,6-tetramethyl-4-piperidyl methacrylate (TMPM) with sulfur monochloride (S2Cl2) according to Scheme S1.† The conversion of TMPM into BiTEMPS methacrylate was confirmed by FTIR spectroscopy through the disappearance of the secondary amine stretch at 3312 cm−1 (Fig. S1†), 1H NMR (Fig. S2†), and elemental analysis. BiTEMPS methacrylate was employed as a dynamic cross-linker and was incorporated into polymer networks via free radical polymerization. Two catalyst-free poly(hexyl methacrylate) (PHMA) networks containing different amounts of dynamic dialkylamino disulfide bonds were synthesized at 25 °C using the azo-based low temperature initiator V-70. The synthesized XLPHMA-2 and XLPHMA-5 networks contained 2 mol% and 5 mol% of BiTEMPS methacrylate, respectively, with respect to the total amounts of HMA monomer and BiTEMPS methacrylate cross-linker. The network synthesis is schematically shown in Fig. 1(a).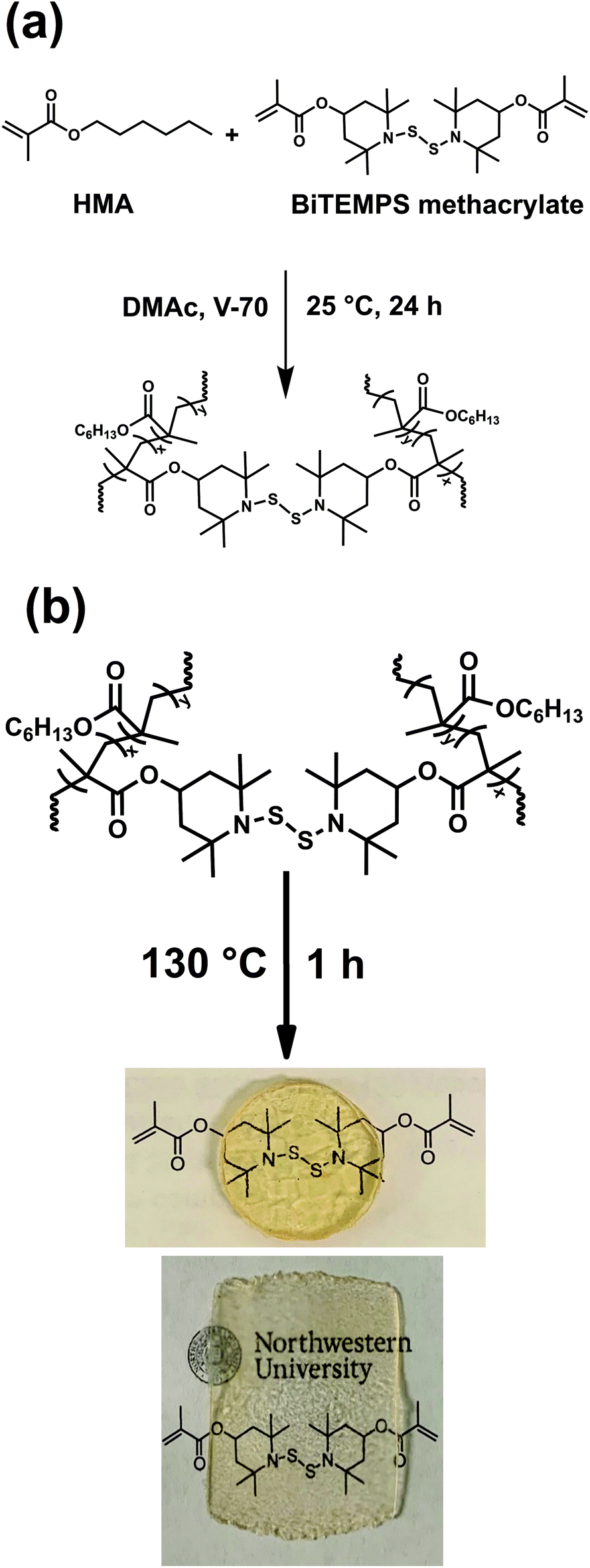 | ||
| Fig. 1 (a) Synthesis of PHMA networks and (b) processing of the networks into disk and film samples. | ||
The network materials were washed with dichloromethane and methanol mixtures and dried before characterization. The cross-linked nature of XLPHMA-2 and XLPHMA-5 networks was confirmed by swelling in toluene (Table S1†). Table S1† shows that XLPHMA-5 swells less and contains higher gel fraction than XLPHMA-2, consistent with a higher degree of cross-linking as a result of the higher cross-linker concentration. As-synthesized XLPHMA-2 and XLPHMA-5 networks were analyzed by DSC. No thermal transition was detected for the as-synthesized XLPHMA-2 sample, but the XLPHMA-5 network showed a Tg of 18 °C (Fig. S3†). We note that linear PHMA synthesized under similar conditions has a Tg of −6 °C.95 This substantial difference in Tg between linear PHMA and XLPHMA-5 is attributed to the relative rigidity of the XLPHMA-5 sample imparted by the cyclic skeleton structure of BiTEMPS.37,95
Both XLPHMA-2 and XLPHMA-5 networks were malleable at 130 °C and were molded into films and disks for further characterization (Fig. 1b). Linear polymethacrylates are known to have excellent optical properties due to their high transparency. The highly transparent appearances of the molded XLPHMA-2 and XLPHMA-5 samples indicates that neither the addition of BiTEMPS methacrylate as cross-linker nor the processing at high T had negative effects on the material optical properties. The molded samples also retained their cross-linked nature as evidenced from the swelling results. Notably, the molded XLPHMA-2 and XLPHMA-5 networks showed lower swelling ratios but higher gel fractions compared to their respective as-synthesized samples (Table S1†). These results suggest that additional cross-linking was achieved via bond rearrangements during the processing at sufficiently higher T.95 Molded XLPHMA-2 and XLPHMA-5 networks were also characterized by DSC with XLPHMA-2 and XLPHMA-5 samples exhibiting Tg values of 7 °C and 18 °C, respectively (Fig. S3†). These results indicate that the network Tg and related properties can be easily tuned via the cross-linker concentration.
Fig. 2 illustrates T-dependent DMA properties for the molded XLPHMA-2 and XLPHMA-5 samples. As shown in Fig. 2a, the storage modulus (E′) curves of both XLPHMA-2 and XLPHMA-5 samples display a rubbery plateau well above their Tgs, a characteristic of cross-linked materials. Additionally, Fig. 2a shows that the rubbery plateau E′ value of XLPHMA-5 network is approximately three times that of XLPHMA-2 network. Based on expectations from Flory's ideal rubber elasticity theory, which indicates that the tensile modulus (E) in the rubbery plateau scales linearly with cross-link density and with absolute T,96 these results indicate that XLPHMA-5 sample has a substantially higher cross-link density than XLPHMA-2. Given that the loss modulus (E′′) is much smaller than the storage modulus E′ in the rubbery plateau regime (recall that tanδ = E′′/E′), the value of E in the rubbery plateau regime can be estimated by the E′ value. Thus, in an ideal elastomer, the E′ value in the rubbery plateau regime should exhibit a linear increase with T. Associative CANs that undergo exclusive exchange reactions where cross-link density remains constant are expected to follow a similar trend. Values of E′ in the rubbery plateau regime of CANs with dual associative and dissociative mechanisms have also been observed to increase with increasing T.28,31 However, as shown in Table S2,† the E′ values in the rubbery plateau regimes for both XLPHMA-2 and XLPHMA-5 networks decrease slightly with increasing T. These results are consistent with the dynamic mechanism in the BiTEMPS disulfide bonds being of a dissociative nature. Although a radical-mediated exchange mechanism of BiTEMPS was previously reported,94 our results suggest that disulfide bonds in BiTEMPS dissociate at a faster rate than their recombination/exchange. While DMA experiments for CANs have been traditionally performed to demonstrate their cross-linked nature or recovery of cross-link density, we note that simple DMA measurements can also be useful in understanding the nature of the dynamic chemistry present in CANs by observing the T-dependence of the rubbery plateau E′.
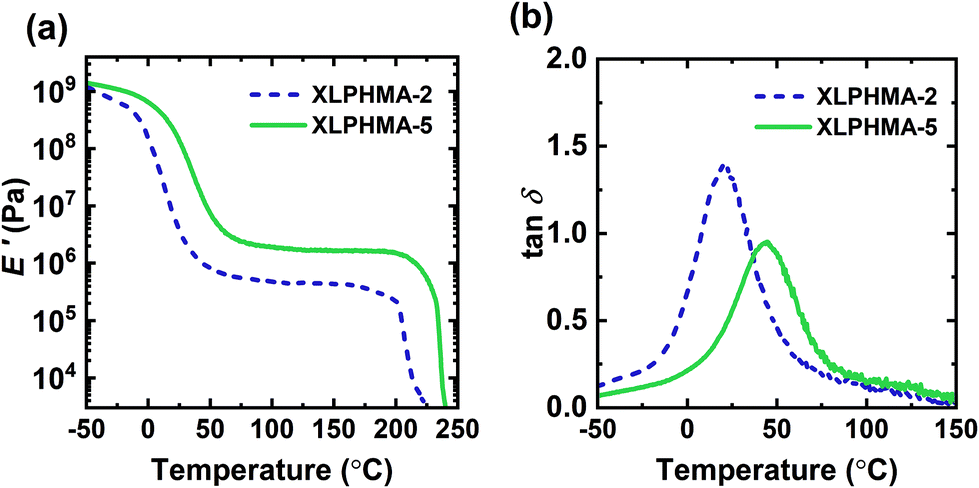 | ||
| Fig. 2 (a) Tensile storage modulus (E′) and (b) damping ratio (tanδ = E′′/E′) of 1st mold XLPHMA-2 and XLPHMA-5 networks. | ||
Typically, the rubbery plateau of dissociative CANs extends up to an approximate “gel-to-sol transition” T where the network cross-links undergo enough dissociation to enable flow similar to thermoplastics.97 To characterize the T of this transition (Tgel), the molded XLPHMA-2 and XLPHMA-5 samples were heated to ∼250 °C in a high-T DMA experiment. The XLPHMA-2 and XLPHMA-5 samples exhibit Tgel values of ∼204 °C and ∼234 °C, respectively. The 30 °C difference in Tgel indicates that the thermal stability of the network nature in these materials can be improved by increasing the cross-linker concentration. Importantly, the existence of an apparent gel-to-sol transition and the flow of the samples at high T confirm the dissociative nature of BiTEMPS dynamic chemistry.
Fig. 2b shows the damping ratio tanδ of the XLPHMA-2 and XLPHMA-5 samples as a function of T. The T at which tanδ is a maximum is sometimes considered as a “shifted” Tg value.98 The tanδ peak for XLPHMA-2 is located at 20 °C, which is substantially lower than that of XLPHMA-5 (45 °C). This large difference in “shifted” Tg values is in line with the difference observed in the Tg values determined by DSC. In addition, XLPHMA-2 showed a higher tanδ peak value than XLPHMA-5. The tanδ peak value is a qualitative measure of the viscoelastic behavior of the material at the peak temperature.99 A higher tanδ peak value indicates larger energy loss and more viscous or liquid-like behavior, whereas a lower value indicates lower energy loss and more elastic behavior.100 Because cross-links restrict the mobility of polymer chains, one would expect the peak value of the tanδ curve of the less cross-linked XLPHMA-2 to be higher than that of the more cross-linked XLPHMA-5 network, which is consistent with our results. As shown in Fig. S4a,† at high temperatures (T > ∼180 °C) where the dynamic dissociation of BiTEMPS cross-links become sufficiently high, tanδ values of both XLPHMA-2 and XLPHMA-5 networks increase as a result of the instability of network sample with decreasing cross-link density at higher T. This is also confirmed by the increase of E′′ values to form secondary E′′ peaks before the samples disintegrate and flow (Fig. S4b†).
Fig. 3 shows DMA results for the 1st molded, 2nd molded, and 3rd molded XLPHMA-5 networks. The storage modulus curves for all molded samples exhibit a quasi-rubbery plateau at T ≥ ∼70 °C with values exceeding 1.0 MPa, consistent with their cross-linked natures. As shown in Table 1, the average E′ value in the quasi-rubbery plateau regime for all molded XLPHMA-5 samples decreases with increasing T, consistent with the dissociative nature of the disulfide bonds in BiTEMPS. Additionally, at a given T in the rubbery plateau region the average E′ values of XLPHMA-5 samples are, within experimental error, independent of molding cycle through three cycles. In accord with Flory's ideal rubber elasticity theory,96 these results indicate that XLPHMA-5 achieves full recovery of cross-link density, within error, after multiple recycling steps. The full recovery of cross-link density of reprocessed XLPHMA-5 networks is further confirmed by the swelling ratio and gel fraction data (Table S1†).
 | ||
| Fig. 3 Tensile storage modulus (E′) and damping ratio (tanδ = E′′/E′) of 1st mold, 2nd mold, and 3rd mold XLPHMA-5 network samples. | ||
The creep performance of the synthesized networks was characterized at 70 °C, well above their Tg values. In the creep experiment, a 3.0 kPa shear stress was applied for 50000 s or 13.9 h during which strain was measured. Upon stress removal, creep recovery was measured for 2 h. Fig. 4a shows creep-recovery curves of the XLPHMA-5 network at 70 °C under different stress levels. XLPHMA-5 exhibited almost no creep over 13.9 h for stress levels ranging from 3 kPa to 10 kPa. For all stress levels tested, after an elastic deformation, the strain remained almost invariant with time, indicating that the network maintained excellent dimensional stability under these conditions. Fig. 4a also shows that the strain scales linearly with stress, which is confirmed by the creep compliance data. It is well known that the creep compliance for a linear viscoelastic material is independent of the applied stress due to the linear relationship between stress and strain at any time for such materials.101Fig. 4b shows that the creep compliance, J(t), curves of XLPHMA-5 at different stresses overlap each other, confirming the linear viscoelastic behavior of XLPHMA-5 network under these conditions.
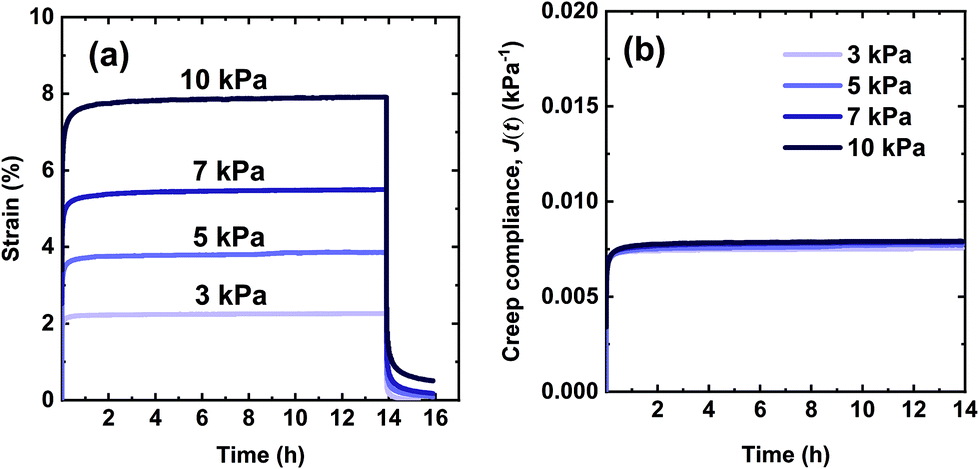 | ||
| Fig. 4 Curves for the XLPHMA-5 network showing (a) creep and creep recovery and (b) creep compliance (J(t) = ε(t)/σ) at 70 °C. | ||
Fig. 5 shows the creep-recovery curve of the XLPHMA-2 network. When the 3 kPa stress was applied, XLPHMA-2 showed an instantaneous strain of 23%, which was measured 2 s after the start of the creep test. This instantaneous strain is significantly higher than the ∼1% instantaneous strain exhibited by the network containing 5 mol% BiTEMPS methacrylate under the same conditions. This large difference in instantaneous strain is qualitatively consistent with the lower cross-link density of the XLPHMA-2 sample. As shown in Fig. 5, the strain exhibited by the XLPHMA-2 sample increased progressively with time for 8 h after which it increased at an accelerated rate. After ∼12 h, the strain increased significantly, reaching 2000% at t = 13.9 h. When the stress was removed, the strain was only weakly recovered with a large residual strain remaining 2 h after stress removal. This large, apparently permanent creep deformation is also reflected in the physical appearance of the XLPHMA-2 sample at the end of the creep-recovery experiment (Fig. S5†). These results reveal that, with sufficient levels of cross-linking units, BiTEMPS dynamic chemistry is very effective in arresting long-term elevated-T creep of CANs. However, the cross-linker content is crucial to improving the creep resistance of these networks. Only CANs that are sufficiently cross-linked, potentially significantly beyond the percolation limit, as is the case with XLPHMA-5, exhibit excellent elevated-T creep resistance.
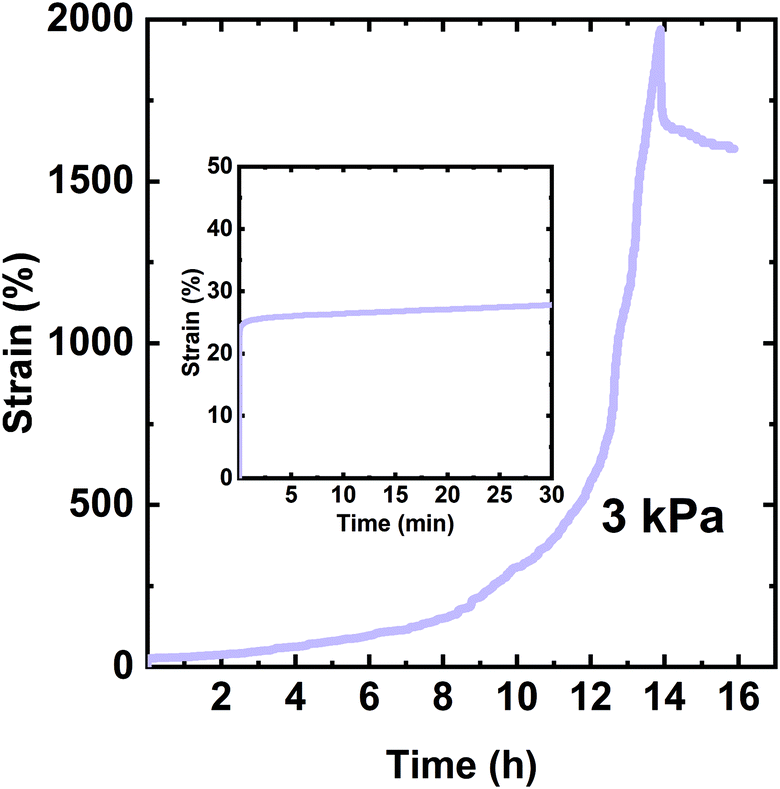 | ||
| Fig. 5 Creep and creep recovery of the XLPHMA-2 network at 70 °C. The inset shows the creep response in the first 30 min of the creep experiment. | ||
It is critical to note that creep response at short times may not be indicative of creep performance at long times. At short times, low levels of covalent cross-links may have a smaller effect on creep as physical chain entanglements can act as temporary cross-links over this timescale.34 The short-term (≤30 min) creep response of the same XLPHMA-2 sample at 70 °C and 3.0 kPa creep stress is shown in the inset in Fig. 5. The XLPHMA-2 exhibited a relatively small creep strain over the 20 min timeframe from 10 to 30 min, Δε = ε30min − ε10min = 0.02 or 2%. However, that short-term behavior in no way reflects the response at long creep time, with a very large creep strain of several hundred percent being exhibited over any selected 20 min timeframe in the final hour (12.9–13.9 h) of the creep experiment. Such differences in short-time and long-time elevated-T creep response may be expected for many CANs because of the slow rates of exchange/dissociation reactions associated with covalent chemistries employed in these materials. Unfortunately, this implies that some studies that have reported good elevated-T creep resistance with creep characterization for only short 10–30 min timescales may not provide a proper assessment of elevated-T creep resistance for real-life applications that require dimensional stability over relatively long time-periods.
Although creep response of specific CANs is often reported at only one or two T values,13,38,40,46–53,55,61,63,64 creep measurements as a function of T are important as they may enable direct determination of the T-dependent viscosity of these materials and thus the viscous flow activation energy.102 To investigate its T-dependence of viscosity, the creep response of XLPHMA-5 was studied at four T values from 70 °C to 130 °C (Fig. 6). The T-dependent viscosity was calculated from the slope of the linear time-dependent region of the creep curves. The viscosity (η), creep strain (Δε), and creep recovery as functions of T are summarized in Table 2. The creep strain of XLPHMA-5 at 70 °C was quantified to be 0.0007 (i.e., 0.07%) after 13.9 h of continuous stress, affirming the network's excellent dimensional stability. The strain at 70 °C was nearly fully recovered less than 30 min after stress removal (Fig. 6c). These results indicate that the BiTEMPS chemistry is extremely slow at 70 °C allowing both creep arrest and full or nearly full strain recovery at this temperature. XLPHMA-5 also maintained its excellent creep resistance at 90 °C; the network exhibits a creep strain of only 0.0038 (i.e., 0.38%). These creep strain values are comparable to values exhibited by static networks.34–36 We note that the disulfide bond in BiTEMPS has been shown to undergo perceptible radical-mediated exchange reactions at 80 °C.94 Thus, we expect that BiTEMPS chemistry was active but at very low levels during 90 °C the creep test. This is also confirmed by the creep-recovery behavior of the network at 90 °C as the strain was incompletely recovered 2 h after stress removal. However, given the negligible creep strain and the low residual strain (0.54%), we believe that the rate of disulfide dissociation is sufficiently low to nearly arrest creep at 90 °C.
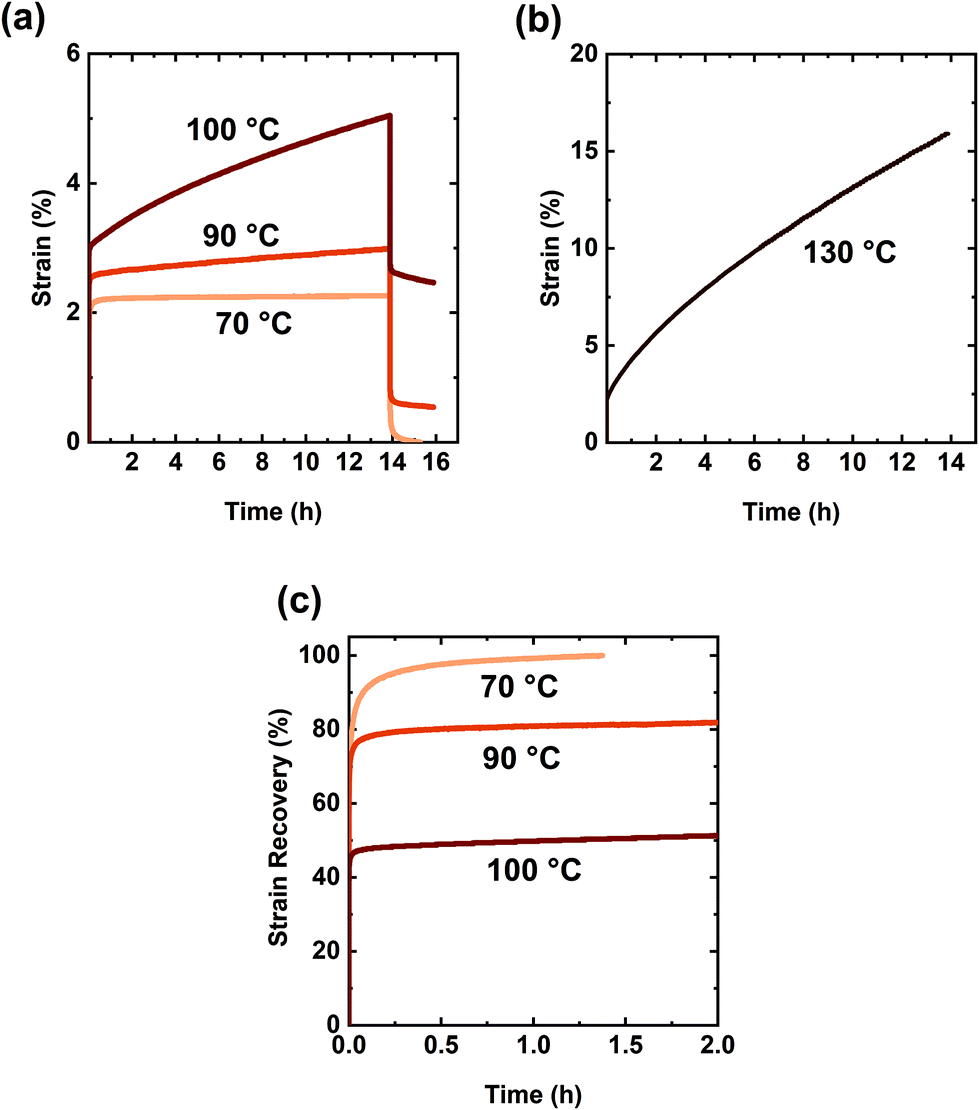 | ||
| Fig. 6 (a) Creep and creep recovery curves of the XLPHMA-5 network at different temperatures under a constant stress of 3 kPa. (b) Creep curve of the XLPHMA-5 network at the processing temperature (130 °C) under a constant stress of 3 kPa. (c) Strain recovery as a function of time for the XLPHMA-5 network at different temperatures after 13.9 h of creep testing under a constant stress of 3 kPa. | ||
At 100 °C, XLPHMA-5 exhibited a creep strain of 0.019 or 1.9%, indicating a faster rate of disulfide dissociation under these conditions. Although BiTEMPS chemistry was unable to arrest creep completely at this T, the response of the network is consistent with strong creep resistance at 100 °C. Upon increasing the T to the 130 °C processing T, the sample exhibited a more pronounced creep strain, which is expected as the network is malleable at this T. One may expect CANs to creep significantly at the processing T. However, although more pronounced, the creep strain was only 12.5% after 13.9 of continuous stress at 130 °C. These results suggest that a significant fraction of the dynamic cross-links in XLPHMA-5 remain linked in this dissociative CAN at the processing T and atmospheric pressure.
Fig. 7 shows an Arrhenius plot of the T-dependent viscosity calculated from the XLPHMA-5 creep data, confirming that the network viscosity of our dissociative CAN can be fit with an Arrhenius relationship over the tested T range (70–130 °C). The viscous flow activation energy (Ea,η) was calculated from the slope of the linear fit to be 106.7 kJ mol−1. It is noteworthy that the Ea,η value is very close to the value reported for the bond dissociation energy of the disulfide bond in BiTEMPS (109.6 kJ mol−1).94 This indicates that, for the conditions of our study, the network creep behavior is dominated by the dynamic chemistry of the cross-linker. These results corroborate the findings we reported recently that indicate that having a dynamic chemistry with a sufficiently high activation energy is a key for arresting elevated-T creep in CANs.47 We also note that studies are warranted to consider how creep resistance may be modified in going from the linear viscoelastic regime to the nonlinear viscoelastic regime at much higher stresses.
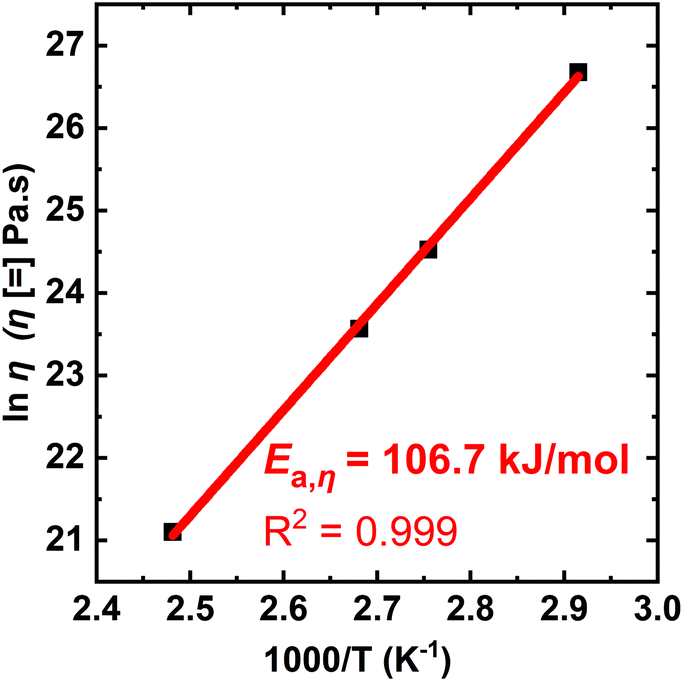 | ||
| Fig. 7 Arrhenius activation energy of viscosity for the XLPHMA-5 network as calculated from creep data taken over a T-range of 70–130 °C. | ||
We also studied the T-dependent stress relaxation behavior of XLPHMA-5 (Fig. 8a). We note that stress relaxation measurements are generally done at sufficiently small strain that the response is in the linear viscoelastic regime which allows for rational comparison with T-dependent creep response in the linear viscoelastic regime, a condition met by our creep studies. Many studies that have reported the stress relaxation of CANs assume that these materials follow the Maxwell model with a single-mode exponential decay and thus a single relaxation time, e.g., ref. 78. Based on this assumption, the relaxation time (τ) is estimated as the time required for the relaxation modulus to decrease to 1/e (37.8%) of its initial value. Using this method, we estimated the T-dependent τ values, which were observed to reasonably follow the Arrhenius relationship (Fig. S7†). The single-mode stress-relaxation activation energy was calculated from the slope of the linear fit to be 108.5 kJ mol−1, which is in relatively close agreement with the activation energy reported for a similar poly(hexyl methacrylate) network cross-linked with a different BiTEMPS-containing cross-linker (105.8 kJ mol−1).103 However, it is known that in general the stress relaxation of CANs does not correspond to a single exponential decay.37,39 This also applies to XLPHMA-5 as the stress relaxation curves of the network do not fit single exponential decays as described by the Maxwell model (Fig. S8a†). The Kohlrausch-Williams-Watts (KWW) stretched exponential decay function, which accounts for a breadth of relaxation times, is widely used to describe stress relaxation in polymers.31,39,104 The KWW decay function can be written as:
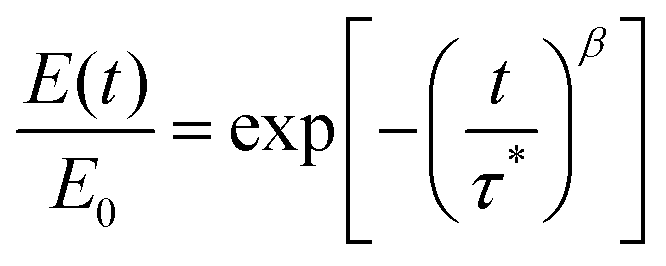 | (2) |
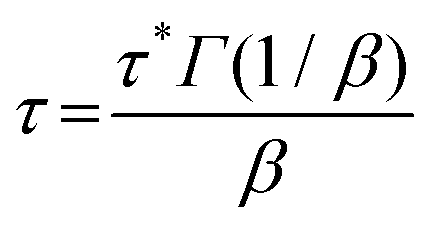 | (3) |
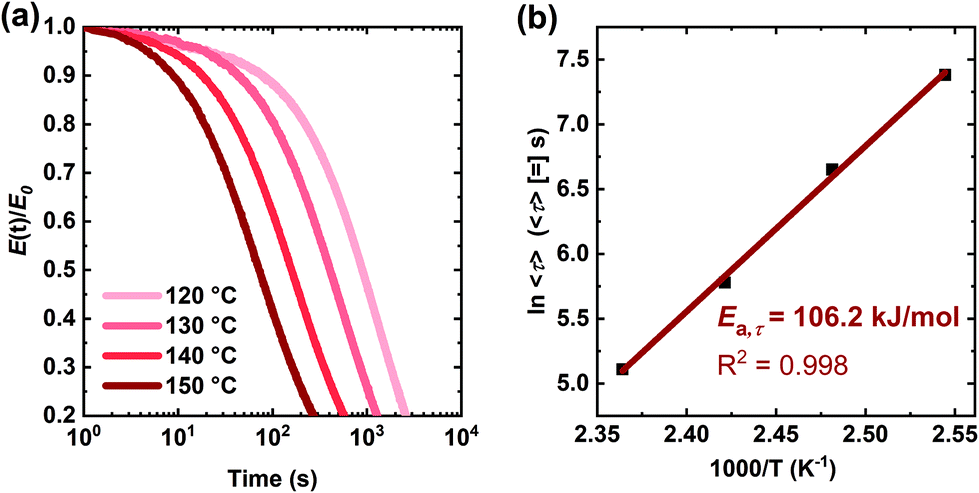 | ||
| Fig. 8 (a) Normalized stress relaxation curves of the XLPHMA-5 network at different temperatures under a strain of 5%. (b) Arrhenius apparent activation energy of stress relaxation associated with average relaxation time for the XLPHMA-5 network. | ||
| T (°C) | τ* (s) | β | <τ> (s) | R 2 |
|---|---|---|---|---|
| 120 | 1367 | 0.76 | 1604 | 0.999 |
| 130 | 650 | 0.75 | 773 | 0.999 |
| 140 | 266 | 0.73 | 323 | 0.998 |
| 150 | 127 | 0.68 | 166 | 0.993 |
The average relaxation time decreases significantly with increasing T, consistent with a change in network structure as a result of more de-cross-linking at higher T. These results are also consistent with the strong T-dependence of BiTEMPS dynamic chemistry. The processing time employed in this study (1 h) is roughly 4.7 times the average relaxation time at the molding temperature (130 °C). Previous study has indicated that three times the average relaxation time is a good empirical molding time for polymers.106 This suggests that the processing time of XLPHMA-5 at 130 °C may potentially be reduced from 1 h to ∼40 min. The values of β associated with the KWW fits were also observed to vary with T. Typically, studies that have analysed the dynamics of amorphous materials via KWW fits have reported a narrower relaxation distribution (higher β values) with increasing T, regardless of whether the relaxation dynamics were associated with α- or β-relaxation processes.107–109 Of course, the <τ> values for either α- or β-relaxation processes would be many orders of magnitude lower than those associated with stress relaxation results such as those in Fig. 8 and Table 3 at T ≥ ∼Tg + 100 °C. Thus, the stress relaxation results for XLPHMA-5 shown in Fig. 8 are not reflective of such polymer dynamics. Importantly, for our stress relaxation studies of the XLPHMA-5 system, we found that the value of β decreases slightly with increasing T, consistent with broader relaxation distribution at higher T over the T-range studied. This trend is likely due to the reduction of the network character of XLPHMA-5 system with increasing T from 120 °C to 150 °C.
The KWW average relaxation times calculated from eqn (3) were also fitted to the Arrhenius equation (Fig. 8b), yielding a stress relaxation activation energy (Ea,τ) of 106.2 kJ mol−1, only slightly lower than the Ea,τ value calculated using the Maxwell model which assumes a single exponential decay response. Notably, the stress relaxation activation energy value determined from the fitting of the stress relaxation data (obtained from 120 °C to 150 °C) to the KWW function is in excellent agreement with the viscous flow activation energy (Ea,η = 106.7 kJ mol−1) determined from the creep experiments (obtained from 70 °C to 130 °C). Thus, for the T-ranges studied, these results indicate that the creep and stress relaxation mechanisms in these networks have the same T-dependence. Furthermore, the values of Ea,η and Ea,τ are in very good agreement with the bond dissociation energy of disulfide bonds in BiTEMPS (BDE = 109.6 kJ mol−1).94 In total, these results provide a strong argument for the notion that, over relevant T ranges, the dynamic mechanism of the disulfide bonds in BiTEMPS dominates the XLPHMA-5 responses associated with elevated-T creep and stress relaxation behaviors.
4. Conclusions
We prepared two reprocessable poly(hexyl methacrylate) networks containing different amounts of the dynamic cross-linker BiTEMPS methacrylate. Using the rubbery plateau E′, we demonstrated the dissociative nature of the BiTEMPS dynamic chemistry. The reprocessability of the synthesized networks at 130 °C, including the full recovery of cross-link density after multiple recycling steps, was also demonstrated. It is worth noting that BiTEMPS methacrylate utilized in this study is the only dynamic cross-linker reported in the literature that could be utilized in the synthesis of addition-type polymer networks made exclusively from vinyl monomers and recovering full cross-link density after reprocessing.95 Furthermore, we have shown that the amount of cross-linker incorporated in the network can have significant impact on the creep response. When BiTEMPS methacrylate content was increased from 2 mol% to 5 mol%, the creep resistance and creep recovery were significantly enhanced. Importantly, the network must contain sufficient dynamic cross-linker to exhibit long-term arrested creep at elevated T. With the addition of 5 mol% of BiTEMPS methacrylate, the XLPHMA-5 network exhibited almost no creep at 70 °C and 90 °C after 13.9 h of continuous 3 kPa shear stress. Based on our results for XLPHMA-2, we have also illustrated the importance of long-term creep characterization of CANs because the excellent short-term creep suppression (10–30 min) at 70 °C in no way reflected the poor long-term creep performance (up to 13.9 h).High-T stress relaxation of the network containing 5 mol% BiTEMPS methacrylate was also characterized. The stress relaxation experimental data in the 120–150 °C T-range were fitted to single exponential decay (Maxwell model) and stretched exponential decay (KWW function). We found that KWW provides a much better fit to the data, indicating that the network stress relaxation mechanism cannot be described by a single-relaxation process. The stress relaxation activation energy (Ea,τ), associated with the average relaxation time calculated from stress relaxation data, and the viscous flow activation energy (Ea,η), associated with the network viscosity as determined from creep data, were found to be in excellent agreement. The values of Ea,τ and Ea,η were also very similar to the bond dissociation energy of the disulfide bond in BiTEMPS, indicating that, for the conditions studied, the creep and stress relaxation responses are predominantly controlled by the BiTEMPS dynamic chemistry.
This work provides a simple method to prepare dissociative CANs that not only exhibit full cross-link density recovery after multiple reprocessing steps but also long-term creep resistance at elevated-T up to 90 °C that is similar to that of conventional, permanently cross-linked polymer networks. The effectiveness of BiTEMPS dynamic chemistry in arresting elevated-T creep offers a potential solution to the technological stumbling block associated with the creep susceptibility of CANs. Combined with the results of another recent study of elevated-T creep resistance in alkoxyamine-based dissociative CANs,47 our results indicate that dynamic chemistries with high activation energy or bond dissociation energy can provide an important route to achieve excellent creep resistance at temperatures a few tens of degrees below the reprocessing T. Further studies are warranted of the utility of other dynamic chemistries, whether dissociative or associative or both, for achieving excellent elevated-T creep resistance in addition-type as well as step-growth-type CANs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support of Northwestern University via discretionary funds associated with a Walter P. Murphy Professorship (J. M. T.) and support of SABIC (M. B. R.). This work made use of the MatCI Facility at Northwestern University, which receives support from the MRSEC Program (NSF DMR-1720139) of the Materials Research Center at Northwestern University.Notes and references
- Y. Yang, X. Ding and M. W. Urban, Prog. Polym. Sci., 2015, 49–50, 34–59 CrossRef CAS.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Adv. Mater., 2017, 29, 1606100 CrossRef PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- M. Podgorski, B. D. Fairbanks, B. E. Kirkpatrick, M. McBride, A. Martinez, A. Dobson, N. J. Bongiardina and C. N. Bowman, Adv. Mater., 2020, 32, 1906876 CrossRef CAS PubMed.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- M. Guerre, C. Taplan, J. M. Winne and F. E. Du Prez, Chem. Sci., 2020, 11, 4855–4870 RSC.
- R. Mo, J. Hu, H. Huang, S. Sheng and X. Y. Zhang, J. Mater. Chem. A, 2019, 7, 3031–3038 RSC.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS.
- Z. Pei, Y. Yang, Q. Chen, Y. Wei and Y. Ji, Adv. Mater., 2016, 28, 156–160 CrossRef CAS PubMed.
- L. Li, X. Chen, K. Jin and J. M. Torkelson, Macromolecules, 2018, 51, 5537–5546 CrossRef CAS.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- M. M. Obadia, A. Jourdain, P. Cassagnau, D. Montarnal and E. Drockenmuller, Adv. Funct. Mater., 2017, 27, 1703258 CrossRef.
- J. J. Lessard, L. F. Garcia, C. P. Easterling, M. B. Sims, K. C. Bentz, S. Arencibia, D. A. Savin and B. S. Sumerlin, Macromolecules, 2019, 52, 2105–2111 CrossRef CAS.
- J. Bai, H. Li, Z. Shi and J. Yin, Macromolecules, 2015, 48, 3539–3546 CrossRef CAS.
- K. K. Oehlenschlaeger, J. O. Mueller, J. Brandt, S. Hilf, A. Lederer, M. Wilhelm, R. Graf, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Adv. Mater., 2014, 26, 3561–3566 CrossRef CAS PubMed.
- L. M. Polgar, M. van Duin, A. A. Broekhuis and F. Picchioni, Macromolecules, 2015, 48, 7096–7105 CrossRef CAS.
- C. Shao, M. Wang, H. Chang, F. Xu and J. Yang, ACS Sustainable Chem. Eng., 2017, 5, 6167–6174 CrossRef CAS.
- H. Otsuka, K. Aotani, Y. Higaki and A. Takahara, J. Am. Chem. Soc., 2003, 125, 4064–4065 CrossRef CAS PubMed.
- Y. Higaki, H. Otsuka and A. Takahara, Macromolecules, 2006, 39, 2121–2125 CrossRef CAS.
- F. Wang, M. Z. Rong and M. Q. Zhang, J. Mater. Chem., 2012, 22, 13076–13084 RSC.
- H. Otsuka, Polym. J., 2013, 45, 879–891 CrossRef CAS.
- C. E. Yuan, M. Z. Rong and M. Q. Zhang, Polymer, 2014, 55, 1782–1791 CrossRef CAS.
- K. Jin, L. Li and J. M. Torkelson, Adv. Mater., 2016, 28, 6746–6750 CrossRef CAS PubMed.
- Y. Jia, Y. Matt, Q. An, I. Wessely, H. Mutlu, P. Theato, S. Bräse, A. Llevot and M. Tsotsalas, Polym. Chem., 2020, 11, 2502–2510 RSC.
- X. Chen, S. Hu, L. Li and J. M. Torkelson, ACS Appl. Polym. Mater., 2020, 2, 2093–2101 CrossRef CAS.
- X. Chen, L. Li, K. Jin and J. M. Torkelson, Polym. Chem., 2017, 8, 6349–6355 RSC.
- S. Hu, X. Chen and J. M. Torkelson, ACS Sustainable Chem. Eng., 2019, 7, 10025–10034 CrossRef CAS.
- X. Chen, L. Li, T. Wei, D. C. Venerus and J. M. Torkelson, ACS Appl. Mater. Interfaces, 2019, 11, 2398–2407 CrossRef CAS PubMed.
- L. Li, X. Chen and J. M. Torkelson, Macromolecules, 2019, 52, 8207–8216 CrossRef CAS.
- L. H. Sperling, Introduction to Physical Polymer Science, John Wiley & Sons, Hoboken, United States, 2006 Search PubMed.
- L. E. Nielsen, J. Macromol. Sci., Part C, 1969, 3, 69–103 CrossRef CAS.
- D. J. Plazek, J. Polym. Sci., Part A-2: Polym. Phys., 1966, 4, 745–763 CrossRef CAS.
- K. Watanabe, Rubber Chem. Technol., 1962, 35, 182–199 CrossRef.
- A. Takahashi, R. Goseki, K. Ito and H. Otsuka, ACS Macro Lett., 2017, 6, 1280–1284 CrossRef CAS.
- L. Chen, L. Zhang, P. J. Griffin and S. J. Rowan, Macromol. Chem. Phys., 2020, 221, 1900440 CrossRef CAS.
- M. K. McBride, B. T. Worrell, T. Brown, L. M. Cox, N. Sowan, C. Wang, M. Podgorski, A. M. Martinez and C. N. Bowman, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 175–198 CrossRef CAS PubMed.
- J. J. Cash, T. Kubo, D. J. Dobbins and B. S. Sumerlin, Polym. Chem., 2018, 9, 2011–2020 RSC.
- A. Breuillac, A. Kassalias and R. Nicolaÿ, Macromolecules, 2019, 52, 7102–7113 CrossRef CAS.
- Y. Spiesschaert, M. Guerre, I. De Baere, W. Van Paepeogem, J. M. Winne and F. E. Du Prez, Macromolecules, 2020, 53, 2485–2495 CrossRef CAS.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- H. Zhang, C. Cai, W. X. Liu, D. D. Li, J. W. Zhang, N. Zhao and J. Xu, Sci. Rep., 2017, 7, 11833 CrossRef.
- X. W. Xu, S. Q. Ma, S. Wang, J. H. Wu, Q. Li, N. Lu, Y. L. Liu, J. T. Yang, J. Feng and J. Zhu, J. Mater. Chem. A, 2020, 8, 11261–11274 RSC.
- Y. J. Liu, Z. H. Tang, D. Wang, S. W. Wu and B. C. Guo, J. Mater. Chem. A, 2019, 7, 26867–26876 RSC.
- L. Li, X. Chen, K. Jin, M. Bin Rusayyis and J. M. Torkelson, Macromolecules, 2021, 54, 1452–1464 CrossRef CAS.
- L. Li, Ph.D. thesis (Dynamic Chemistries in Non-linear Polymer Systems: From Applications for Sustainability to Fundamental Theories), Northwestern University, 2019.
- Z. Yang, Q. Wang and T. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 21691–21699 CrossRef CAS.
- G. Lopez, L. Granado, G. Coquil, A. Larez-Sosa, N. Louvain and B. Ameduri, Macromolecules, 2019, 52, 2148–2155 CrossRef CAS.
- B. Zhang, Z. A. Digby, J. A. Flum, E. M. Foster, J. L. Sparks and D. Konkolewicz, Polym. Chem., 2015, 6, 7368–7372 RSC.
- W. Liu, D. F. Schmidt and E. Reynaud, Ind. Eng. Chem. Res., 2017, 56, 2667–2672 CrossRef CAS.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS.
- Y. Zhou, R. Groote, J. G. P. Goossens, R. P. Sijbesma and J. P. A. Heuts, Polym. Chem., 2019, 10, 136–144 RSC.
- F. Caffy and R. Nicolay, Polym. Chem., 2019, 10, 3107–3115 RSC.
- M. O. Saed, A. Gablier and E. M. Terentejv, Adv. Funct. Mater., 2020, 30, 1906458 CrossRef CAS.
- Q. Li, S. Ma, S. Wang, Y. Liu, M. Abu Taher, B. Wang, K. Huang, X. Xu, Y. Han and J. Zhu, Macromolecules, 2020, 53, 1474–1485 CrossRef CAS.
- Y. Yang, F.-S. Du and Z.-C. Li, Polym. Chem., 2020, 11, 1860–1870 RSC.
- C. A. Tretbar, J. A. Neal and Z. Guan, J. Am. Chem. Soc., 2019, 141, 16595–16599 CrossRef CAS PubMed.
- H. Zhang, D. Wang, W. Liu, P. Li, J. Liu, C. Liu, J. Zhang, N. Zhao and J. Xu, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2011–2018 CrossRef CAS.
- P. Chakma, Z. A. Digby, J. Via, M. P. Shulman, J. L. Sparks and D. Konkolewicz, Polym. Chem., 2018, 9, 4744–4756 RSC.
- S. Wang, S. Ma and Q. Li, et al. , Macromolecules, 2020, 53, 2919–2931 CrossRef CAS.
- D. W. Hanzon, X. He, H. Yang, Q. Shi and K. Yu, Soft Matter, 2017, 13, 7061–7073 RSC.
- J. Ma, X. Mu, C. N. Bowman, Y. Sun, M. L. Dunn, H. J. Qi and D. Fang, J. Mech. Phys. Solids, 2014, 70, 84–103 CrossRef CAS.
- Q. Li, S. Ma, N. Lu, J. Qiu, J. Ye, Y. Liu, S. Wang, Y. Han, B. Wang, X. Xu, H. Feng and J. Zhu, Green Chem., 2020, 22, 7769–7777 RSC.
- S. Debnath, S. K. Tiwary and U. Ojha, ACS Appl. Polym. Mater., 2021, 3, 2166–2177 CrossRef CAS.
- N. Lu, Q. Li, S. Ma, B. Wang, X. Xu, S. Wang, J. Ye, J. Qiu and J. Zhu, Eur. Polym. J., 2021, 147, 110291 CrossRef CAS.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444–2450 CrossRef CAS PubMed.
- A. Carmine, Y. Domoto, N. Sakai and S. Matile, Chem. – Eur. J., 2013, 19, 11558–11563 CrossRef CAS PubMed.
- Z. Q. Lei, H. P. Xiang, Y. J. Yuan, M. Z. Rong and M. Q. Zhang, Chem. Mater., 2014, 26, 2038–2046 CrossRef CAS.
- A. Rekondo, R. Martin, A. Ruiz de Luzuriaga, G. Cabañero, H. J. Grande and I. Odriozola, Mater. Horiz., 2014, 1, 237–240 RSC.
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861–1872 RSC.
- L. Imbernon, E. K. Oikonomou, S. Norvez and L. Leibler, Polym. Chem., 2015, 6, 4271–4278 RSC.
- H. P. Xiang, H. J. Qian, Z. Y. Lu, M. Z. Rong and M. Q. Zhang, Green Chem., 2015, 17, 4315–4325 RSC.
- A. Ruiz de Luzuriaga, R. Martin, N. Markaide, A. Rekondo, G. Cabañero, J. Rodríguez and I. Odriozola, Mater. Horiz., 2016, 3, 241–247 RSC.
- I. Azcune and I. Odriozola, Eur. Polym. J., 2016, 84, 147–160 CrossRef CAS.
- L. Zhang, L. Chen and S. J. Rowan, Macromol. Chem. Phys., 2017, 218, 1600320 CrossRef.
- D. J. Fortman, R. L. Snyder, D. T. Sheppard and W. R. Dichtel, ACS Macro Lett., 2018, 7, 1226–1231 CrossRef CAS.
- M. Zhang, F. Zhao, W. Xin and Y. Luo, ChemistrySelect, 2020, 5, 4608–4618 CrossRef CAS.
- Q. Liu, Y. Liu, H. Zheng, C. Li, Y. Zhang and Q. Zhang, J. Polym. Sci., 2020, 58, 1092–1104 CrossRef CAS.
- L. Li, X. Chen and J. M. Torkelson, ACS Appl. Polym. Mater., 2020, 2, 4658–4665 CrossRef CAS.
- S. Sunner, Acta Chem. Scand., 1955, 9, 837–846 CrossRef CAS.
- J. A. Kerr, Chem. Rev., 1966, 66, 465–500 CrossRef CAS.
- I. Park, S. S. Sheiko, A. Nese and K. Matyjaszewski, Macromolecules, 2009, 42, 1805–1807 CrossRef CAS.
- D. Sakamaki, S. Ghosh and S. Seki, Mater. Chem. Front., 2019, 3, 2270–2282 RSC.
- H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef.
- A. V. Tobolsky, W. J. MacKnight and M. Takahashi, J. Phys. Chem., 1964, 68, 787–790 CrossRef CAS.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975–3980 CrossRef CAS PubMed.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- S. Nevejans, N. Ballard, J. I. Miranda, B. Reck and J. M. Asua, Phys. Chem. Chem. Phys., 2016, 18, 27577–27583 RSC.
- W. C. Danen and D. D. Newkirk, J. Am. Chem. Soc., 1976, 98, 516–520 CrossRef CAS.
- B. Maillard and K. U. Ingold, J. Am. Chem. Soc., 1976, 98, 520–523 CrossRef CAS.
- A. Takahashi, R. Goseki and H. Otsuka, Angew. Chem., Int. Ed., 2017, 56, 2016–2021 CrossRef CAS.
- M. Bin Rusayyis and J. M. Torkelson, Macromolecules, 2020, 53, 8367–8373 CrossRef CAS.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, Ithaca, United States, 1953 Search PubMed.
- B. R. Elling and W. R. Dichtel, ACS Cent. Sci., 2020, 6, 1488–1496 CrossRef CAS PubMed.
- K. P. Menard, Dynamic mechanical analysis: A practical introduction, CRC Press, Boca Raton, United States, 2008 Search PubMed.
- D. J. T. Hill, M. C. S. Perera, P. J. Pomery and H. K. Toh, Polymer, 2000, 41, 9131–9137 CrossRef CAS.
- J. F. McCabe and H. Arikawa, J. Dent. Res., 1998, 77, 1874–1880 CrossRef CAS PubMed.
- C. A. Tweedie and K. J. Van Vliet, J. Mater. Res., 2006, 21, 1576–1589 CrossRef CAS.
- A. Jourdain, R. Asbai, O. Anaya, M. M. Chehimi, E. Drockenmuller and D. Montarnal, Macromolecules, 2020, 53, 1884–1900 CrossRef CAS.
- M. Aiba, T. Koizumi, M. Futamura, K. Okamoto, M. Yamanaka, Y. Ishigaki, M. Oda, C. Ooka, A. Tsuruoka, A. Takahashi and H. Otsuka, ACS Appl. Polym. Mater., 2020, 2, 4054–4061 CrossRef CAS.
- Q. Ge, Y. Kai, Y. Ding and H. J. Qi, Soft Matter, 2012, 8, 11098–11105 RSC.
- J. C. Hooker and J. M. Torkelson, Macromolecules, 1995, 23, 7683–7692 CrossRef.
- M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, New York, United States, 2003 Search PubMed.
- A. Dhinojwala, J. C. Hooker and J. M. Torkelson, J. Non-Cryst. Solids, 1994, 172–174, 286–296 CrossRef CAS.
- D. B. Hall, A. Dhinojwala and J. M. Torkelson, Phys. Rev. Lett., 1997, 79, 103–106 CrossRef CAS.
- Z. Wang, B. A. Sun, H. Y. Bai and W. H. Wang, Nat. Commun., 2014, 5, 5823 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00187f |
| This journal is © The Royal Society of Chemistry 2021 |