K2S2O8-mediated coupling of 6-amino-7-aminomethyl-thiazolino-pyridones with aldehydes to construct amyloid affecting pyrimidine-fused thiazolino-2-pyridones†
Received
11th August 2021
, Accepted 28th October 2021
First published on 29th October 2021
Abstract
We herein present the synthesis of diversely functionalized pyrimidine fused thiazolino-2-pyridones via K2S2O8-mediated oxidative coupling of 6-amino-7-(aminomethyl)-thiazolino-2-pyridones with aldehydes. The developed protocol is mild, has wide substrate scope, and does not require transition metal catalyst or base. Some of the synthesized compounds have an ability to inhibit the formation of Amyloid-β fibrils associated with Alzheimer's disease, while others bind to mature amyloid-β and α-synuclein fibrils.
Introduction
Nitrogen fused heterocycles have attracted considerable interest among synthetic chemists because of their broad range of pharmacological properties.1–12 In particular, the pyrimidine is an important heterocyclic motif which is present as a core skeleton in a variety of drugs that possess a wide range of pharmacological activities, including anticancer,13,14 adrenoreceptor antagonists,15,16 antitubercular,17 antimalarial,18 anxiolytic19 and Amyloid-β aggregation inhibitory properties20etc. (Fig. 1). The nucleotide bases and other biomolecules also contain this structural motif. Therefore, the development of different synthetic strategies for the preparation of substituted pyrimidines has received great attention.
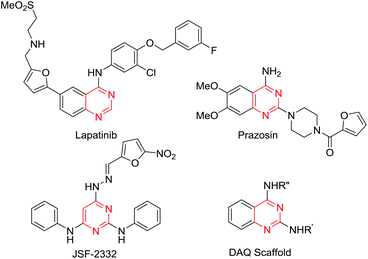 |
| | Fig. 1 Structures of the drugs lapatinib (breast cancer and solid tumour inhibitor), adrenoreceptors antagonists, JSF-2332 (antitubercular agent) and DAQ (amyloid aggregation inhibitor). The pyrimidine moiety in their core structures is highlighted in red. | |
The thiazolino fused 2-pyridone central fragment have proven to be a privileged scaffold. By changing the substitution pattern and thus fine-tuning the properties of the compounds, numerous analogues have been made over the years, with varying biological activities.1,2 Some analogues substituted with smaller alkyl groups at the C-8 position have proven to have interesting antibacterial properties.21 Rigidification of the central fragment by substituting it with bulkier aryl groups, resulted in analogues which inhibit the formation of amyloids, both bacterial curli as well as human Aβ-peptides (compound A, Fig. 2).8,9 Annulation of bicyclic thiazolino-2-pyridones with different nitrogen heterocycles is another strategy to rigidify the central fragment (compound B–E).21–23 This ring extension approach has resulted in compounds capable of modulating the formation of amyloid fibrils,21 which is a property of diagnostic and therapeutic value. Recently, through the inter- and intramolecular Povarov reaction, we constructed pyridine/chromeno-fused 2-pyridones where some analogues (e.g. compound E and F) were shown to bind to Amyloid-β 1–40 (Aβ40) and α-Synuclein (α-Syn) amyloid fibrils.23,24
 |
| | Fig. 2 Thiazolino-2-pyridone based compounds capable of modulating formation (compound A–D) and binding to (compound E and F) α-synuclein and amyloid-β fibrils. | |
Thus, small changes in the substitution pattern or in the core structure of ring fused 2-pyridones can potentially alter their biological properties significantly. We envisioned that ‘Scaffold hopping’ by replacing the pyridine ring with a pyrimidine ring in the tricyclic framework (Fig. 3) could change or improve the amyloid affecting properties. The modification would not alter essential features of the central scaffold, but the extra nitrogen could result in additional interactions, such as being an acceptor for hydrogen bonding.
 |
| | Fig. 3 Scaffold hopping of the tricyclic central fragment. | |
Being equipped with various reactive chemical functionalities, the thiazolino-2-pyridone is potentially a sensitive scaffold to modify, especially under basic and/or oxidative conditions. Hence, a synthetically simple and mild protocol where other reactive functionalities remain untouched is highly desired to install the pyrimidine unit into the thiazolino-2-pyridone scaffold. Oxidative condensation of 2-aminobenzylamines with aldehydes is an attractive strategy to construct the pyrimidine ring.25 K2S2O8 is a mild oxidant, which has been utilized in oxidative coupling reactions to construct nitrogen containing heterocycles.26–29 Herein, we report K2S2O8-mediated aerobic oxidative coupling of 6-amino-7-(aminomethyl)-thiazolino-2-pyridones with aldehydes for the synthesis of a novel tricyclic pyrimidine fused 2-pyridone scaffold. Rewardingly, the scaffold modification resulted in compounds with interesting amyloid modulating properties.
Results and discussion
Our studies began with the synthesis of C-6 and C-7 substituted thiazolino fused 2-pyridones 6a–e as substrates (Scheme 1). An acyl ketene-imine cyclocondensation between thiazolines 1a–d and Meldrum's acid derivative 2a–b afforded thiazolo-2-pyridones 3a–e. Subsequent azidation gave 4a–e, which was then nitrated (5a–e) and finally reduced to produce 6-amino-7-(aminomethyl)-thiazolino-2-pyridones 6a–e in good yields.
 |
| | Scheme 1 Synthesis of 6-amino-7-(aminomethyl)-thiazolino-2-pyridones 6a–6e. Reagents and conditions: (a) TFA (1.0 equiv.), DCE, MWI at 120 °C, 3 min. (b) NaN3 (1.6 equiv.), DMF, rt, 30 min. (c) NaNO2 (1.02 equiv.), TFA (12 equiv.), DCM, rt, 12 h. (d) Pd/C (0.2 equiv.), MeOH, H2 (40 bar), rt, 12 h. | |
Heating 6-amino-7-(aminomethyl)-thiazolino-2-pyridone 6a (1 equiv.) with 4-NO2-benzaldehyde (1.5 equiv.) in MeCN at 80 °C without oxidant resulted in formation of the tetrahydropyrimidine intermediate but the desired product 7a was not observed (Table 1, entry 1). The intermediate was found to be unstable as it underwent oxidation spontaneously but very slowly. Encouraged by the formation of this intermediate, we next investigated the reaction in presence of different oxidants. When the reaction was conducted under air and oxygen, only 10% and 15% of 7a was formed, respectively. Stronger oxidants such as I2, mCPBA and (NH4)2S2O8 resulted in complex mixtures as expected, without the formation of 7a. Only traces of 7a was formed in the presence of CAN or IBX and although oxone or AgOAc provided the desired product, the yields were still low (entries 9 and 10). To our delight, when the reaction was carried out using K2S2O8 under an oxygen atmosphere, 67% of 7a was isolated (entry 11). The yield could even be improved to 79% by performing the reaction with 3 equiv. of K2S2O8 (entry 13).
Table 1 Development of the reaction conditions for synthesis of pyrimidine fused thiazolino-2-pyridones 7a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|

|
| Entry |
Oxidant (equiv.) |
7ab (% yield) |
|
Reagents and conditions: 6a (0.34 mmol, 1.0 equiv.), 4-NO2-benzaldehyde (0.51 mmol, 1.5 equiv.), K2S2O8 (1.02 mmol, 3.0 equiv.), O2, MeCN (3 mL).
Isolated yields.
|
| 1 |
None |
0 |
| 2 |
Air |
10 |
| 3 |
O2 |
15 |
| 4 |
O2, I2 (3) |
0 |
| 5 |
O2, mCPBA (3) |
0 |
| 6 |
O2, (NH4)2S2O8 (3) |
0 |
| 7 |
O2, CAN (3) |
Trace |
| 8 |
O2, IBX (3) |
Trace |
| 9 |
O2, Oxone (3) |
20 |
| 10 |
O2, AgOAc(3) |
28 |
| 11 |
O2, K2S2O8 (1) |
67 |
| 12 |
O2, K2S2O8 (2) |
75 |
| 13 |
O2, K2S2O8 (3) |
79 |
To investigate substrate scope and generate enough diversity for later structure activity relationship studies, different aldehydes and substituted 2-pyridones were used under the established reaction conditions (Scheme 2). Aliphatic aldehydes, benzaldehyde, naphthaldehyde, aryl aldehydes bearing electron-withdrawing/donating groups and heterocyclic aromatic aldehydes were all well tolerated and furnished the desired pyrimidine fused thiazolino-2-pyridones in 48–79% yield. Previous studies have confirmed that the carboxylic acid group is essential for amyloid modulating properties.23 Thus, all methyl carboxylates were hydrolysed to the corresponding carboxylic acid with LiOH. Saponification resulted in compounds 8a–x in 42–90% yield.
 |
| | Scheme 2 Substrate scope for the synthesis of pyrimidine fused thiazolino-2-pyridones, 7a–x under the established reaction conditions. | |
Further, to construct pyrimidine fused thiazolino-2-pyridone analogues efficiently and to allow late stage fine-tuning of the substitution pattern, C-10 unsubstituted pyrimidine fused thiazolino-2-pyridone 7x was brominated with NBS in 80% yield. The Suzuki coupling of intermediate 9 with aryl/heteroaryl boronic acids afforded 7y–ad in good yields (Scheme 3). The corresponding carboxylic acids 8y–ad were subsequently generated by hydrolysis of the corresponding methyl esters.
 |
| | Scheme 3 Synthesis of C-10 substituted pyrimidine fused thiazolino-2-pyridones via bromination and Suzuki coupling. | |
Next, we investigated the suitability of the reaction for preparation of di-aryl substituted pyrimidine fused thiazolino-2-pyridones 10a–i (Scheme 4). For this, substrate 6e was used, and it was combined with various substituted aryl aldehydes, as shown in Scheme 4. This reaction also proceeded well with electron withdrawing (10a, 10b, 10c), electron-donating (10d, 10e), bicyclic (10e, 10g, 10i), as well as heteroaryl (10h) aldehyde substrates. Saponification eventually provided the desired carboxylic acids 11a–i.
 |
| | Scheme 4 Substrate scope for the synthesis of disubstituted pyrimidine fused thiazolino-2-pyridones, 10a–i. | |
To demonstrate the generality of this mild method, 2-(aminomethyl)-aniline and aryl aldehydes were allowed to react under the established conditions. As expected, these mild reaction conditions proved general and pyrimidines 13a–d were isolated in 52–85% yields (Scheme 5).
 |
| | Scheme 5 Substrate scope for the synthesis of 2-substituted quinazolines, 13a–d. | |
Mechanistically, 6-amino-7-(aminomethyl)-thiazolino-2-pyridone 6a undergoes condensation reaction with aldehyde to give tetrahydro pyrimidine intermediate 14. Under heating K2S2O8 decomposes to radical anions30 which oxidize the intermediate 14 to furnish pyrimidine fused thiazolino-2-pyridone 7a (Scheme 6).
 |
| | Scheme 6 Possible reaction mechanism for the K2S2O8 mediated aerobic oxidative coupling of 6-amino-7-(aminomethyl)-thiazolino-2-pyridones with aldehydes. | |
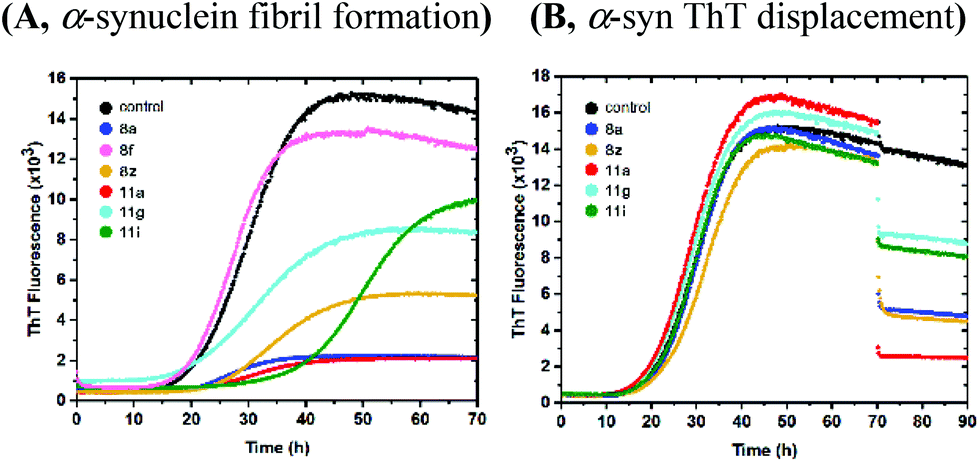
The obtained carboxylic acids 8a–ad and 11a–i were first evaluated for their ability to modulate the formation of α-Syn fibrils using a Thioflavin T (ThT) fluorescence assay31 (Fig. 4 and S1†). In this assay, the effects on fibril formation is observed as changes of the lag phase duration. Further, the ability to bind α-Syn fibrils and displace fibril bound ThT is indicated by a reduced ThT fluorescence amplitude in comparison to the control experiments, where no peptidomimetic compound was included. Compound 8a, 8z and 11a (Fig. 4A) that were equipped with the 4-nitrophenyl group as R3-substituent displayed a moderate to strong ability to bind α-Syn fibrils. Conversely, 8f with a p-tolyl group shows no significant ability to bind α-Syn fibrils. Interestingly, compound 11g with a 2-naphthyl group as R3-substituent, also displayed moderate fibril binding, while compound 11i carrying a 1-naphthyl group as R3-substituent inhibits the amyloid fibril formation to some degree, seen as an extension of the lag time. The R1 substituent had a minor influence on the binding ability of the compounds to α-Syn fibrils. Of the nine different R1 substituents investigated in combination with the 4-nitrophenyl group as R3 substituent, cyclopropyl seems to be superior to the others in terms of amyloid fibril binding (8a and 11a). To verify that the reduced fluorescence indeed is an effect of ThT displacement rather than modulation of amyloid fibril formation, selected compounds were added to already formed α-Syn fibrils (Fig. 4B and S2†). In addition, samples taken after complete fibrilization with compound 8a was visualized with TEM and found to contain fibrils that were indistinguishable from the control (Fig. S4†).
 |
| | Fig. 4 Evaluation of compounds 8a, 8f, 8z, 11a, 11g and 11i for their effect on α-synuclein fibril formation and mature fibril binding. (A) Compounds were included from the start of the reaction. (B) Compound were added to mature fibrils after 70 hours of incubation. | |
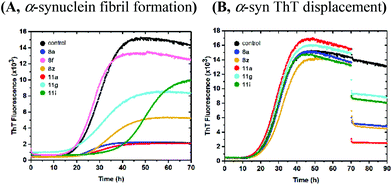
In similar fibrillation assays, compounds 8a–d and 11a–i were evaluated for their ability to affect Aβ40 fibrils, and modulate the fibril formation (Fig. 5 and S4, S5†). Again, an interesting SAR was observed with respect to the R3-substitutents. Compound 8a and 8z were found to bind to Aβ40 fibrils (Fig. 5A). Compound 8f, which had no effect on α-Syn, inhibited the formation of Aβ40 fibrils. Compound 11g with a 2-naphthyl group as R3-substituent was found to be a potent inhibitor of Aβ40 fibril formation, as the lag time was extended beyond the duration of the experiments (70 h), while compound 11i with a 1-naphthyl group was found to be a very week accelerator of Aβ40 fibril formation (Fig. 5A). Further to confirm the modulating effect of compounds, a ThT displacement assay was performed. To distinguish between modulating and binding activities, compounds were added to mature Aβ40 fibrils (Fig. 5B) using the same approach as for α-synuclein. Indeed, compound 8f and 11g were found to be true inhibitors as ThT fluorescence signals were not dropped when the compound was added to mature Aβ fibrils (Fig. 5B). Thus, compound 8f and 11g were found to affect Aβ40 fibrils selectively, which previously has been an issue in designing small molecule inhibitors of amyloid fibril formation.32
 |
| | Fig. 5 Evaluation of compounds 8a, 8f, 8z, 11g and 11i for their effect on Aβ-fibril formation and fibril binding. (A) Compounds were included from the start of the reaction. (B) Compounds were added to mature fibrils after 47 hours of incubation, and the incubation was continued. | |
We were delighted by the potent effect of 11g as an inhibitor of Aβ40 amyloid fibril formation. To explore the structure activity relationship further, the pyridine fused analogues 15a–b were prepared by employing a three-component procedure published previously (Scheme 7).23 The pyridine fused thiazolino 2-pyridones 15a–b display a similar structure activity relationship as their pyrimidine counterparts (compound 11g and 11i). While the 2-naphthyl substituted compound 15a inhibits Aβ40 amyloid formation (Fig. 6B) as effectively as 11g, the 1-naphthyl substituted 15b rather accelerate the formation of Aβ40 amyloid fibrils (Fig. 6B). 15b also accelerates α-synuclein fibril formation slightly, (Fig. 6A), while 15a instead slightly extend the lag phase of α-synuclein amyloid formation (Fig. 6A).
 |
| | Scheme 7 Synthesis of the pyridine fused polyheterocycles 15a–b by three component Povarov reactions, according to established procedure.23 | |
 |
| | Fig. 6 Evaluation of compounds 15a–b for their effect against α-synuclein (A) and Aβ40 fibrils in vitro (B). | |
Next, a set of active compounds were evaluated for their cytotoxicity in HeLa cells using a Neutral Red assay. All the selected compounds were found to have minimal effect on cell viability at the tested concentrations and are less toxic compared to positive control curcumin which is a known amyloid affector (Fig. S7†). The compound 11g started showing some toxicity when tested at 100 μM, but it is still less toxic compared to curcumin.
While many pharmaceuticals do contain nitroaryl motifs, this functionality is often avoided in drug development due to its genotoxic potential.33–35
Since several of the most active amyloid fibril binding 2-pyridones presented above are equipped with the 4-nitrophenyl group, we were prompted to evaluate their mutagenicity by the Ames Salmonella/mammalian-microsome mutagenicity spot test.36 Compound 8a and 11a, which bind to amyloid fibrils, were found to be mutagenic (Fig. S6A†). However, to our delight, none of the active analogues including 11g that were lacking the p-nitro phenyl substituent induced mutations in any of the four Salmonella typhimurium strains tested; TA98, TA100, TA1535 and TA1537 (Fig. S6B†).
Conclusion
A series of tricyclic pyrimidine fused thiazolino-2-pyridones were prepared from readily available aldehydes and 6-amino-7-(aminomethyl)-thiazolino-2-pyridones in a single step using inexpensive K2S2O8 as oxidant. This is to the best of our knowledge a new heterocyclic scaffold and several of these new pyrimidine fused thiazolino-2-pyridone carboxylic acids display an ability to bind α-synuclein and Aβ40 fibrils. Compound 8f inhibit and 11g strongly inhibit Aβ40 fibril formation in vitro. Thus, these new compounds have great potential to be developed as tool compounds for further investigations of neurodegenerative diseases such as Alzheimer's and Parkinson's disease. Both nitrophenyl analogues 8a and 11a, which were strong binders to α-Syn amyloid fibrils, were mutagenic. Still, the new collection of pyrimidine fused 2-pyridones contained some non-mutagenic analogues where the 2-naphthyl substituted analogue 11g stood out as very interesting. This analogue displayed weaker binding to α-Syn and Aβ40 amyloid fibrils but in addition had a strong inhibiting effect on Aβ40 amyloid fibril formation. Further studies to develop active compounds as diagnostic/therapeutic agents are currently underway.
Experimental section
General information
Unless stated, all reagents and solvents were used as received from commercial suppliers. All reactions were carried out under an inert atmosphere with dry solvents under anhydrous conditions, unless otherwise indicated. 4 Å molecular sieves (MS) were activated at 300 °C under a vacuum for 4 h. Microwave reactions were performed in sealed vessels using a Biotage Initiator microwave synthesizer; temperatures were monitored by an internal IR probe. TLC was performed on purchased aluminium backed silica gel plates (median pore size 60 Å, fluorescent indicator 254 nm) and detected with UV light at 254 and 366 nm. Flash column chromatography was performed using silica gel (0.063–0.200 mesh). Automated flash column chromatography was performed using a BiotageIsolera One system and purchased prepacked silica gel cartridges (Biotage SNAP Cartridge, KP-Sil). Preparative HPLC was performed with a Gilson instrument using a Phenomenex column (250 × 21.2 mm; Gemini 5 µm NX-C18, 110 Å). The optical rotation was measured with a Rudolph Autopol IV polarimeter 343 at 22 °C and 589 nm. [α] is reported in deg ml g−1 dm−1, concentrations (c) are given in g per 100 mL. IR spectra were recorded on a Bruker Alpha-t spectrometer. The samples were prepared as KBr pellets or between NaCl plates; absorbances are given in reciprocal cm. 1H, 13C, and 19F NMR spectra were recorded on a Bruker Avance III 400 MHz spectrometer with a BBO-F/H Smartprobe, or a Bruker Avance III HD 600 MHz spectrometer with a CP TCI HCN, 5 mm cryoprobe, at 298 K, unless another temperature is given. All spectrometers were operated by Topspin 3.5.7. Resonances are given in ppm relative to TMS and calibrated to solvent residual signals (CDCl3) δH = 7.26 ppm; δC = 77.16 ppm; (CD3)2SO δH = 2.50 ppm, δC = 39.51 ppm. The following abbreviations are used to indicate splitting patterns: s = singlet; d = doublet; dd = double doublet; t = triplet; m = multiplet; bs = broad singlet. LC-MS was conducted on a Micromass ZQ mass spectrometer using ES+ ionization unless otherwise stated. HRMS was performed on a mass spectrometer with ESI-TOF (ES+). Amyloid formation was probed by thioflavin T (ThT) fluorescence, with a FLUOstar Omega instrument (BMG Labtech, Germany), using excitation and emission filters of 440 and 480 nm, respectively.
General procedure for the preparation of Meldrum's acid derivative 2a-b.
Chloroacetic acid (26.22 g, 0.27 mol), DMAP (35.60 g, 0.29 mol) and Meldrum's acid (40.0 g, 0.27 mol) were dissolved in DCM (300 mL) at 0 °C. DCC (62.98 g, 0.30 mol) dissolved in DCM (50 mL) was added dropwise to the mixture over 40 min. The reaction mixture was then left stirring at room temperature overnight and subsequently quenched with 6% (aq.) KHSO4(200 mL). The resulting precipitate was filtered off. The filtrate was washed five times with 6% KHSO4, (400 mL) followed by two times with brine (200 mL), then dried over anhydrous Na2SO4, filtrated, and concentrated, the crude product was used without further purification. The spectral data of 2a–2b is provided in ESI.†
General procedure for synthesis of (3a–3e).
In a microwave reaction tube equipped with a magnetic stirrer, thiazoline (34.67 mmol, 1.0 equiv.) and Meldrum's acid derivative (19.12 g, 86.69 mmol, 2.5 equiv.) was dissolved in 1,2-dichloroethane (150 mL). TFA (2.70 mL, 35.36 mmol, 1.02 equiv.) was added, the tube was sealed and heated to 120 °C under microwave irradiations for 3 min. The reaction mixture was cooled to room temp.diluted with DCM (100 mL) and washed with saturated aqueous sodium bicarbonate solution (100 mL) followed by brine (100 mL). The aqueous phases were re-extracted with DCM (2 mL each). The organic phases were combined, dried, filtered and evaporated. The compound was purified with flash column chromatography. The spectral data of 3a–3e is provided in ESI.†
General procedure for synthesis of (4a–4e).
To a stirred solution of 3a (8.84 mmol, 1.0 equiv.) in dry DMF (5 mL) was added sodium azide (14.14 mmol, 1.6 equiv.) The mixture was stirred under an atmosphere of nitrogen at rt for 30 min or until full consumption of starting material was indicated by TLC. The reaction mixture was then diluted with water (100 mL) and extracted with DCM (200 mL) dried over Na2SO4, and concentrated. The compound was purified with flash column chromatography. The spectral data of 4a–4e is provided in ESI.†
General procedure for synthesis of 6-nitro-7-(azidomethyl)-thiazolino-2-pyridones (5a–5c and 5e).
The 2-pyridone of general structure 4 (11.75 mmol, 1.0 equiv.) was dissolved in DCM (25 mL) in an open RBF. Air was bubbledthrough the solution for 5–10 minutes. NaNO2 was added to the stirred solution. The reaction mixture was left for another 5–10 min before theair source was removed and TFA was added dropwise over 10–15 min at rt. The reaction mixture was again purged with air bubbleing the solution for 5–10 min. After removal of the air source the reaction was left stirring in the open RBF. The reaction was then quenched with saturated aqueous NaHCO3 (20 mL) and extracted with DCM. The combined organic layers were dried over anhydrous sodium sulphate, filtered and evaporated. The compound was purified with flash column chromatography.
For synthesis of 5d, the compound 4d (1.0 mmol, 1.0 equiv.) was dissolved in acetic anhydride (3.4 mL) and cooled to −40 °C. Acetic anhydride (1.7 mL) was cooled on ice, and HNO3 (65% aq) (0.10 mL, 1.5 mmol, 1.5 equiv.) was added slowly. The diluted acid was transferred to a dropping funnel and added slowly to the stirred solution. The reaction mixture was allowed to warm to 0 °C and stirred until completion was indicated by TLC (EtOAc). The reaction mixture was then quenched with methanol (10 mL) at 0 °C and transferred to a separation funnel with ice-cold NaHCO3 (saturated aqueous) (10 mL) and EtOAc (10 mL). The phases were separated, and the aqueous phase was extracted once more with EtOAc (10 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered, and evaporated. The crude product was purified with flash column chromatography (100 g SNAP Cartridge; ethyl acetate in heptane, 20–100% in 8 CV): brown solid of 5d, 2.01 g (48%). The spectral data of 5a–5e is provided in ESI.†
General procedure for synthesis of 6-amino-7-(aminomethyl)-thiazolino-2-pyridones (6a–6e).
The 2-pyridone of general structure 5 (3.55 mmol, 1.0 equiv.) was dissolved in MeOH (15 mL) in a sealed tube. The solution was purged with nitrogen for 5 min Pd/C (0.71 mmol, 0.2 equiv.) was added to the solution. The reaction mixture was again purged for 5 min. The tube was then inserted in a high pressure reactor. The reaction mixture was then stirred under hydrogen gas (40 bar) at room temp. over night. When TLC (neutral alumina plate, (5% MeOH in DCM) indicated completion. Then solution was filtered through a pad of Celite® and evaporated. The crude product was used for the next step without further purification. The spectral data of 6a–6e is provided in ESI.†
Synthesis of pyrimidine fused 2-pyridone from aldehydes and 6-amino-7-(aminomethyl)-thiazolino-2-pyridones (7a–x, 10a–i and intermediate 14).
A solution of 6-amino-7-(aminomethyl)-2-pyridones (0.34 mmol, 1.0 equiv.), aldehyde (0.51 mmol, 1.5 equiv.) in CH3CN (2 mL) was stirred at 80 °C until the condensation was found complete by TLC analysis (about 2 h). To the same solution was added K2S2O8 (1.02 mmol, 3.0 equiv.), and the mixture was stirred at 80 °C for 12 hours under an O2 atmosphere (balloon). After completion of the reaction (monitored by TLC), the mixture was diluted with DCM (5 mL), washed with brine (3 mL), dried, over anhydrous sodium sulphate, filtered, and evaporated. The resulting residue was purified by silica gel column chromatography to give the desired product. The identity and purity of the product was confirmed by 1H and 13C NMR spectroscopic analysis. The spectral data of 7a is shown below, whereas for 7b-x, 10a-i, 14, please see the ESI.† The experimental procedures and spectral data for 9, 7a–7ad, 10a-i are also provided in ESI.†
Methyl (R)-5-cyclopropyl-2-(4-nitrophenyl)-10-oxo-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylate (7a).
The compound was prepared by following general procedure. The product was purified by automated flash column chromatography (25 g SNAP Cartridge) eluting with 0–80% ethyl acetate in heptane, and 100 mg of 6a was converted to 114 mg (79%) of 7a, isolated as a bright yellow solid; [α]25D −104 (c 0.19, CHCl3); IR (KBr): ν 1746, 1672, 1631, 1588, 1502, 1340, 1227, 736 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.70 (s, 1H), 8.77 (d, J = 8.7 Hz, 2H), 8.32 (d, J = 8.7 Hz, 2H), 5.79 (d, J = 7.5 Hz, 1H), 3.82 (s, 3H), 3.80–3.73 (m, 1H), 3.62 (d, J = 11.5 Hz, 1H), 1.87–1.81(m, 1H), 1.15 (dd, J = 13.4, 6.2 Hz, 2H), 0.76 (t, J = 15.1 Hz, 2H); 13C{1H} NMR (151 MHz, CDCl3) δ 168.0, 159.1, 158.9, 156.2, 149.3, 144.5, 144.4, 142.7, 129.9, 129.3, 123.8, 106.7, 63.1, 53.6, 31.7, 9.0, 7.5, 7.4; HRMS (ESI-TOF) m/z [M + H]+ calcd for C20H17N4O5S+ 425.0920, found 425.0920.
General procedure for preparation of acids (8a–8ad, 11a–11i) and (14a).
The ester was dissolved in THF (2 mL) and LiOH (0.1 M aq; 1.4 equiv.) was added while stirring. The reaction mixture was left stirring at r.t. while monitored with TLC (EtOAc). Upon completion, the reaction was quenched with HCl (1 M aq; 1.5 equiv.). The mixture was evaporated and the watery residue was partitioned between chloroform (10 mL) and brine (5 mL). The phases were separated and the aqueous phase was extracted with another portion of CHCl3 (10 mL). The organic phases were combined, dried over anhydrous sodium sulphate, filtered and evaporated. The residue was re-dissolved in DMSO (1 mL) and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min) The fractions containing the desired product were combined and diluted with H2O (1:1) and freeze-dried.
(R)-5-Cyclopropyl-2-(4-nitrophenyl)-10-oxo-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8a).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 50 mg of 7a was converted to 40 mg (83%) of 8a, isolated as a dark yellow solid; [α]25D −64 (c 0.24, DMSO); IR (KBr): ν 1721, 1649, 1580, 1565, 1521, 1500, 1443, 1337 cm−1; 1H NMR (600 MHz, DMSO) δ 13.75 (s, 1H), 9.75 (s, 1H), 8.68 (d, J = 9.0 Hz, 2H), 8.43 (d, J = 9.0 Hz, 2H), 5.68 (dd, J = 8.7, 1.3 Hz, 1H), 3.89 (dd, J = 11.7, 8.7 Hz, 1H), 3.67 (dd, J = 11.7, 1.2 Hz, 1H), 1.91–1.86 (m, 1H), 1.14–1.09 (m, 2H), 0.75–0.61 (m, 2H); 13C {1H}NMR (151 MHz, DMSO) δ 169.3, 157.9, 157.2, 156.6, 148.7, 145.7, 143.6, 142.6, 129.8, 128.7, 124.2, 105.3, 63.1, 31.4, 8.7, 7.2, 7.0; HRMS (ESI-TOF) m/z [M + H]+ calcd for C19H15N4O5S+ 411.0758, found 411.0749.
(R)-2-(4-Cyanophenyl)-5-cyclopropyl-10-oxo-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8b).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 50 mg of 7b was converted to 32 mg (66%) of 8b, isolated as a bright yellow solid; [α]25D −28 (c 0.23, DMSO); IR (KBr): ν 1722, 1632, 1495, 1444, 1383, 765 cm−1; 1H NMR (600 MHz, DMSO) δ 13.74 (s, 1H), 9.74 (s, 1H), 8.61 (d, J = 8.3 Hz, 2H), 8.05 (d, J = 8.4 Hz, 2H), 5.66 (d, J = 8.6 Hz, 1H), 3.88 (dd, J = 11.6, 8.8 Hz, 1H), 3.66 (d, J = 11.6 Hz, 1H), 1.91–1.87 (m, 1H), 1.14–1.09 (m, 2H), 0.74–0.62 (m, 2H); 13C{1H} NMR (151 MHz, DMSO) δ 169.2, 157.9, 157.5, 156.6, 145.6, 143.6, 141.0, 132.9, 129.7, 128.2, 118.7, 112.8, 105.2, 63.1, 31.4, 8.7, 7.2, 7.0; HRMS (ESI-TOF) m/z [M + H]+ calcd for C20H15N4O3S+ 391.0865, found 391.0862.
(R)-5-Cyclopropyl-2-(4-fluorophenyl)-10-oxo-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8c).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 50 mg of 7c was converted to 43 mg (89%) of 8c, isolated as a light yellow solid; [α]25D −35 (c 0.22, DMSO); IR (KBr): ν 2987, 2911, 1758, 1626, 1599, 1566, 1500, 1445, 1389, 1340, 1223, 856, 817, 767 cm−1; 1H NMR (600 MHz, DMSO) δ 13.68 (s, 1H), 9.69 (s, 1H), 8.52–8.49 (m, 2H), 7.41 (t, J = 8.8 Hz, 2H), 5.65 (d, J = 8.6 Hz, 1H), 3.87 (dd, J = 11.7, 8.7 Hz, 1H), 3.64 (d, J = 11.7 Hz, 1H), 1.89–1.87 (m, 1H), 1.13–1.08 (m, 2H), 0.73–0.61 (m, 2H); 13C{1H}NMR (151 MHz, DMSO) δ 169.3, 164.8 (d, J = 248.4 Hz), 158.5, 158.1, 156.4, 144.1, 143.7, 133.4 (d, J = 2.5 Hz), 130.0 (d, J = 8.7 Hz), 129.1, 115.9 (d, J = 21.8 Hz), 105.2, 63.0, 31.3, 8.7, 7.2, 7.0; 19F{1H} NMR (376 MHz, DMSO) δ −110.4; HRMS (ESI-TOF) m/z [M + H]+ calcd for C19H15FN3O3S+ 384.0813, found 384.0810.
(R)-5-Cyclopropyl-2-(2,4-difluorophenyl)-10-oxo-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic Acid (8d).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 30 mg of 7d was converted to 20 mg (69%) of 8d, isolated as a yellow solid; [α]25D −32 (c 0.29, DMSO); IR (KBr): ν 1740, 1680, 1597, 1566, 1445, 1389, 1240, 1223, 856, 767 cm−1; 1H NMR (400 MHz, DMSO) δ 9.71 (s, 1H), 8.23–8.11 (m, 1H), 7.49–7.38 (m, 1H), 7.29 (td, J = 8.4, 2.3 Hz, 1H), 5.58 (d, J = 8.2 Hz, 1H), 3.83 (dd, J = 11.5, 8.6 Hz, 1H), 3.66 (s, 1H), 1.87 (dd, J = 9.4, 4.1 Hz, 1H), 1.17–1.01 (m, 2H), 0.67 (dd, J = 33.4, 4.9 Hz, 2H); 13C{1H}NMR (151 MHz, DMSO) δ 169.2, 163.3 (dd, J = 250.1, 11.9 Hz), 160.9 (dd, J = 257.5, 12.6 Hz), 157.9, 157.2 (d, J = 5.1 Hz), 156.1, 145.1, 143.7, 133.3 (d, J = 10.2 Hz), 128.9, 123.7 (d, J = 13.3 Hz), 112.0 (dd, J = 21.4, 3.7 Hz), 105.4, 105.0 (dd, J = 61.8, 35.7 Hz), 64.6, 31.7, 8.7, 7.2, 7.0; 19F {1H}NMR (376 MHz, DMSO) δ −107.2, −108.9; HRMS (ESI-TOF) m/z [M + H]+ calcd for C19H14F2N3O3S+ 402.0742, found 402.0745.
(R)-5-Cyclopropyl-10-oxo-2-(3-(trifluoromethyl)phenyl)-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8e).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 30 mg of 7e was converted to 17 mg (59%) of 8e, isolated as a bright yellow solid; [α]25D −8 (c 0.18, DMSO); IR (KBr): ν 1743, 1642, 1580, 1508, 1437, 1260, 1222, 1123, 763 cm−1; 1H NMR (600 MHz, DMSO) δ 9.75 (s, 1H), 8.75 (d, J = 12.1 Hz, 2H), 7.94 (d, J = 7.8 Hz, 1H), 7.85 (t, J = 7.8 Hz, 1H), 5.67 (d, J = 8.7 Hz, 1H), 3.91–3.87 (m, 1H), 3.66 (d, J = 11.7 Hz, 1H), 1.90–1.87 (m, 1H), 1.15–1.10 (m, 2H), 0.74–0.62 (m, 2H); 13C{1H}NMR (151 MHz, DMSO) δ 169.2, 158.1, 157.7, 156.6, 145.0, 143.6, 137.8, 131.4, 130.3 (q, J = 34.7 Hz), 130.0, 129.8, 127.2 (q, J = 3.5 Hz), 123.7 (q, J = 272.3 Hz), 121.5, 105.3, 63.0, 31.3, 8.7, 7.2, 7.0; 19F {1H}NMR (376 MHz, CDCl3) δ −62.5; HRMS (ESI-TOF) m/z [M + H]+ calcd for C20H15F3N3O3S+ 434.0781, found 434.0776.
(R)-5-Cyclopropyl-10-oxo-2-(p-tolyl)-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8f).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 30 mg of 7f was converted to 23 mg (80%) of 8f, isolated as a yellow solid; [α]25D −23 (c 0.20, DMSO); IR (KBr): ν 1748, 1644, 1580, 1500, 1443, 1409, 1228 cm−1; 1H NMR (600 MHz, DMSO) δ 13.65 (s, 1H), 9.68 (s, 1H), 8.37 (d, J = 7.9 Hz, 2H), 7.38 (d, J = 7.9 Hz, 2H), 5.65 (d, J = 8.6 Hz, 1H), 3.92–3.83 (m, 1H), 3.64 (d, J = 11.7 Hz, 1H), 2.40 (s, 3H), 1.89–1.87 (m, 1H), 1.13–1.08 (m, 2H), 0.73–0.61 (m, 2H); 13C{1H}NMR (151 MHz, DMSO) δ 169.4, 159.5, 158.2, 156.3, 143.7, 143.6, 140.7, 134.2, 130.0, 129.5, 128.9, 105.3, 62.8, 31.2, 21.0, 8.7, 7.2, 7.0; HRMS (ESI-TOF) m/z [M + H]+ calcd for C20H18N3O3S+ 380.1063, found 380.1058.
(R)-5-Cyclopropyl-2-(4-methoxyphenyl)-10-oxo-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d] pyrimidine-8-carboxylic acid (8g).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 30 mg of 7g was converted to 18 mg (62%) of 8g, isolated as a bright yellow solid; [α]25D −51 (c 0.20, DMSO); IR (KBr): ν 1745, 1641, 1581, 1501, 1440, 1385, 1303, 1255, 1183 cm−1; 1H NMR (600 MHz, DMSO) δ 13.67 (s, 1H), 9.63 (s, 1H), 8.41 (d, J = 8.9 Hz, 2H), 7.11 (d, J = 8.9 Hz, 2H), 5.63 (dd, J = 8.6, 1.1 Hz, 1H), 3.87 (s, 1H), 3.86 (s, 3H), 3.62 (dd, J = 11.7, 1.1 Hz, 1H), 1.87–1.84 (m, 1H), 1.10–106 (m, 2H), 0.72–0.59 (m, 2H); 13C{1H}NMR (151 MHz, DMSO) δ 169.4, 161.6, 159.4, 158.2, 156.2, 143.8, 143.2, 129.4, 128.3, 128.5, 114.2, 105.3, 62.9, 55.4, 31.2, 8.7, 7.2, 7.0; HRMS (ESI-TOF) m/z [M + H]+ calcd for C20H18N3O4S+ 396.1018, found 396.1013.
(R)-2-(Benzo[d][1,3]dioxol-5-yl)-5-cyclopropyl-10-oxo-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8h).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 50 mg of 7h was converted to 32 mg (66%) of 8h, isolated as a bright yellow solid; [α]25D −27 (c 0.30, DMSO); IR (KBr): ν 2932, 2589, 1742, 1639, 1591, 1560, 1522, 1491, 1467, 1250 cm−1; 1H NMR (600 MHz, DMSO) δ 9.63 (s, 1H), 8.08 (dd, J = 8.2, 1.6 Hz, 1H), 7.90 (d, J = 1.5 Hz, 1H), 7.10 (d, J = 8.2 Hz, 1H), 6.14 (s, 2H), 5.66–5.58 (m, 1H), 3.85 (dd, J = 11.7, 8.7 Hz, 1H), 3.63 (dd, J = 11.7, 1.3 Hz, 1H), 1.86 (td, J = 8.1, 4.1 Hz, 1H), 1.12–1.06 (m, 2H), 0.72–0.60 (m, 2H); 13C{1H}NMR (151 MHz, DMSO) δ 169.3, 159.0, 158.2, 156.2, 149.7, 148.0, 143.7, 143.6, 131.2, 128.7, 122.7, 108.6, 107.2, 105.3, 101.7, 63.1, 31.4, 8.7, 7.2, 7.0; HRMS (ESI-TOF) m/z [M + H]+ calcd for C20H16N3O5S+ 410.0805, found 410.0802.
(R)-5-Cyclopropyl-10-oxo-2-phenyl-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8i).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 30 mg of 7i was converted to 16 mg (55%) of 8i, isolated as a yellow solid; [α]25D −39 (c 0.21, DMSO); IR (KBr): ν 1737, 1635, 1589, 1559, 1518, 1464, 1442, 1413, 1229 cm−1; 1H NMR (600 MHz, DMSO) δ 9.67 (s, 1H), 8.46 (d, J = 6.5 Hz, 2H), 7.56 (t, J = 7.7 Hz, 3H), 5.65 (dd, J = 8.6, 1.0 Hz, 1H), 3.88 (dd, J = 11.7, 8.7 Hz, 1H), 3.64 (dd, J = 11.7, 1.2 Hz, 1H), 1.88–1.84 (m, 1H), 1.12–1.07 (m, 2H), 0.72–0.67 (m, 2H); 13C{1H} NMR (151 MHz, DMSO) δ 169.4, 159.4, 158.2, 156.3, 144.0, 143.7, 136.8, 130.8, 129.1, 128.8, 127.7, 105.3, 62.9, 31.3, 8.7, 7.2, 7.0; HRMS (ESI-TOF) m/z [M + H]+ calcd for C19H16N3O3S+ 366.0912, found 366.0902.
(R)-5-Cyclopropyl-2-(naphthalen-2-yl)-10-oxo-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8j).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 50 mg of 7j was converted to 43 mg (90%) of 8j, isolated as a dark yellow solid; [α]25D −10 (c 0.20, DMSO); IR (KBr): ν 1749, 1644, 1582, 1494, 1437, 1390, 1341, 1221, 772 cm−1; 1H NMR (600 MHz, DMSO) δ 13.69 (s, 1H), 9.74 (s, 1H), 9.05 (s, 1H), 8.58 (dd, J = 8.6, 1.5 Hz, 1H), 8.16–8.13 (m, 1H), 8.09 (d, J = 8.7 Hz, 1H), 8.01–7.98 (m, 1H), 7.62–7.57 (m, 2H), 5.67 (d, J = 8.0 Hz, 1H), 3.88 (dd, J = 11.6, 8.7 Hz, 1H), 3.69–3.63 (m, 1H), 1.93–1.85 (m, 1H), 1.14–1.09 (m, 2H), 0.76–0.62 (m, 2H); 13C{1H}NMR (151 MHz, DMSO) δ 169.3, 159.4, 158.2, 156.4, 144.2, 143.8, 134.3, 134.2, 132.8, 129.2, 129.1, 127.8, 127.5, 126.7, 124.6, 105.3, 63.0, 31.3, 8.8, 7.3, 7.0; HRMS (ESI-TOF) m/z [M + H]+ calcd for C23H18N3O3S+ 416.1069, found 416.1080.
(R)-5-Cyclopropyl-10-oxo-2-(thiophen-3-yl)-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8k).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 50 mg of 7k was converted to 28 mg (58%) of 8k, isolated as a light yellow solid; [α]25D −27 (c 0.17, DMSO); IR (KBr): ν 1756, 1725, 1631, 1582, 1538, 1501, 1451, 1391, 1222, 756 cm−1; 1H NMR (600 MHz, DMSO) δ 13.70 (s, 1H), 9.63 (s, 1H), 8.46–8.36 (m, 1H), 7.91–7.83 (m, 1H), 7.71 (dd, J = 4.9, 3.1 Hz, 1H), 5.63 (d, J = 8.6 Hz, 1H), 3.86 (dd, J = 11.6, 8.8 Hz, 1H), 3.63 (d, J = 11.6 Hz, 1H), 1.86 (dd, J = 9.4, 4.0 Hz, 1H), 1.13–1.07 (m, 2H), 0.73–0.60 (m, 2H); 13C{1H} NMR (151 MHz, DMSO) δ 169.3, 158.1, 157.1, 156.3, 143.7, 143.6, 140.9, 128.7, 128.3, 127.5, 127.0, 105.3, 63.0, 31.3, 8.7, 7.2, 7.0; HRMS (ESI-TOF) m/z [M + H]+ calcd for C19H14N3O3S2+ 372.0477, found 372.0478.
(R)-5-Cyclopropyl-2-(furan-2-yl)-10-oxo-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8l).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 35 mg of 7l was converted to 22 mg (52%) of 8l, isolated as a yellow solid; [α]25D −39 (c 0.27, DMSO); IR (KBr): ν 1740, 1678, 1632, 1581, 1502, 1442, 1406, 1278 cm−1; 1H NMR (400 MHz, DMSO) δ 9.59 (s, 1H), 7.98 (s, 1H), 7.35 (d, J = 3.3 Hz, 1H), 6.74 (dd, J = 3.3, 1.7 Hz, 1H), 5.64 (d, J = 8.5 Hz, 1H), 3.87 (dd, J = 11.8, 8.7 Hz, 1H), 3.63 (d, J = 11.8 Hz, 1H), 1.91–1.81 (m, 1H), 1.13–1.01 (m, 2H), 0.65 (dt, J = 10.9, 7.5 Hz, 2H); 13C{1H}NMR (100 MHz, DMSO) δ 169.3, 158.0, 156.8, 153.1, 151.7, 146.0, 144.0, 143.5, 128.7, 113.6, 112.7, 105.4, 63.0, 31.3, 8.7, 7.2, 7.0; HRMS (ESI-TOF) m/z [M + H]+ calcd for C17H14N3O4S+ 356.0707, found 356.0707.
(R)-5-Cyclopropyl-10-oxo-2-(pyridin-4-yl)-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8m).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 40 mg of 7m was converted to 25 mg (65%) of 8m, isolated as a bright yellow solid; [α]25D −8 (c 0.18, DMSO); IR (KBr): ν 1637, 1556, 1493, 1443, 1383, 761 cm−1; 1H NMR (400 MHz, DMSO) δ 9.74 (s, 1H), 8.79 (s, 2H), 8.31 (d, J = 5.7 Hz, 2H), 5.66 (d, J = 7.5 Hz, 1H), 3.91–3.84 (m, 1H), 3.65 (d, J = 11.7 Hz, 1H), 1.87 (dd, J = 9.3, 4.0 Hz, 1H), 1.17–1.05 (m, 2H), 0.67 (dd, J = 37.1, 4.2 Hz, 2H); 13C{1H}NMR (100 MHz, DMSO) δ 169.3, 157.9, 157.3, 156.6, 150.6, 145.7, 143.9, 143.6, 130.2, 121.4, 105.3, 63.1, 31.4, 8.7, 7.3, 7.0; HRMS (ESI-TOF) m/z [M + H]+ calcd for C18H15N4O3S+ 367.0865, found 367.0868.
(R)-2,5-Dicyclopropyl-10-oxo-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8n).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 50 mg of 7n was converted to 36 mg (75%) of 8n, isolated as a pale yellow solid; [α]25D −10 (c 0.25, DMSO); IR (KBr): ν 1736, 1663, 1630, 1589, 1509, 1461, 1406, 1227, 1030 cm−1; 1H NMR (600 MHz, DMSO) δ 13.63 (s, 1H), 9.47 (s, 1H), 5.59 (d, J = 8.6 Hz, 1H), 3.83 (dd, J = 11.7, 8.7 Hz, 1H), 3.60 (d, J = 11.7 Hz, 1H), 2.36–2.31 (m, 1H), 1.89–1.75 (m, 1H), 1.13–1.04 (m, 6H), 0.73–0.51 (m, 2H); 13C{1H} NMR (151 MHz, DMSO) δ 169.4, 167.3, 158.1, 156.0, 143.4, 142.3, 128.2, 105.1, 63.8, 31.2, 18.0, 10.5, 8.7, 7.1, 6.9; HRMS (ESI-TOF) m/z [M + H]+ calcd for C16H16N3O3S+ 330.0912, found 330.0908.
(R)-2-Cyclohexyl-5-cyclopropyl-10-oxo-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8o).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 30 mg of 7o was converted to 18 mg (62%) of 8o, isolated as a pale yellow solid; [α]25D −6 (c 0.18, DMSO); IR (KBr) ν 3429, 2863, 1752, 1616, 1579, 1554, 1521, 1493, 1466, 1411, 1344, 1218 cm−1; 1H NMR (400 MHz, DMSO) δ 9.56 (s, 1H), 5.58 (d, J = 8.2 Hz, 1H), 3.81 (dd, J = 11.7, 8.7 Hz, 1H), 3.60 (d, J = 11.7 Hz, 1H), 2.94 (dd, J = 9.2, 5.8 Hz, 1H), 1.96 (d, J = 11.7 Hz, 2H), 1.82 (dd, J = 7.3, 4.6 Hz, 3H), 1.74–1.54 (m, 3H), 1.47–1.35 (m, 2H), 1.32–1.23 (m, 1H), 1.13–1.02 (m, 2H), 0.69–0.54 (m, 2H); 13C {1H}NMR (100 MHz, DMSO) δ 169.7, 169.4, 158.2, 155.9, 143.4, 143.1, 128.5, 104.9, 63.1, 46.5, 31.6, 31.5, 31.3, 25.7, 8.7, 7.1, 6.9; HRMS (ESI-TOF) m/z [M + H]+ calcd for C19H22N3O3S+ 372.1376, found 372.1378.
(R)-5-Cyclopropyl-10-oxo-2-propyl-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8p).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 30 mg of 7p was converted to 12 mg (42%) of 8p, isolated as a pale yellow solid; [α]25D −8 (c 0.26, DMSO); IR (KBr) ν 1748, 1665, 1600, 1491, 1219 cm−1; 1H NMR (600 MHz, DMSO) δ 9.54 (s, 1H), 5.57 (d, J = 8.3 Hz, 1H), 3.81 (dd, J = 11.5, 8.8 Hz, 1H), 3.60 (d, J = 11.6 Hz, 1H), 2.96 (t, J = 7.5 Hz, 2H), 1.84–1.80 (m, 3H), 1.09–1.04 (m, 2H), 0.93 (d, J = 7.4 Hz, 3H), 0.63–0.57 (m, 2H); 13C{1H} NMR (151 MHz, DMSO) δ 169.4, 166.4, 163.1, 158.2, 155.8, 143.4, 128.4, 104.9, 63.1, 40.4, 31.4, 21.5, 13.7, 8.7, 7.1, 6.9; HRMS (ESI-TOF) m/z [M + H]+ calcd for C17H18N3O3S+ 332.1063, found 332.1054.
(R)-2-(4-Nitrophenyl)-10-oxo-5-phenyl-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8q).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 50 mg of 7q was converted to 34 mg (70%) of 8q, isolated as a yellow solid; [α]25D −37 (c 0.24, DMSO); IR (KBr): ν 1646, 1523, 1501, 1438, 1375, 1342, 1222 cm−1; 1H NMR (600 MHz, DMSO) δ 8.94 (s, 1H), 8.67 (dd, J = 8.8, 2.0 Hz, 2H), 8.43–8.41 (m, 2H), 7.58 (t, J = 7.7 Hz, 3H), 7.52 (t, J = 7.4 Hz, 2H), 5.75 (d, J = 8.4 Hz, 1H), 3.90–3.87 (m, 1H), 3.64 (d, J = 11.6 Hz, 1H); 13C {1H}NMR (151 MHz, DMSO) δ 169.1, 157.7, 157.4, 156.2, 148.8, 145.2, 143.5, 142.5, 133.5, 130.1, 129.3, 128.8, 128.7, 128.6, 124.2, 107.3, 64.0, 31.6; HRMS (ESI-TOF) m/z [M + H]+ calcd for C22H15N4O5S+ 447.0758, found 447.0748.
(R)-2-(Naphthalen-2-yl)-10-oxo-5-phenyl-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8r).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 50 mg of 7r was converted to 35 mg (72%) of 8r, isolated as a bright yellow solid; [α]25D −11 (c 0.22, DMSO); IR (KBr): ν 1736, 1642, 1585, 1512, 1494, 1437, 1383, 1219, 1194 cm−1; 1H NMR (600 MHz, DMSO) δ 13.84 (s, 1H), 9.02 (s, 1H), 8.92 (s, 1H), 8.56 (dd, J = 8.7, 1.3 Hz, 1H), 8.13 (d, J = 7.5 Hz, 1H), 8.09 (d, J = 8.7 Hz, 1H), 7.98 (d, J = 7.6 Hz, 1H), 7.61–7.58 (m, 5H), 7.52 (t, J = 7.4 Hz, 2H), 5.78 (d, J = 8.5 Hz, 1H), 3.91–3.88 (m, 1H), 3.63 (d, J = 8.0 Hz, 1H); 13C {1H}NMR (151 MHz, DMSO) δ 169.3, 159.6, 158.0, 156.0, 143.6, 143.5, 134.2, 133.6, 133.0, 130.1, 129.3, 129.1, 128.7, 128.4, 128.0, 127.7, 126.5, 126.7, 124.6, 107.5, 63.7, 31.4; HRMS (ESI-TOF) m/z [M + H]+ calcd for C26H18N3O3S+ 452.1063, found 452.1061.
(R)-5-Methoxy-10-oxo-2-phenyl-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8s).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 30 mg of 7s was converted to 14 mg (49%) of 8s, isolated as a yellow solid; [α]25D −253 (c 0.15, DMSO); IR (KBr): ν 1745, 1638, 1562, 1509, 1459, 1422, 1308, 1218 cm−1; 1H NMR (600 MHz, DMSO) δ 9.44 (s, 1H), 8.48 (dd, J = 7.8, 1.6 Hz, 2H), 7.59–7.56 (m, 3H), 5.66 (d, J = 7.9 Hz, 1H), 3.95–3.92 (m, 1H), 3.85 (s, 3H), 3.72 (dd, J = 11.6, 0.9 Hz, 1H); 13C {1H}NMR (151 MHz, DMSO) δ 169.2, 160.0, 157.3, 153.5, 143.6, 136.8, 136.6, 131.0, 129.1, 128.9, 127.7, 124.9, 63.4, 61.0, 32.1; HRMS (ESI-TOF) m/z [M + H]+ calcd for C17H14N3O4S+ 356.0705, found 356.0702.
(R)-5-Methoxy-2-(4-nitrophenyl)-10-oxo-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8t).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 50 mg of 7t was converted to 28 mg (58%) of 8t, isolated as a bright yellow solid; [α]25D −36 (c 0.39, DMSO); IR (KBr): ν 1748, 1649, 1631, 1524, 1504, 1339, 1220 cm−1; 1H NMR (600 MHz, DMSO) δ 9.50 (s, 1H), 8.70 (d, J = 9.0 Hz, 2H), 8.44 (d, J = 9.0 Hz, 2H), 5.56 (d, J = 7.7 Hz, 1H), 3.92–3.87 (m, 1H), 3.85 (s, 3H), 3.74 (d, J = 11.2 Hz, 1H); 13C{1H} NMR (151 MHz, DMSO) δ 168.6, 157.6, 157.1, 153.7, 153.6, 148.7, 143.4, 142.7, 137.8, 128.8, 125.5, 124.2, 64.5, 61.0, 32.6; HRMS (ESI-TOF) m/z [M + H]+ calcd for C17H13N4O6S+ 401.0556, found 401.0549.
(R)-2-(2,4-Difluorophenyl)-5-methoxy-10-oxo-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8u).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 40 mg of 7u was converted to 22 mg (57%) of 8u, isolated as a light yellow solid; [α]25D −76 (c 0.33, DMSO); IR (KBr): ν 1746, 1636, 1507, 1457, 1422, 1328, 1218 cm−1; 1H NMR (600 MHz, DMSO) δ 9.46 (s, 1H), 8.18 (dd, J = 8.8, 6.9 Hz, 1H), 7.44 (td, J = 9.3, 4.8 Hz, 1H), 7.30 (td, J = 8.4, 2.3 Hz, 1H), 5.62 (d, J = 7.9 Hz, 1H), 3.92 (dd, J = 11.5, 8.5 Hz, 1H), 3.85 (s, 3H), 3.74–3.72 (m, 1H); 13C{1H}NMR (151 MHz, DMSO) δ 168.9, 163.1 (dd, J = 250.3, 11.9 Hz), 160.9 (dd, J = 257.7, 12.6 Hz), 157.8, 157.1, 153.4, 143.4, 136.5, 133.3 (d, J = 10.2 Hz), 128.8, 124.8, 122.6 (dd, J = 9.5, 3.6 Hz), 112.0 (dd, J = 21.4, 3.6 Hz), 105.3 (d, J = 26.2 Hz), 63.7, 61.0, 32.2; 19F {1H}NMR (356 MHz, DMSO) δ −108.6 to −108.9 (m), −109.2 (dd, J = 19.3, 9.5 Hz); HRMS (ESI-TOF) m/z [M + H]+ calcd for C17H12F2N3O4S+ 392.0511, found 392.0506.
(R)-5-Methoxy-10-oxo-2-(p-tolyl)-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8v).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 20 mg of 7v was converted to 10 mg (52%) of 8v, isolated as a light yellow solid; [α]25D −33 (c 0.42, DMSO); IR (KBr): ν 1744, 1631, 1507, 1458, 1422, 1328, 1219 cm−1; 1H NMR (600 MHz, DMSO) δ 9.40 (s, 1H), 8.37 (d, J = 8.1 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H), 5.55 (d, J = 7.7 Hz, 1H), 3.87 (d, J = 8.4 Hz, 1H), 3.84 (s, 3H), 3.71 (d, J = 11.3 Hz, 1H), 2.40 (s, 3H); 13C{1H} NMR (151 MHz, DMSO) δ 168.9, 159.9, 157.3, 153.3, 143.6, 140.8, 135.6, 134.2, 129.5, 128.8, 127.7, 124.6, 64.3, 60.9, 32.5, 21.1; HRMS (ESI-TOF) m/z [M + H]+ calcd for C18H16N3O4S+ 370.0856, found 370.0853.
(R)-5-Methoxy-2-(4-methoxyphenyl)-10-oxo-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8w).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 30 mg of 7w was converted to 21 mg (73%) of 8w, isolated as a yellow solid; [α]25D −16 (c 0.40, DMSO); IR (KBr): ν 1736, 1645, 1505, 1460, 1431, 1373, 1253 cm−1; 1H NMR (600 MHz, DMSO) δ 9.37 (s, 1H), 8.42 (d, J = 8.9 Hz, 2H), 7.12 (d, J = 9.0 Hz, 2H), 5.57 (d, J = 8.0 Hz, 1H), 3.90–3.87 (m, 1H), 3.86 (s, 3H), 3.83 (s, 3H), 3.70 (d, J = 11.4 Hz, 1H); 13C {1H}NMR (151 MHz, DMSO) δ 168.8, 161.7, 159.8, 157.4, 153.3, 143.6, 135.0, 129.5, 129.4, 129.0, 124.3, 114.3, 63.8, 60.9, 55.4, 32.3; HRMS (ESI-TOF) m/z [M + H]+ calcd for C18H16N3O5S+ 386.0805, found 386.0798.
(R)-2-(4-Nitrophenyl)-10-oxo-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8x).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 35 mg of 7x was converted to 21 mg (62%) of 8x, isolated as a yellow solid; [α]25D −24 (c 0.29, DMSO); IR (KBr): ν 1748, 1671, 1587, 1523, 1436, 1348 cm−1; 1H NMR (400 MHz, DMSO) δ 9.38 (s, 1H), 8.66 (d, J = 8.8 Hz, 2H), 8.41 (d, J = 8.8 Hz, 2H), 6.81 (s, 1H), 5.65 (d, J = 8.0 Hz, 1H), 3.96 (dd, J = 11.7, 8.6 Hz, 1H), 3.71 (d, J = 11.8 Hz, 1H); 13C {1H}NMR (100 MHz, DMSO) δ 169.1, 158.4, 158.3, 157.5, 148.7, 145.9, 142.8, 142.7, 130.0, 129.6, 128.7, 124.1, 94.3, 63.1, 32.1; HRMS (ESI-TOF) m/z [M + H]+ calcd for C16H11N4O5S+ 371.0445, found 371.0451.
(R)-2-(4-Nitrophenyl)-10-oxo-5-(3-(trifluoromethyl)phenyl)-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8y).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 30 mg of 7y was converted to 15 mg (52%) of 8y, isolated as a bright yellow solid; [α]25D −213 (c 0.15, DMSO); IR (KBr): ν 1753, 1679, 1639, 1524, 1502, 1445, 1341, 1169 cm−1; 1H NMR (400 MHz, DMSO) δ 8.96 (s, 1H), 8.68 (d, J = 8.9 Hz, 2H), 8.44 (d, J = 8.9 Hz, 2H), 7.88 (dd, J = 25.2, 7.3 Hz, 4H), 5.80 (d, J = 8.2 Hz, 1H), 3.93 (d, J = 2.5 Hz, 1H), 3.68 (d, J = 11.3 Hz, 1H); 13C{1H}NMR (100 MHz, DMSO) δ 169.1, 157.8, 157.6, 156.1, 148.8, 145.7, 143.5, 142.4, 134.7, 134.6, 129.6 (dd, J = 52.4, 21.4 Hz), 128.8, 128.5, 126.8, 125.5 (dd, J = 18.3, 3.5 Hz), 124.2, 122.7, 106.1, 63.8, 31.7; 19F {1H}NMR (376 MHz, DMSO) δ −61.0 to −61.1 (m); HRMS (ESI-TOF) m/z [M + H]+ calcd for C23H14F3N4O5S+ 515.0632, found 515.0634.
(R)-5-(4-Fluorophenyl)-2-(4-nitrophenyl)-10-oxo-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8z).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 25 mg of 7z was converted to 15 mg (62%) of 8z, isolated as a light yellow solid; [α]25D −72 (c 0.18, DMSO); IR (KBr): ν 1739, 1650, 1605, 1580, 1526, 1509, 1489, 1339, 1221, 734 cm−1; 1H NMR (400 MHz, DMSO) δ 8.94 (s, 1H), 8.67 (d, J = 8.8 Hz, 2H), 8.43 (d, J = 8.8 Hz, 2H), 7.54 (s, 2H), 7.42 (t, J = 8.8 Hz, 2H), 5.78 (d, J = 7.9 Hz, 1H), 3.93–3.88 (m, 1H), 3.66 (d, J = 11.4 Hz, 1H); 13C{1H} NMR (100 MHz, DMSO) δ 169.1, 163.4, 160.9, 157.6 (d, J = 28.6 Hz), 156.3, 149.8, 145.4, 143.5, 142.5, 132.5 (d, J = 8.7 Hz), 129.8 (d, J = 9.5 Hz), 128.8, 128.7, 124.2, 116.3 (d, J = 21.7 Hz), 106.4, 63.9, 31.6; 19F{1H}NMR (376 MHz, DMSO) δ −112.6; HRMS (ESI-TOF) m/z [M + H]+ calcd for C22H14FN4O5S+ 465.0663, found 465.0666.
(R)-2,5-Bis(4-nitrophenyl)-10-oxo-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8aa).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 30 mg of 7aa was converted to 19 mg (65%) of 8aa, isolated as a dark yellow solid; [α]25D −23 (c 0.22, DMSO); IR (KBr): ν 1752, 1677, 1635, 1601, 1520, 1441, 1345 cm−1; 1H NMR (400 MHz, DMSO) δ 9.03 (s, 1H), 8.69 (d, J = 8.9 Hz, 2H), 8.45–8.42 (m, 4H), 7.83 (d, J = 6.6 Hz, 2H), 5.79 (d, J = 7.9 Hz, 1H), 3.96–3.91 (m, 1H), 3.70 (d, J = 11.2 Hz, 1H); 13C{1H}NMR (100 MHz, DMSO) δ 169.0, 157.7, 156.2, 148.8, 147.3, 145.9, 143.5, 142.4, 140.6, 131.8, 128.8, 128.7, 128.2, 124.4, 124.2, 105.7, 64.0, 31.9; HRMS (ESI-TOF) m/z [M + H]+ calcd for C22H14N5O7S+ 492.0608, found 492.0604.
(R)-5-(4-(Methylsulfonyl)phenyl)-2-(4-nitrophenyl)-10-oxo-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8ab).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 30 mg of 7ab was converted to 14 mg (48%) of 8ab, isolated as a yellow solid; [α]25D −13 (c 0.20, DMSO); IR (KBr): ν 1752, 1681, 1634, 1521, 1501, 1440, 1342, 1150 cm−1; 1H NMR (400 MHz, DMSO) δ 9.00 (s, 1H), 8.69 (d, J = 8.8 Hz, 2H), 8.44 (d, J = 8.9 Hz, 2H), 8.13 (d, J = 8.7 Hz, 2H), 7.65–7.60 (m, 2H), 5.81 (d, J = 7.9 Hz, 1H), 3.94 (dd, J = 11.7, 8.7 Hz, 1H), 3.70 (d, J = 11.1 Hz, 1H), 3.34 (s, 3H); 13C{1H} NMR (100 MHz, DMSO) δ 169.1, 157.7, 156.2, 148.8, 145.6, 143.5, 142.4, 140.8, 138.7, 132.1, 131.5, 131.3, 128.8, 127.9, 124.2, 106.1, 64.8, 43.4, 31.7; HRMS (ESI-TOF) m/z [M + H]+ calcd for C23H17N4O7S2+ 525.0533, found 525.0525.
(R)-5-(Benzo[d][1,3]dioxol-5-yl)-2-(4-nitrophenyl)-10-oxo-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8ac).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 35 mg of 7ac was converted to 17 mg (50%) of 8ac, isolated as a bright yellow solid; [α]25D −102 (c 0.16, DMSO); IR (KBr): ν 1751, 1680, 1649, 1567, 1339, 1230 cm−1; 1H NMR (400 MHz, DMSO) δ 9.00 (s, 1H), 8.68 (d, J = 8.9 Hz, 2H), 8.43 (d, J = 8.9 Hz, 2H), 7.03 (dd, J = 58.6, 25.1 Hz, 3H), 6.15 (s, 2H), 5.77 (d, J = 8.3 Hz, 1H), 3.89 (s, 1H), 3.64 (d, J = 11.5 Hz, 1H); 13C{1H}NMR (100 MHz, DMSO) δ 169.2, 157.7, 157.4, 156.5, 148.7, 147.9, 147.6, 145.2, 143.4, 142.5, 142.4, 128.9, 128.7, 124.2, 123.9, 110.4, 109.0, 107.1, 101.5, 63.8, 31.4; HRMS (ESI-TOF) m/z [M + H]+ calcd for C23H15N4O7S+ 491.0656, found 491.0646.
(R)-2-(4-Nitrophenyl)-10-oxo-5-(thiophen-3-yl)-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (8ad).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 30 mg of 7ad was converted to 20 mg (69%) of (8ad), isolated as a bright yellow solid; [α]25D −16 (c 0.33, DMSO); IR (KBr): ν 1752, 1680, 1632, 1529, 1443, 1258 cm−1; 1H NMR (400 MHz, DMSO) δ 9.06 (s, 1H), 8.68 (d, J = 8.9 Hz, 2H), 8.42 (d, J = 8.9 Hz, 2H), 7.85–7.74 (m, 2H), 7.32 (d, J = 4.8 Hz, 1H), 5.68 (d, J = 8.3 Hz, 1H), 3.88–3.83 (m, 1H), 3.64 (s, 1H); 13C{1H}NMR (151 MHz, DMSO) δ 168.9, 157.7, 157.4, 156.4, 148.8, 145.8, 143.4, 142.5, 133.2, 129.0, 128.8, 128.6, 127.6, 126.7, 124.2, 102.5, 64.6, 31.8; HRMS (ESI-TOF) m/z [M + H]+ calcd for C20H13N4O5S2+ 453.0322, found 453.0324.
(R)-5-Cyclopropyl-2-(4-nitrophenyl)-10-oxo-4-phenyl-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido [3,4-d]pyrimidine-8-carboxylic acid (11a).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 25 mg of 10a was converted to 22 mg (90%) of 11a, isolated as a dark yellow solid; [α]25D −128 (c 0.28, DMSO); IR (KBr): ν 1746, 1678, 1339, 1565, 1525, 1503, 1431, 1343, 1219 cm−1; 1H NMR (400 MHz, DMSO) δ 8.78–8.63 (m, 2H), 8.50–8.34 (m, 2H), 7.81–7.67 (m, 2H), 7.62–7.45 (m, 3H), 5.71 (dd, J = 8.7, 1.8 Hz, 1H), 3.91 (dd, J = 11.8, 8.8 Hz, 1H), 3.65 (dd, J = 11.8, 1.8 Hz, 1H), 1.24 (dd, J = 13.1, 6.9 Hz, 1H), 0.32–0.16 (m, 2H), 0.15–0.03 (m, 2H); 13C{1H}NMR (100 MHz, DMSO) δ 169.3, 164.8, 157.7, 155.7, 148.6, 148.1, 145.4, 142.5, 140.4, 129.6, 129.5, 128.7, 128.6, 127.8, 124.1, 105.9, 63.6, 31.3, 15.0, 9.9, 9.6; HRMS (ESI-TOF) m/z [M + H]+ calcd for C25H19N4O5S+ 487.1076, found 487.1084.
(R)-2-(4-Cyanophenyl)-5-cyclopropyl-10-oxo-4-phenyl-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (11b).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 25 mg of 10b was converted to 21 mg (87%) of (11b), isolated as a dark yellow solid; [α]25D −115 (c 0.26, DMSO); IR (KBr): ν 2226, 1752, 1665, 1590, 1553, 1457, 1441, 1216, 1180, 749 cm−1; 1H NMR (400 MHz, DMSO) δ 8.61 (d, J = 8.4 Hz, 2H), 8.02 (d, J = 8.4 Hz, 2H), 7.73 (dd, J = 5.0, 2.2 Hz, 2H), 7.52 (d, J = 5.5 Hz, 3H), 5.70 (d, J = 8.7 Hz, 1H), 3.91 (dd, J = 11.7, 8.9 Hz, 1H), 3.64 (d, J = 11.7 Hz, 1H), 1.28–1.18 (m, 1H), 0.32–0.13 (m, 2H), 0.09 (d, J = 4.8 Hz, 2H); 13C{1H}NMR (100 MHz, DMSO) δ 169.3, 164.7, 157.8, 156.0, 147.7, 145.4, 140.8, 140.4, 132.8, 129.6, 129.5, 128.5, 128.2, 127.8, 118.7, 112.7, 105.9, 63.5, 31.2, 15.0, 9.9, 9.6; HRMS (ESI-TOF) m/z [M + H]+ calcd for C26H19N4O3S+ 467.1172, found 467.1168.
(R)-5-Cyclopropyl-2-(4-fluorophenyl)-10-oxo-4-phenyl-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (11c).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 25 mg of 10c was converted to 14 mg (58%) of 11c, isolated as a dark yellow solid; [α]25D −8 (c 0.26, DMSO); IR (KBr): ν 1748, 1672, 1638, 1574, 1484, 1428, 1235, 1221, 1151 cm−1; 1H NMR (400 MHz, DMSO) δ 8.53 (dd, J = 9.0, 5.7 Hz, 2H), 7.75 (dd, J = 6.5, 2.8 Hz, 2H), 7.53 (dd, J = 5.1, 1.8 Hz, 3H), 7.40 (t, J = 8.9 Hz, 2H), 5.67 (dd, J = 8.7, 1.7 Hz, 1H), 3.89 (dd, J = 11.8, 8.7 Hz, 1H), 3.63 (dd, J = 11.8, 1.8 Hz, 1H), 1.24 (td, J = 7.8, 4.0 Hz, 1H), 0.30–0.16 (m, 2H), 0.15–0.10 (m, 2H); 13C{1H}NMR (100 MHz, DMSO) δ 169.3, 165.1, 164.6, 162.6, 157.9, 156.9, 146.6, 145.5, 140.6, 133.2, (d, J = 2.8 Hz), 130.1 (d, J = 8.9 Hz), 129.5, 127.8, 127.2, 115.8 (d, J = 21.9 Hz), 105.7, 63.6, 31.2, 15.1, 9.9, 9.6; 19F {1H}NMR (376 MHz, DMSO) δ −110.4; HRMS (ESI-TOF) m/z [M + H]+ calcd for C25H19FN3O3S+ 460.1131, found 460.1138.
(R)-5-Cyclopropyl-10-oxo-4-phenyl-2-(p-tolyl)-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (11d).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 25 mg of 10d was converted to 18 mg (74%) of 11d, isolated as a dark yellow solid; [α]25D −16 (c 0.32, DMSO); IR (KBr): ν 1748, 1674, 1574, 1535, 1499, 1482, 1427, 1217 cm−1; 1H NMR (400 MHz, DMSO) δ 8.39 (d, J = 8.2 Hz, 2H), 7.74 (dd, J = 6.5, 2.9 Hz, 2H), 7.56–7.51 (m, 3H), 7.38 (d, J = 8.1 Hz, 2H), 5.61 (d, J = 8.0 Hz, 1H), 3.85 (dd, J = 11.4, 8.8 Hz, 1H), 3.62 (dd, J = 11.6, 1.5 Hz, 1H), 2.40 (s, 3H), 1.22 (dd, J = 12.3, 6.6 Hz, 1H), 0.28–0.15 (m, 2H), 0.12 (dd, J = 8.7, 3.4 Hz, 2H); 13C{1H} NMR (151 MHz, DMSO) δ 169.2, 164.4, 158.0, 157.8, 146.4, 145.6, 140.8, 140.5, 134.0, 129.5, 129.4, 129.3, 127.7, 127.7, 127.6, 105.6, 64.0, 31.5, 21.1, 15.0, 9.9, 9.6; HRMS (ESI-TOF) m/z [M + H]+ calcd for C26H22N3O3S+ 456.1382, found 456.1387.
(R)-2-(Benzo[d][1,3]dioxol-5-yl)-5-cyclopropyl-10-oxo-4-phenyl-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (11e).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 25 mg of 10e was converted to 14 mg (58%) of 11e, isolated as a dark yellow solid; [α]25D −13 (c 0.30, DMSO); IR (KBr): ν 1752, 1663, 1592, 1459, 1221, 1130 cm−1; 1H NMR (600 MHz, DMSO) δ 8.09 (dd, J = 8.2, 1.5 Hz, 1H), 7.91 (d, J = 1.5 Hz, 1H), 7.78–7.62 (m, 2H), 7.52–7.50 (m, 3H), 7.09 (d, J = 8.2 Hz, 1H), 6.13 (s, 2H), 5.52 (d, J = 7.5 Hz, 1H), 3.80 (d, J = 8.8 Hz, 1H), 3.62 (d, J = 10.5 Hz, 1H), 1.23–1.19 (m, 1H), 0.22–0.19 (m, 2H), 0.16–0.07 (m, 2H); 13C{1H} NMR (151 MHz, DMSO) δ 168.9, 164.1, 157.9, 157.1, 149.5, 147.9, 145.6, 140.8, 131.1, 129.5, 129.3, 127.7, 127.4, 124.2, 122.6, 108.5, 107.2, 105.1, 101.7, 63.1, 31.9, 15.0, 9.9, 9.7; HRMS (ESI-TOF) m/z [M + H]+ calcd for C26H20N3O5S+ 486.1118, found 486.1123.
(R)-5-Cyclopropyl-10-oxo-2,4-diphenyl-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (11f).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 25 mg of 10f was converted to 17 mg (70%) of 11f, isolated as a dark yellow solid; [α]25D −15 (c 0.19, DMSO); IR (KBr): ν 1751, 1673, 1574, 1534, 1503, 1427, 1215 cm−1; 1H NMR (400 MHz, DMSO) δ 8.49 (dd, J = 7.8, 1.8 Hz, 2H), 7.75 (dd, J = 6.4, 2.9 Hz, 2H), 7.58–7.51 (m, 6H), 5.68 (dd, J = 8.7, 1.8 Hz, 1H), 3.89 (dd, J = 11.8, 8.7 Hz, 1H), 3.63 (dd, J = 11.8, 1.8 Hz, 1H), 1.27–1.21 (m, 1H), 0.21 (ddd, J = 11.3, 10.0, 5.2 Hz, 2H), 0.15–0.08 (m, 2H); 13C{1H} NMR (100 MHz, DMSO) δ 169.3, 164.6, 158.0, 158.8, 146.8, 145.5, 140.5, 136.6, 130.7, 129.5, 129.4, 128.8, 127.9, 127.8, 127.7, 105.8, 63.4, 31.2, 15.0, 9.9, 9.6; HRMS (ESI-TOF) m/z [M + H]+ calcd for C25H20N3O3S+ 442.1225, found 442.1234.
(R)-5-Cyclopropyl-2-(naphthalen-2-yl)-10-oxo-4-phenyl-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (11g).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 25 mg of 10g was converted to 16 mg (66%) of 11g, isolated as a dark yellow solid; [α]25D −5 (c 0.21, DMSO); IR (KBr): ν 1746, 1671, 1631, 1575, 1484, 1426, 1218 cm−1; 1H NMR (400 MHz, DMSO) δ 9.05 (s, 1H), 8.61 (dd, J = 8.7, 1.6 Hz, 1H), 8.15 (dd, J = 5.7, 3.4 Hz, 1H), 8.09 (d, J = 8.8 Hz, 1H), 8.02–7.98 (m, 1H), 7.81–7.75 (m, 2H), 7.63–7.59 (m, 2H), 7.55 (dd, J = 6.2, 2.7 Hz, 3H), 5.73–5.63 (m, 1H), 3.89 (dd, J = 11.6, 8.7 Hz, 1H), 3.64 (dd, J = 11.7, 1.6 Hz, 1H), 1.28–1.22(m, 1H), 0.30–0.18 (m, 2H), 0.16–0.11 (m, 2H); 13C{1H}NMR (151 MHz, DMSO) δ 169.3, 164.6, 158.0, 157.7, 146.7, 145.6, 140.7, 134.2, 134.1, 132.8, 129.6, 129.5, 129.0, 128.3, 127.0, 127.8, 127.7, 126.8, 124.8, 105.8, 63.8, 31.3, 15.1, 9.9, 9.6; HRMS (ESI-TOF) m/z [M + H]+ calcd for C29H22N3O3S+ 492.1382, found 492.1384.
(R)-5-Cyclopropyl-10-oxo-4-phenyl-2-(thiophen-3-yl)-7,8-dihydro-10H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (11h).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 25 mg of 10h was converted to 15 mg (62%) of 11h, isolated as a dark yellow solid; [α]25D −4 (c 0.25, DMSO); IR (KBr): ν 1750, 1670, 1573, 1533, 1481, 1438, 1218 cm−1; 1H NMR (400 MHz, DMSO) δ 8.42 (dd, J = 3.0, 1.2 Hz, 1H), 7.88 (dd, J = 5.0, 1.2 Hz, 1H), 7.69 (ddd, J = 8.1, 5.6, 2.3 Hz, 3H), 7.52 (dd, J = 5.1, 1.8 Hz, 3H), 5.66 (dd, J = 8.6, 1.7 Hz, 1H), 3.88 (s, 1H), 3.62 (dd, J = 11.8, 1.8 Hz, 1H), 1.26–1.19 (m, 1H), 0.25–0.13 (m, 2H), 0.14–0.06 (m, 2H); 13C{1H}NMR (100 MHz, DMSO) δ 169.4, 164.5, 157.9, 155.7, 146.1, 145.5, 140.7, 140.6, 129.5, 129.4, 128.2, 127.8, 127.5, 127.4, 127.1, 105.8, 63.5, 31.9, 15.0, 9.9, 9.6; HRMS (ESI-TOF) m/z [M + H]+ calcd for C23H18N3O3S2+ 448.0784, found 448.0791.
(R)-5-Cyclopropyl-2-(naphthalen-1-yl)-10-oxo-4-phenyl-7,8-dihydro-10H thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (11i).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 30 mg of 10i was converted to 20 mg (69%) of 11i, isolated as a bright yellow solid; [α]25D −9 (c 0.23, DMSO); IR (KBr): ν 1744, 1663, 1642, 1571, 1534, 1499, 1354, 1294, 778 cm−1; 1H NMR (400 MHz, DMSO) δ 9.00–8.89 (m, 1H), 8.28 (dd, J = 7.2, 1.1 Hz, 1H), 8.10 (d, J = 8.2 Hz, 1H), 8.06–8.01 (m, 1H), 7.75 (dd, J = 6.6, 2.9 Hz, 2H), 7.70–7.65 (m, 1H), 7.59 (dd, J = 6.9, 3.0 Hz, 2H), 7.54–7.48 (m, 3H), 5.64 (d, J = 7.4 Hz, 1H), 3.87 (dd, J = 11.6, 8.7 Hz, 1H), 3.64 (dd, J = 11.6, 1.5 Hz, 1H), 1.25 (dd, J = 13.5, 7.0 Hz, 1H), 0.28–0.11 (m, 4H); 13C{1H}NMR (100 MHz, DMSO) δ 169.2, 164.2, 159.9, 158.0, 147.2, 145.3, 140.7, 134.5, 133.7, 130.6, 130.5, 129.5, 129.4, 128.5, 127.8, 127.3, 126.1, 126.0, 125.3, 105.4, 64.1, 31.6, 15.0, 9.9, 9.7; HRMS (ESI-TOF) m/z [M + H]+ calcd for C29H22N3O3S+ 492.1376, found 492.1380.
General procedure for synthesis of 2-substituted quinazolines (13a–d).
A solution of 2-aminobenzylamines (0.34 mmol, 1.0 equiv.), aldehydes (0.51 mmol, 1.5 equiv.) in CH3CN (2 mL) was stirred at 80 °C until the condensation was found complete by TLC analysis (about 2 h). To the same solution was then added K2S2O8 (1.02 mmol, 3.0 equiv.), and the mixture was stirred at 80 °C for 12 hours more under O2 atmosphere (balloon). After completion of the reaction (monitored by TLC), the mixture was diluted with DCM (5 mL), washed with brine (3 mL), dried, over anhydrous sodium sulfate, filtered, and evaporated. The resulting residue was purified by silica gel column chromatography to give the desired product. The identity and purity of the product was confirmed by 1H and 13C NMR spectroscopic analysis.
2-(4-Nitrophenyl)quinazoline (13a)37.
The product was prepared by following the general procedure and purified by automated flash column chromatography (25 g SNAP Cartridge) eluting with 0–30% ethyl acetate in heptane, and 42 mg of 12 was converted to 73 mg (85%) of 13a, isolated as a brown solid; IR (KBr): ν 1618, 1605, 1584, 1554, 1346 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.52 (s, 1H), 8.82 (d, J = 8.9 Hz, 2H), 8.37 (d, J = 8.9 Hz, 2H), 8.13 (d, J = 8.4 Hz, 1H), 7.97 (dd, J = 12.6, 4.3 Hz, 2H), 7.73–7.68 (m, 1H); 13C {1H}NMR (100 MHz, CDCl3) δ 160.8, 158.9, 150.7, 149.4, 143.8, 134.9, 129.6, 129.0, 128.5, 127.4, 124.0, 123.9; HRMS (ESI-TOF) m/z [M + H]+ calcd for C14H10N3O2+ 252.0777, found 252.0777.
2-(Benzo[d][1,3]dioxol-5-yl)quinazoline (13b)38.
The product was prepared by following the general procedure and purified by automated flash column chromatography (25 g SNAP Cartridge) eluting with 0–30% ethyl acetate in heptane, and 42 mg of 12 was converted to 44 mg (52%) of 13b, isolated as a yellow solid; IR (KBr): ν 1678, 1617, 1568, 1453, 1277 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.40 (s, 1H), 8.23 (dd, J = 8.2, 1.7 Hz, 1H), 8.11 (d, J = 1.7 Hz, 1H), 8.05–7.99 (m, 1H), 7.91–7.84 (m, 2H), 7.57 (td, J = 7.3, 1.0 Hz, 1H), 6.95 (d, J = 8.2 Hz, 1H), 6.05 (s, 2H); 13C{1H}NMR (100 MHz, CDCl3) δ 160.7, 160.5, 150.9, 150.0, 148.3, 134.2, 132.7, 128.6, 127.2, 127.1, 123.7, 123.5, 108.9, 108.5, 101.6; HRMS (ESI-TOF) m/z [M + H]+ calcd for C15H11N2O2+ 251.0821, found 251.0821.
2-(Naphthalen-2-yl)quinazoline (13c)39.
The product was prepared by following the general procedure and purified by automated flash column chromatography (25 g SNAP Cartridge) eluting with 0–30% ethyl acetate in heptane, and 42 mg of 12 was converted to 72 mg (83%) of 13c, isolated as a white solid; IR (KBr): ν 1619, 1598, 1566, 1476, 1409 cm−1; 1H NMR (600 MHz, CDCl3) δ 9.44 (s, 1H), 9.17 (s, 1H), 8.76 (dd, J = 8.6, 1.6 Hz, 1H), 8.11 (d, J = 8.4 Hz, 1H), 8.05 (dd, J = 7.8, 4.6 Hz, 1H), 7.99 (d, J = 8.6 Hz, 1H), 7.92–7.88 (m, 1H), 7.87–7.82 (m, 2H), 7.55–7.50 (m, 3H); 13C{1H} NMR (151 MHz, CDCl3) δ 160.9, 160.5, 150.8, 135.4, 134.7, 134.2, 133.5, 129.3, 129.0, 128.6, 128.3, 127.8, 127.3, 126.3, 125.5, 123.6; HRMS (ESI-TOF) m/z [M + H]+ calcd for C18H13N2+ 257.1079, found 257.1081.
2-(Furan-2-yl)quinazoline (13d)37.
The product was prepared by following the general procedure and purified by automated flash column chromatography (25 g SNAP Cartridge) eluting with 0–30% ethyl acetate in heptane, and 42 mg of 12 was converted to 51 mg (77%) of 13d, isolated as a yellow solid; IR (KBr): ν 1617, 1589, 1582, 1497, 1339 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.29 (s, 1H), 8.01(d, J = 9.2 Hz, 1H), 7.80 (ddd, J = 12.4, 7.0, 3.5 Hz, 2H), 7.64–7.62 (m, 1H), 7.48 (ddd, J = 8.0, 7.0, 0.9 Hz, 1H), 7.39 (dd, J = 2.8, 0.8 Hz, 1H), 6.54 (dd, J = 3.6, 1.6 Hz, 1H); 13C{1H}NMR (100 MHz, CDCl3) δ 160.7, 154.0, 152.5, 150.3, 145.3, 134.5, 128.3, 127.2, 123.3, 114.1, 112.3; HRMS (ESI-TOF) m/z [M + H]+ calcd for C12H9N2O+ 197.0715, found 197.0718.
(R)-5-Cyclopropyl-2-(4-nitrophenyl)-10-oxo-2,3,4,7,8,10-hexahydro-1H-thiazolo[3′,2′:1,6]pyrido[3,4-d]pyrimidine-8-carboxylic acid (14a).
The compound was prepared by following general procedure and purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), 25 mg of 14 was converted to 8 mg (33%) of 14a, isolated as a yellow solid; [α]25D −81 (c 0.21, DMSO); IR (KBr): ν 1722, 1642, 1587, 1509, 1498, 1448, 1422, 1400, 1249 cm−1; 1H NMR (400 MHz, DMSO) δ 8.22 (dd, J = 8.7, 6.0 Hz, 2H), 7.69 (dd, J = 16.0, 8.7 Hz, 2H), 5.71 (dd, J = 52.1, 3.1 Hz, 1H), 5.42 (d, J = 8.6 Hz, 1H), 5.30 (dd, J = 23.4, 2.9 Hz, 1H), 3.91 (dd, J = 17.6, 10.4 Hz, 1H), 3.73–3.67 (m, 1H), 3.52 (d, J = 16.5 Hz, 1H), 3.46 (d, J = 11.9 Hz, 2H), 1.57–1.41 (m, 1H), 0.77 (dd, J = 8.0, 2.0 Hz, 2H), 0.54–0.28 (m, 2H); 13C{1H}NMR (100 MHz, DMSO) δ 169.8, 155.2, 150.6, 146.8, 130.1, 128.9, 128.4, 124.5, 123.3, 110.6, 65.6, 62.6, 54.1, 31.3, 9.9, 6.8, 6.4; HRMS (ESI-TOF) m/z [M + H]+ calcd for C19H19N4O5S+ 415.1076, found 415.1077.
(R)-10-Cyclopropyl-7-(naphthalen-2-yl)-5-oxo-9-phenyl-2,3-dihydro-5H-thiazolo[2,3-g][1,7]naphthyridine-3-carboxylic acid (15a).
(R)-Methyl 6-amino-8-cyclopropyl-5-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyridine-3-carboxylate (100 mg, 0.4 mmol) was used to prepare (R)-methyl 10-cyclopropyl-7-(naphthalen-2-yl)-5-oxo-9-phenyl-2,3-dihydro-5H-thiazolo[2,3-g][1,7]naphthyridine-3-carboxylate (105 mg, 55%) according to the general procedure described earlier.23 The product was purified by automated flash column chromatography (25 g SNAP Cartridge) eluting with 0–80% ethyl acetate in heptane, isolated as a dark yellow solid. 30 mg of the methyl ester was hydrolysed as described,23 purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), to give the title compound (18 mg, 62%) isolated as a bright yellow solid; [α]25D −29 (c 0.13, DMSO); IR (KBr): ν 1744, 1670, 1586, 1556, 1511, 1487, 1469, 1440, 1255, 804 cm−1; 1H NMR (400 MHz, DMSO) δ 8.84 (s, 1H), 8.47 (dd, J = 8.7, 1.7 Hz, 1H), 8.26 (s, 1H), 8.08–8.05 (m, 2H), 7.99–7.96 (m, 1H), 7.59–7.47 (m, 7H), 5.65 (dd, J = 8.7, 1.9 Hz, 1H), 4.00–3.66 (m, 1H), 3.58 (dd, J = 11.7, 1.9 Hz, 1H), 1.14 (dd, J = 9.4, 4.0 Hz, 1H), 0.21–0.08 (m, 4H); 13C{1H}NMR (100 MHz, DMSO) δ 169.6, 158.4, 152.4, 147.0, 145.4, 141.2, 140.6, 135.1, 133.4, 133.1, 132.4, 129.3, 128.7, 127.8, 127.6, 126.9, 126.7, 126.6, 126.3, 126.0, 124.5, 63.3, 31.1, 15.7, 10.5, 10.4; HRMS (ESI-TOF) m/z [M + H]+ calcd for C30H24N3O3S+ 491.1424, found 491.1423.
(R)-10-Cyclopropyl-7-(naphthalen-1-yl)-5-oxo-9-phenyl-2,3-dihydro-5H-thiazolo[2,3-g][1,7]naphthyridine-3-carboxylic acid (15b).
(R)-Methyl 6-amino-8-cyclopropyl-5-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyridine-3-carboxylate (100 mg, 0.4 mmol) was used to prepare(R)-methyl 10-cyclopropyl-7-(naphthalen-1-yl)-5-oxo-9-phenyl-2,3-dihydro-5H-thiazolo[2,3-g][1,7]naphthyridine-3-carboxylate (108 mg, 57%) according to the general procedure described earlier.23 The product was purified by automated flash column chromatography (25 g SNAP Cartridge) eluting with 0–80% ethyl acetate in heptane, isolated as a dark yellow solid. 30 mg of the methyl ester was hydrolysed as described,23 purified with preparative HPLC (30–100% MeCN in water + 0.75% HCOOH; gradient in 40 min, 100% for 10 min), to give the title compound (22 mg, 75%) isolated as a bright yellow solid; [α]25D −25 (c 0.15, DMSO); IR (KBr): ν 1743, 1671, 1649, 1631, 1586, 1555, 1511, 1487, 1453, 804 cm−1; 1H NMR (400 MHz, DMSO) δ 13.58 (s, 1H), 8.32 (d, J = 8.0 Hz, 1H), 8.03 (t, J = 7.2 Hz, 2H), 7.82–7.71 (m, 2H), 7.65–7.60 (m, 1H), 7.59–7.47 (m, 4H), 7.47–7.38 (m, 3H), 5.66 (dd, J = 8.5, 1.5 Hz, 1H), 3.88 (dd, J = 11.7, 8.7 Hz, 1H), 3.59 (dd, J = 11.7, 1.5 Hz, 1H), 1.17 (dd, J = 9.4, 3.8 Hz, 1H), 0.23–0.01 (m, 4H); 13C{1H}NMR (100 MHz, DMSO) δ 170.2, 158.9, 155.4, 147.0, 146.0, 141.5, 141.0, 137.2, 134.4, 132.5, 131.1, 130.1, 129.6, 128.8, 128.4, 127.2, 126.5, 126.0, 125.9, 107.2, 63.8, 31.6, 16.2, 11.0, 10.8; HRMS (ESI-TOF) m/z [M + H]+ calcd for C30H24N3O3S+ 491.1424, found 491.1423.
Conflicts of interest
F.A. has ownership interests in Quretech Bio AB. Other authors have no competing financial interest.
Acknowledgements
FA is grateful to the Swedish Research Council (2017-02339; 2017-00695 and 2018-04589), the Knut and Alice Wallenberg foundation (KAW 2013.0031), the Göran Gustafsson foundation, the Kempe Foundation (SMK-1755), the Swedish Foundation for Strategic Research (SB12-0070), the National Institutes of Health (R01AI134847-01A1), the Erling Perssons Stiftelse and the Michael J Fox foundation for financial support. This project has been supported under the framework of the JPIAMR – Joint Programming Initiative on Anti-microbial Resistance.
References
- I. Horvath, C. F. Weise, E. K. Andersson, E. Chorell, M. Sellstedt, C. Bengtsson, A. Olofsson, S. J. Hultgren, M. Chapman, M. Wolf-Watz, F. Almqvist and P. Wittung-Stafshede, J. Am. Chem. Soc., 2012, 134, 3439–3444 CrossRef CAS PubMed.
- J. S. Pinkner, H. Remaut, F. Buelens, E. Miller, V. Aberg, N. Pemberton, M. Hedenstrom, A. Larsson, P. Seed, G. Waksman, S. J. Hultgren and F. Almqvist, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17897–17902 CrossRef CAS PubMed.
- E. Chorell, J. S. Pinkner, G. Phan, S. Edvinsson, F. Buelens, H. Remaut, G. Waksman, S. J. Hultgren and F. Almqvist, J. Med. Chem., 2010, 53, 5690–5695 CrossRef CAS PubMed.
- V. Aberg and F. Almqvist, Org. Biomol. Chem., 2007, 5, 1827–1834 RSC.
- P. Engström, K. S. Krishnan, B. D. Ngyuen, E. Chorell, J. Normark, J. Silver, R. J. Bastidas, M. D. Welch, S. J. Hultgren, H. Wolf-Watz, R. H. Valdivia, F. Almqvist and S. Bergström, mBio, 2014, 6, e02304–14 Search PubMed.
- J. A. Good, C. Andersson, S. Hansen, J. Wall, K. S. Krishnan, A. Begum, C. Grundstrom, M. S. Niemiec, K. Vaitkevicius, E. Chorell, P. Wittung-Stafshede, U. H. Sauer, A. E. Sauer-Eriksson, F. Almqvist and J. Johansson, Cell Chem. Biol., 2016, 23, 404–414 CrossRef PubMed.
- V. Aberg, F. Norman, E. Chorell, A. Westermark, A. Olofsson, A. E. Sauer-Eriksson and F. Almqvist, Org. Biomol. Chem., 2005, 3, 2817–2823 RSC.
- L. Cegelski, J. S. Pinkner, N. D. Hammer, C. K. Cusumano, C. S. Hung, E. Chorell, V. Aberg, J. N. Walker, P. C. Seed, F. Almqvist, M. R. Chapman and S. J. Hultgren, Nat. Chem. Biol., 2009, 5, 913–919 CrossRef CAS PubMed.
- E. K. Andersson, C. Bengtsson, M. L. Evans, E. Chorell, M. Sellstedt, A. E. Lindgren, D. A. Hufnagel, M. Bhattacharya, P. M. Tessier, P. Wittung-Stafshede, F. Almqvist and M. R. Chapman, Chem. Biol., 2013, 20, 1245–1254 CrossRef CAS PubMed.
- I. Horvath, M. Sellstedt, C. Weise, L. M. Nordvall, G. Krishna Prasad, A. Olofsson, G. Larsson, F. Almqvist and P. Wittung-Stafshede, Arch. Biochem. Biophys., 2013, 532, 84–90 CrossRef CAS PubMed.
- J. A. D. Good, J. Silver, C. Núñez-Otero, W. Bahnan, K. S. Krishnan, O. Salin, P. Engström, R. Svensson, P. Artursson, Å. Gylfe, S. Bergström and F. Almqvist, J. Med. Chem., 2016, 59, 2094–2108 CrossRef CAS PubMed.
- J. A. D. Good, M. Kulén, J. Silver, K. S. Krishnan, W. Bahnan, C. Núñez-Otero, I. Nilsson, E. Wede, E. de Groot, Å. Gylfe, S. Bergström and F. Almqvist, J. Med. Chem., 2017, 60, 9393–9399 CrossRef CAS PubMed.
- S. Mahboobi, A. Sellmer, M. Winkler, E. Eichhorn, H. Pongratz, T. Ciossek, T. Baer, T. Maier and T. Beckers, J. Med. Chem., 2010, 53, 8546–8555 CrossRef CAS PubMed.
- J. B. Bharate, N. McConnell, G. Naresh, L. Zhang, N. R. Lakkaniga, L. Ding, N. P. Shah, B. Frett and H. Y. Li, Sci. Rep., 2018, 8, 3722 CrossRef PubMed.
- A. Antonello, P. Hrelia, A. Leonardi, G. Marucci, M. Rosini, A. Tarozzi, V. Tumiatti and C. Melchiorre, J. Med. Chem., 2005, 48, 28–31 CrossRef CAS PubMed.
- M. Rosini, A. Antonello, A. Cavalli, M. L. Bolognesi, A. Minarini, G. Marucci, E. Poggesi, A. Leonardi and C. Melchiorre, J. Med. Chem., 2003, 46, 4895–4903 CrossRef CAS PubMed.
- D. Inoyama, S. D. Paget, R. Russo, S. Kandasamy, P. Kumar, E. Singleton, J. Occi, M. Tuckman, M. D. Zimmerman, H. P. Ho, A. L. Perryman, V. Dartois, N. Connell and J. S. Freundlich, Antimicrob. Agents Chemother., 2018, 62(3), e02063–17 CrossRef CAS PubMed.
- S. Madapa, Z. Tusi, A. Mishra, K. Srivastava, S. K. Pandey, R. Tripathi, S. K. Puri and S. Batra, Bioorg. Med. Chem., 2009, 17, 222–234 CrossRef CAS PubMed.
- G. Guerrini, G. Ciciani, S. Daniele, L. Di Cesare Mannelli, C. Ghelardini, C. Martini and S. Selleri, Bioorg. Med. Chem., 2017, 25, 1901–1906 CrossRef CAS PubMed.
- T. Mohamed, A. Shakeri, G. Tin and P. P. Rao, ACS Med. Chem. Lett., 2016, 7, 502–507 CrossRef CAS PubMed.
- P. Singh, E. Chorell, K. S. Krishnan, T. Kindahl, J. Åden, P. Wittung-Stafshede and F. Almqvist, Org. Lett., 2015, 17, 6194–6197 CrossRef CAS PubMed.
- M. Sellstedt and F. Almqvist, Org. Lett., 2008, 10, 4005–4007 CrossRef CAS PubMed.
- P. Singh, D. E. Adolfsson, J. Ådén, A. G. Cairns, C. Bartens, K. Brännström, A. Olofsson and F. Almqvist, J. Org. Chem., 2019, 84, 3887–3903 CrossRef CAS PubMed.
- D. E. Adolfsson, M. Tyagi, P. Singh, A. Deuschmann, J. Ådén, A. L. Gharibyan, S. W. Jayaweera, A. E. G. Lindgren, A. Olofsson and F. Almqvist, J. Org. Chem., 2020, 85, 14174–14189 CrossRef CAS PubMed.
- M. Saha, P. Mukherjee and A. R. Das, Tetrahedron Lett., 2017, 58, 2044–2049 CrossRef CAS.
- Z. Yang, X. Chen, S. Wang, J. Liu, K. Xie, A. Wang and Z. Tan, J. Org. Chem., 2012, 77, 7086–7091 CrossRef CAS PubMed.
- J. B. Gary, A. K. Cook and M. S. Sanford, ACS Catal., 2013, 3, 700–703 CrossRef CAS.
- H. Wang, L.-N. Guo and X.-H. Duan, Org. Lett., 2013, 15, 5254–5257 CrossRef CAS PubMed.
- D. Ma, W. Chen, G. Hu, Y. Zhang, Y. Gao, Y. Yin and Y. Zhao, Green Chem., 2016, 18, 3522–3526 RSC.
- A. Kumar, R. A. Maurya and D. Saxena, Mol. Diversity, 2010, 14, 331–341 CrossRef CAS PubMed.
- E. Chorell, E. Andersson, M. L. Evans, N. Jain, A. Gotheson, J. Aden, M. R. Chapman, F. Almqvist and P. Wittung-Stafshede, PLoS One, 2015, 10, 0140194 CrossRef PubMed.
- D. Goyal, S. Shuaib, S. Mann and B. Goyal, ACS Comb. Sci., 2017, 19, 55–80 CrossRef CAS PubMed.
- K. Nepali, H.-Y. Lee and J.-P. Liou, J. Med. Chem., 2019, 62, 2851–2893 CrossRef CAS PubMed.
- P. Wardman, Environ. Health Perspect., 1985, 64, 309–320 CrossRef CAS PubMed.
- D. Olender, J. Żwawiak and L. Zaprutko, Pharmaceuticals, 2018, 11(54), 1–29 Search PubMed.
- B. N. Ames, J. McCann and E. Yamasaki, Mutat. Res., 1975, 31, 347–364 CAS.
- B. Han, X.-L. Yang, C. Wang, Y.-W. Bai, T.-C. Pan, X. Chen and W. Yu, J. Org. Chem., 2012, 77, 1136–1142 CrossRef CAS PubMed.
- Y.-Y. Peng, Y. Zeng, G. Qiu, L. Cai and V. W. Pike, J. Heterocycl. Chem., 2010, 47, 1240–1245 CrossRef CAS.
- Z. Ma, T. Song, Y. Yuan and Y. Yang, Chem. Sci., 2019, 10, 10283–10289 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. 1H NMR, 13C NMR, and 19F NMR for all new compounds and fibril binding assays for carboxylic acids (PDF). See DOI: 10.1039/d1ob01580j |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Jörgen
Ådén
a,
Anna
Gharibyan
b,
Dan E.
Adolfsson
a,
Sanduni Wasana
Jayaweera
b,
Pardeep
Singh
a,
Katarina
Vielfort
c,
Mohit
Tyagi
a,
Mari
Bonde
a,
Sven
Bergström
c,
Anders
Olofsson
b and
Fredrik
Almqvist
a,
Jörgen
Ådén
a,
Anna
Gharibyan
b,
Dan E.
Adolfsson
a,
Sanduni Wasana
Jayaweera
b,
Pardeep
Singh
a,
Katarina
Vielfort
c,
Mohit
Tyagi
a,
Mari
Bonde
a,
Sven
Bergström
c,
Anders
Olofsson
b and
Fredrik
Almqvist