Synthesis, C–H bond functionalisation and cycloadditions of 6-styryl-1,2-oxathiine 2,2-dioxides†
Received
10th June 2021
, Accepted 29th June 2021
First published on 29th June 2021
Abstract
A series of 6-styryl-1,2-oxathiine 2,2-dioxides have been efficiently obtained by a two-step protocol from readily available (1E,4E)-1-(dimethylamino)-5-arylpenta-1,4-dien-3-ones involving a regioselective sulfene addition and subsequent Cope elimination. Pd-Mediated direct C–H bond functionalisation of the 6-styryl-1,2-oxathiine 2,2-dioxides and a wider selection of 5,6-diaryl substituted 1,2-oxathiine 2,2-dioxides proceeded smoothly to afford C-3 (hetero)aryl substituted analogues and the results are contrasted with those of a complementary bromination – Suzuki cross-coupling sequence. Whilst the cycloaddition of benzyne, derived from in situ fluoride initiated decomposition of 2-(trimethylsilyl)phenyl trifluoromethanesulfonate, to the substituted 1,2-oxathiine 2,2-dioxides resulted in low yields of substituted naphthalenes, the addition of 4-phenyl-1,2,4-triazoline-3,5-dione to the 6-styryl-1,2-oxathiine 2,2-dioxides afforded novel 5,9-dihydro-1H-[1,2]oxathiino[5,6-c][1,2,4]triazolo[1,2-a]pyridazine-1,3(2H)-dione 8,8-dioxides through a silica-mediated isomerisation of the initial [4 + 2] adducts.
Introduction
The 1,2-oxathiine 2,2-dioxide unit is a useful but little studied six-membered heterocyclic ring system.1–4 Transformation of the 1,2-oxathiine 2,2-dioxide unit into five-membered heterocyclic rings, pyrazoles and pyrroles, has been accomplished upon condensation with hydrazines5 and 2,4-disubstituted furans result from thermolysis.6 Indeed thermolysis of 4,6-dimethyl-1,2-oxathiine 2,2-dioxide provides facile access to 2,4-dimethylfuran from which the intricate side chain of mycolactones A and B7 and the A and C ring moieties of taxol8 have been obtained. Terphenylenes result from the cycloaddition of acetylenes to 1,2-oxathiine 2,2-dioxides under forcing conditions with the expulsion of SO3.9,10 Recently, the 1,2-oxathiine 2,2-dioxide unit has attracted interest in materials application including in energy storage/battery technology,11 as photoresists for high resolution lithography12 and as a core unit in a photochromic dithienylethene.13 The synthesis, chemistry and applications of 1,2-oxathiine 2,2-dioxides has recently been reviewed.14
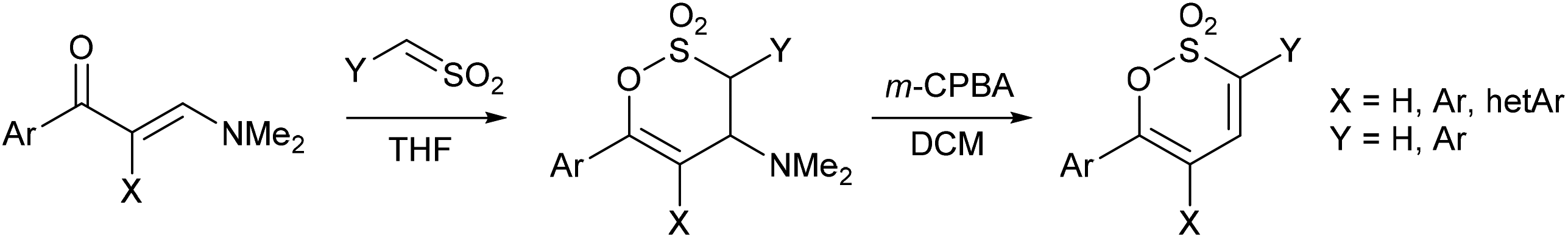
We have previously reinvestigated the relatively scarcely employed addition of sulfenes to enaminoketones15–18 as a route to diversely substituted 4-dialkylamino-3,4-dihydro-1,2-oxathiine 2,2-dioxides and successfully eliminated the dialkylamine moiety to afford a series of unsaturated mono-, di- and tri-(hetero)aryl substituted 1,2-oxathiine 2,2-dioxides (Scheme 1).13,19
 |
| | Scheme 1 | |
In this work we explore the selectivity of the addition of sulfenes to α,β-unsaturated enaminoketones derived from either (E)-4-arylbut-3-en-2-ones or 4-phenylbut-3-yn-2-one to afford unsaturated 1,2-oxathiine 2,2-dioxides bearing either a C-6 arylethenyl or arylethynyl moiety after Cope elimination of the amine function from the intermediate 3,4-dihydro 1,2-oxathiine 2,2-dioxides. Further functionalisation of the oxathiine dioxide ring is examined by transition metal-mediated cross-coupling and cycloaddition chemistry leading to diversely substituted derivatives.
Results and discussion
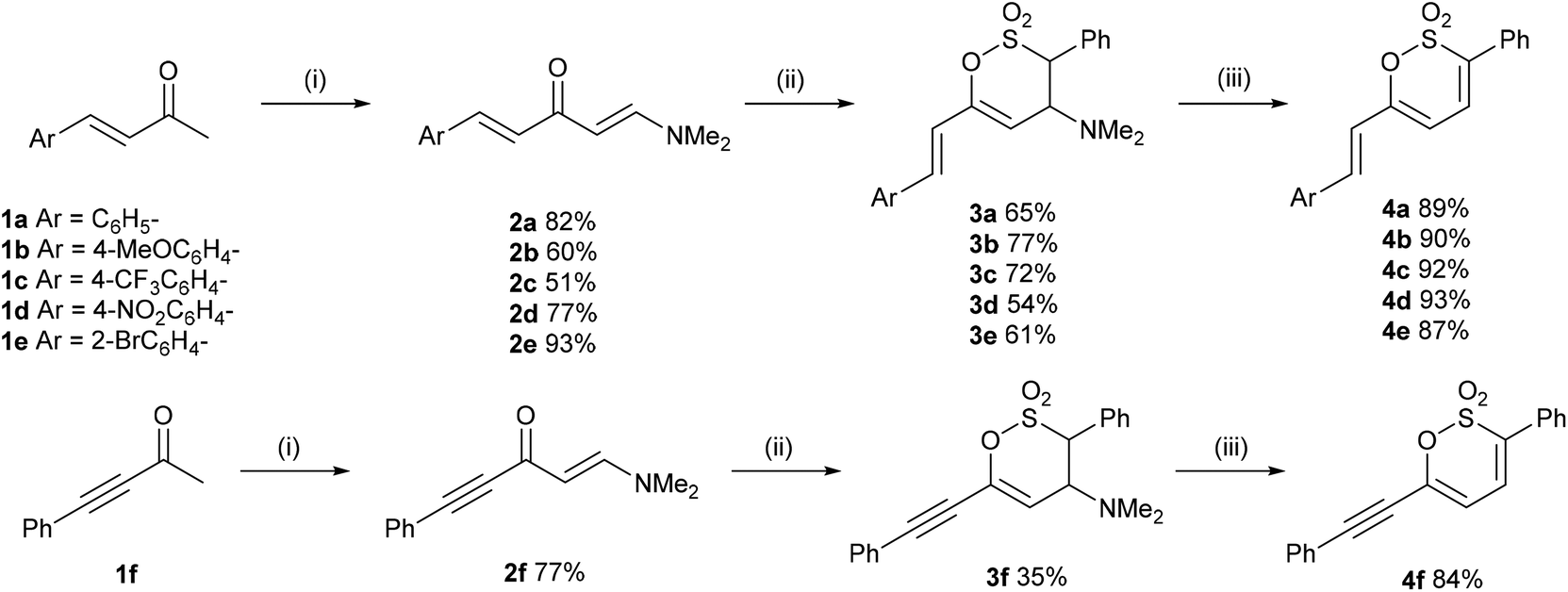
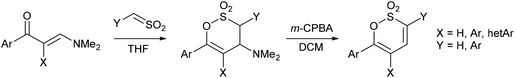
Heating the (E)-4-(4-aryl)but-3-en-2-ones 1a–e in PhMe containing excess DMFDMA readily afforded the (1E,4E)-1-(dimethylamino)-5-phenylpenta-1,4-dien-3-ones 2a–e in 51–93% yield. The geometry of each C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C unit within the molecule was readily established by 1H NMR spectroscopy with the styryl moiety affording a doublet for 4-H at ca. δ 6.8 and for 5-H at ca. δ 7.6 with J ∼ 15.8 Hz and the dimethylaminoethene unit affording a doublet for 2-H at ca. δ 5.3 and for 1-H δ 7.8 with J ∼ 12.4 Hz. (E)-1-(Dimethylamino)-5-phenylpent-1-en-4-yn-3-one 2f was obtained in a similar manner from 4-phenylbut-3-yn-2-one 1f in 77% yield (Scheme 2).
C unit within the molecule was readily established by 1H NMR spectroscopy with the styryl moiety affording a doublet for 4-H at ca. δ 6.8 and for 5-H at ca. δ 7.6 with J ∼ 15.8 Hz and the dimethylaminoethene unit affording a doublet for 2-H at ca. δ 5.3 and for 1-H δ 7.8 with J ∼ 12.4 Hz. (E)-1-(Dimethylamino)-5-phenylpent-1-en-4-yn-3-one 2f was obtained in a similar manner from 4-phenylbut-3-yn-2-one 1f in 77% yield (Scheme 2).
 |
| | Scheme 2 Reagents and conditions: (i) Either DMFDMA, reflux or DMFDMA, L-proline (10 mol%), reflux; (ii) Et3N, PhCH2SO2Cl, THF, 0 °C–rt; (iii) m-CPBA, DCM, 0 °C–rt. | |
Generating phenylsulfene in situ using our established protocol19 of the dropwise addition of excess phenylmethanesulfonyl chloride to a cold (−10–0 °C) stirred solution of the enaminoketone 1a–f, in anhydrous THF containing excess Et3N gave the 6-styryl, 3a–e, and 6-phenyethynyl, 3f, 4-dimethylamino-3,4-dihydro-1,2-oxathiine 2,2-dioxides in 35–77% yield. In the 1H NMR spectra of 3a–f the alkene proton, 5-H, resonates at ca. δ 5.6 and appears as a doublet (J4,5 ∼ 2.5 Hz), 4-H appears at ca. δ 4.4 as a dd (J ∼ 2.5, 11.3 Hz) and 3-H, adjacent to the SO2 moiety, appears as a doublet (J3,4 ∼ 11.3 Hz) at ca. δ 4.5. The magnitude of the 3J coupling between 3-H and 4-H suggests a trans-diaxial arrangement in accord with data observed for simple 3,6-diaryl substituted 4-dimethylamino-3,4-dihydro-1,2-oxathiine 2,2-dioxides where J3,4 ∼ 11.3 Hz.19 The styryl moiety of 3a–f remains intact, since the sulfene addition is regiospecific to the enaminoketone moiety, and affords the expected pair of doublets δ ∼6.6 and 7.1 with J = 15.9 Hz.
Treatment of 3a–f with m-CPBA in cold DCM readily effected the Cope elimination of the 4-dimethylamino function to afford 4a–f in 84–93% yield, without any epoxidation of the styryl function. In the 1H NMR spectra of 4a–f, 4-H, conjugated with the SO2 moiety, resonated further downfield (∼δ 6.9) than 5-H (∼δ 6.2) and each appeared as a doublet with J4,5 = 6.6–7.4 Hz, typical of a cis-diene unit. The styryl moiety of 4a–f predictably afforded a pair of doublets δ ∼6.6 and 7.3 with J = 15.8 Hz.
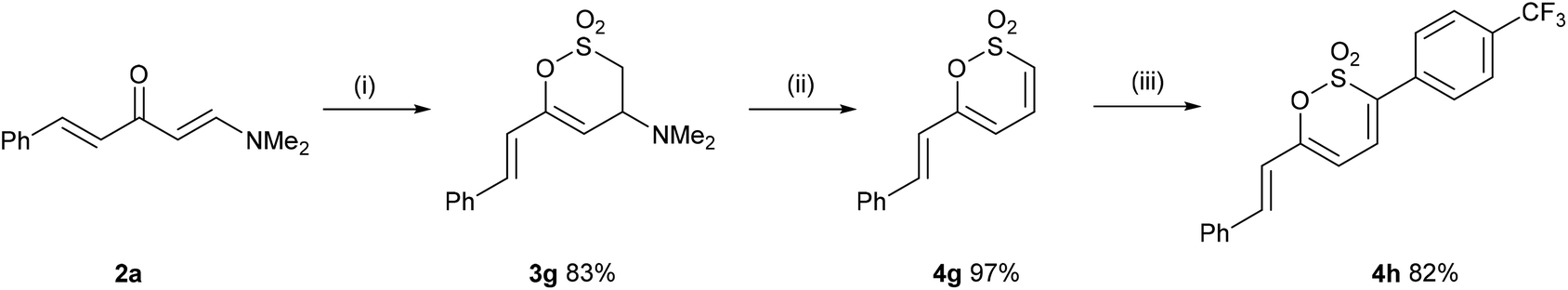
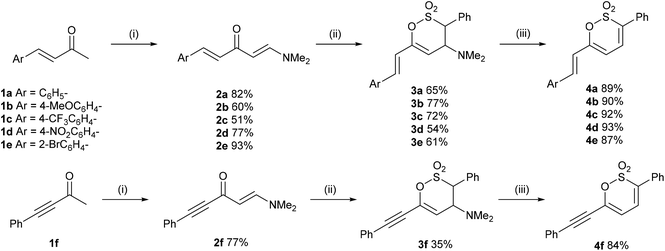
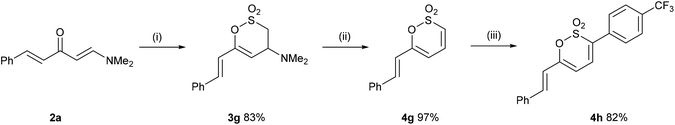
In a complementary strategy to 3-aryl-6-styryl-1,2-oxathiine 2,2-dioxides, the addition of sulfene, generated from MeSO2Cl and Et3N, to 2a to afford the 3,4-dihydro-1,2-oxathiine 2,2-dioxide 3g which was subsequently transformed into the known 1,2-oxathiine 2,2-dioxide 4g in an overall yield of 81% (Scheme 3) which is comparable to that reported by Bisetty et al.20 The signal for 3-H in the 1H NMR spectrum of 4g (d6-DMSO) resonated at δ 7.26 and exhibited typical cis-alkene coupling with J3,4 = 10 Hz; 4-H appeared as a dd at δ 7.26 and 5-H as a doublet (J4,5 = 6.7 Hz) at δ 6.31. The styryl group exhibited the predicted doublets at δ 7.09 and 7.21 with J = 16 Hz.
 |
| | Scheme 3 Reagents and conditions: (i) Et3N, MeSO2Cl, THF, 0 °C–rt; (ii) m-CPBA, DCM, 0 °C–rt; (iii) AgOAc (1.1 eq.), Pd(PPh3)4 (10 mol%), 4-I-C6H4CF3 (3 eq.), DMF, 80 °C, N2. | |
As a consequence of the similarity between the key functional group of 4g (sultone) to 2H-pyrones and coumarins (lactones), we were inspired by direct C–H functionalisation strategies,21 in particular the work of Pereira et al., for the direct C-3 arylation of coumarins.22 Thus, heating 4g in DMF containing AgOAc (1.1 eq.), Pd(PPh3)4 (10 mol%) and 4-iodobenzotrifluoride (3 eq.) overnight gave 4h in 82% yield.
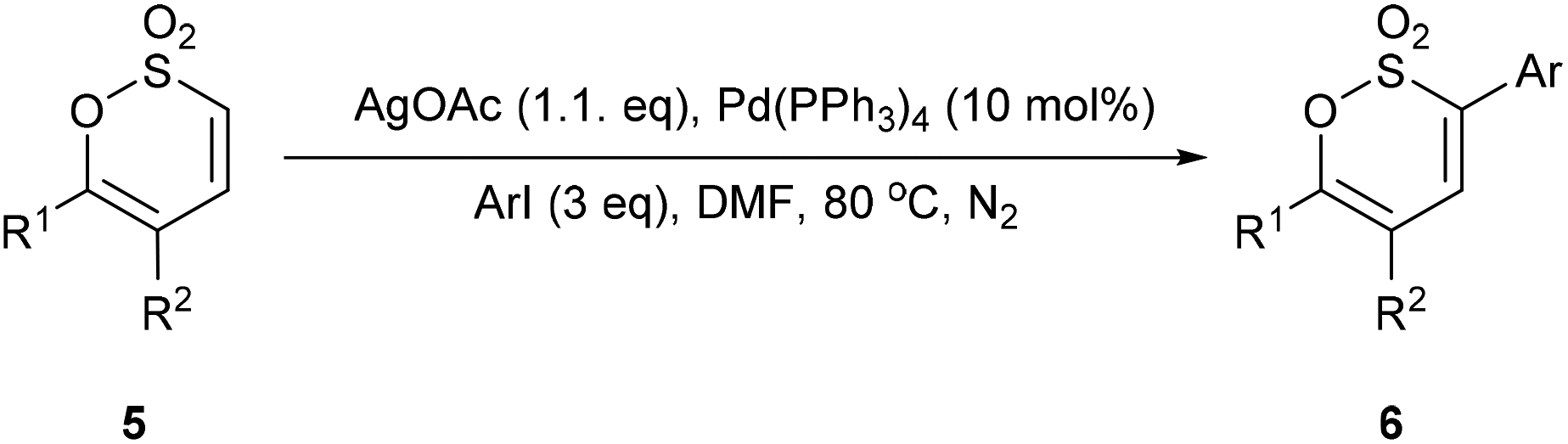
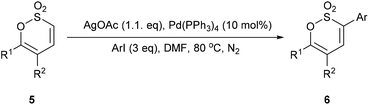
Encouraged by the success of this alternate strategy to functionalise C-3 of the 1,2-oxathiine 2,2-dioxide moiety we selected three mono- and bis-aryl substituted 1,2-oxathiine 2,2-dioxides prepared in our previous study.19 Employing the aforementioned direct C–H bond functionalisation reaction conditions, a series of 3-aryl 1,2-oxathiine 2,2-dioxides were obtained (Table 1) in good to moderate, though as yet un-optimised, yields. This ring functionalisation protocol is a useful tool for the direct introduction of, in particular electron deficient hetero(aryl) groups at C-3 which otherwise require incorporation into the starting arylmethanesulfonyl chloride, aryl sulfene precursor, of which there are relatively few available inexpensive examples.
Table 1 Direct C–H bond arylation of 5a–c
|
|
| Substrate |
R1 |
R2 |
Product |
Ar |
Yielda (%) |
|
Yields are unoptimized.
Compound 6f is slightly air sensitive and becomes red upon standing.
|
|
5a
|
Ph |
Ph |
6a
|
Ph |
39 |
|
5a
|
Ph |
Ph |
6b
|
4-MeOC6H4 |
26 |
|
5a
|
Ph |
Ph |
6c
|
4-CF3C6H4 |
57 |
|
5a
|
Ph |
Ph |
6d
|
4-Pyridyl |
68 |
|
5b
|
Ph |
H |
6e
|
Ph |
32 |
|
5c
|
4-MeOC6H4 |
4-MeOC6H4 |
6f
|
4-Pyridyl |
33b |
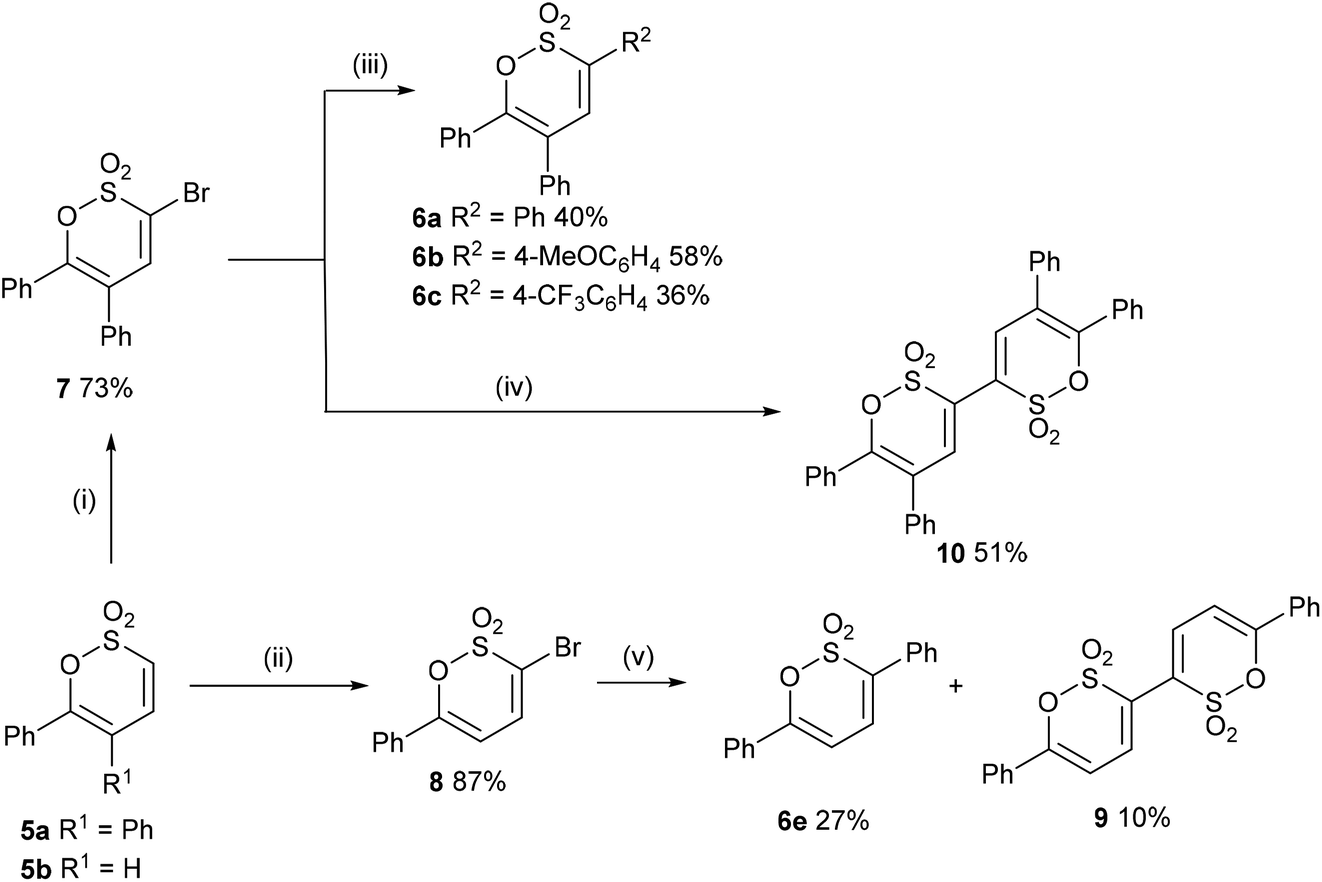
In a complementary exploration of C-3 functionalisation 5a was brominated (Br2, CHCl3, 72 h, reflux) to afford 7 in 73% yield. Guided by reaction conditions employed by Wu et al., for the 3-arylation of 3-bromopyran-2-ones23 the Suzuki cross-coupling of 7 with arylboronic acids gave 6a (40%), 6b (58%) and 6c (36%) (unoptimized yields); it is noteworthy that the highest yield for the Suzuki cross-coupling reaction was recorded with the electron rich 4-methoxyphenylboronic acid and complements the C–H bond functionalisation where the best yield was noted for electron deficient coupling partners. Bromination of 5b was marginally more effective, though did require the addition of pyridine (0.25 eq.) to assist with the elimination step, affording 8 in 87% yield. Suzuki cross-coupling of the latter afforded 6e in 27% together with 6,6′-diphenyl-[3,3′-bi(1,2-oxathiine)] 2,2,2′,2′-tetraoxide 9 (10%) as a consequence of homo-coupling (Scheme 4). In the 1H NMR spectrum of 9 5,5′-H appears as a distinct doublet at δ 6.63 with J = 7.4 Hz; the signal for 4,4′-H was coincidental with the signal for the aromatic ring protons.
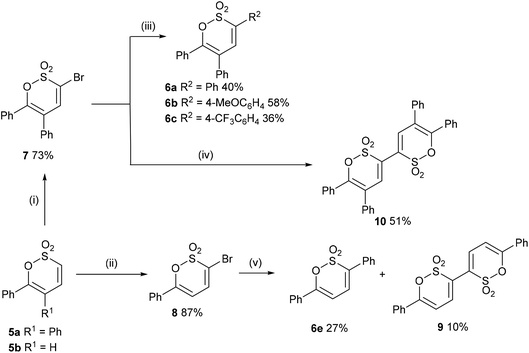 |
| | Scheme 4 Reagents and conditions: (i) Br2, CHCl3, reflux, 72 h; (ii) Br2, pyridine (0.25 eq.), CHCl3, reflux; (iii) ArB(OH)2 (1.5 eq.), Pd(OAc)2 (5 mol%), PCy3 (10 mol%) (K2HPO4·3H2O (3 eq.), DMAc/MeOH, N2; (iv) B2pin2, Pd(dppf)Cl2, KOAc, 1,4-dioxane, 80 °C, 1.5 h, N2; (v) PhB(OH)2 (1.5 eq.), Pd(OAc)2 (5 mol%), PCy3 (10 mol%), K2HPO4·3H2O (3 eq.), DMAc, N2. | |
The one step direct C–H functionalisation protocol was more effective for the preparation of 6a, 6c, and 6e whereas the bromination – Suzuki cross-coupling protocol afforded 6b in better yield albeit requiring greater synthetic effort.
Attempts to effect a Suzuki–Miyaura borylation of 7 employing B2Pin2 and Pd(dppf)Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24 in order to afford a 3-borylated adduct for exploitation in alternative Suzuki cross-coupling reactions with aryl halides were unsuccessful and whilst no borylated product could be identified the formation of the homo-coupled oxathiine 10 (δ4,4′-H 7.56) was optimised to 51%. From the present study it would appear that the 3-bromo-1,2-oxathiine 2,2-dioxide unit is susceptible to homo-coupling. Indeed, homo-coupling of aryl halides has been observed during borylation reactions and utilised to good effect for example in the total synthesis of Pusilatin A25 and biindolyls.26
24 in order to afford a 3-borylated adduct for exploitation in alternative Suzuki cross-coupling reactions with aryl halides were unsuccessful and whilst no borylated product could be identified the formation of the homo-coupled oxathiine 10 (δ4,4′-H 7.56) was optimised to 51%. From the present study it would appear that the 3-bromo-1,2-oxathiine 2,2-dioxide unit is susceptible to homo-coupling. Indeed, homo-coupling of aryl halides has been observed during borylation reactions and utilised to good effect for example in the total synthesis of Pusilatin A25 and biindolyls.26
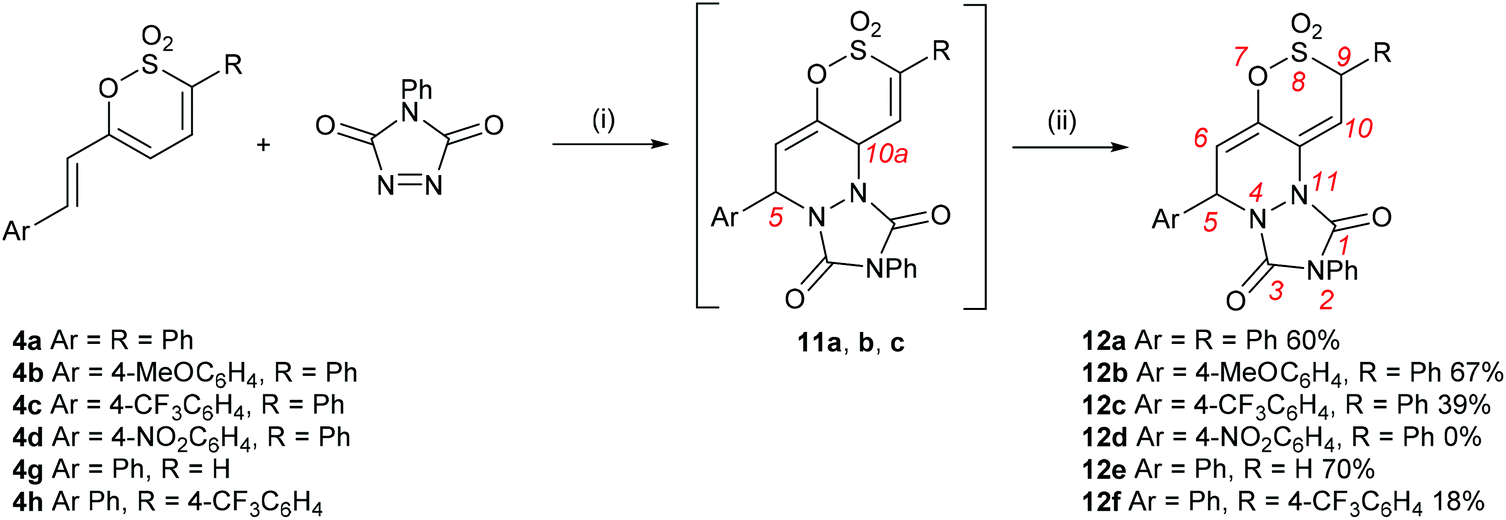
Metz et al., have previously examined the cycloaddition of acetylenic dienophiles e.g. DMAD ethyl propiolate and phenylacetylene27 to the fixed diene unit of 4,6-diaryl-1,2-oxathiine 2,2-dioxides and noted that the cycloaddition required forcing conditions e.g. for DMAD either 150 °C (100 W microwave) for 30 min or 25 °C, 1300 MPa for 3 days.28 We were interested to examine the cycloaddition of the reactive hetero dienophile, 4-phenyl-1,2,4-triazoline-3,5-dione (PTAD), to the di- and tri-phenyl substituted oxathiine 2,2-dioxides 5a and 6a, respectively and in particular the addition of PTAD to the series of 4a–d, g and h in anticipation that the latter would readily add to the diene unit comprised of the C5–C6 CC unit and the 6-styryl substituent to afford a novel tricyclic ring system. PTAD has attracted much interest over the years29,30 and there has been a resurgence in its use as a consequence of its excellent dienophile character31–34 and as a component in new electrophilic substitution ‘click’ reactions.35
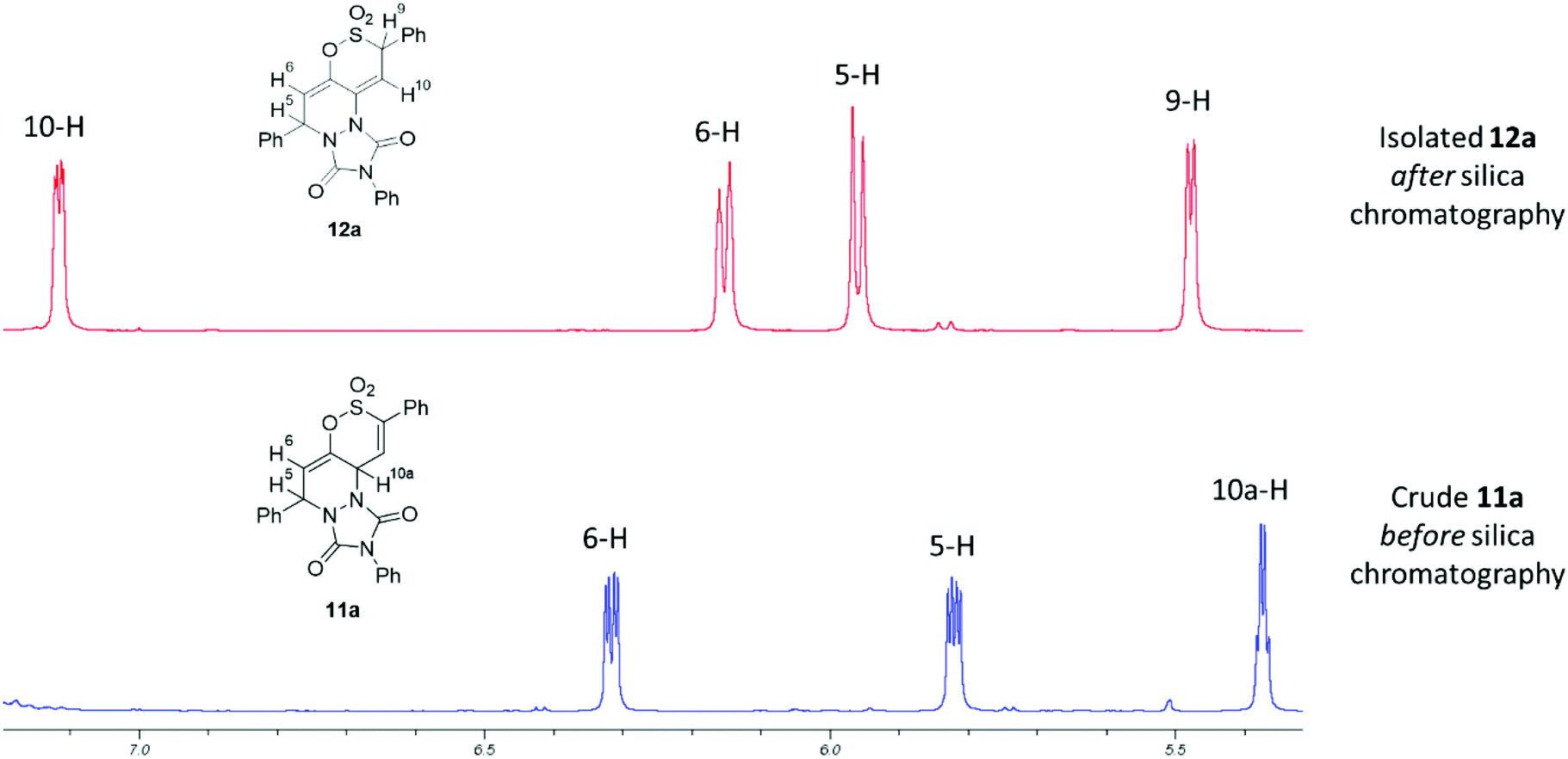
Prolonged heating of a mixture of PTAD and either 5a or 6a in 1,2-DCE failed to afford any cycloadduct, confirming the experimental observations by Metz et al.,27,28 that cycloadditions to the 1,2-oxathiine 2,2-dioxide ‘diene’ unit require forcing conditions. Encouragingly, the wine-red colour of a solution of PTAD and the 6-styryl-1,2-oxathiine 2,2-dioxide 4a in 1,2-DCE gradually discharged overnight suggestive of consumption of the PTAD. Examination of the 1H NMR spectrum of the resulting crude product indicated the formation of the expected [4 + 2] adduct, 5,10a-dihydro-1H-[1,2]oxathiino[5,6-c][1,2,4]triazolo[1,2-a]pyridazine-1,3(2H)-dione 8,8-dioxide, 11a, by the presence of three well separated signals at δ 5.37 (10a-H), δ 5.81 (5-H) and δ 6.31 (6-H) with the signal for 10-H coincidental with the aromatic signals. A pair of cross-peak contours between 5-H and 10a-H were also observed on the NOESY spectrum of 11a, pointing to a syn disposition between these two protons in agreement with the suprafacial fashion of the hetero Diels–Alder addition of PTAD. Rather unexpectedly, 11a rearranged upon a quick clean up by short column chromatography on silica to afford the isomeric 5,9-dihydro-1H-[1,2]oxathiino[5,6-c][1,2,4]triazolo[1,2-a]pyridazine-1,3(2H)-dione 8,8-dioxide 12a in 60% yield (Scheme 5). The 1H NMR spectrum of 12a presented four well-resolved signals at δ 5.48 (ddd, J = 0.8, 1.0, 3.6 Hz, 9-H), δ 5.96 (ddd, J = 0.5, 0.8, 6.1 Hz, 5-H), δ 6.14 (ddd, J = 1.0, 1.7, 6.1 Hz, 6-H) and δ 7.11 (ddd, J = 0.5, 1.7, 3.6 Hz, 10-H) (see ESI†). Although no conformational information about these protons could be inferred via NOESY, their multiplicity and coupling constants (J = 1.7 Hz for 6-H and 10-H, J = 0.8 Hz for 5-H and 9-H) attested to 6-H and 10-H being appended on the terminal positions of a diene fragment, with 5-H and 9-H on their periphery and at a distance where no long-range coupling can occur between them. The spectra of the initial isolated crude adduct 11a and rearranged isolated product 12a are presented in Fig. 1.
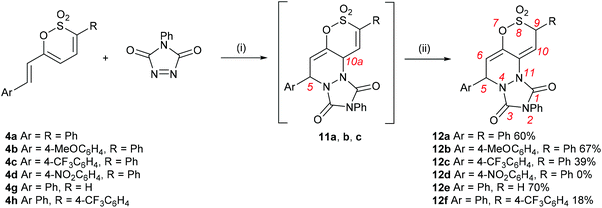 |
| | Scheme 5 Reagents and conditions: (i) 1,2-DCE, rt and or reflux; (ii) column chromatography on silica, CH2Cl2. | |
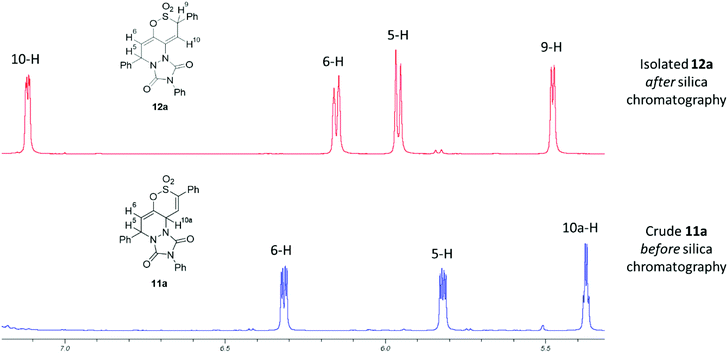 |
| | Fig. 1
1H NMR spectra (selected chemical shift range) of initial adduct (11a) and isolated product (12a) after elution from silica. | |
Additional NMR evidence for the proposed structure 12a was obtained via 2D-NMR experiments that provided more information regarding the position of 9-H at δ 5.48. In particular, the HSQC spectrum of 12a suggested that 9-H is situated on a C atom that resonates at δ 63.07. Utilising this information, the HMBC spectrum of the compound presented a long-range correlation of this carbon with the adjacent proton at δ 7.11 (10-H), as well as one with the aromatic protons at circa δ 7.56; no such correlation was observed with either of the other protons of the tricyclic core [δ 5.96 (5-H) and δ 6.14 (6-H)].
The addition of PTAD proceeded in a similar manner with 4b and 4c to afford the analogous 5,9-dihydro-adducts 12b and c, respectively. In each case a small amount of the crude adduct was purified by trituration and characterised to establish the identity of the initial [4 + 2] adduct prior to rearrangement upon column chromatography silica. Indeed, stirring a solution of 11c in EtOAc containing suspended chromatography silica readily effected the rearrangement to afford 12c in quantitative yield in a matter of minutes. The trend of decreasing yield of adduct with increasing electron withdrawing power of the styryl unit was further confirmed by the failure of 4d, bearing a 4-nitrostyryl group, to afford any adduct. Interestingly, the addition of PTAD to 4h, in which the Ph and 4-CF3C6H4 groups are inverted relative to those in 4c, proceeded to afford 12f in 18% yield, further supporting the observation that the presence of electron withdrawing substituents is detrimental to the efficiency of the addition process.
The addition of excess PTAD to 4g, in which the 1,2-oxathiine ring is unsubstituted at C-3, only gave the mono-adduct 12e in 70% after stirring in 1,2-DCE overnight followed by routine treatment with chromatography silica. The 1H NMR spectrum of 12e further supports the silica-mediated double bond migration and affords a multiplet at δ 4.24 accounting for the diastereotopic 9-H pair; 5-H and 6-H appear at δ 5.91 and δ 6.08, respectively and 10-H at δ 6.96.
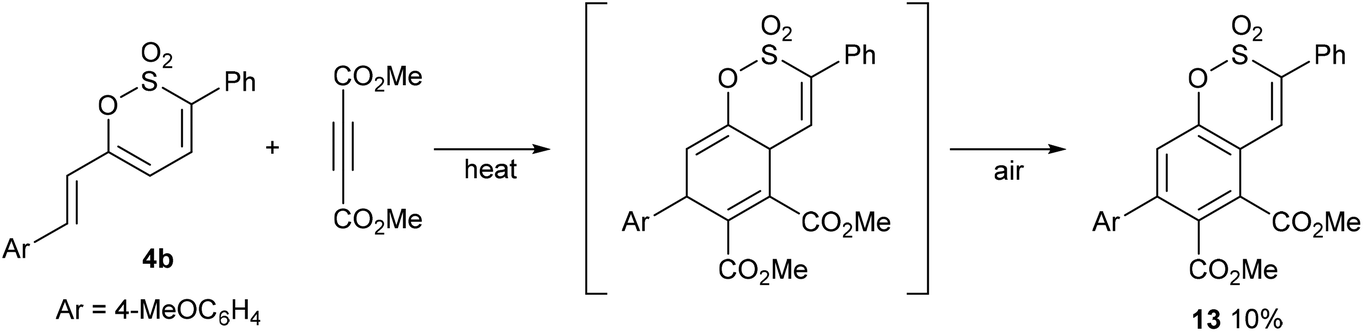
Continuing our initial survey of cycloadditions to the 6-styryl substituted 1,2-oxathiines, a mixture of 4b and DMAD was heated overnight. Elution of the resulting dark, multi-component reaction mixture from silica gave the aromatised adduct 13 in 10% yield as the only identifiable product; providing an alternative, albeit low yielding, de novo route to a diversely substituted benzoxathiine 2,2-dioxide (Scheme 6).
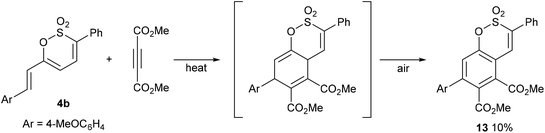 |
| | Scheme 6 | |
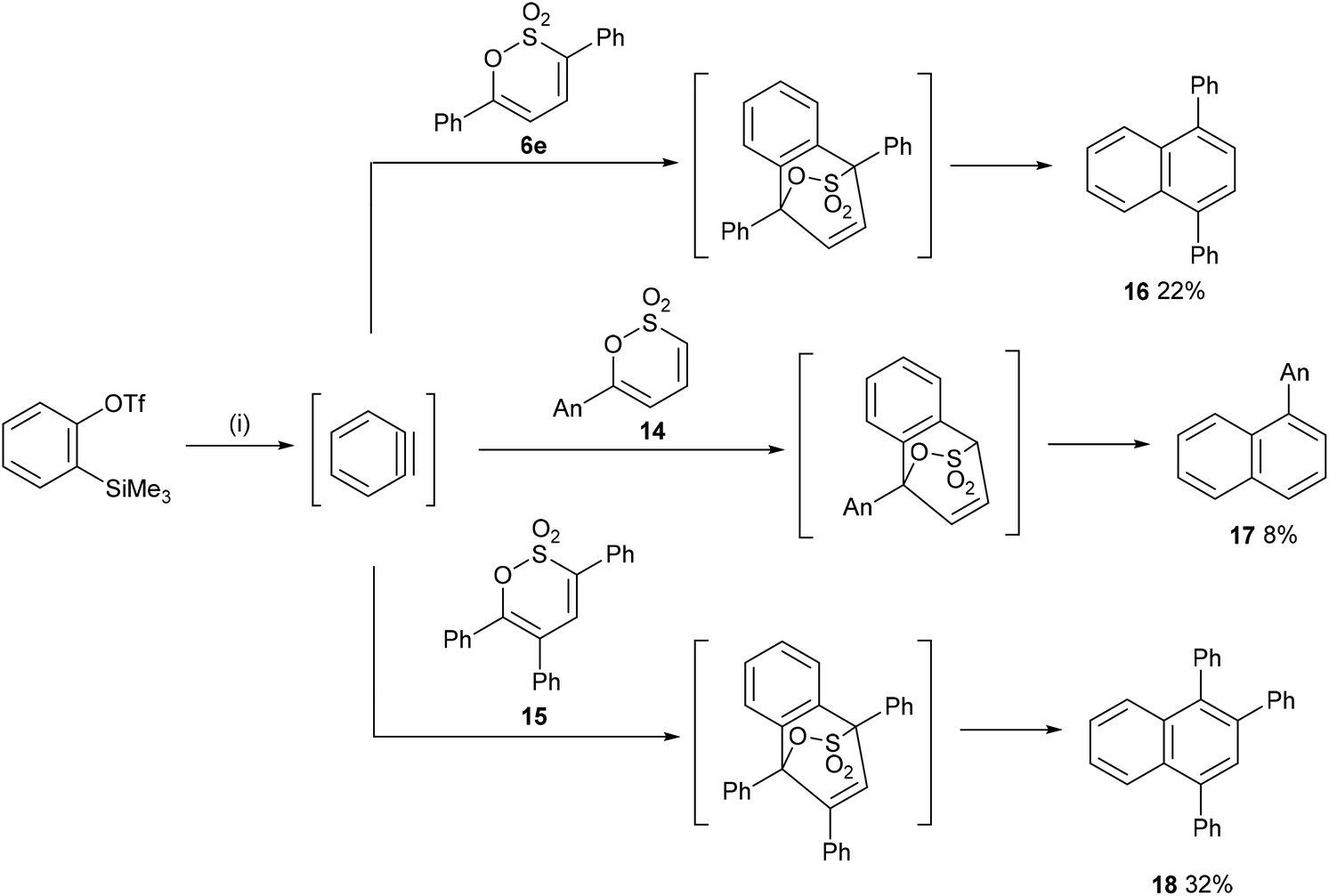
In a final exploration of the reactivity of the substituted 1,2-oxathiine 2,2-dioxides, benzyne, generated in situ from the action of excess CsF on 2-(trimethylsilyl)phenyl trifluoromethanesulfonate,36 was added to 6e, 14 and 15 to afford 1,4-diphenylnaphthalene 16 (22%), 1-(4-methoxyphenyl)naphthalene 17 (8%) and 1,2,4-triphenylnaphthalene 18 (32%), respectively (Scheme 7). Unfortunately, addition of benzyne to 6-styryl-1,2-oxathiines 4a,b gave a complex reaction product from which no pure compound could be isolated.
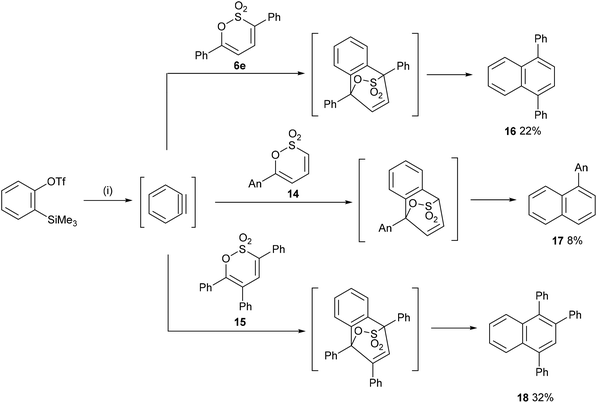 |
| | Scheme 7 Reagents and conditions: (i) CsF, MeCN, rt. | |
In summary, a small library of 3-aryl-1,2-oxathiine 2,2-dioxides with 6-arenyl and 6-arynyl substituents was constructed starting from α-methyl ketones via our established 3-step protocol, highlighting the suitability of these steps in furnishing 1,2-oxathiine 2,2-dioxides with unsaturated moieties on the 6-position. Attempts at coupling selected aryl substituted 1,2-oxathiine 2,2-dioxides with aryl iodides in a C–H activation coupling reaction allowed the functionalisation of the 3-position; the protocol also allowed for the introduction of a pyridyl group, thus avoiding the use of an expensive sulfonyl halide precursor in a potentially challenging sulfene addition. Bromination of selected aryl substituted 1,2-oxathiine 2,2-dioxides proceeded with moderate efficiency and the brominated analogues were coupled to aryl boronic acids in a Suzuki cross-coupling method to assess the efficiency of this method in functionalising the 1,2-oxathiine ring scaffold. Interestingly, the two Pd-catalytic routes presented complimentary reactivity profiles, with C–H activation coupling appearing to be more suitable for appending electron deficient aryl groups and the Suzuki cross-coupling more compatible for incorporating electron rich aryl groups. An additional aspect made apparent during these attempts was the unexpected ability of 1,2-oxathiine 2,2-dioxides to undergo homo-coupling to give bis-heterocyclic analogues. Utilising the styryl analogues in cycloadditions proved to be of special interest in the case of PTAD, as the cycloadducts resulting from the addition of PTAD readily rearrange in the presence of silica towards dienic regioisomers, which presents an unusual case of structure alteration during purification. Finally, a preliminary exploration of benzyne addition towards naphthalene derivatives met with limited success, with further work required to optimise the reaction conditions and ascertain potential substitution effects.
Experimental
Equipment and materials
Unless otherwise stated, reagents and solvents were purchased from major chemical catalogue companies and were used as supplied. Routine 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded on a Bruker Avance DPX400 in CDCl3. Occasional 1H NMR spectra were also recorded on a Bruker Avance 300 MHz instrument in CDCl3. Chemical shifts are provided in parts per million (ppm) using either the residual solvent peak or TMS as the internal reference. Coupling constants (J) are provided in Hz and where applicable, in order to resolve close signals and extract valuable coupling information, the raw FID data was processed using a Gaussian multiplication in place of the more usual exponential multiplication. All FT-IR spectra were recorded on a Nicolet 380 FTIR spectrophotometer equipped with a diamond ATR attachment (neat sample). Flash column chromatography was performed on chromatography silica gel (Fluorochem, 40-63 micron particle size distribution). All final compounds were homogeneous by TLC using a range of eluent systems of differing polarity (Merck TLC aluminium sheets silica gel 60 F254 (cat. no. 105554)). High resolution mass spectra were recorded on an Agilent 6210 1200 SL TOF spectrometer within the IPOS centre at the University of Huddersfield.
The following styrylketones and enaminoketones were obtained by standard procedures and possessed spectroscopic and physical data in agreement with literature data: (E)-4-(4-methoxyphenyl)but-3-en-2-one,37 (E)-4-(4-nitrophenyl)but-3-en-2-one,38 (E)-4-(4-(4-trifluoromethyl)phenyl)but-3-en-2-one,39 (E)-4-(2-bromophenyl)but-3-en-2-one40 and (1E,4E)-1-(dimethylamino)-5-phenylpenta-1,4-dien-3-one.41
Preparation of enaminoketones
(1E,4E)-1-(Dimethylamino)-5-phenylpenta-1,4-dien-3-one 2a.
4-Phenyl-3-buten-2-one (5.00 g, 34.2 mmol) was dissolved in DMFDMA (11.4 mL, 85.5 mmol, 2.5 eq.) under a N2 atmosphere and heated to 80 °C, before the addition of L-proline (0.39 g, 3.42 mmol, 10% mol). The reaction mixture was stirred at 80 °C overnight and then cooled. The volatile components were removed by rotary evaporation and the crude solid was triturated with Et2O to afford the title compound as a pale yellow-brown solid (5.65 g, 82%); mp 90–95 °C (lit. mp 94–96 °C (ref. 41)); νmax (neat): 3020, 2916, 1639, 1616 (CO), 1577, 1549, 1496, 1484, 1432, 1365, 1345, 1275, 1219, 1200, 1158, 1116, 1030 cm−1; δH (400 MHz, CDCl3) 2.88 (s, 3H, N(CH3)2), 3.12 (s, 3H, N(CH3)2), 5.27 (d, J = 12.5 Hz, 1H, OCCHCHNMe2), 6.79 (d, J = 15.8 Hz, 1H, PhCHCHCO), 7.29–7.37 (m, 3H, Ar–H), 7.53–7.55 (m, 2H, Ar–H), 7.55 (d, J = 15.7 Hz, 1H, PhCHCHCO), 7.75 (d, J = 12.5 Hz, 1H, OCCHCHNMe2); δC (100 MHz, CDCl3) 37.27, 44.99, 96.39, 127.88, 128.31, 128.71, 129.23, 135.82, 138.49, 153.46, 186.32.
(1E,4E)-1-(Dimethylamino)-5-(4-methoxyphenyl)penta-1,4-dien-3-one 2b.
(E)-4-(4-Methoxyphenyl)but-3-en-2-one (6.93 g, 39.3 mmol) was dissolved in PhMe (50 mL) and DMFDMA (15.6 mL, 118.0 mmol, 3 eq.) and L-proline (0.45 g, 10% mol) under a N2 atmosphere. The mixture was refluxed for 5 d and then cooled to room temperature whereupon the volatile components were removed by rotary evaporation. The resulting solid was triturated with Et2O to afford the product as a yellow solid (5.46 g, 60%); mp = 100–102 °C (lit. mp 99–101 °C (ref. 42)); νmax (neat): 2996, 2909, 2837, 1653, 1610, 1597, 1573, 1531, 1509, 1434, 1417, 1405, 1355, 1300, 1269, 1247, 1170, 1144, 1084, 1026 cm−1; δH (400 MHz, CDCl3) 2.89 (br s, 3H, NCH3), 3.08 (br s, 3H, NCH3), 3.82 (s, 3H, CH3O), 5.25 (d, J = 12.5 Hz, 1H, 2-H), 6.67 (d, J = 15.9 Hz, 1H, 4-H), 6.87–6.89 (m, 2H, Ar–H), 7.48–7.50 (m, 2H, Ar–H), 7.53 (d, J = 15.9 Hz, 1H, 5-H), 7.73 (d, J = 12.5 Hz, 1H, 1-H); δC (100 MHz, CDCl3) 37.43, 44.90, 55.33, 96.40, 114.17, 126.16, 128.55, 129.40, 138.26, 153.14, 160.61, 186.54; found [M + H]+ = 232.1334, C14H17NO2 requires [M + H]+ = 232.1335.
(1E,4E)-1-(Dimethylamino)-5-(4-(trifluoromethyl)phenyl)penta-1,4-dien-3-one 2c.
(E)-4-(4-(Trifluoromethyl)phenyl)but-3-en-2-one was dissolved in DMFDMA (7.75 mL, 58.3 mmol, 2.5 eq.) and PhMe (15 mL) under a N2 atmosphere. The mixture was heated to reflux overnight and then cooled to room temperature whereupon the volatile components were removed by rotary evaporation. The obtained crude solid mass was crushed into a fine powder and triturated with 1:1 Et2O/P.E. to yield the product as a yellow solid (3.17 g, 51%); mp = 131–133 °C (lit. mp 133–134 °C (ref. 42)); νmax (neat): 3062, 2916, 2803, 1653, 1613 (CO), 1537, 1422, 1360, 1318, 1264, 1161, 1111, 1089, 1063, 1009 cm−1; δH (400 MHz, CDCl3) 2.91 (s, 3H, NCH3), 3.16 (s, 3H, NCH3), 5.28 (d, J = 12.4 Hz, 1H, 2-H), 6.85 (d, J = 15.7 Hz, 1H, 4-H), 7.58 (d, J = 15.7 Hz, 1H, 5-H), 7.59–7.66 (m, 4H, Ar–H), 7.79 (d, J = 12.4 Hz, 1H, 1-H); δC (100 MHz, CDCl3) 37.29, 45.13, 96.42, 124.05 (q, J = 272 Hz), 125.65 (q, J = 3.8 Hz), 127.93, 130.63, 130.65 (q, J = 32.3 Hz), 136.61, 139.35, 153.86, 185.55; δF (376.5 MHz, CDCl3) −62.66; found [M + H]+ = 270.1100, C14H14F3NO requires [M + H]+ = 270.1103.
(1E,4E)-1-(Dimethylamino)-5-(4-nitrophenyl)penta-1,4-dien-3-one 2d.
(E)-4-(4-Nitrophenyl)but-3-en-2-one (1.00 g, 5.2 mmol) was dissolved in PhMe (25 mL) and DMFDMA (1.73 mL, 13.0 mmol, 2.5 eq.) under a N2 atmosphere. The reaction mixture was refluxed for 6h and then cooled to room temperature whereupon the volatile components were removed by rotary evaporation. The crude product was filtered through a silica column (neat CHCl3 to 5% MeOH/CHCl3) to afforded the title compound as an orange solid after trituration with Et2O (0.99 g, 77%); mp = 152–154 °C; νmax (neat): 3035, 2912, 1650, 1614 (CO), 1593, 1537, 1505, 1436, 1424, 1355, 1335, 1302, 1265, 1089 cm−1; δH (400 MHz, CDCl3) 2.91 (s, 3H, NCH3), 3.17 (s, 3H, NCH3), 5.28 (d, J = 12.2 Hz, 1H, 2-H), 6.89 (d, J = 15.9 Hz, 1H, 4-H), 7.58 (d, J = 15.9 Hz, 1H, 5-H), 7.65–7.68 (m, 2H, Ar–H), 7.80 (d, J = 12.2 Hz, 1H, 1-H), 8.20–8.22 (m, 2H, Ar–H); δC (100 MHz, CDCl3) 37.35, 45.18, 96.54, 124.05, 128.31, 132.38, 135.54, 142.37, 147.79, 154.12, 184.94; found [M + H]+ = 247.1082, C13H14N2O3 requires [M + H]+ = 247.1080.
(1E,4E)-1-(2-Bromophenyl)-5-(dimethylamino)penta-1,4-dien-3-one 2e.
(E)-4-(2-Bromophenyl)but-3-en-2-one was dissolved in DMFDMA (7.40 mL, 55.5 mmol, 2.5 eq.) under a N2 atmosphere and the reaction mixture was heated to reflux overnight and then cooled to room temperature. The volatile components were removed by rotary evaporation to afford the product as a pale brown oil (5.77 g, 93%); νmax (neat): 2908, 2803, 1654, 1610 (CO), 1541, 1463, 1416, 1351, 1258, 1217, 1199, 1082, 1020 cm−1; δH (400 MHz, CDCl3) 2.90 (s, 3H, NCH3), 3.14 (s, 3H, NCH3), 5.31 (d, J = 12.5 Hz, 1H, 2-H), 6.71 (d, J = 15.8 Hz, 1H, 4-H), 7.14–7.19 (m, 1H, Ar–H), 7.27–7.30 (m, 1H, Ar–H), 7.57–7.63 (m, 2H, Ar–H), 7.75 (d, J = 12.5 Hz, 1H, 1-H), 7.86 (d, J = 15.8 Hz, 1H, 5-H); δC (100 MHz, CDCl3) 37.27, 45.08, 95.90, 125.26, 127.51, 127.62, 130.16, 131.42, 133.27, 135.95, 136.83, 153.71, 186.03; found [M + H]+ = 280.0328, C13H1479BrNO requires [M + H]+ = 280.0331.
(E)-1-(Dimethylamino)-5-phenylpent-1-en-4-yn-3-one 2f.
4-Phenylbut-3-yn-2-one (3.03 mL, 20.8 mmol, 1.0 eq.) was dissolved in PhMe (20 mL) and DMFDMA (4.15 mL, 31.2 mmol, 1.5 eq.) under a N2 atmosphere. The resulting mixture was heated at reflux overnight. Upon cooling the volatile components were removed under reduced pressure to afford a viscous crude oil which upon trituration with Et2O afforded the title compound as a pale brown powder (3.18 g, 77%); mp = 80–82 °C, (lit. mp = 86–87 °C (ref. 43)); νmax (neat): 1621 (CO), 1558, 1488, 1441, 1405, 1343, 1308, 1267, 1207, 1193, 1178, 1115, 1025 cm−1; δH (400 MHz, CDCl3) 2.89 (s, 3H, NCH3), 3.17 (s, 3H, NCH3), 5.33 (d, J = 12.6 Hz, 1H, COCH), 7.33–7.41 (m, 3H, Ar–H), 7.54–7.57 (m, 2H, Ar–H), 7.74 (d, J = 12.6 Hz, 1H, CHNMe); δC (100 MHz, CDCl3) 37.36, 45.36, 86.53, 87.79, 102.17, 121.38, 128.45, 129.55, 132.40, 158.22, 174.78; found [M + H]+ = 200.1068, C13H13NO requires [M + H]+ = 200.1073.
Preparation of 3,4-dihydro-1,2-oxathiine 2,2-dioxides
(E)-4-(Dimethylamino)-3-phenyl-6-styryl-3,4-dihydro-1,2-oxathiine 2,2-oxide 3a.
A solution of phenylmethanesulfonyl chloride (8.27 g, 43.4 mmol, 3.5 eq.) in anhydrous THF (35 mL) was added dropwise to a cold (−10 °C) stirred solution of (1E,4E)-1-(dimethylamino)-5-phenylpenta-1,4-dien-3-one (2.5 g, 12.4 mmol) and Et3N (6.05 mL, 43.4 mmol, 3.5 eq.) in anhydrous THF (35 mL) under nitrogen, over 10 min, with the rate of addition such that the temperature was maintained at or below 0 °C. Upon completion of the addition the reaction mixture was warmed to room temperature overnight, before being filtered to remove the precipitated Et3N·HCl. Removal of the solvent gave the crude product which was purified by column chromatography (alumina, 20% to 30% EtOAc/hexane) to yield the title product as a pale yellow solid (2.87 g, 65%); mp = 124–126 °C (from EtOAc/hexane); νmax (neat): 2830, 2793, 1651, 1496, 1468, 1454, 1368 (O-SO2), 1315, 1268, 1228, 1191, 1170 (O-SO2), 1073, 1039 cm−1; δH (400 MHz, CDCl3) 2.25 (s, 6H, N(CH3)2), 4.38 (dd, J = 2.4, 11.2 Hz, 1H, 4-H), 4.53 (d, J = 11.2 Hz, 1H, 3-H), 5.50 (d, J = 2.6 Hz, 1H, 5-H), 6.55 (d, J = 15.9 Hz, 1H, PhCHCH), 7.04 (d, J = 15.9 Hz, 1H, PhCHCH), 7.32–7.39 (m, 3H, Ar–H), 7.44–7.46 (m, 5H, Ar–H), 7.52–7.55 (m, 2H, Ar–H); δC (100 MHz, CDCl3) 41.24, 63.35, 64.58, 108.09, 119.62, 127.06, 128.77, 128.83, 129.23, 129.26, 129.75, 129.76, 131.72, 135.59, 149.60; found [M + H]+ = 356.1323, C20H21NO3S requires [M + H]+ = 356.1315.
(E)-4-(Dimethylamino)-6-(4-methoxystyryl)-3-phenyl-3,4-dihydro-1,2-oxathiine 2,2-dioxide 3b.
A solution of phenylmethanesulfonyl chloride (10.30 g, 54.0 mmol, 2.5 eq.) in anhydrous THF (60 mL) and DCM (10 mL) was subsequently added dropwise over 20 min to a cold (−5 °C) stirred solution of (1E,4E)-1-(dimethylamino)-5-(4-methoxyphenyl)penta-1,4-dien-3-one (5.00 g, 21.6 mmol) and Et3N (7.53 mL, 54.0 mmol, 2.5 eq.) in anhydrous THF (60 mL) and DCM (15 mL) under nitrogen. Upon completion of the addition, the reaction mixture was stirred for 2 h at 0 °C, before warming to room temperature overnight and then filtered to remove the precipitated Et3N·HCl. Removal of the solvent gave the crude product which was purified by column chromatography (alumina, 20% to 50% EtOAc/hexane) to afford the title compound as a yellow solid (6.39 g, 77%); mp = 125–127 °C (from EtOAc/hexane); νmax (neat): 2976, 2938, 2831, 2781, 1601, 1510, 1495, 1453, 1372 (O-SO2), 1319, 1251, 1226, 1169 (O-SO2), 1109, 1015 cm−1; δH (400 MHz, CDCl3) 2.25 (s, 6H, N(CH3)2), 3.83 (s, 3H, OCH3), 4.39 (dd, J = 2.2, 11.1 Hz, 1H, 4-H), 4.52 (d, J = 11.1 Hz, 1H, 3-H), 5.43 (d, J = 2.5 Hz, 1H, 5-H), 6.43 (d, J = 15.9 Hz, 1H, HCCHC–O), 6.88–6.91 (m, 2H, Ar–H), 6.99 (d, J = 15.9 Hz, 1H, AnCHCH), 7.38–7.41 (m, 2H, Ar–H), 7.44–7.45 (m, 3H, Ar–H), 7.53–7.55 (m, 2H, Ar–H); δC (100 MHz, CDCl3) 41.18 (2 × C), 55.35, 63.32, 64.61, 106.79, 114.29, 117.42, 128.35, 128.44, 129.21, 129.36, 129.72, 129.74, 131.32, 149.89, 160.18; found [M + H]+ = 386.1417, C21H23NO4S requires [M + H]+ = 386.1424.
(E)-4-(Dimethylamino)-3-phenyl-6-(4-(trifluoromethyl)styryl)-3,4-dihydro-1,2-oxathiine 2,2-dioxide 3c.
A solution of phenylmethanesulfonyl chloride (3.80 g, 20.0 mmol, 1.8 eq.) in anhydrous THF (50 mL) was added dropwise over 10 min to a cold (−10 °C) stirred solution of (1E,4E)-1-(dimethylamino)-5-(4-(trifluoromethyl)phenyl)penta-1,4-dien-3-one (3.00 g, 11.1 mmol) and Et3N (2.77 mL, 20.0 mmol, 1.8 eq.) in anhydrous THF (50 mL) under nitrogen. The resulting mixture was stirred for 2 h at −10 °C and was warmed to room temperature overnight and then filtered to remove the precipitated Et3N·HCl. Removal of the solvent gave the crude product which was purified by column chromatography (basic alumina, 30% to 50% EtOAc/hexane) to afford the title compound as an off-white powder after trituration with Et2O (3.40 g, 72%); mp = 128–129 °C (from EtOAc/hexane); νmax (neat): 2980, 2948, 2359, 2341, 1652, 1613, 1496, 1455, 1414, 1376 (O-SO2), 1321, 1267, 1221, 1188, 1163 (O-SO2), 1105, 1063, 1044, 1013 cm−1; δH (400 MHz, CDCl3) 2.26 (s, 6H, N(CH3)2), 4.39 (dd, J = 2.7, 11.1 Hz, 1H, 4-H), 4.54 (d, J = 11.1 Hz, 1H, 3-H), 5.58 (d, J = 2.7 Hz, 1H, 5-H), 6.63 (d, J = 15.9 Hz, 1H, F3CC6H4CH), 7.05 (d, J = 15.8 Hz, 1H, CHCHC–O), 7.45–7.46 (m, 3H, Ar–H), 7.53–7.55 (m, 4H, Ar–H), 7.61–7.63 (m, 2H, Ar–H); δC (100 MHz, CDCl3) 41.22, 63.35, 64.63, 109.77, 122.02, 124.04 (q, J = 270 Hz), 125.79 (q, J = 3.7 Hz), 127.17, 129.07, 129.27, 129.73, 129.85, 130.13, 130.34 (q, J = 32 Hz), 139.04, 149.16; δF (376.5 MHz, CDCl3) −62.64; found [M + H]+ = 424.1191, C21H20F3NO3S requires [M + H]+ = 424.1192.
(E)-4-(Dimethylamino)-6-(4-nitrostyryl)-3-phenyl-3,4-dihydro-1,2-oxathiine 2,2-dioxide 3d.
A solution of phenylmethanesulfonyl chloride (1.27 g, 6.7 mmol, 1.8 eq.) in anhydrous THF (15 mL) was added dropwise over 10 min to a cold (−10 °C) stirred solution of (1E,4E)-1-(dimethylamino)-5-(4-nitrophenyl)penta-1,4-dien-3-one (0.92 g, 3.7 mmol) and Et3N (0.90 mL, 6.7 mmol, 1.8 eq.) in anhydrous THF (15 mL) and DCM (20 mL) under nitrogen. The resulting mixture was stirred at −5 °C for 2 h and was warmed to room temperature over 2 h. Removal of the solvent afforded a sticky solid mass which was purified by column chromatography (basic alumina, 20% EtOAc/hexane to neat EtOAc) to afford the title product as a pale yellow solid after trituration with Et2O (0.80 g, 54%); mp = 156–158 °C (from EtOAc/hexane); νmax (neat): 2787, 2359, 2341, 1619, 1590, 1506, 1498, 1456, 1377 (O-SO2), 1335, 1267, 1226, 1189, 1139 (O-SO2), 1107, 1072, 1053, 1038 cm−1; δH (400 MHz, CDCl3) 2.26 (s, 6H, N(CH3)2), 4.40 (dd, J = 2.5, 11.3 Hz, 1H, 4-H), 4.55 (d, J = 11.3 Hz, 1H, 3-H), 5.64 (d, J = 2.5 Hz, 1H, 5-H), 6.70 (d, J = 15.8 Hz, 1H, O2NC6H4CH), 7.07 (d, J = 15.8 Hz, 1H, CHCHC–O), 7.45–7.54 (m, 5H, Ar–H), 7.58–7.60 (m, 2H, Ar–H), 8.22–8.24 (m, 2H, Ar–H); δC (100 MHz, CDCl3) 41.25, 63.30, 64.69, 123.78, 124.22, 124.30, 127.56, 128.90, 129.02, 129.24, 129.32, 129.70, 129.95, 141.92, 147.55; found [M + H]+ = 401.1168, C20H20N2O5S requires [M + H]+ = 401.1169.
(E)-6-(2-Bromostyryl)-4-(dimethylamino)-3-phenyl-3,4-dihydro-1,2-oxathiine 2,2-dioxide 3e.
A solution of phenylmethanesulfonyl chloride (7.45 g, 39.2 mmol, 2.2 eq.) in anhydrous THF (60 mL) was added dropwise over 15 min to a cold (−5 °C) stirred solution of (1E,4E)-1-(2-bromophenyl)-5-(dimethylamino)penta-1,4-dien-3-one (5.00 g, 17.8 mmol) and Et3N (5.46 mL, 39.2 mmol, 2.2 eq.) in anhydrous THF (60 mL) under nitrogen. Upon completion of the addition the mixture was stirred for 2 h at −5 °C and then warmed to room temperature overnight and then filtered to remove the precipitated Et3N·HCl. Removal of the solvent gave the crude product which was purified by column chromatography (basic alumina, 30% EtOAc/hexane) to afford the title product as a pale-yellow powder after washing with Et2O (4.68 g, 61%); mp = 128–130 °C (from EtOAc/hexane); νmax (neat): 2978, 2936, 2863, 2837, 2794, 2359, 2343, 1657, 1496, 1458, 1437, 1364 (O-SO2), 1337, 1310, 1280, 1259, 1207, 1186 (O-SO2), 1171, 1149, 1092, 1076, 1046, 1022 cm−1; δH (400 MHz, CDCl3) 2.26 (s, 6H, N(CH3)2), 4.37 (dd, J = 2.4, 11.4 Hz, 1H, 4-H), 4.55 (d, J = 11.4 Hz, 1H, 3-H), 5.55 (d, J = 2.4 Hz, 1H, 5-H), 6.50 (d, J = 15.4 Hz, 1H, BrC6H4CH), 7.14–7.18 (m, 1H, Ar–H), 7.28–7.32 (m, 1H, Ar–H), 7.34 (d, J = 15.4 Hz, 1H, CHCHC–O), 7.45–7.46 (m, 3H, Ar–H), 7.53–7.55 (m, 3H, Ar–H), 7.55–7.61 (m, 1H, Ar–H); δC (100 MHz, CDCl3) 41.23, 63.33, 64.58, 109.35, 122.49, 124.63, 126.95, 127.55, 129.21, 129.24, 129.75, 129.78, 129.80, 130.35, 133.37, 135.68, 149.41; found [M + H]+ = 434.0421, C20H2079BrNO3S requires [M + H]+ = 434.0423.
4-(Dimethylamino)-3-phenyl-6-(phenylethynyl)-3,4-dihydro-1,2-oxathiine 2,2-dioxide 3f.
A solution of phenylmethanesulfonyl chloride (5.73 g, 30.0 mmol, 3.0 eq.) in anhydrous THF (30 mL) was added dropwise to a cold (−10 °C) stirred solution of (E)-1-(dimethylamino)-5-phenylpent-1-en-4-yn-3-one (2.00 g, 10.0 mmol) and Et3N (4.20 mL, 30.0 mmol, 3.0 eq.) in anhydrous THF (30 mL). Upon completion of the addition the reaction mixture was stirred at −10 °C for 2 h and warmed room temperature overnight and then filtered to remove the precipitated Et3N·HCl. Removal of the solvent gave the crude product which was purified by column chromatography (alumina, 20% EtOAc/petroleum spirit (40–60 °C)) to afford the title product as an off-white solid after trituration with Et2O (1.23 g, 35%); mp = 128–130 °C (from EtOAc/petroleum spirit (40–60 °C)); νmax (neat): 2779, 2214, 1647, 1488, 1455, 1442, 1366 (O-SO2), 1316, 1299, 1280, 1258, 1228, 1182 (O-SO2), 1168, 1108, 1071, 1050, 1032 cm−1; δH (400 MHz, CDCl3) 2.27 (s, 6H, N(CH3)2), 4.35 (dd, J = 2.7, 11.3 Hz, 1H, 4-H), 4.50 (d, J = 11.3 Hz, 1H, 3-H), 5.90 (d, J = 2.7 Hz, 1H, 5-H), 7.35–7.42 (m, 3H, Ar–H), 7.44–7.45 (m, 3H, Ar–H), 7.51–7.53 (m, 4H, Ar–H); δC (100 MHz, CDCl3) 41.28, 63.53, 64.85, 80.78, 91.69, 114.60, 120.93, 128.54, 128.87, 129.24, 129.68, 129.71, 129.84, 131.92, 135.18; found [M + H]+ = 354.1157, C20H19NO3S requires [M + H]+ = 354.1158.
(E)-4-(Dimethylamino)-6-styryl-3,4-dihydro-1,2-oxathiine 2,2-dioxide 3g.
A solution of methanesulfonyl chloride (4.7 mL, 60.8 mmol) in anhydrous THF (35 mL) was added dropwise to a cold (−10 °C) stirred solution of (1E,4E)-1-(dimethylamino)-5-phenylpenta-1,4-dien-3-one (8.16 g, 40.5 mmol) and Et3N (8.40 mL, 60.8 mmol) in anhydrous THF (100 mL) under nitrogen, over 15 min, with the rate of addition such that the temperature was maintained at or below 0 °C. Upon completion of the addition the reaction mixture was warmed to room temperature and stirred overnight, before being filtered to remove the precipitated Et3N·HCl. Removal of the solvent gave the crude product which was purified by column chromatography using 80% EtOAc in hexane produced a brown solid (9.40 g, 83%); mp = 132–134 °C; νmax (neat) 1364, 1347, 1167, 749, 686, 632, 571, 529; δH (300 MHz, CDCl3) 2.35 (s, 6H, NMe2), 3.26 (dd, J = 11.4, 13.3 Hz, 1H, 3-H), 3.50 (ddd, J = 1.0, 7.3, 13.3 Hz, 1H, 3-H), 4.13 (dddd, J = 2.6, 7.16, 8.9, 10.3 Hz, 1H, 2-H), 5.36 (d, J = 2.6 Hz, 1H, 4-H), 6.49 (d, J = 15.9 Hz, 1H, PhCH), 7.00 (d, J = 15.9 Hz, 1H, CHCHC–O), 7.30–7.45 (5H, m, Ar–H); δC (100 MHz, CDCl3) 40.64, 43.07, 59.47, 108.59, 119.69, 127.04, 128.80, 131.55, 135.53, 149.62; found [M + H]+ = 279.0923, C14H17NO3S requires [M + H]+ = 279.0928.
Preparation of 1,2-oxathiine 2,2-doxides
(E)-3-Phenyl-6-styryl-1,2-oxathiine 2,2-dioxide 4a.
A suspension of m-CPBA (0.69 g, 2.76 mmol, 1.4 eq.) in DCM (15 mL) was added dropwise to a cold (0–5 °C) stirred solution of (E)-4-(dimethylamino)-3-phenyl-6-styryl-3,4-dihydro-1,2-oxathiine 2,2-oxide (0.70 g, 1.97 mmol) in DCM (25 mL). Upon completion of the addition the reaction mixture was warmed to room temperature over 2 h and then washed with H2O (20 mL), aq. Na2SO3 (0.7 M, 2 × 15 mL), aq. NaOH(aq) (1 M, 15 mL) and H2O (15 mL). The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure to afford the target compound as a bright yellow solid (0.54 g, 89%); mp = 155–158 °C; νmax (neat): 3025, 1627, 1602, 1540, 1494, 1448, 1356 (O-SO2), 1291, 1263, 1199, 1169 (O-SO2), 1069, 1032 cm−1; δH (400 MHz, CDCl3) 6.06 (d, J = 7.1 Hz, 1H, 5-H), 6.67 (d, J = 16.0 Hz, HCCHC–O), 7.33–7.42 (m, 4H, Ar–H/PhHCCH), 7.43–7.47 (m, 3H, Ar–H), 7.50–7.53 (m, 2H, Ar–H), 7.60–7.63 (m, 2H, Ar–H); δC (100 MHz, CDCl3) 105.45, 118.63, 127.57, 127.60, 128.98, 129.00, 129.04, 129.64, 129.84, 130.25, 134.85, 135.18, 135.65, 155.07; found [M + Na]+ = 333.0562, C18H14O3S requires [M + Na]+ = 333.0564.
(E)-6-(4-Methoxystyryl)-3-phenyl-1,2-oxathiine 2,2-dioxide 4b.
A suspension of m-CPBA (3.14 g, 12.7 mmol, 1.4 eq.) in DCM (50 mL) was added dropwise over 15 min to a cold (0–5 °C) stirred solution of (E)-4-(dimethylamino)-6-(4-methoxystyryl)-3-phenyl-3,4-dihydro-1,2-oxathiine 2,2-dioxide (3.50 g, 9.1 mmol) in DCM (75 mL). Upon completion of the addition the reaction mixture was warmed to room temperature over 2 h and then washed with H2O (20 mL), aq. Na2SO3 (0.7 M, 2 × 15 mL), aq. NaOH(aq) (1 M, 15 mL) and H2O (15 mL). The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure to afford the title compound as an orange solid (2.78 g, 90%); mp = 205–207 °C; νmax (neat): 2962, 1631, 1598, 1571, 1540, 1509, 1444, 1361 (O-SO2), 1311, 1298, 1251, 1171 (O-SO2), 1151, 1070, 1026 cm−1; δH (400 MHz, CDCl3) 3.85 (s, 3H, OCH3), 6.05 (d, J = 7.4 Hz, 1H, 5-H), 6.53 (d, J = 15.8 Hz, 1H, HCCHC–O), 6.87 (d, J = 7.4 Hz, 1H, 4-H), 6.91–6.93 (m, 2H, Ar–H), 7.31 (d, J = 15.8 Hz, 1H, AnCHCH), 7.43–7.47 (m, 5H, Ar–H), 7.60–7.62 (m, 2H, Ar–H); δC (100 MHz, CDCl3) 55.42, 104.48, 114.46, 116.35, 127.51, 128.01, 129.01, 129.18, 129.23, 129.67, 130.38, 134.11, 135.40, 155.52, 160.91; found [M]˙+ = 340.0761, C19H16O4S requires [M]˙+ = 340.0763.
(E)-3-Phenyl-6-(4-(trifluoromethyl)styryl)-1,2-oxathiine 2,2-dioxide 4c.
A suspension of m-CPBA (1.64 g, 7.1 mmol, 1.5 eq.) in DCM (50 mL) was added dropwise over 10 min to a cold (0–5 °C) stirred solution of (E)-4-(dimethylamino)-3-phenyl-6-(4-(trifluoromethyl)styryl)-3,4-dihydro-1,2-oxathiine 2,2-dioxide (2.00 g, 4.7 mmol) in DCM (50 mL). Upon completion of the addition the reaction mixture was warmed to room temperature over 2 h and then washed with H2O (75 mL), aq. Na2SO3 (0.7 M, 2 × 50 mL), aq. NaOH(aq) (1 M, 50 mL) and H2O (60 mL). The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure to afford the title compound as pale yellow crystals (1.64 g, 92%); mp = 220–221 °C (from DCM); νmax (neat): 3039, 1631, 1610, 1542, 1514, 1492, 1359 (O-SO2), 1323, 1266, 1263, 1164 (O-SO2), 1119, 1108, 1067, 1032, 1014 cm−1; δH (400 MHz, CDCl3) 6.12 (d, J = 7.1 Hz, 1H, 3-H), 6.73 (d, J = 15.6 Hz, 1H, F3CC6H4CH), 6.88 (d, J = 6.1 Hz, 1H, 4-H), 7.35 (d, J = 15.6 Hz, 1H, CHCHC–O), 7.45–7.47 (m, 3H, Ar–H), 7.59–7.65 (m, 6H, Ar–H); δC (100 MHz, CDCl3) 106.65, 120.99, 123.92 (q, J = 271 Hz), 125.92 (q, J = 3.2 Hz), 127.62, 127.63, 128.63, 129.09, 130.05, 130.06, 131.01 (q, J = 33 Hz), 133.62, 135.80, 138.53, 154.31; δF (376.5 MHz, CDCl3) −62.64; found [M]˙+ = 378.0532, C19H13F3O3S requires [M]˙+ = 378.0537.
(E)-6-(4-Nitrostyryl)-3-phenyl-1,2-oxathiine 2,2-dioxide 4d.
A solution of m-CPBA (0.52 g, 2.3 mmol, 1.5 eq.) in DCM (25 mL) was added dropwise over 10 min to a cold (0–5 °C) stirred solution of (E)-4-(dimethylamino)-6-(4-nitrostyryl)-3-phenyl-3,4-dihydro-1,2-oxathiine 2,2-dioxide (0.60 g, 1.5 mmol) in DCM (25 mL). Upon completion of the addition the reaction mixture was warmed to room temperature overnight. Consecutive washes of the reaction mixture with H2O (60 mL), aq. Na2SO3 (0.7 M, 2 × 50 mL), aq. NaOH (1 M, 50 mL) and H2O (50 mL) afforded an organic layer which was dried over anhydrous Na2SO4. Removal of the solvent under reduced pressure gave the title compound as yellow crystals (0.49 g, 93%); mp = 230–232 °C; νmax (neat): 3060, 1607, 1590, 1514, 1446, 1339 (O-SO2), 1259, 1168 (O-SO2), 1104, 1060, 1034, 1009 cm−1; δH (400 MHz, CDCl3) 6.18 (d, J = 7.2 Hz, 1H, 5-H), 6.80 (d, J = 15.8 Hz, 1H, O2NC6H4CH), 6.90 (d, J = 7.2 Hz, 1H, 4-H), 7.37 (d, J = 15.8 Hz, 1H, CHCHC–O), 7.46–7.48 (m, 3H, Ar–H), 7.61–7.66 (m, 4H, Ar–H), 8.24–8.26 (m, 2H, Ar–H); δc (100 MHz, CDCl3) 107.58, 122.69, 124.31, 127.66, 128.00, 128.41, 129.13, 129.91, 130.22, 132.52, 136.45, 141.36, 147.90, 153.88; found [M]˙+ = 355.0504, C18H13NO5S requires [M]˙+ = 355.0514.
(E)-6-(2-Bromostyryl)-3-phenyl-1,2-oxathiine 2,2-dioxide 4e.
A suspension of m-CPBA (2.39 g, 10.4 mmol, 1.5 eq.) in DCM (60 mL) was added dropwise over 15 min to a cold (0–5 °C) stirred solution of (E)-6-(2-bromostyryl)-4-(dimethylamino)-3-phenyl-3,4-dihydro-1,2-oxathiine 2,2-dioxide (3.00 g, 6.9 mmol) in DCM (60 mL). Upon completion of the addition the reaction mixture was warmed to room temperature over 2 h. Consecutive washes of the reaction mixture with H2O (60 mL), aq. Na2SO3 (0.7 M, 2 × 50 mL), aq. NaOH (1 M, 50 mL) and H2O (50 mL) afforded an organic layer which was dried over anhydrous Na2SO4. Removal of the solvent under reduced pressure gave the title compound as yellow crystals (2.34 g, 87%); mp = 142–145 °C (from DCM); νmax (neat): 1627, 1539, 1435, 1359 (O-SO2), 1304, 1252, 1174 (O-SO2), 1069, 1022 cm−1; δH (400 MHz, CDCl3) 6.10 (d, J = 6.6 Hz, 1H, 5-H), 6.61 (d, J = 15.6 Hz, 1H, BrC6H4CH), 6.87 (d, J = 6.6 Hz, 1H, 4-H), 7.19–7.20 (m, 1H, Ar–H), 7.30–7.32 (m, 1H, Ar–H), 7.44–7.47 (m, 3H, Ar–H), 7.57–7.68 (m, 5H, Ar–H/CHCHC–O); δC (100 MHz, CDCl3) 106.29, 121.38, 125.17, 127.16, 127.63, 127.65, 128.75, 129.05, 129.94, 130.19, 130.50, 133.59, 133.77, 135.18, 135.47, 154.65; found [M]˙+ = 387.9768, C18H1379BrO3S requires [M]˙+ = 387.9769.
3-Phenyl-6-(phenylethynyl)-1,2-oxathiine 2,2-dioxide 4f.
A suspension of m-CPBA (1.00 g, 4.35 mmol, 1.4 eq.) in DCM (15 mL) was added dropwise to a cold (0–5 °C) stirred solution of 4-(dimethylamino)-3-phenyl-6-(phenylethynyl)-3,4-dihydro-1,2-oxathiine 2,2-dioxide (1.10 g, 3.10 mmol) in DCM (15 mL). The resulting reaction mixture was warmed to room temperature overnight and then washed with H2O (40 mL), aq. Na2SO3 (0.7 M, 40 mL), aq. NaOH (1 M, 30 mL) and H2O (30 mL). The organic layer was dried over anhydrous Na2SO4 and the solvent removed under reduced pressure gave the crude solid which was triturated with Et2O to afford the title compound as a yellow solid (0.81 g, 84%); mp = 133–135 °C; νmax (neat): 2203, 1622, 1546, 1488, 1441, 1361 (O-SO2), 1280, 1270, 1251, 1177 (O-SO2), 1155, 1126, 1063, 1031 cm−1; δH (400 MHz, CDCl3) 6.38 (d, J = 7.1 Hz, 1H, 5-H), 6.80 (d, J = 7.1 Hz, 1H, 4-H), 7.37–7.47 (m, 6H, Ar–H), 7.53–7.56 (m, 2H, Ar–H), 7.58–7.62 (m, 2H, Ar–H); δC (100 MHz, CDCl3) 81.14, 98.52, 111.43, 120.50, 127.69, 128.02, 128.66, 129.09, 129.87, 130.23, 130.27, 132.05, 137.37, 139.80; found [M]˙+ = 308.0491, C18H12O3S requires [M]˙+ = 308.0502.
(E)-6-Styryl-1,2-oxathiine 2,2-dioxide 4g.
A suspension of m-CPBA (10.31 g, 46.0 mmol, 77%) in DCM (75 mL) was added dropwise to a cold (0–5 °C) stirred solution of (E)-4-(dimethylamino)-6-styryl-3,4-dihydro-1,2-oxathiine 2,2-oxide (8.56 g, 30.67 mmol) in DCM (50 mL). Upon completion of the addition the reaction mixture was warmed to room temperature over 2 h and then washed with H2O (2 × 50 mL), aq. Na2SO3 (0.7 M, 2 × 30 mL), aq. NaOH(aq) (1 M, 30 mL) and H2O (50 mL). The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure to afford the target compound as a pale brown solid after trituration with Et2O/hexane (6.95 g, 97% yield); mp = 108–110 °C; νmax (neat) 1599, 1372, 1357, 1155, 846, 772, 669, 565, 504.9, 475 cm−1; δH (400 MHz, CDCl3) 5.95 (d, J = 6.8 Hz, 1H, 5-H), 6.61 (dd, J = 10.4 Hz, 1H, 3-H), 6.62 (d, J = 15.6 Hz, 1H, PhCH), 6.87 (d, J = 6.8, 10.4, 1H, 4-H), 7.32–7.41 (4H, m, Ar–H, CHCHCO), 7.48–7.51 (m, 2H, ArH); δC (100 MHz, CDCl3) 103.80, 118.70, 119.22, 127.66, 128.99, 129.78, 134.47, 134.97, 136.22, 156.35; found [M + H]+ = 234.0347. C12H10O3S requires [M + H]+ = 234.0352.
Preparation of bromo substituted 1,2-oxathiine 2,2-dioxides
3-Bromo-3,6-diphenyl-1,2-oxathiine 2,2-dioxide 7.
A solution of Br2 (2.81 mL, 17.6 mmol) in CHCl3 (50 mL) was added dropwise over 30 min to a stirred solution of 5,6-diphenyl-1,2-oxathiine 2,2-dioxide (5.00 g, 17.6 mmol) in CHCl3 (200 mL) at room temperature. The resulting solution was stirred at 70 °C for 72 h, before being cooled and diluted with CHCl3 (100 mL) and washed with an aq. Na2S2O3 solution (250 mL) and H2O (2 × 150 mL). Removal of the dried (anhydrous Na2SO4) organic layer gave the product after trituration with 9:1 Et2O/EtOAc as an off-white solid (4.68 g, 73%); mp = 137–138 °C (from EtOAc/hexane); νmax (neat): 3041, 1621, 1546, 1489, 1446, 1377 (O-SO2), 1339, 1272, 1188 (O-SO2), 1157, 1124, 1108, 1008, 1000 cm−1; δH (400 MHz, (CD3)2CO): δ 7.30–7.43 (m, 10H, Ar–H), 7.62 (s, 1H, 4-H); δC (100 MHz, (CD3)2CO): 110.58, 119.64, 128.48, 128.56, 129.13, 129.19, 129.23, 130.68, 130.80, 134.54, 140.21, 152.09; found [M + Na]+ = 384.9500, C16H1179BrO3S requires [M + Na]+ = 384.9504.
3-Bromo-6-phenyl-1,2-oxathiine 2,2-dioxide 8.
A solution of Br2 (0.78 mL, 15.2 mmol, 1.1 eq.) in CHCl3 (50 mL) was added dropwise over 20 min to a stirred solution of 6-phenyl-1,2-oxathiine 2,2-dioxide (2.87 g, 13.8 mmol) in CHCl3 (100 mL) at room temperature. Upon completion of the addition the mixture the reaction mixture was set to reflux (70 °C) for 16 h before cooling to room temperature. Pyridine (0.28 mL, 3.5 mmol, 0.25 eq.) was added and the mixture was heated at reflux for 15 min. The reaction mixture was diluted with CHCl3 (100 mL) and washed with aq. Na2S2O3 solution (150 mL), aq. HCl (1 M, 100 mL) and H2O (100 mL). The obtained organic layer was dried with anhydrous Na2SO4 and the solvent was removed to afford the crude product, which was triturated with 8:2 petroleum ether:EtOAc to afford the title compound as an off-white solid (3.43 g, 87%) mp = 130°–132 °C (from CHCl3); νmax (neat): 3037, 2918, 2849, 1626, 1542, 1494, 1447, 1365 (O-SO2), 1261, 1179 (O-SO2), 1070, 1035, 1006 cm−1; δH (400 MHz, CDCl3) 6.33 (d, J = 7.3 Hz, 1H, 4-H), 7.10 (d, J = 7.3 Hz, 1H, 5-H), 7.44–7.52 (m, 3H, Ar–H), 7.69–7.71 (m, 2H, Ar–H); δC (100 MHz, CDCl3) 101.57, 110.98, 125.55, 129.12, 130.07, 131.53, 135.09, 159.43; found [M + Na]+ = 308.9186, C10H779BrO3S requires [M + Na]+ = 308.9191.
Suzuki cross-coupling reactions
3,5,6-(Triphenyl)-1,2-oxathiine 2,2-dioxide 6a.
A suspension of 3-bromo-5,6-diphenyl-1,2-oxathiine 2,2-dioxide (0.40 g, 1.1 mmol, 1.0 eq.), PhB(OH)2 (0.20 g, 1.65 mmol, 1.5 eq.), Pd(OAc)2 (0.012 g, 0.06 mmol, 5% mol), PCy3 (0.031 g, 0.11 mmol, 10% mol) and K2HPO4·3H2O (0.75 g, 3.3 mmol, 3.0 eq.) in DMAc (10 mL) under nitrogen were heated to 75 °C for 48 h. The solvent was removed azeotropically with consecutive portions of PhMe and the resulting crude was purified by short column chromatography (silica, neat DCM), to afford the title compound as pale yellow microneedles (0.16 g, 40%); mp = 158–160 °C (lit. mp = 159–160 °C (ref. 13)); νmax (neat) 3060, 1620, 1540, 1445, 1368 (O-SO2), 1350, 1187 (O-SO2), 1129, 1012, 972, 852, 773, cm−1; δH (CDCl3, 400 MHz) 7.03 (s, 1H, 4-H), 7.26–7.39 (m, 10H, Ar–H), 7.47–7.51 (m, 3H, Ar–H), 7.68–7.73 (m, 2H, Ar–H); δC (CDCl3, 100 MHz) 118.99, 127.75, 128.23, 128.38, 129.07, 129.14, 129.21, 129.29, 129.90, 129.96, 130.28, 131.12, 133.99, 134.13, 135.81, 152.12.
3-(4-Methoxyphenyl)-5,6-diphenyl-1,2-oxathiine 2,2-dioxide 6b.
3-Bromo-5,6-diphenyl-1,2-oxathiine 2,2-dioxide (0.40 g, 1.1 mmol, 1.0 eq.), 4-MeOC6H4B(OH)2 (0.25 g, 1.7 mmol, 1.5 eq.), PCy3 (0.031 g, 0.11 mmol, 10% mol) and K2HPO4·3H2O (0.75 g, 3.3 mmol, 3 eq.) were charged into a dried flask under nitrogen. A suspension of Pd(OAc)2 (0.012 g, 0.06 mmol, 5% mol) in DMAc (7.5 mL) and MeOH (7.5 mL) was added and the reaction mixture was heated at 60 °C overnight. The cooled reaction mixture was poured into H2O (15 mL) and the product was extracted with EtOAc (4 × 20 mL). The combined extracts were washed with aq. NaHCO3 (40 mL) and brine (40 mL), dried over anhydrous Na2SO4 and the solvent was removed to afford the crude product which was purified by a short column (silica, neat DCM) to afford the title compound as a pale-yellow solid after trituration with Et2O (0.25 g, 58%); mp = 147–148 °C (from DCM); νmax (neat): 1604, 1510, 1444, 1364 (O-SO2), 1305, 1272, 1254, 1228, 1180 (O-SO2), 1127, 1035, 1007 cm−1; δH (CDCl3, 400 MHz) 3.85 (s, 3H, OCH3), 6.91 (s, 1H, 4-H), 6.96–6.98 (m, 2H, An–H), 7.22–7.34 (m, 2H, Ar–H), 7.60–7.62 (m, 2H, An–H); δC (CDCl3, 100 MHz) 55.46, 114.54, 119.06, 122.15, 128.16, 128.27, 129.13, 129.16, 129.22 (2C), 130.09, 131.21, 132.26, 133.87, 135.98, 151.50, 161.03; found [M + Na]+ = 413.0813, C23H18O4S requires [M + Na]+ = 413.0824.
5,6-Diphenyl-3-(4-trifluoromethylphenyl)-1,2-oxathiine 2,2 dioxide 6c.
A solution of 3-bromo-5,6-diphenyl-1,2-oxathiine 2,2-dioxide (0.40 g, 1.1 mmol, 1.0 eq.), 4-CF3C6H4B(OH)2 (0.31 g, 1.7 mmol, 1.5 eq.), Pd(OAc)2 (0.012 g, 0.06 mmol, 5% mol), PCy3 (0.031 g, 0.11 mmol, 10% mol) and K2HPO4·3H2O (0.75 g, 3.3 mmol, 3 eq.) in DMAc (7.5 mL) and MeOH (7.5 mL) under nitrogen was heated to 60 °C overnight. The cooled reaction mixture was poured into H2O (20 mL) and extracted with EtOAc (2 × 40 mL). The combined extracts were washed with aq. NaHCO3 (40 mL), brine (40 mL), dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The crude solid was purified by a short column (silica, neat DCM) to afford the title compound as pale-yellow crystals (0.17 g, 36%); mp = 160–162 °C (lit mp = 161–163 °C (ref. 19)); νmax (neat) 3062, 1616, 1574, 1555, 1487, 1444, 1366 (O-SO2), 1351, 1322, 1277, 1172 (O-SO2), 1116, 1067, 1036, 1014, 1006 cm−1; δH ((CD3)2CO, 400 MHz) 7.33–7.43 (m, 10H, Ar–H), 7.54 (s, 1H, 4-H), 7.88 (app. d, J = 8 Hz, 2H, Ar–H), 8.02 (app. d, J = 8 Hz, 2H, Ar–H); δC ((CD3)2CO, 100 MHz) 119.00, 123.75 (q, J = 271 Hz), 126.04 (q, J = 3.8 Hz), 128.09, 128.33, 128.53, 129.07, 129.32, 130.62, 130.83, 131.71 (q, J = 33 Hz), 132.64, 133.45, 135.42, 135.63, 153.04; δF (CDCl3, 376 MHz) −63.3.
3,6-Diphenyl-1,2-oxathiine 2,2-dioxide 6e.
A suspension of 3-bromo-6-phenyl-1,2-oxathiine 2,2-dioxide (0.40 g, 1.4 mmol, 1.0 eq.), PhB(OH)2 (0.25 g, 2.1 mmol, 1.5 eq.), Pd(OAc)2 (0.015 g, 0.07 mmol, 5% mol), PCy3 (0.039 g, 0.14 mmol, 10% mol) and K2HPO4·3H2O (0.95 g, 4.2 mmol, 3.0 eq.) in DMAc (10 mL) under nitrogen was heated to 75 °C for 48 h. The cooled reaction mixture was poured into H2O (40 mL) and extracted with EtOAc (5 × 20 mL). The organic extracts were washed with H2O (100 mL), brine (100 mL) and dried over anhydrous Na2SO4. Removal of the solvent under reduced pressure afforded the crude product which was subjected to column chromatography (silica, 50% DCM/petroleum ether) to give fraction 1: 3,6-diphenyl-1,2-oxathiine 2,2-dioxide 6e (0.11 g, 27%) as a pale yellow solid mp 129–131 °C (lit. mp = 130–132 °C (ref. 19)); νmax (neat) 3063, 1632, 1558, 1494, 1448, 1353 (O-SO2), 1335, 1264, 1173 (O-SO2), 1072, 1031, 1009 cm−1; δH (CDCl3, 400 MHz) 6.58 (d, J = 7.1 Hz, 1H, 5-H), 6.96 (d, J = 7.1 Hz, 1H, 4-H), 7.50–7.47 (m, 6H, Ar–H), 7.67–7.64 (m, 2H, Ar–H), 7.83–7.77 (m, 2H, Ar–H); δC (CDCl3, 100 MHz) 101.60, 125.49, 127.66, 128.98, 129.01, 129.05, 129.87, 130.12, 130.65, 131.13, 134.48, 156.02.
Fraction 2: 6,6′-diphenyl-[3,3′-bi(1,2-oxathiine)] 2,2,2′,2′-tetraoxide 9 (0.02 g, 10%) as an orange solid; mp = 227–230 °C; νmax (neat): 1614, 1537, 1490, 1445, 1376 (O-SO2), 1340, 1309, 1266, 1237, 1183 (O-SO2), 1064, 1032, 1004 cm−1; δH (CDCl3, 400 MHz) 6.63 (d, J = 7.4 Hz, 2H, 5,5′-H), 7.47–7.51 (m, 8H, Ar–H/4,4′-H), 7.76–7.77 (m, 2H, Ar–H); δC (CDCl3, 100 MHz) 102.06, 124.24, 125.81, 129.18, 130.04, 131.61, 131.90, 157.17; found [M + Na]+ = 437.0126, C20H14O6S2 requires [M + Na]+ = 437.0132.
CH-activated coupling protocol
3,5,6-(Triphenyl)-1,2-oxathiine 2,2-dioxide 6a.
5,6-Diphenyl-1,2-oxathiine 2,2-dioxide (0.40 g, 1.41 mmol, 1.0 eq.), Pd(PPh3)4 (0.16 g, 0.14 mmol, 10% mol) and AcOAg (0.26 g, 1.6 mmol, 1,1 eq.) were charged into a flame dried flask under N2. DMF (5 mL) was added, followed by PhI (0.47 mL, 4.22 mol, 3.0 eq.) and the resulting suspension was heated to 80 °C overnight. The cooled reaction mixture was poured into H2O (10 mL) and extracted with EtOAc (4 × 15 mL). The combined organic extracts were washed with H2O (50 mL), dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The crude product was subjected to short column chromatography (silica, neat DCM) to afford the title compound after trituration with Et2O as a yellow solid (0.20 g, 39.2%). Physical and spectroscopic data were identical to previously prepared material.13
3-(4-Methoxyphenyl)-5,6-diphenyl-1,2-oxathiine 2,2-dioxide 6b.
5,6-Diphenyl-1,2-oxathiine 2,2-dioxide (0.20 g, 0.7 mmol, 1.0 eq.), p-MeOC6H4I (0.49 g, 2.1 mmol, 3.0 eq.) and AcOAg (0.13 g, 0.8 mmol, 1.1 eq.) were charged into a flame dried flask under N2. After the addition of DMF (5 mL), Pd(PPh3)4 (0.08 g, 0.07 mmol, 10% mol) the reaction mixture was heated to 80 °C overnight. Upon cooling the reaction mixture was diluted with EtOAc (80 mL) and washed with H2O (60 mL) and brine (60 mL). The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The crude product was subjected to short column chromatography (silica, 20% EtOAc/petroleum ether) to afford the target compound which was recrystallised from EtOAc/petroleum ether as a yellow solid (0.07 g, 26%). Physical and spectroscopic data were identical to previously prepared material.
5,6-Diphenyl-3-((4-trifluoromethy)phenyl)-1,2-oxathiine 2,2 dioxide 6c.
5,6-Diphenyl-1,2-oxathiine 2,2-dioxide (0.20 g, 0.7 mmol), 4-CF3C6H4I (0.31 mL, 2.1 mmol, 3.0 eq.) and AgOAc (0.13 g, 0.8 mmol, 1.1 eq.) were charged into a flame-dried flask under N2. DMF (5 mL) and Pd(PPh3)4 (0.08 g, 0.07 mmol, 10% mol) were added and the reaction mixture was heated to 80 °C for 2 h. Upon cooling the reaction mixture was filtered through celite and the celite was washed with EtOAc (2 × 50 mL). Removal of the solvent gave the crude product which was triturated with Et2O to afford the title product as a pale yellow solid (0.17 g, 57%). Physical and spectroscopic data were identical to previously prepared material.19
5,6-Diphenyl-3-(pyridin-4-yl)-1,2-oxathiine 2,2-dioxide 6d.
5,6-Diphenyl-1,2-oxathiine 2,2-dioxide (0.20 g, 0.7 mmol), 4-iodopyridine (0.43 g, 2.1 mmol, 3.0 eq.) and AgOAc (0.13 g, 0.8 mmol, 1.1 eq.) were charged into a flame-dried flask under N2. DMF (10 mL) was added followed by Pd(PPh3)4 (0.08 g, 0.07 mmol, 10% mol) and the reaction mixture was heated to 80 °C for 1 h. Upon cooling the reaction mixture was filtered through celite and the celite was washed with EtOAc (2 × 50 mL). Removal of the solvent gave a crude mixture that was purified by column chromatography (silica, 40% to 60% EtOAc/petroleum ether) to yield the title compound as a pale yellow solid (0.17 g, 68%); mp = 144–146 °C; vmax (neat): 1592, 1558, 1540, 1371 (O-SO2), 1181 (O-SO2), 1133, 1073 cm−1; δH (CDCl3, 400 MHz) 7.25–7.29 (m, 4H, Ar–H), 7.33–7.38 (m, 6H, Ar–H), 7.57–7.58 (m, 2H, 3,5-Py-H), 8.70–8.71 (m, 2H, 2,6-Py-H); δC (CDCl3, 100 MHz) 118.95, 121.25, 128.33, 128.64, 129.05, 129.37 (2 × C), 130.68, 130.78, 131.38, 135.24, 136.76, 137.47, 150.55, 153.70; found [M + H]+ = 362.0843, C21H15NO3S requires [M + H]+ = 362.0845.
5,6-Bis(4-methoxyphenyl)-3-(pyridin-4-yl)-1,2-oxathiine 2,2-dioxide 6f.
5,6-Bis(4-methoxyphenyl)-1,2-oxathiine 2,2-dioxide (0.50 g, 1.5 mmol), 4-iodopyridine (0.89 g, 4.4 mmol, 3.0 eq.) and AgOAc (0.27 g, 1.6 mmol, 1.1 eq.) were charged into a flame-dried flask under N2. DMF (5 mL) was added, followed by Pd(PPh3)4 (0.17 g, 0.15 mmol, 10% mol) and the reaction mixture was heated to 80 °C for 0.5 h. Upon cooling the reaction mixture was filtered through celite and the celite was washed with EtOAc (2 × 50 mL). Removal of the solvent gave a crude mixture that was purified by column chromatography (silica, neat EtOAc to 10% MeOH/EtOAc) to afford the title compound after trituration with EtOAc as a pale red solid which darkens upon standing (0.20 g, 33%); mp = 134–138 °C (decomp.); vmax (neat): 1600, 1591, 1568, 1531, 1514, 1499, 1460, 1444, 1413, 1366 (O-SO2), 1302, 1292, 1251, 1173 (O-SO2), 1137, 1117, 1107, 1071, 1025, 1015 cm−1; δH (CDCl3, 400 MHz) 3.80 (s, 3H, OMe), 3.83 (s, 3H, OMe), 6.76–6.78 (m, 2H, Ar–H), 6.89–6.91 (m, 2H, Ar–H), 7.19 (s, 1H, 4-H), 7.19–7.21 (m, 2H, Ar–H), 7.30–7.32 (m, 2H, Ar–H), 7.57–7.58 (m, 2H, 3,5-Py-H), 8.69–8.71 (m, 2H, 2,6-Py-H); δC (CDCl3, 100 MHz) 55.36, 55.40, 113.83, 114.83, 117.21, 121.19, 123.08, 127.59, 130.06, 130.24, 131.07, 137.24, 137.98, 150.21, 153.61, 159.66, 161.40; found [M + H]+ = 422.1054, C23H19NO5S requires [M + H]+ = 422.1060.
(E)-6-Styryl-3-(4-(trifluoromethyl)phenyl)-1,2-oxathiine 2,2-dioxide 4h.
(E)-6-Styryl-1,2-oxathiine 2,2-dioxide (0.50 g, 2.1 mmol), 4-CF3C6H4I (0.94 mL, 6.4 mmol, 3.0 eq.) and AgOAc (0.39 g, 2.3 mmol, 1.1 eq.) were charged into a flame-dried flask under N2. DMF (15 mL) was added followed by Pd(PPh3)4 (0.24 g, 0.2 mmol, 10% mol) and the reaction mixture was heated to 80 °C for 2 h. Upon cooling the reaction mixture was filtered through celite and the celite was washed with EtOAc (2 × 50 mL). Removal of the solvent gave a crude mixture that was triturated with Et2O/DCM (1:1) to afford the title compound as an orange solid (0.65 g, 82%); mp = 192–195 °C; vmax (neat): 1631, 1616, 1543, 1353, 1326 (O-SO2), 1263, 1195, 1170 (O-SO2), 1153, 1111, 1064, 1017 cm−1; δH (CDCl3, 400 MHz) 6.10 (d, J = 7.1 Hz, 1H, 5-H), 6.68 (d, J = 15.9 Hz, 1H, PhCHCH), 6.96 (d, J = 7.1 Hz, 1H, 4-H), 7.36–7.42 (m, 4H, ArH/PhCHCH), 7.51–7.53 (m, 2H, ArH), 7.69–7.74 (m, 4H, ArH); δC (CDCl3, 100 MHz) 105.25, 118.38, 123.74 (q, J = 272 Hz), 126.02 (q, J = 3.7 Hz), 127.72, 127.84, 129.03, 129.92, 130.64, 131.54 (q, J = 33 Hz), 133.23, 133.75, 134.97, 136.57, 155.93; δF (376.5 MHz, CDCl3) −62.90; found [M]˙+ = 378.0530, C19H13F3O3S requires [M]˙+ = 378.0532.
3,6-Diphenyl-1,2-oxathiine 2,2-dioxide 6e.
6-Phenyl-1,2-oxathiine 2,2-dioxide (0.21 g, 1.0 mmol), AcOAg (0.18 g, 1.1 mmol, 1.1 eq.) and PhI (0.33 mL, 3.0 mmol, 3.0 eq.) were charged into a flame dried flask under N2. After the addition of DMF (5 mL) and Pd(PPh3)4 (0.12 g, 0.1 mmol, 10% mol) the reaction mixture was heated to 80 °C overnight. Upon cooling the reaction mixture was poured into H2O (10 mL) and extracted with EtOAc (5 × 20 mL). The combined organic extracts were washed with H2O (80 mL), dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The crude product was subjected to short column chromatography (silica, 20% to 50% EtOAc in petroleum ether) to afford the title compound after recrystallisation from EtOAc/petroleum ether as a pale yellow solid (0.09 g, 32%). Physical and spectroscopic data were identical to previously prepared material.19
Attempted Suzuki–Miyaura borylation
5,5′,6,6′-Tetraphenyl-[3,3′-bi(1,2-oxathiine)] 2,2,2′,2′-tetraoxide 10.
A suspension of 3-bromo-3,6-diphenyl-1,2-oxathiine 2,2-dioxide (0.5 g, 1.38 mmol), B2pin2 (0.16 g, 0.7 mmol, 0.5 eq.), KOAc (0.34 g, 3.5 mmol, 2.5 eq.), Pd(dppf)Cl2 (0.12 g, 0.14 mmol, 10% mol) in degassed 1,4-dioxane (20 mL) under a N2 atmosphere was stirred at 80 °C for 1.5 h. Upon cooling the mixture was diluted with H2O (30 mL) and the biphasic mixture extracted with EtOAc (5 × 50 mL) and the combined extracts washed with brine (2 × 50 mL), dried over anhydrous Na2SO4. Removal of the solvent gave a crude product which was purified by column chromatography (silica, neat DCM) to afford the title compound as a yellow-orange solid (0.20 g, 51%); mp = 190 °C (darkening and decomp); vmax (neat): 3062, 1609, 1533, 1486, 1442, 1364 (O-SO2), 1329, 1317, 1284, 1191 (O-SO2), 1154, 1126, 1102, 1073, 1035, 1008 cm−1; δH (CDCl3, 400 MHz) 7.23–7.38 (m, 20H, ArH), 7.56 (s, 2H, 2 × 4-H); δH (CDCl3, 400 MHz) 119.69, 123.78, 128.33, 128.67, 129.10, 129.36, 129.45, 130.55, 130.89, 134.83, 136.75, 153.46; found [M + Na]+ = 589.0749, C32H22O6S2 requires [M + Na]+ = 589.0758.
Preparation of phenyltriazolinedione adducts
2,5,9-Triphenyl-5,9-dihydro-1H-[1,2]oxathiino[5,6-c][1,2,4]triazolo[1,2-a]pyridazine-1,3(2H)-dione 8,8-dioxide 12a.
A solution of PTAD (0.32 g, 1.6 mmol, 1.2 eq.) in 1,2-DCE (10 mL) was added dropwise to a stirred solution of (E)-3-phenyl-6-styryl-1,2-oxathiine 2,2-dioxide (0.4 g, 1.3 mmol) in 1,2-DCE (10 mL) and the resulting reaction mixture was stirred overnight at room temperature. Removal of the solvent gave the crude product which was eluted through a short column (silica, neat DCM) and the resulting pale yellow solid was triturated with Et2O afford the product (0.38 g, 60%) as an off-white solid; mp = 146–147 °C (from DCM); vmax (neat): 3073, 1768, 1712, 1620, 1494, 1455, 1412, 1384 (O-SO2), 1304, 1220, 1165 (O-SO2), 1148, 1103, 1030 cm−1; δH (CDCl3, 400 MHz) 5.48 (ddd, J = 0.8, 1.0, 3.6 Hz, 1H, 9-H), 5.96 (ddd, J = 0.5, 0.8, 6.1 Hz, 1H, 5-H), 6.14 (ddd, J = 1.0, 1.7, 6.1 Hz, 1H, 6-H), 7.11 (ddd, J = 0.5, 1.7, 3.6 Hz, 1H, 10-H), 7.33–7.57 (m, 15H, ArH); δC (100 MHz, CDCl3) 56.05, 63.07, 106.93, 112.26, 125.53, 126.60, 128.53, 128.75, 129.25, 129.35 (2 × C), 129.39, 129.92, 130.01, 130.20, 130.28, 133.31, 142.17, 148.20, 148.95; HRMS: Found [M + H]+ = 486.1121, C26H19N3O5S requires [M + H]+ = 486.1125.
5-(4-Methoxyphenyl)-2,9-diphenyl-5,9-dihydro-1H-[1,2]oxathiino[5,6-c][1,2,4]triazolo[1,2-a] pyridazine-1,3(2H)-dione 8,8-dioxide 12b.
A solution of PTAD (0.33 g, 1.9 mmol, 1.6 eq.) in 1,2-DCE (15 mL) was added dropwise to a stirred solution of (E)-6-(4-methoxystyryl)-3-phenyl-1,2-oxathiine 2,2-dioxide (0.40 g, 1.2 mmol) in 1,2-DCE (25 mL) at room temperature and the resulting reaction mixture was stirred at room temperature overnight and then heated at reflux for 1.5 h. Removal of the solvent gave the crude product which was eluted through a short column (silica, neat DCM) and the resulting solid was triturated with Et2O to afford the title product as a pale pink solid (0.40 g, 67%); mp = 158–160 °C (from DCM); vmax (neat): 3063, 1770, 1712, 1608, 1512, 1504, 1491, 1442, 1401, 1381 (O-SO2), 1307, 1262, 1240, 1170 (O-SO2), 1144, 1103, 1089, 1020 cm−1; δH (CDCl3, 400 MHz) 3.82 (s, 3H, OMe), 5.48 (dd, J = 1.1, 3.5 Hz, 1H, 9-H), 5.92 (dd, J = 0.5, 6.1 Hz, 1H, 5-H), 6.14 (ddd, J = 1.1, 1.6, 6.1 Hz, 1H, 6-H), 6.94–6.96 (m, 2H, ArH), 7.06–7.09 (ddd, J = 0.5, 1.6, 3.5 Hz, 1H, 10-H), 7.34–7.55 (m, 12H, ArH); δC (CDCl3, 100 MHz) 55.37, 55.64, 63.09, 106.73, 112.53, 114.59, 114.60, 125.14, 125.52, 126.67, 128.70, 129.22, 129.33, 129.42, 129.92, 130.04, 130.25 (2 × C), 142.12, 148.20, 149.12, 160.76; HRMS: Found [M]˙+ = 515.1148, C27H21N3O6S requires [M]˙+ = 515.1151.
2,9-Diphenyl-5-(4-(trifluoromethyl)phenyl)-5,9-dihydro-1H-[1,2]oxathiino[5,6-c][1,2,4]triazolo[1,2-a]pyridazine-1,3(2H)-dione 8,8-dioxide 12c.
A solution of PTAD (0.28 g, 1.6 mmol, 2.0 eq.) in 1,2-DCE (8 mL) was added dropwise to a stirred solution of (E)-3-phenyl-6-(4-(trifluoromethyl)styryl)-1,2-oxathiine 2,2-dioxide (0.30 g, 0.8 mmol) in 1,2-DCE (8 mL) at room temperature and then the resulting mixture was heated at 50 °C overnight. Removal of the solvent gave the crude product which was purified by column chromatography (silica, 20% to 30% EtOAc/petroleum spirit) to afford the title compound after trituration with Et2O as colourless crystals (0.17 g, 39%); mp = 173–176 °C (EtOAc/P.E.); vmax (neat): 1776, 1709, 1620, 1599, 1495, 1405, 1388, 1323 (O-SO2), 1166 (O-SO2), 1115, 1067, 1018 cm−1; δH (CDCl3, 400 MHz) 5.49 (dd, J = 1.0, 3.6 Hz, 1H, 9-H), 6.01 (dd, J = 0.5, 6.2 Hz, 1H, 5-H), 6.14 (ddd, J = 1.0, 1.5, 6.2 Hz, 1H, 6-H), 7.14 (ddd, J = 0.5, 1.5, 3.6 Hz, 1H, 10-H), 7.35–7.54 (m, 10H, ArH), 7.63–7.74 (m, 4H, ArH); δC (CDCl3, 100 MHz) 55.30, 63.11, 107.66, 111.34, 123.65 (q, J = 273 Hz), 125.46, 126.41 (q, J = 3.5 Hz), 127.93, 128.90, 128.98, 129.12, 129.32, 129.40, 129.90, 130.00, 130.38, 132.13 (q, J = 33 Hz), 137.18, 142.68, 148.07, 148.85; δF (376.5 MHz, CDCl3) −62.89 (s, 3F); HRMS: Found [M + Na]+ = 576.0815, C27H18F3N3O5S requires [M + Na]+ = 576.0819.
2,5-Diphenyl-5,9-dihydro-1H-[1,2]oxathiino[5,6-c][1,2,4]triazolo[1,2-a]pyridazine-1,3(2H)-dione 8,8-dioxide 12e.
A solution of PTAD (0.25 g, 1.4 mmol, 1.1 eq.) in 1,2-DCE (10 mL) was added dropwise to a stirred solution of (E)-6-styryl-1,2-oxathiine 2,2-dioxide (0.30 g, 1.3 mmol) in 1,2-DCE (15 mL) at room temperature overnight. Further PTAD (0.23 g, 1.3 mmol) was added with stirring at room temperature continued for 3 h and then the mixture was stirred at 50 °C for a further 2 h. Removal of the solvent gave a crude product which was purified by a short column (silica, 0% to 10% EtOAc/DCM) to afford a solid that was triturated with Et2O to yield the title compound as an off-white solid (0.37 g, 70%); mp = 196–198 °C (EtOAc/DCM); vmax (neat): 1762, 1704, 1621, 1497, 1458, 1402, 1384 (O-SO2), 1305, 1291, 1272, 1182, 1172 (O-SO2), 1157, 1142, 1102, 1086, 1032 cm−1; δH (CDCl3, 400 MHz) 4.24 (ddd, J = 0.7, 4.7, 17.6 Hz, 1H, syn-9-H), 4.28 (ddd, J = 0.7, 5.5, 17.6 Hz, 1H, anti-9-H), 5.93 (d, J = 6.0 Hz, 1H, 5-H), 6.10 (ddd, J = 0.7, 1.7, 6.0 Hz, 1H, 6-H), 6.98 (ddd, J = 1.7, 4.7, 5.5 Hz, 1H, 10-H), 7.36–7.46 (m, 10H, ArH); δC (CDCl3, 100 MHz) 46.92, 56.05, 101.24, 112.35, 125.58, 126.08, 128.42, 128.76, 129.26, 129.27, 129.94, 130.20, 133.37, 142.09, 148.26, 148.83; HRMS: Found [M + H]+ = 410.0800, C20H15N3O5S requires [M + H]+ = 410.0802.
2,5-Diphenyl-9-(4-(trifluoromethyl)phenyl)-5,9-dihydro-1H-[1,2]oxathiino[5,6-c][1,2,4]triazolo[1,2-a]pyridazine-1,3(2H)-dione 8,8-dioxide 12f.
A solution of PTAD (0.11 g, 0.6 mmol, 1.1 eq.) in 1,2-DCE (7 mL) was added dropwise to a solution of (E)-6-styryl-3-(4-(trifluoromethyl)phenyl)-1,2-oxathiine 2,2-dioxide (0.20 g, 0.5 mmol) in 1,2-DCE (10 mL) at room temperature. After stirring overnight at room temperature, a further portion of PTAD (0.05 g, 0.3 mmol) was added and the reaction mixture was heated at 50 °C for 72 h. Removal of the solvent gave the crude product which was purified by column chromatography (silica, 20% EtOAc/petroleum spirit) to afford an off-white solid that was triturated with Et2O to yield the title product as a white powder (0.09 g, 18%); mp = 156–157 °C (EtOAc/petroleum spirit); vmax (neat): 1770, 1715, 1410, 1388 (O-SO2), 1328, 1184, 1167 (O-SO2), 1133, 1103, 1069 cm−1; δH (CDCl3, 400 MHz) 5.53 (dd, J = 1.0, 3.5 Hz, 1H, 9-H), 5.98 (dd, J = 0.5, 5.9 Hz, 1H, 5-H), 6.20 (ddd, J = 1.0, 1.5, 5.9 Hz, 1H, 6-H), 7.08 (ddd, J = 0.5, 1.5, 3.5 Hz, 1H, 10-H), 7.33–7.51 (m, 10H, ArH), 7.69–7.71 (m, 2H, ArH), 7.75–7.77 (m, 2H, ArH); δC (CDCl3, 100 MHz) 56.06, 62.56, 105.71, 112.91, 123.64 (q, J = 274 Hz), 125.50, 126.32 (q, J = 3.7 Hz), 126.98, 128.48, 128.82, 129.27, 129.39, 130.10, 130.13, 130.41, 132.62 (q, J = 33 Hz), 133.14, 133.47, 141.98, 148.20, 148.89; δF (376.5 MHz, CDCl3) −62.94 (s, 3F); HRMS: Found [M + H]+ = 554.0994, C27H18F3N3O5S requires [M + H]+ = 554.0996.
PTAD addition reactions to 6-styryl-1,2-oxathiine 2,2-dioxides for purpose of characterization of the silica sensitive initial adducts – isolation of 5-(4-methoxyphenyl)-2,9-diphenyl-5,10a-dihydro-1H-[1,2]oxathiino[5,6-c][1,2,4] triazolo[1,2-a]pyridazine-1,3(2H)-dione 8,8-dioxide 11b.
A solution of PTAD (0.19 g, 1.1 mmol, 1.2 eq.) in 1,2-DCE (5 mL) was added dropwise to a stirred solution of (E)-6-(4-methoxystyryl)-3-phenyl-1,2-oxathiine 2,2-dioxide (0.30 g, 0.9 mmol) in 1,2-DCE (10 mL) at room temperature and the mixture was stirred at room temperature overnight. Removal of the solvent and trituration of the resulting solid with AcMe afforded the title compound as a white powder; mp = 164–168 °C; vmax (neat): 1717, 1599, 1503, 1409, 1372 (O-SO2), 1256, 1180 (O-SO2), 1139, 1089, 1026 cm−1; δH (CDCl3, 400 MHz) 5.36 (q, J = 2.3 Hz, 1H, 10a-H), 5.78 (dd, J = 2.2, 5.1 Hz, 1H, 5-H), 6.31 (dd, J = 2.2, 5.1 Hz, 1H, 6-H), 6.88–6.91 (m, 2H, ArH), 7.35–7.50 (m, 11H, ArH/10-H), 7.68–7.71 (m, 2H, ArH); δC (CDCl3, 100 MHz) 54.45, 54.86, 55.31, 114.35, 116.66, 125.24, 125.86, 126.96, 128.16, 128.55, 129.20, 129.21, 129.31, 130.39, 130.40, 130.81, 141.45, 141.85, 149.99, 154.64, 160.58; HRMS: Found [M − H]− = 514.1062, C27H21N3O6S requires [M − H]− = 514.1067.
Isolation of 2,9-diphenyl-5-(4-(trifluoromethyl)phenyl)-5,10a-dihydro-1H-[1,2]oxathiino [5,6-c][1,2,4]triazolo [1,2-a]pyridazine-1,3(2H)-dione 8,8-dioxide 11c.
A solution of PTAD (0.20 g, 1.2 mmol, 1.1 eq.) in 1,2-DCE (8 mL) was added dropwise to a stirred solution of (E)-3-phenyl-6-(4-(trifluoromethyl)styryl)-1,2-oxathiine 2,2-dioxide (0.40 g, 1.1 mmol) in 1,2-DCE (10 mL) at room temperature and the mixture was stirred at room temperature overnight. Removal of the solvent gave a solid which was triturated with AcMe and n-pentane to afford the title compound as a white powder; mp = 169–172 °C; vmax (neat): 1780, 1721, 1495, 1411, 1377, 1325 (O-SO2), 1256, 1182, 1141 (O-SO2), 1111, 1090, 1058, 1021 cm−1; δH (CDCl3, 400 MHz) 5.40 (q, J = 2.3 Hz, 1H, 10a-H), 5.88 (dd, J = 5.0, 2.3 Hz, 1H, 5-H), 6.33 (dd, J = 5.0, 2.0 Hz, 1H, 6-H), 7.35–7.55 (m, 9H, ArH/10-H), 7.63–7.70 (m, 6H, ArH); δC (CDCl3, 100 MHz) 54.54, 54.59, 115.72, 123.72 (q, J = 272 Hz, CF3), 125.18, 126.16 (q, J = 3.6 Hz, o-ArC-CF3), 126.55, 128.12, 128.75, 129.11, 129.27, 129.28, 129.40, 130.17, 130.95, 131.92 (q, J = 33 Hz, C-CF3), 137.54, 141.76, 142.67, 149.85, 154.50; δF (376.5 MHz, CDCl3) −62.90 (s, 3F); HRMS: Found [M − H]− = 552.0846, C27H18F3N3O5S requires [M − H]− = 552.0843.
Dimethyl 7-(4-methoxyphenyl)-3-phenylbenzo[e][1,2]oxathiine-5,6-dicarboxylate 2,2-dioxide 13.
A mixture of DMAD (0.12 mL, 0.97 mmol, 1.1 eq.) and (E)-6-(4-methoxystyryl)-3-phenyl-1,2-oxathiine 2,2-dioxide (0.30 g, 0.88 mmol, 1.0 eq.) was heated to reflux overnight. Upon cooling the complex reaction mixture was subjected to flash column chromatography (using DCM, 0% to 10% MeOH in DCM). The fractions with an Rf = 0.9 (10% MeOH/DCM) were combined to afford the title product after trituration from petroleum spirit/Et2O 8:2 as an off-white solid (0.04 g, 10.0%) mp = 214–216 °C; νmax (neat): 2953, 1747, 1728 (CO), 1719 (CO), 1609, 1516, 1491, 1438, 1387, 1373 (O-SO2), 1329, 1275, 1247, 1217, 1180 (O-SO2), 1152, 1112, 1021 cm−1; δH (CDCl3, 400 MHz) 3.65 (s, 3H, CH3O), 3.86 (s, 3H, CO2CH3), 3.93 (s, 3H, CO2CH3), 6.96–6.98 (m, 2H, Ar–H), 7.28–7.30 (m, 2H, Ar–H), 7.41 (s, 1H, 8-H), 7.50–7.51 (m, 3H, Ar–H), 7.63 (s, 1H, 4-H), 7.67–7.69 (m, 2H, Ar–H); δC (CDCl3, 100 MHz) 52.70, 53.38, 55.37, 114.23, 117.62, 122.12, 127.18, 129.15, 129.32, 129.91, 130.27, 130.53, 130.76, 131.58, 138.58, 144.08, 151.29, 160.18, 165.94, 167.71; HRMS: Found [M + Na]+ = 503.0770, C25H20O8S requires [M + Na]+ = 503.0777.
Addition of benzyne to substituted 1,2-oxathiine 2,2-dioxides
1,4-Diphenylnaphthalene 16.
A solution of 2-(trimethylsilyl)phenyl trifluoromethanesulfonate (0.48 mL, 2.0 mmol, 1.1 eq.) in anhydrous MeCN (5 mL) was added dropwise to a stirred suspension of 3,6-diphenyl-1,2-oxathiine 2,2-dioxide (0.5 g, 1.8 mmol) and CsF (0.68 g, 4.5 mmol, 2.5 eq.) in anhydrous MeCN (25 mL) at room temperature under a N2 atmosphere. Upon completion of the addition the reaction mixture was stirred at room temperature for 2 h and then heated to reflux for 1 h. The cooled mixture was diluted with H2O (50 mL) and extracted with EtOAc (2 × 25 mL). The combined organic extracts were washed with brine (30 mL), dried over anhydrous Na2SO4 and the solvent removed. The resulting crude product was purified by column chromatography (silica, 0% to 3% EtOAc/petroleum spirit), to afford a pale-yellow solid that was triturated with n-pentane to afford the title compound as an off-white solid (0.11 g, 22%); mp = 126–129 °C [lit. mp = 132–133 °C (ref. 44)]; vmax (neat): 3052, 1598, 1573, 1512, 1488, 1469, 1446, 1384, 1238, 1154, 1071, 1028 cm−1; δH (CDCl3, 400 MHz) 7.43–7.57 (m, 14H, ArH), 7.96–8.00 (m, 2H, ArH); δC (CDCl3, 100 MHz) 125.87, 126.41, 126.48, 127.29, 128.32, 130.17, 131.92, 139.84, 140.83.
1-(4-Methoxyphenyl)naphthalene 17.
2-(Trimethylsilyl)phenyl trifluoromethanesulfonate (0.56 mL, 2.3 mmol, 1.1 eq.) was added dropwise via syringe to a stirred suspension of 6-(4-methoxyphenyl)-1,2-oxathiine 2,2-dioxide (0.5 g, 2.1 mmol) and CsF (0.51 g, 3.4 mmol, 1.6 eq.) in anhydrous MeCN (25 mL) at room temperature under a N2 atmosphere and the mixture was stirred at room temperature overnight. The reaction mixture was diluted with H2O (50 mL) and extracted with EtOAc (2 × 25 mL). The combined organic extracts were washed with brine (30 mL), dried over anhydrous Na2SO4 and the solvent removed to afford the crude product which was eluted from silica with 10% EtOAc/petroleum spirit to afford the title adduct after trituration with n-pentane as an off-white solid (0.04 g, 8%); mp = 111–114 °C [lit. mp = 112–113 °C (ref. 45)]; vmax (neat): 2991, 2954, 2831, 1607, 1513, 1504, 1461, 1451, 1437, 1422, 1393, 1282, 1239, 1207, 1173, 1143, 1107, 1057, 1030 cm−1; δH (CDCl3, 400 MHz) 3.89 (s, 3H, OMe), 7.02–7.04 (m, 2H, ArH), 7.39–7.53 (m, 6H, ArH), 7.83–7.85 (m, 1H, ArH), 7.89–7.93 (m, 2H, ArH); δC (CDCl3, 100 MHz) 55.38, 113.72, 125.42, 125.72, 125.94, 126.08, 126.92, 127.35, 128.27, 131.13, 131.83, 133.13, 133.84, 139.91, 158.94.
1,2,4-Triphenylnaphthalene 18.
A solution of 3,5,6-(triphenyl)-1,2-oxathiine 2,2-dioxide (0.50 g, 1.4 mmol) and 2-(trimethylsilyl)phenyl trifluoromethanesulfonate (0.36 mL, 1.5 mmol, 1.1 eq.) in anhydrous MeCN (10 mL) was added dropwise to a stirred suspension of CsF (0.53 g, 3.5 mmol, 2.5 eq.) in anhydrous MeCN (15 mL) at room temperature under a N2 atmosphere. The resulting reaction mixture was stirred at room temperature for 3.5 h before being poured into H2O (25 mL) and extracted with EtOAc (2 × 25 mL). The combined organic extracts were washed with brine (30 mL) dried over anhydrous Na2SO4 and the solvents removed under reduced pressure. The crude product was eluted from silica with 10% Et2O/petroleum spirit to afford the title product, after washing with n-pentane, as an off-white solid (0.16 g, 32%); mp = 159–160 °C [lit. mp = 159–161 °C (ref. 46)]; vmax (neat): 3053, 3019, 1599, 1573, 1491, 1439, 1419, 1379, 1251, 1209, 1141, 1070, 1056, 1032 cm−1; δH (CDCl3, 400 MHz) 7.13–7.35 (m, 10H, ArH), 7.40–7.61 (m, 8H, ArH), 7.74–7.76 (m, 1H, ArH), 7.99–8.01 (m, 1H, ArH); δC (CDCl3, 100 MHz) 125.76, 126.04, 126.07, 126.26, 126.79, 127.20, 127.37, 127.61, 127.88, 128.33, 130.15, 130.20, 130.94, 131.56, 133.06, 137.14, 137.89, 139.06, 139.79, 140.61, 141.84.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- S. Mondal, Chem. Rev., 2012, 112, 5339–5355 CrossRef CAS PubMed.
-
M. Sainsbury, Chapter 40: Compounds containing a Six-membered Ring having two Hetero-atoms from Group VIB of the Periodic Table: Dioxanes, Oxathianes and Dithianes, in Rodd's Chemistry of Carbon Compounds, 2nd edn, 1964, vol. IV, pp. 375–426 Search PubMed.
-
M. J. Cook, Chapter 2.26: Six-membered Rings with More than One Oxygen or Sulfur Atom, in Comprehensive Heterocyclic Chemistry, ed. A. J. Boulton and A. McKillop, 1984, vol. 3, pp. 943–994 Search PubMed.
-
E. Kleinpeter, Chapter 8.10: 1,2-Dioxins, Oxathiins, Dithiins, and their Benzo Derivatives, in Comprehensive Heterocyclic Chemistry III, ed. A. Aitken, 2008, vol. 8, pp. 677–738 Search PubMed.
- K. A. Ali, A. Jäger and P. Metz, ARKIVOC, 2016, iii, 15–22 Search PubMed.
- Th. Morel and P. E. Verkade, Recl. Trav. Chim. Pays-Bas, 1951, 70, 35–49 CrossRef CAS.
- R. P. van Summeren, B. L. Feringa and A. J. Minnaard, Org. Biomol. Chem., 2005, 3, 2524–2533 RSC.
- O. Arjona, M. L. León and J. Plumet, J. Org. Chem., 1999, 64, 272–275 CrossRef CAS PubMed.
- J. Gaitzsch, V. Rogachev, P. Metz, M. S. Yusubov, V. D. Filimonov and O. Kataeva, J. Sulfur Chem., 2009, 30, 4–9 CrossRef CAS.
- J. Gaitzsch, V. Rogachev and P. Metz, Synthesis, 2014, 46, 0531–0536 CrossRef.
-
M. Zhang and C. Han, US Patent, US20180294483A1, 2018 Search PubMed;
K. Hatta, N. Shimosaka, M. Machida, M. Aoki and M. Miyamoto, PCT Int. Appl, WO2015107910A1, 2015 Search PubMed.
-
J. H. Ko, Y. J. Ha, C. H. Lee, Y. M. Lim and J. A. Ahn, Republic of Korea Patent, KR2009084547A, 2009 Search PubMed;
T. Aoai and A. Endo, Japanese Patent, JP2004144933A, 2004 Search PubMed;
F. Urano, M. Nakahata, H. Fujie and K. Ono, Japanese Patent, JP03223865A, 1991 Search PubMed.
- S. Aiken, C. D. Gabbutt, B. M. Heron, C. R. Rice and D. Zonidis, Org. Biomol. Chem., 2019, 17, 9578–9584 RSC.
-
O. D. C. C. de Azevedo, B. M. Heron and D. Zonidis, Chap. 3 (Synthesis, Reactivity and Applications of 1,2-Oxathiine 2,2-dioxides), in Targets in Heterocyclic Systems, Italian Chemical Society, vol. 24, 2020 Search PubMed.
- G. Opitz and E. Tempel, Angew. Chem., Int. Ed. Engl., 1964, 3, 754–755 CrossRef.
- W. E. Truce, D. J. Abraham and P. Son, J. Org. Chem., 1967, 32, 990–997 CrossRef CAS PubMed.
- P. Schenone, G. Bignardi and S. Morasso, J. Heterocycl. Chem., 1972, 9, 1341–1346 CrossRef CAS.
- B. Zwanenburg, Product class 3: thioaldehyde and thioketone S,S-dioxides and oxyimides (sulfenes and derivatives), Sci. Synth., 2004, 27, 123–134 CAS.
- S. Aiken, K. Anozie, O. D. C. C. de Azevedo, L. Cowen, R. J. L. Edgar, C. D. Gabbutt, B. M. Heron, P. A. Lawrence, A. J. Mills, C. R. Rice, M. W. J. Urquhart and D. Zonidis, Org. Biomol. Chem., 2019, 17, 9585–9604 RSC.
- P. Singh, K. Bisetty and M. P. Mahajan, S. Afr. J. Chem., 2009, 62, 47–55 CAS.
- H. M. L. Davies and D. Morton, J. Org. Chem., 2016, 81, 343–350 CrossRef CAS PubMed; I. A. Stepek and K. Itami, ACS Mater. Lett., 2020, 2, 951–974 CrossRef; A. M. Prendergast and G. P. McGlacken, Eur. J. Org. Chem., 2018, 6068–6082 CrossRef.
- S. Martins, P. S. Branco, M. C. de la Torre, M. A. Sierra and A. Pereira, Synlett, 2010, 2918–2922 CAS.
- X. Lei, L. Gao, Q. Ding, Y. Peng and J. Wu, Org. Biomol. Chem., 2011, 9, 6265–6270 RSC.
- T. Ishiyama, M. Murata and N. Miyaura, J. Org. Chem., 1995, 60, 7508–7510 CrossRef CAS.
- T. Yamada, H. Takiguchi, K. Ohmori and K. Suzuki, Org. Lett., 2018, 20, 3579–3582 CrossRef CAS PubMed.
- H. A. Duong, S. Chua, P. B. Huleatt and C. L. L. Chai, J. Org. Chem., 2008, 73, 9177–9180 CrossRef CAS PubMed.
- J. Gaitzsch, V. O. Rogachev, M. Zahel and P. Metz, Synthesis, 2014, 46, 0531–0536 CrossRef.
- J. Gaitzsch, V. O. Rogachev, P. Metz, M. S. Yusubov, V. D. Filimonov and O. Kataeva, J. Sulfur Chem., 2009, 30, 4–9 CrossRef CAS.
- K. De Bruycker, S. Billiet, H. A. Houck, S. Chattopadhyay, J. M. Winne and F. E. Du Prez, Chem. Rev., 2016, 116, 3919–3974 CrossRef CAS PubMed; C. J. Moody, Adv. Heterocycl. Chem., 1982, 30, 1–45 CrossRef; S. Rádl, Adv. Heterocycl. Chem., 1996, 67, 119–205 CrossRef; D. Craig, S. R. J. Spreadbury and A. J. P. White, Chem. Commun., 2020, 56, 9803–9806 RSC.
- C. D. Gabbutt, J. D. Hepworth and B. M. Heron, Tetrahedron, 1995, 51, 13277–13290 CrossRef CAS.
- L. Liao, R. An, H. Li, Y. Xu, J.-J. Wu and X. Zhao, Angew. Chem., 2020, 59, 11010–11019 CrossRef CAS PubMed.
- S. Akiyama, S. Nomura, K. Kubota and H. Ito, J. Org. Chem., 2020, 85, 4172–4181 CrossRef CAS PubMed.
- I. Dissanayake, J. D. Hart, E. C. Becroft, C. J. Sumby and C. G. Newton, J. Am. Chem. Soc., 2020, 142, 13328–13333 CrossRef CAS PubMed.
- Z. Zhou, J. Chen, H. Chen and W. Kong, Chem. Sci., 2020, 11, 10204–10211 RSC.
- P. Mondal, G. Jana, P. K. Behera, P. K. Chattaraj and N. K. Singha, Macromolecules, 2020, 53, 8313–8323 CrossRef CAS.
- Y. Himeshima, T. Sonoda and H. Kobayashi, Chem. Lett., 1983, 1211–1214 CrossRef CAS; J.-A. Garcia-López and M. F. Greaney, Chem. Soc. Rev., 2016, 45, 6766–6798 RSC; A. V. Dubrovskiy, N. A. Markina and R. C. Larock, Org. Biomol. Chem., 2013, 11, 191–218 RSC.
- W. M. Weber, L. A. Hunsaker, S. F. Abcouwer, L. M. Deck and D. L. Vander Jagt, Biorg. Med. Chem., 2005, 13, 3811–3820 CrossRef CAS.
- S. Paul and M. Gupta, Synth. Commun., 2005, 35, 213–222 CrossRef CAS.
- K. R. Buszek and N. Brown, Org. Lett., 2007, 9, 707–710 CrossRef CAS PubMed.
- P. S.-W. Leung, Y. Teng and P. H. Toy, Org. Lett., 2010, 12, 4996–4999 CrossRef CAS.
- D. Kumar, D. N. Kommi, P. Chopra, M. I. Ansari and A. K. Chakraborti, Eur. J. Org. Chem., 2012, 6407–6413 CrossRef CAS.
- S. Zhou, B.-W. Yan, S.-X. Fan, J.-S. Tian and T.-P. Loh, Org. Lett., 2018, 20, 3975–3979 CrossRef CAS.
- X. Liang, P. Guo, W. Yang, M. Li, C. Jiang, W. Sun, T.-P. Loh and Y. Jiang, Chem. Commun., 2020, 56, 2043–2046 RSC.
- J. G. Smith and R. B. McCall, J. Org. Chem., 1980, 45, 3982–3986 CrossRef CAS.
- G. A. Molander and Fl. Beaumard, Org. Lett., 2010, 12, 4022–4025 CrossRef CAS.
- R. C. Larock and Q. Tian, J. Org. Chem., 1998, 63, 2002–2009 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob01125a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
B. Mark
Heron
,
B. Mark
Heron