
Direct radical functionalization methods to access substituted adamantanes and diamondoids
William K.
Weigel
III
ab,
Hoang T.
Dang
a,
Abigail
Feceu
b and
David B. C.
Martin
 *b
*b
aChemistry, University of Iowa, Iow City, Iowa, USA
bUniversity of California Riverside, Riverside, California, USA. E-mail: david-martin@uiowa.edu
First published on 9th October 2021
Abstract
Adamantane derivatives have diverse applications in the fields of medicinal chemistry, catalyst development and nanomaterials, owing to their unique structural, biological and stimulus-responsive properties, among others. The synthesis of substituted adamantanes and substituted higher diamondoids is frequently achieved via carbocation or radical intermediates that have unique stability and reactivity when compared to simple hydrocarbon derivatives. In this review, we discuss the wide range of radical-based functionalization reactions that directly convert diamondoid C–H bonds to C–C bonds, providing a variety of products incorporating diverse functional groups (alkenes, alkynes, arenes, carbonyl groups, etc.). Recent advances in the area of selective C–H functionalization are highlighted with an emphasis on the H-atom abstracting species and their ability to activate the particularly strong C–H bonds that are characteristic of these caged hydrocarbons, providing insights that can be applied to the C–H functionalization of other substrate classes.
 William K. Weigel III | William K. Weigel III completed his B.S. in Biochemistry at California State Polytechnic University, Pomona before beginning his graduate studies at the University of California, Riverside in 2015. Under the supervision of Prof. Dave Martin, his research was focused on synthesizing anacardic acid derivatives for the treatment of cancer and the selective aminoalkylation of adamantanes and higher diamonoids, completing his PhD in Chemistry in 2020. He is currently a postdoc in the lab of Prof. Raphael Franzini at the University of Utah and is studying bioorthogonal reactions. |
 Hoang T. Dang | Hoang T. Dang was born in Vietnam and raised in Garden Grove, California. He received his B.S. in Biochemistry in 2017 at California State University, Long Beach and he conducted undergraduate research with Prof. Kensaku Nakayama and Prof. Valerie Schmidt. Hoang is currently pursuing a Ph.D. in Chemistry at the University of Iowa, where he works with Prof. Dave Martin on developing photocatalytic C–H activation methods. His research interests include photochemical metal-catalyzed and organocatalysis strategies to accomplish classic organic reactions (H-atom transfer, cycloadditions) under mild conditions. |
 Abigail Feceu | Abigail Feceu was born in Southern California, USA and she obtained her Bachelor of Science degree in Chemistry from California State Polytechnic University, Pomona. In 2019, she received her Ph.D. from the University of California, Riverside under the supervision of Prof. Dave Martin. Her research focused on photochemical methodologies for adamantane functionalization and natural product synthesis. In 2020, Abigail moved to Wuppertal, Germany and joined the research group of Prof. L. J. Gooßen at Ruhr-Universität Bochum as a post-doctoral researcher. |
 David B. C. Martin | David B. C. Martin grew up in Calgary, Canada and received his Bachelor of Science degree in Chemistry from the University of British Columbia. From 2005–06, he worked at the Merck-Frosst Centre for Therapeutic Research in the Medicinal Chemistry Department. In 2011, Dave obtained his Ph.D. from the University of California, Irvine where he worked with Prof. Chris Vanderwal on the application of Zincke aldehydes toward the synthesis of Strychnos alkaloids, including a short synthesis of strychnine. After pursuing post-doctoral research in the lab of Prof. David MacMillan in the area of photoredox catalysis, Dave spent five years at the University of California, Riverside before moving to the University of Iowa in 2019. His research group develops new (photo-)catalytic transformations for bond activation and strategies for the synthesis of biologically active targets. |
1. Introduction
1.1. Background
Adamantanes are a fascinating class of polycyclic hydrocarbons that have unique physical and chemical properties.1 As members of the larger family of diamondoids, these caged hydrocarbons are so named because they are nanoscale substructures of the sp3-hybridized diamond lattice and are characterized by a high degree of symmetry (Fig. 1).2,3 Adamantane (1, C10H16), the simplest member of the diamondoids, has Td symmetry and thus has only two distinct C–H positions: four equivalent tertiary bridgehead positions connected by six methylene bridges giving rise to its tricyclic cage-like form. Just as diamond is a rigid and mechanically strong allotrope of carbon, diamondoids are rigid and thermodynamically stable molecules of the general formula C4n+6H4n+12, ranging from the structurally defined molecules diamantane (2), triamantane (3), pentamantane etc. to the more loosely defined “nanodiamonds”, which have even been detected in interstellar space.4 Diamondoids from all size regimes continue to be studied for their unique physical and spectroscopic properties and applications in both medicinal and materials applications.5–7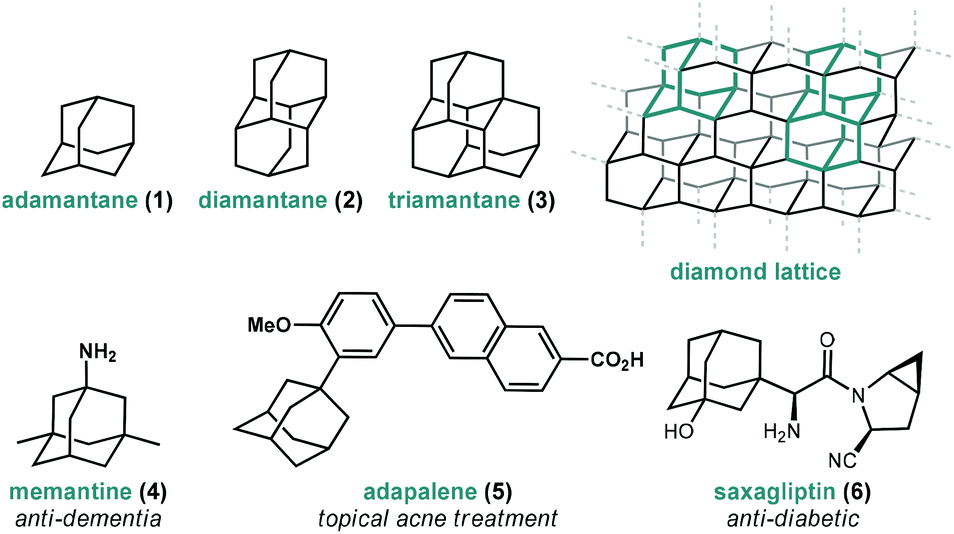 | ||
| Fig. 1 Structure of three simple diamondoids, the diamond lattice and three clinically approved small molecule drugs containing adamantane. | ||
Of particular significance is the parent member adamantane (1) which, due to its non-planar three-dimensional shape and hydrophobic nature, appears in a number of clinically approved drugs (e.g., memantine 4), sometimes as a compact lipophilic substituent used to improve binding properties (e.g., adapalene 5, saxagliptin 6).8–13 Adamantane is also frequently used as a bulky alkyl substituent on ligand frameworks14 such as the first stable crystalline N-heterocyclic carbene IAd (13, Fig. 2) reported by Arduengo,15 Jacobsen's chiral chromium catalyst for hetero-Diels–Alder reactions (14),16 a Z-selective Grubbs olefin metathesis catalyst (20),17 and Davies’ PTAD ligand for Rh(II) dimers.18 Furthermore, the rigid, saturated (and therefore non-conducting) framework of adamantane is commonly used as a scaffold for connecting unsaturated linkers and chromophores in optical materials and nanoscale frameworks (e.g., 18, 21).19–26 These myriad important applications were enabled by several synthetic milestones in the synthesis and substitution of adamantane which make it the readily available starting material it is today.
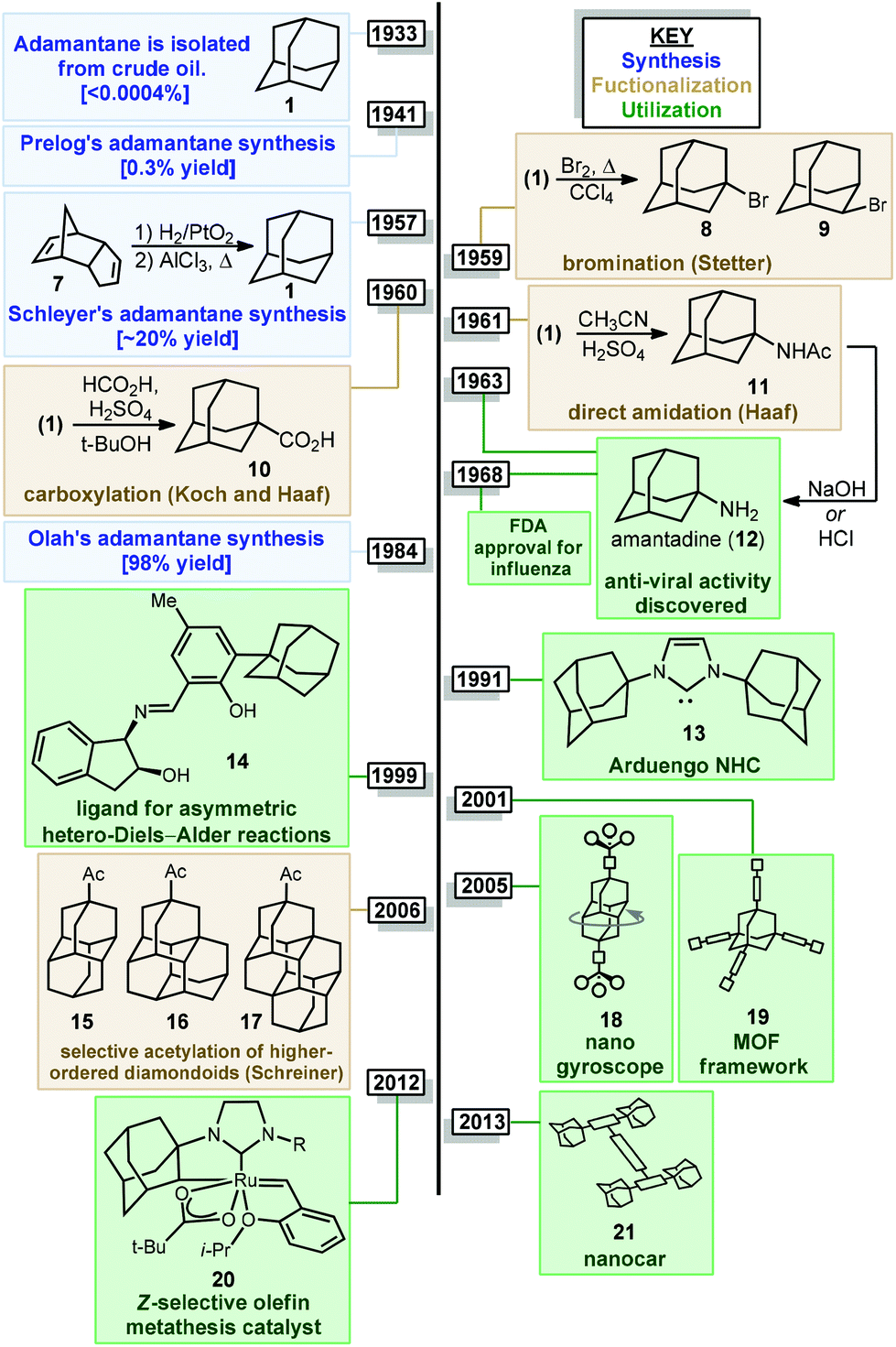 | ||
| Fig. 2 Milestones in the synthesis, functionalization, and utilization of adamantane. | ||
1.2. History
The caged structure of adamantane was hypothesized in 1924 by H. Decker in a report discussing the structure of diamond, and he dubbed this molecule “decaterpene”.27 Adamantane was then discovered in 1933 by isolation from petroleum sources, although its isolation was extremely limited due to its low natural abundance (∼0.0004% petroleum content).28 Significant interest in the structure of these unusual hydrocarbons in the 1930s led to the first reported synthesis of adamantane by Prelog and Seiwerth in 1941.29,30 Using classical enolate alkylation reactions for ring closure, Wolff–Kishner reductions and a final double decarboxylation process, they achieved a 1.5% yield of adamantane over the final steps from the previously synthesized Meerwein's ester 22, Scheme 1.31 Improvements to this route reported by Stetter increased the yield to 6.5%, however the overall strategy still suffered from the multiple steps necessary to remove the functional groups used to assemble the cage.32,33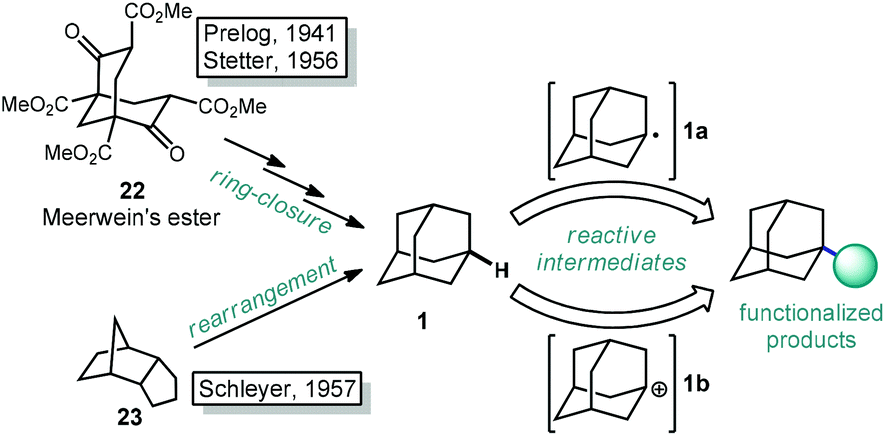 | ||
| Scheme 1 Synthetic routes to adamantane and its functionalized derivatives. | ||
An alternative route to making adamantane was discovered by Paul von Ragué Schleyer in 1957 using a Lewis acid-promoted rearrangement of tetrahydrodicyclopentadiene (23). Believed to bear resemblance to the geosynthetic origin of adamantane in petroleum, this groundbreaking isomerization approach was able to improve the yield to 20–40% (Fig. 2).32,34 The mechanism of this transformation is proposed to involve a complex series of cationic 1,2 bond migrations and hydride shifts, terminated by the addition of a hydride nucleophile.35,36 This process is thought to be thermodynamically controlled and proceeds due to the high stability of adamantane (and the 1-adamantyl carbocation 1b) relative to precursor 23 (and all isomeric carbocations leading to 1b).37,38 Related methods have been applied by Schleyer to the synthesis of diamantane (2), which is also known as congressane after its appearance as the Congress Emblem of the 1963 London IUPAC meeting.39,40 Further improvements using superacid catalysis by Olah and coworkers allow the conversion of less stable polycyclic hydrocarbon isomers to diamondoids, providing the most practical method to access diamantane, triamantane and higher diamondoids.41–43 These methods have been commercialized, facilitating the use of diamondoids in new applications and exploration of their conversion to substituted derivatives.44,45
Interest in adamantanes surged again in the 1960s when the first evidence of antiviral activity was reported for a simple amine derivative. In 1961, Haaf et al. reported an amidation/hydrolysis sequence to form 1-aminoadamantane,46,47 also known as amantadine (12, Fig. 2), which was found to display potent anti-influenza A properties in 1964.48,49 Rimantadine, a chiral aminoalkylated adamantane with anti-viral activity, was similarly introduced in the 1990s. Ultimately, the use of aminoadamantanes to treat influenza was discontinued due to the development of widespread resistance mechanisms. Shortly after amantadine was discovered, the two-carbon homolog memantine (4) was synthesized and patented by Eli Lilly and Company as an anti-diabetic.50 Memantine would later be repurposed as an anti-dementia drug for the treatment of Alzheimer's disease, with FDA approval in 2003, and is still used for this application.51 Examples of substituted adamantanes in other disease areas include Adapalene (5),52 an anti-acne medication, and saxagliptin (6), a DPP-4 inhibitor used in the treatment of diabetes.
With the exception of 1-aminoadamantanes, which can be directly synthesized via a Ritter or nitration reaction, the synthesis of substituted adamantanes typically relies on halogenated intermediates that can be converted to the corresponding radical 1a or carbocation 1b for substitution (Scheme 1). The tertiary carbocation 1b is unusually stable due to hyperconjugation with filled bonding orbitals within the rigid cage while radical 1a is destabilized due to its inability to relax to a more stable pyramidal conformation. For the bromination of adamantane, the ratio of 1-bromoadamantane (8) and 2-bromoadamantane (9) depends on the reagents employed, with a phase-transfer catalyst system reported by Schreiner and Fokin providing nearly complete selectivity for the more substituted product.53,54 Similarly, different conditions allow diamantane to be converted to a mixture of medial 3°-bromide (24), apical 3°-bromide (25) and 2°-bromide products (26, Scheme 2). Substitution of these alkyl halides can be accomplished via SN1-type substitution with a variety of carbon and heteroatom nucleophiles and by conversion to the corresponding radical by traditional methods. These methods, which are reliable and provide access to discrete substitution patterns by isolation of the appropriate halide intermediate, nonetheless require multistep sequences and the separation of byproducts at each step to provide the desired diamondoid products.
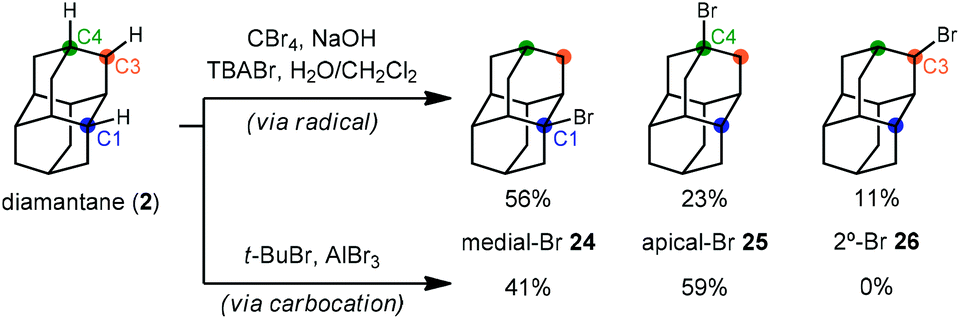 | ||
| Scheme 2 Effect of reaction conditions on apical and medial selectivity in diamantane bromination. | ||
Alternatively, the direct substitution of adamantane has long been a test-bed for direct C–H functionalization methods, particularly because of the unusually high bond dissociation energies (BDEs) of this class of molecules (96 and 99 kcal mol−1 for 2° and 3° C–H bonds, respectively).55 As a result, H-atom abstractors with high reactivity are required, including O-centered radicals derived from peroxides and photoexcited diaryl ketone species.56,57 A central challenge that emerges is the issue of selectivity between non-equivalent 2° and 3° positions of adamantane, higher order diamondoids (e.g., 2, 3; Fig. 1) and substituted derivatives (e.g., drug molecules 5, 6). This challenge has placed limitations on the complexity of reaction partners, where multiple different types of C–H bonds would lead to complex mixtures of substitution products. Solutions to this challenge have involved the careful study of mechanistic details and identification of key features such as polar effects and polarizability that enable the selective apical arylation and acetylation of higher diamondoids as reported by Albini and Schreiner (see 15–17, Fig. 2), both discussed in greater detail below. Based on these mechanistic insights, the direct conversion of diamondoid C–H bonds to C–C substituted products can be carried out in a selective manner, and often provides the most efficient access to these versatile building blocks. Over 60 years after Schleyer's synthesis, the range of applications of adamantanes and higher order diamondoids appears only limited by a chemist's creativity and their ability to produce substituted derivatives in a controlled, selective and efficient manner.
1.3. Scope of the review
Traditional methods for the conversion of diamondoid C–H bonds to C–halide, C–O, C–N and other C–X bonds are numerous and have been reviewed elsewhere.58–66 Likewise, reactions for the use of halo- and hydroxyadamantanes as intermediates for substitution have been the focus of previous literature summaries. This review will focus on direct methods for the conversion of diamondoid C–H to C–C bonds proceeding via radical intermediates. These transformations, spanning over 50 years of published work, are organized according to the reaction classes and this review will highlight the diverse species used for the key H-atom abstraction step. While many papers describe a single example of adamantane as a substrate, particular attention will be given to reports that focus on the functionalization of diamondoids as a class of substrates.2. Acylations
Acyladamantanes, molecules featuring a direct C–C bond between adamantane and the carbonyl group of a ketone, ester or aldehyde, are one of the earliest derivatives to be made by a direct C–H to C–C radical functionalization. Acyladamantanes are useful intermediates that can be further modified (e.g., by reductive amination) and while many traditional methods have been applied for their synthesis, most rely on a pre-functionalized adamantyl substrate and/or harsh conditions for accomplishing the necessary introduction of the acyl group. Therefore, direct methods for forging C–C bonds to adamantanes that eliminate the need for pre-functionalization are desirable. By accessing the reactive nucleophilic adamantyl radical, direct acylations of adamantane are achievable and fall into three main categories described in this section: chlorocarbonylation, acetylation and formylation.2.1. Chlorocarbonylation
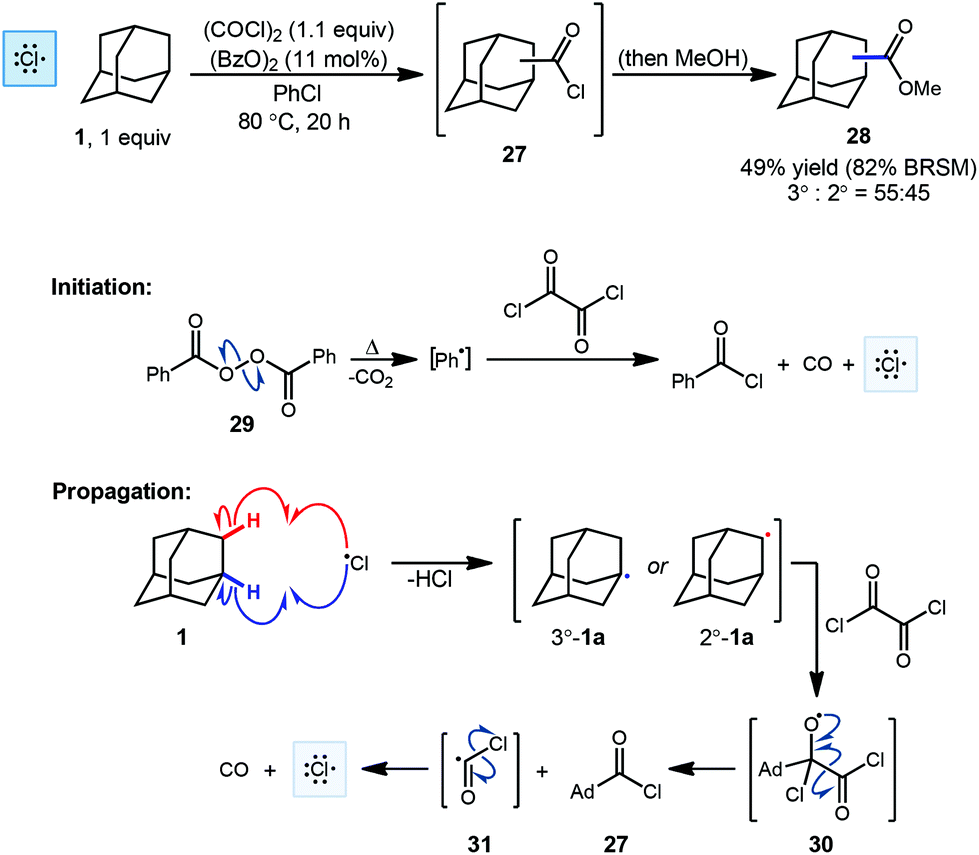
One of the first examples of radical-mediated C–H to C–C functionalization was described by Tabushi and coworkers in 1968 (Scheme 3).67 This seminal report involved the radical chlorocarbonylation of adamantane using oxalyl chloride and a benzoyl peroxide radical initiator, leading to acylated intermediate 27 as illustrated below. The adamantyl radical 1a is generated using chlorine radical which is initially produced via homolysis of peroxide 29 and then propagated according to the mechanism shown (30 → 31 → Cl˙). Following methanolysis of 27, a mixture of methyl 1- and 2-adamantanecarboxylates (28, 49% yield, 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :45 rr) was obtained. The yield reported by the authors was 82% BRSM (based on the recovered starting material, in this case, adamantane). After statistical correction, this distribution of regioisomers corresponds to a reactivity ratio of 3.67:1 favoring the tertiary adamantyl positions. This preferential reactivity of the tertiary C–H bonds of adamantanes was also observed in prior radical halogenations by Smith and Williams, a critical process that helped showcase the potential utility of radical-based methods early on.68 One factor that leads to the preferential reactivity of the 3° position is reduced steric hindrance, where approach to the 2°-C–H groups leads to significant 1,3-diaxial interactions that are absent at the 3° position.
:45 rr) was obtained. The yield reported by the authors was 82% BRSM (based on the recovered starting material, in this case, adamantane). After statistical correction, this distribution of regioisomers corresponds to a reactivity ratio of 3.67:1 favoring the tertiary adamantyl positions. This preferential reactivity of the tertiary C–H bonds of adamantanes was also observed in prior radical halogenations by Smith and Williams, a critical process that helped showcase the potential utility of radical-based methods early on.68 One factor that leads to the preferential reactivity of the 3° position is reduced steric hindrance, where approach to the 2°-C–H groups leads to significant 1,3-diaxial interactions that are absent at the 3° position.
 | ||
| Scheme 3 Benzoyl peroxide initiated chlorocarbonylation using oxalyl chloride. | ||
Investigating this system further, Tabushi later showed that using adamantanes with different substituents at the 1-position altered the ratio of 3°:2° chlorocarbonylation products on a steric basis (OMe>CO2Me>Me>CN>H).69 This product ratio ranged from 1.2:1 for unsubstituted adamantane (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H) to 5.7:1 (ROMe). Interestingly, these substituent effects were drastically different from those observed in prior radical halogenations by Owens et al. using trichloromethyl radical.70 While Tabushi saw correlation only to steric size, Owens found that the same substituents influenced the regioselectivity through both steric and polar effects according to a linear free-energy relationship expressed by the Taft equation.
H) to 5.7:1 (ROMe). Interestingly, these substituent effects were drastically different from those observed in prior radical halogenations by Owens et al. using trichloromethyl radical.70 While Tabushi saw correlation only to steric size, Owens found that the same substituents influenced the regioselectivity through both steric and polar effects according to a linear free-energy relationship expressed by the Taft equation.
2.2. Acetylation
In a related investigation by Tabushi and coworkers, they demonstrated in 1973 that adamantane can be selectively acetylated using an excess of diacetyl under irradiation to give 1-acetyladamantane (32) as the sole regioisomer (Scheme 4).71,72 The C–H abstracting species is believed to be the triplet-excited state of diacetyl since no acetylation occurred in the presence of pyrene, a known quencher of diacetyl phosphorescence via energy transfer.73 Despite also being a quencher, the presence of oxygen actually increased the rate of acetylation and the production of the 2-acetyladamantane (33) and 1-adamantanol, the autooxidation product. The increase in photoacetylation rate and loss of selectivity is attributed to the generation of reactive methyl radicals which form in a decarboxylative process after oxygen and diacetyl react (eqn (1)).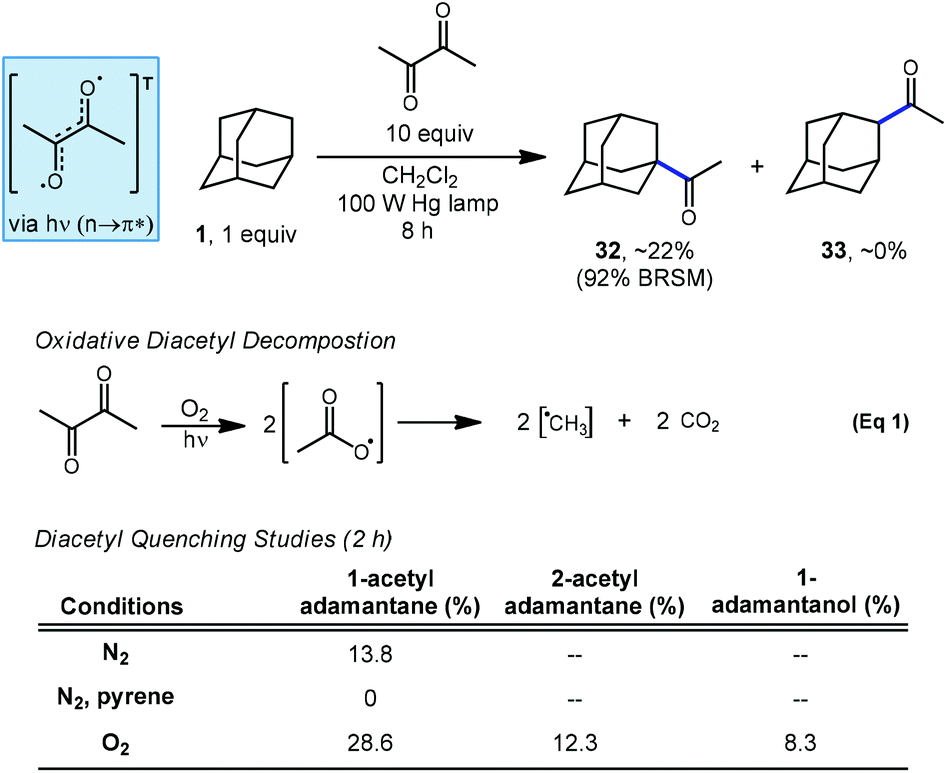 | ||
| Scheme 4 Photoacetylation of adamantane using diacetyl. | ||
Tabushi also showed that the photoacetylation of diamantane proceeded selectively at the methine positions using diacetyl under conditions similar to those used for adamantane.74 However, while adamantane features four equivalent methine positions, higher ordered diamondoids possess two or more nonequivalent 3° positions. In the case of diamantane featuring two apical and six medial positions (see positions labeled C1 and C4 in Scheme 2), photoacetylation using diacetyl favored mono-acetylation at the apical position over the medial position in a 5.5:1 ratio. Acetylation of the methylene positions and bis-addition products were not observed.
Schreiner and coworkers later showed that apical selectivity during photoacetylation under similar conditions is maintained in even higher ordered tri-, tetra-, and pentamantanes despite an increase in the number of possible medial positions (see Fig. 3).75 For instance, despite the unique number of 3°-C–H positions increasing from two in diamantane 15 to four in triamantane 16 and [121]tetramantane 17, acetylation of the apical position is still favored over the other medial positions in an ∼5:1 ratio. Even higher apical selectivity is observed in the acetylation products for [1(2)3]tetramantane 34 and [123]tetramantane 35 (despite 35 having six unique C–H positions).76 [1(2,3)4]pentamantane (36) showed near complete apical selectivity and resulted in a significant amount of bis acetylated product as well. The selectivity in these reactions is largely governed by the relative steric accessibility of the apical positions over the more hindered medial positions.
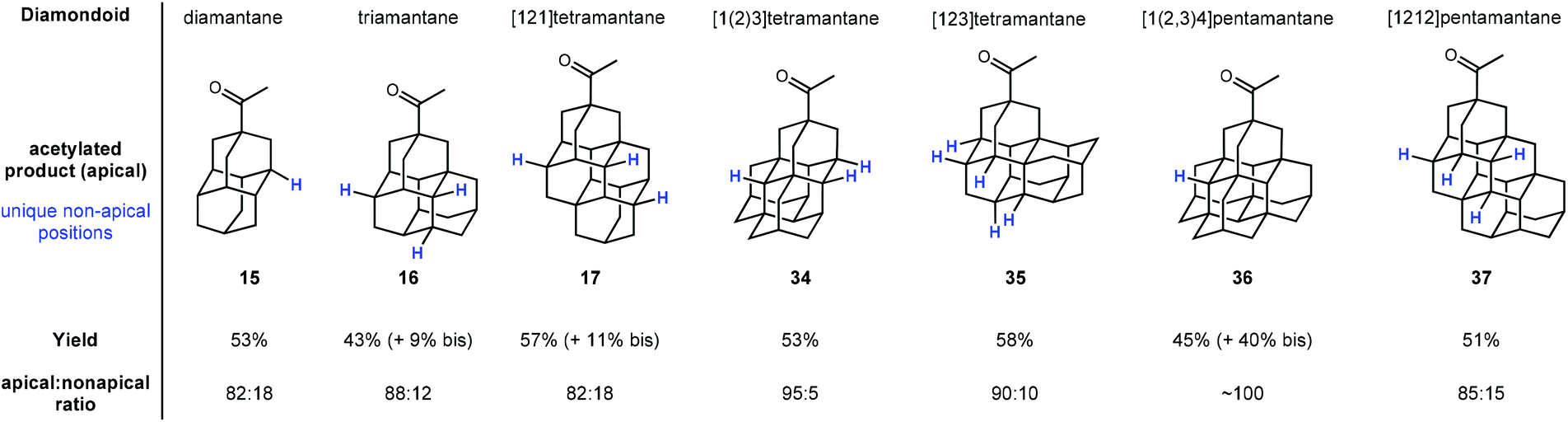 | ||
| Fig. 3 Apical photoacetylation products in higher-ordered diamondoids. Other unique non-apical tertiary C–H positions are indicated in blue. | ||
2.3. Formylation
Recently, Kamijo et al. have reported a method for formylating unactivated C(sp3)–H bonds in many substrates including adamantane through the intermediacy of an O-benzylaldoxime 40 (Scheme 5a).77 Although the formylated product is not produced directly in a single step, the overall transformation introduces a carbonyl group, similar to other reactions in this section. In this reaction, 4-benzoylpyridine (4-BzPy) is used with a mercury lamp to generate a photoexcited triplet (4-BzPyT) which acts as the C–H abstractor for adamantane (Scheme 5b). The triplet is produced through light-dependent n to π* transition between the singlet ground (S0) and excited state (S1) followed by intersystem crossing (ISC) to the excited triplet (T1), a common manifold for other ketone and quinone catalysts. While the resulting radical species 42 could in principle turn over in a photocatalytic process by transferring a hydrogen atom or a proton/electron stepwise to regenerate the ketone 4-BzPy, they are typically used in a stoichiometric amount and likely form a 1,2-diol dimer byproduct.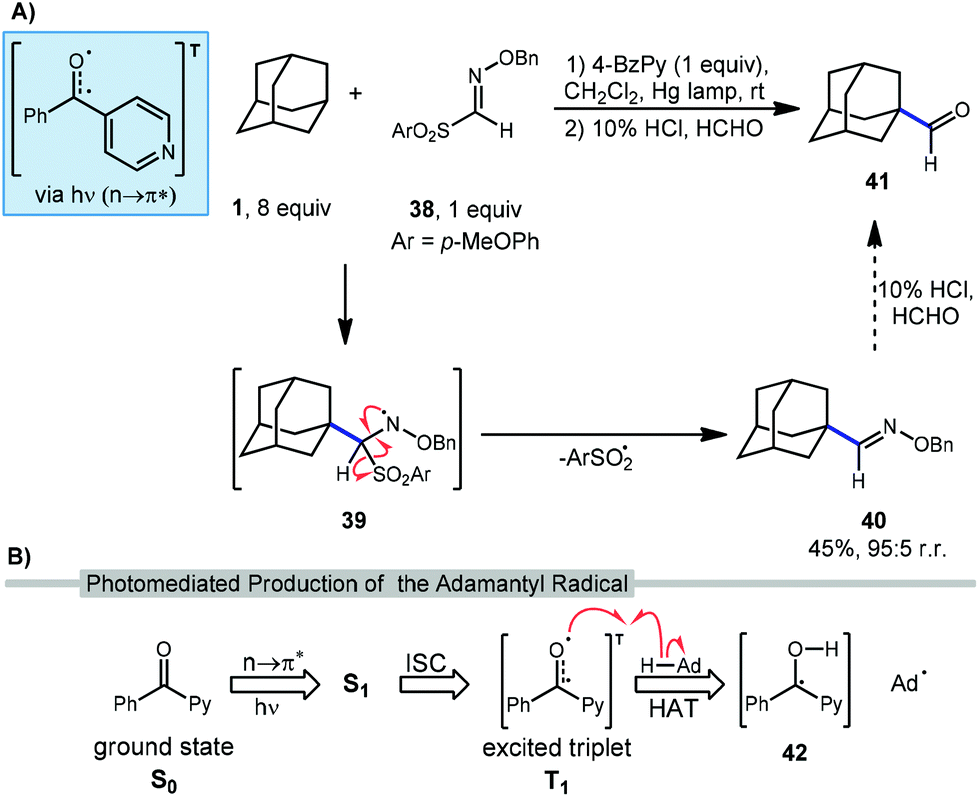 | ||
| Scheme 5 Light-enabled formylation of adamantane using sulfonyl aldoxime. | ||
After HAT with 4-BzPyT, Kamijo revealed that the resulting adamantyl radical can readily intercept aldoxime 38 to give amino radical 39. Fragmentation of this intermediate results in loss of arylsulfonyl radical to form the O-benzylaldoxime product 40 in 45% isolated yield high regioselectivity for the methine position (95:5). This aldoxime can then be converted to the formylated product 41 using a solution of formaldehyde in aqueous HCl. While this final hydrolysis step was not performed on adamantyl product 40, it is expected to proceed smoothly based on data for other substrates in this work.
2.4. Esterification and amidation
In 2018, Doyle and coworkers used nickel and photoredox catalysis to enable direct C–C bond formation between unactivated alkanes and chloroformate 43 to form ester products.78 Adamantanecarboxylate ester 44 was synthesized in 46% yield with a slight preference for the less substituted regioisomer (2°:3° = 1.3:1) using the conditions shown in Scheme 6. The chloroformate is thought to undergo an oxidative addition with LnNi(0) 45 to form LnNi(II)(Cl)(CO2R) 46 followed by SET with the excited iridium photocatalyst to produce a Ni(III) species 47. Photolytic cleavage liberates a chlorine radical which performs HAT with adamantane to generate the adamantyl radical (48 → 49). Addition of the adamantyl radical forms nickel complex 50 which undergoes reductive elimination to give the ester product and the resulting Ni(I) species 51 is turned over by single electron reduction from Ir(II) species 52.
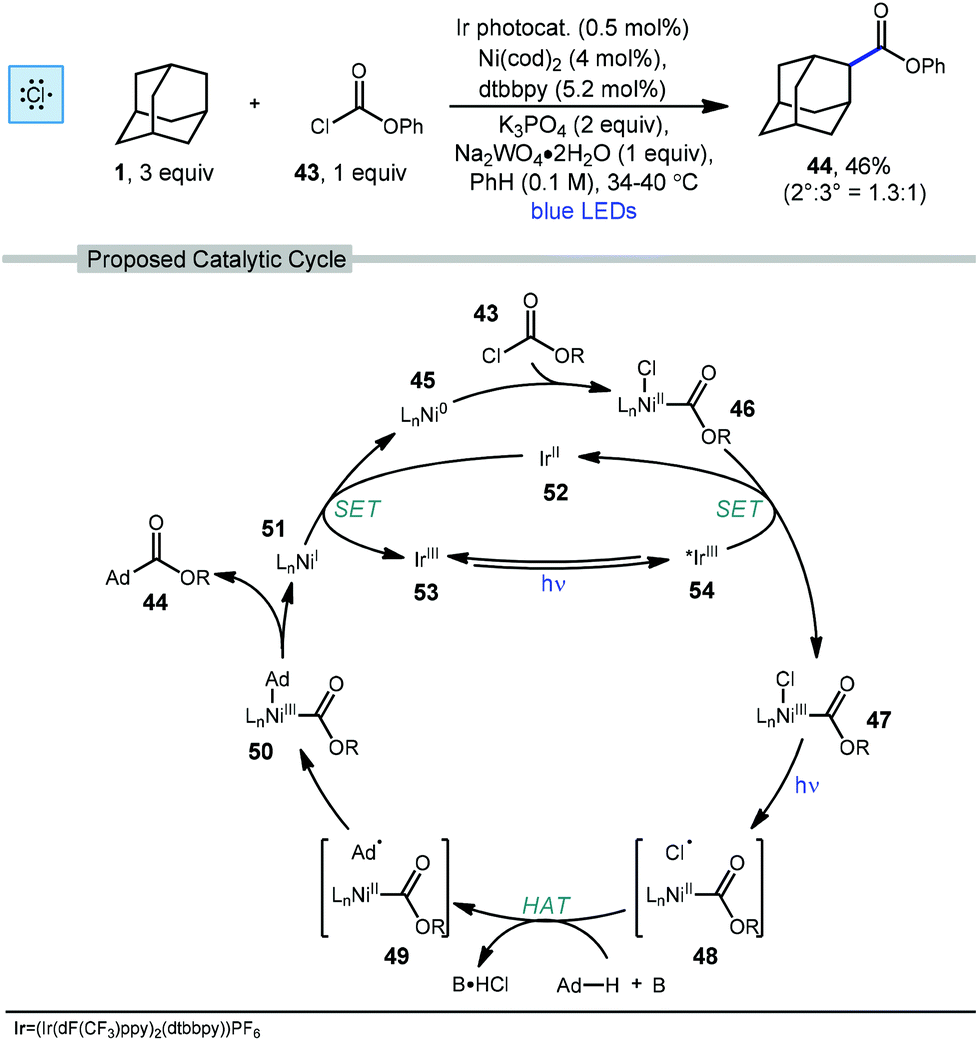 | ||
| Scheme 6 Esterification of adamantane with chloroformate 43 using dual Ni/Ir photoredox catalysis. | ||
As seen earlier in this section with respect to chlorocarbonylation, this reaction also demonstrates the tendency of chlorine radical to perform HAT with low regioselectivity. The slight preference for the formation of the ester product at the bridge position may occur due to reversible radical capture by nickel and a bias for reductive elimination of the secondary alkyl fragment. This is thought to occur in other nickel-catalyzed reactions involving the capture of 2° and 3° adamantyl radicals (see Scheme 44).
Li and coworkers have recently reported an amidation of adamantane and other alkanes using aromatic isocyanides and di-tert-butylperoxide (DTBP) (Scheme 7).79 Three isocyanides with differing aryl substituents were used with adamantane to afford amides 56–58 in low to moderate yields (33–53%). The reaction is thought to first involve the ferrocene-catalyzed formation of tert-butoxyl radical 59 which produces the adamantyl radical via HAT. Addition of the adamantyl radical into the isocyanide forms iminyl radical intermediate 60 that is then trapped by oxygen as hydroperoxide 61. Finally, homolytic O–O bond scission to oxygen-centered radical 62 is followed by hydrogen abstraction and tautomerization to give the final amide product. The products were isolated as single regioisomers, however one would also expect the formation of the isomeric 3° product based on the typical selectivity of the tert-butoxyl radical in related reactions (e.g., 2:1, Scheme 45).
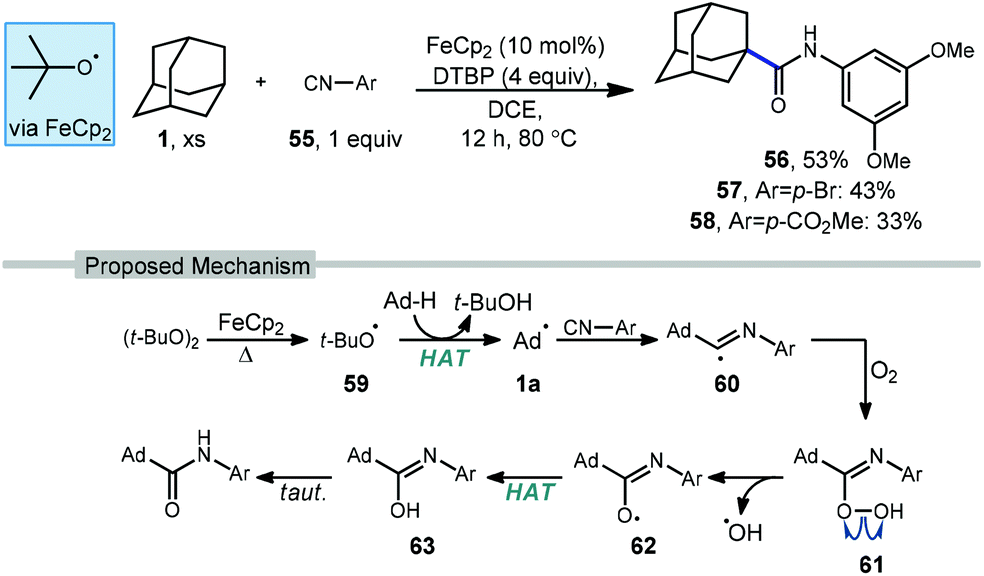 | ||
| Scheme 7 Iron-catalyzed radical amidation of adamantane via aryl isocyanides. | ||
3. Carbonylation with carbon monoxide
Carbonylation reactions of adamantane using carbon monoxide (CO) are employed to access acyladamantane products, similar to the acylative transformations described in the previous section. However, the processes described below primarily involve the direct, metal-free reaction of adamantyl radical 1a with CO in solution to give the adamantyl acyl radical 64 (Scheme 8, top). Various strategies for arriving at this acyl radical have been developed and then combined with different radical acceptors to obtain carbonylated products 65. The adamantyl radical can also be coupled to CO through transition metal-catalyzed processes such as insertion (66 → 67; Scheme 8, bottom) and reductive elimination (67 → 68) to provide carboxylate derivatives. These carbonylative transformations are presented together in this section.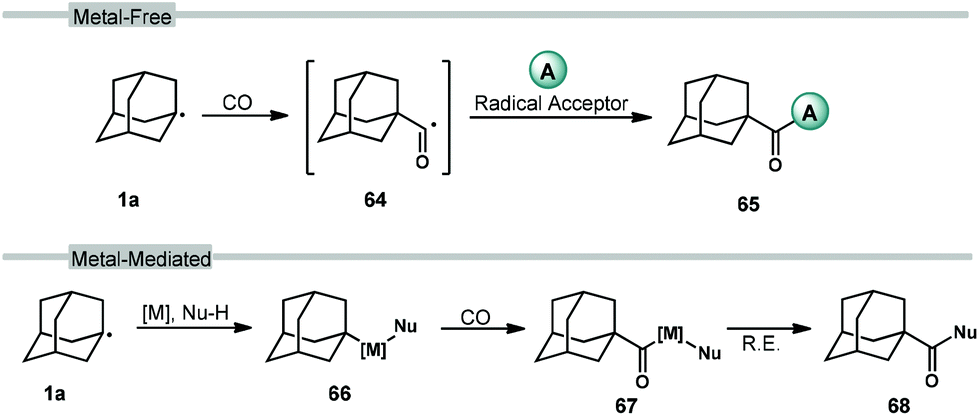 | ||
| Scheme 8 General scheme for carbonylations of the adamantyl radical using carbon monoxide. | ||
3.1. Carboxylation using NHPI
The C–H functionalization of saturated hydrocarbons using carbon monoxide, although attractive, is quite challenging due to a general lack of selectivity and low conversion of starting material.80–82 Work done by Ishii in 1998 showed that oxidation of polycyclic alkanes in the presence of air and carbon monoxide can produce carboxylated products using a catalytic free radical mechanism (Scheme 9).83N-Hydroxyphthalimide (NHPI, 69) is used as a catalyst in the presence of O2 to generate the (phthalimide-N-oxyl (PINO, 70) radical without photoactivation and under mild conditions. However, selectivity is quite poor when using adamantane as the hydrocarbon and a mixture of carbonylated and oxygenated products 71–76 were obtained.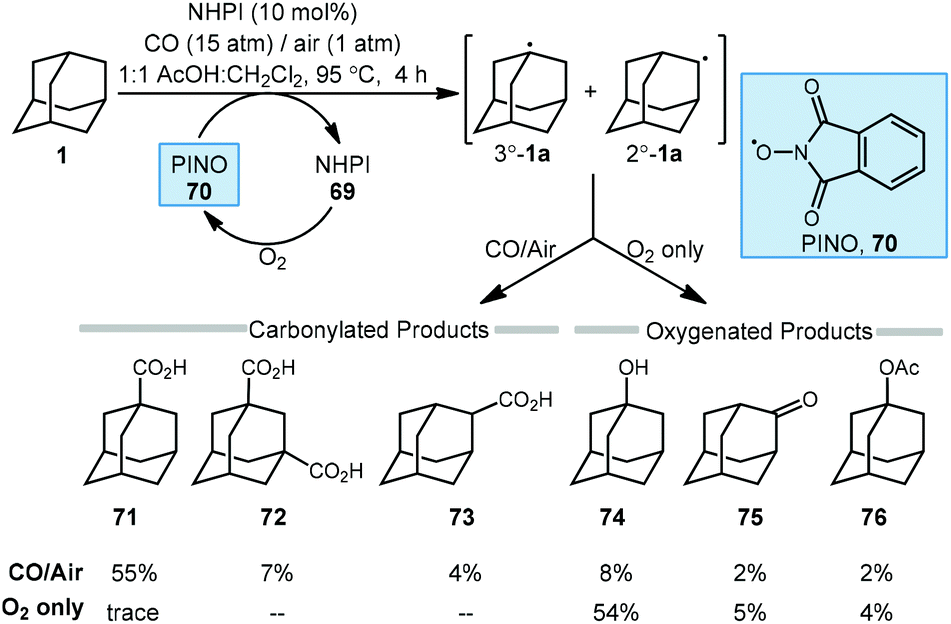 | ||
| Scheme 9 Oxidative carbonylation of adamantane using the phthalimide-N-oxyl radical. | ||
A likely mechanism begins with generating the reactive intermediate PINO from NHPI in the presence of O2. The PINO radical will abstract a hydrogen at either the secondary or tertiary position of adamantane to give the 3°-1a and 2°-1a adamantyl radicals. These radicals are intercepted with CO forming the acyl radical 64 and upon subsequent reaction with O2, eventually generates carboxylic acids 71–73. Interestingly, when 1-adamantanecarbocylic acid (71) is used as the starting material, the dicarboxylate 72 was formed in 57% yield. Alternatively, the adamantyl radicals can instead react directly with O2 to give the oxygenated products 1-adamantanol (74), 2-adamantanone (75), and 1-acetoxyadamantane (76). When only O2 is used, these oxygenated products are favored. Overall, the reaction selectivity is not significantly impacted by other changes in conditions (e.g., solvent and temperature).
3.2. Photocatalytic carbonylation
In 2011, Ryu and Albini reported a three-component coupling reaction between adamantane, carbon monoxide, and Michael acceptors using photocatalytic methods (Scheme 10).84 This multi-component process generated unsymmetrical ketones with tetrabutylammonium decatungstate (TBADT, 78) as the photocatalyst and a xenon lamp as the UV light source. A l:1.3 mixture of the 1- and 2-adamantane ketosuccinates (79) was obtained, although the yield was relatively low (35%) compared to other cyclic hydrocarbon substrates.
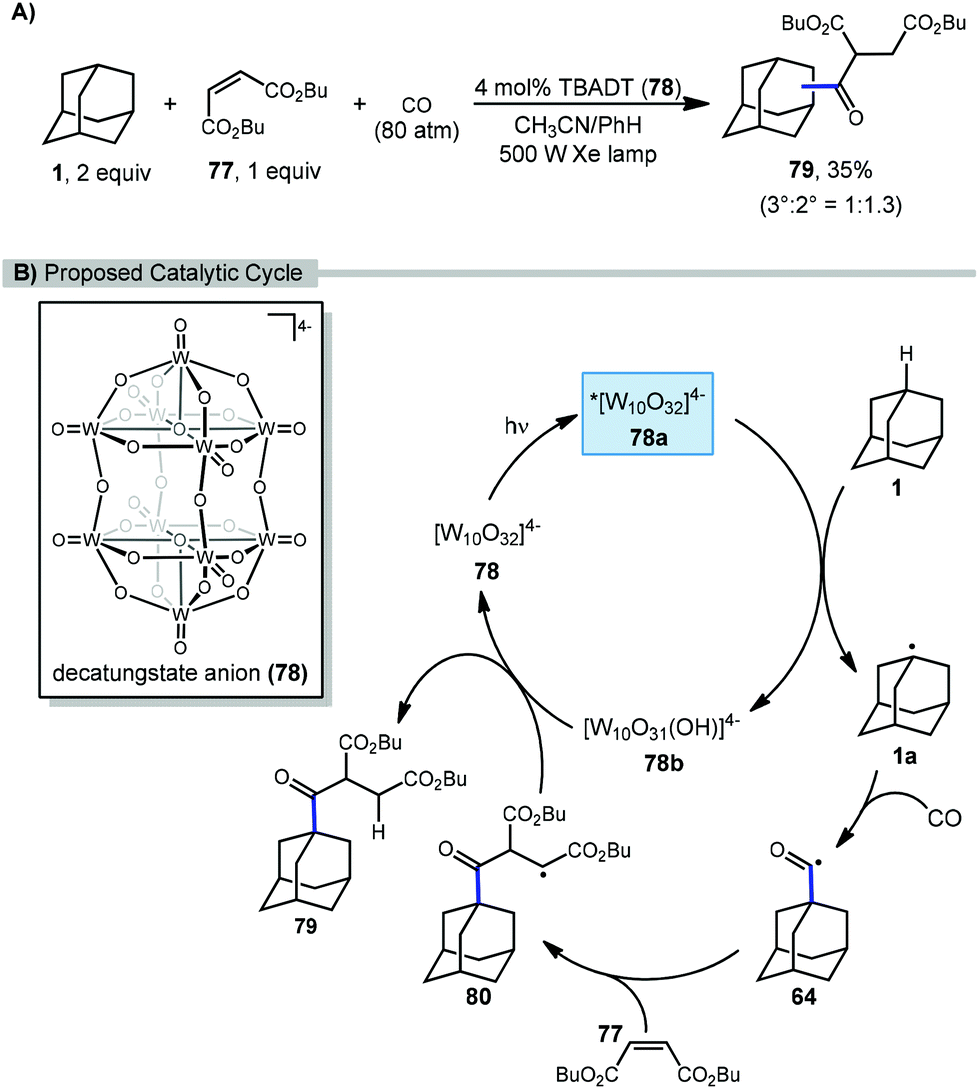 | ||
| Scheme 10 Three-component synthesis of adamantyl ketones using decatungstate photocatalysis. | ||
The proposed catalytic cycle for the C–H functionalization of adamantane using a tandem carbonylation-addition process is illustrated in Scheme 10. Upon irradiation of TBADT, the excited photocatalyst 78a abstracts a hydrogen from adamantane generating adamantyl radical 1a. Carbonylation results in the acyl radical 64 followed by a Giese addition to give succinyl radical 80. Reduction of intermediate 80 by the reduced photocatalyst 78b turns over the catalyst and produces the desired functionalized adamantane product 79, either via HAT or an electron-transfer/proton-transfer mechanism.
3.3. Transition metal-catalyzed carbonylation
The use of transition metals for C–H activation methods that include carbonylation is an attractive route for generating products containing carbonyl groups. However, commonly used second- and third-row transition metal catalysts (e.g., Ru, Rh) are expensive and require complex organometallic preparations, limiting their use.85–87 First row transition metals like nickel and copper are excellent alternatives due to their low toxicity, low cost and relative abundance.88With this in mind, Wu et al. reported the first copper-catalyzed carbonylative C–H activation in 2016 using a variety of alkanes and amides to generate imides.89 Wu employed a Cu(I) species and DTBP to selectively functionalize adamantane at the tertiary position with N-methylacetamide (81), likely via the tert-butoxyl radical, generating the imide 82 in 31% yield (Scheme 11, top). The acylation of adamantane required a cosolvent and proceeded in lower yield than other hydrocarbons.
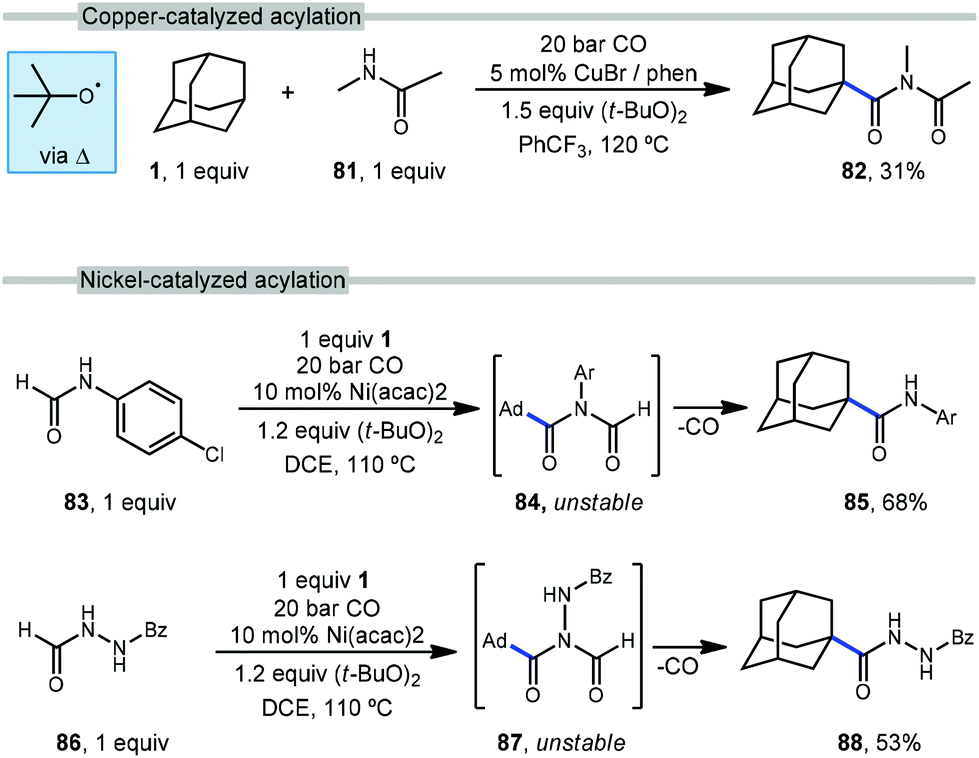 | ||
| Scheme 11 Metal-catalyzed adamantane amidations using carbon monoxide. | ||
In addition to copper, nickel catalysis has also previously been reported for the activation of inert C(sp3)–H bonds.90 Recently, Yufeng Li et al. developed a Ni-catalyzed carbonylation of unreactive alkanes such as adamantane with formamides to obtain amide products.91Scheme 11 (bottom) illustrates two selective carbonylations employing adamantane. The first reaction uses an equimolar amount of formanilide 83 to give the amide product 85 in modest 68% yield while the second utilizes N-formylbenzohydrazide (86) which gave the corresponding diacylhydrazine 88 in 53% yield. Similar to the work done by Wu and coworkers with copper, DTBP is homolytically cleaved with the help of nickel to form the key reactive intermediate, the tert-butoxyl radical. However, unlike Wu's work, the Ni-catalyzed insertion of CO generates imide intermediates (84, 87) which are unstable and quickly undergo loss of the formyl group as CO to give the observed amide or hydrazide product.
Carbon monoxide can also be used for the synthesis of adamantyl esters. Lei and coworkers reported a palladium-catalyzed oxidative carbonylation method for hydrocarbons including adamantane using benzyl alcohol (89) and carbon monoxide to generate one carbon-homologated esters (Scheme 12).92 The reaction proceeds at relatively low CO pressure (5 bar) to give ester product 90 in good yield (68%) with preference for activation at the bridgehead position of adamantane (3°:2° = 3:1). Once the adamantyl radical is generated by HAT, it is proposed to intercept a Pd–CO complex to form an acyl palladium species (potentially in equilibrium with acyl radical) that reacts with benzyl alcohol to afford the product.
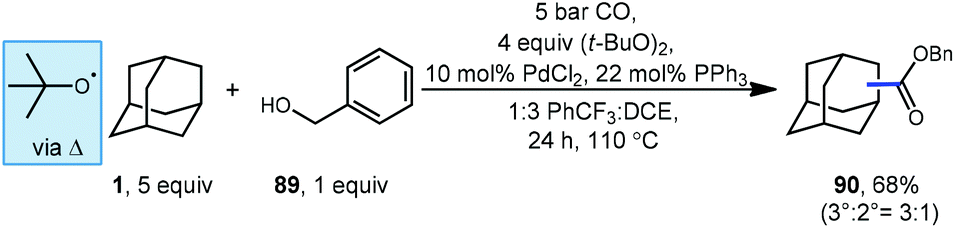 | ||
| Scheme 12 Esterification of adamantane using palladium-catalyzed oxidative carbonylation. | ||
3.4. Metal-free oxidative radical carbonylation
Lei et al. have also developed a metal-free oxidative carbonylation of alkanes via direct C–H bond activation using DTBP in the presence of carbon monoxide and benzyl alcohol to generate esters (Scheme 13).93 A variety of alkanes including adamantane underwent efficient conversion to corresponding benzyl ester products like 90. While the reaction proceeded in good yield, the regioselectivity of the H-atom abstraction step was rather low, giving a 2:1 ratio of 3°:2° product in 77% yield. Like many examples in this Review, the key HAT step here is facilitated by the tert-butoxyl radical, and the nucleophilic adamantyl radical is proposed to react directly with CO. This approach offers a convenient metal-free alternative to palladium-catalyzed method discussed in the previous section; however, it requires a significantly higher pressure of CO to intercept the radical intermediate.
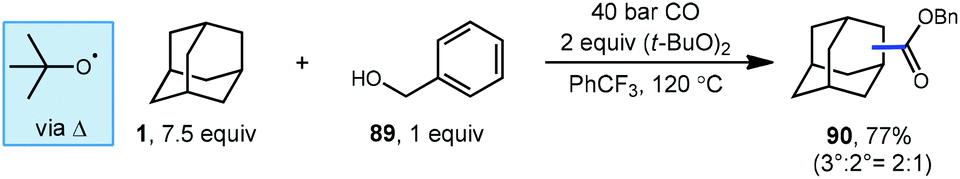 | ||
| Scheme 13 Metal-free esterification of adamantane using DTBP. | ||
4. Alkylation via alkene addition reactions
The addition of the adamantyl radical to an alkene acceptor is a convenient method of effecting adamantane alkylation. In this category, Giese-type reactions, which involve trapping of a carbon centered radical with an electrophilic alkene (Scheme 14), are frequently used. The resonance-stabilized radical intermediate, commonly an α-acyl radical, can undergo reduction and protonation (91 → 92) or further reactions such as chain propagation or tandem addition steps which lead to a diverse array of transformations.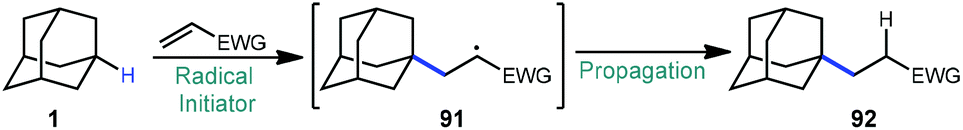 | ||
| Scheme 14 General Giese reaction involving adamantane. | ||
Adamantane alkylation has traditionally been accomplished using pre-functionalized halo-adamantanes together with organometallic methods such as Wurtz and Grignard reactions or via Lewis acids such as FeCl3 and AlCl3.58,94,95 However, avoiding the need for preactivated starting materials through direct C–H activation of adamantane is an attractive option both in terms of atom economy and step count. As such, there are many examples of Giese-type additions to alkenes that proceed by a radical C–H abstraction to directly generate the necessary adamantyl radical.
4.1. First peroxide-catalyzed alkene additions
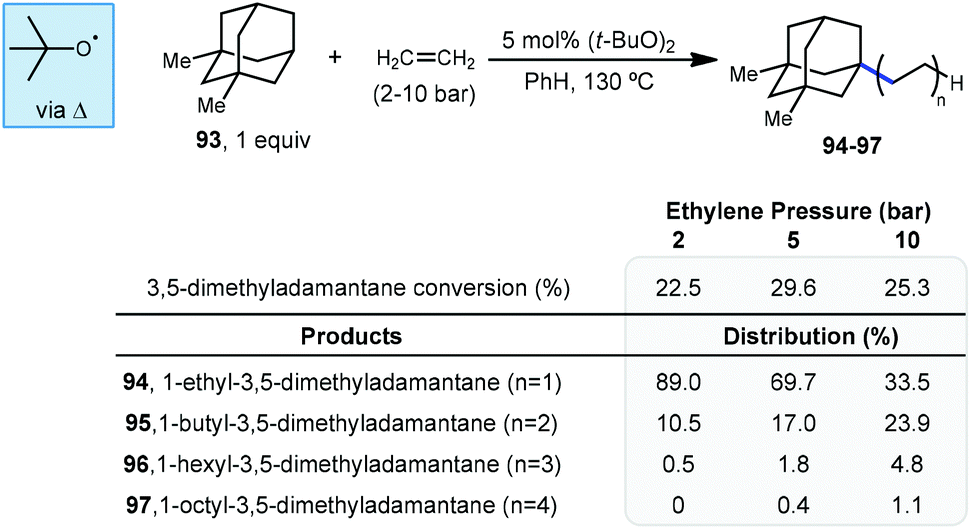
In 1974, the earliest report of a direct C–H alkylation of adamantanes was published by Tabushi and Fukunishi, wherein they described the reaction of 1,3-dimethyladamantane (93) with ethylene gas using 5 mol% DTBP for initiation (Scheme 15).96 With 2 bar ethylene, 1-alkylated 3,5-dimethyladamantane products 94–97 were formed in 22.5% yield (with the remainder being unreacted adamantane) with nearly exclusive regioselectivity for the bridgehead position. Among the products, the ethyl product 94 was predominantly formed (89%) although smaller quantities of the telomeric butyl and hexyl products (95 and 96) were also generated. These telomeric products suggest a radical chain mechanism is likely where the radical adds to the alkene and the resulting 1° alkyl radical abstracts from the next molecule of adamantane or (at a slower rate) adds to another alkene. While higher pressures of ethylene only led to slightly higher conversion to products, they resulted in a noticeably higher distribution of oligomer products. Adamantane (1) also underwent alkylation to form the ethyl and butyl products (∼11% yield) but increasing the ethylene pressure beyond 5 bar led to near quantitative recovery of starting material. Other alkenes such as 1-hexene, 1-octene, cyclohexene and cyclooctene were also reacted with 1,3-dimethyladamantane to give moderate yields of 1:1 alkylation adducts (up to 26% yield). This is the only example of an alkylation reaction with simple unactivated alkenes as radical acceptors.
 | ||
| Scheme 15 Alkylation of dimethyladamantane via radical addition to ethylene. | ||
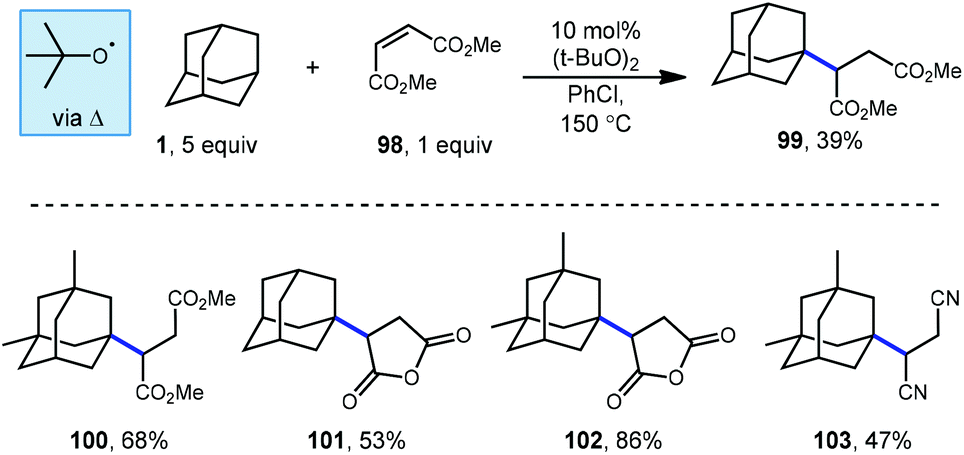
Tabushi and Fukinishi would later go on to report that peroxide catalysis could also be used with different electron-withdrawn olefins to give alkylated adamantane products 99–103 (Scheme 16) in moderate to good yields.97 The reactive species responsible for the production of the adamantyl radical in these reactions is once again tert-butoxyl radical and is generated at elevated temperatures using DTBP.
 | ||
| Scheme 16 Early DTBP-catalyzed Giese reactions with electron-deficent olefins. | ||
4.2. Photochemical alkylation
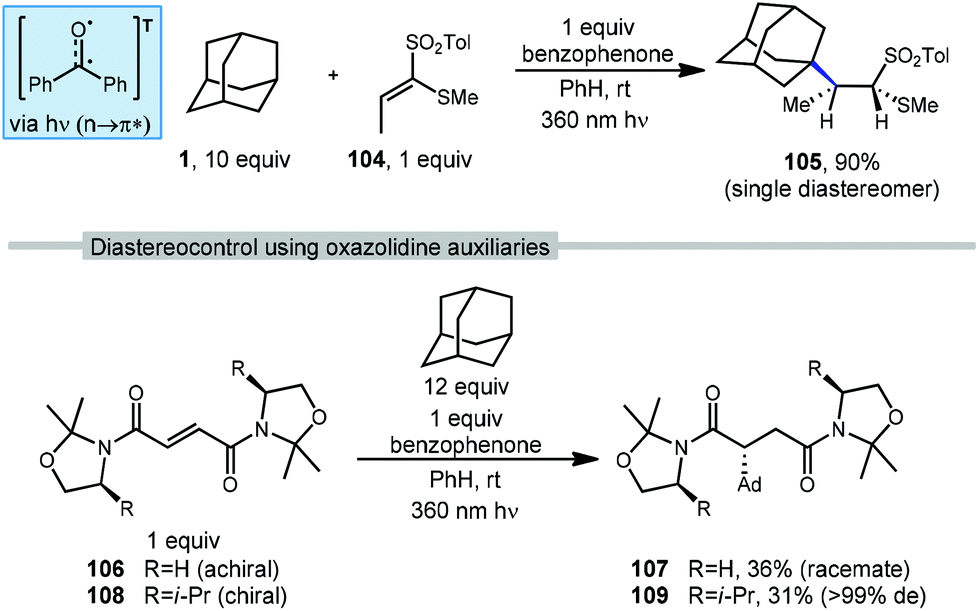
There are several examples of photochemical adamantane alkylation reactions that occur under mild conditions and tolerate a variety of electron-deficient alkene reaction partners. These reactions are enabled by the inclusion of a photosensitive additive, the properties of which change significantly between ground state and excited state, facilitating electron transfer or H-atom transfer. Compounds such as benzophenone necessitate the use of high energy UV light, however, other photocatalysts and dual catalyst systems have been developed that function under visible light.In 2000, Albini and coworkers developed a photochemical radical alkylation to access α-thiosulfones from ketene dithioacetal S,S-dioxides (Scheme 17, top).98 α-Thiosulfones are convenient synthetic intermediates that can be used for several key transformations, such as desulfurization99 and pyrolysis to obtain carbonyl derivatives.100 With stoichiometric amounts of benzophenone, one equivalent of (E)-1-(methylthio)-1-[(4-methylphenyl)-sulfonyl]propene (104), excess adamantane and UV light, the thiosulfone product 105 was obtained in 90% yield as a single diastereomer determined by NOE spectroscopy. The stereoselectivity was dependent on the steric bulk of the alkyl radical; cyclohexane was alkylated in 89% yield but with lower diastereoselectivity (d.r. = 82:18). Alkylation was selective for the tertiary bridgehead position and trace amounts of phenyladamantane, bis-adamantane, 1-adamantanol, and 2-adamantanone were also observed. These oxygenated trace products were the result of reaction with residual oxygen.
 | ||
| Scheme 17 Diastereoselective alkene additions using benzophenone. | ||
Albini et al. used similar conditions to study radical additions to fumaric acid diamides adorned with oxazolidine chiral auxiliaries to generate functionalized products with a high degree of diastereoselectivity.101 As shown in Scheme 17 (bottom), chiral and achiral oxazolidines were tested with excess adamantane and benzophenone as photosensitizer. Irradiation at 360 nm resulted in the triplet excited state of benzophenone which provides the 3° adamantyl radical and addition to achiral fumaryloxazolidine (RH, 106) gave the amidated adamantane product (107, 36% yield) as a racemate. The use of a substituted chiral oxazolidine (Ri-Pr, 108) provided the reaction with a good degree of stereocontrol and the amide product 109 was obtained as a single diastereomer (31% yield, >99 de).
A common and particularly reactive class of electron-deficient olefins are the α,β-unsaturated nitriles. These substrates commonly participate in Giese additions to produce adducts possessing nitrile moieties that map onto natural products and other metabolites of interest.102 However, accessing alkyl nitriles directly through traditional methods has limitations due to toxicity of the organometallics and cyanide salts required.103,104 Albini's photochemical benzophenone platform provides a much safer route to the alkylation of these alkenyl nitriles.105 Using this approach, cyanoethyl adamantanes were synthesized using excess adamantane, one equivalent of acrylonitrile (110) and one equivalent of photosensitizer (Scheme 18, top). The resulting products were formed as a mixture of the two adamantyl regioisomers (111, 50% yield) in a 6:1 ratio. The yield was determined by gas chromatography and the reported isolated yield was somewhat lower.
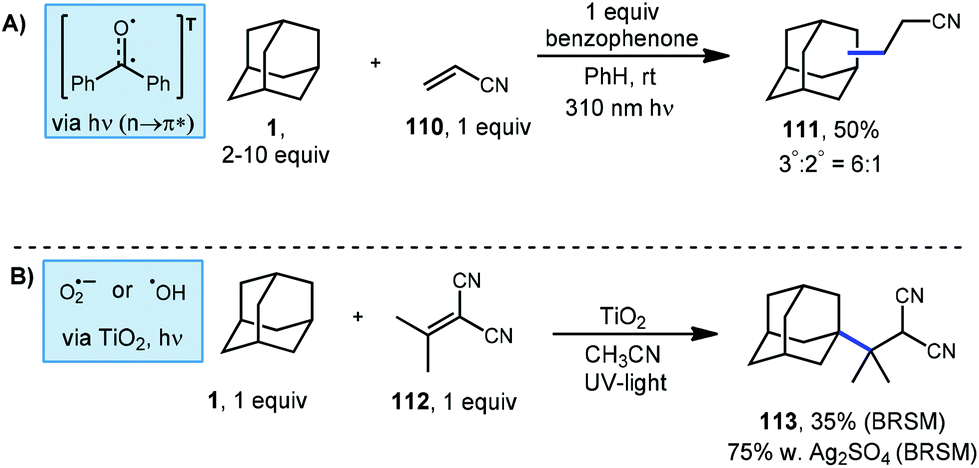 | ||
| Scheme 18 Photochemically mediated radical additions of adamantane to α,β-unsaturated nitriles. | ||
Albini also tested TiO2, a known heterogeneous photo-oxidant in the presence of oxygen or water.106 As shown in Scheme 18 (bottom), isopropylidene malononitrile and adamantane were treated with TiO2 and irradiated under UV light. A yield of 35% of product 113 was obtained based on recovered starting material. Oxygenated products were also observed which were attributed to reaction with oxygen. A higher yield of 75% was obtained when silver sulfate was included as an oxidant.
Albini and coworkers continued to study new methods for photochemical C–H functionalization of hydrocarbons and in 2006, they reported two additional methods to synthesize β-alkylated ketones using UV activation (Scheme 19).107 The adamantyl radical was generated via HAT using either stochiometric benzophenone or catalytic TBADT. When using benzophenone as the abstractor, only 1-adamantyl derivatives were observed and cyclic ketone products 115 and 116 were generated with yields of 65% and 40%, respectively. The high performance of adamantane at low concentration is explained by relative rates of radical addition to the enone. In a report for radical addition to a similar electron deficient olefin, the rate of addition by adamantyl radical (k = 3.0 × 108 M−1 s−1) was found to be approximately two orders of magnitude faster than cyclohexyl radical (k = 3.3 × 106 M−1 s−1).108
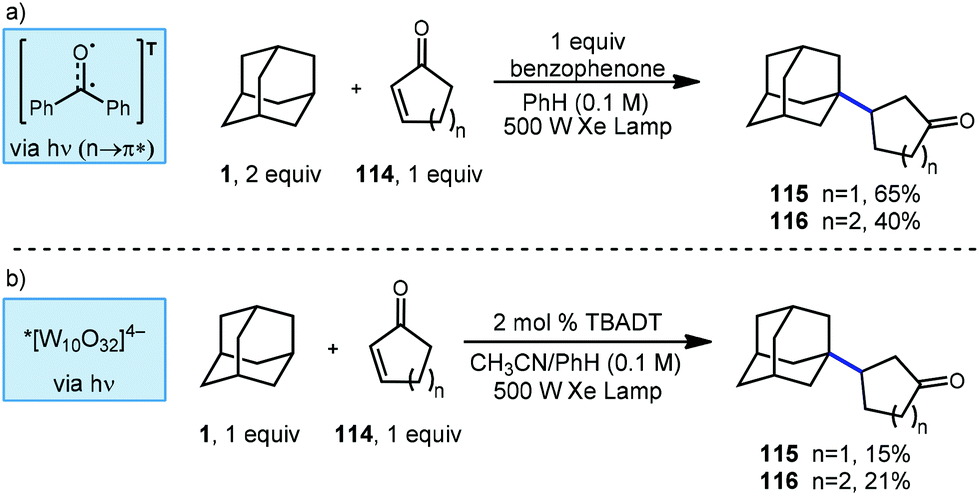 | ||
| Scheme 19 Light-driven radical additions to cyclic enones. | ||
Switching the photocatalyst to TBADT resulted in lower yields, with cyclopentanone product 115 giving 15% yield and the cyclohexanone product 116 giving 21% yield. The authors do not comment on the selectivity of the reaction with TBADT; however, based on low the selectivity of TBADT in other similar reactions (Scheme 10), both regioisomers would be expected to be present. The proposed reaction mechanism involves a direct H-atom abstraction by the excited photocatalyst as opposed to energy transfer or electron transfer pathways.
In 2019, our research group demonstrated a photochemical alkylation strategy using a broad range of alkenes and a variety of adamantanes with remarkable selectivity (>20:1 r.r.) for the 3° position (Scheme 20a).109 Inspired by previous work done by MacMillan and coworkers involving the functionalization of α-hydroxy C–H bonds,110 this approach uses a dual catalytic system involving an iridium photocatalyst (Ir(dF(CF3)ppy)2(d(CF3)bpy)PF6135 (see Scheme 21) in tandem with a quinuclidine-based HAT catalyst. Optimal results were obtained using a novel quinuclidinol-derived HAT co-catalyst 136 with a more electron-withdrawing sulfonate group. When these catalysts are used together under blue light irradiation, adamantane reacts with electron-deficient alkenes such as dehydroalanine derivative 117 to give the alkylated product 118 in 89% yield (Scheme 20a).
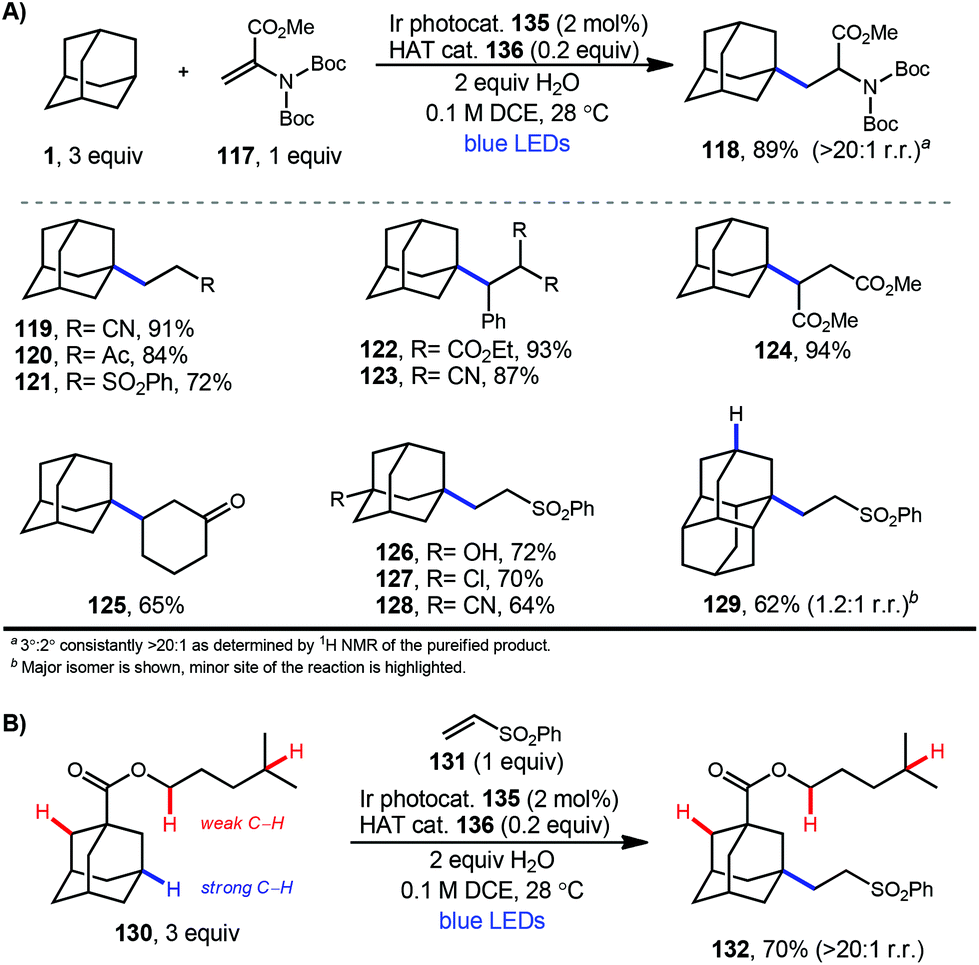 | ||
| Scheme 20 Site-selective alkylation of adamantane using dual photoredox catalysis. | ||
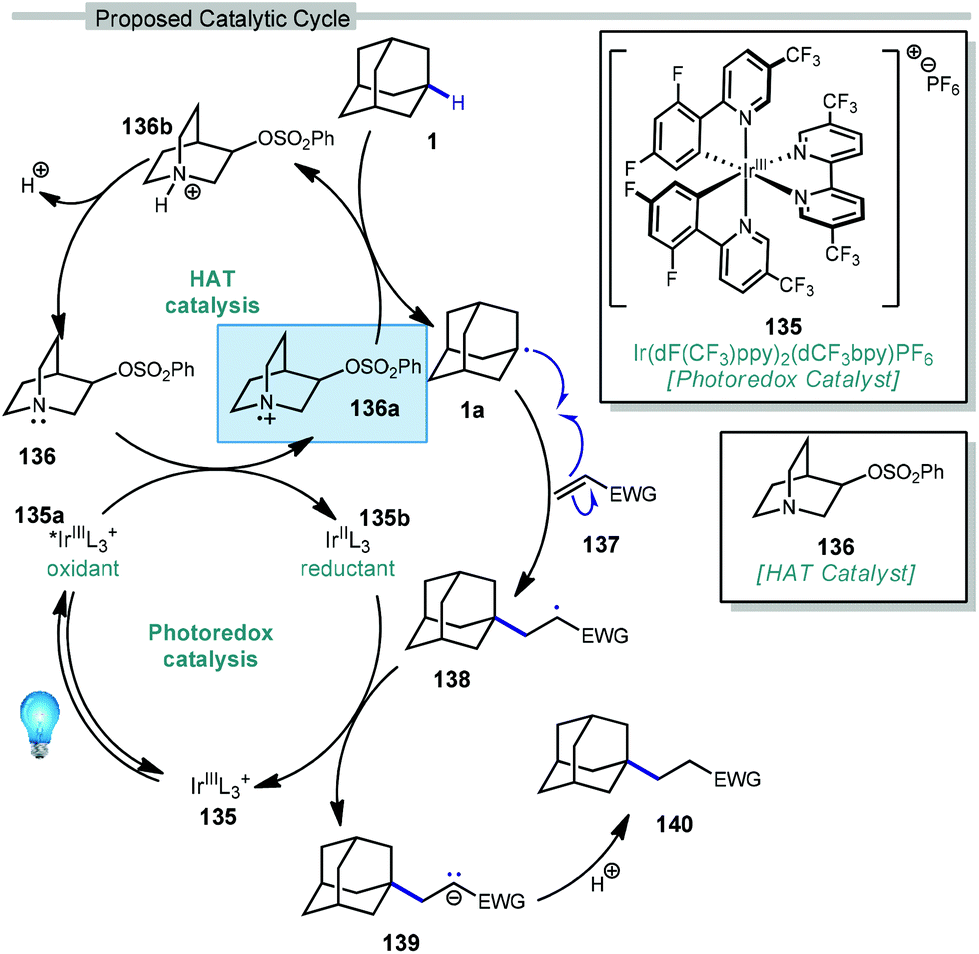 | ||
| Scheme 21 Proposed dual catalytic cycle for adamantane alkene additions. | ||
Alkylation proceeded smoothly for alkenes featuring a number of electron-withdrawing groups such as ketones, esters, nitriles, and sulfones (119–125, 65–94% yield). The robustness of the reaction also enabled a wide of adamantanes to perform well in this reaction (typically 60–75% yield). These included adamantanes substituted at the 1-position 126–128 (alkyl, aryl, hydroxyl, halide, nitrile, acetyl), diamantane 129, 2-adamantanone, and drug derivatives such as N-Boc amantadine and N-Boc memantine, which all reacted with high regioselectivity.
In this system, an electrophilic quinuclidinium radical cation (136a) is produced that is well suited for HAT from hydridic C–H bonds like those found at the methine positions of adamantane. Notably, this favorable polarity matching in the transition state of the HAT step enables the selective functionalization of stronger C–H bonds over weaker ones with impressive regioselectivity. Complex adamantane substrates such as 130 (Scheme 20b) featuring multiple weaker C–H bonds were alkylated with phenyl vinyl sulphone 131 to give the 3° adamantyl product 132 in 70% yield and >20:1 r.r. The reaction also showed resilience during direct competition experiments between equimolar amounts of adamantane and many other diverse substrates. These substrates featured a range of C–H bonds (e.g., 2° and 3° alkyl, benzyl, amidyl, formyl, etc.) including large polyfunctional substrates (e.g., limonin and progesterone) and were added without significantly competing with or diminishing yields of adamantane product 121.
A comparison of this catalyst system with other photocatalytic HAT systems described in this Review was carried out, as shown in Table 1. The intermolecular competition between adamantane and octanal only favored the reaction of adamantane with the quinuclidine catalyst, whereas all other catalysts were less efficient and led to ketone 134 as the major product. This remarkable selectivity is attributed to the preference for the most electron-rich “hydridic” C–H bond being activated, enhanced by the positive charge on abstractor 136a and the optimized electron-withdrawing nature of the sulfonate substituent.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst system | Yield 121 | Yield 134 | Selectivity |
| Ir-1=(Ir(dF(CF3)ppy)2(d(CF3)bpy)PF6; Q-1=quinuclidin-3-yl benzenesulfonate; PT = 5,7,12,14-pentac-enetetrone; TBADT =tetrabutylammonium decatungstate; Ir-2=(Ir(dF(CF3)ppy)2(dtbbpy)PF6; Acr =Nicewicz acridinium photocatalyst. |
||||
| 1 | Ir-1(135), Q-1 (136) | 63% | 14% |
4.5:1 |
| 2 | PT (187) | 13% | 26% | 1:2 |
| 3 | TBADT (78) | 8% | 41% | 1:5.1 |
| 4 | Ir-2, NBu4OBz | 7% | 34% | 1:4.9 |
| 5 | Acr (150), HPO42− | 7% | 7% | 1:1 |
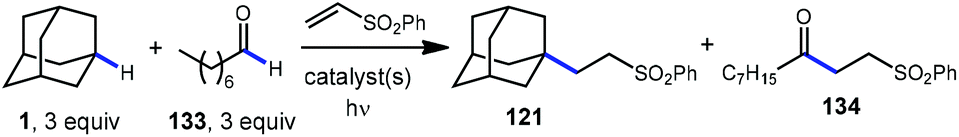
The driving force for the process is the formation of the relatively strong N–H bond in quinuclidinium 136b following the HAT event with adamantane (N–H BDE ∼100 kcal mol−1 or higher).111 This effect was leveraged by installing an electron withdrawing substituent on quinuclidine to further increase the N–H BDE and increase the efficiency of the reaction in terms of both yield and reaction time.
Another iridium photoredox-catalyzed alkylation, which uses chlorine radical to generate the adamantyl radical, was reported a few months prior to the quinuclidine-based method discussed above. While most implementations of iridium(III) photocatalysts use relatively inert counter ions such as hexafluorophosphate, Barriault and coworkers showed that using a catalyst containing a chloride counter ion, [Ir(dF(CF3)ppy)2(dtbbpy)]Cl, chlorine radical can be generated. This catalyst was used to alkylate adamantane using dimethyl maleate (98, Scheme 22) to form the adamantyl succinate 99 in 61% yield (3°:2° = 62:38).112 The reaction is thought to proceed similarly to the dual catalytic cycle depicted in Scheme 21 with chlorine radical serving in place of quinuclidine radical cation 136a to perform HAT, leading to the diminished regioselectivity. Nonetheless, this strategy was effective for a variety of other hydrocarbons, ethers, aldehydes and heteroatom-activated starting materials.
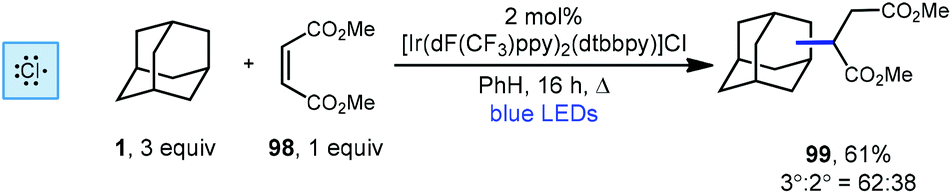 | ||
| Scheme 22 Radical addition to dimethylmaleate using an iridium photocatalyst. | ||
Nicewicz and Alexanian have also recently developed a general strategy for organic photoredox-catalyzed C–H abstraction from an array of aliphatic substrates followed by trapping with a variety of radical acceptors using an acridinium photocatalyst, phosphate salt and blue light (Scheme 23).113 This general process reported in 2018 allowed for the direct diversification of C–H bonds into new C–N, C–S, C–X and C–C bonds. The methodology uses a highly oxidizing acridinium photocatalyst 150 to oxidatively generate oxygen-centered phosphate radicals capable of doing C–H abstraction of aliphatic compounds including adamantane. When using an aryl-adamantyl Adapalene precursor with 3-butenone (141) as the radical acceptor, ketone product 148 was formed in 45% yield. This reactivity extended beyond Giese type reactions to non-alkene radical acceptors such as 4-(trifluoromethyl)benzenesulfonyl azide (142) and N-fluorobenzenesulfonimide (NFSI, 143). N-Phthalimidyl memantine underwent fluorination to give fluorinated product 146 in 86% yield while azidation to provide 145 was possible in 55% yield. Unsubstituted adamantane was also converted to the azide in 75% yield.
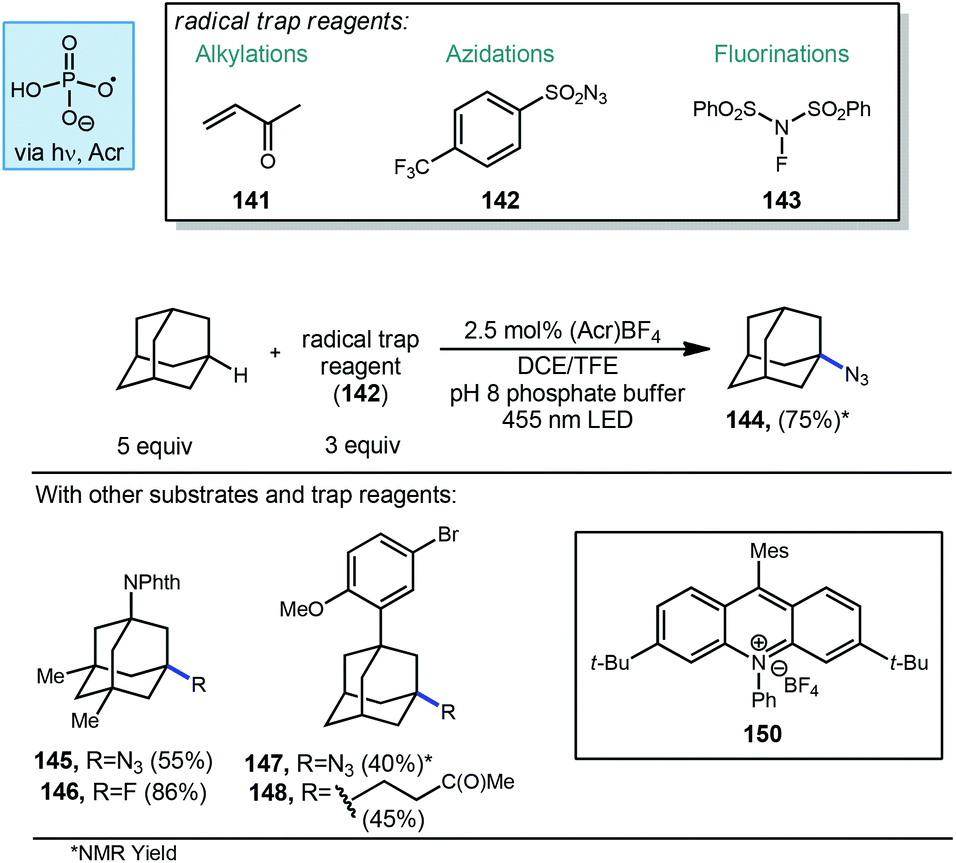 | ||
| Scheme 23 Photochemical C–H functionalizations of adamantane using acridinium catalyst 150. | ||
Recently, Knowles, Alexanian and coworkers have also demonstrated a method for functionalizing unactivated C(sp3)–H bonds using a phosphate and a photocatalyst. Unlike the work from Nicewicz discussed above involving a phosphate radical created through single-electron oxidation, a thorough mechanistic investigation indicates a mechanism that proceeds via multisite-proton-coupled electron transfer (MS-PCET).114 Using an iridium photocatalyst together with a monobasic phosphate salt, adamantane and its substituted derivatives were alkylated with 1,1-bis(phenylsulfonyl)ethylene (152, Scheme 24) in moderate to good yield (153–155, 58–80%). A protected memantine derivative was also alkylated in 40% yield. NMR titration experiments and crystallographic evidence indicate the formation of a noncovalent 1:1 Ir(III)-phosphate complex via hydrogen bonding between 3,3′-bipyridyl positions and the phosphate oxygen (see 156). The reaction is belived to be dependant on the formation of this complex since virtually no alkylation was observed when a competitive binder (fluoride ion) was included or when an Ir(III) catalyst with fluorinated 3,3′-bipyridyl positions was used.
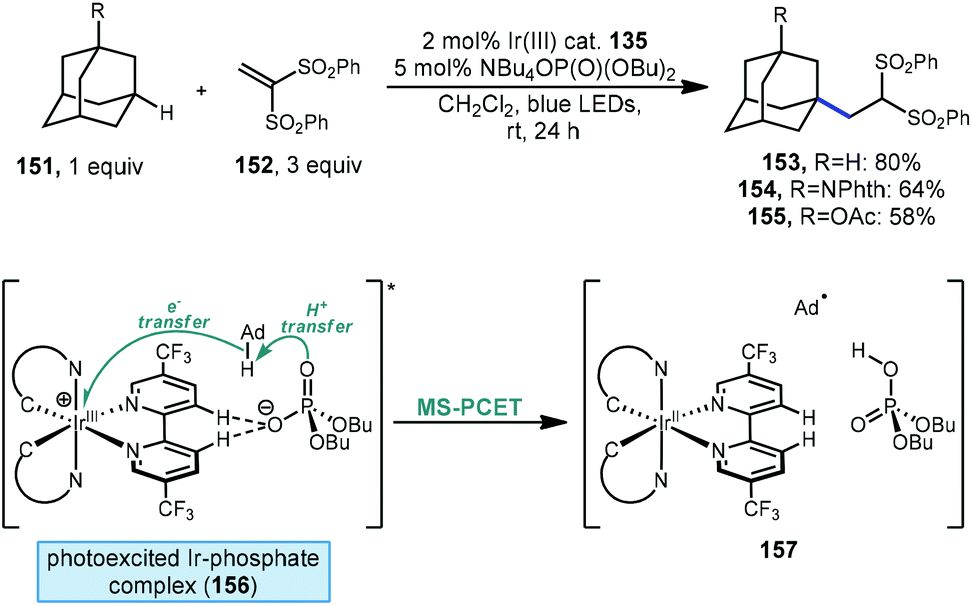 | ||
| Scheme 24 Alkene addition via photocatalytically induced proton-coupled electron transfer. | ||
Similar to HAT, PCET generates the adamantyl radical via homolytic C–H bond cleavage. However, unlike HAT, which involves the movement of a proton and an electron together to a single location (one orbital), MS-PCET involves the movement of the proton and electron to two separate orbitals. Quenching studies suggest a mechanism involving the excitation of the pre-associated Ir(III)-phosphate complex followed by a concerted PCET process with the adamantane substrate to give the adamantyl radical, the reduced Ir(II) photocatalyst and the protonated phosphate base (see 157). Once generated, the adamantyl radical proceeds to react with the olefin and Ir(II) species in a manner similar to the process shown in Scheme 21.
4.3. Additional non-photochemical alkylations
A catalytic oxyalkylation of alkenes with adamantanes and other alkanes was reported by Ishii et al. in 2001 using the PINO radical as the reactive intermediate and a cobalt co-catalyst in the presence of molecular oxygen.115 This reaction resulted in oxyalkylated products in both the hydroxylated and ketone oxidation state (Scheme 25a). Under optimized conditions, methyl fumarate (158) and adamantane gave 98% conversion with a 78% yield (71/29 ratio of hydroxy product 159 and ketone product 160. A variety of alkenes were also tested using dimethyl adamantane with yields ranging from modest to excellent however in most cases, a mixture of products was observed (Scheme 25b, 161–165).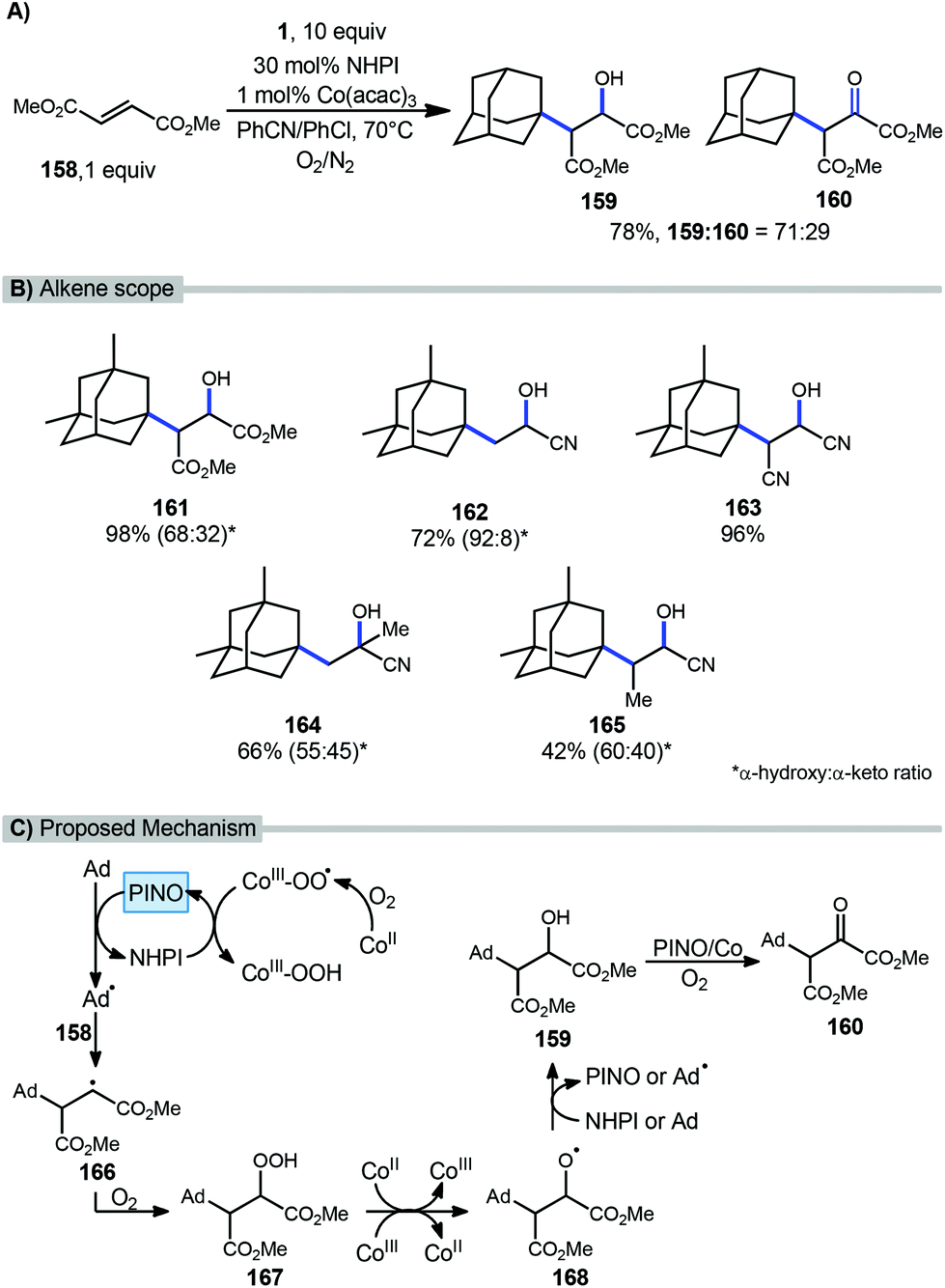 | ||
| Scheme 25 Cobalt-catalyzed oxyalkylation of alkenes with adamantane. | ||
A proposed mechanism is summarized in Scheme 25c. These reactions are thought to be initiated by single-electron transfer (SET) to Co(acac)3 to give a Co(II) species that undergoes reaction with O2 to form a Co(III)-O2 complex. HAT from NHPI to this cobalt complex generates the incipient PINO radical. PINO then goes on to generate the adamantyl radical via another HAT process. Adamantyl radical addition into the alkene 158 affords alkyl radical intermediate 166 which is quickly trapped by O2 leading to hydroperoxide 167. This hydroperoxide undergoes decomposition by Co ions giving the alkoxy radical intermediate 168. Additional HAT from either NHPI or adamantane yields the hydroxylated product 159. Further oxidation to α-keto ester 160 is then possible in the presence of O2 with the assistance of Co(acac)3 and PINO.
Roberts and coworkers reported a fascinating metal-free radical cascade reaction using allyloxyl intermediate 176 as the key radical chain-propagating reagent (Scheme 26).116 In this work, the reaction of interest involved excess adamantane, N-methylmaleimide as the acceptor (169, NMM), “acetal reagent A” 170, and di-tert-butyl hyponitrite (TBHN) as a radical initiator. With adamantane, the final ester product 171 was obtained in 50% yield as a 79:21 regiochemical mixture favoring the tertiary position. Product formation proceeds as part of a complex radical cascade enabled by acetal reagent A.
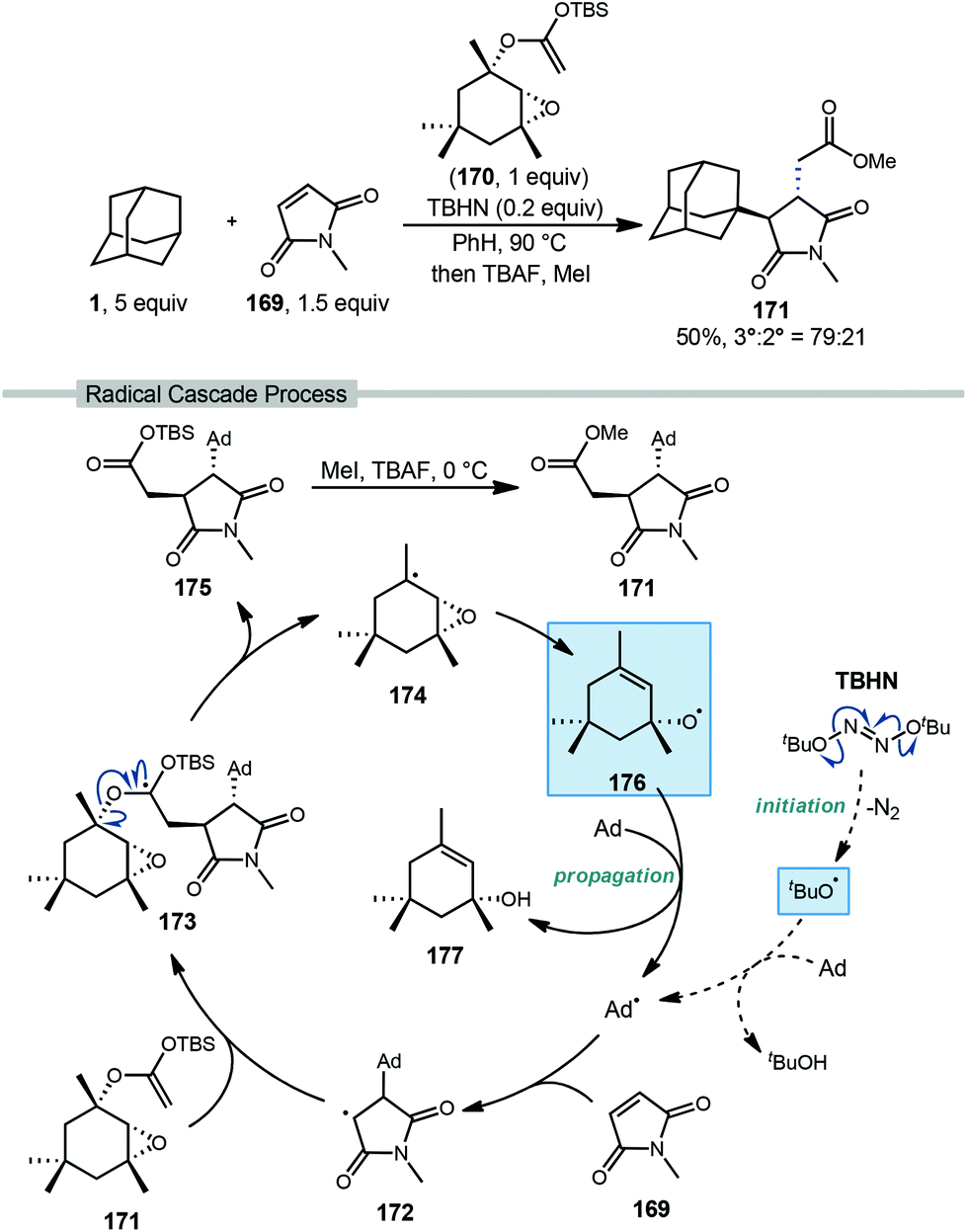 | ||
| Scheme 26 Three-component coupling of adamantane via polarity-matched alkene additions reported by Roberts. | ||
5. Addition–fragmentation processes with alkene and alkyne reagents
The addition–fragmentation sequence (Scheme 27) is well represented in the realm of radical chemistry and has been reported in a variety of cases involving the adamantyl radical.117 After C–H abstraction, the resulting adamantyl radical 1a then adds into an appropriate radical acceptor such as an alkene leading to a β-adamantyl radical intermediate 178 which undergoes fragmentation to an alkenylated adamantane product 179. Alkynes, which are valuable building blocks in organic synthesis and as important structural compounds in both material science and chemical biology,118,119 also behave similarly but instead involve fragmentation of the analogous vinyl radical (180).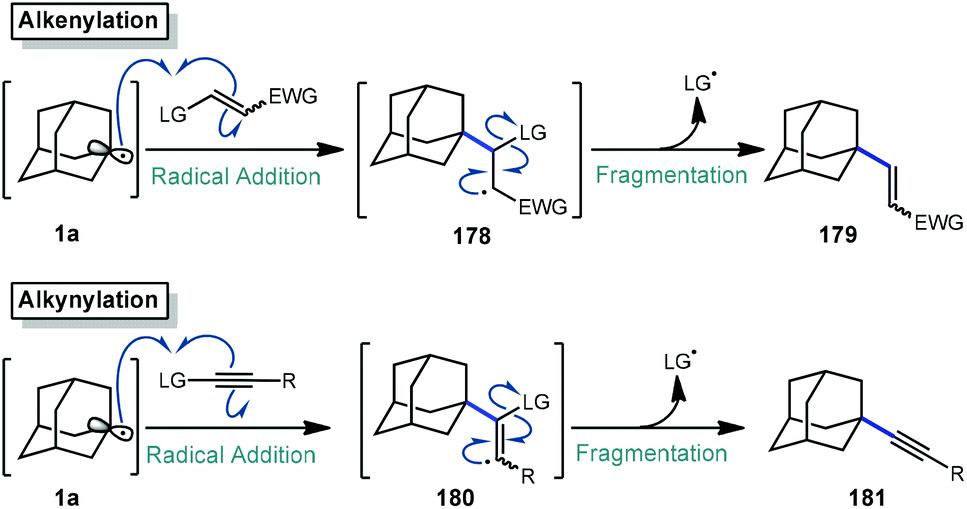 | ||
| Scheme 27 General addition-fragmentantion sequences. | ||
The reactions discussed in this section involve addition into electron-deficient alkene or alkyne acceptors to accomplish alkenylative, allylative, or alkynylative transformations (Scheme 27). Commonly, radical acceptors feature an electron-withdrawing group to aid in stabilizing radical intermediate 178 (or 180) as well as a leaving group that promotes a facile fragmentation immediately following addition. These reactions may be accomplished by both metal-free and metal-mediated radical processes. We note that the formylation reaction discussed in section 2.3 proceeds via a similar fragmentation following addition to a CN radical acceptor.
5.1. Decarboxylative alkenylation
Sun et al. demonstrated that styrenyl adamantanes (185, Scheme 28) can be made using cinnamic acid (182) in the presence of DTBP and heat.120 In this case, homolytic thermolysis of DTBP results in tert-butoxyl radical that acts as the C–H abstractor giving rise to the reactive adamantyl radical. This reaction is then thought to proceed via initial radical addition to form benzylic radical 184, followed by loss of carbon dioxide and an additional H-atom. This addition–fragmentation process is selective for the tertiary adamantyl position, favoring it over the secondary position and formed the styrenyl product in an 83:17 ratio, consistent with other HAT processes involving the tert-butoxyl radical.
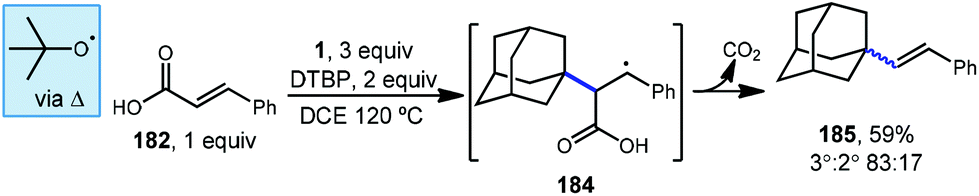 | ||
| Scheme 28 Decarboxylative alkenylation of adamantane using cinnamic acid. | ||
5.2. Photochemical allylation
Allylation of adamantanes has been demonstrated using 1,2-bis(phenylsulfonyl)propene (186, Scheme 29) as an allylating agent.121 The adamantyl radical is generated using 5,7,12,14-pentacenetetraone (PT, 187) with visible light irradiation. The excited state 187a, with biradical character similar to photoexcited benzophenone, is a competent C–H abstractor, presumably leading to semiquinone form 187b.122–124 Adamantyl radical 1a then intercepts the electrophilic sulfonylated propene to generate radical intermediate 194. Fragmentation and transfer of a H-atom from the PT catalyst results in allylated adamantane, 188 and benzenesulfinic acid as a byproduct. Under these conditions, 188 was isolated in 63% yield with the allylated methine being favored in a 9:1 ratio over the methylene position. These conditions also were tolerated by a range of substituted adamantanes. Using substituted adamantanes 189–193 generally favored C–H functionalization at one of the remaining methine positions at a ratio of >95:5.
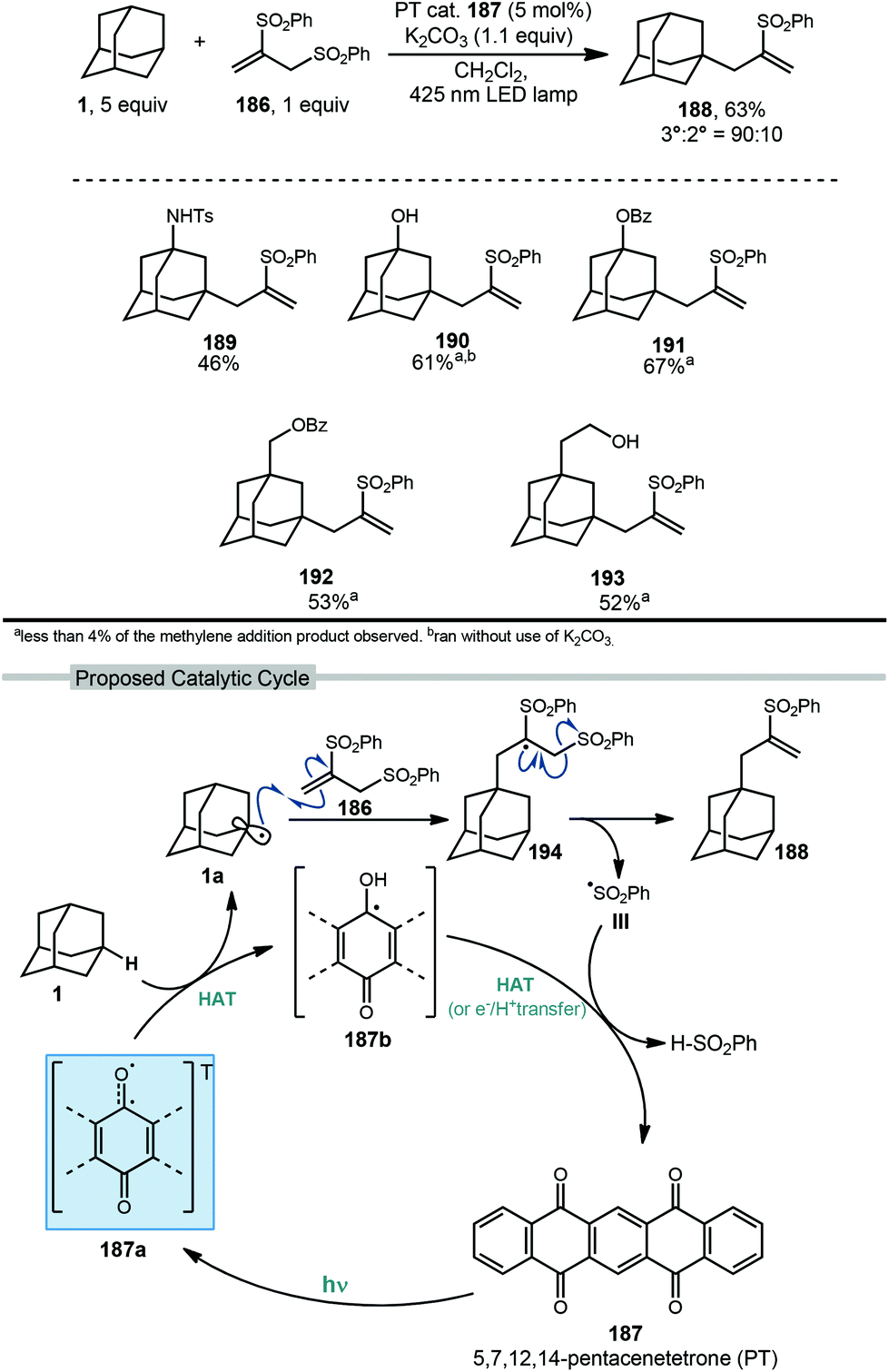 | ||
| Scheme 29 5,7,12,14-Pentacenetetrone-catalyzed allylation using 1,2-bis(phenylsulfonyl)propene 186. | ||
Wang and coworkers recently developed a method for the directed allylation of adamantane using a tethered amidyl radical via intramolecular 1,5-HAT (202, Scheme 30).125 When the oxyarylamide group is tethered at the 2° position of adamantane 195, allylation using sulfone 196 occurred at the 3° position (via 3° C–H abstraction) to give allyl adamantane 197 in 73% yield. Moving the tether to the 3° position of adamantane 198 led to allylation at the 2° position (2° C–H abstraction) in 67% yield, indicating that in both cases the highly favored 1,5-HAT was driving the selectivity.
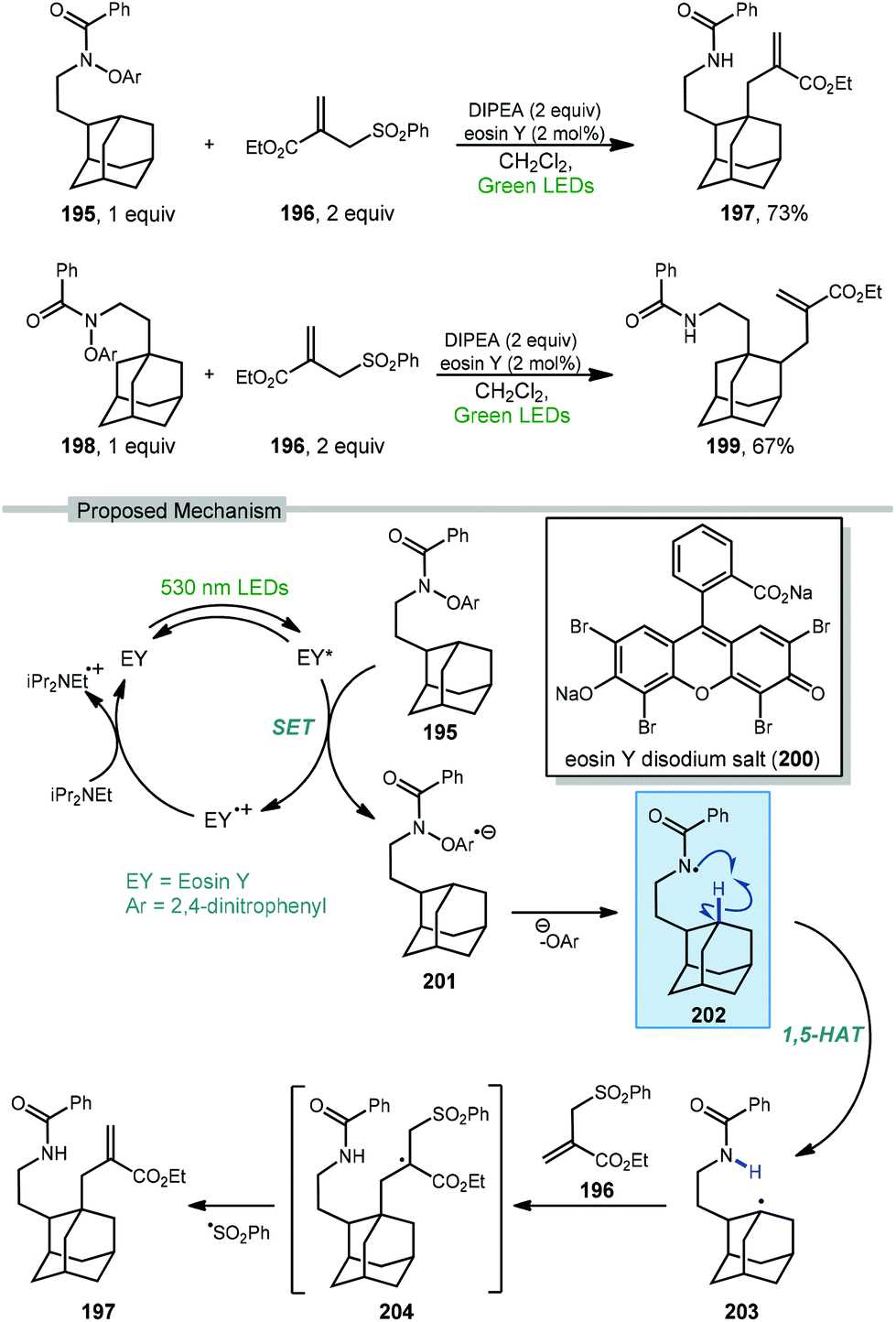 | ||
| Scheme 30 Directed photocatalytic allylation of adamantane via tethered amidyl radical intermediates. | ||
The amidyl radical is generated photocatalytically using green LEDs to excite eosin Y (EY, 200) which undergoes SET to the prefunctionalized adamantane featuring di-nitroaryloxy amide 195. The resulting radical anion, 201, then fragments to give the key amidyl radical 202. Similar to a Hofmann–Löffler–Freytag reaction, this remote amidyl radical was used to generate the adamantyl radical 203via a 1,5 C–H abstraction.126 Interception of the allyl sulfone by the adamantyl radical and subsequent fragmentation of 204 gives the 3° allylated adamantane.
5.3. Photochemical alkenylation
Addition/fragmentation sequences also work well in the preparation of alkenylated adamantyl derivatives through the formation of a new C(sp3)–C(sp2) bond. In 2014, Inoue and Kamijo used benzophenone for the photochemical alkenylation of substituted adamantanes using trans-1,2-bis(phenylsulfonyl)ethylene 206 to the give corresponding alkenylated sulfonyl-adamantanes (207–209, Scheme 31).127 The formation of these vinyladamantanes proceeded with high selectivity for the methine position with good yields in MeCN under irradiation from a 100 W mercury lamp. The presence of proximal heteroatoms (X=O, N) typically activates adjacent C(sp3)–H bonds through hyperconjugation with the nearby lone pair.56 C(sp3)–H bonds of heteroatom-substituted carbons are therefore typically selectively abstracted over other aliphatic positions as seen in the reaction with ambroxide (210). Interestingly, oxa- and azaadamantanes (211 and 212, respectively) were the only exceptions to this rule according to Inoue and Kamijo, presumably because no hyperconjugation between the lone pair and C–H bond can occur within the rigid cage-like structure of these hetero-adamantanes.128
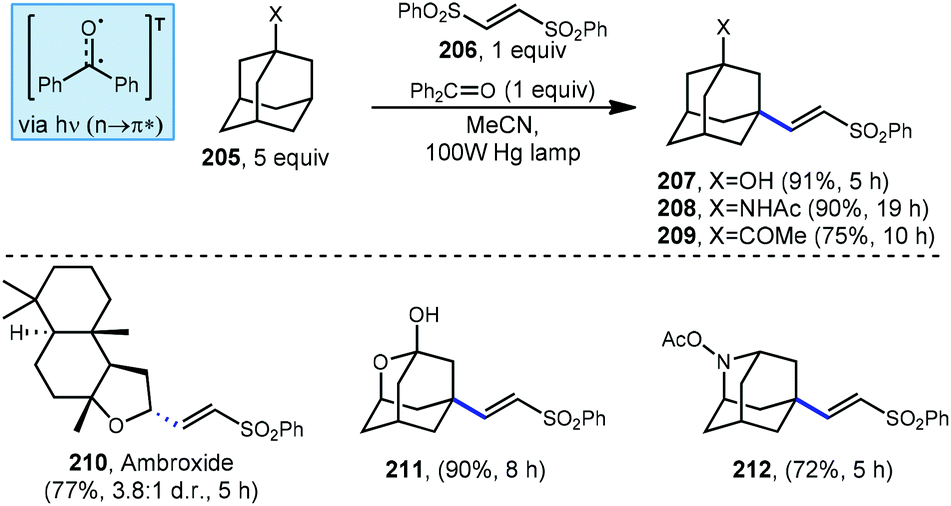 | ||
| Scheme 31 Photochemical alkenylations of substituted adamantanes using benzophenone. | ||
5.4. Metal-catalyzed alkenylation
A copper-catalyzed radical addition/cyclization of various unactivated cycloalkanes including adamantane with alkynyl-phosphine oxide 213 to give cyclic alkenylated products was reported by Zhao et al. in 2018 (Scheme 32).129 The cyclic benzo[b]phosphole oxide product 214 forms from two sequential C–H abstractions and the formation of two new C–C bonds. The initial tert-butoxyl radical is formed after the Cu(I)-assisted homolysis of DTBP with heating. Addition of the adamantyl radical into electron-deficient alkyne 213 results in the key alkenyl radical intermediate 215. This radical can then follow two regiochemically divergent paths both leading to the same product. In path A, ortho-addition of the alkenyl radical leads to delocalized radical 216. In Path B, ipso-addition of the alkenyl radical results in the 4-membered spirocyclic intermediate 217 which opens to give phosphorus-centered radical 218, followed by ortho-readdition to give aryl radical 216. By either proposed pathway, radical 216 can re-aromatize via oxidation with Cu(II)(Ot-Bu) to give the product and regenerate Cu(I).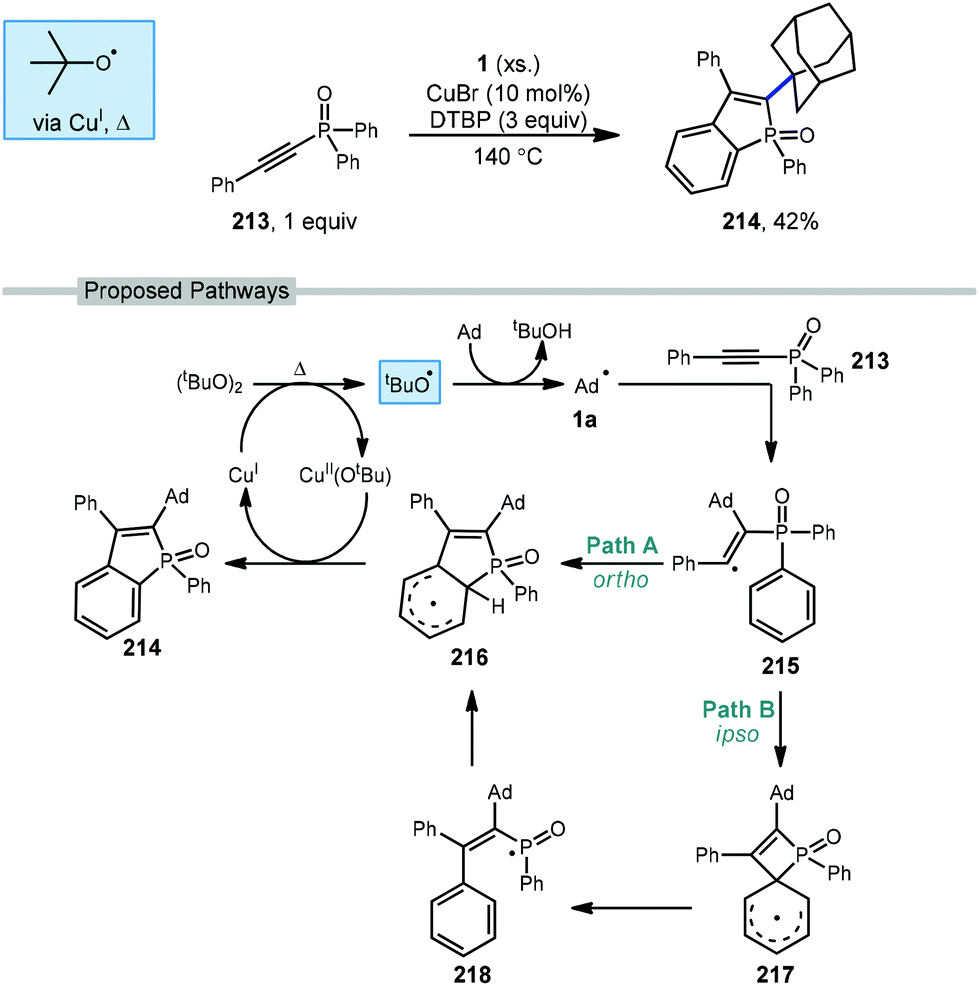 | ||
| Scheme 32 Copper-catalyzed alkenylative cyclization using alkynyl-phosphine oxide 213. | ||
5.5. Alkynylation
An early example of a metal-free C–H alkynylation was reported by Fuchs using alkynyl triflone 219 to functionalize a variety of C–H bonds, including those on adamantane (Scheme 33).130,131 Using AIBN and heat for initiation, the tertiary alkynylated adamantane 220 was obtained in 60% yield. The authors note the possibility of two mechanistic pathways depending on the regiochemistry of the initial adamantyl radical addition to the alkyne. In the case of β-addition (path A), sulfonyl vinyl radical 221 undergoes homolytic fragmentation to give vinylidene carbene 222 and (trifluoromethyl)sulfonyl radical 223. Once formed, the vinylidene carbene undergoes a Fritsch–Buttenberg–Wiechell rearrangement to the alkynylated product.132 Alternatively, α-addition to the alkyne (or a 1,2-shift from 221) affords β-sulfonyl vinyl radical 224. This intermediate can then eject (trifluoromethyl)sulfonyl radical to form the substitution product. In both cases the transient sulfonyl radical undergoes further fragmentation to sulfur dioxide and trifluoromethyl radical which propagates the reaction by C–H abstraction, forming fluoroform as the byproduct.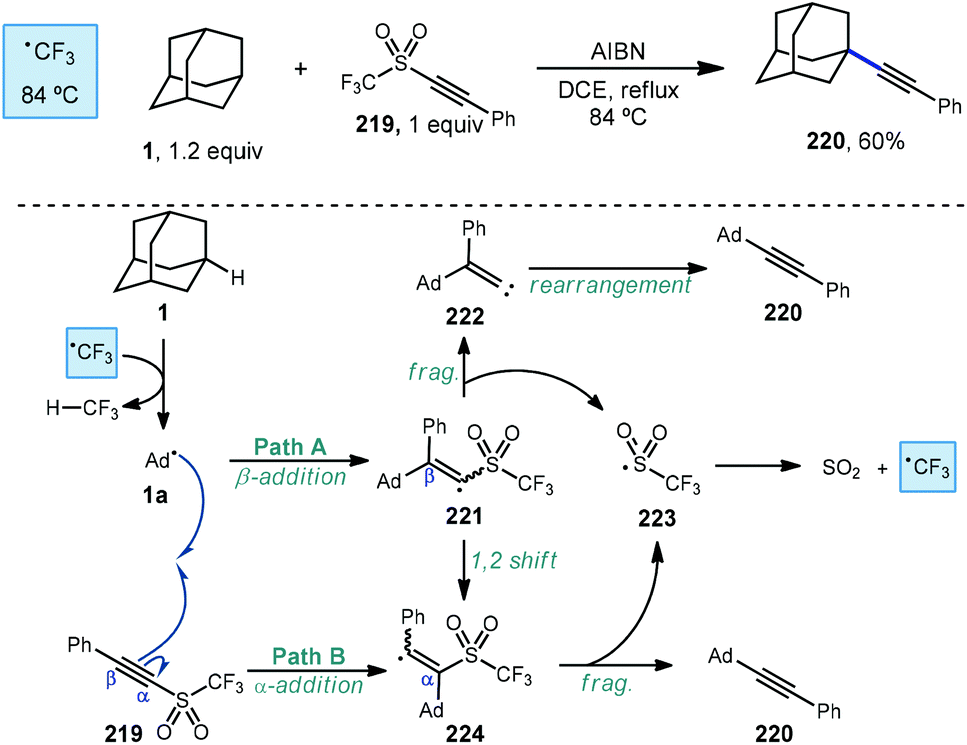 | ||
| Scheme 33 Adamantane alkynylation using alkynyl-triflone 219. | ||
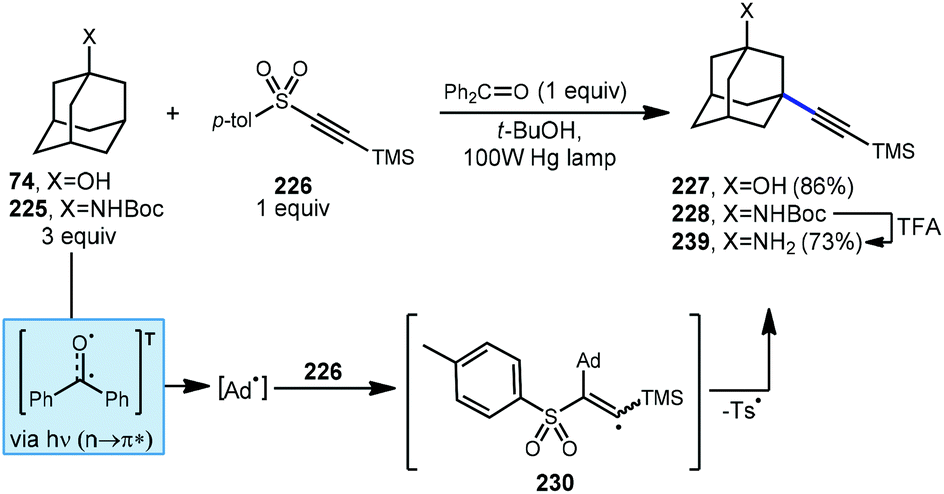
Alkynylative transformations of substituted adamantanes using metal-free photocatalytic methods have also been demonstrated using tosyl-(trimethylsilyl)acetylene 227 with benzophenone and a mercury lamp (Scheme 34).133 Upon irradiation with UV light, the triplet excited state is generated which then abstracts a hydrogen atom from the tertiary position of adamantane, generating the adamantyl radical. This intermediate then reacts with the electron-deficient alkyne and subsequent release of the toluenesulfinyl radical from alkenyl intermediate 231 generates the alkynylated adamantyl product, similar to the Fuchs example above. Under these conditions, 1-adamantanol (74) was alkynylated in 86% yield and N-Boc-protected amantadine was alkynylated and deprotected to the free amine using TFA in 73% yield over 2 steps (225 → 228 → 229).
 | ||
| Scheme 34 Photochemical alkynylation of substituted adamantanes using tosyl-(trimethylsilyl)acetylene 226. | ||
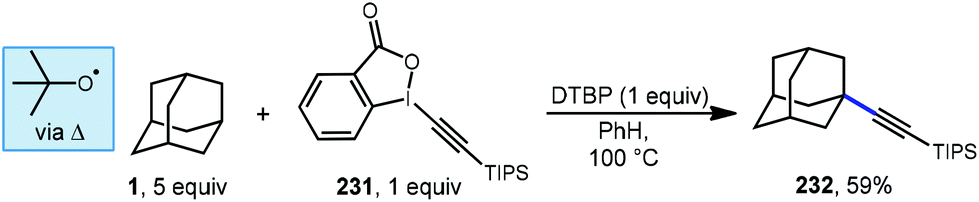
Xu and coworkers investigated an efficient method for the direct alkynylation of substrates containing unactivated C(sp3)–H bonds under metal-free conditions using an ethynyl benziodoxolone (EBX) 231 (Scheme 35).134 When adamantane was used, C–H functionalization was selective for the tertiary position and a 59% yield was reported for the adamantyl product 232. The authors did not quantify the regioselectivity for adamantane but the same reaction with methylcyclohexane reacted at the tertiary position selectively (>25:1 r.r.). The reaction was completely suppressed by the radical trapping agent TEMPO, and the alkyl-TEMPO product was observed, suggesting the reaction proceeds via a radical intermediate. A plausible mechanism begins with thermal homolytic cleavage of DTBP to generate the tert-butoxyl radical, which reacts with adamantane giving the adamantyl radical. This is followed by addition to the triple bond of the EBX reagent with subsequent β-elimination to give the final alkynylated product.
 | ||
| Scheme 35 Adamantane alkynylation using EBX reagent 231. | ||
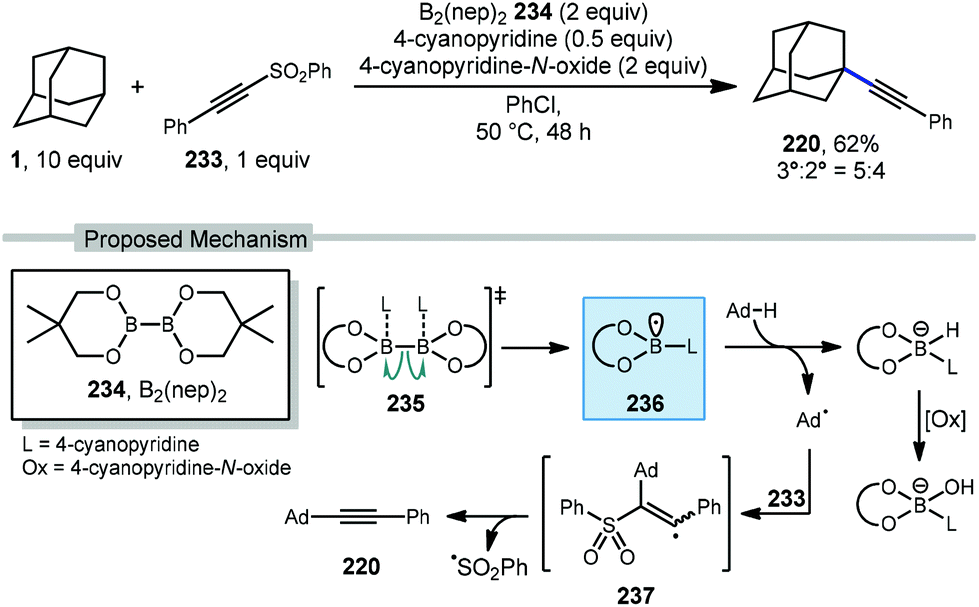
Recently, Tang and coworkers reported an alkynylation of adamantane using a pyridine-boryl radical (Scheme 36).135 Using bis(neopentyl glycolato)-diboron 234, 4-cyanopyridine is added to facilitate ligand-induced B–B homolysis to generate the key boryl radical (see 235 → 236). The adamantyl radical is then produced which adds into ((phenylethynyl)sulfonyl)benzene (233). The resulting vinyl radical intermediate 237 then fragments with loss of sulfonyl radical to afford the alkyne product 220. This alkynylation approach gave the final product in 62% yield with only marginal preference for the tertiary adamantyl position (3°:2° = 5:4). The inclusion of 4-cyanopyridine-N-oxide as oxidant was needed for best results; however, control experiments using cyclohexane without the use of any oxidant still proceeded in 12% yield suggesting that oxygen may also function as an oxidant to some degree.
 | ||
| Scheme 36 Boryl radical-mediated adamantane alkynylation. | ||
6. Arylation
Aryl adamantanes appear both as scaffolds in nanomaterials and in bioactive compounds, such as the marketed acne medication Adapalene (5), known by the generic name differin. These compounds can be constructed by non-radical methods such as transition metal catalysis or by radical-mediated C–H arylation, which will be discussed here.6.1. Photooxidations using TCB
In 1996, Albini et al. used 1,2,4,5-tetracyanobenzene (TCB) to arylate adamantane in a UV-promoted process.136,137 The mono-arylated product 239 (Scheme 37a) was predominantly formed by reaction exclusively at the tertiary position while the di-arylated product 240 was formed to a lesser degree. Under UV-light, Albini had previously shown that TCB in its singlet excited state is a strong oxidant capable of inducing SET from a variety of alkanes.138 After oxidation, a solvent-caged adamantyl radical cation/TCB radical anion pair is generated (see 241, Scheme 37b). The adamantyl radical cation then either loses a proton to solvent or, in the case of a substituted adamantane, may undergo substituent elimination to create the tertiary adamantyl radical. The C–C cleavage that occurs with substituent elimination is an endergonic process while deprotonation to form the adamantyl radical is exergonic (approx. −17.1 kcal mol−1) and is therefore the favored route to the adamantyl radical. After its formation, the adamantyl radical then quickly recombines with the radical anion of TCB to form anion 242 (analogous to a Meisenheimer complex) and rearomatization through the loss of cyanide then gives the arylated adamantyl product.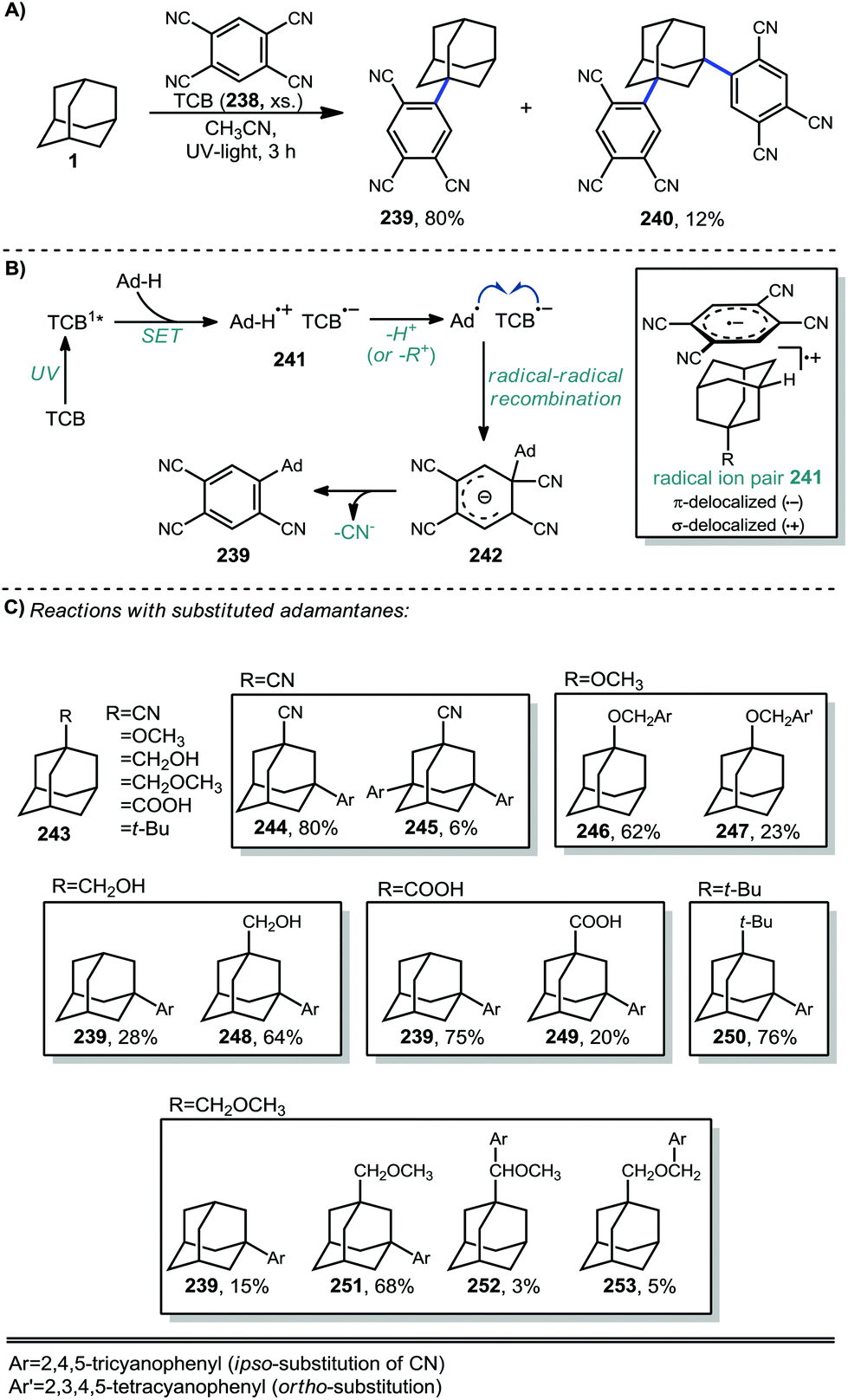 | ||
| Scheme 37 Light-driven arylation of adamantanes using TCB. | ||
Several substituted adamantanes (243) were subjected to the same conditions, resulting in the formation of a variety of products formed through mono- or bis- C–H arylation of the cage, arylation with loss of the substituent, or C–H arylation of the substituent itself (Scheme 37c). Arylation of 1-methoxyadamantane occurred on the methoxy substituent rather than on the adamantyl moiety, giving two benzylic ether products. The expected 2,4,5-tricyanophenyl substitution product 246 was predominantly produced (63% yield) but interestingly, an appreciable amount of ortho-functionalization also occurred to form 2,3,5,6-tetracyanophenyl product 247 (23% yield). 1-Adamantanecarboxylic acid proceeded mostly with decarboxylation to the monoaryl adamantane product 239 with a minor amount retaining the carboxy group (249).
More recent work by Schreiner, Fokin, and coworkers showed that arylation with TCB on higher order diamondoids (254–257) proceeds with 100% apical selectivity (Fig. 4A).75,139,140 This remarkable selectivity surpasses the vast majority of reactions of higher order diamondoids such as acetylation (see Fig. 3). Delocalization of the radical cation through the Sigma framework, as represented in blue in the electrostatic potential surface in Fig. 4B, leads to elongation and weakening of the apical C–H bonds (Fig. 4C), which are more easily deprotonated to give apical radicals selectively. The UV-promoted arylation was demonstrated up to pentamantane with high selectivity. Virtually no functionalization reactions beyond pentamantane have been reported in the literature, perhaps due to difficulty in accessing sufficient quantities of pure starting materials.
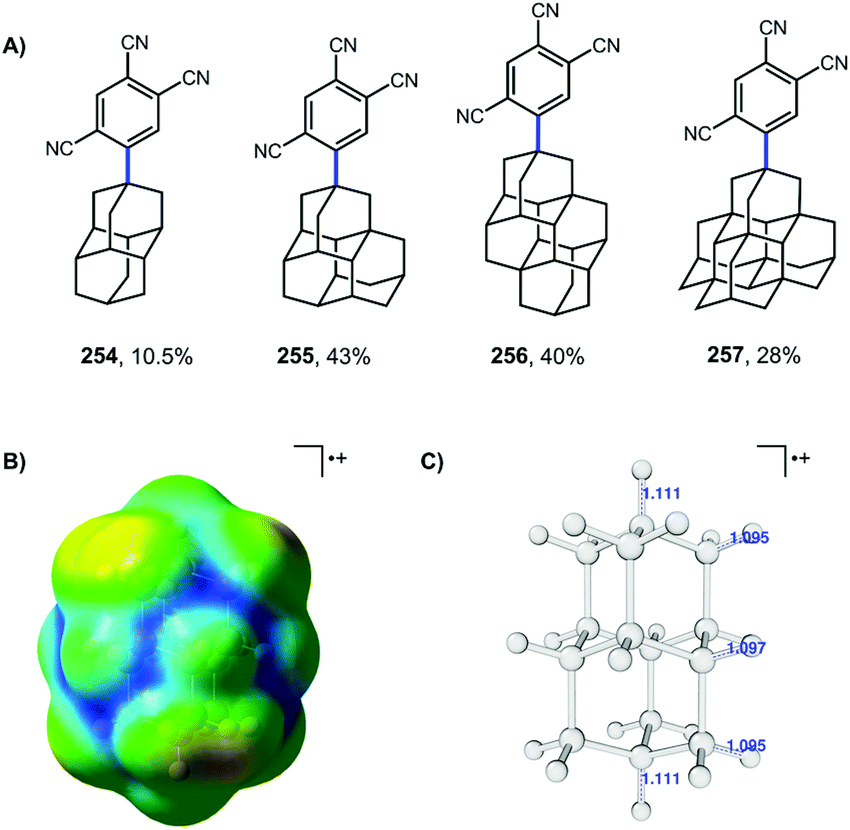 | ||
| Fig. 4 (A) TCB arylation of higher order diamondoids. (B) Electrostatic potential surfaces for the diamantyl radical cation (symmetrically σ-delocalized). (C) C–H bond lengths (Å) for diamondoid radical cations are longest at their geometric center (apical postions). Structures optimized in Gaussian by the authors using B3LYP/6-31(d). | ||
6.2. Minisci-type arylations
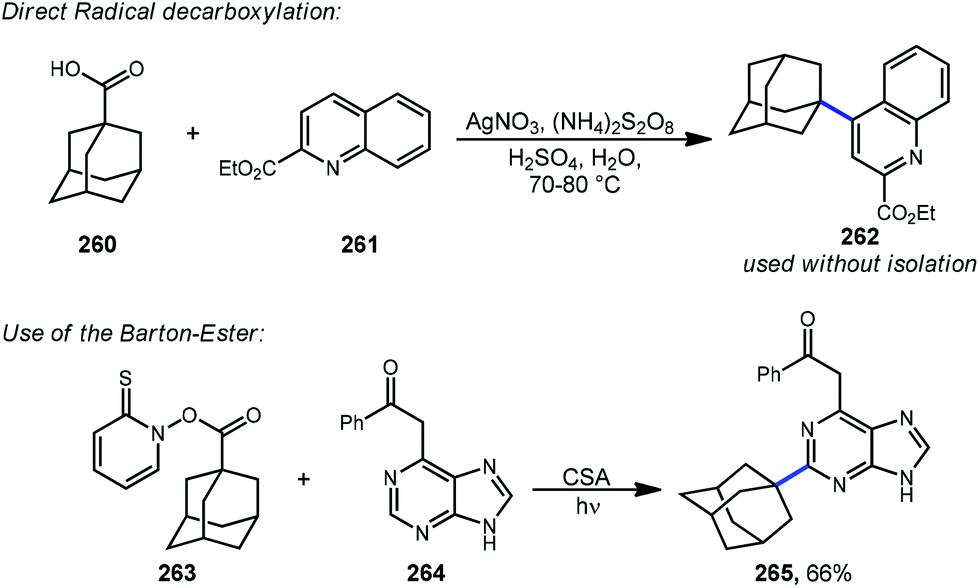
Minisci reactions like the one shown in Scheme 38 are characterized by the addition an alkyl radical to an electron-deficient heteroaromatic compound, leading to the formation of a new C(sp3)–C(sp2) bond.141 Heteroaromatic adamantane-containing nucleoside analogs have been known to possess antiherpetic63 and antifolate142 activity and can be synthesized using Minisci-type reactions. Traditionally these reactions have applied stoichiometric ammonium persulfate and silver nitrate to induce an oxidative decarboxylation to generate the alkyl radical.143 This decarboxylative approach using 1-adamantanecarboxylic acid (260) was taken in the preparation of 262, an adamantane containing anti-tuberculosis precursor as shown in Scheme 39.144 Adamantane was also used in early work by Barton for the alkylation of caffeine 264 and other purines.145,146 Since then, several methods have been developed that are metal-free or do not rely on pre-functionalized adamantanes to arrive at the adamantyl radical intermediate. | ||
| Scheme 38 General Minisci-type radical addition. | ||
 | ||
| Scheme 39 Traditional Minisci approaches. | ||
In 2013, Antonchick and coworkers developed a Minisci-type arylation reaction using a selective oxidative cross-coupling approach.147 One example, shown in Scheme 40, shows the arylation of adamantane using 4,7-dichloroquinoline (266) in the presence of phenyliodine bis(trifluoroacetate) (PIFA) as oxidant and NaN3, as a critical additive. The key H-atom abstractor is likely the azide radical and the corresponding arylated product 267 was obtained in 84% yield as a single isomer. Overall, Antonchick et al. used this metal-free oxidative cross-coupling approach of heteroarenes and a variety of alkanes to obtain arylated products under mild, non-acidic conditions, with short reaction times and in high yields.
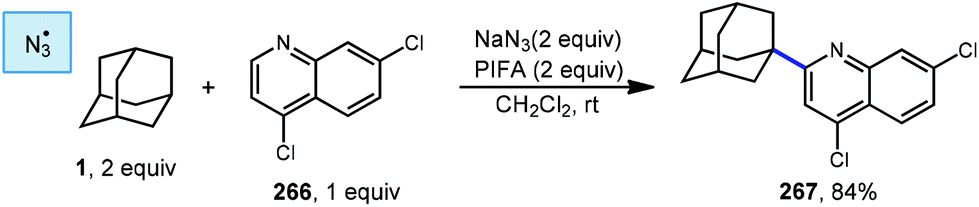 | ||
| Scheme 40 Oxidative Minisci arylation using sodium azide. | ||
Kwong et al. and Guo et al. have also shown that DTBP can enable the oxidative cross coupling of adamantane with indoles148 (268 → 269) and purine nucleosides149 (270 → 271) under similar reaction conditions (Scheme 41). Both methods were developed using neat cycloalkanes or cyclic ethers, however adamantane, a solid, requires an aromatic solvent (e.g., xylene or benzene). In both cases, an excess of five equivalents of adamantane was enough for the reaction to proceed in moderate to good yield.
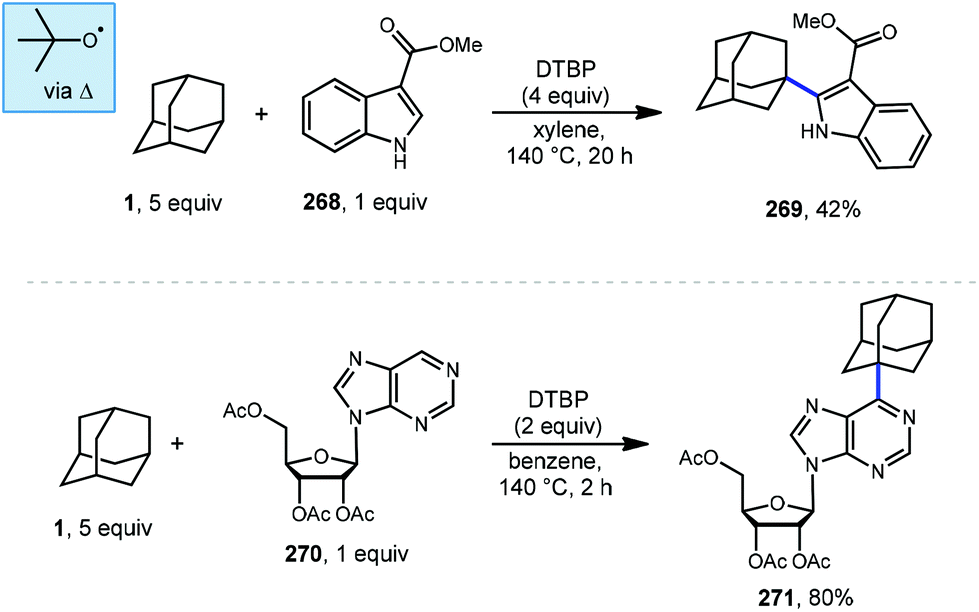 | ||
| Scheme 41 Minisci reactions of indoles and purines using DTBP. | ||
More recently in 2017, Li et al. also found the tert-butoxyl radical was effective for oxidative cross couplings of adamantane with oxazoles and thiazoles (Scheme 42).150 The tert-butoxyl radical in this case does not originate from DTBP and instead comes from the homolysis of tert-butylperoxybenzoate (TBPB) and uses catalytic cobalt(II) chloride to assist in radical generation, presumably through the formation of a cobalt(III) benzoate. This approach afforded the both the adamantyl oxazole 273 and thiazole 274 products in moderate yield.
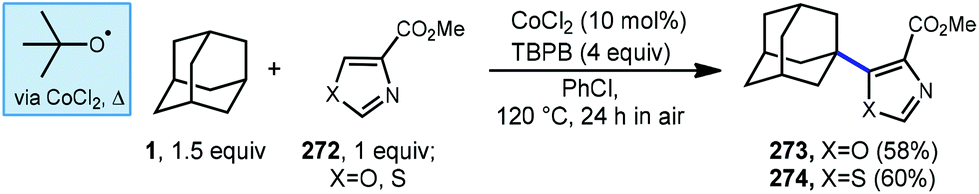 | ||
| Scheme 42 Cobalt-assisted Minisci reactions of oxazoles and thiazoles. | ||
Togo et al. showed that the introduction of heteroaromatic bases onto hydrocarbons can also be accomplished using benzoyl peroxide in the absence transition metals and under irradiation free conditions.151 When adamantane and lepidine (275) were treated with trifluoroacetic acid in benzene under irradiation with benzoyl peroxide, the desired alkylated quinoline product 276 was obtained in 75% yield (Scheme 43a). Other heteroaromatic bases also produced the arylated product in good yields (277–279; 63–86%). In this reaction, thermal homolysis of benzoyl peroxide generates benzoyloxy radical (or phenyl radical after thermal decarboxylation) which then abstracts a hydrogen from adamantane generating the adamantyl radical. Nucleophilic radical addition with a protonated heteroaromatic base and subsequent oxidation (formal loss of H-atom) forms the protonated product. Lepidine alkylation with adamantane has also been reported by Jin and coworkers using hydrogen peroxide and blue light as a source of hydroxyl radical (Scheme 43b).152
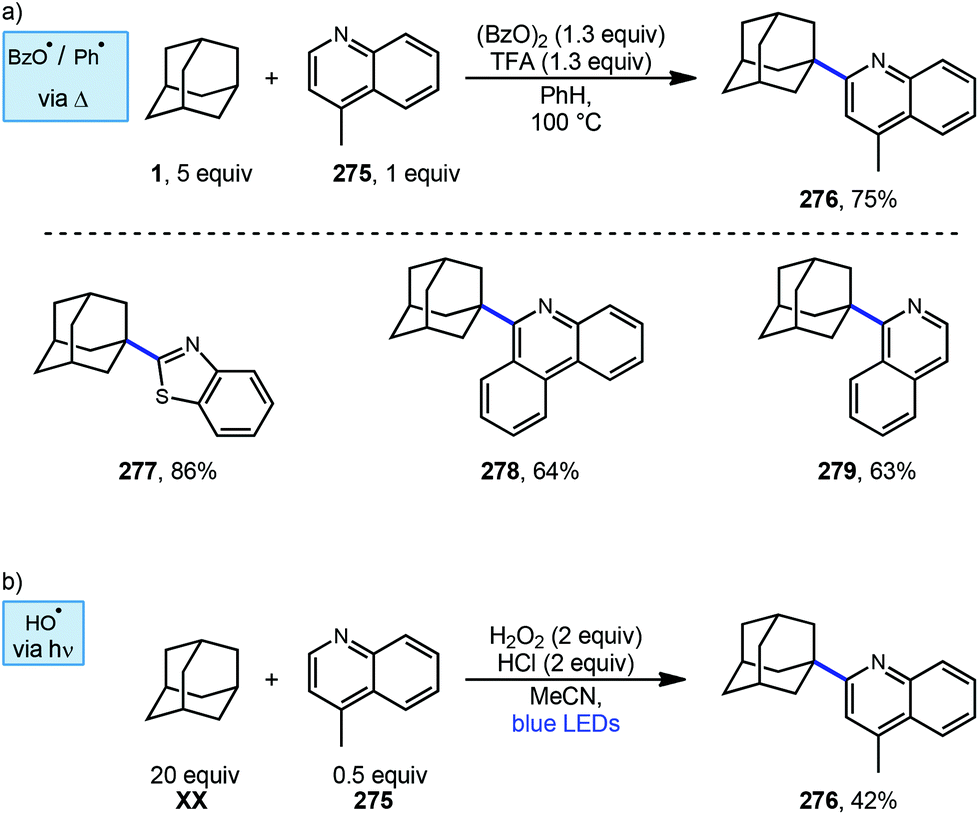 | ||
| Scheme 43 Minisci reactions involving lepidine (275) and other heteroaromatic bases. | ||
6.3. Dual-catalytic arylation
In 2018, another direct arylation was performed by MacMillan and coworkers using a dual catalytic system of 1 mol% tetrabutylammonium decatungstate and 5 mol% nickel bipyridine complex (Scheme 44).153 Adamantane derivatives underwent arylation predominantly at the methylene position for both 2-adamantanone (75) and bromo-adamantanone (281) to give a 53% and 48% yield, respectively.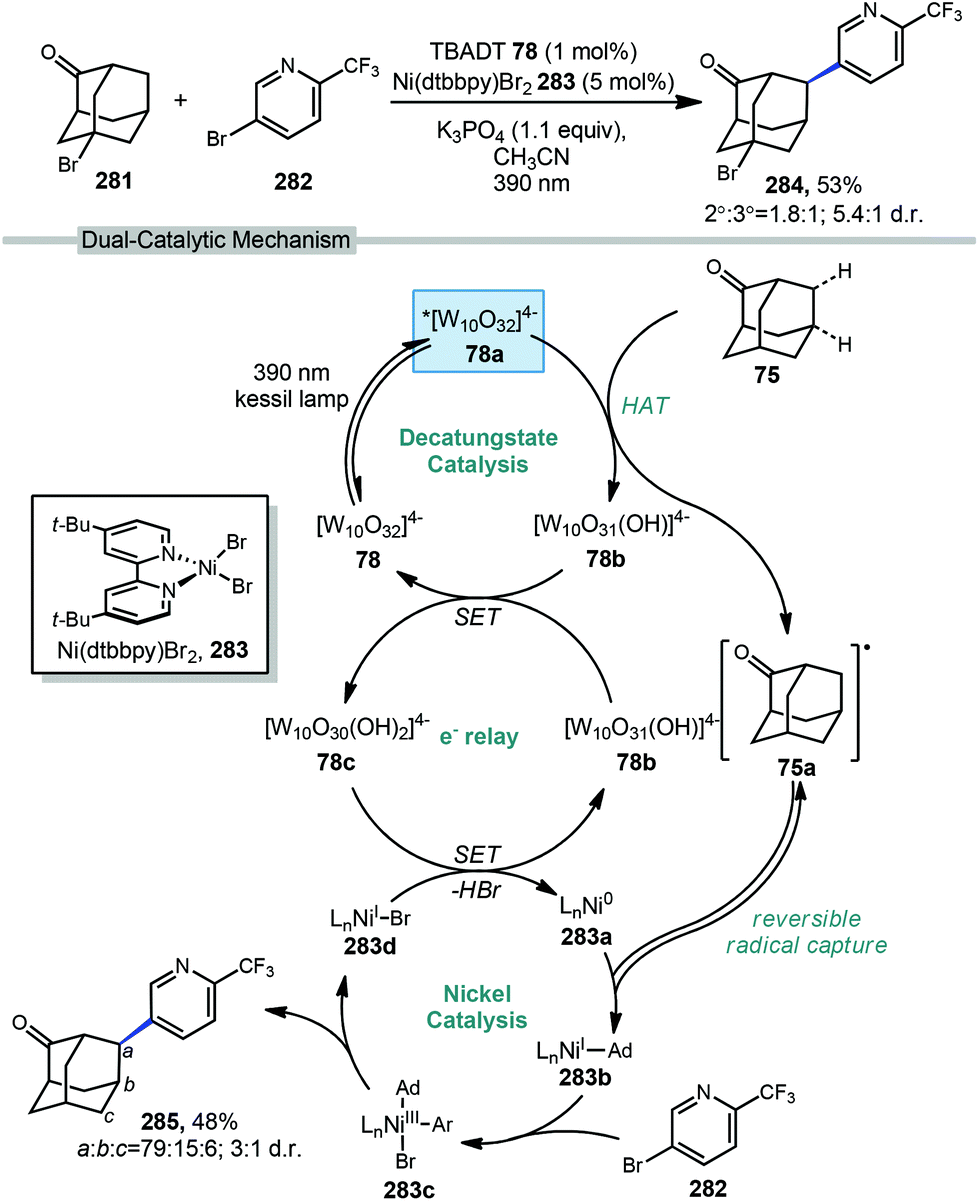 | ||
| Scheme 44 Photochemically driven nickel-catalyzed arylation of adamantanones. | ||
Interestingly, MacMillan and coworkers observed selective arylation at the methylene position of two adamantanone substrates despite existing precedent that decatungstate-catalyzed functionalization occurs in a 5:1 ratio favoring the methine position of adamantane.154 This reversal in regioselectivity can be rationalized by the deactivating effect of the ketone on the adjacent 3°-positions and reversible radical capture during nickel-catalysis, with the selectivity being imparted by a preference for reductive elimination of the 2° aryl adamantane product.155 Notably, in this work, the authors report both regioselectivity and diastereoselectivity (at the new methine stereocenter) in these functionalized adamantyl products. This was achieved by careful separation of the individual regio- and stereoisomers from the crude reaction mixture.
The proposed mechanism begins with the photoexcitation of TBADT followed by intersystem crossing to give the triplet excited state (78 → 78a). This is followed by HAT from a hydrocarbon like 2-adamantanone (75) by the photoexcited catalyst, providing the singly reduced decatungstate 78b and the adamantyl radical 75a. MacMillan proposes a disproportionation event involving the singly reduced decatungstate to regenerate the active HAT catalyst and form a doubly reduced decatungstate species 78c. The alkyl radical is captured by Ni(0) leading to an alkyl Ni(I) intermediate (238a→238b). Oxidative addition of the aryl bromide then gives the key Ni(III) intermediate 283c which undergoes reductive elimination to yield the desired cross-coupled aryl product 285. A final SET step and loss of HBr between the active nickel catalyst and double-reduced TBADT catalyst complete both catalytic cycles.
6.4. Cascade reactions involving aromatic systems
DTBP and copper have been used together with adamantane for accomplishing arylative transformations that occur through a radical cascade process. While an adamantyl radical is not directly involved in arylation, these reactions involve radical addition into an acceptor which triggers a subsequent proximity-induced intramolecular aromatic substitution to give arylated adamantane products.Yadav and Yadav reported the use of isothiocyanates (286, 287) for the formation of adamantyl benzothiazoles (288, 289) using excess DTBP and catalytic copper(II) diacetate.156 In this process, DTBP is homolytically converted to an oxyradical through either thermal or Cu-mediated decomposition and then generates the adamantyl radical. The adamantyl radical adds into the isothiocyanate to form resonance-stabilized thiyl radical intermediate 290 and then cyclizes to the cyclohexadienyl radical 291. Oxidation by Cu(II) or tert-butoxyl radical to the cyclohexadienyl cation 292 then follows and deprotonation with tert-butoxide gives the final benzothiazole product. The yield and distribution of 3° and 2° adamantyl products was slightly dependent on the substitution of the isothiocyanate with para-nitro derivative 289 leading to lower yield and regioselectivity.
Similarly, Zhu et al. demonstrated a related cascade process under similar copper-catalyzed conditions using aryl isocyanides (Scheme 46).157 Adamantane was used with 2-isocyanobiphenyl 293 to form a 2:1 mixture of adamantyl phenanthridines 294 in 51% yield. This powerful transformation enables dual C–C bond formation to the isocyanide carbon using both C(sp3)–H and C(sp2)–H functionalizations.
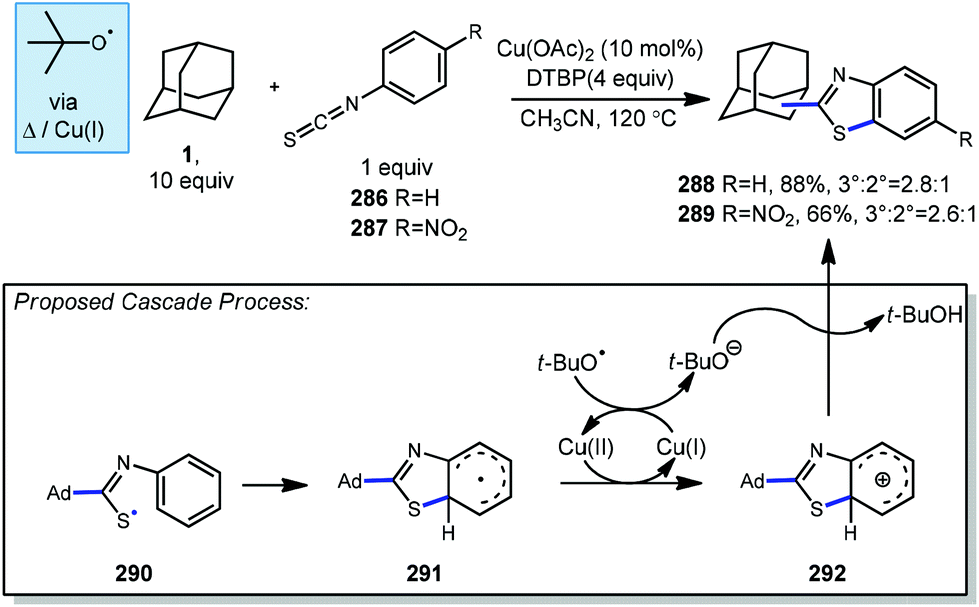 | ||
| Scheme 45 Cascade arylations involving isothiocyanates. | ||
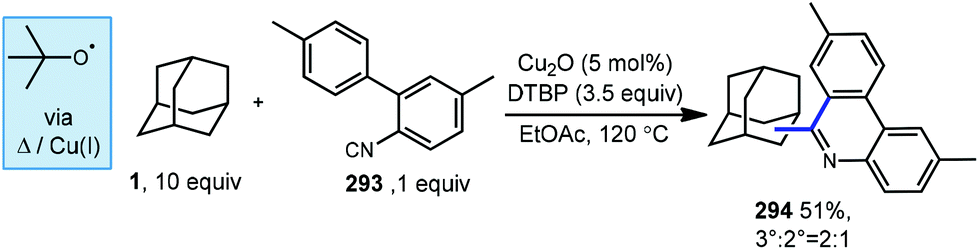 | ||
| Scheme 46 Copper-catalyzed annulation involving aryl isocyanides. | ||
Li and coworkers reported another cascade with adamantane using N-methyl-N-phenylmethylacrylamide (295, Scheme 47) with Cu2O and dicumyl peroxide (DCP) to afford cyclized 3,3-dialkyloxindole 296 in 68% yield and a 6:1 ratio favoring the tertiary adamantyl position.158 The mechanism of this reaction begins with a Giese-type addition to the alkene, followed by cyclization and rearomatization analogous to the mechanism in Scheme 45. DCP was chosen over DTBP by the authors due to better results with it during optimization using cyclohexane. A similar cascade process using DTBP has been reported by Liang et al. with N-allylanilides 297 to give 3,3-dialkyl-indolines 298 and 299 selectively at the tertiary adamantyl position.159
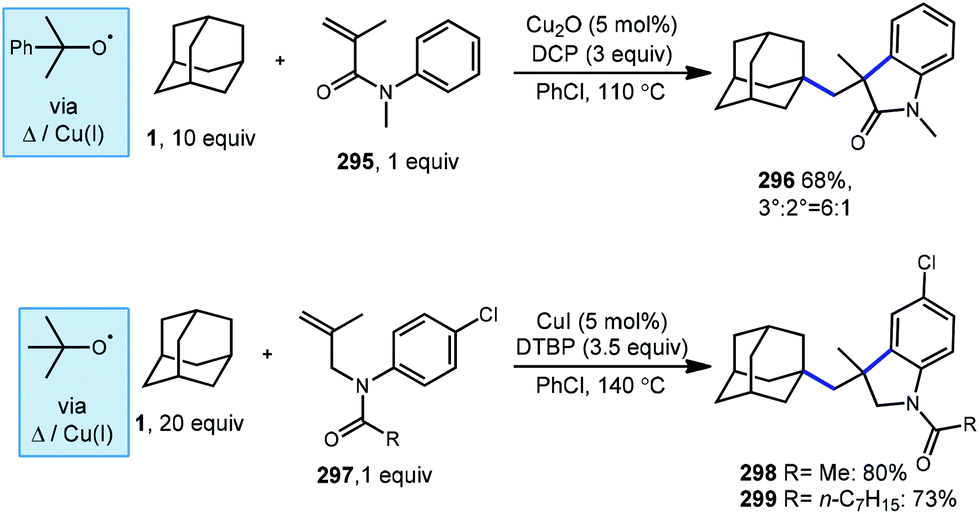 | ||
| Scheme 47 Cascade annulation of adamantanes involving Cu(II) and peroxide. | ||
7. Introduction of nitrogen-containing groups
7.1. Cyanation
Classically, cyanation reactions are usually thought of in terms of the nucleophilic character of simple cyanides such as KCN adding to an electrophilic reaction partner. However, over the years, a variety of electrophilic cyanation reagents have been developed160 and pair well with the nucleophilic character of the adamantyl radical.Inoue and coworkers showed that using benzophenone as a photomediator in conjunction with p-toluenesulfonyl cyanide (300, TsCN), adamantane and adamantanol could be cyanated in high yield (Scheme 48).161,162 Both cyanoadamantane products 301 and 302 were obtained as the methine addition products. In this reaction, the oxyl radical generated from excitation of benzophenone abstracts a hydrogen from the tertiary position on adamantane giving an adamantyl radical. This nucleophilic radical then goes on to react with TsCN generating the product and a sulfinyl radical. Catalytic turnover occurs with HAT by the sulfiniyl radical from the ketyl intermediate giving sulfinic acid and the renewed photocatalyst. Although benzophenone was used stoichiometrically with adamantane, the reaction proceeded when used in substoichiometric quantities with other substrates such as 1,2-dioxane.
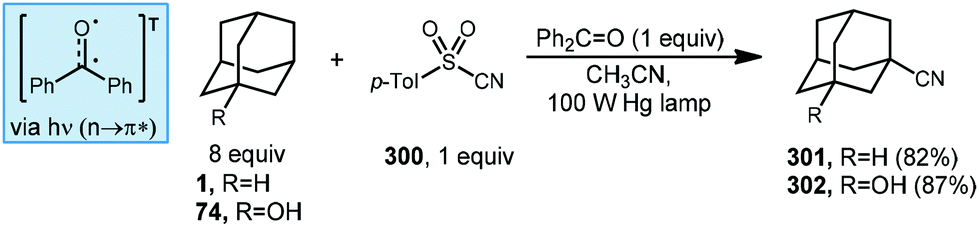 | ||
| Scheme 48 Benzophenone-mediated cyanation of adamantanes. | ||
Schreiner et al. have also found that using TsCN as an electrophilic cyanide source with a NHPI/PINO system effectively cyanated a variety of adamantanes and diamantanes (Scheme 49).163 A few known reaction conditions were tried in order to access the PINO radical such as azobisisobutyronitrile (AIBN), Co(II/III) salts, and ceric ammonium nitrate (CAN). CAN was found to be superior in this regard, and initially gave the 1-cyanoadamantane product 301 in 42% yield. However, the formation of significant amounts of 1-nitroadamantane as a side product was also observed with the use of CAN. The addition of base suppressed the formation of the undesired nitro product with lithium carbonate giving the best result. Under this optimized TsCN/NHPI/CAN/Li2CO3 system, the cyanoadamantane was obtained in 77% yield. Other substituted adamantanes were obtained with varying degrees of success (303–314). Methyl adamantane 303 was obtained 71% yield, while di- and trimethyl adamantanes 307 and 308 were only isolated with yields of 33% and 28%, respectively. This drop in reactivity was also observed by Olah and coworkers in the cyanations of methylated bromoadamantanes using Lewis acid catalysis.164
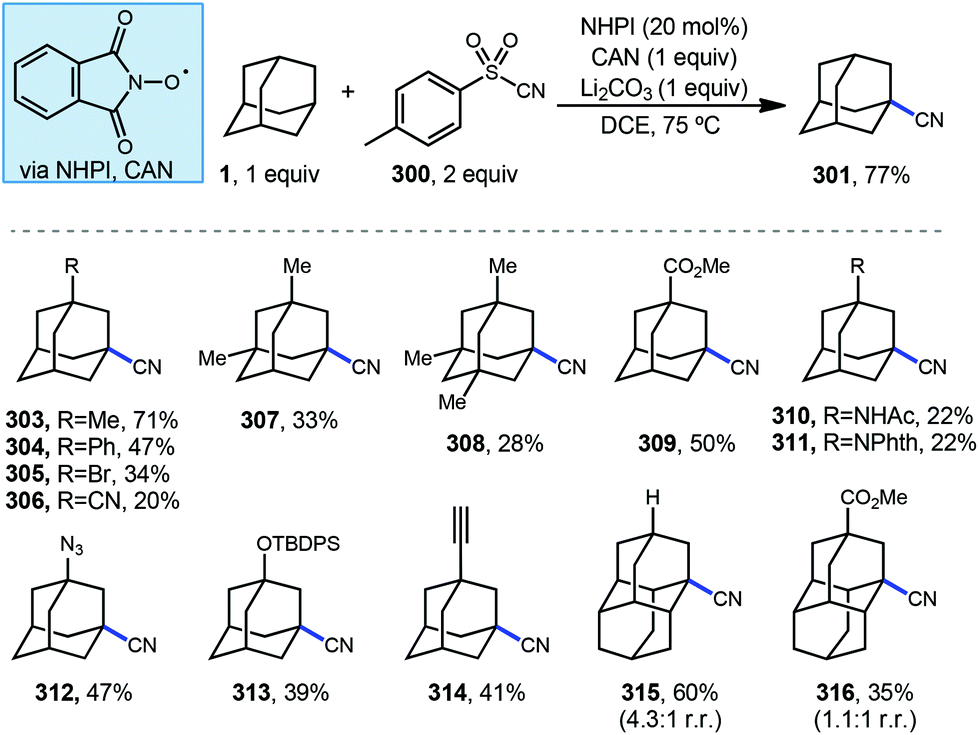 | ||
| Scheme 49 HAT-mediated cyanation of diamondoids via PINO radical. | ||
A range of other useful adamantyl nitriles were accessible including acetamides, phthalimides, azides, alkynes, and silyl ethers. Diamantane 315 was also cyanated in a 4.3:1 ratio favoring the medial position, while the carboxylate derivative 316 was was formed as a mixture of medial regioisomers (1.1:1 r.r.). Notable among these reaction conditions is the use of only one equivalent of adamantane. Typically, most adamantane C–H activation reactions will use higher amounts of the adamantane in order to suppress further C–H activation of the product. Here, however, installation of the electron withdrawing cyano group deactivates the cage to further cyanation, as evidenced by the difference in yield between the formation of mono- and di-cyanated products 301 and 306.
In 2018 Ma, Zhang and coworkers reported a metal-free cyanation reaction using a hypervalent iodine reagent (317, Scheme 50).165 With 20 mol% of TBPB as a catalytic initiator, C(sp3)–H bonds in a very broad scope of cyclic hydrocarbons, ethers and amine derivatives underwent direct cyanation at elevated temperature, including adamantane in 76% yield as determined by 1H NMR. The initial HAT occurs with the tert-butoxyl or phenyl radical (formed through decarboxylation of the benzoyloxy radical), while the chain-carrying radical is likely the aroyloxy radical derived from 2-iodobenzoic acid. Spin-trapping and competition experiments were consistent with a radical mechanism for hydrocarbons and iminium ion generation with amine substrates.
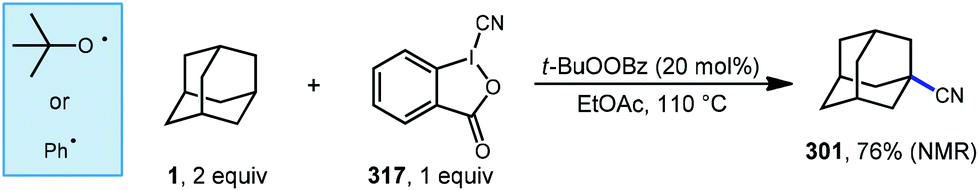 | ||
| Scheme 50 Cyanation of adamantane using a hypervalent iodine reagent. | ||
7.2. Aminoalkylation
DTBP was also used for the α-alkylation of α-amino carbonyls 318 with adamantane and other alkanes by Cheng et al. (Scheme 51).166 The tert-butoxyl radical is thought to both facilitate the formation of the imine intermediate 318a and generate the adamantyl radical that adds into the imine, necessitating the use of excess peroxide. Aminoalkylated product 319 was isolated in 46% yield as the tertiary regioisomer.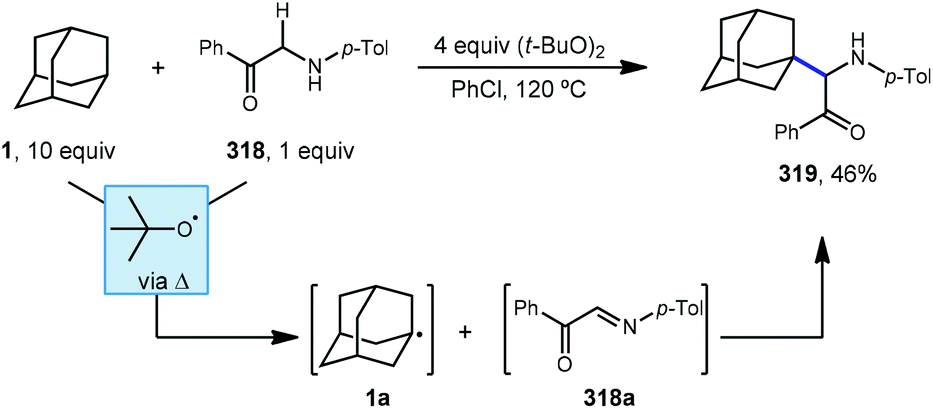 | ||
| Scheme 51 Aminoalkylation of adamantane using DTBP. | ||
In 2020, our group described an aminoalkylation strategy with adamantanes using a dual photocatalytic/HAT system (described earlier in section 4.2).167 A variety of protected imines and hydrazones were used as radical acceptors to generate aminoadamantyl products (Scheme 52) in moderate to high yields with excellent selectivity for tertiary adamantyl position. Electron-deficient hydrazones 320–322 and N-tosylimines 323–325 with electron-withdrawing aryl groups (Y=CO2Me, CN, F) worked better than those bearing neutral or donating groups (Y=H, OMe). A trisubstituted cyclic sulfonylimine demonstrated this reaction's ability to form even the sterically congested C–C bond in sulfonamide product 326 in 83% yield. This cyclic sulfonylimine also reacted well with substituted adamantanes 327–329 such as 1-adamantanol, although other electron-withdrawing substituents (R2=Cl, Ac) were less effective.
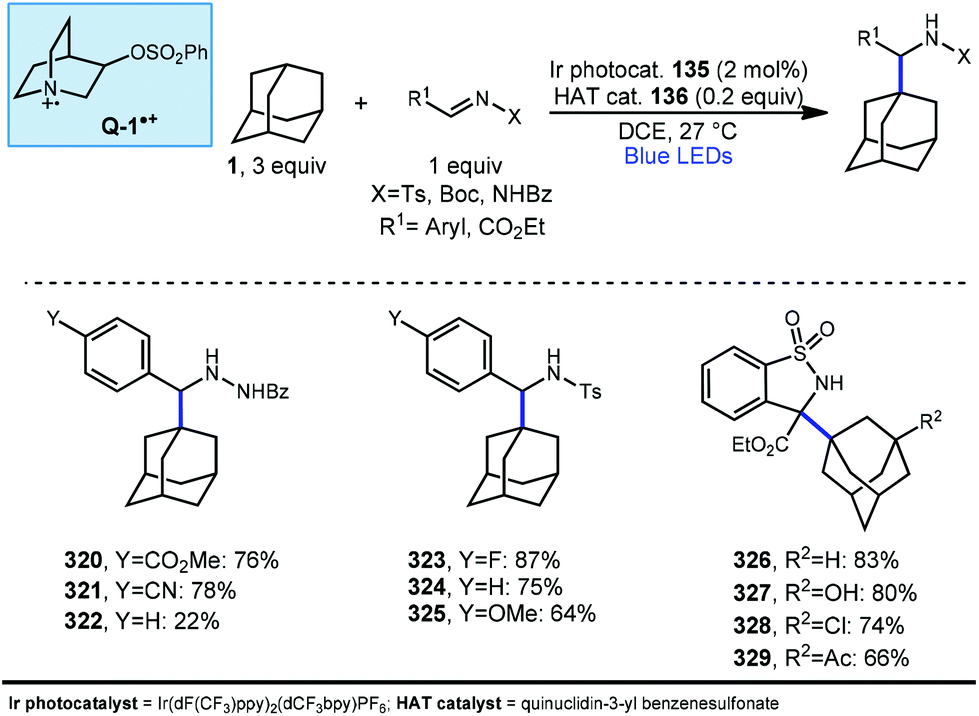 | ||
| Scheme 52 Aminoalkylation of adamantanes via dual photoredox/HAT catalysis. | ||
The use of the N-mesitylsulfinyl group as a chiral auxiliary provided good stereocontrol for the radical addition of adamantane to sulfinimine substrate 330 (X=Mes, Y=Ph) and the sulfinamide product 331 was obtained in 73% yield and >20:1 d.r. (Scheme 53). Other sulfinyl groups such as tert-butyl and para-tolyl were less effective at asymmetric induction than the mesityl group (see reactions with glyoxalate-derived sulfinimines 332–334). In contrast to the protected imines in Scheme 52, the chiral sulfinimines performed better using PT catalyst 187 (see Scheme 29) as the direct HAT photocatalyst, which still displayed excellent regioselectivity and resulted in less degradation of the starting material under the reaction conditions.
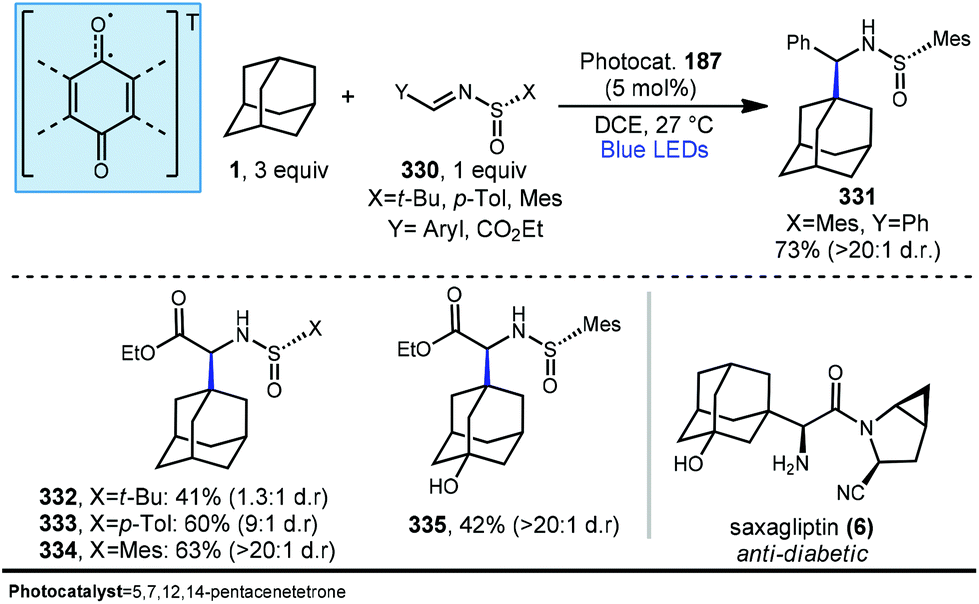 | ||
| Scheme 53 Asymmetric aminoalkylation of adamantanes. | ||
Deprotection of the aminoalkylated products to their free amines with SmI2 or TFA leads to useful drug-like pharmacophores and building blocks. In particular r, after deprotection of the sulfinyl group, glyoxylate-derived chiral sulfinimines enabled direct preparation of an enantiopure adamantyl glycine from 334 and the 3-hydroxyadamantyl core of saxagliptin from precursor 335.
8. Summary and outlook
The functionalization of adamantanes and higher diamondoids has garnered continued interest from the late 1950s when they became readily available to the present day. As part of this sustained effort, adamantanes have served as test substrates for a variety of C–H functionalization reactions, particularly due to the unusually strong C(sp3)–H bonds, providing a challenging molecule in terms of both reactivity and regioselectivity. Furthermore, derivatized adamantanes have served as important synthetic targets in their own right, particularly due to their versatile use as architectural linkers and as a “lipophilic bullet” in medicinal chemistry applications, as it was described by Schreiner and coworkers.10 Beginning with the early work of Schleyer and Haaf, increased access to adamantane derivatives has driven new discoveries in biological and chemical function, and new applications have in turn offered the motivation for more versatile synthetic methods. This interest in diamondoids within synthesis and catalysis disciplines continues to reinforce the need for novel regioselective methods.As we have summarized in this review, key to the selectivity between different C–H bonds in the context of radical reactions is the nature of the H-atom abstracting species (Fig. 5). Upon examining the many reactions described above, certain trends become apparent. For adamantane itself, selectivity for the sterically more accessible 3°-C–H bond over the 2°-position ranges from >20:1 in case of the quinuclidine-derived radical cation 136a, the PINO radical 70, and photoexcited TCB (via oxidation/deprotonation mechanism) to approximately 2–5:1 in the case of various examples of tert-butoxyl radical-mediated reactions, to ∼1:1.3 in the case of decatungstate as reported by Albini and Ryu. The range for HAT using an excited carbonyl triplet (i.e., benzophenone, diacetyl, quinones) also favors the 3°-position but varies considerably between medium to high selectivity based on the abstractor and reaction conditions used. With 2-adamantanone substrates, the selectivity of decatungstate reported by MacMillan and coworkers shifted further up to 1:4, favoring the 2° position, which is at least in part due to a proposed reversible activation process followed by a selective product-forming reductive elimination from nickel. Evidently, the inherent selectivity of each abstracting species can be estimated, but varies from reaction to reaction based on the nature of the next C–C (or C–X) bond-forming step and whether the key C–H abstracting step is reversible. Furthermore, some reports do not quantify any minor products, so one cannot conclusively determine a regioisomeric ratio in these cases. These factors complicate a rigid assignment of selectivity for each species, but lead to the ranges shown in Fig. 5. The pioneering work of Schreiner and colleagues has provided a detailed look into the regioselectivity of many different processes (some of which are outside the scope of this review, i.e., C–X bond formation), including comparing experimental C1:C2 ratios to calculated transition state energies and similar comparisons for kinetic isotope effect experiments.
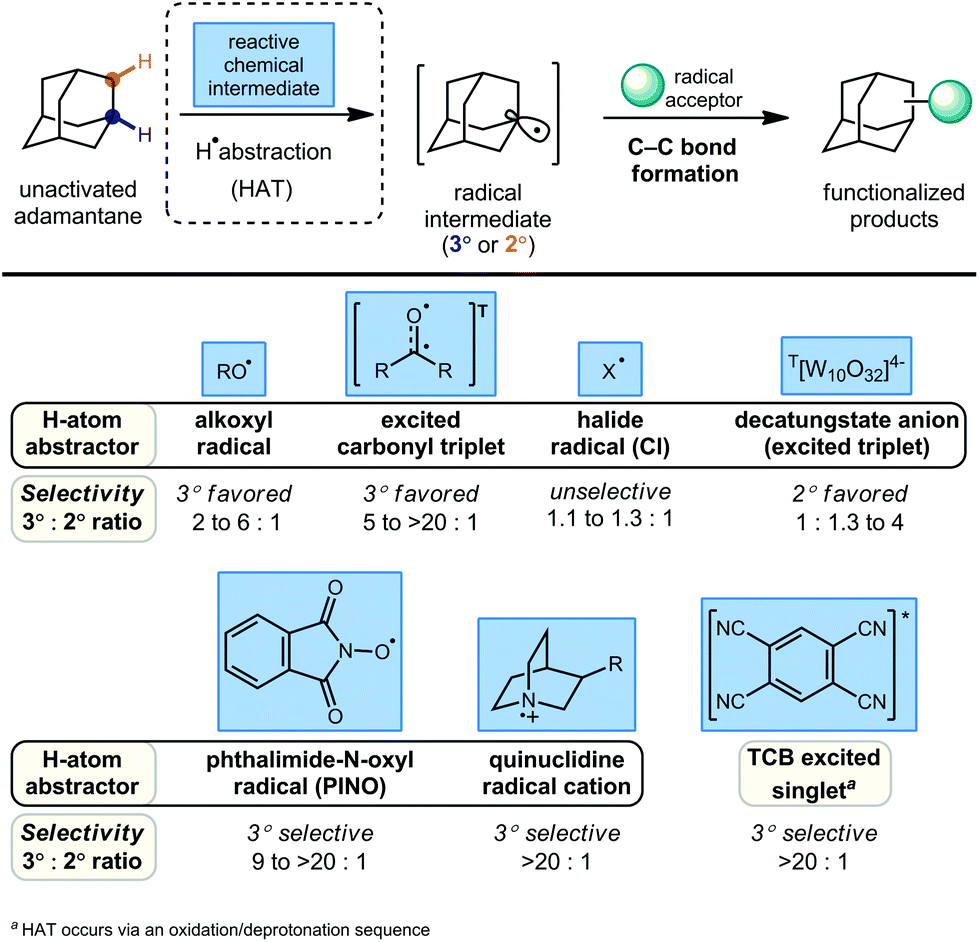 | ||
| Fig. 5 Summary of regioselectivity profiles for the most common H-atom abstractors. | ||
A full description of regioselectivity with higher order diamondoids is similarly complicated by limited data, particularly the small number of reports of functionalizations beyond diamantane. In general, the 3°-positions react preferentially, but the ratio between apical and medial positions varies based on the abstracting species. Schreiner and Fokin have reported the regioselective acetylation of diamantane with 4.6:1 selectivity for the apical position (see section 2.2). Similar apical selectivity observed across several tri- to pentamantane substrates points to polarizability in the C–H abstraction step with triplet diacetyl being the most important contributor. The photochemical arylation described by Albini through an oxidation/deprotonation mechanism with TCB was applied to diamantane, which also resulted in selective apical functionalization (11% yield, single isomer), and impressively, Schreiner and Fokin demonstrated this apical selectivity applies to even higher-ordered diamondoids. This outcome is predicted based on elongation (and acidification) of the apical C–H bonds in the radical cation that results from photooxidation. Schreiner has also calculated barriers for C1, C3 and C4-functionalizations of diamantane with different activation reagents, further highlighting the role of polarizability and other factors in the apical-selective acetylation and charge-transfer effects in medial-selective PINO-catalyzed substitutions. For a longer description of the selective functionalization of diamantane in other C–H to C–X transformations not covered here, the work of Schreiner and Fokin should be highlighted.140
In the larger context of C–H activation chemistry, documenting the role of the effects described above can allow a better understanding of existing methods and inspire the design of new C–H functionalization systems. The unusual properties of adamantane and diamantane allow us to probe the dominant factor for selectivity in each case, distinguishing between sterics, charge-transfer effects and polarizability. These insights may guide not only the selection of appropriate methods for a given diamondoid target, but also the application of these methods to other substrate classes beyond the diamondoids. Given the continued use of adamantanes as linkers in nanomaterials, as “lipophilic bullets” in the optimization of therapeutics and as bulky, electron-rich substituents in a variety of ligand-design applications, we expect that interest in the new methods for the selective functionalization of diamondoids will continue to attract the attention and innovative minds of organic chemists for the foreseeable future.
Conflicts of interest
There are no conflicts to declare.References
- R. C. Fort, Adamantane: The Chemistry of Diamond Molecules, Marcel Dekker, New York, 1976, vol. 5 Search PubMed.
- H. Schwertfeger, A. A. Fokin and P. R. Schreiner, Angew. Chem., Int. Ed., 2008, 47, 1022 CrossRef CAS PubMed.
- J. E. P. Dahl, J. M. Moldowan, T. M. Peakman, J. C. Clardy, E. Lobkovsky, M. M. Olmstead, P. W. May, T. J. Davis, J. W. Steeds, K. E. Peters, A. Pepper, A. Ekuan and R. M. K. Carlson, Angew. Chem., Int. Ed., 2003, 42, 2040 CrossRef CAS.
- H.-C. Chang, J. Phys.: Conf. Ser., 2016, 728, 062004 CrossRef.
- C. Tyborski, T. Hückstaedt, R. Gillen, T. Otto, N. A. Fokina, A. A. Fokin, P. R. Schreiner and J. Maultzsch, Carbon, 2020, 157, 201 CrossRef CAS.
- T. Rander, T. Bischoff, A. Knecht, D. Wolter, R. Richter, A. Merli and T. Möller, J. Am. Chem. Soc., 2017, 139, 11132 CrossRef CAS.
- M. Vörös, T. Demjén, T. Szilvási and A. Gali, Phys. Rev. Lett., 2012, 108, 267401 CrossRef PubMed.
- A. Milanese, E. Gorincioi, M. Rajabi, G. Vistoli and E. Santaniello, Bioorg. Chem., 2011, 39, 151 CrossRef CAS PubMed.
- D. J. Augeri, J. A. Robl, D. A. Betebenner, D. R. Magnin, A. Khanna, J. G. Robertson, A. Wang, L. M. Simpkins, P. Taunk, Q. Huang, S.-P. Han, B. Abboa-Offei, M. Cap, L. Xin, L. Tao, E. Tozzo, G. E. Welzel, D. M. Egan, J. Marcinkeviciene, S. Y. Chang, S. A. Biller, M. S. Kirby, R. A. Parker and L. G. Hamann, J. Med. Chem., 2005, 48, 5025 CrossRef CAS.
- L. Wanka, K. Iqbal and P. R. Schreiner, Chem. Rev., 2013, 113, 3516 CrossRef CAS PubMed.
- T. P. Stockdale and C. M. Williams, Chem. Soc. Rev., 2015, 44, 7737 RSC.
- J. Joubert, W. J. Geldenhuys, C. J. Van der Schyf, D. W. Oliver, H. G. Kruger, T. Govender and S. F. Malan, ChemMedChem, 2012, 7, 375 CrossRef CAS.
- J. G. Henkel, J. T. Hane and G. Gianutsos, J. Med. Chem., 1982, 25, 51 CrossRef CAS.
- K. A. Agnew-Francis and C. M. Williams, Adv. Synth. Catal., 2016, 358, 675 CrossRef CAS.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef CAS.
- A. G. Dossetter, T. F. Jamison and E. N. Jacobsen, Angew. Chem., Int. Ed., 1999, 38, 2398 CrossRef CAS.
- B. K. Keitz, K. Endo, P. R. Patel, M. B. Herbert and R. H. Grubbs, J. Am. Chem. Soc., 2012, 134, 693 CrossRef CAS.
- R. P. Reddy, G. H. Lee and H. M. L. Davies, Org. Lett., 2006, 8, 3437 CrossRef CAS PubMed.
- Q. Li, C. Jin, P. A. Petukhov, A. V. Rukavishnikov, T. O. Zaikova, A. Phadke, D. H. LaMunyon, M. D. Lee and J. F. W. Keana, J. Org. Chem., 2004, 69, 1010 CrossRef CAS.
- Q. Li, A. V. Rukavishnikov, P. A. Petukhov, T. O. Zaikova, C. Jin and J. F. W. Keana, J. Org. Chem., 2003, 68, 4862 CrossRef CAS.
- K. Nasr, N. Pannier, J. V. Frangioni and W. Maison, J. Org. Chem., 2008, 73, 1056 CrossRef CAS.
- O. Plietzsch, C. I. Schilling, M. Tolev, M. Nieger, C. Richert, T. Muller and S. Bräse, Org. Biomol. Chem., 2009, 7, 4734 RSC.
- H. Lim and J. Y. Chang, Macromolecules, 2010, 43, 6943 CrossRef CAS.
- Q. Fang, S. Gu, J. Zheng, Z. Zhuang, S. Qiu and Y. Yan, Angew. Chem., Int. Ed., 2014, 53, 2878 CrossRef CAS.
- Q. Fang, J. Wang, S. Gu, R. B. Kaspar, Z. Zhuang, J. Zheng, H. Guo, S. Qiu and Y. Yan, J. Am. Chem. Soc., 2015, 137, 8352 CrossRef CAS PubMed.
- P. Kahl, J. P. Wagner, C. Balestrieri, J. Becker, H. Hausmann, G. J. Bodwell and P. R. Schreiner, Angew. Chem., Int. Ed., 2016, 55, 9277 CrossRef CAS.
- H. Decker, Angew. Chem., Int. Ed. Engl., 1924, 37, 781 CrossRef.
- S. Landa and V. Macháček, Collect. Czech. Chem. Commun., 1933, 5, 1 CrossRef CAS.
- V. Prelog and R. Seiwerth, Berichte Dtsch. Chem. Ges. B Ser., 1941, 74, 1644 CrossRef.
- V. Prelog and R. Seiwerth, Berichte Dtsch. Chem. Ges. B Ser., 1941, 74, 1769 CrossRef.
- H. Meerwein, F. Kiel, G. Klösgen and E. Schoch, J. Prakt. Chem., 1922, 104, 161 CrossRef CAS.
- R. C. Fort and P. von R. Schleyer, Chem. Rev., 1964, 64, 277 CrossRef CAS.
- H. Stetter, O.-E. Bänder and W. Neumann, Chem. Ber., 1956, 89, 1922 CrossRef CAS.
- P. von R. Schleyer, J. Am. Chem. Soc., 1957, 79, 3292 CrossRef.
- P. von R. Schleyer and M. M. Donaldson, J. Am. Chem. Soc., 1960, 82, 4645 CrossRef.
- P. von R. Schleyer, P. Grubmüller, W. F. Maier, O. Vostrowsky, L. Skattebøl and K. H. Holm, Tetrahedron Lett., 1980, 21, 921 CrossRef CAS.
- M. A. McKervey, Chem. Soc. Rev., 1974, 3, 479 RSC.
- R. P. Kirchen, T. S. Sorensen and S. M. Whitworth, Can. J. Chem., 1993, 71, 2016 CrossRef CAS.
- C. Cupas, P. von R. Schleyer and D. J. Trecker, J. Am. Chem. Soc., 1965, 87, 917 CrossRef CAS.
- T. M. Gund, E. Osawa, V. Z. Williams and P. von R. Schleyer, J. Org. Chem., 1974, 39, 2979 CrossRef CAS.
- O. Farooq, S. M. F. Farnia, M. Stephenson and G. A. Olah, J. Org. Chem., 1988, 53, 2840 CrossRef CAS.
- G. A. Olah, G. K. S. Prakash, J. G. Shih, V. V. Krishnamurthy, G. D. Mateescu, G. Liang, G. Sipos, V. Buss, T. M. Gund and P. von R. Schleyer, J. Am. Chem. Soc., 1985, 107, 2764 CrossRef CAS.
- M. A. Mckervey, Tetrahedron, 1980, 36, 971 CrossRef CAS.
- J. E. Dahl, S. G. Liu and R. M. K. Carlson, Science, 2003, 299, 96 CrossRef CAS PubMed.
- J. E. P. Dahl, J. M. Moldowan, Z. Wei, P. A. Lipton, P. Denisevich, R. Gat, S. Liu, P. R. Schreiner and R. M. K. Carlson, Angew. Chem., Int. Ed., 2010, 49, 9881 CrossRef CAS.
- W. Haaf, Angew. Chem., 1961, 73, 144 CrossRef CAS.
- W. Haaf, Chem. Ber., 1964, 97, 3234 CrossRef CAS.
- W. L. Davies, R. R. Grunert, R. F. Haff, J. W. McGahen, E. M. Neumayer, M. Paulshock, J. C. Watts, T. R. Wood, E. C. Hermann and C. E. Hoffmann, Science, 1964, 144, 862 CrossRef CAS.
- G. Hubsher, M. Haider and M. S. Okun, Neurology, 2012, 78, 1096 CrossRef CAS PubMed.
- A. Witt, N. Macdonald and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2004, 3, 109 CrossRef CAS PubMed.
- S. K. Sonkusare, C. L. Kaul and P. Ramarao, Pharmacol. Res., 2005, 51, 1 CrossRef CAS PubMed.
- H. Baldwin, G. Webster, L. Stein Gold, V. Callender, F. E. Cook-Bolden and E. Guenin, Am. J. Clin. Dermatol., 2021, 22, 315 CrossRef PubMed.
- P. R. Schreiner, O. Lauenstein, I. V. Kolomitsyn, S. Nadi and A. A. Fokin, Angew. Chem., Int. Ed., 1998, 37, 1895 CrossRef CAS.
- P. R. Schreiner, O. Lauenstein, E. D. Butova, P. A. Gunchenko, I. V. Kolomitsin, A. Wittkopp, G. Feder and A. A. Fokin, Chem. – Eur. J., 2001, 7, 4996 CrossRef CAS.
- G. H. Kruppa and J. L. Beauchamp, J. Am. Chem. Soc., 1986, 108, 2162 CrossRef CAS PubMed.
- A. A. Fokin and P. R. Schreiner, Chem. Rev., 2002, 102, 1551 CrossRef CAS PubMed.
- Y. Ando and K. Suzuki, Chem. – Eur. J., 2018, 24, 15955 CrossRef CAS.
- R. I. Khusnutdinov and N. A. Shchadneva, Russ. Chem. Rev., 2019, 88, 800 CrossRef CAS.
- M.-G. A. Shvekhgeimer, Russ. Chem. Rev., 1996, 65, 555 CrossRef.
- E. I. Bagrii, A. I. Nekhaev and A. L. Maksimov, Pet. Chem., 2017, 57, 183 CrossRef CAS.
- L. I. Kas'yan and V. A. Pal'chikov, Russ. J. Org. Chem., 2010, 46, 1 CrossRef.
- M. A. Gunawan, J.-C. Hierso, D. Poinsot, A. A. Fokin, N. A. Fokina, B. A. Tkachenko and P. R. Schreiner, New J. Chem., 2013, 38, 28 RSC.
- E. A. Shokova and V. V. Kovalev, Russ. J. Org. Chem., 2012, 48, 1007 CrossRef CAS.
- R. Hrdina, Synthesis, 2019, 51, 629 CrossRef CAS.
- V. V. Sevost'yanova, M. M. Krayushkin and A. G. Yurchenko, Russ. Chem. Rev., 1970, 39, 817 CrossRef.
- I. K. Moiseev, N. V. Makarova and M. N. Zemtsova, Russ. Chem. Rev., 1999, 68, 1001 CrossRef CAS.
- I. Tabushi, J. Hamuro and R. Oda, J. Org. Chem., 1968, 33, 2108 CrossRef CAS.
- G. W. Smith and H. D. Williams, J. Org. Chem., 1961, 26, 2207 CrossRef CAS.
- I. Tabushi, T. Okada, Y. Aoyama and R. Oda, Tetrahedron Lett., 1969, 10, 4069 CrossRef.
- P. H. Owens, G. J. Gleicher and L. M. Smith, J. Am. Chem. Soc., 1968, 90, 4122 CrossRef CAS.
- I. Tabushi, S. Kojo and Z. Yoshida, Tetrahedron Lett., 1973, 14, 2329 CrossRef.
- I. Tabushi, S. Kojo and K. Fukunishi, J. Org. Chem., 1978, 43, 2370 CrossRef CAS.
- K. Sandros and H. L. J. Bäckström, Acta Chem. Scand., 1962, 16, 958 CrossRef CAS.
- I. Tabushi, S. Kojo, P. von R. Schleyer and T. M. Gund, J. Chem. Soc., Chem. Commun., 1974, 591 RSC.
- P. R. Schreiner, N. A. Fokina, B. A. Tkachenko, H. Hausmann, M. Serafin, J. E. P. Dahl, S. Liu, R. M. K. Carlson and A. A. Fokin, J. Org. Chem., 2006, 71, 6709 CrossRef CAS.
- A. A. Fokin, P. A. Gunchenko, A. A. Novikovsky, T. E. Shubina, B. V. Chernyaev, J. E. P. Dahl, R. M. K. Carlson, A. G. Yurchenko and P. R. Schreiner, Eur. J. Org. Chem., 2009, 5153 CrossRef CAS.
- S. Kamijo, G. Takao, K. Kamijo, M. Hirota, K. Tao and T. Murafuji, Angew. Chem., Int. Ed., 2016, 55, 9695 CrossRef CAS.
- L. K. G. Ackerman, J. I. M. Alvarado and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 14059 CrossRef CAS PubMed.
- H. Yuan, Z. Liu, Y. Shen, H. Zhao, C. Li, X. Jia and J. Li, Adv. Synth. Catal., 2019, 361, 2009 CrossRef CAS.
- D. H. R. Barton and D. Doller, Acc. Chem. Res., 1992, 25, 504 CrossRef CAS.
- J. Sommer and J. Bukala, Acc. Chem. Res., 1993, 26, 370 CrossRef CAS.
- B. A. Arndtsen, R. G. Bergman, T. A. Mobley and T. H. Peterson, Acc. Chem. Res., 1995, 28, 154 CrossRef CAS.
- S. Kato, T. Iwahama, S. Sakaguchi and Y. Ishii, J. Org. Chem., 1998, 63, 222 CrossRef CAS.
- I. Ryu, A. Tani, T. Fukuyama, D. Ravelli, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2011, 50, 1869 CrossRef CAS.
- Q. Liu, H. Zhang and A. Lei, Angew. Chem., Int. Ed., 2011, 50, 10788 CrossRef CAS.
- X.-F. Wu and H. Neumann, ChemCatChem, 2012, 4, 447 CrossRef CAS.
- X.-F. Wu, H. Neumann and M. Beller, Chem. Rev., 2013, 113, 1 CrossRef CAS PubMed.
- S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234 CrossRef CAS.
- Y. Li, K. Dong, F. Zhu, Z. Wang and X.-F. Wu, Angew. Chem., Int. Ed., 2016, 55, 7227 CrossRef CAS PubMed.
- L. C. M. Castro and N. Chatani, Chem. Lett., 2015, 44, 410 CrossRef CAS.
- Z. Han, D. Chaowei, L. Lice, M. Hongfei, B. Hongzhong and L. Yufeng, Tetrahedron, 2018, 74, 3712 CrossRef.
- L. Lu, R. Shi, L. Liu, J. Yan, F. Lu and A. Lei, Chem. – Eur. J., 2016, 22, 14484 CrossRef CAS.
- L. Lu, D. Cheng, Y. Zhan, R. Shi, C.-W. Chiang and A. Lei, Chem. Commun., 2017, 53, 6852 RSC.
- P. von R. Schleyer, E. Osawa and Z. Majerski, J. Org. Chem., 1971, 36, 205 CrossRef CAS.
- United States, US3437701A, 1969.
- I. Tabushi and K. Fukunishi, J. Org. Chem., 1974, 39, 3748 CrossRef CAS.
- K. Fukunishi and I. Tabushi, Synthesis, 1988, 826 CrossRef CAS.
- A. M. González-Cameno, M. Mella, M. Fagnoni and A. Albini, J. Org. Chem., 2000, 65, 297 CrossRef.
- K. Ogura, A. Kayano, N. Sumitani, M. Akazome and M. Fujita, J. Org. Chem., 1995, 60, 1106 CrossRef CAS.
- B. Wladislaw, L. Marzorati and R. B. Uchôa, Synthesis, 1986, 964 CrossRef CAS.
- G. Campari, M. Fagnoni, M. Mella and A. Albini, Tetrahedron: Asymmetry, 2000, 11, 1891 CrossRef CAS.
- P. J. Scheuer, Acc. Chem. Res., 1992, 25, 433 CrossRef CAS.
- D. P. Curran and S. Hadida, J. Am. Chem. Soc., 1996, 118, 2531 CrossRef CAS.
- H. I. Tashtoush and R. Sustmann, Chem. Ber., 1992, 125, 287 CrossRef CAS.
- A. M. Cardarelli, M. Fagnoni, M. Mella and A. Albini, J. Org. Chem., 2001, 66, 7320 CrossRef CAS PubMed.
- L. Cermenati, D. Dondi, M. Fagnoni and A. Albini, Tetrahedron, 2003, 59, 6409 CrossRef CAS.
- D. Dondi, A. M. Cardarelli, M. Fagnoni and A. Albini, Tetrahedron, 2006, 62, 5527 CrossRef CAS.
- A. L. J. Beckwith and J. S. Poole, J. Am. Chem. Soc., 2002, 124, 9489 CrossRef CAS PubMed.
- H.-B. Yang, A. Feceu and D. B. C. Martin, ACS Catal., 2019, 9, 5708 CrossRef CAS.
- J. L. Jeffrey, J. A. Terrett and D. W. C. MacMillan, Science, 2015, 349, 1532 CrossRef CAS PubMed.
- W.-Z. Liu and F. G. Bordwell, J. Org. Chem., 1996, 61, 4778 CrossRef CAS PubMed.
- S. Rohe, A. O. Morris, T. McCallum and L. Barriault, Angew. Chem., Int. Ed., 2018, 57, 15664 CrossRef CAS PubMed.
- K. A. Margrey, W. L. Czaplyski, D. A. Nicewicz and E. J. Alexanian, J. Am. Chem. Soc., 2018, 140, 4213 CrossRef CAS PubMed.
- C. M. Morton, Q. Zhu, H. Ripberger, L. Troian-Gautier, Z. S. D. Toa, R. R. Knowles and E. J. Alexanian, J. Am. Chem. Soc., 2019, 141, 13253 CrossRef CAS PubMed.
- T. Hara, T. Iwahama, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2001, 66, 6425 CrossRef CAS PubMed.
- Y. Cai, H.-S. Dang and B. P. Roberts, Tetrahedron Lett., 2004, 45, 4405 CrossRef CAS.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58 CrossRef CAS PubMed.
- Z.-Z. Zhang, B. Liu, C.-Y. Wang and B.-F. Shi, Org. Lett., 2015, 17, 4094 CrossRef CAS PubMed.
- W. Liu, Z. Chen, L. Li, H. Wang and C.-J. Li, Chem. – Eur. J., 2016, 22, 5888 CrossRef CAS.
- J. Ji, P. Liu and P. Sun, Chem. Commun., 2015, 51, 7546 RSC.
- S. Kamijo, K. Kamijo, K. Maruoka and T. Murafuji, Org. Lett., 2016, 18, 6516 CrossRef CAS PubMed.
- M. W. Forkner, L. L. Miller and S. F. Rak, Synth. Met., 1990, 36, 65 CrossRef CAS.
- J. E. Almlof, M. W. Feyereisen, T. H. Jozefiak and L. L. Miller, J. Am. Chem. Soc., 1990, 112, 1206 CrossRef CAS.
- I. Baxter, D. W. Cameron and R. B. Titman, J. Chem. Soc. C, 1971, 1253 RSC.
- K. Wu, L. Wang, S. Colón-Rodríguez, G.-U. Flechsig and T. Wang, Angew. Chem., Int. Ed., 2019, 58, 1774 CrossRef CAS.
- M. E. Wolff, Chem. Rev., 1963, 63, 55 CrossRef CAS.
- Y. Amaoka, M. Nagatomo, M. Watanabe, K. Tao, S. Kamijo and M. Inoue, Chem. Sci., 2014, 5, 4339 RSC.
- V. Malatesta and K. U. Ingold, J. Am. Chem. Soc., 1981, 103, 609 CrossRef CAS.
- D. Ma, J. Pan, L. Yin, P. Xu, Y. Gao, Y. Yin and Y. Zhao, Org. Lett., 2018, 20, 3455 CrossRef CAS PubMed.
- J. Gong and P. L. Fuchs, J. Am. Chem. Soc., 1996, 118, 4486 CrossRef CAS.
- J. Xiang, W. Jiang and P. L. Fuchs, Tetrahedron Lett., 1997, 38, 6635 CrossRef CAS.
- P. J. Stang and J. A. Bjork, J. Chem. Soc., Chem. Commun., 1978, 1057 RSC.
- T. Hoshikawa, S. Kamijo and M. Inoue, Org. Biomol. Chem., 2012, 11, 164 RSC.
- Z.-F. Cheng, Y.-S. Feng, C. Rong, T. Xu, P.-F. Wang, J. Xu, J.-J. Dai and H.-J. Xu, Green Chem., 2016, 18, 4185 RSC.
- J.-B. Han, H. H. San, A. Guo, L. Wang and X.-Y. Tang, Adv. Synth. Catal., 2021, 363, 2366 CrossRef CAS.
- M. Mella, M. Freccero, T. Soldi, E. Fasani and A. Albini, J. Org. Chem., 1996, 61, 1413 CrossRef CAS.
- M. Mella, M. Freccero and A. Albini, Tetrahedron, 1996, 52, 5533 CrossRef CAS.
- A. Albini, M. Mella and M. Freccero, Tetrahedron, 1994, 50, 575 CrossRef CAS.
- A. A. Fokin, P. R. Schreiner, N. A. Fokina, B. A. Tkachenko, H. Hausmann, M. Serafin, J. E. P. Dahl, S. Liu and R. M. K. Carlson, J. Org. Chem., 2006, 71, 8532 CrossRef CAS PubMed.
- A. A. Fokin, B. A. Tkachenko, P. A. Gunchenko, D. V. Gusev and P. R. Schreiner, Chem. – Eur. J., 2005, 11, 7091 CrossRef CAS PubMed.
- M. A. J. Duncton, MedChemComm, 2011, 2, 1135 RSC.
- I. Kavai, L. H. Mead, J. Drobniak and S. F. Zakrzewski, J. Med. Chem., 1975, 18, 272 CrossRef CAS PubMed.
- J. M. Anderson and J. K. Kochi, J. Am. Chem. Soc., 1970, 92, 1651 CrossRef CAS.
- A. Nayyar, V. Monga, A. Malde, E. Coutinho and R. Jain, Bioorg. Med. Chem., 2007, 15, 626 CrossRef CAS PubMed.
- D. H. R. Barton, B. Garcia, H. Togo and S. Z. Zard, Tetrahedron Lett., 1986, 27, 1327 CrossRef CAS.
- E. Castagnino, S. Corsano, D. H. R. Barton and S. Z. Zard, Tetrahedron Lett., 1986, 27, 6337 CrossRef CAS.
- A. P. Antonchick and L. Burgmann, Angew. Chem., Int. Ed., 2013, 52, 3267 CrossRef CAS PubMed.
- Q. Yang, P. Y. Choy, Y. Wu, B. Fan and F. Y. Kwong, Org. Biomol. Chem., 2016, 14, 2608 RSC.
- D.-C. Wang, R. Xia, M.-S. Xie, G.-R. Qu and H.-M. Guo, Org. Biomol. Chem., 2016, 14, 4189 RSC.
- X. Wang, B. Lei, L. Ma, L. Zhu, X. Zhang, H. Zuo, D. Zhuang and Z. Li, Chem. – Asian J., 2017, 12, 2799 CrossRef CAS PubMed.
- L. Zhou and H. Togo, Eur. J. Org. Chem., 2019, 1627 CrossRef CAS.
- H. Zhao, Z. Li and J. Jin, New J. Chem., 2019, 43, 12533 RSC.
- I. B. Perry, T. F. Brewer, P. J. Sarver, D. M. Schultz, D. A. DiRocco and D. W. C. MacMillan, Nature, 2018, 560, 70 CrossRef CAS PubMed.
- L. P. Ermolenko and C. Giannotti, J. Chem. Soc., Perkin Trans. 2, 1996, 1205 RSC.
- O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander and M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 4896 CrossRef CAS PubMed.
- A. K. Yadav and L. D. S. Yadav, Org. Biomol. Chem., 2015, 13, 2606 RSC.
- Z.-Q. Zhu, T.-T. Wang, P. Bai and Z.-Z. Huang, Org. Biomol. Chem., 2014, 12, 5839 RSC.
- Z. Li, Y. Zhang, L. Zhang and Z.-Q. Liu, Org. Lett., 2014, 16, 382 CrossRef CAS.
- D. Liang, B. Huo, Y. Dong, Y. Wang, Y. Dong, B. Wang and Y. Ma, Chem. – Asian J., 2019, 14, 1932 CrossRef CAS PubMed.
- J. Schörgenhumer and M. Waser, Org. Chem. Front., 2016, 3, 1535 RSC.
- S. Kamijo, T. Hoshikawa and M. Inoue, Org. Lett., 2011, 13, 5928 CrossRef CAS PubMed.
- T. Hoshikawa, S. Yoshioka, S. Kamijo and M. Inoue, Synthesis, 2013, 45, 874 CrossRef CAS.
- J.-P. Berndt, F. R. Erb, L. Ochmann, J. Beppler and P. R. Schreiner, Synlett, 2019, 30, 493 CrossRef CAS.
- G. A. Olah, O. Farooq and G. K. S. Prakash, Synthesis, 1985, 1140 CrossRef CAS.
- M.-X. Sun, Y.-F. Wang, B.-H. Xu, X.-Q. Ma and S.-J. Zhang, Org. Biomol. Chem., 2018, 16, 1971 RSC.
- H. Peng, J.-T. Yu, Y. Jiang, H. Yang and J. Cheng, J. Org. Chem., 2014, 79, 9847 CrossRef CAS PubMed.
- W. K. Weigel, H. T. Dang, H.-B. Yang and D. B. C. Martin, Chem. Commun., 2020, 56, 9699 RSC.
| This journal is © The Royal Society of Chemistry 2022 |