
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and stability of 1-aminoalkylphosphonic acid quaternary ammonium salts†
Anna
Brol
 and
Tomasz K.
Olszewski
*
and
Tomasz K.
Olszewski
*
Faculty of Chemistry, Wrocław University of Science and Technology, ul. Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland. E-mail: tomasz.olszewski@pwr.edu.pl
First published on 7th May 2021
Abstract
An effective protocol for the quaternization of simple 1-aminoalkylphosphonic acids under basic conditions using Me2SO4 as a convenient alkylating agent is reported. During the course of the reaction, phosphonic acid quaternary ammonium derivatives, along with their corresponding monoesters are formed. Subsequent direct acidic hydrolysis of the crude reaction mixture leads to the desired novel N,N,N-trialkyl-N-(1-phosphonoalkyl)ammonium salts with overall yields of up to 88%. The developed protocol is general in scope and the products are purified by simple crystallization to give stable solids. Novel quaternary ammonium salts bearing a phosphonic group are generally unreactive in acidic and alkaline media. However, some of them undergo Hofmann elimination and substitution reactions in the presence of a base.
Introduction
Quaternary ammonium compounds are well known to have various applications,1 and are often employed as surfactants, fabric softeners,2 electrolytes3 and supramolecular structures,4 and also in organic synthesis as solvents, catalysts or starting materials.5 However, their biological activity as herbicides and pesticides6 and most importantly their activity against a wide range of bacteria, viruses, yeasts, and fungi are the most likely reason for the growing interest in this class of compounds.7 Among the known quaternary ammonium compounds, the organophosphorus based derivatives represent an interesting subclass endowed with equally interesting properties.8 Simple N,N,N-trialkyl-N-(1-phosphonoalkyl)-ammonium salts, as well as their longer alkyl chain homologs comprising of a pentavalent phosphorus atom (RP(O)(OH)2) attached to a sp3 carbon atom spacer adjacent to a quaternary ammonium group, have found applications as petroleum recovery agents,9 fungicides10 and for the treatment of abnormal calcium and phosphate metabolism.11 They are also successfully used as scale inhibitors,12 phase transfer catalysts (PTC)13 and antimicrobial agents (Fig. 1).14 It is worth mentioning that there has recently been a high demand for non-leaching antimicrobial materials and monolayer coatings containing biocidal quaternary ammonium compounds with anchors, such as phosphonates, allowing for attachment to metal oxide surfaces to further enhance infection control efforts. This application is particularly important in institutional and hospital environments in which metal surfaces often come into contact with potentially pathogenic organisms.8,15 Owing to the important applications of phosphonic acid quaternary ammonium compounds, their preparation in an effective manner plays a crucial role. The synthesis of long chain substituted phosphonic acid quaternary ammonium compounds is quite well described in the literature and is based on three step combination of the Arbuzov reaction followed by a quaternization involving reaction between a tertiary amine and an alkyl halide (the Menshutkin reaction)16 and finally bis-dealkylation of the resulting quaternary ammonium phosphonate esters (Scheme 1a).15 Surprisingly there are very few structures of simple N,N,N-trialkyl-N-(1-phosphonoalkyl)ammonium salts reported in the literature and their syntheses are rarely described. Quinlan reported the quaternization of various cyclic aminophosphonic acids with methyl iodide by heating for 8 h in ethanol, leading to the desired quaternary ammonium derivatives, however, no yields or spectroscopic characterisation data were provided (Scheme 1b).17 Medved and Kabachnik reported that the quaternization of (1-amino-1-methylethyl)phosphonic acid with methyl iodide by heating in methanol in a sealed tube at 130 °C does not produce the desired product. In turn heating above that temperature led to decomposition of the starting material and isolation of tetramethylammonium iodide and phosphoric acid (Scheme 1c).18 The authors briefly mentioned the use of dimethyl sulfate, but the product of quaternization was isolated with only a 27% yield. In turn, Redmore demonstrated that quaternization of N,N-dialkylaminomethylphosphonic acid esters can be performed with methyl iodide, and subsequent dealkylation of the phosphonic acid esters leads to the desired phosphonic acid quaternary ammonium derivatives, however, the yields were not reported and the description concerns only derivatives of the simplest acid, aminomethylphosphonic acid (Scheme 1d).12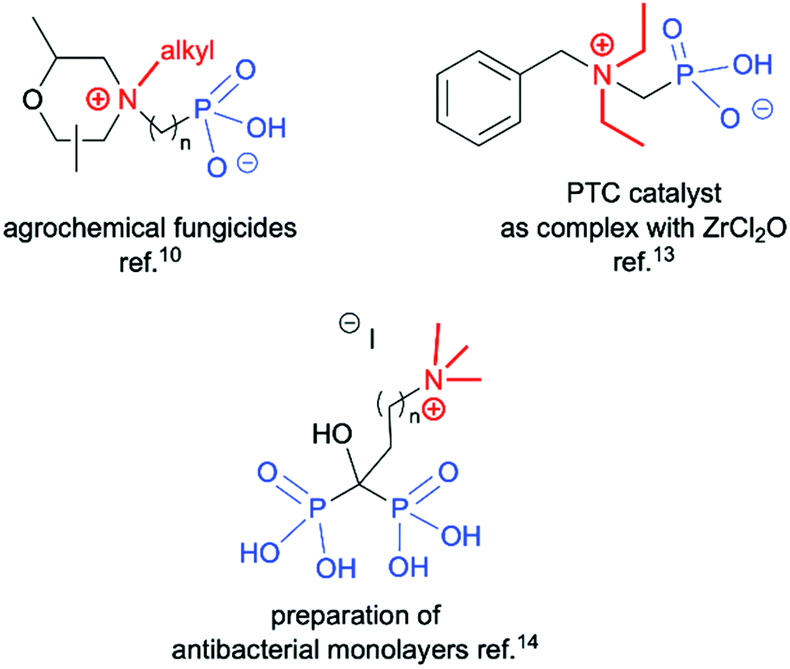 | ||
| Fig. 1 Representative structures of organophosphorus based quaternary ammonium compounds with potential applications. | ||
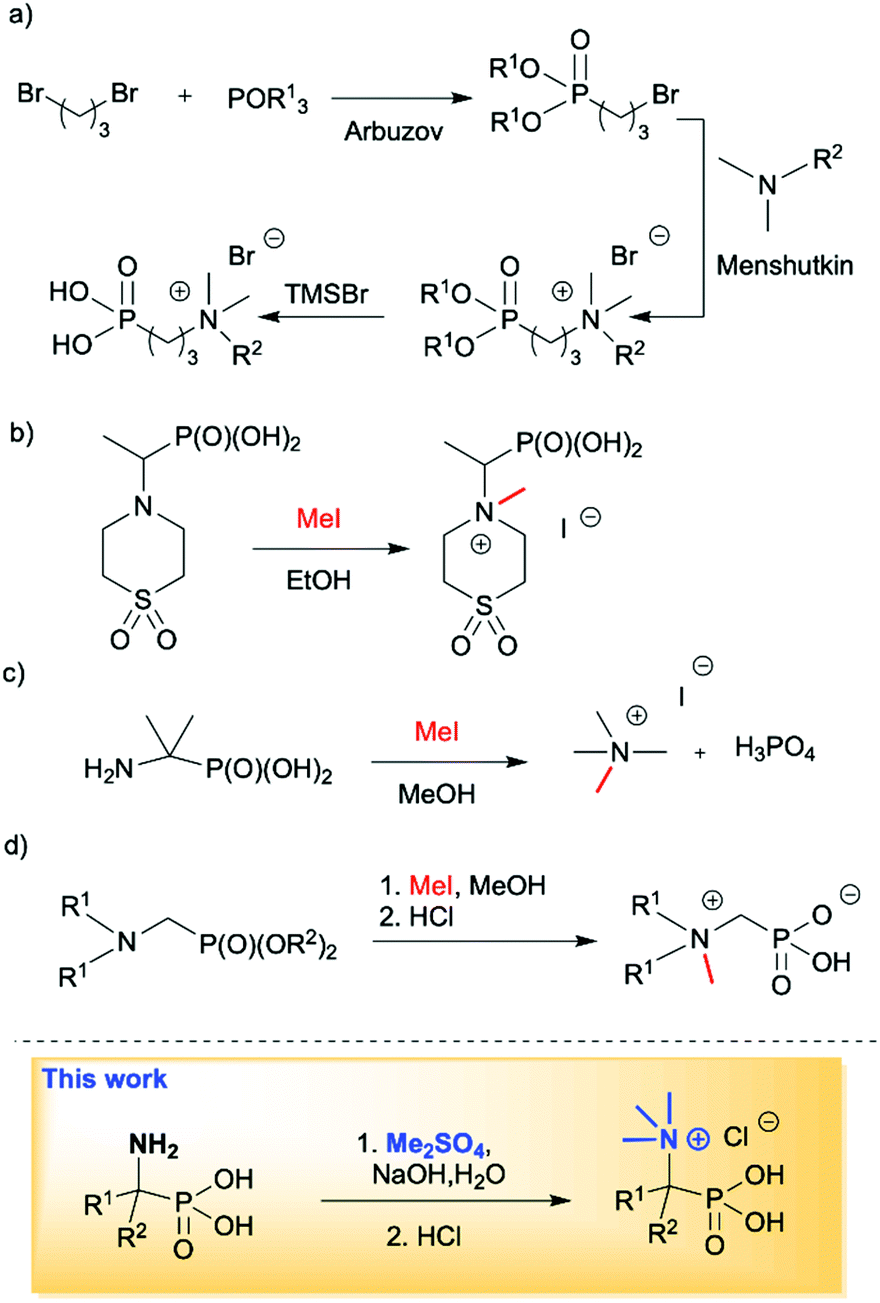 | ||
| Scheme 1 Known methods leading to phosphonic acid quaternary ammonium compounds. | ||
Our continuous interest in the preparation of novel organophosphorus compounds and their applications19 prompted us to investigate effective methods leading to novel phosphonic acid quaternary ammonium compounds. Herein, we report the results of our study on the development of an effective protocol for quaternization of easily available 1-aminoalkylphosphonic acids with dimethyl sulphate, as an effective alkylating reagent, under basic conditions and leading, after dealkylation under acidic conditions, to novel phosphonic acid quaternary ammonium compounds with high yields. Additionally, we present the results of our studies on the stability of this subclass of quaternary ammonium compounds in acidic and alkaline media.
Results and discussion
Synthesis of novel N,N,N-trimethyl-N-(1-phosphonoalkyl)ammonium salts
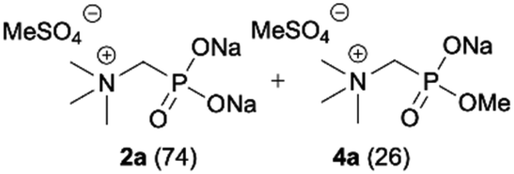
We started our experimentations by examining whether the procedures found in the literature can be applied for the quaternization of 1-aminoalkylphosphonic acids (Scheme 1). Quinlan reported the quaternization of cyclic aminophosphonic acids (bearing a tertiary nitrogen atom) by heating in ethanol and water with simple alkylating agents such as benzyl chloride or methyl iodide, however, this method failed in our case.17 Only the starting material was recovered when 1-(dimethylamino)ethylphosphonic acid was used as a substrate, even when the reaction medium was alkalized (Scheme 2a). Therefore, we decided to change the starting material to 1-aminophosphonic acid 1a, bearing a free amino group and to use methyl iodide or dimethyl sulfate (Me2SO4) as alkylating agents to examine the protocol reported by Kabachnik and Medved (Scheme 2b–e).18 Preliminary experiments with MeI performed in a mixture of boiling ethanol and water for 4 h did not produce the desired product of quaternization (Scheme 2b). Alkalization of the reaction mixture with NaOH and the use of an excess of volatile methyl iodide led to quaternization of the nitrogen atom with a 91% yield (Scheme 2c). In turn, with dimethyl sulfate and neutral conditions, only product 3 was observed as a result of the mono-alkylation of the phosphonic acid group (Scheme 2d). However, in alkaline medium, the desired phosphonic acid quaternary ammonium salt was generated and accompanied by the product of mono-alkylation [O-alkylation] 4a (Scheme 2e). The latter could be easily dealkylated under acidic conditions and thus converted into the desired phosphonic acid quaternary ammonium salt.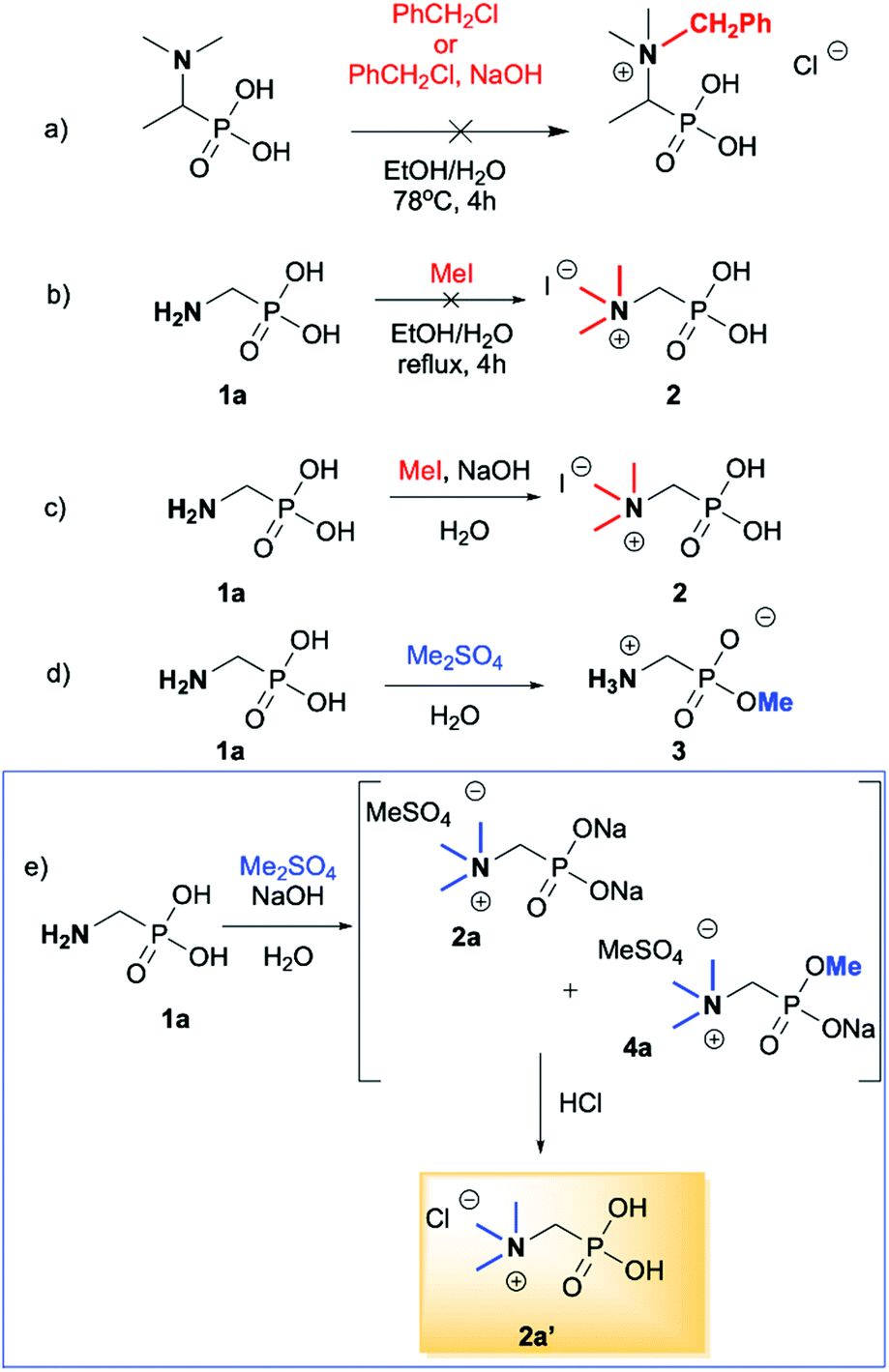 | ||
| Scheme 2 Evaluation of the reaction conditions of the quaternization reaction. | ||
Despite the fact that both methods gave positive results, we decided to use the protocol employing less volatile dimethyl sulfate. We found the iodide ion unacceptable for further research as it exhibits strong reducing properties and its removal requires laborious operations (precipitation of AgI with Ag2O or the use of ion-exchange resins) while methyl sulfates are easier to remove by hydrolysis and the precipitation of BaSO4.
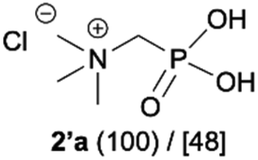
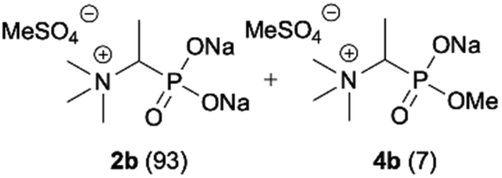
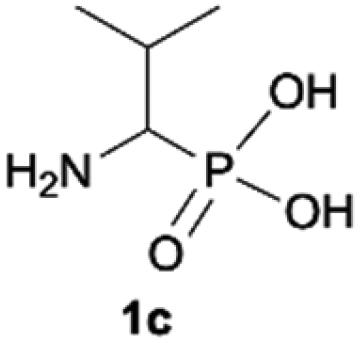
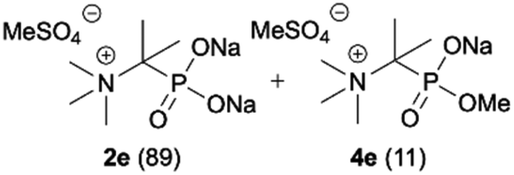
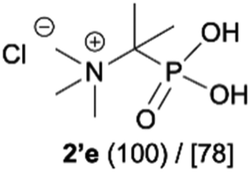
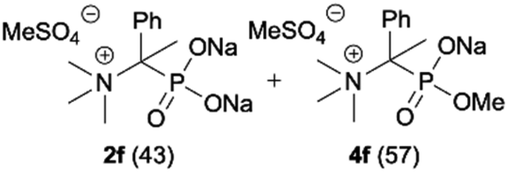
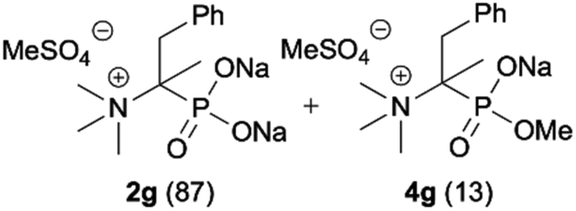
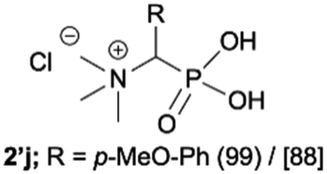
During optimization of the quaternization reaction we observed that an excess of a base and dimethyl sulfate were required to ensure successful quaternization. Furthermore, the mono-alkylated by-product was indeed easily dealkylated in the presence of HCl and converted to the desired phosphonic acid quaternary ammonium salt. Under these conditions a set of the desired phosphonic acid quaternary ammonium compounds was obtained with yields of up to 88%. The developed protocol was found to be quite general and structurally different, 1-aminoalkylphosphonic acids 1a–j undergoes successful quaternization with only slight modification of the optimized conditions (Table 1). In general, the 1-aminoalkylphosphonic acids 1 were reacted with NaOH and Me2SO4 in aqueous solution in a ratio of 1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5.0:4.0 over 48 h at room temperature (Table 1). After that time, for simple unhindered substrates a full conversion of the starting material was observed and the reaction led predominantly to the desired phosphonic acid quaternary ammonium compounds 2 (Table 1, entries 1, 2, 4, 5, 7 and 10), for example, phosphonic analogues of glycine (Table 1, entry 1), alanine (Table 1, entry 2) or phenylglycine (Table 1, entry 4), and only around 20% of the monoesters 4 were detected. However, in the case of more sterically hindered, labile substituents present in substrates 1, for example, the phosphonic analogue of the valine (Table 1, entry 3) reaction could not reach completion unless an additional portion of NaOH and Me2SO4 was added (Table 1, entries 3, 6, 8, 9).20 This could be explained by the fact that the rising steric hindrance in the substrate results in a reduced N-alkylation reaction rate, while the reaction rate of Me2SO4 hydrolysis to MeOH and MeSO4H remains the same. After completion of the quaternization reaction the mixture composed of phosphonic acid quaternary ammonium compounds 2 and the corresponding monoesters 4 were directly submitted for dealkylation under acidic conditions to produce the desired phosphonic acid quaternary ammonium compounds 2′ accompanied by MeOH and H2SO4. We have found that all the prepared compounds were stable under acidic conditions and heating with 4 M HCl (4 h, 100 °C) did not lead to any decomposition. H2SO4 was conveniently removed by the addition of BaCl2 and separation of the precipitated BaSO4. Subsequent removal of NaCl, evaporation of the filtrate and crystallization of the crude product from EtOH and Et2O resulted in isolation of the pure phosphonic acid quaternary ammonium compounds 2′a–j with good to high yields (40–88%). In comparison with the protocol reported by Kabachnik and Medved,18 our method leads to significantly higher yields of the isolated quaternary compounds 2′.
:5.0:4.0 over 48 h at room temperature (Table 1). After that time, for simple unhindered substrates a full conversion of the starting material was observed and the reaction led predominantly to the desired phosphonic acid quaternary ammonium compounds 2 (Table 1, entries 1, 2, 4, 5, 7 and 10), for example, phosphonic analogues of glycine (Table 1, entry 1), alanine (Table 1, entry 2) or phenylglycine (Table 1, entry 4), and only around 20% of the monoesters 4 were detected. However, in the case of more sterically hindered, labile substituents present in substrates 1, for example, the phosphonic analogue of the valine (Table 1, entry 3) reaction could not reach completion unless an additional portion of NaOH and Me2SO4 was added (Table 1, entries 3, 6, 8, 9).20 This could be explained by the fact that the rising steric hindrance in the substrate results in a reduced N-alkylation reaction rate, while the reaction rate of Me2SO4 hydrolysis to MeOH and MeSO4H remains the same. After completion of the quaternization reaction the mixture composed of phosphonic acid quaternary ammonium compounds 2 and the corresponding monoesters 4 were directly submitted for dealkylation under acidic conditions to produce the desired phosphonic acid quaternary ammonium compounds 2′ accompanied by MeOH and H2SO4. We have found that all the prepared compounds were stable under acidic conditions and heating with 4 M HCl (4 h, 100 °C) did not lead to any decomposition. H2SO4 was conveniently removed by the addition of BaCl2 and separation of the precipitated BaSO4. Subsequent removal of NaCl, evaporation of the filtrate and crystallization of the crude product from EtOH and Et2O resulted in isolation of the pure phosphonic acid quaternary ammonium compounds 2′a–j with good to high yields (40–88%). In comparison with the protocol reported by Kabachnik and Medved,18 our method leads to significantly higher yields of the isolated quaternary compounds 2′.
|
|
|||
|---|---|---|---|
| Entry | Substrate | Quaternization reaction composition 2c (%) + 4c (%) | Dealkylation product 2′ conversionc (%)/yieldd [%] |
| a Reaction conditions for quaternization: 1-aminoalkylphosphonic acid 1 (5 mmol), aq. NaOH (25 mmol), Me2SO4 (20 mmol), 48 h at room temperature. Dealkylation was performed with aqueous 12 M HCl at 100 °C for 4 h. b Six equivalents of both NaOH and Me2SO4 were used. c Calculated based on 31P NMR spectra of crude reaction mixture. d Yield of the pure isolated product. | |||
| 1 |
|
|
|
| 2 |
|
|
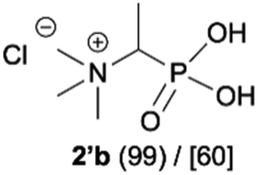
|
| 3b |
|
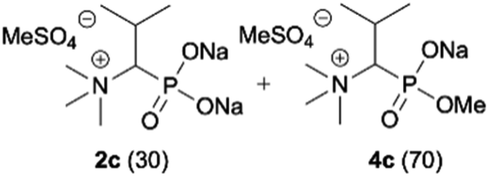
|
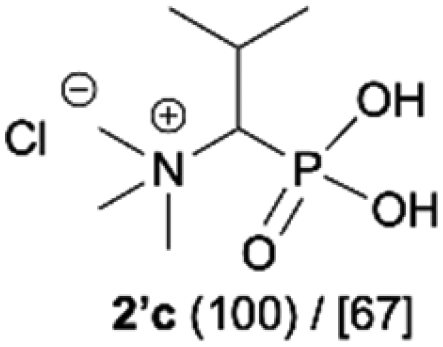
|
| 4 |
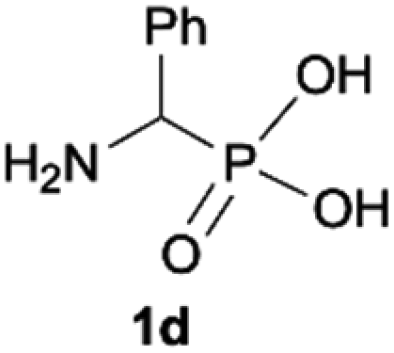
|
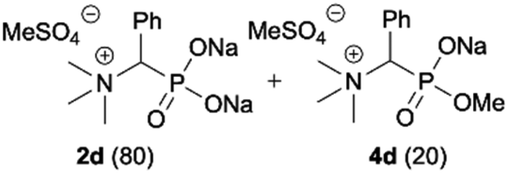
|
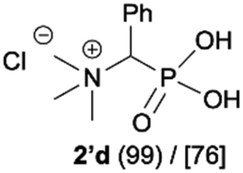
|
| 5 |
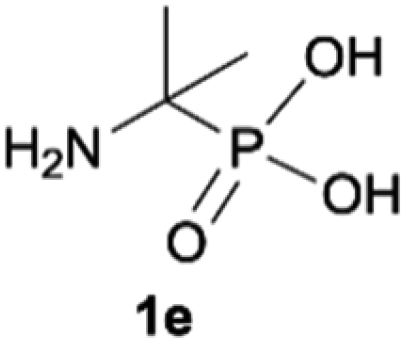
|
|
|
| 6b |
|
|
|
| 7 |
|
|
|
| 8b |
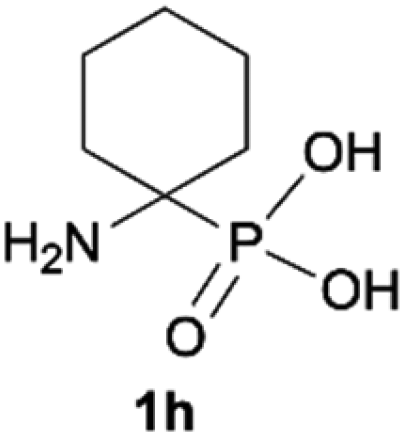
|
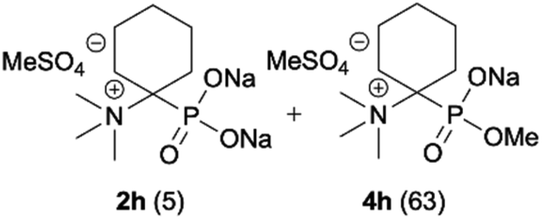
|
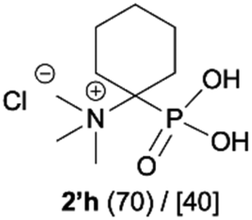
|
| 9 |
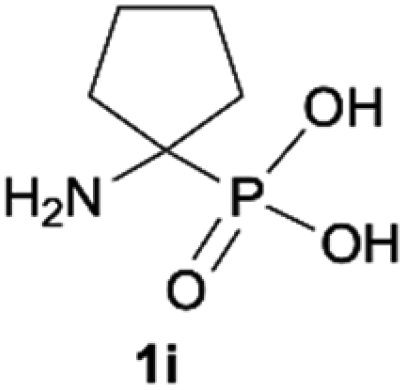
|
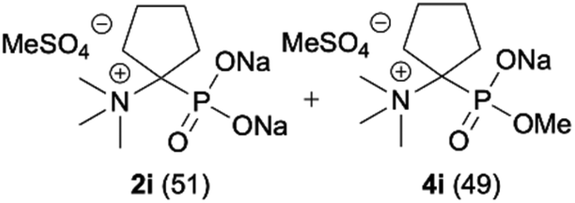
|
|
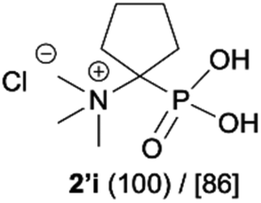
| 10 |
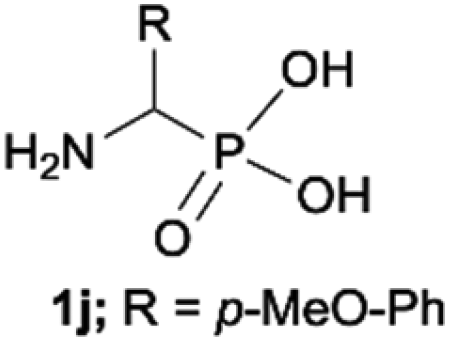
|
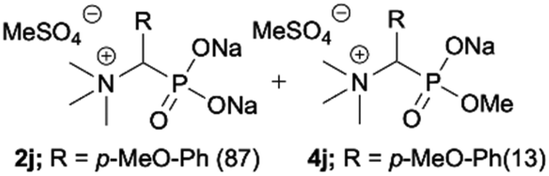
|
|
Stability studies of the novel N,N,N-trimethyl-N-(1-phosphonoalkyl) ammonium salts
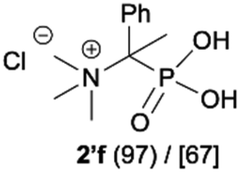
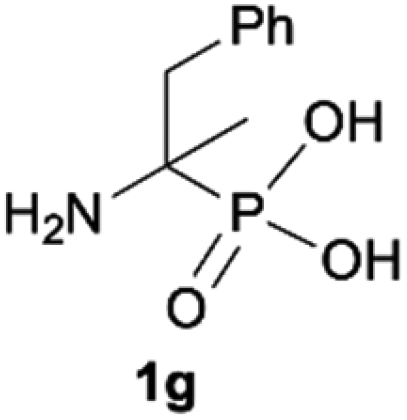
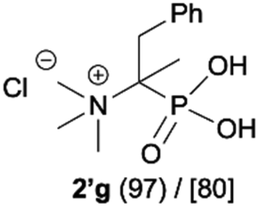
As most of the phosphonic acid quaternary ammonium compounds 2′ synthesised herein have not been described in the literature previously, their stability in alkaline media was tested after their isolation. Quaternary ammonium compounds 2′a–2′e were stable under prolonged exposure to NaOH (100 °C, 35 h, aq. 3.3 M NaOH). However, in the case of compounds having β-protons such as 2′f, 2′g and 2′h decomposition products were clearly observed under these conditions, such as olefins and hydroxyphosphonates, as a result of Hofmann elimination and additional nucleophilic substitution (Scheme 3).21 Likewise, for compounds 2′i and 2′j, detectable by NMR, traces of decomposition products were also observed (1% and 7% respectively).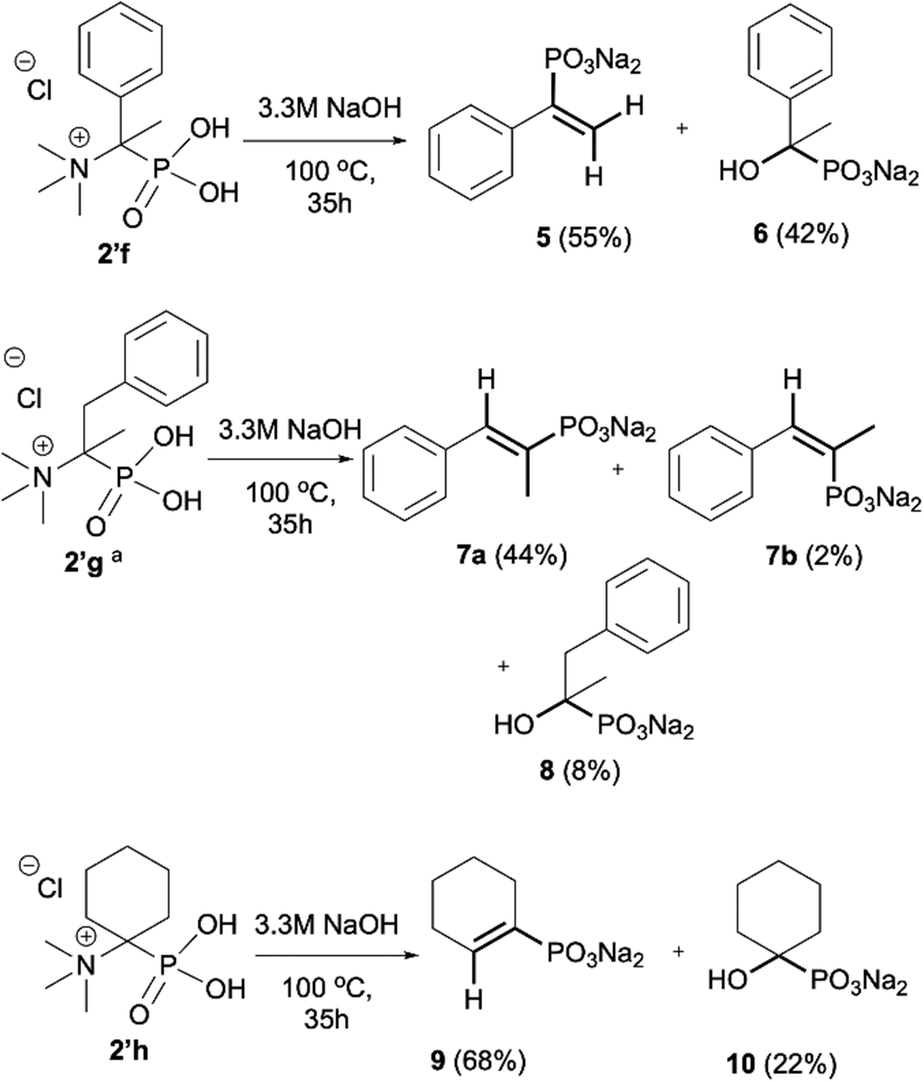 | ||
| Scheme 3 Degradation of quaternary ammonium compounds 2′f, 2′g and 2′h in the presence of NaOH (100 °C, 35 h). Conversion calculated based on the 31P NMR spectra of the crude reaction mixture. aConversion of 2′ was 57%. | ||
The Hofmann elimination is often used in organic synthesis for the transformation of quaternary ammonium salts into olefins,22 however, scarce reports in the literature describe Hofmann elimination in the case of the quaternary ammonium phosphonates23 and to the best of our knowledge, this is the first report describing Hofmann elimination and nucleophilic substitution in the case of quaternary ammonium phosphonic acids.
The formation of elimination products 5, 7a, 7b and 9 from phosphonic acid quaternary ammonium compounds 2′f, 2′g and 2′h was unambiguously confirmed using NMR analysis and compared with the literature data.24,25 The presence of olefins was confirmed by 31P NMR registered without decoupling from 1H and the formation of the corresponding multiplets, for example, for olefin 5 (δP 10.41, doublet of doublets, 3JHP(cis) = 18.3 Hz, 3JHP(trans) = 36.0 Hz),24a for 7a (δP 14.90, doublet of quartets, 3JHP(cis) = 20.6 Hz, 3JHP = 13.1 Hz) and 7b (δP 14.90, doublet of quartets, 3JHP(trans) = 37.4 Hz, 3JHP = 10.7 Hz)24b and for 9 (δP 10.23, doublet, 3JHP(cis) = 19.6 Hz).24a This assignment was additionally confirmed using 1H NMR in which signals from the olefinic protons clearly appeared, for example, for olefin 5 (δH 5.54, JHH = 1.2 Hz, 3JHP(cis) = 18.3 Hz; 5.34, JHH = 1.2 Hz, 3JHP(trans) = 36.7 Hz),24a for 7a (δH 6.72, 3JHP(cis) = 20.8 Hz) and 7b (δH 6.41, 3JHP(trans) = 37.6 Hz)24b and for 9 (δH 5.88, 3JHP(cis) = 19.0 Hz).24a
Additionally, we have clearly observed the presence of hydroxy phosphonates 6, 8, 10 as a result of the nucleophilic substitution.25,26 From the literature, the classical examples of nucleophilic substitution under the conditions of Hofmann elimination include transformation of gramine methiodide into 3-hydroxymethylindole26a or the synthesis of hydroxymethyl-ferrocene.26b This process is believed to follow the SN2 mechanism (Scheme 4, left).26d However, in our case, salts 2′f, 2′g and 2′h are significantly crowded, not only owing to the presence of bulky trimethylamine groups, but also because of the tetrahedral, negatively charged phosphonic group. Taking into consideration these steric and electronic effects, nucleophilic access (OH−) to both the β-protons and α-carbon will be difficult. These arguments, along with similar examples described previously in the literature for non-phosphorylated compounds,27 led us to propose an alternative mechanism, in which a shift from SN2 to SN1 occurs and the first step is the dissociation of the C–N bond in compound 2′′f and the formation of the carbocation 11 (Scheme 4). The carbocation 11 may in turn undergo one of the typical reactions, namely the addition of a nucleophile to form 1-hydroxyalkylphosphonic acid 6 or the elimination of a proton to yield a derivative of vinylphosphonic acid 5. The SN1 mechanism seems to be plausible, as it explains the formation of both reaction products. Additionally, carbocations derived from 2′f, 2′g and 2′h may be stabilized by delocalization or hyperconjugation, unlike carbocations derived from other salts 2′ (Scheme 4, right).
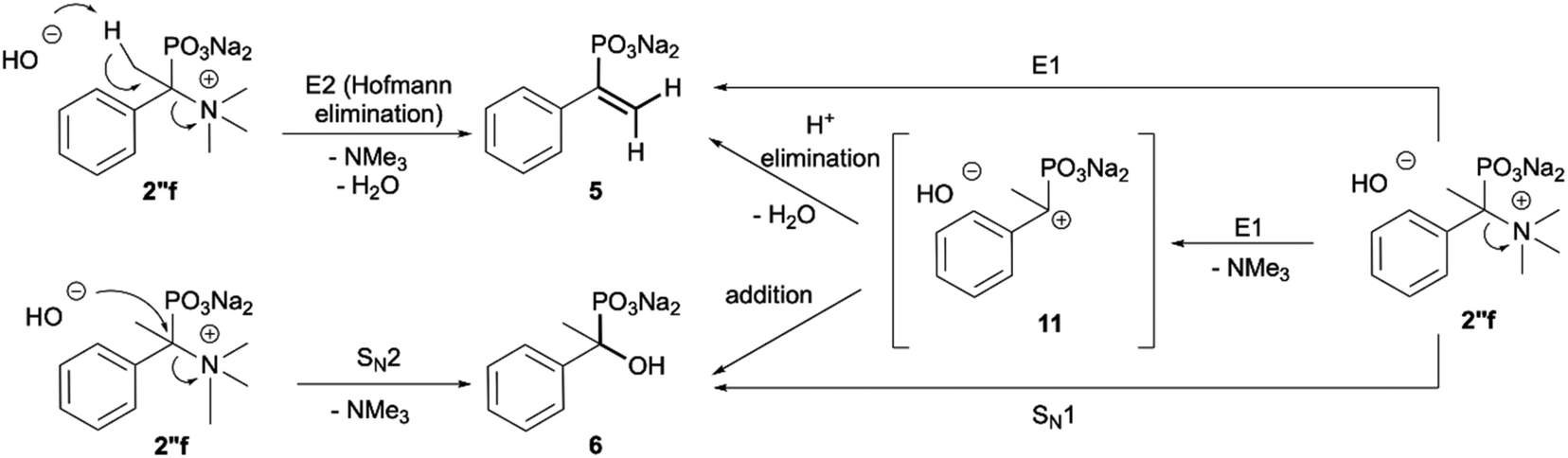 | ||
| Scheme 4 Classical (E2 and SN2; left) and carbocationic (right) mechanisms of degradation of the phosphonic acid quaternary ammonium compounds using the example of 2′′f. | ||
Conclusions
We have reported a simple and high yielding two-step one-pot method for the quaternization of 1-aminoalkylphosphonic acids 2′, for example, phosphonic analogues of glycine, alanine, phenylalanine and valine, with the use of dimethyl sulfate under aqueous alkaline conditions. Although quaternization is accompanied by unwanted alkylation of the phosphonic acid group, the undesired monoesters undergo easy dealkylation after treatment of the crude reaction mixture with aqueous HCl. The desired quaternary ammonium compounds 2′ were conveniently isolated and purified by crystallization. Most of the obtained compounds were synthesised and fully characterised for the first time. All the obtained compounds were stable under acidic conditions. In the presence of NaOH, most of the synthesized salts were unreactive; however, compounds 2′f, 2′g and 2′h underwent elimination resulting in the formation of the corresponding olefins and, surprisingly, nucleophilic substitution yielding 1-hydroxyalkylphosphonates. To the best of our knowledge, this is the first report describing Hofmann elimination and nucleophilic substitution in the case of quaternary ammonium phosphonic acids. A carbocation with a phosphonic group was proposed to be the reactive intermediate formed during the course of the degradation of the phosphonic acid quaternary ammonium compounds.Experimental section
Materials and methods
Solvents and NaOH were purchased from Chempur and Stanlab and used without purification. Dimethyl sulfate and deuterium oxide were purchased from Sigma-Aldrich. Reactions that required heating were performed on a heating mantle or in heating block apparatus with external temperature control. The 1H, 13C{1H} and 31P NMR spectra were collected on a Jeol 400yh instrument (400 MHz for 1H NMR, 162 MHz for 31P NMR and 100 MHz for 13C NMR) and were processed with dedicated software (Delta 5.0.5). NMR experiments recorded in D2O were referenced to the respective residual 1H signal of the solvent. Multiplicities are reported using the following abbreviations: s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). The reported J values are those observed from the splitting patterns in the spectrum and may not reflect the true coupling constant values. High resolution mass spectra were collected using electrospray ionization on a Waters LCT Premier XE TOF instrument. As all compounds 2′ melted with vigorous decomposition the melting/decomposition points were determined using a constant 5 °C min−1 rate using Digimelt Apparatus.Synthesis of starting materials
1-Aminoalkylphosphonic acids 1a–1j were obtained using Sorokas’ protocol28 and the reaction of an appropriate carbonyl compound with acetamide, acetyl chloride and PCl3 in acetic acid.Experimental procedures
Caution: Dimethyl sulfate is extremely toxic. Contact with the liquid or inhaling the vapor should be avoided. This reagent should be handled with great caution and all actions should be performed under a fume hood.Synthesis of quaternary ammonium methylsulfates 2 and 4
Method A: The appropriate 1-aminoalkylphosphonic acid 1 (5.0 mmol) was dissolved in NaOH solution (25 mmol, 1.00 g in 8 ml of water) and stirred for 10 min. Subsequently, Me2SO4 (20 mmol, 2.52 g, 1.89 ml) was added dropwise over 3 min. The initially two-phase mixture was homogenized after 60 min. Stirring was continued for 48 h at 20 °C. The progress of the reaction was controlled by means of 31P NMR.Method B: The appropriate 1-aminoalkylphosphonic acid 1 (5.0 mmol) was dissolved in NaOH solution (30 mmol, 1.20 g in 10 ml of water) and stirred for 10 min. Subsequently, Me2SO4 (30 mmol, 3.78 g, 2.84 ml) was added dropwise over 3 min. The initially two-phase mixture was homogenized after 60 min. Stirring was continued for 48 h at 20 °C. The progress of the reaction was controlled by means of 31P NMR. If the conversion was not satisfactory, another portion of the NaOH solution (10 mmol, 0.40 g, in 2 ml of water) was added to a stirred solution and after 10 minutes Me2SO4 (10 mmol, 1.26 g, 0.95 ml) was added dropwise. The progress was monitored by means of 31P NMR. This step was repeated until the result was satisfactory (full conversion of the substrate).
Hydrolysis of quaternary ammonium methylsulfates 2 and 4 and isolation of 2′a–2′j
The reaction mixture was added to aq. 12 M HCl (1 ml of acid per 1 ml of mixture) and subsequently refluxed for 4 h. After that, the reaction was cooled down and an aqueous solution of BaCl2 (1.0 mmol per 1.0 mmol of Me2SO4) was added dropwise. After 1 h, the precipitated BaSO4 was removed by centrifugation (5000 rpm, 5 min), washed with water and centrifuged again. The combined aqueous layers were evaporated under reduced pressure to dryness. EtOH (10 ml, 99.8%) was added to the resulting semisolid residue and the mixture was refluxed for 3 min. After cooling, the precipitated NaCl was removed by suction and washed with EtOH (99.8%) (4 × 3 ml). The collected filtrates were evaporated under reduced pressure, yielding the crude phosphonic acid quaternary ammonium derivatives 2′. The crude products 2′ were purified by crystallization from EtOH (99.8%) (1.5 ml of EtOH per 1.0 g of crude product) and precipitated by the addition of Et2O (4.5 to 6.0 ml) and cooling at −20 °C. The precipitated products were filtered off, washed with cold Et2O (4 × 2 ml) and dried in vacuo.Testing the stability of compounds 2′ in alkaline medium
A solution of N,N,N-trialkyl-N-(1-phosphonoalkyl)ammonium salt 2′ (0.15 mmol) in 3.3 M NaOH in D2O (2.0 mmol, 0.60 ml) was heated at 100 °C for 35 h. An inorganic solid was precipitated in the test tube. Subsequently, 1H and 31P NMR spectra were recorded. Afterwards, reference materials (corresponding hydroxy-phosphonates) were added, and the spectra were recorded again. In the case of salt 2′h, a crude product containing 6%mol of phosphonic acid, was used.Identification of degradation products
The degradation products (olefins) were identified by comparison of the chemical shifts in the 1H and 31P NMR spectra with values reported in the literature for exact or similar structures and the formation of 1-hydroxyalkylphosphonic acids 6 and 10 was confirmed by the addition of prepared standards.Synthesis of 1-hydroxyalkylphosphonic acids used as reference materials (standards)
1-Hydroxy-1-(phenyl)ethylphosphonic acid (6)30 was obtained using the method previously reported by Sekines’ et al.,31 starting from tris(trimethylsilyl)phospite and acetophenone (1.20 g, 10.0 mmol). Crude 6 (contaminated with 12%mol of phosphonic acid and 5%mol of phosphoric acid) was obtained (1.59 g, 72% yield) as a white solid. Crystallization attempts failed, therefore crude 6 was used as a reference material. 1H NMR (17 mg, 0.55 ml D2O): δ 7.36–7.44 (m, 2H), 7.14–7.29 (m, 3H), 1.62 (d, J = 15.3 Hz, 3H). 31P NMR (17 mg, 0.55 ml D2O): δ 23.51 (quartet, J = 15.3 Hz). 13C{1H} NMR (27 mg, 0.55 ml D2O): δ 141.3, 128.3, 128.3, 127.7 (d, J = 2.9 Hz), 126.0, 125.9, 73.2 (d, J = 159.7 Hz), 24.0 (d, J = 3.5 Hz). HRMS (TOF-ES+) calcd for C8H11O4P [M + H]+m/z: 203.0473, found: 203.0478.1-Hydroxycyclohexylphosphonic acid (10)24a was synthesized according to Goldeman and Sorokas’ protocol and subsequently dealkylated by heating in 8M HCl (100 °C, 4 hours).32 Compound 10 was obtained (1.20 g, 70% isolated yield) as a white solid. 1H NMR (17 mg, 0.55 ml D2O): δ 1.58–1.69 (m, 2H), 1.30–1.55 (m, 7H), 0.97–1.14 (m, 1H). 31P NMR (17 mg, 0.55 ml D2O): δ 27.3 (broad s). 13C{1H} NMR (22 mg, 550uL D2O): δ 70.7 (d, J = 163.2 Hz), 30.4, 30.4, 24.7, 19.6, 19.5. HRMS (TOF-ES+) calcd for C6H13NO4P [M + H]+m/z: 181.0630, found: 181.0632.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the Polish Ministry of Science and Education for the Wrocław University of Science and Technology.Notes and references
- (a) F. Bureš, Top. Curr. Chem., 2019, 377, 14 Search PubMed; (b) C. W. Weston, J. R. Papcun and M. Dery, Ammonium compounds, in Kirk–Othmer encyclopedia of chemical technology, Wiley, NY, 2003, vol. 2, pp. 711–762 Search PubMed.
- (a) S. Mehan, V. K. Aswal and J. Kohlbrecher, Langmuir, 2014, 30, 9941–9950 Search PubMed; (b) S. Mishra and V. K. Tyagi, J. Oleo Sci., 2007, 56, 269–276 Search PubMed.
- (a) M. Watanabe, M. L. Thomas, S. Zhang, U. Kazuhide, Y. Tomohiro and D. Kaoru, Chem. Rev., 2017, 117, 7190–7239 Search PubMed; (b) V. Selvamani, V. Suryanarayanan, D. Velayutham and S. Gopukumar, J. Solid State Electrochem., 2016, 20, 2283–2293 Search PubMed.
- (a) J. Přech, P. Pizarro, D. P. Serrano and J. Čejka, Chem. Soc. Rev., 2018, 47, 8263–8830 Search PubMed; (b) R. Bai, Q. Sun, N. Wang, Y. Zou, G. Guo, S. Iborra, A. Corma and J. Yu, Chem. Mater., 2016, 28, 6455–6458 Search PubMed.
- For recent reviews see: (a) T. Nakamura, K. Okuno, R. Nishiyori and S. Shirakawa, Chem. – Asian J., 2020, 15, 463–472 Search PubMed; (b) D. Kristofikova, V. Modrock, M. Meciarova and R. Sebesta, ChemSusChem, 2020, 13, 2828–2858 Search PubMed; (c) L. Roiser, K. Zielke and M. Waser, Asian J. Org. Chem., 2018, 7, 852–864 Search PubMed; (d) D. Qian and J. Sun, Chem. – Eur. J., 2018, 25, 3740–3751 Search PubMed; (e) K. C. Lethesh, W. Dehaen and K. Binnemans, RSC Adv., 2014, 4, 4472–4477 Search PubMed.
- (a) M. Pateiro-Moure, M. Arias-Estevez and J. Simal-Gandara, Environ. Sci. Technol., 2013, 47, 4984–4998 Search PubMed; (b) T. Nardin, C. Barnaba, F. Abballe, G. Trenti, M. Malacarne and R. Larcher, J. Sep. Sci., 2017, 40, 3928–3937 Search PubMed.
- For selected articles see: (a) P. I. Hora, S. G. Pati, P. J. McNamara and W. A. Arnold, Environ. Sci. Technol. Lett., 2020, 7, 622–631 Search PubMed; (b) P. Makvandi, R. Jamaledin, M. Jabbari, N. Nikfarjam and A. Borzacchiello, Dent. Mater., 2018, 34, 851–867 Search PubMed; (c) M. C. Jennings, K. P. C. Minbiole and W. M. Wuest, ACS Infect. Dis., 2015, 1, 288–303 Search PubMed; (d) J. Pernak, Przemysł Chem., 2013, 92, 1653–1656 Search PubMed.
- C. Queffélec, M. Petit, P. Janvier, A. Knight and B. Bujoli, Chem. Rev., 2012, 112, 3777–3807 Search PubMed.
- B. Sillion, L. Germanaud, S. Brunel and P. Le Perchec, Oil Gas Sci. Technol., 1986, 41, 773–785 Search PubMed.
- D. Zanke, J. Boehm, B. Leuner, H. Lyr and E. Nega, N-alkyl-phosphonic dimethylmorpholinio salts, processes for their preparation and their use as fungicides, DE 4115434A1, 1992 Search PubMed.
- (a) F. H. Ebetino, S. M. Kaas and M. D. Francis, Preparation of quaternary nitrogen-containing phosphonate compounds for treating abnormal calcium and phosphate metabolism, WO 9324499, 1993 Search PubMed; (b) F. H. Ebetino, M. D. Francis and S. M. Kaas, Quaternary nitrogen-containing, ring-containing phosphonate compounds, pharmaceutical compositions, and methods for treating abnormal calcium and phosphate metabolism, US 5753634, 1998 Search PubMed.
- D. Redmore, Quternery aminomethyl phosphonates as scale inhibitors, US 4420399, 1982 Search PubMed.
- X. K. Fu and S. Y. Wen, Synth. Commun., 1995, 25, 2435–2442 Search PubMed.
- (a) B. Denizot, F. Hindre and D. Portet, WO2006/053910, 2006; (b) Y. Song, Novel DXR inhibitors for antimicrobial therapy, WO 2013158846, 2013 Search PubMed.
- L. M. Porosa, G. Wolfaardt, A. J. Lough and D. A. Foucher, Curr. Microw. Chem., 2015, 2, 69–82 Search PubMed , and the references cited therein.
- For the seminal work of Menshutkin see: (a) N. Z. Menshutkin, Phys. Chem., 1890, 5, 589–600 Search PubMed. For more recent articles see: (b) Y.-Y. Chang, T.-L. Ho and W.-S. Chung, J. Org. Chem., 2014, 79, 9970–9978 Search PubMed; (c) O. Acevedo and W. L. Jorgensen, J. Phys. Chem. B, 2010, 114, 8425–8430 Search PubMed; (d) K. J. Stanger, J.-J. Lee and B. D. Smith, J. Org. Chem., 2007, 72, 9663–9668 Search PubMed.
- (a) P. M. Quinlan, Quaternary ammonium salts of 1,4-thiazin alkanephosphonic acids, US 3446156, 1982 Search PubMed; (b) P. M. Quinlan, Di-(quaternary ammonium) salts of 1,4-thiazine alkanephosphonic acids, US 4264768, 1981 Search PubMed.
- (a) T. Y. Medved and M. I. Kabachnik, Izv. Akad. Nauk SSSR, Ser. Khim., 1955, 1049–1052 Search PubMed; (b) T. Y. Medved and M. I. Kabachnik, Russ. Chem. Bull., 1955, 4, 961–964 Search PubMed. Compound 2′e (as betaine) was characterised by elemental analysis.
- (a) J. Iwanejko, M. Sowiński, E. Wojaczyńska, T. K. Olszewski and M. Górecki, RSC Adv., 2020, 10, 14618–14629 Search PubMed; (b) E. Żymańczyk-Duda, N. Dunal, M. Brzezińska-Rodak, A. Osiewała, T. K. Olszewski, M. Klimek-Ochab and M. Serafin-Lewańczuk, Bioorg. Chem., 2019, 93, 102751 Search PubMed; (c) E. Wolińska, K. Hałdys, J. Góra, T. K. Olszewski, B. Boduszek and R. Latajka, Chem. Biodiversity, 2019, 16, e1900167 Search PubMed; (d) J. Iwanejko, A. Brol, B. M. Szyja, M. Daszkiewicz, E. Wojaczyńska and T. K. Olszewski, Org. Biomol. Chem., 2019, 17, 7352–7359 Search PubMed; (e) T. K. Olszewski, Tetrahedron: Asymmetry, 2015, 26, 393–399 Search PubMed; (f) E. Pieta, E. Podstawka-Proniewicz, B. Boduszek, T. K. Olszewski, M. Nattich-Rak and Y. Kim, Appl. Surf. Sci., 2015, 335, 167–183 Search PubMed; (g) E. Podstawka-Proniewicz, E. Pieta, K. Zborowski, A. Kudelski, B. Boduszek, T. K. Olszewski, Y. Kim and L. M. Proniewicz, J. Phys. Chem. A, 2014, 118, 5614–5625 Search PubMed; (h) T. K. Olszewski, J. Galezowska, B. Boduszek and H. Kozlowski, Eur. J. Org. Chem., 2007, 3539–3540 Search PubMed; (i) B. Boduszek, T. K. Olszewski, W. Goldeman and M. Konieczna, Phosphorus, Sulfur Silicon Relat. Elem., 2006, 181, 787–795 Search PubMed.
- To ensure full conversion, the reaction mixture was heated at 60 °C for 4 h but without any influence on increasing conversion. When a stoichiometric amount of alkylating reagent was used for unhindered substrate 1a, a mixture of 2a, 4a, and di- and mono-methylated compounds, and their monoesters were obtained. Moreover, in alkaline solutions dimethyl sulfate undergoes hydrolysis to methanol and methyl sulfate or sulfuric acid. Therefore, an excess of Me2SO4 is required to reach full conversion.
- For the seminal work on Hofmann elimination see: (a) A. W. Hofmann, Ann. Chem. Pharm., 1851, 78, 253–286 Search PubMed. For more recent examples see: (b) K. Banert, M. Hagedorn, M. Heck, R. Hertel, A. Ihle, I. Mueller, T. Pester, T. Shoker and P. R. Rablen, J. Org. Chem., 2020, 85, 13630–13643 Search PubMed; (c) K. Banert, M. Heck, A. Ihle, J. Kronawitt, T. Pester and T. Shoker, J. Org. Chem., 2018, 83, 5138–5148 Search PubMed; (d) A. C. Cope and A. S. Mehta, J. Am. Chem. Soc., 1963, 85, 1949–1952 Search PubMed.
- For recent examples see: (a) H. Mao, P.-M. Wang and J. Xu, Tetrahedron, 2021, 81, 131913 Search PubMed; (b) E. Tayama, K. Hirano and S. Baba, Tetrahedron, 2020, 76, 131064 Search PubMed; (c) T. Abe, H. Shimizu, S. Takada, T. Tanaka, M. Yoshikawa and K. Yamada, Org. Lett., 2018, 20, 1589–1592 Search PubMed; (d) L. Yang, L. Wei and J.-P. Wan, Chem. Commun., 2018, 54, 7475–7478 Search PubMed; (e) H. Shadmon, A. Basu, L. H. Eckhard, A. J. Domb and N. Beyth, New J. Chem., 2018, 42, 1542 Search PubMed; (f) N. Yamamoto, T. Okada, Y. Harada, N. Kutsumura, S. Imaide, T. Saitoh, H. Fujii and H. Nagase, Tetrahedron, 2017, 73, 5751–5758 Search PubMed.
- (a) M. Hédi and Z. Hédi, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 25–33 Search PubMed; (b) K. Troev and D. M. Roundhill, Phosphorus, Sulfur Silicon Relat. Elem., 1988, 37, 243–245 Search PubMed; (c) E. M. Georgiev, R. Tsevi, V. Vassileva, K. Troev and D. M. Roundhill, Phosphorus, Sulfur Silicon Relat. Elem., 1994, 88, 139–145 Search PubMed; (d) V. Vassileva, E. M. Georgiev, K. Troev and D. M. Roundhill, Phosphorus, Sulfur Silicon Relat. Elem., 1994, 92, 101–107 Search PubMed.
- Identification of degradation products was made by comparision of chemical shifts and coupling constants with exact or very similar compounds described in: (a) G. L. Kenyon and F. H. Westheimer, J. Am. Chem. Soc., 1966, 88, 3557–3561 Search PubMed; (b) L. L. Khemchyan, J. V. Ivanova, S. S. Zalesskiy, V. P. Ananikov, I. P. Beletskaya and A. A. Starikova, Adv. Synth. Catal., 2014, 356, 771–780 Search PubMed.
- For more details see ESI.†.
- (a) E. Leete and L. Marion, Can. J. Chem., 1953, 31, 775–784 Search PubMed; (b) D. Lednicer, T. A. Mashburn and Ch. R. Hauser, Org. Synth., 1960, 40, 52 Search PubMed; (c) A. C. Cope and E. R. Trumbull, in Organic Reactions, John Wiley and Sons, Inc., New York, 1960, ch. 11, vol. 350–352, pp. 317–493 Search PubMed; (d) R. J. Baumgarten, J. Chem. Educ., 1968, 45, 122 Search PubMed; (e) Q. Xia, L. Qiu, S. Yu, H. Yang, L. Li, Y. Ye, Z. Gu and L. Ren, Environ. Sci. Technol., 2019, 53, 14430–14440 Search PubMed.
- (a) E. D. Hughes, C. K. Ingold and C. S. Patel, J. Chem. Soc., 1933, 526–530 Search PubMed; (b) G. Norcross and H. T. Openshaw, J. Chem. Soc., 1949, 1174–1179 Search PubMed; (c) L. Vivier, V. Dominguez, G. Perot and S. Kasztelan, J. Mol. Catal., 1991, 67, 267–275 Search PubMed.
- M. Soroka, Liebigs Ann. Chem., 1990, 4, 331–334 Search PubMed.
- Compound 2′a described as chloride and characterised only by melting point and elemental analysis: (a) T. Y. Medwed and M. I. Kabachnik, Izv. Akad. Nauk SSSR, Ser. Khim., 1951, 620–623 ( Chem. Abstr. , 1952 , 7996 ) Search PubMed. As betaine for spectroscopic characterisation see: (b) Y. S. Abdel-Ghany, M. A. Ihnat, D. D. Miller, C. M. Kunin and H. H. Tong, J. Med. Chem., 1993, 36, 784–789 Search PubMed.
- Compound 6 characterised by 31P NMR: Z. H. Kudzin, J. Drabowicz, M. Sochacki and W. Wisniewski, Phosphorus, Sulfur Silicon Relat. Elem., 1994, 92, 77–94 Search PubMed.
- M. Sekine, I. Yamamoto, A. Hashizume and T. Hata, Chem. Lett., 1977, 6, 485–488 Search PubMed.
- W. Goldeman and M. Soroka, Synthesis, 2006, 3019–3024 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob00703c |
| This journal is © The Royal Society of Chemistry 2021 |