
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWell-defined polyvinylpyridine-block-polystyrene diblock copolymers via RAFT aqueous-alcoholic dispersion polymerization: synthesis and isoporous thin film morphology†
Katharina
Nieswandt
 a,
Prokopios
Georgopanos
*a and
Volker
Abetz
*ab
a,
Prokopios
Georgopanos
*a and
Volker
Abetz
*ab
aHelmholtz-Zentrum Geesthacht, Institute of Membrane Research, Max-Planck-Straße 1, 21502 Geesthacht, Germany. E-mail: prokopios.georgopanos@hzg.de; volker.abetz@hzg.de
bInstitute of Physical Chemistry, University of Hamburg, Martin-Luther-King-Platz 6, 20146 Hamburg, Germany
First published on 15th March 2021
Abstract
In this work, the synthesis and characterization of polyvinylpyridine-polystyrene (PVP-b-PS) diblock copolymers via reversible addition–fragmentation chain transfer (RAFT) dispersion polymerization is presented. A series of poly(4-vinylpyridine) (P4VP) and poly(2-vinylpyridine) (P2VP) homopolymers were prepared by RAFT bulk polymerization at 80 °C using either a carboxylic acid functionalized trithiocarbonate or a non-functionalized trithiocarbonate RAFT agent. The P4VP and P2VP macroRAFT agents acted as stabilizers in the subsequent RAFT-mediated polymerization-induced self-assembly (PISA), when they were chain extended via RAFT aqueous-alcoholic dispersion polymerization of styrene at 70 °C. Following this protocol, high styrene conversions between 86–99% were achieved, leading to a series of well-defined, high molecular weight PVP-b-PS diblock copolymers with narrow molecular weight distributions as confirmed by proton nuclear magnetic resonance (1H NMR) spectroscopy and gel permeation chromatography (GPC). The bulk and surface morphologies of the diblock copolymers were investigated via transmission electron microscopy (TEM), atomic force microscopy (AFM), and scanning electron microscopy (SEM). The solution-cast and spin-coated thin films of the P4VP-b-PS and P2VP-b-PS diblock copolymers exhibited microphase-separated structures with spherical or cylindrical order according to their compositions. Spin-coated PVP-b-PS diblock copolymer films with weight fractions of the minority PVP block ranging from 17–24% were converted into porous surfaces by a controlled alignment and swelling strategy, exploiting the distinct selectivities of different solvents for the individual blocks.
Introduction
Over the past decades, controlled reversible deactivation radical polymerization (RDRP) has been developed into a versatile and widely used technique to synthesize well-defined polymers.1–3 Nowadays, it is an established method of choice to design precisely structured block copolymers via radical polymerization.4,5 Initially, RDRP techniques, including the extensively studied atom transfer radical polymerization (ATRP), nitroxide-mediated polymerization (NMP) and reversible addition–fragmentation chain transfer (RAFT) polymerization, were conducted as homogeneous polymerizations in bulk, organic solvents, or water.6–8 Although such polymerizations are still of high interest today, the well-controlled polymerization of slowly-propagating radicals, such as resonance-stabilized styrenics, appeared challenging for decades.9 Since a long time, polystyrene latexes are produced mostly by free radical heterogeneous polymerizations stabilized and controlled by surfactants as emulsifiers.10–12 Those polymer colloids dispersed in aqueous systems are formed by free radical emulsion (or ‘latex’) polymerizations.13 Commonly, they are stabilized by a surfactant, which stabilizes the emulsion at the beginning and the monomer-swollen micelles as well as the latex particles towards the end of the polymerization.7,14 However, on the one hand, surfactants may separate from the particles, which may lead to particle coagulation. On the other hand, it is difficult to remove the surfactant completely from the final polymer. Surfactant-free emulsion polymerization systems were developed in the 1970s in order to avoid the adverse side effects that arose from surfactant-controlled emulsion polymerizations.15 This polymerization technique, which gained importance during the Second World War, has developed into a global industry.13 Nowadays, the combination of RDRP methods with emulsion or dispersion polymerization techniques is a feasible approach to produce research and industrially relevant copolymers with predetermined molecular weights and architectures.16,17 These methods provide efficient heat transfer, low viscosity during the entire reaction time and enable the production of well-defined polymers.14,16–18Dispersion polymerization initially involves a completely homogeneous solution.19–22 However, the resultant polymer is not soluble in the polymerization medium.7,23 In RAFT dispersion polymerization, a previously synthesized soluble homopolymer, a so-called macromolecular chain transfer agent (macroRAFT agent), is used as the stabilizer of the following dispersion polymerization. Initially, the second block grows continuously in the homogenous phase. With increasing length of the second block, the forming amphiphilic diblock copolymer chains become increasingly insoluble in the reaction medium. Once a critical block length exceeds the critical micelle degree of polymerization (CMDP), the amphiphilic diblock copolymers self-assemble into sterically stabilized spherical micelles.7 This phase separation process is called polymerization-induced self-assembly (PISA) and is caused by the insolubility of the second block in the polymerization medium during the continuous chain extension.24–28 Especially for the commonly slow polymerization of monomers like styrene,29 PISA has proven to be a viable approach to accelerate RAFT polymerizations.30–32 Over the years, PISA has evolved into a versatile technique for the efficient synthesis of block copolymer nanoparticles.33–36 In recent years, numerous polymerization protocols for aqueous emulsions, aqueous dispersions or non-aqueous polymerizations have been developed. In particular, RAFT formulations in alcoholic media have received considerable attention.37–39 PISA formulations in pure alcoholic media often showed incomplete monomer conversions and slow reaction rates. Several research groups investigated the influence of water, as co-solvent, on alcoholic PISA formulations. Charleux et al. studied the polymerization of benzyl methacrylate (BzMA) in dispersion polymerizations when using a poly(methacrylic acid-co-poly(ethylene oxide) monomethyl ether methacrylate) (P(MAA-b-PEOMA)) macroRAFT agent.40 Similarly, Zhang et al. investigated the dispersion polymerization of styrene in ethanol/water using a 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (BDMAT) RAFT agent.41 Their results showed that the use of water as co-solvent in alcoholic media significantly increased the polymerization rates. Based on these findings, Armes et al. examined the chain extension of poly(2-(dimethylamino)ethyl methacrylate) (PDMA) macroRAFT agent with benzyl methacrylate (BzMA) in ethanol/water mixtures and also found a significant increase in reaction rates.42 They attributed this increase to the insolubility of the monomer in water, which then preferentially enters the growing particles, increasing the local monomer concentration and thus increasing the rate of polymerization.40,42 Furthermore, water as a co-solvent reduces the solubility of the growing polymer chains, which lowers the CMDP. The initial (slow) homogeneous polymerization turns more rapidly into a faster dispersion polymerization.40–42
During the past years, there has been a growing interest in nanostructured materials for technological applications, such as nanostructured membranes, templates and data storage media. Amongst others, near-monodisperse diblock copolymers, which consist of two incompatible blocks, were brought into focus.43–47 Sriprom et al. studied the microphase separation of diblock copolymers synthesized via RAFT polymerization, containing one monodisperse (poly(methyl methacrylate) (PMMA) block and one polydisperse (poly(butyl acrylate) (PBA) block, in thin films.48 The PMMA-b-PBA diblock copolymers microphase-separated into regularly ordered domains with high reproducibility and long-range order. However, they observed that an increase in dispersity led to a shift of the morphological transitions to more asymmetric volume fractions, in comparison to a monodisperse system. Essentially the same observations were previously reported by Hillmyer and co-workers.49 Their predictions suggested that the domain spacing increased as the dispersity of one block was increased, while the dispersity of the other block remained constantly narrow.
In order to control the orientation of the block copolymer microdomains precisely, numerous techniques have been developed.50–53 Especially methods that rely on external stimuli are important since they do not alter the chemistry of the block copolymer components on a molecular level.54 External constraints that eliminate orientational and topological defects of self-assembled block copolymers could be temperature gradients, solvent annealing or shear.55 In particular, extensively studied solvent vapor annealing presents an effective and non-destructive way to influence the orientation of block copolymer microdomains in thin films.47,51,56,57
A convenient approach to synthesize medium molecular weight poly(3-vinylpyridine)-b-polystyrene via RAFT emulsion polymerization and their thin film morphology were presented before.18 In the present study, poly(4-vinylpyridine) (P4VP) and poly(2-vinylpyridine) (P2VP) were employed as macroRAFT agents. Here, a two-step synthetic route to poly(4-vinylpyridine)-b-polystyrene and poly(2-vinylpyridine)-b-polystyrene is described via a RAFT aqueous-alcoholic dispersion polymerization, which takes advantage of the PISA approach. The first block contains 4VP or 2VP polar moieties, which are expected to stabilize the dispersion polymerization and to enable PISA. The two isomers P2VP and P4VP are characterized in their chemical behavior by the functionality of nitrogen within the aromatic system.58 Simple alcohols such as methanol tend to dissolve both isomers quite well due to the formation of hydrogen bonds, whereas polystyrene is not dissolved in common alcohols. The RAFT dispersion polymerization used in this work takes advantage of these different solubilities. The trithiocarbonate terminated P4VP/P2VP block is synthesized via RAFT bulk polymerization in the first step at 80 °C, followed by a surfactant-free RAFT dispersion polymerization of styrene at 70 °C in an 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 methanol/water mixture. For this purpose, either a carboxylic acid functionalized trithiocarbonate or a non-functionalized trithiocarbonate RAFT agent were used to investigate the effect of the end group functionality on PISA and the dispersion stability. Since PISA is a promising way to generate accurately tunable diblock copolymer compositions and thus a variety of accessible diblock copolymer morphologies, the synthesized P4VP-b-PS and P2VP-b-PS (for the sake of simplicity PVP-b-PS) diblock copolymers were investigated with respect to their film formation behavior. The PVP-b-PS diblock copolymers were characterized by gel permeation chromatography (GPC) and proton nuclear magnetic resonance (1H NMR) spectroscopy. Their bulk and film surface morphologies were analyzed via transmission electron microscopy (TEM), atomic force microscopy (AFM) and scanning electron microscopy (SEM), respectively. To further investigate the effects on the surface morphology of the diblock copolymer in the thin films, thermal annealing was combined with solvent annealing. Distinct selectivities of different solvents for the individual blocks made it possible to obtain different types of porous surface structures.
:20 methanol/water mixture. For this purpose, either a carboxylic acid functionalized trithiocarbonate or a non-functionalized trithiocarbonate RAFT agent were used to investigate the effect of the end group functionality on PISA and the dispersion stability. Since PISA is a promising way to generate accurately tunable diblock copolymer compositions and thus a variety of accessible diblock copolymer morphologies, the synthesized P4VP-b-PS and P2VP-b-PS (for the sake of simplicity PVP-b-PS) diblock copolymers were investigated with respect to their film formation behavior. The PVP-b-PS diblock copolymers were characterized by gel permeation chromatography (GPC) and proton nuclear magnetic resonance (1H NMR) spectroscopy. Their bulk and film surface morphologies were analyzed via transmission electron microscopy (TEM), atomic force microscopy (AFM) and scanning electron microscopy (SEM), respectively. To further investigate the effects on the surface morphology of the diblock copolymer in the thin films, thermal annealing was combined with solvent annealing. Distinct selectivities of different solvents for the individual blocks made it possible to obtain different types of porous surface structures.
Experimental
Materials
Experiments were carried out with Ultrapure water (resistivity >18.2 MΩ cm−1) obtained from a Millipore (Merck, Germany) Direct-Q® UV water purification system. 2,2′-Azobis(2-methylpropionitrile) (AIBN, 98%, Sigma-Aldrich, Germany, stored at 4 °C), 4-cyano-4-[(dodecylsulfanylthiocarbonyl)-sulfanyl]pentanoic acid (CDTPA, 97%, Sigma-Aldrich, stored at 4 °C), 2-cyano-2-propyl dodecyl trithiocarbonate (CPDTC, 97%, Sigma-Aldrich, stored at 4 °C), chloroform (CHCl3, ≥99.8%, VWR, Germany), N,N-dimethylacetamide (DMAc, ≥99.9%, Sigma-Aldrich), methanol (99.9%, Sigma-Aldrich), ethanol (≥99.8%, Sigma-Aldrich), 2-propanol (≥99.5%, Sigma-Aldrich), tetrahydrofuran (THF, 99.8%, Merck), 1,4-dioxane (≥99.5%, Sigma-Aldrich), N,N-dimethylformamide (DMF, >99.5%, Merck) and iodine (I2, 99.5%, VWR) were used as received. Styrene (99%, Sigma-Aldrich, contained methyl ether hydroquinone as an inhibitor, stored at 4° C) was freshly percolated through a column of basic aluminum oxide (>98%, Sigma-Aldrich) prior to use to remove the inhibitor. 4-Vinylpyridine (4VP) (97%, Sigma-Aldrich) and 2-vinylpyridine (2VP) (97%, Sigma-Aldrich) were dried over CaH2 (≥95%, Sigma-Aldrich) overnight and distilled under reduced pressure prior to use.![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n,app = 20 kDa, (theoretical number average molecular weight as calculated by 1H NMR: n,th = 20 kDa), molecular weight dispersity: Đ = 1.08.
n,app = 20 kDa, (theoretical number average molecular weight as calculated by 1H NMR: n,th = 20 kDa), molecular weight dispersity: Đ = 1.08.
The RAFT polymerization of 4VP with CPDTC was performed similarly. Monomer conversion: 62%, GPC: n,app = 16 kDa (n,th = 18 kDa), Đ = 1.08.
n,app = 104 kDa (n,th = 119 kDa), Đ = 1.29.
n,app = 21 kDa (n,th = 25 kDa), Đ = 1.26. The RAFT polymerization of 2VP with CPDTC was performed accordingly. Monomer conversion: 53%, GPC: n,app = 21 kDa (n,th = 25 kDa), Đ = 1.25.
n,app = 109 kDa (n,th = 93 kDa), Đ = 1.06.
Characterization
The conversion of 4VP was determined in CDCl3 by 1H NMR from the integral ratio of the aromatic P4VP signal at 8.48–8.09 ppm and the monomer signal at 8.55 ppm. The conversion of styrene in the dispersion polymerization, determined in THF-d8, was calculated from the decrease of the integral of the monomer peaks. Therefore a reference sample was taken prior to and at the end of the polymerization. The aromatic P4VP signal at 8.48–8.09 ppm was used as a reference. The conversion in P2VP and P2VP-b-PS experiments was estimated analogously.
Additional, thermal annealing of the samples was conducted at a temperature Tannealing = 180 °C in vacuum for 15 h. For the topography microphase reconstruction two different methods were followed: (a) the spin-coated samples were exposed for 10 min in 1,4-dioxane vapor at room temperature followed by dip-coating in ethanol for 5 min and drying under vacuum at room temperature for 10 h and (b) the samples were dip-coated in dimethylformamide for 3 s, exposed to air for 5 min and subsequently immersed in water for 3 h followed by air exposure for 10 h.
Results and discussion
For RAFT-mediated dispersion polymerization, the choice of the macroRAFT agent and solvent are crucial in order to polymerize a certain monomer. The macroRAFT agent acts as a steric stabilizer and plays a key role in keeping the system stable and obtaining monodisperse block copolymers. In the present work, RAFT dispersion polymerizations of styrene were conducted using PVP macroRAFT agents. The PVP-b-PS diblock copolymers were prepared via a two-step RAFT polymerization (Fig. 1). Either CDTPA or CPTTC-terminated PVP was synthesized first via bulk polymerization and then used as the macroRAFT agent in the dispersion polymerization of styrene, forming the desired PVPk-PSm diblock copolymers with k and m being the mean degrees of polymerization (DPs) of PVP and PS, respectively. An 80:20 methanol/water mixture was selected as the continuous phase for these RAFT dispersion polymerization formulations.
 | ||
| Fig. 1 (a) Exemplary synthetic route to P4VPk-PSm diblock copolymers using the CDTPA RAFT agent. Synthesis of the P4VP-CDTPA macroRAFT agent by RAFT bulk polymerization followed by the synthesis of P4VPk-PSmvia surfactant-free RAFT dispersion polymerization in methanol/water (80/20, v/v). The synthesis of P2VPk-PSm was conducted accordingly with a P2VP-CDTPA macroRAFT precursor; (b) chemical structure of the two trithiocarbonate RAFT agents used in this study. | ||
RAFT bulk polymerization and surfactant-free RAFT dispersion polymerization
The RAFT bulk polymerizations were stopped at monomer conversions <70% to maintain a high fidelity of the trithiocarbonate functionality and avoid the occurrence of an excess of termination products from coupling reactions.60,61 Moreover, early termination facilitates further processing by prevention of excessive viscosity of the bulk sample. The two homopolymers P4VP and P2VP vary in molecular weight as well as the RAFT end-group. One aim of this study was to identify suitable RAFT agents for the subsequent PISA with styrene through RAFT-mediated dispersion polymerization. Table 1 summarizes the PVP homopolymers and their respective analytical data.| Homopolymera |
n,app
[kDa] |
w,app
[kDa] |
Conv. [%] |
n,th
[kDa] |
Đ | RAFT agent |
|---|---|---|---|---|---|---|
|
a Superscripts denote the mean DPs as determined by 1H NMR spectroscopy.
b The apparent molecular weights n,app and w,app were determined by DMAc GPC calibrated with PS standards.
c
n,th was calculated as follows: n,th = [monomer]/[RAFT] × Mmonomer × monomer conversion + MRAFT. M: molecular weight.
|
||||||
| P4VP186 | 20 | 22 | 69 | 20 | 1.08 | CDTPA |
| P4VP168 | 16 | 18 | 62 | 18 | 1.08 | CPDTC |
| P2VP234 | 21 | 27 | 53 | 25 | 1.26 | CDTPA |
| P2VP234 | 21 | 26 | 53 | 25 | 1.24 | CDTPA |
| P2VP234 | 21 | 26 | 53 | 25 | 1.25 | CPDTC |
| P2VP215 | 19 | 23 | 52 | 23 | 1.20 | CDTPA |
| P2VP186 | 20 | 25 | 46 | 20 | 1.27 | CDTPA |
For the following RAFT dispersion polymerization (Fig. 1), the P4VP macroRAFT agent was dissolved in an 80:20 methanol/water mixture. The comparison of the Hansen solubility parameters shows that the solubility of styrene is higher in methanol than in water (Table S1†). However, the styrene solubility in the chosen reaction medium is kept sufficiently high to justify a true dispersion polymerization mechanism, which initially proceeds homogeneously until a critical PS block length is exceeded. More importantly, water as a co-solvent significantly worsens the solvency for the growing polystyrene chains.41 This lowers the critical mean DP required for micellar nucleation. The initial (slow) homogeneous polymerization more rapidly turns into a faster dispersion polymerization, in which the polymerization occurs within the monomer-swollen micelles.40,42
Characterization of the PVP-b-PS diblock copolymers via1H NMR and GPC
P4VP/P2VP served as macroRAFT agent and macro-stabilizer in the RAFT dispersion polymerization of styrene in methanol/water (80/20, v/v) and AIBN as the initiator of the reaction. Styrene, as well as the P4VP macroRAFT agent and AIBN are soluble in the 80:20 methanol/water mixture. Thus, the polymerization, or more precisely the chain extension of PVP with styrene, starts homogeneously until a critical PS block length is exceeded and the PVP-b-PS chains start to self-assemble into micelles. In the dispersion polymerization of styrene, a quantitative monomer conversion (93–99%) was reached, as indicated by 1H NMR. The apparent number-average molecular weights (n,app) of the PVP-b-PS diblock copolymers were obtained via GPC. The GPC curves of the P4VP-b-PS diblock copolymers reveal that the curves corresponding to the diblock copolymers significantly shift towards higher molecular weights compared to the respective macroRAFT agents (Fig. 2). Additionally, the GPC traces show dispersities of Đ = 1.14–1.29, which indicate well-controlled RAFT dispersion polymerizations. This is further evidenced by the observation of hardly any homopolymer impurities in the GPC traces. The PS fraction in the P4VP-b-PS diblock copolymers is approximately 79–83% by weight as obtained by 1H NMR. The respective diblock copolymers as well as 1H NMR and GPC data are depicted in Table 2.
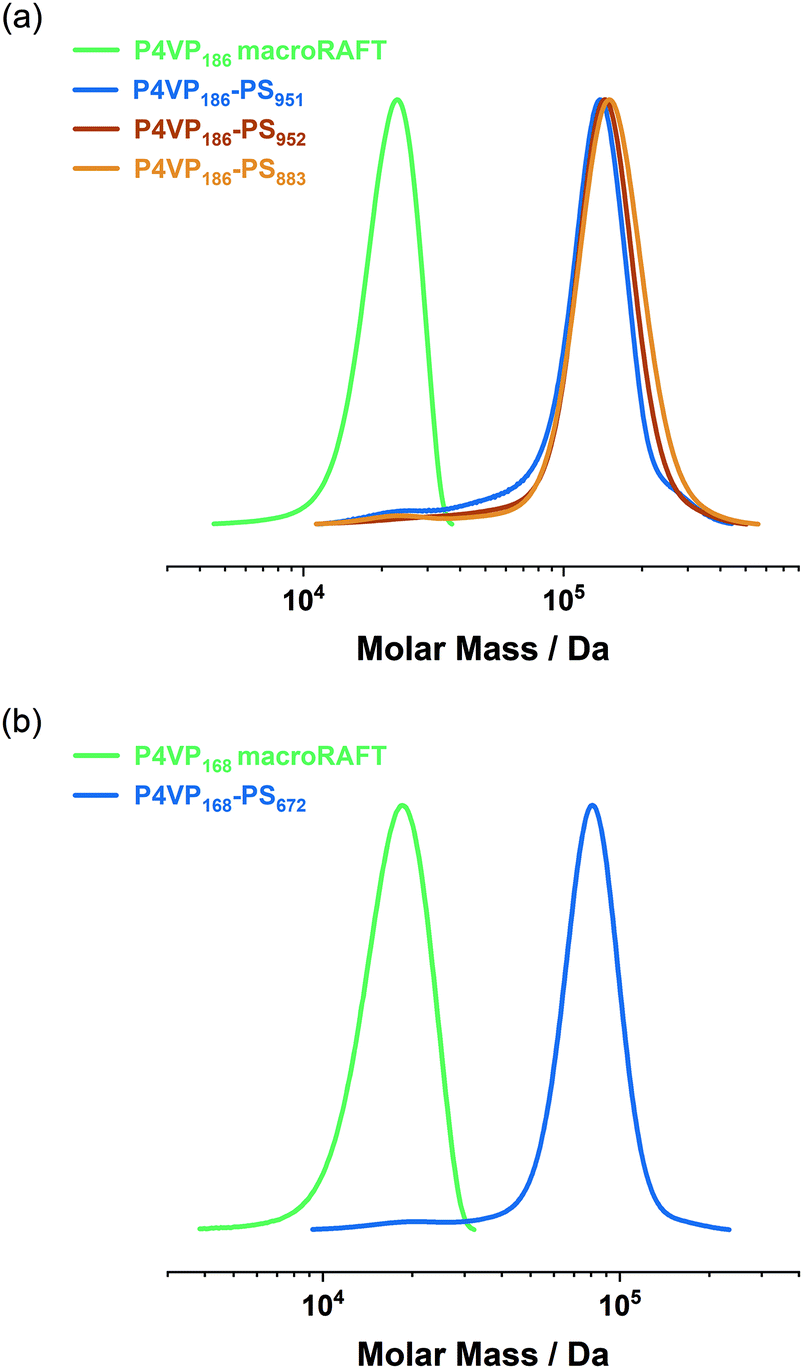 | ||
| Fig. 2 GPC curves recorded for (a) a P4VP186 macroRAFT agent and the corresponding P4VP186-PSm diblock copolymers. The shift of the diblock copolymer curves towards higher molecular weights indicates the successful RAFT dispersion polymerization, (b) a P4VP168 macroRAFT agent and the corresponding P4VP168-PSm diblock copolymer. A well-controlled RAFT dispersion polymerization is indicated by low dispersities (Đ = 1.14–1.29) of the final P4VP-b-PS diblock copolymers. | ||
| PVP-PS compositiona |
n,app
[kDa] |
w,app
[kDa] |
Conv. [%] |
n,th
[kDa] |
Đ | f PVP [wt%] | RAFT agent |
|---|---|---|---|---|---|---|---|
|
a Superscripts denote the mean DPs as determined by 1H NMR spectroscopy.
b Determined by DMAc GPC calibrated with PS standards. For P2VPk-PSm, the GPC curve peaks were analysed separately and assigned as precursor and diblock copolymer peak.
c
n,th was calculated as follows: n,th = [monomer]/[macroRAFT] × Mmonomer × monomer conversion + MmacroRAFT.
|
|||||||
| P4VP186-PS951 | 104 | 135 | 99 | 119 | 1.29 | 17 | CDTPA |
| P4VP186-PS952 | 122 | 147 | 99 | 120 | 1.20 | 17 | CDTPA |
| P4VP168-PS672 | 70 | 79 | 99 | 88 | 1.14 | 21 | CPDTC |
| P4VP186-PS883 | 125 | 155 | 93 | 112 | 1.24 | 19 | CDTPA |
| P2VP234-PS768 | 137 | 152 | 87 | 105 | 1.11 | 24 | CDTPA |
| P2VP234-PS663 | 130 | 144 | 87 | 94 | 1.11 | 27 | CDTPA |
| P2VP234-PS739 | 136 | 150 | 87 | 102 | 1.11 | 24 | CDTPA |
| P2VP234-PS566 | 119 | 131 | 86 | 84 | 1.10 | 29 | CPDTC |
| P2VP215-PS672 | 109 | 116 | 91 | 93 | 1.06 | 24 | CDTPA |
| P2VP215-PS595 | 92 | 97 | 86 | 85 | 1.06 | 27 | CDTPA |
The RAFT agent, which contains a dodecyl end-group on the ω-terminal-end of the polymer chain and a carboxylic acid group as well as a cyano group on the α-terminal end, is slightly more hydrophilic compared to the RAFT agent with similar chain length but only a cyano-group on the α-terminal end. To quantify this statement, the n-octanol–water partition coefficient logP, a measure of the hydrophilicity of a chemical compound, is determined. It is negative for polar substances and positive for non-polar substances. The values obtained were calculated logP (ClogP) values using ChemDraw 13.0 (PerkinElmer, United States of America). The ClogP for CDTPA was calculated as 6.76, while the ClogP for CPDTC was determined to be 7.64. Hence, CDTPA is slightly more hydrophilic than CPDTC. Surprisingly, exchanging the carboxylic acid on the α-terminal end with a less hydrophilic residue does not significantly influence PISA. Hence, the RAFT dispersion polymerizations of styrene with CDTPA- or CPDTC-terminated macroRAFT agents exhibit similar monomer conversions. The difference between the monomers 4VP and 2VP, especially regarding the molar masses and dispersities achieved in the homopolymerizations, is probably more decisive than the choice of RAFT agent regarding the two trithiocarbonates to be compared. If 4VP is replaced by 2VP, an isomer that differs only in the position of the nitrogen atom, the homopolymerization results in approximately 10% lower monomer conversions. From GPC traces (Fig. 3), it becomes evident that the results for P2VP-b-PS are similar compared to the P4VP-b-PS experiments. However, for P2VP234-PS663 and P2VP234-PS739 there are a residual amount of P2VP234 macroRAFT agent in the diblock copolymer GPC traces. This homopolymer impurity, which accounts for approximately 5% of the total polymer, can probably be attributed to unreacted macroRAFT agent chains. For other P2VP215-PS672 and P2VP215-PS595 diblock copolymers, hardly any macroRAFT residues were visible in the GPC traces. When evaluating the chromatograms quantitatively, however, it becomes apparent that the P2VP-b-PS diblock copolymer peaks on their own show very low dispersities (Đ = 1.06–1.11) and, due to styrene conversions of almost 90%, as indicated by 1H NMR, in most cases, also exhibit high molecular weights of over 100 kDa (Table 2).
 | ||
| Fig. 3 GPC curves recorded for (a) a P2VP234 macroRAFT agent and the corresponding P2VP234-PSm diblock copolymers; (b) a P2VP215 macroRAFT agent and the corresponding P2VP215-PSm diblock copolymers. | ||
To summarize, the chosen RAFT aqueous-alcoholic dispersion polymerization method is suitable for producing high molecular weight diblock copolymers under simple reaction conditions in acceptable reaction times. This synthetic route, which does not require surfactants or other additives, not only demonstrates the preparation of PVP-b-PS diblock copolymers with molecular weights above 130 kDa (n,app), but also enables the synthesis of relatively narrow molecular weight distributed polymers with monomer conversions of 86–99%. Hence, this RAFT-mediated PISA approach enables the synthesis of diblock copolymers in molecular weight ranges and qualities that are normally only obtained by anionic polymerization methods.62–64
Morphological investigation of PVP-b-PS diblock copolymers
Fig. 4 shows TEM images of the P4VP-b-PS and P2VP-b-PS diblock copolymers. For P4VP186-PS951 with a P4VP weight fraction of 17% P4VP and an n,th of 119 kDa a spherical morphology of P4VP spheres in a continuous PS matrix was identified. The sample P4VP168-PS672, with an increased P4VP weight fraction of 21% and a lower n,th of 88 kDa, shows the coexistence of hexagonally arranged cylinders and spheres (Fig. 4c and d). Areas with perpendicularly and parallel cut cylinders are observed elongated and oriented in a mutual direction. Considering the different sample positions and sections, spherical areas were identified as directly adjacent to the cylindrical areas. Due to their size and arrangement, the perpendicularly cut cylinders could be easily distinguished from the spheres, which are smaller and do not show a hexagonal arrangement. For a P2VP215-PS672 bulk film, with a P2VP weight fraction of 24% and a total n,th of 93 kDa, a cylindrical morphology with hexagonally arranged P2VP cylinders was observed, with the cylinders found parallel and perpendicularly oriented to the plane (Fig. 4e and f). The results show, that the homopolymer impurities do not alter the morphologies of the PVP-b-PS diblock copolymers.
 | ||
| Fig. 4 TEM images of the PVP-b-PS bulk films. The bright areas correspond to the polystyrene microphase in the TEM images. (a and b) TEM image of a P4VP186-PS951 bulk structure; (c and d) TEM image of a P4VP168-PS672 bulk structure; (e and f) TEM image of a P2VP215-PS672 bulk structure. | ||
Fig. 5 shows six AFM QNM height images of pristine and annealed spin-coated diblock copolymer films. Pristine spin-coated films of the diblock copolymers show a relatively flat but microphase-separated surface, with the respective morphology depending on both layer thickness and copolymer composition. However, all of the pristine sample films show not perfectly delimited microstructures. The PS phase (Fig. 5a, d and g) appears bright and the PVP phase dark. Upon thermal annealing, the chain mobility is increased and the surface is flattened (Fig. S3 and S4†). Additional AFM images were taken after the two-step solvent annealing process. The AFM height image of a thin spin-coated P4VP186-PS951 film, with a P4VP weight fraction of 17%, shows distinct spherical P4VP microdomains in a continuous PS matrix after 1,4-dioxane vapor annealing, which enhanced the microphase separation and diminished defects in the microstructure of the diblock copolymer. The PS phase (Fig. 5b, e and h) appears dark and PVP bright, due to swelling of the phase. Subsequent dip-coating in ethanol generated a porous spherical microstructure due to the collapse of the P4VP microphase. The PS phase (Fig. 5c, f and i) appears bright, the pores appear dark and the PVP phase, which is collapsed, appears as a bright ring surrounding the pores. For a chosen diblock copolymer, with a higher P4VP weight fraction of 21% (P4VP168-PS672) and a lower n,th of 88 kDa, a cylindrical microstructure was found in AFM height images after solvent annealing. Dip-coating in ethanol produced a porous cylindrical surface microstructure. Regarding P2VP-b-PS, a diblock copolymer with a P2VP weight fraction of 24% and a total n,th of 93 kDa (P2VP215-PS672), shows a porous cylindrical microstructure after the two-step solvent annealing process. Fig. 6 shows SEM images of the PVP-b-PS diblock copolymer thin films. The images were taken of the pristine films, after therma l annealing followed by 1,4-dioxane vapor annealing and after final dip-coating in ethanol. The PS phase (Fig. 6) appears dark and the PVP phase bright. The brighter rings surrounding the pores (Fig. 6c, f and i) can be assigned to the electron-rich iodine, which selectively stained the PVP phase.71 Due to its high atomic number (53), iodine leads to strong backscattering of electrons.59 Although SEM and AFM produce similar representations of the thin film surface structure, SEM enabled to capture a large area view of the surface structure at once, while AFM only allowed to view 1 μm × 1 μm areas. Hence, SEM images confirmed that large-scale porous surface structures were created by the surface reconstruction annealing process. In order to further investigate the impact of solvent annealing, to control the alignment of the thin film topography, as well as to obtain a modified isoporous topographical structure, a further annealing technique was developed. Based on results from the structure formation of isoporous membranes by non-solvent induced phase separation (NIPS),72,73 a process partially imitating NIPS was followed. To investigate the effects of solvent exchange on the topography, the thin film on the Si-wafer was first dip-coated in a solvent {selective for one block} and then immersed into a non-solvent for both blocks. More precisely, the thermally annealed diblock copolymer thin film was dip-coated in DMF for 3 seconds, dried for 5 minutes in the presence of air and finally immersed into a water bath for 3 hours. Dry DMF was found to completely dissolve the thin film within 3 s. Therefore, water was added to DMF in amounts of 5–12 vol%. A DMF/water mixture of 92/8 v/v led to the best results regarding pore formation and was used for further experiments. Dip-coating in DMF, which is a good solvent for both blocks of the diblock copolymer but a selective solvent for the PVP block, {according to the solubility parameters of the solvents and blocks74,75} leads to highly swollen, soft PVP domains. Upon immersion in water, a non-solvent for both blocks, the initial microstructure is conserved and the PVP chains collapse with their domains forming open pores.72 According to the previous assumption, the AFM height image of a DMF/water annealed P4VP186-PS951 film shows an isoporous spherical microstructure with more uniformly arranged pores (Fig. 7) compared to a 1,4 dioxane/ethanol annealed P4VP186-PS951 film (Fig. 5 and 6). The diblock copolymer (Fig. 7) appears grey (SEM) or bright (AFM) while the pores appear dark.
 | ||
| Fig. 5 Surface topography studies via QNM AFM height images (1 μm × 1 μm) of spin-coated thin films of (a) P4VP186-PS951 pristine, (b) P4VP186-PS951 thermally annealed at 180 °C followed by 10 min 1,4-dioxane vapor annealing, (c) P4VP186-PS951 after thermal annealing at 180 °C followed by 10 min 1,4-dioxane vapor annealing and subsequent dip-coating in ethanol for 5 min; (d) P4VP168-PS672 pristine; (e) P4VP168-PS672 thermally and 1,4-dioxane vapor annealed; (f) P4VP168-PS672 dip-coated in ethanol; (g) P2VP215-PS672 pristine; (h) P2VP215-PS672 thermally and 1,4-dioxane vapor annealed; (i) P2VP215-PS672 dip-coated in ethanol. The PS phase appears bright. | ||
 | ||
| Fig. 6 SEM images (EsB detector) of spin-coated thin films of a 2 wt% PVP-b-PS CHCl3 solution prior to and after thermal combined with solvent annealing. (a) P4VP186-PS951 pristine spin-coated thin film, (b) P4VP186-PS951 thermally annealed at 180 °C followed by 10 min 1,4-dioxane vapor annealing, (c) P4VP186-PS951 after thermal annealing at 180 °C followed by 10 min 1,4-dioxane vapor annealing and subsequent dip-coating in ethanol for 5 min; (d) P4VP168-PS672 pristine; (e) P4VP168-PS672 thermally and 1,4-dioxane vapor annealed; (f) P4VP168-PS672 dip-coated in ethanol; (g) P2VP215-PS672 pristine; (h) P2VP215-PS672 thermally and 1,4-dioxane vapor annealed; (i) P2VP215-PS672 dip-coated in ethanol. PS appears as dark grey, P4VP as light grey and the pores appear black. In (c), (f) and (i) the brighter rings around the pores can be assigned to the P4VP phase stained by the electron rich iodine. | ||
 | ||
| Fig. 7 AFM and SEM studies of spin-coated thin films of a 2 wt% PVP-b-PS CHCl3 solution. The films were dip-coated in DMF for 3 s, exposed to air for 5 min and immersed in water for 3 h. Surface topography via QNM AFM height images (1 μm × 1 μm) and SEM images (SE detector) of the isoporous thin films. The diblock copolymer appears grey (SEM) or bright (AFM) while the pores appear dark. (a) AFM image and (b) SEM image of a DMF/water annealed P4VP186-PS951 thin film; (c) AFM image and (d) SEM image of a DMF/water annealed P2VP215-PS672 thin film. | ||
In order to correlate the different porous structures obtained by the two different solvent annealing techniques quantitatively, the pore sizes were determined from SEM images of samples sputter coated with 0.5 nm platinum using the software Imagic IMS (Imagic Bildverarbeitung AG, Switzerland). Measurements yielded mean pore diameters for a DMF/water annealed P4VP186-PS951 film and a 1,4 dioxane/ethanol annealed P4VP186-PS951 film to be 17.6 nm ± 4.0 nm and 11.7 nm ± 5.5 nm, respectively. Hence, the mean pore diameter of the DMF/water annealed P4VP186-PS951 film was 1.5 times larger than the mean pore diameter determined for the 1,4 dioxane/ethanol annealed P4VP186-PS951 film. Furthermore, the DMF/water annealed P4VP186-PS951 film shows a lower standard deviation of the mean pore diameter and a higher order in terms of pore diameter distribution. The median pore diameter is 17.7 nm for the DMF/water annealed sample and 10.5 nm for the 1,4-dioxane/ethanol annealed sample, which for the latter shows a significantly larger deviation from the mean pore diameter and thus a more inhomogeneous pore size distribution. A similar pore diameter deviation was found for P2VP215-PS672 when comparing the two annealing processes. The mean pore diameter determined for the P2VP215-PS672 thin film after DMF/water annealing (Fig. 7) was 21.3 nm ± 3.5 nm (median pore diameter 21.8 nm) and 15.4 nm ± 6 nm (median pore diameter 14.0 nm) for the 1,4-dioxane/ethanol annealed film (Fig. 6). Thus, DMF/water annealing led to a 1.4 times larger mean pore diameter and a narrower pore size distribution.
Regarding the self-assembly properties, the PVP-b-PS diblock copolymers of this work are well comparable with the self-assembly properties of PS-b-PVP diblock copolymers obtained via anionic polymerization, previously mentioned in literature. Gallei et al. and Jung et al. investigated the structure formation of isoporous membranes of PS-b-P4VP and PS-b-P2VP, respectively.76,77 The examined diblock copolymers had similar molecular weights and block compositions in the region wherein PVP is expected to form spherical or cylindrical domains in bulk.
Conclusions
In this work, the synthesis of high molecular weight P4VP-b-PS and P2VP-b-PS diblock copolymers was performed in a two-step process of a RAFT bulk polymerization followed by a surfactant-free RAFT dispersion polymerization. A series of P4VP and P2VP homopolymers with dispersities ranging from 1.08–1.27 were prepared via RAFT bulk polymerization at 80 °C using two different trithiocarbonate RAFT agents. The choice of RAFT agent did not show significant effects on monomer conversion or dispersity.A PVP macroRAFT agent was chain extended via RAFT aqueous-alcoholic dispersion polymerization of styrene at 70 °C and 20% w/w solids content, reaching quantitative conversion within 24 h as indicated by 1H NMR. A series of PVP-b-PS diblock copolymers was prepared with high n between 70 and 137 kDa and low dispersities as evaluated by 1H NMR and GPC. The proposed particle formation mechanism is the polymerization-induced self-assembly (PISA), which involves the in situ formation of micellar objects after initial chain growth in the aqueous-alcoholic phase. In the bulk state the block copolymers microphase separate into regularly ordered cylindrical or spherical morphologies, as revealed by TEM.
Spin-coated PVP-b-PS diblock copolymer films with weight fractions of the minority PVP block ranging from 17–24% were converted to porous structures by a selective alignment and swelling strategy and investigated via AFM and SEM.
A comparison of two annealing procedures aiming to obtain isoporous thin films was performed. The first approach combined thermal annealing with 1,4-dioxane vapor annealing, followed by dip-coating in ethanol. The second technique comprised thermal annealing, followed by dip-coating in DMF and immersion into water. Both procedures led to porous structures, but with structural differences. DMF/water annealed P4VP186-PS951 and P2VP215-PS672 films showed an isoporous spherical surface microstructure with more uniformly arranged pores compared to the corresponsing films annealed in 1,4 dioxane/ethanol. Moreover, measurements revealed that the mean pore diameter of the filmes annealed in DMF/water were approximately 1.5 times larger with a lower standard deviation than the mean pore diameter of the corresponding films annealed in 1,4 dioxane/ethanol. The modified porous topographical structures were obtained by distinct selectivities of the different solvents for the individual blocks. Hence, the study showed a variable pore alignment and pore swelling by solvent annealing with different solvent combinations. When annealing with DMF/water, isoporous thin films could be prepared when using either P2VP-b-PS or P4VP-b-PS diblock copolymers.
Conflicts of interest
There are no conflicts to declare.Author contributions
K.N. and P.G.: conceptualization; K.N. and P.G.: methodology, investigation, data curation, formal analysis, validation; P.G. and V.A.: supervision. V.A.: resources, K.N. and P.G.: writing – original draft, K.N., P.G. and V. A.: writing – review & editing.Acknowledgements
The authors thank Maren Brinkmann, Thomas Emmler and Silvio Neumann for GPC and NMR measurements. Special thanks to Evgeni Sperling for sample preparation and AFM measurements as well as to Martin Held, Anke-Lisa Höhme and Erik Schneider for SEM and TEM sample preparation, measurements and discussions. Financial support by a collaborative project of the Helmholtz Association and Russian Science Foundation (grant number HRSF-0075) is gratefully acknowledged. Open Access funding is provided by the University of Hamburg.Notes and references
- E. Rizzardo and D. H. Solomon, Aust. J. Chem., 2012, 65, 945–969 CrossRef CAS.
- A. D. Jenkins, R. G. Jones and G. Moad, Pure Appl. Chem., 2009, 82, 483–491 Search PubMed.
- K. Matyjaszewski, B. S. Sumerlin and N. V. Tsarevsky, ACS Publ., 2012, 1100, 1–13 CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2008, 2477–2479 RSC.
- C. L. McCormick and A. B. Lowe, Acc. Chem. Res., 2004, 37, 312–325 CrossRef CAS.
- T. G. Ribelli, D. Konkolewicz, S. Bernhard and K. Matyjaszewski, J. Am. Chem. Soc., 2014, 136, 13303–13312 CrossRef CAS PubMed.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2013, 4, 873–881 RSC.
- V. Delplace, E. Guégain, S. Harrisson, D. Gigmes, Y. Guillaneuf and J. Nicolas, Chem. Commun., 2015, 51, 12847–12850 RSC.
- M. R. Hill, R. N. Carmean and B. S. Sumerlin, Macromolecules, 2015, 48, 5459–5469 CrossRef CAS.
- W. D. Harkins, J. Am. Chem. Soc., 1947, 69, 1428–1444 CrossRef CAS.
- G. Lichti, R. G. Gilbert and D. H. Napper, J. Polym. Sci., Polym. Chem. Ed., 1983, 21, 269–291 CrossRef CAS.
- W. D. Harkins, J. Polym. Sci., 1950, 5, 217–251 CrossRef CAS.
- C. Barner-Kowollik, Handbook of RAFT Polymerization, John Wiley & Sons, Weinheim, 2008 Search PubMed.
- R. G. Gilbert, Emulsion polymerization: a mechanistic approach, Academic Press, London, 1995 Search PubMed.
- J. Goodwin, J. Hearn, C. Ho and R. Ottewill, Colloid Polym. Sci., 1974, 252, 464–471 CrossRef CAS.
- F. Lauterbach and V. Abetz, Soft Matter, 2020, 16, 2321–2331 RSC.
- M. Semsarilar and S. Perrier, Nat. Chem., 2010, 2, 811–820 CrossRef CAS PubMed.
- K. Nieswandt, P. Georgopanos, C. Abetz, V. Filiz and V. Abetz, Materials, 2019, 12, 3145 CrossRef CAS PubMed.
- J. Rieger, C. Grazon, B. Charleux, D. Alaimo and C. Jérôme, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2373–2390 CrossRef CAS.
- Y. Li and S. P. Armes, Angew. Chem., 2010, 122, 4136–4140 CrossRef.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS PubMed.
- G. Delaittre, M. Save and B. Charleux, Macromol. Rapid Commun., 2007, 28, 1528–1533 CrossRef CAS.
- R. Arshady, Colloid Polym. Sci., 1992, 270, 717–732 CrossRef CAS.
- N. P. Truong, J. F. Quinn, M. R. Whittaker and T. P. Davis, Polym. Chem., 2016, 7, 4295–4312 RSC.
- J. Yeow and C. Boyer, Adv. Sci., 2017, 4, 1700137 CrossRef PubMed.
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 350–355 RSC.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. Armes, Langmuir, 2012, 28, 914–922 CrossRef CAS PubMed.
- M. Semsarilar and V. Abetz, Macromol. Chem. Phys., 2020, 222, 2000311 CrossRef.
- T. Arita, M. Buback, O. Janssen and P. Vana, Macromol. Rapid Commun., 2004, 25, 1376–1381 CrossRef CAS.
- C.-Q. Huang and C.-Y. Pan, Polymer, 2010, 51, 5115–5121 CrossRef CAS.
- W.-D. He, X.-L. Sun, W.-M. Wan and C.-Y. Pan, Macromolecules, 2011, 44, 3358–3365 CrossRef CAS.
- W. Cai, W. Wan, C. Hong, C. Huang and C. Pan, Soft Matter, 2010, 6, 5554–5561 RSC.
- T. T. Dao, L. Vezenkov, G. Subra, V. Ladmiral and M. Semsarilar, Polym. Chem., 2021, 12, 113–121 RSC.
- N. P. Truong, C. Zhang, T. A. Nguyen, A. Anastasaki, M. W. Schulze, J. F. Quinn, A. K. Whittaker, C. J. Hawker, M. R. Whittaker and T. P. Davis, ACS Macro Lett., 2018, 7, 159–165 CrossRef CAS.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., 2020, 132, 8444–8470 CrossRef.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- Y. Pei and A. B. Lowe, Polym. Chem., 2014, 5, 2342–2351 RSC.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Polym. Chem., 2014, 5, 3466–3475 RSC.
- W. M. Wan, X. L. Sun and C. Y. Pan, Macromol. Rapid Commun., 2010, 31, 399–404 CrossRef CAS PubMed.
- X. Zhang, J. Rieger and B. Charleux, Polym. Chem., 2012, 3, 1502–1509 RSC.
- F. Huo, X. Wang, Y. Zhang, X. Zhang, J. Xu and W. Zhang, Macromol. Chem. Phys., 2013, 214, 902–911 CrossRef CAS.
- E. Jones, M. Semsarilar, P. Wyman, M. Boerakker and S. Armes, Polym. Chem., 2016, 7, 851–859 RSC.
- G. G. du Sart, I. Vukovic, Z. Vukovic, E. Polushkin, P. Hiekkataipale, J. Ruokolainen, K. Loos and G. ten Brinke, Macromol. Rapid Commun., 2011, 32, 366–370 CrossRef PubMed.
- I. Tokarev, R. Krenek, Y. Burkov, D. Schmeisser, A. Sidorenko, S. Minko and M. Stamm, Macromolecules, 2005, 38, 507–516 CrossRef CAS.
- A. Knoll, R. Magerle and G. Krausch, J. Chem. Phys., 2004, 120, 1105–1116 CrossRef CAS PubMed.
- J. N. Albert and T. H. Epps III, Mater. Today, 2010, 13, 24–33 CrossRef CAS.
- E. B. Gowd, M. Böhme and M. Stamm, IOP Conf. Ser.: Mater. Sci. Eng., 2010, 14, 012015 Search PubMed.
- W. Sriprom, M. James, S. Perrier and C. Neto, Macromolecules, 2009, 42, 3138–3146 CrossRef CAS.
- N. A. Lynd and M. A. Hillmyer, Macromolecules, 2005, 38, 8803–8810 CrossRef CAS.
- S. Park, J.-Y. Wang, B. Kim, J. Xu and T. P. Russell, ACS Nano, 2008, 2, 766–772 CrossRef CAS PubMed.
- K. Fukunaga, H. Elbs, R. Magerle and G. Krausch, Macromolecules, 2000, 33, 947–953 CrossRef CAS.
- E. B. Gowd, B. Nandan, M. K. Vyas, N. C. Bigall, A. Eychmüller, H. Schlörb and M. Stamm, Nanotechnology, 2009, 20, 415302 CrossRef PubMed.
- J. K. Bosworth, M. Y. Paik, R. Ruiz, E. L. Schwartz, J. Q. Huang, A. W. Ko, D.-M. Smilgies, C. T. Black and C. K. Ober, ACS Nano, 2008, 2, 1396–1402 CrossRef CAS PubMed.
- C. Liedel, C. W. Pester, M. Ruppel, V. S. Urban and A. Böker, Macromol. Chem. Phys., 2012, 213, 259–269 CrossRef CAS.
- S. Sakurai, Polymer, 2008, 49, 2781–2796 CrossRef CAS.
- A. Sidorenko, I. Tokarev, S. Minko and M. Stamm, J. Am. Chem. Soc., 2003, 125, 12211–12216 CrossRef CAS PubMed.
- S. H. Kim, M. J. Misner, T. Xu, M. Kimura and T. P. Russell, Adv. Mater., 2004, 16, 226–231 CrossRef CAS.
- J. G. Kennemur, Macromolecules, 2019, 52, 1354–1370 CrossRef CAS.
- A. Boyde, F. A. Mccorkell, G. K. Taylor, R. J. Bomphrey and M. Doube, Microsc. Res. Tech., 2014, 77, 1044–1051 CrossRef CAS PubMed.
- B. Yu, J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3544–3557 CrossRef CAS.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC.
- S. Förster, M. Zisenis, E. Wenz and M. Antonietti, J. Chem. Phys., 1996, 104, 9956–9970 CrossRef.
- M. Antonietti, S. Heinz, M. Schmidt and C. Rosenauer, Macromolecules, 1994, 27, 3276–3281 CrossRef CAS.
- W. Zha, C. D. Han, D. H. Lee, S. H. Han, J. K. Kim, J. H. Kang and C. Park, Macromolecules, 2007, 40, 2109–2119 CrossRef CAS.
- D. H. Lee, H. Cho, S. Yoo and S. Park, J. Colloid Interface Sci., 2012, 383, 118–123 CrossRef CAS PubMed.
- T. Xu, J. Stevens, J. Villa, J. T. Goldbach, K. W. Guarini, C. T. Black, C. J. Hawker and T. P. Russell, Adv. Funct. Mater., 2003, 13, 698–702 CrossRef CAS.
- S. Park, J.-Y. Wang, B. Kim and T. P. Russell, Nano Lett., 2008, 8, 1667–1672 CrossRef CAS PubMed.
- S. Park, B. Kim, J. Y. Wang and T. P. Russell, Adv. Mater., 2008, 20, 681–685 CrossRef CAS.
- E. J. Crossland, S. Ludwigs, M. A. Hillmyer and U. Steiner, Soft Matter, 2007, 3, 94–98 RSC.
- R. Olayo-Valles, S. Guo, M. Lund, C. Leighton and M. A. Hillmyer, Macromolecules, 2005, 38, 10101–10108 CrossRef CAS.
- Z. Zhang, M. M. Rahman, C. Abetz, B. Bajer, J. Wang and V. Abetz, Macromol. Rapid Commun., 2019, 40, 1800729 CrossRef PubMed.
- V. Abetz, Macromol. Rapid Commun., 2015, 36, 10–22 CrossRef CAS PubMed.
- M. Radjabian and V. Abetz, Prog. Polym. Sci., 2020, 102, 101219 CrossRef CAS.
- J. Brandrup, H. Immergut and A. Grulke, Polymer handbook, John Wiley & Sons, New York, 1999 Search PubMed.
- L. Oss-Ronen, J. Schmidt, V. Abetz, A. Radulescu, Y. Cohen and Y. Talmon, Macromolecules, 2012, 45, 9631–9642 CrossRef CAS.
- M. Gallei, S. Rangou, V. Filiz, K. Buhr, S. Bolmer, C. Abetz and V. Abetz, Macromol. Chem. Phys., 2013, 214, 1037–1046 CrossRef CAS.
- A. Jung, S. Rangou, C. Abetz, V. Filiz and V. Abetz, Macromol. Mater. Eng., 2012, 297, 790–798 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00074h |
| This journal is © The Royal Society of Chemistry 2021 |