One-pot synthesis of N-substituted benzannulated triazoles via stable arene diazonium salts†
Received
19th May 2021
, Accepted 21st June 2021
First published on 22nd June 2021
Abstract
A mild and effective one-pot synthesis of 1,2,3-benzotriazin-4(3H)-ones and benzothiatriazine-1,1(2H)-dioxide analogues has been developed. The method involves the diazotisation and subsequent cyclisation of 2-aminobenzamides and 2-aminobenzenesulfonamides via stable diazonium salts, prepared using a polymer-supported nitrite reagent and p-tosic acid. The transformation was compatible with a wide range of aryl functional groups and amide/sulfonamide-substituents and was used for the synthesis of pharmaceutically important targets. The synthetic utility of the one-pot diazotisaton–cyclisation process was further demonstrated with the preparation of an α-amino acid containing 1,2,3-benzotriazin-4(3H)-one.
Introduction
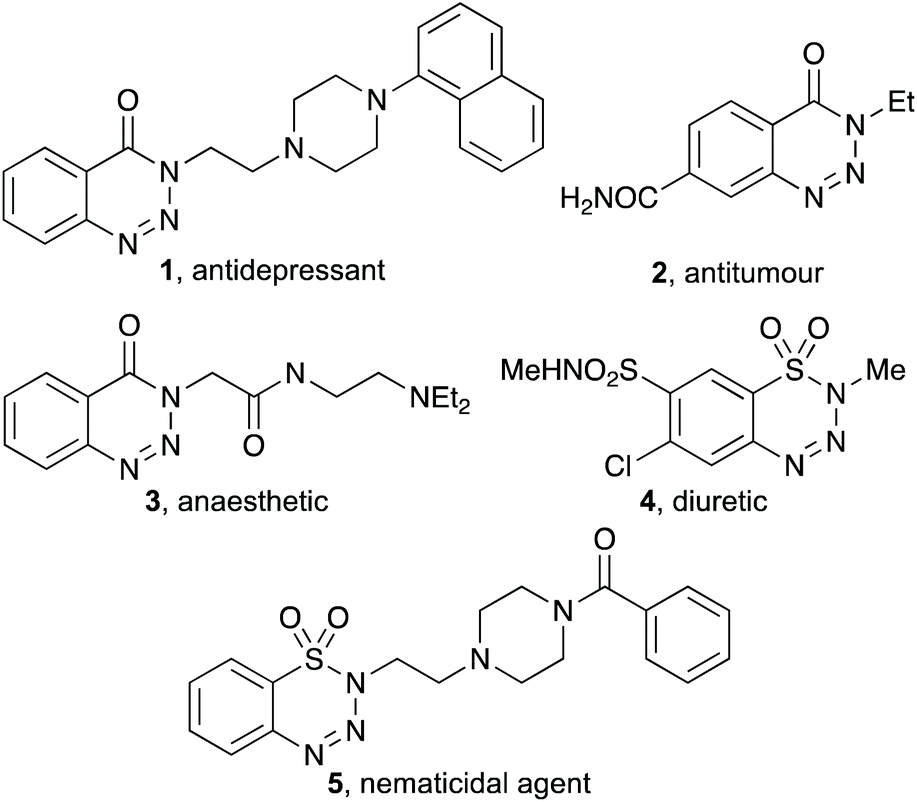
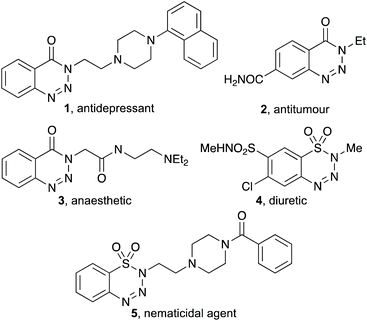
Benzotriazin-4(3H)-ones and benzothiatriazine-1,1(2H)-dioxides are privileged heterocyclic moieties in medicinal chemistry and are key components of a wide range of pharmaceutically-relevant compounds (Fig. 1).1,2 For example, benzotriazin-4(3H)-one 1 bearing a naphthalene-substituted piperazine side-chain was found to be a high affinity agent for the 5-HT1A receptor.2d Benzotriazin-4(3H)-one 3 is a potent anaesthetic with an IC50 value comparable to lidocaine,2c while benzothiatriazine-1,1(2H)-dioxide 4 is a well-known diuretic agent.2a Benzannulated triazoles have also been widely used for other applications, such as rubber components, dyes and as photoluminescence agents.1 Furthermore, these benzannulated heterocycles are useful synthetic precursors in organic chemistry.1 The groups of Murakami,3 Liu,4 and Cheng5 have shown that these compounds can undergo nickel-catalysed denitrogenative insertion reactions, with alkenes, alkynes and allenes, while palladium-catalysed denitrogenation and subsequent reaction with isocyanides6a or alkynes6b have allowed the preparation of 3-isoindolin-1-ones and o-alkynylated benzamides, respectively. Other nickel-catalysed denitrogentative substitution processes7 and visible-light-activated annulation reactions have also been reported.8 Specific reactions of benzothiatriazine-1,1(2H)-dioxides have been described for the synthesis of biaryl sultams, using thermal,9 visible light10 or acid-promoted11 methods.
 |
| | Fig. 1 Biologically active benzotriazin-4(3H)-ones and benzothiatriazine-1,1(2H)-dioxides. | |
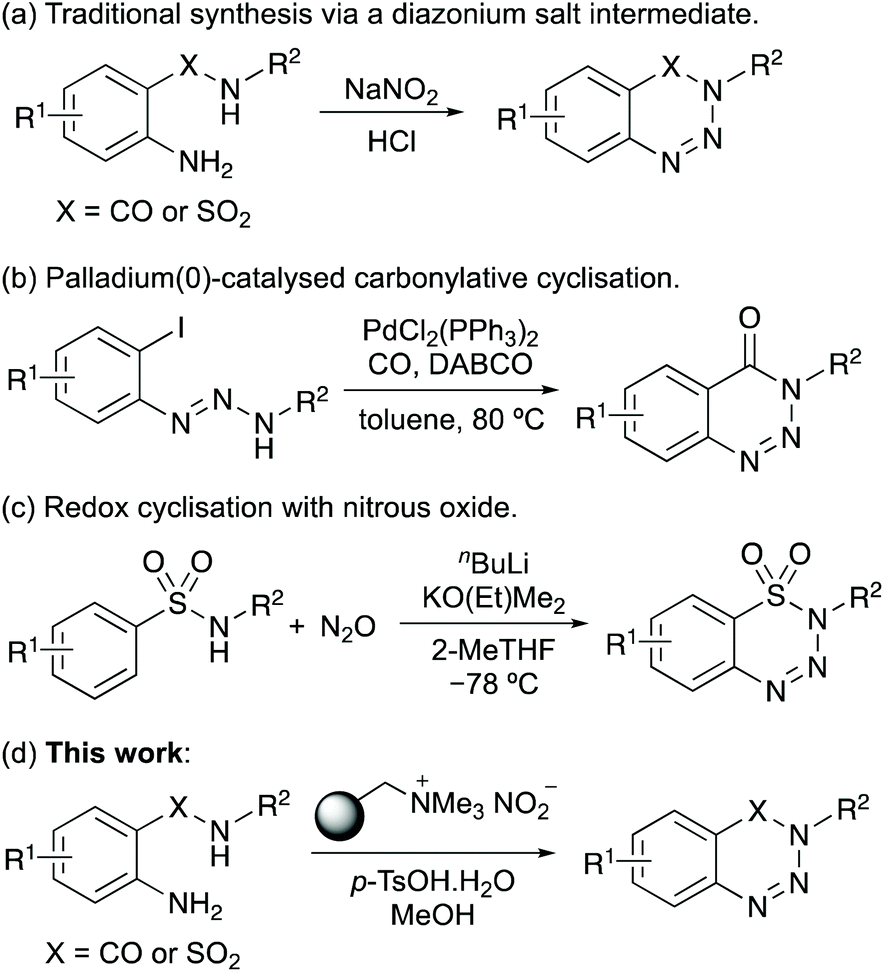
The importance of 1,2,3-benzotriazin-4(3H)-ones and 1,2,3-benzothiatriazine-1,1(2H)-dioxides have resulted in the development of a wide range of methods for their synthesis.1 Traditionally, these compounds were prepared from 2-aminobenzamides or 2-aminobenzenesulfonamides by reaction with sodium nitrite and hydrochloric acid (Scheme 1a).2b,12 Due to the harsh acidic conditions that limits substrate scope, unstable diazonium salts and the use of sodium nitrite that can lead to the release of toxic nitrogen oxides, milder reagents and reaction conditions for this transformation have been reported.13 In addition, new approaches have been developed for the preparation of 1,2,3-benzotriazin-4(3H)-ones and 1,2,3-benzothiatriazine-1,1(2H)-dioxides.14 For example, Chandrasekhar and Sankararaman reported the preparation of N-substituted 1,2,3-benzotriazin-4(3H)-ones via the palladium(0)-catalysed carbonylation of 1,3-diaryltriazenes (Scheme 1b),15 while Cui and co-workers prepared both series of compounds by a redox cyclisation of amides and sulfonamides with nitrous oxide (Scheme 1c).16 The Song group have reported the preparation of 1,2,3-benzotriazin-4(3H)-ones by the oxidative rearrangement of 3-aminoindazoles.17
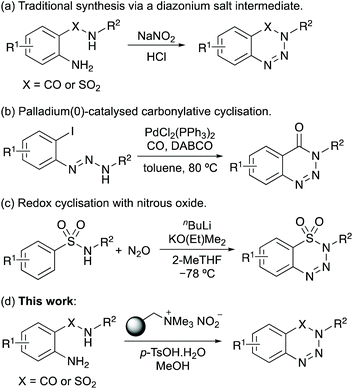 |
| | Scheme 1 Methods for the synthesis of benzotriazin-4(3H)-ones and benzothiatriazine-1,1(2H)-dioxides. | |
Despite the wide range of approaches now developed for the synthesis of these heterocycles, there is still a need for a mild and general approach from readily available starting materials, that avoids the requirement of harsh acidic or strongly oxidising conditions and precious transition metal catalysis. In recent years, issues associated with standard diazonium salt formation (NaNO2, HCl) of aromatic compounds have been overcome with the use of polymer-supported nitrite reagents and milder acids.18 For example, Filimonov and co-workers demonstrated that aryl diazonium tosylate salts could be prepared using nitrite supported on a tetraalkylammonium functionalised resin, such as Amberlyst A-26, in the presence of p-tosic acid.18c,d As well as the mild reaction conditions, the aryl diazonium tosylate salts were found to have high thermal and aging stability. Our group have exploited this safe and mild approach for aryl diazonium salt formation in one-pot processes for (radio)iodination,19 Heck–Matsuda reactions,20 and the synthesis of benzotriazoles.21 We now report the one-pot synthesis of 1,2,3-benzotriazin-4(3H)-ones and 1,2,3-benzothiatriazine-1,1(2H)-dioxides via stable aryl diazonium salts, by reaction of readily available 2-aminobenzamides and 2-aminobenzenesulfonamides with a polymer-supported nitrite reagent and p-tosic acid (Scheme 1d). As well as demonstrating the scope of these processes, we describe the application of this transformation for the synthesis of various biologically active compounds, including the synthesis of an α-amino acid containing 1,2,3-benzotriazin-4(3H)-one.
Results and discussion
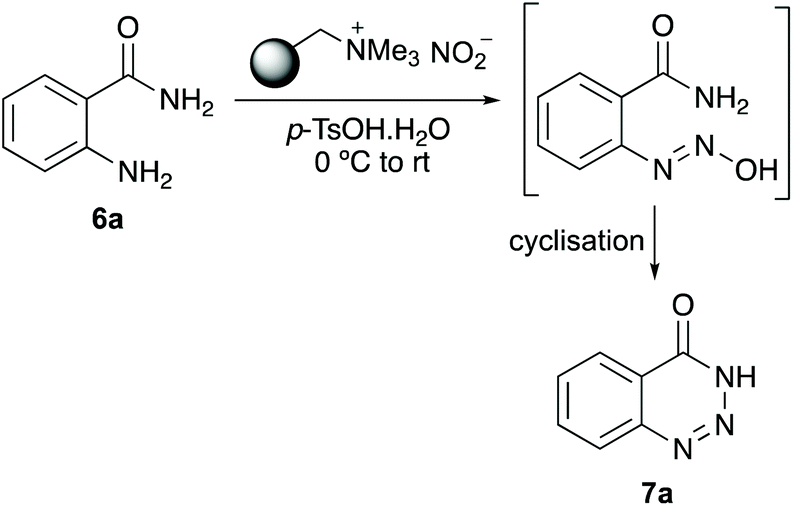
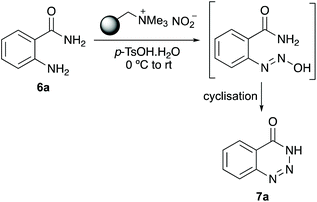
The study began with the optimisation of a one-pot diazotisation and cyclisation of 2-aminobenzamide (6a) for the preparation of 1,2,3-benzotriazin-4(3H)-one (7a) (Table 1). Following our previous work,19–21 a polymer-supported nitrite reagent, readily prepared by the ion exchange of Amberlyst A-26 with an aqueous solution of sodium nitrite, was used in combination with p-tosic acid.18c,d The reaction was initially trialled using one equivalent of both reagents and methanol as the reaction solvent (entry 1). While the transformation proceeded at room temperature, a reaction time of 48 hours was required for completion and gave 7a in 39% yield (entry 1). Using three equivalents of both reagents led to a substantial improvement, generating 7a in 87% yield after 2 hours (entry 2). A further increase in the number of equivalent of reagents resulted in a faster, but less efficient process (entry 3). A solvent screen was also performed (entries 4–6), and while these reactions gave 7a in good yields, methanol was deemed the optimal solvent. It should be noted that as well as avoiding toxic reagents and harsh conditions, an advantage of this process is the simple work-up procedure and purification of products. On reaction completion, the polymer resin is removed by filtration (and recycled), and the product then purified by flash column chromatography. By comparison, if the polymer-supported nitrite reagent is replaced with sodium nitrite in this process, while the reaction is completed in a similar time, the requirement of performing a work-up involving aqueous washes to remove p-TsOH, results in the isolation of 7a in 56% yield (entry 7).
Table 1 Optimisation of the one-pot diazotisation and cyclisation of 2-aminobenzamide (6a)
|
|
| Entry |
Reagentsa (equiv.) |
Solvent |
Time (h) |
Yieldb (%) |
|
Polymer supported nitrite and p-tosic acid.
Isolated yield.
Reaction was performed using sodium nitrite.
|
| 1 |
1 |
MeOH |
48 |
39 |
| 2 |
3 |
MeOH |
2 |
87 |
| 3 |
6 |
MeOH |
1 |
62 |
| 4 |
3 |
MeCN |
2 |
55 |
| 5 |
3 |
THF |
2 |
73 |
| 6 |
3 |
EtOAc |
2 |
65 |
| 7c |
3 |
MeOH |
2.5 |
56 |
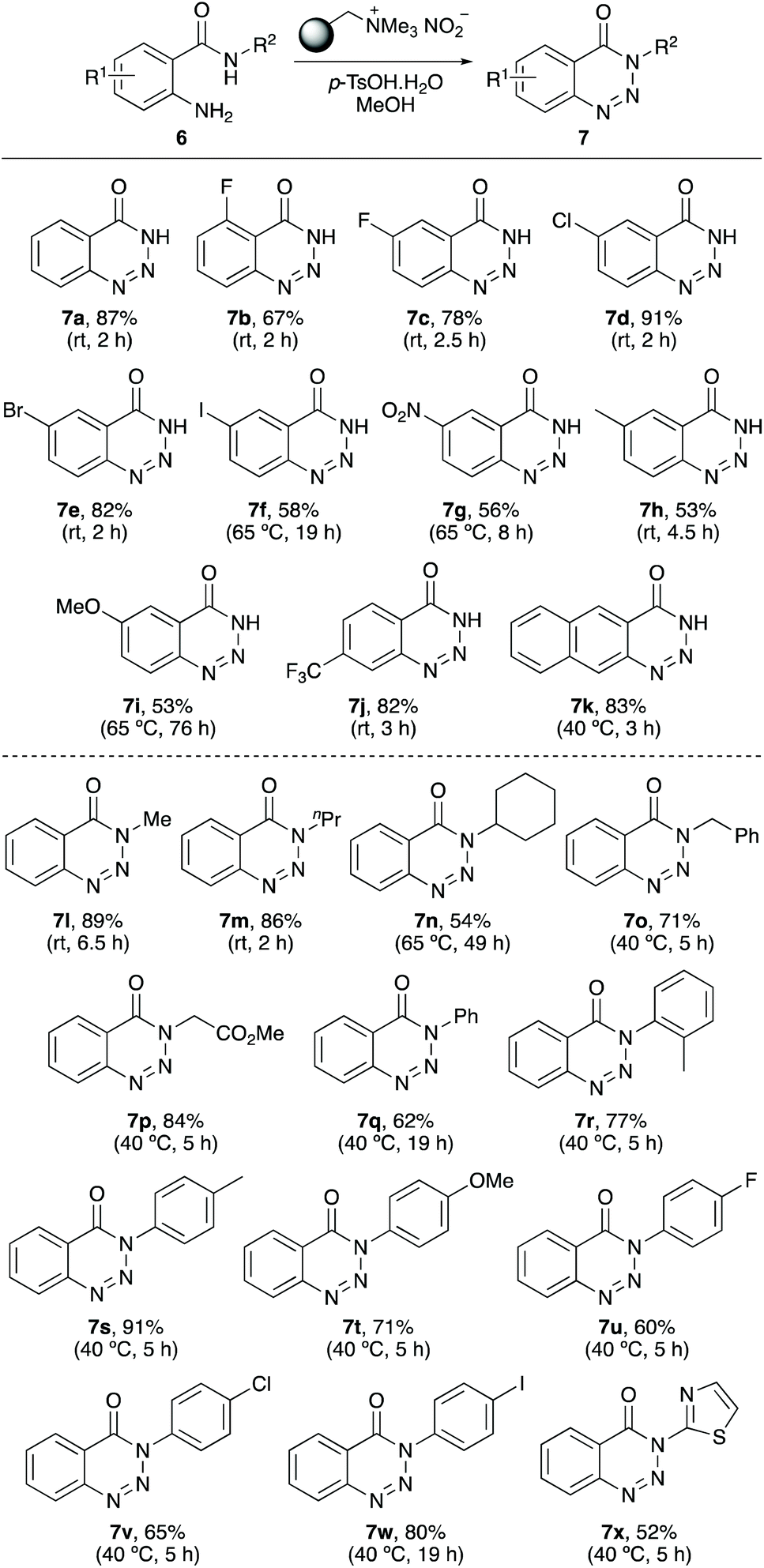
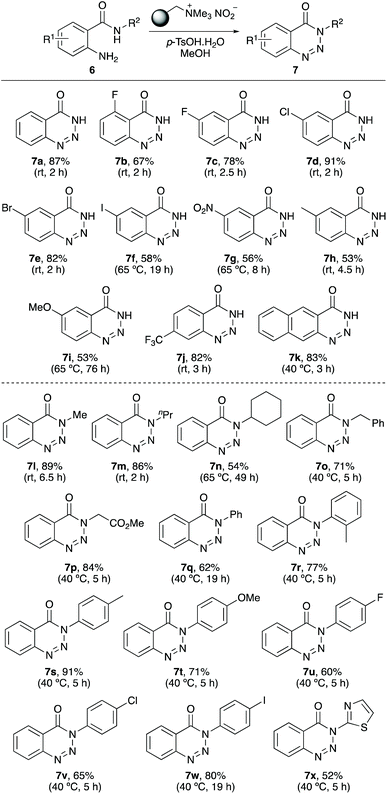
Following optimisation, the scope of the reaction was explored for the preparation of other N-unsubstituted 1,2,3-benzotriazin-4(3H)-ones (Scheme 2). 2-Aminobenzamides 6a–6k bearing a range of o-, m- and p-substituted functional groups were found to be substrates for the reaction, providing the corresponding 1,2,3-benzotriazin-4(3H)-ones 7a–7k in 53–91% yields. For the majority of the 2-aminobenzamides, the transformation proceeded at room temperature in reaction times of 2–4.5 h. Only methoxy-substituted 2-aminobenzamide 6i required both elevated temperatures (65 °C) and a long reaction time (76 h). This is likely due to the reduced electrophilic nature of the diazo intermediate prior to cyclisation.
 |
| | Scheme 2 Reaction scope for the synthesis of 1,2,3-benzotriazin-4(3H)-ones. | |
The next stage of the project investigated the application of the one-pot method for the synthesis of N-substituted 1,2,3-benzotriazin-4(3H)-ones (Scheme 2). Unlike most of the simple 2-aminobenzamides used in the first part of this study that were commercially available, many of the N-alkyl and N-aryl substituted benzamides required synthesis. These were prepared in one-step by the reaction of isatoic anhydride with various amines.22,23 Reaction of N-alkyl benzamides with polymer-supported nitrite reagent and p-tosic acid, under optimised conditions gave the corresponding N-alkyl 1,2,3-benzotriazin-4(3H)-ones 7l–7p in 54–89% yields. With these compounds, it was found that cyclisation of more sterically hindered secondary amides required more forcing conditions. For example, N-cyclohexyl benzamide 6n required a reaction time of 49 hours and a reaction temperature of 65 °C, while a t-butyl analogue demonstrated the limitation of this approach with no reaction observed. N-Aryl benzamides also required slightly elevated temperatures (40 °C) but all substrates investigated were converted to the corresponding N-aryl 1,2,3-benzotriazin-4(3H)-ones (7q–7x). The process was efficient for the preparation of 1,2,3-benzotriazin-4(3H)-ones bearing both electron-rich and electron-deficient N-aryl substituents and was also compatible with N-heterocyclic moieties (e.g.7x). The synthesis of 7r demonstrates that ortho-substituted N-aryl groups are also tolerated during the cyclisation step.
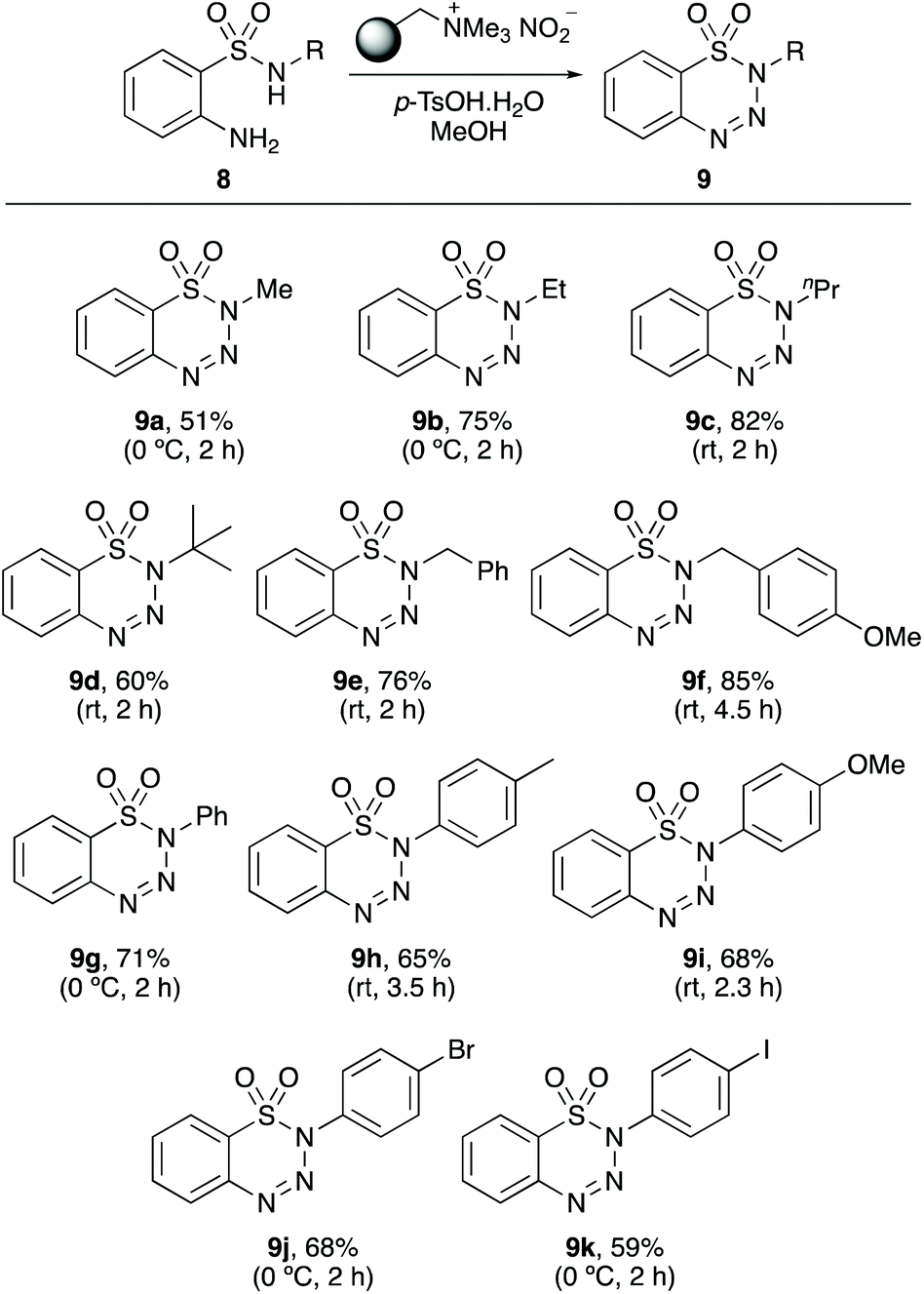
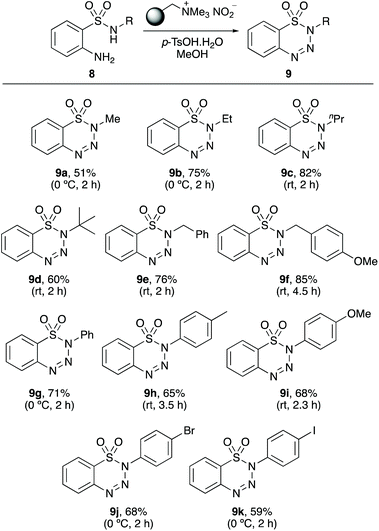
The research programme then examined the scope of the one-pot diazotisation and cyclisation for the preparation of N-substituted 1,2,3-benzothiatriazine-1,1(2H)-dioxides 9 (Scheme 3). The N-substituted benzenesulfonamides 8 were prepared in two steps by reaction of 2-nitrobenzenesulfonyl chloride with an amine, under basic conditions, followed by reduction of the nitro-group with tin dichloride.23 The N-substituted 2-aminobenzenesulfonamides 8 were found to be more reactive to diazotisation and cyclisation than the corresponding benzamides 6. Reactions proceeded at 0 °C or room temperature and with relatively short reaction times (2–4.5 h). For 2-amino-N-methylbenzenesulfonamide (8a) and N-ethyl analogue 8b, room temperature reactions led to denitrogenation of the diazo intermediates and isolation of the deaminated N-alkylbenzenesulfonamides as the major products. However, performing these reactions at 0 °C, allowed clean transformations and the isolation of 8a and 8b in 51% and 75% yields, respectively. Reaction of the other N-alkylbenzenesulfonamides proceeded smoothly at room temperature and gave 1,2,3-benzothiatriazine-1,1(2H)-dioxides 9c–9f, in 60–85% yields. A demonstration of the increased reactivity of this class of compound was the synthesis of t-butyl analogue 9d. While the corresponding 2-aminobenzamide gave no product, 9d was isolated in 60% yield. Similarly, the reaction of 2-amino-N-arylbenzenesulfonamides also proceeded at 0 °C or room temperature and gave the corresponding N-aryl 1,2,3-benzothiatriazine-1,1(2H)-dioxides 9g–9k in 59–71% yields. Several of the compounds prepared in this part of the study are used for the production of materials. For example, 9a, 9d and 9g are blowing agents for the formation of cellular rubber,24 while 9a and 9g are also used as catalysts for free-radical polymerisation.25
 |
| | Scheme 3 Reaction scope for the synthesis of 1,2,3-benzothiatriazine-1,1(2H)-dioxides. | |
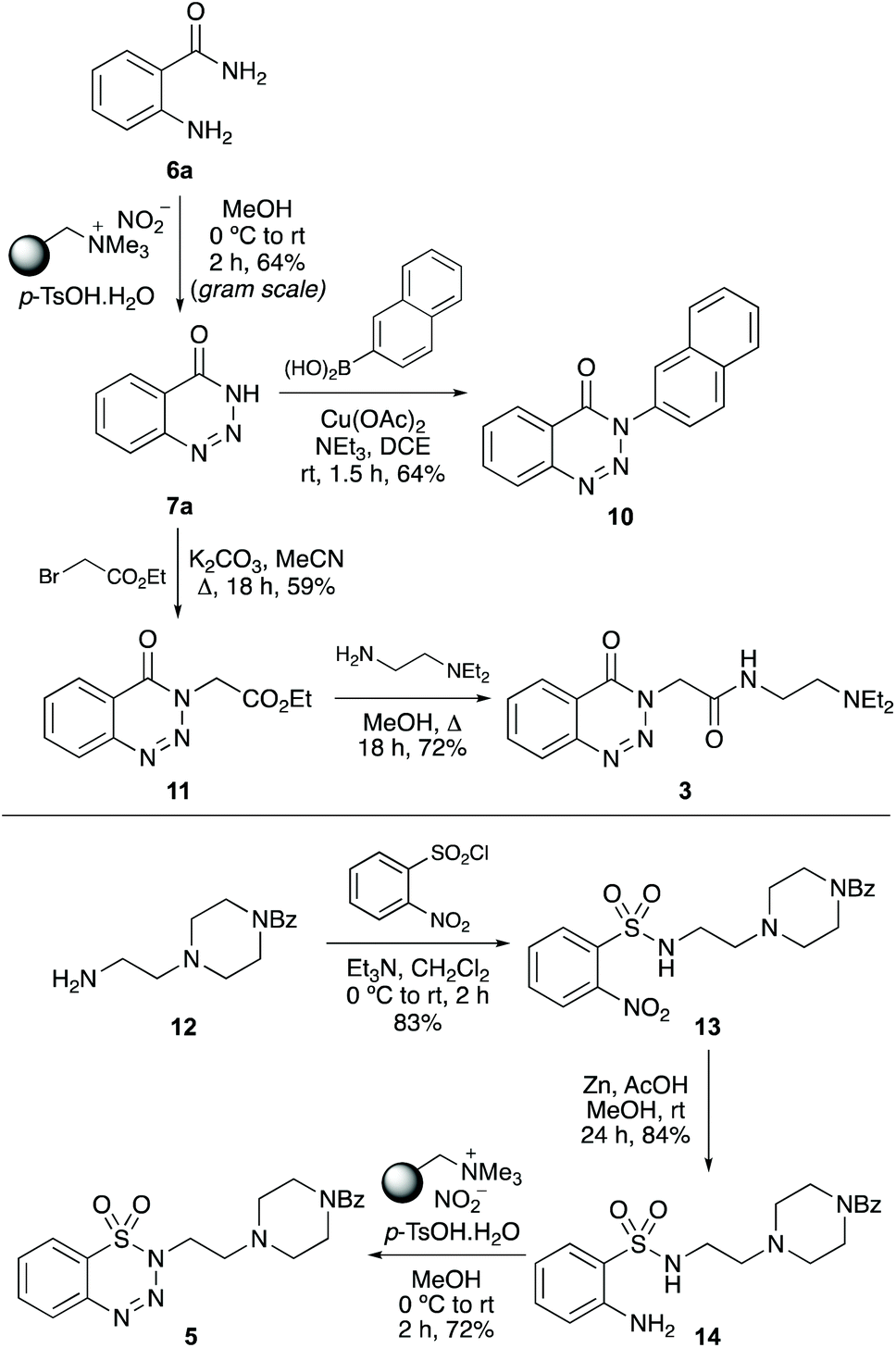
We next examined the application of the one-pot process for the synthesis of biologically active benzannulated triazoles. Initially, the optimised conditions for diazotisation and cyclisation (Table 1, entry 2) were used for the gram scale synthesis of 1,2,3-benzotriazin-4(3H)-one (7a). This gave 7a in 64% yield (Scheme 4). Copper(II)-catalysed N-arylation of 7a with naphthalene-2-boronic acid under basic conditions completed the two-step synthesis of chorismate mutase inhibitor 10.26 1,2,3-Benzotriazin-4(3H)-one (7a) was also used for the three-step synthesis of anaesthetic compound 3, which has similar activity to lidocaine.2cN-Alkylation of 7a with ethyl bromoacetate, followed by aminolysis with N,N-diethylethylenediamine gave 3 in good overall yield. As well as the synthesis of biologically active 1,2,3-benzotriazin-4(3H)-ones, the one-pot method was also used for a new synthesis of benzothiatriazine-1,1(2H)-dioxide 5, a compound with nematicidal activity and a potential pesticide with low-toxicity.2e Initially, amine 12 was prepared in two steps.23N-Benzoylpiperazine was alkylated with N-(2-bromoethyl)phthalimide and this was followed by removal of the phthalimide protecting group with hydrazine. Reaction of 12 with 2-nitrobenzenesulfonyl chloride and nitro-group reduction with zinc/acetic acid gave sulfonamide 14 in high yields. Application of the one-pot diazotisation and cyclisation method with 14, proceeded at room temperature and after a reaction time of 2 hours, gave nematicidal agent 5 in 72% yield.
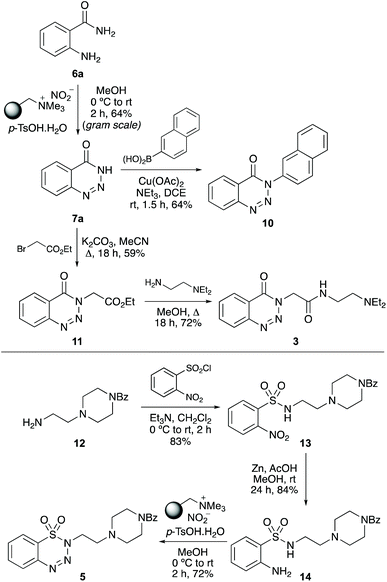 |
| | Scheme 4 Synthesis of biologically active benzannulated triazoles 3, 5 and 10. | |
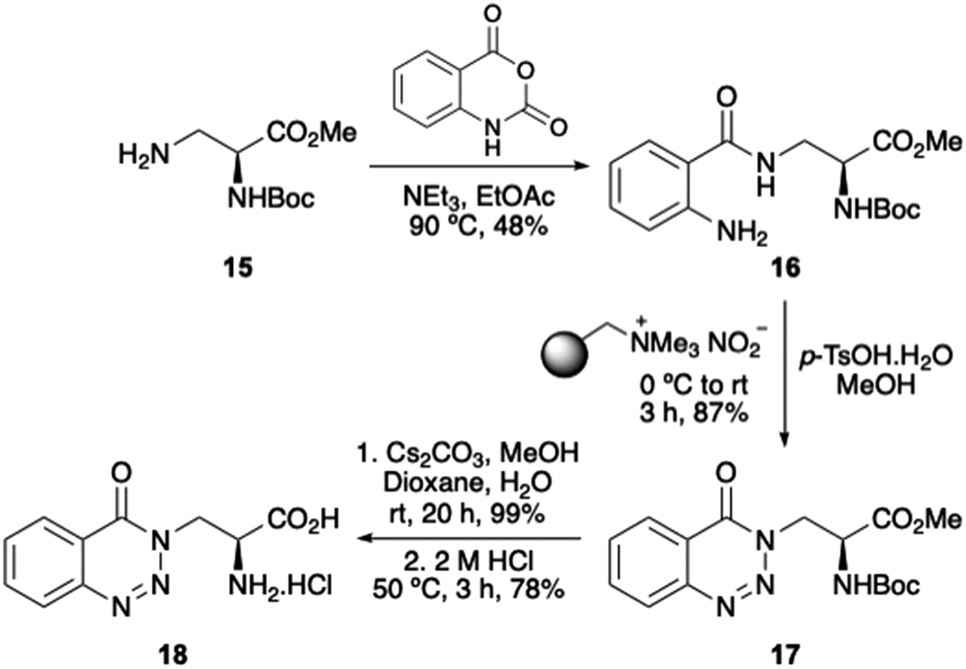
We have an interest in the development of novel α-amino acids that can be used as biological probes.27 Thus, as a proof-of-concept experiment, we were interested in exploring whether the one-pot diazotisation and cyclisation procedure could be used for the preparation of an α-amino acid bearing a 1,2,3-benzotriazin-4(3H)-one side-chain. Key intermediate 16 was prepared by the reaction of known L-3-aminoalanine derivative 1528 with isatoic anhydride (Scheme 5). Diazotisation and cyclisation of 16 was complete in 3 hours and gave 1,2,3-benzotriazin-4(3H)-one 17 in 87% yield. Despite the use of p-tosic acid, no Boc-group deprotection was observed under the mild diazotisation reaction conditions. Ester hydrolysis with caesium carbonate at room temperature, followed by acid-mediated removal of the Boc-protecting group gave parent amino acid 18.
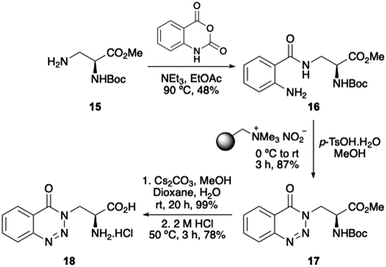 |
| | Scheme 5 Synthesis of α-amino acid 18. | |
Conclusions
In summary, a mild and general one-pot process has been developed for the synthesis of both 1,2,3-benzotriazin-4(3H)-ones and 1,2,3-benzothiatriazine-1,1(2H)-dioxides via stable aryl diazonium tosylate salts. The use of a polymer-supported nitrite reagent and p-tosic acid facilitated the rapid diazotisation and cyclisation of 2-aminobenzamides and 2-aminobenzenesulfonamides, yielding the target compounds in good yields. The process was found to be compatible for a wide range of substrates,29 bearing N-aryl or N-alkyl substituted amides or sulfonamides. The mild nature of this transformation allowed the preparation of various agents for rubber and polymer production, as well as the synthesis of medicinally important compounds. The use of this process for the preparation of a benzotriazin-4(3H)-one derived amino acid serves as a platform for future work and the synthesis of more complex analogues for chemical biology applications.
Experimental
All reagents and starting materials were obtained from commercial sources and used as received. All dry solvents were purified using a PureSolv 500 MD solvent purification system. Brine refers to a saturated aqueous solution of sodium chloride. Flash column chromatography was performed using Merck Geduran Si 60 (35–70 μm) silica gel. Merck aluminium-backed plates pre-coated with silica gel 60F254 were used for thin layer chromatography and were visualised with a UV lamp or by staining with KMnO4 or ninhydrin. 1H NMR spectra were recorded on a Bruker DPX spectrometer at either 400 or 500 MHz and data are reported as follows: chemical shift in ppm relative to tetramethylsilane as the internal standard, multiplicity (integration, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet or overlap of non-equivalent resonances). 13C NMR spectra were recorded on a Bruker DPX spectrometer at either 101 or 126 MHz and data are reported as follows: chemical shift in ppm relative to tetramethylsilane or the solvent as internal standard (CDCl3: δ 77.0 ppm, CD3OD: δ 49.0 ppm, DMSO-d6: δ 39.5 ppm), multiplicity with respect to hydrogen (deduced from DEPT experiments, C, CH, CH2 or CH3). Infrared spectra were recorded on a Shimadzu FTIR-84005 spectrometer; wavenumbers are indicated in cm−1. Mass spectra were recorded using a JEOL JMS-700 Spectrometer or a Bruker microTOFq High Resolution Mass Spectrometer. Melting points were determined on a Gallenkamp melting point apparatus and are reported uncorrected.
General procedure for preparation of polymer supported nitrite resin
To a stirred solution of sodium nitrite (5.50 g, 80.0 mmol) in water (200 mL) was added Amberlyst A26 hydroxide form resin (10.0 g, 40.0 mmol). The reaction mixture was stirred at rt for 1 h. The polymer-supported resin was filtered and washed with water until the filtrate was of neutral pH. The content of the polymer-supported nitrite was 3.5 mmol of NO2 per g.18
General Procedure for synthesis of 1H-benzotriazinones
To a stirred solution of the 2-aminobenzamides (1 equiv.) in methanol (10 mL mmol−1) at 0 °C was added polymer-supported nitrite (containing 3.0 equiv. of NO2) and p-toluenesulfonic acid monohydrate (3 equiv.). The reaction mixture was stirred for 1 h at 0 °C. The reaction mixture was then warmed to room temperature and stirred until completion. The resin was filtered and washed with methanol (20 mL mmol−1). The reaction mixture was concentrated in vacuo. Purification by flash column chromatography gave the 1H-benzotriazinones.
1,2,3-Benzotriazin-4(3H)-one (7a)30.
The reaction was carried out as described in the general procedure using 2-aminobenzamide (6a) (0.303 g, 2.20 mmol), polymer-supported nitrite (1.89 g, containing 6.60 mmol of NO2) and p-toluenesulfonic acid monohydrate (1.26 g, 6.60 mmol). The reaction mixture was stirred at 0 °C for 1 h, warmed to room temperature and stirred for 1 h. Purification by flash column chromatography, eluting with 35% ethyl acetate in hexane gave 1,2,3-benzotriazin-4(3H)-one (7a) (0.294 g, 87%) as a white solid. Mp 212–215 °C (lit.30 215–217 °C); δH (400 MHz, DMSO-d6) 7.91 (1H, br t, J 7.7 Hz, 7-H), 8.08 (1H, br t, J 7.7 Hz, 6-H), 8.18 (1H, br d, J 7.7 Hz, 8-H), 8.22 (1H, br d, J 7.7 Hz, 5-H), 14.94 (1H, br s, NH); δC (101 MHz, DMSO-d6) 120.2 (C), 124.3 (CH), 127.8 (CH), 132.6 (CH), 135.5 (CH), 144.2 (C), 155.6 (C); m/z (EI) 147 (M+. 100%), 104 (22), 92 (69), 76 (72), 63 (83).
5-Fluoro-1,2,3-benzotriazin-4(3H)-one (7b).
The reaction was carried out as described in the general procedure using 6-fluoro-2-aminobenzamide (6b) (0.200 g, 1.30 mmol), polymer-supported nitrite (1.11 g, containing 3.89 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.740 g, 3.89 mmol). The reaction mixture was stirred at 0 °C for 1 h, warmed to room temperature and stirred for 1 h. Purification by flash column chromatography, eluting with 70% diethyl ether in hexane gave 5-fluoro-1,2,3-benzotriazin-4(3H)-one (7b) (0.144 g, 67%) as a white solid. Mp 200–204 °C; νmax/cm−1 (neat) 3161 (NH), 2975 (CH), 2160, 2012, 1700 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1480, 1262, 815; δH (500 MHz, DMSO-d6) 7.68 (1H, dd, J 10.5, 9.0 Hz, 6-H), 8.00 (1H, br d, J 8.0 Hz, 8-H), 8.07 (1H, ddd, J 10.5, 8.0, 5.0 Hz, 7-H), 14.97 (1H, br s, NH); δC (126 MHz, DMSO-d6) 109.9 (C, 2JCF = 8.8 Hz), 118.6 (CH, d, 2JCF = 20.2 Hz), 124.0 (CH, 4JCF = 3.8 Hz), 136.5 (CH, 3JCF = 10.1 Hz), 145.7 (C), 152.7 (C, 3JCF = 1.3 Hz), 158.7 (C, d, 1JCF = 264.6 Hz); m/z (ESI) 166.0410 (MH+. C7H5FN3O requires 166.0411).
O), 1480, 1262, 815; δH (500 MHz, DMSO-d6) 7.68 (1H, dd, J 10.5, 9.0 Hz, 6-H), 8.00 (1H, br d, J 8.0 Hz, 8-H), 8.07 (1H, ddd, J 10.5, 8.0, 5.0 Hz, 7-H), 14.97 (1H, br s, NH); δC (126 MHz, DMSO-d6) 109.9 (C, 2JCF = 8.8 Hz), 118.6 (CH, d, 2JCF = 20.2 Hz), 124.0 (CH, 4JCF = 3.8 Hz), 136.5 (CH, 3JCF = 10.1 Hz), 145.7 (C), 152.7 (C, 3JCF = 1.3 Hz), 158.7 (C, d, 1JCF = 264.6 Hz); m/z (ESI) 166.0410 (MH+. C7H5FN3O requires 166.0411).
6-Fluoro-1,2,3-benzotriazin-4(3H)-one (7c)17.
The reaction was carried out as described in the general procedure using 2-amino-5-fluorobenzamide (6c) (0.150 g, 0.970 mmol), polymer-supported nitrite (0.836 g, containing 2.92 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.556 g, 2.92 mmol). The reaction mixture was stirred at 0 °C for 1 h, warmed to room temperature and stirred for 1.5 h. Purification by flash column chromatography, eluting with 30% ethyl acetate in hexane gave 6-fluoro-1,2,3-benzotriazin-4(3H)-one (7c) (0.125 g, 78%) as a white solid. Spectroscopic data were consistent with the literature.17 Mp 206–208 °C; δH (400 MHz, DMSO-d6) 7.91–8.00 (2H, m, 5-H and 7-H), 8.31 (1H, ddd, J 8.8, 5.0, 0.4 Hz, 8-H), 15.04 (1H, br s, NH); δC (101 MHz, DMSO-d6) 109.5 (CH, 2JCF = 24.0 Hz), 122.4 (C, 3JCF = 9.4 Hz), 123.9 (CH, d, 2JCF = 24.4 Hz), 131.6 (CH, d, 3JCF = 9.5 Hz), 141.5 (C, d, 4JCF = 2.2 Hz), 155.0 (C, d, 4JCF = 3.1 Hz), 163.2 (C, d, 1JCF = 253.4 Hz); m/z (EI) 165 (M+. 20%), 122 (10), 94 (21), 78 (88), 63 (100).
6-Chloro-1,2,3-benzotriazin-4(3H)-one (7d)31.
The reaction was carried out as described in the general procedure using 2-amino-5-chlorobenzamide (6d) (0.150 g, 0.880 mmol), polymer-supported nitrite (0.759 g, containing 2.65 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.504 g, 2.65 mmol). The reaction mixture was stirred at 0 °C for 1 h, warmed to room temperature and stirred for 1 h. Purification by flash column chromatography, eluting with 35% ethyl acetate in hexane gave 6-chloro-1,2,3-benzotriazin-4(3H)-one (7d) (0.145 g, 91%) as a white solid. Mp 190–192 °C (lit.31 195–196 °C); δH (400 MHz, DMSO-d6) 8.12 (1H, dd, J 8.7, 2.4 Hz, 7-H), 8.18 (1H, d, J 2.4 Hz, 5-H), 8.22 (1H, d, J 8.7 Hz, 8-H), 15.11 (1H, br s, NH); δC (101 MHz, DMSO-d6) 121.7 (C), 123.5 (CH), 130.2 (CH), 135.6 (CH), 136.9 (C), 142.8 (C), 154.6 (C); m/z (EI) 181 (M+. 18%), 138 (7), 110 (11), 78 (88), 63 (100).
6-Bromo-1,2,3-benzotriazin-4(3H)-one (7e)32.
The reaction was carried out as described in the general procedure using 2-amino-5-bromobenzamide (6e) (0.150 g, 0.700 mmol), polymer-supported nitrite (0.601 g, containing 2.10 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.399 g, 2.10 mmol). The reaction mixture was stirred at 0 °C for 1 h, warmed to room temperature and stirred for 1 h. Purification by flash column chromatography, eluting with 25% ethyl acetate in hexane gave 6-bromo-1,2,3-benzotriazin-4(3H)-one (7e) (0.129 g, 82%) as a white solid. Spectroscopic data were consistent with the literature.32 Mp 207–209 °C; δH (400 MHz, DMSO-d6) 8.12 (1H, d, J 8.6 Hz, 8-H), 8.24 (1H, dd, J 8.6, 2.2 Hz, 7-H), 8.31 (1H, d, J 2.2 Hz, 5-H), 15.10 (1H, br s, NH); δC (101 MHz, DMSO-d6) 121.7 (C), 125.6 (C), 126.5 (CH), 130.1 (CH), 138.3 (CH), 142.9 (C), 154.3 (C); m/z (EI) 227 (M+. 23%), 225 (23), 184 (22), 182 (21), 156 (17), 154 (18), 78 (78), 63 (100).
6-Iodo-1,2,3-benzotriazin-4(3H)-one (7f).
The reaction was carried out as described in the general procedure using 2-amino-5-iodobenzamide (6f) (0.150 g, 0.570 mmol), polymer-supported nitrite (0.497 g, containing 1.72 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.372 g, 1.72 mmol). The reaction mixture was stirred for 1 h at 0 °C and then heated under reflux for 18 h. Purification by flash column chromatography, eluting with 40% ethyl acetate in hexane gave 6-iodo-1,2,3-benzotriazin-4(3H)-one (7f) (0.0910 g, 58%) as a white solid. Mp 127–130 °C; νmax/cm−1 (neat) 3071 (NH), 2945 (CH), 1672 (CO), 1227, 1188; δH (400 MHz, CD3OD) 7.90 (1H, d, J 8.6 Hz, 8-H), 8.37 (1H, dd, J 8.5, 2.0 Hz, 7-H), 8.64 (1H, d, J 2.0 Hz, 5-H); δC (101 MHz, CD3OD) 99.5 (C), 122.8 (C), 130.5 (CH), 134.6 (CH), 145.2 (C), 145.6 (CH), 156.6 (C); m/z (ESI) 295.9288 (MNa+. C7H4IN3NaO requires 295.9291).
6-Nitro-1,2,3-benzotriazin-4(3H)-one (7g)33.
The reaction was carried out as described in the general procedure using 2-amino-5-nitrobenzamide (6g) (0.0800 g, 0.442 mmol), polymer-supported nitrite (0.378 g, containing 1.32 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.252 g, 1.32 mmol). The reaction mixture was stirred for 1 h at 0 °C and heated under reflux for 7 h. Purification by flash column chromatography, eluting with 60% diethyl ether in hexane gave 6-nitro-1,2,3-benzotriazin-4(3H)-one (7g) (0.0480 g, 56%) as a yellow solid. Mp 189–194 °C (lit.33 194 °C); δH (400 MHz, DMSO-d6) 8.42 (1H, dd, J 8.9, 0.5 Hz, 8-H), 8.76 (1H, dd, J 8.9, 2.6 Hz, 7-H), 8.82 (1H, dd, J 2.6, 0.5 Hz, 5-H), 15.42 (1H, br s, NH); δC (101 MHz, DMSO-d6) 120.3 (CH), 121.1 (C), 129.4 (CH), 130.1 (CH), 146.4 (C), 148.5 (C), 154.8 (C); m/z (EI) 192 (M+. 28%), 149 (28), 84 (82), 66 (100).
6-Methyl-1,2,3-benzotriazin-4(3H)-one (7h)34.
The reaction was carried out as described in the general procedure using 2-amino-5-methylbenzamide (6h) (0.100 g, 0.660 mmol), polymer-supported nitrite (0.573 g, containing 2.00 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.380 g, 2.00 mmol). The reaction mixture was stirred at 0 °C for 1 h, warmed to room temperature and stirred for 3.5 h. Purification by flash column chromatography, eluting with 30% ethyl acetate in petroleum ether gave 6-methyl-1,2,3-benzotriazin-4(3H)-one (7h) (0.0570 g, 53%) as a white solid. Mp 219–220 °C (lit.34 217–218 °C); δH (400 MHz, DMSO-d6) 2.54 (3H, s, 6-CH3), 7.89 (1H, dd, J 8.2, 1.6 Hz, 7-H), 8.01 (1H, br s, 5-H), 8.07 (1H, d, J 8.2 Hz, 8-H), 14.86 (1H, br s, NH); δC (101 MHz, DMSO-d6) 21.3 (CH3), 120.1 (C), 123.6 (CH), 127.8 (CH), 136.7 (CH), 142.6 (C), 143.4 (C), 155.6 (C); m/z (EI) 161 (M+. 8%), 104 (8), 89 (6), 78 (82), 63 (100).
6-Methoxy-1,2,3-benzotriazin-4(3H)-one (7i).
The reaction was carried out as described in the general procedure using 5-methoxy-2-aminobenzamide (6i) (0.100 g, 0.600 mmol), polymer-supported nitrite (0.515 g, containing 1.80 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.342 g, 1.80 mmol). The reaction mixture was stirred for 1 h at room temperature and then heated under reflux for 75 h. Purification by flash column chromatography, eluting with 40% ethyl acetate in hexane gave 6-methoxy-1,2,3-benzotriazin-4(3H)-one (7i) (0.0560 g, 53%) as a white solid. Mp 190–192 °C; νmax/cm−1 (neat) 3159 (NH), 2959 (CH), 1662 (CO), 1296, 1141, 895, 833; δH (400 MHz, DMSO-d6) 3.96 (3H, s, 6-OCH3), 7.55 (1H, d, J 2.8 Hz, 5-H), 7.62 (1H, dd, J 9.0, 2.8 Hz, 7-H), 8.12 (1H, d, J 9.0 Hz, 8-H), 14.79 (1H, br s, NH); δC (101 MHz, DMSO-d6) 56.2 (CH3), 104.2 (CH), 122.0 (C), 124.6 (CH), 130.1 (CH), 139.4 (C), 155.6 (C), 161.9 (C); m/z (EI) 177.0534 (M+. C8H7N3O2 requires 177.0538), 134 (12%), 106 (38), 78 (79), 63 (100).
7-Trifluoromethyl-1,2,3-benzotriazin-4(3H)-one (7j).
The reaction was carried out as described in the general procedure using 2-amino-4-trifluorobenzamide (6j) (0.150 g, 0.730 mmol), polymer-supported nitrite (0.630 g, containing 2.20 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.418 g, 2.20 mmol). The reaction mixture was stirred at 0 °C for 1 h, warmed to room temperature and stirred for 2 h. Purification by flash column chromatography, eluting with 30% ethyl acetate in petroleum ether gave 7-trifluoromethyl-1,2,3-benzotriazin-4(3H)-one (7j) (0.130 g, 82%) as a white solid. Mp 185–187 °C; νmax/cm−1 (neat) 3136 (NH), 1694 (CO), 1651 (CC), 1319, 1134, 864; δH (400 MHz, DMSO-d6) 8.20 (1H, br d, J 8.3 Hz, 6-H), 8.40 (1H, br d, J 8.3 Hz, 5-H), 8.57 (1H, br s, 8-H), 15.25 (1H, br s, NH); δC (101 MHz, DMSO-d6) 123.1 (C, q, 1JCF = 273.4 Hz), 123.1 (C), 125.2 (CH, q, 3JCF = 3.2 Hz), 126.3 (CH), 128.1 (CH, q, 3JCF = 3.2 Hz), 134.8 (C, d, 2JCF = 33.1 Hz), 143.9 (C), 154.8 (C); m/z (EI) 215.0301 (M+. C8H4F3N3O requires 215.0306), 172 (6%), 160 (8), 144 (9), 78 (81), 63 (100).
3H-Naphtho[2,3-d][1,2,3]triazin-4-one (7k).
The reaction was carried out as described in the general procedure using 2-aminonaphthalene-3-carboxamide (6k) (0.0330 g, 0.177 mmol), polymer-supported nitrite (0.152 g, containing 0.532 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.101 g, 0.532 mmol). The reaction mixture was stirred at 0 °C for 1 h and heated to 40 °C for 2 h. Purification by flash column chromatography, eluting with 80% ethyl acetate in hexane with 1% triethylamine gave 3H-naphtho[2,3-d][1,2,3]triazin-4-one (7k) (0.0290, 83%) as a white solid. Mp 254–257 °C; νmax/cm−1 (neat) 3006 (NH), 2850 (CH), 2325, 2072, 1823, 1667 (CO), 1203, 747; δH (400 MHz, DMSO-d6) 7.73–7.86 (2H, m, 7-H and 8-H), 8.27–8.38 (2H, m, 6-H and 9-H), 8.84 (1H, s, 10-H), 8.91 (1H, s, 5-H), 14.64 (1H, s, NH); δC (101 MHz, DMSO-d6) 117.7 (C), 125.8 (CH), 127.9 (CH), 128.9 (CH), 129.1 (CH), 129.3 (CH), 129.4 (CH), 133.7 (C), 135.7 (C), 140.5 (C), 155.8 (C); m/z (ESI) 198.0662 (MH+. C11H8N3O requires 198.0662).
N-Methyl-1,2,3-benzotriazin-4(3H)-one (7l)16.
The reaction was carried out as described in the general procedure using 2-amino-N-methylbenzamide (6l) (0.150 g, 1.00 mmol), polymer-supported nitrite (0.850 g, containing 3.00 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.571 g, 3.00 mmol). The reaction mixture was stirred at 0 °C for 1 h, warmed to room temperature and stirred for 5.5 h. Purification by flash column chromatography, eluting with 25% ethyl acetate in petroleum ether gave N-methyl-1,2,3-benzotriazin-4(3H)-one (7l) (0.144 g, 89%) as a white solid. Mp 115–117 °C (lit.16 118–120 °C); δH (500 MHz, DMSO-d6) 3.93 (3H, s, 3-CH3), 7.89–7.95 (1H, m, 6-H), 8.05–8.10 (1H, m, 7-H), 8.19 (1H, dd, J 8.1, 1.0 Hz, 8-H), 8.24 (1H, dd, J 8.1, 1.0 Hz, 5-H); δC (126 MHz, DMSO-d6) 37.0 (CH3), 119.2 (C), 124.3 (CH), 127.9 (CH), 132.8 (CH), 135.2 (CH), 143.9 (C), 155.1 (C); m/z (EI) 161 (M+. 6%), 78 (82), 63 (100).
N-Propyl-1,2,3-benzotriazin-4(3H)-one (7m)35.
The reaction was carried out as described in the general procedure using 2-amino-N-propylbenzamide (6m) (0.150 g, 0.840 mmol), polymer-supported nitrite (0.721 g, containing 2.52 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.479 g, 2.52 mmol). The reaction mixture was stirred at 0 °C for 1 h, warmed to room temperature and stirred for 1 h. Purification by flash column chromatography, eluting with 20% ethyl acetate in hexane gave N-propyl-1,2,3-benzotriazin-4(3H)-one (7m) (0.136 g, 86%) as a white solid. Mp 50–55 °C (lit.35 56–57 °C); δH (500 MHz, DMSO-d6) 0.92 (3H, t, J 7.3 Hz, 3′-H3), 1.83 (2H, sextet, J 7.3 Hz, 2′-H2), 4.33 (2H, t, J 7.3 Hz, 1′-H2), 7.88–7.93 (1H, m, 6-H), 8.04–8.09 (1H, m, 7-H), 8.16 (1H, br d, J 8.0 Hz, 8-H), 8.22 (1H, dd, J 8.0, 1.0 Hz, 5-H); δC (126 MHz, DMSO-d6) 10.9 (CH3), 22.7 (CH2), 50.7 (CH2), 119.2 (C), 124.5 (CH), 127.9 (CH), 132.8 (CH), 135.3 (CH), 143.6 (C), 154.7 (C); m/z (ESI) 212 (MNa+. 100%).
N-Cyclohexyl-1,2,3-benzotriazin-4(3H)-one (7n)13c.
The reaction was carried out as described in the general procedure using 2-amino-N-cyclohexylbenzamide (6n) (0.150 g, 0.690 mmol), polymer-supported nitrite (0.590 g, containing 2.06 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.392 g, 2.06 mmol). The reaction mixture was stirred at room temperature for 1 h and then heated under reflux for 48 h. Purification by flash column chromatography, eluting with 25% ethyl acetate in hexane gave N-cyclohexyl-1,2,3-benzotriazin-4(3H)-one (7n) (0.0850 g, 54%) as a yellow solid. Mp 130–133 °C (lit.13c 129–131 °C); δH (500 MHz, CD3OD) 1.30–1.42 (1H, m, CHH), 1.49–1.62 (2H, m, CH2), 1.76–1.84 (1H, m, CHH), 1.92–2.07 (6H, m, 3 × CH2), 4.95–5.06 (1H, m, 1′-H), 7.89 (1H, td, J 8.1, 1.0 Hz, 6-H), 8.02–8.07 (1H, m, 7-H), 8.15 (1H, br d, J 8.1 Hz, 8-H), 8.31 (1H, dd, J 8.1, 1.0 Hz, 5-H); δC (126 MHz, CD3OD) 26.5 (CH2), 26.9 (2 × CH2), 32.9 (2 × CH2), 58.2 (CH), 120.6 (C), 126.0 (CH), 129.0 (CH), 133.7 (CH), 136.4 (CH), 145.2 (C), 156.6 (C); m/z (EI) 229 (M+. 68%), 172 (43), 158 (69), 148 (75), 130 (40), 105 (76), 78 (100), 63 (100).
N-(Benzyl)-1,2,3-benzotriazin-4(3H)-one (7o)14.
The reaction was carried out as described in the general procedure using 2-amino-N-(4-benzyl)benzamide (6o) (0.0500 g, 0.221 mmol), polymer-supported nitrite (0.190 g, containing 0.663 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.126 g, 0.663 mmol). The reaction mixture was stirred at 0 °C for 1 h and then heated to 40 °C for 4 h. Purification by flash column chromatography, eluting with 30% ethyl acetate in hexane gave N-(benzyl)-1,2,3-benzotriazin-4(3H)-one (7o) (0.0370 g, 71%) as a white solid. Mp 115–119 °C (lit.14 118–120 °C); δH (400 MHz, CDCl3) 5.63 (2H, s, PhCH2), 7.26–7.37 (3H, m, 3′-H, 4′-H and 5′-H), 7.50–7.55 (2H, m, 2′-H and 6′-H), 7.74–7.80 (1H, m, 6-H), 7.89–7.95 (1H, m, 7-H), 8.14 (1H, br d, J 8.2 Hz, 8-H), 8.33 (1H, dd, J 8.0, 1.0 Hz, 5-H); δC (101 MHz, CDCl3) 53.5 (CH2), 120.2 (C), 125.3 (CH), 128.3 (CH), 128.5 (CH), 128.9 (2 × CH), 129.0 (2 × CH), 132.5 (CH), 134.9 (CH), 135.9 (C), 144.5 (C), 155.5 (C); m/z (ESI) 260 (MNa+. 100%).
N-(Methoxycarbonylmethyl)-1,2,3-benzotriazin-4(3H)-one (7p)13c.
To a stirred solution of 2-amino-N-(methoxycarbonylmethyl)benzamide (6p) (0.127 g, 0.601 mmol) in methanol (6 mL) was added polymer-supported nitrite (0.516 g, containing 1.80 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.342 g, 1.80 mmol). The reaction mixture was stirred at 0 °C for 1 h and then heated to 40 °C for 4 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. The reaction mixture was diluted in ethyl acetate (20 mL) and washed with water (4 × 20 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo to give N-(methoxycarbonylmethyl)-1,2,3-benzotriazin-4(3H)-one (7p) (0.110 g, 84%) as a white solid. Mp 128–130 °C (lit.13c 128–130 °C); δH (400 MHz, CDCl3) 3.81 (3H, s OCH3), 5.21 (2H, s, CH2CO2CH3), 7.80–7.86 (1H, m, 6-H), 7.95–8.02 (1H, m, 7-H), 8.19 (1H, br d, J 8.1 Hz, 8-H), 8.37 (1H, dd, J 7.9, 1.1 Hz, 5-H); δC (101 MHz, CDCl3) 50.9 (CH2), 52.8 (CH3), 119.9 (C), 125.4 (CH), 128.8 (CH), 132.8 (CH), 135.3 (CH), 144.5 (C), 155.7 (C), 167.7 (C); m/z (ESI) 242 (MNa+. 100%).
N-Phenyl-1,2,3-benzotriazin-4(3H)-one (7q)13c.
The reaction was carried out as described in the general procedure using 2-amino-N-phenylbenzamide (6q) (0.0500 g, 0.236 mmol), polymer-supported nitrite (0.202 g, containing 0.707 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.135 g, 0.707 mmol). The reaction mixture was stirred at 0 °C for 1 h and then heated to 40 °C for 18 h. Purification by flash column chromatography, eluting with 25% ethyl acetate in hexane gave N-phenyl-1,2,3-benzotriazin-4(3H)-one (7q) (0.0330 g, 62%) as a white solid. Mp 150–153 °C (lit.13c 150–152 °C); δH (400 MHz, CDCl3) 7.49 (1H, tt, J 7.4, 1.3 Hz, 4′-H), 7.53–7.59 (2H, m, 3′-H and 5′-H), 7.63–7.68 (2H, m, 2′-H and 6′-H), 7.83–7.88 (1H, m, 6-H), 7.96–7.03 (1H, m, 7-H), 8.23 (1H, br d, J 8.1 Hz, 8-H), 8.45 (1H, dd, J 7.9, 1.1 Hz, 5-H); δC (101 MHz, CDCl3) 120.6 (C), 125.8 (CH), 126.2 (2 × CH), 128.7 (CH), 129.1 (CH), 129.2 (2 × CH), 132.9 (CH), 135.2 (CH), 139.0 (C), 143.9 (C), 155.4 (C); m/z (ESI) 246 (MNa+. 100%).
N-(2′-Methylphenyl)-1,2,3-benzotriazin-4(3H)-one (7r)13b.
The reaction was carried out as described in the general procedure using 2-amino-N-(2′-methylphenyl)benzamide (6r) (0.100 g, 0.442 mmol), polymer-supported nitrite (0.379 g, containing 1.33 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.252 g, 1.33 mmol). The reaction mixture was stirred at 0 °C for 1 h and then heated to 40 °C for 4 h. Purification by flash column chromatography, eluting with 100% dichloromethane gave N-(2′-methylphenyl)-1,2,3-benzotriazin-4(3H)-one (7r) (0.0810 g, 77%) as a white solid. Mp 158–160 °C (lit.13b 153–155 °C); δH (500 MHz, CDCl3) 2.21 (3H, s, 2′-CH3), 7.33–7.49 (4H, m, 3′-H, 4′-H, 5′-H and 6′-H), 7.86 (1H, br t, J 7.5 Hz, 6-H), 8.01 (1H, br t, J 7.5, 7-H), 8.25 (1H, br d, J 7.5 Hz, 8-H), 8.45 (1H, br d, J 7.5 Hz, 5-H); δC (126 MHz, CDCl3) 17.8 (CH3), 120.4 (C), 125.6 (CH), 127.1 (CH), 127.8 (CH), 128.7 (CH), 129.9 (CH), 131.2 (CH), 132.8 (CH), 135.2 (CH), 135.6 (C), 138.0 (C), 144.1 (C), 155.2 (C); m/z (ESI) 260 (MNa+. 100%).
N-(4′-Methylphenyl)-1,2,3-benzotriazin-4(3H)-one (7s)14.
The reaction was carried out as described in the general procedure using 2-amino-N-(4′-methylphenyl)benzamide (6s) (0.285 g, 1.26 mmol), polymer-supported nitrite (1.09 g, containing 3.78 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.719 g, 3.78 mmol). The reaction mixture was stirred at 0 °C for 1 h and then heated to 40 °C for 4 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. The reaction mixture was diluted in dichloromethane (20 mL) and washed with 1 M aqueous sodium hydroxide (6 × 20 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo to give N-(4′-methylphenyl)-1,2,3-benzotriazin-4(3H)-one (7s) (0.272 g, 91%) as an orange solid. Mp 137–140 °C (lit.14 139–141 °C); δH (400 MHz, CDCl3) 2.45 (3H, s, 4′-CH3), 7.33–7.39 (2H, m, 3′-H and 5′-H), 7.50–7.56 (2H, m, 2′-H and 6′-H), 7.82–7.87 (1H, m, 6-H), 7.96–8.01 (1H, m, 7-H), 8.22 (1H, br d, J 8.1 Hz, 8-H), 8.44 (1H, dd, J 7.9, 1.4 Hz, 5-H); δC (101 MHz, CDCl3) 21.4 (CH3), 120.6 (C), 125.8 (CH), 126.0 (2 × CH), 128.6 (CH), 129.8 (2 × CH), 132.8 (CH), 135.1 (CH), 136.5 (C), 139.2 (C), 143.9 (C), 155.5 (C); m/z (ESI) 260 (MNa+. 100%).
N-(4′-Methoxyphenyl)-1,2,3-benzotriazin-4(3H)-one (7t)14.
The reaction was carried out as described in the general procedure using 2-amino-N-(4′-methoxyphenyl)benzamide (6t) (0.0500 g, 0.206 mmol), polymer-supported nitrite (0.177 g, containing 0.619 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.118 g, 0.619 mmol). The reaction mixture was stirred at 0 °C for 1 h and then heated to 40 °C for 4 h. Purification by flash column chromatography, eluting with 70% diethyl ether in hexane gave N-(4′-methoxyphenyl)-1,2,3-benzotriazin-4(3H)-one (7t) (0.0370 g, 71%) as a yellow solid. Mp 147–150 °C (lit.14 151–153 °C); δH (400 MHz, CDCl3) 3.88 (3H, s, 4′-OCH3), 7.02–7.09 (2H, m, 3′-H and 5′-H), 7.52–7.60 (2H, m, 2′-H and 6′-H), 7.84 (1H, td, J 8.0, 1.0 Hz, 6-H), 7.94–8.01 (1H, m, 7-H), 8.21 (1H, br d, J 8.1 Hz, 8-H), 8.43 (1H, dd, J 8.0, 1.0 Hz, 5-H); δC (101 MHz, CDCl3) 55.7 (CH3), 114.4 (2 × CH), 120.5 (C), 125.7 (CH), 127.4 (2 × CH), 128.6 (CH), 131.9 (C), 132.8 (CH), 135.1 (CH), 143.9 (C), 155.5 (C), 160.0 (C); m/z (ESI) 276 (MNa+. 100%).
N-(4′-Fluorophenyl)-1,2,3-benzotriazin-4(3H)-one (7u)14.
The reaction was carried out as described in the general procedure using 2-amino-N-(4′-fluorophenyl)benzamide (6u) (0.0500 g, 0.217 mmol), polymer-supported nitrite (0.187 g, containing 0.652 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.124 g, 0.652 mmol). The reaction mixture was stirred at 0 °C for 1 h and then heated to 40 °C for 4 h. Purification by flash column chromatography, eluting with 30% ethyl acetate in hexane gave N-(4′-fluorophenyl)-1,2,3-benzotriazin-4(3H)-one (7u) (0.0320 g, 60%) as a white solid. Mp 136–139 °C (lit.14 138–140 °C); δH (400 MHz, CDCl3) 7.20–7.28 (2H, m, 2′-H and 6′-H), 7.61–7.68 (2H, m, 3′-H and 5′-H), 7.82–7.89 (1H, m, 6-H), 7.96–8.01 (1H, m, 7-H), 8.22 (1H, br d, J 8.0 Hz, 8-H), 8.43 (1H, dd, J 8.0, 1.1 Hz, 5-H); δC (101 MHz, CDCl3) 116.1 (2 × CH, d, 2JC–F = 23.0 Hz), 120.4 (C), 125.8 (CH), 128.0 (2 × CH, d, 3JC–F = 8.8 Hz), 128.7 (CH), 133.0 (CH), 134.9 (C, d, 4JC–F = 3.3 Hz), 135.3 (CH), 143.8 (C), 155.4 (C), 162.7 (C, d, 1JC–F = 249.1 Hz); m/z (ESI) 264 (MNa+. 100%).
N-(4′-Chlorophenyl)-1,2,3-benzotriazin-4(3H)-one (7v)13b.
The reaction was carried out as described in the general procedure using 2-amino-N-(4′-chlorophenyl)benzamide (6v) (0.150 g, 0.608 mmol), polymer-supported nitrite (0.522 g, containing 1.82 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.347 g, 1.82 mmol). The reaction mixture was stirred at 0 °C for 1 h and then heated to 40 °C for 4 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. The reaction mixture was diluted in ethyl acetate (20 mL) and washed with 1 M aqueous sodium hydroxide (6 × 20 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo to give N-(4′-chlorophenyl)-1,2,3-benzotriazin-4(3H)-one (7v) (0.102 g, 65%) as a yellow solid. Mp 174–176 °C (lit.13b 173–175 °C); δH (400 MHz, CDCl3) 7.50–7.55 (2H, m, 2′-H and 6′-H), 7.61–7.67 (2H, m, 3′-H and 5′-H), 7.84–7.90 (1H, m, 6-H), 7.98–8.03 (1H, m, 7-H), 8.23 (1H, br d, J 8.2 Hz, 8-H), 8.44 (1H, dd, J 7.9, 1.2 Hz, 5-H); δC (101 MHz, CDCl3) 120.4 (C), 125.8 (CH), 127.4 (2 × CH), 128.8 (CH), 129.4 (2 × CH), 133.1 (CH), 135.0 (C), 135.4 (CH), 137.4 (C), 143.7 (C), 155.3 (C); m/z (ESI) 280 (MNa+. 100%).
N-(4′-Iodophenyl)-1,2,3-benzotriazin-4(3H)-one (7w)14.
The reaction was carried out as described in the general procedure using 2-amino-N-(4′-iodophenyl)benzamide (6w) (0.0900 g, 0.266 mmol), polymer-supported nitrite (0.229 g, containing 0.799 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.152 g, 0.799 mmol). The reaction mixture was stirred at 0 °C for 1 h and then heated to 40 °C for 18 h. Purification by flash column chromatography, eluting with 25% ethyl acetate in hexane gave N-(4′-iodophenyl)-1,2,3-benzotriazin-4(3H)-one (7w) (0.0740 g, 80%) as a white solid. Mp 187–190 °C (lit.14 191–193 °C); δH (400 MHz, CDCl3) 7.42–7.47 (2H, m, 2′-H and 6′-H), 7.83–7.92 (3H, m, 6-H, 3′-H and 5′-H), 7.97–8.04 (1H, m, 7-H), 8.23 (1H, br d, J 8.1 Hz, 8-H), 8.44 (1H, dd, J 7.9, 1.0 Hz, 5-H); δC (101 MHz, CDCl3) 94.5 (C), 120.4 (C), 125.8 (CH), 127.8 (2 × CH), 128.8 (CH), 133.1 (CH), 135.4 (CH), 138.4 (2 × CH), 138.7 (C), 143.7 (C), 155.2 (C); m/z (ESI) 372 (MNa+. 100%).
N-(Thiazol-2′-yl)-1,2,3-benzotriazin-4(3H)-one (7x)13c.
The reaction was carried out as described in the general procedure using N-(2-aminobenzoyl)-2-aminothiazole (6x) (0.147 g, 0.670 mmol), polymer-supported nitrite (0.578 g, containing 2.01 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.387 g, 2.01 mmol). The reaction mixture was stirred at 0 °C for 1 h and then heated to 40 °C for 4 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. The reaction mixture was diluted in ethyl acetate (20 mL) and washed with 1 M aqueous sodium hydroxide (6 × 20 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo to give N-(thiazol-2′-yl)-1,2,3-benzotriazin-4(3H)-one (7x) (0.0800 g, 52%) as an orange solid. Mp 170–173 °C (lit.13c 174–176 °C); δH (400 MHz, CDCl3) 7.44 (1H, d, J 3.5 Hz, 5′-H), 7.89 (1H, d, J 3.5 Hz, 4′-H), 7.88–7.95 (1H, m, 6-H), 8.02–8.09 (1H, m, 7-H), 8.32 (1H, br d, J 8.1 Hz, 8-H), 8.51 (1H, dd, J 7.9, 1.4 Hz, 5-H); δC (101 MHz, CDCl3) 119.1 (CH), 119.4 (C), 126.0 (CH), 129.5 (CH), 133.8 (CH), 135.9 (CH), 140.2 (CH), 142.6 (C), 154.3 (C), 156.3 (C); m/z (ESI) 253 (MNa+. 100%).
General procedure for synthesis of 2H-benzothiatriazin-1,1-dioxides
To a stirred solution of 2-aminobenzenesulfonamides (1 equiv.) in methanol (10 mL mmol−1) at 0 °C was added polymer-supported nitrite (containing 3.0 equiv. of NO2) and p-toluenesulfonic acid monohydrate (3 equiv.). The reaction mixture was stirred at 0 °C until completion or stirred at 0 °C for 1 h and then warmed to room temperature and stirred until completion. The resin was filtered and washed with methanol (20 mL mmol−1). The reaction mixture was concentrated in vacuo. Purification by flash column chromatography gave the 2H-benzothiatriazin-1,1-dioxides.
N-Methyl-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9a)16.
To a stirred solution of 2-amino-N-(methyl)benzenesulfonamide (8a) (0.0840 g, 0.451 mmol) in methanol (6 mL) was added polymer-supported nitrite (0.387 g, containing 1.35 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.257 g, 1.35 mmol). The reaction mixture was stirred at 0 °C for 2 h. The reaction mixture was diluted with ethyl acetate (30 mL) and washed with water (4 × 30 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography, eluting with 50% diethyl ether in hexane gave N-methyl-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9a) (0.0450 g, 51%) as a yellow solid. Mp 70–72 °C (lit.16 68–72 °C); δH (400 MHz, CDCl3) 3.91 (3H, s, NCH3), 7.79 (1H, td, J 7.7, 1.2 Hz, 7-H), 7.87–7.92 (1H, m, 6-H), 8.02–8.07 (2H, m, 5-H and 8-H); δC (101 MHz, CDCl3) 34.0 (CH3), 120.6 (CH), 125.4 (C), 129.5 (CH), 132.8 (CH), 134.3 (CH), 141.8 (C); m/z (ESI) 220 (MNa+. 100%).
N-Ethyl-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9b)36.
To a stirred solution of 2-amino-N-(ethyl)benzenesulfonamide (8b) (0.0500 g, 0.250 mmol) in methanol (2.5 mL) was added polymer-supported nitrite (0.215 g, containing 0.749 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.142 g, 0.749 mmol). The reaction mixture was stirred at 0 °C for 2 h. The reaction mixture was diluted with ethyl acetate (30 mL) and washed with water (4 × 30 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography, eluting with 25% ethyl acetate in hexane gave N-ethyl-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9b) (0.0410 g, 75%) as a dark orange solid. Spectroscopic data were consistent with the literature.36 Mp 75–78 °C; δH (400 MHz, CDCl3) 1.59 (3H, t, J 7.2 Hz, 2′-H3), 4.34 (2H, q, J 7.2 Hz, 1′-H2), 7.77 (1H, td, J 7.8, 1.1 Hz, 6-H), 7.88 (1H, td, J 7.8, 1.4 Hz, 7-H), 8.01 (1H, dd, J 7.8, 1.4 Hz, 5-H), 8.03 (1H, dd, J 7.8, 1.1 Hz, 8-H); δC (101 MHz, CDCl3) 16.0 (CH3), 43.7 (CH2), 120.4 (CH), 125.7 (C), 129.3 (CH), 132.6 (CH), 134.1 (CH), 141.8 (C); m/z (ESI) 234 (MNa+. 100%).
N-Propyl-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9c).
To a stirred solution of 2-amino-N-(propyl)benzenesulfonamide (8c) (0.0650 g, 0.303 mmol) in methanol (3 mL) was added polymer-supported nitrite (0.261 g, containing 0.910 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.173 g, 0.910 mmol). The reaction mixture was stirred at 0 °C for 1 h, warmed to room temperature and stirred for a further 1 h. The reaction mixture was diluted with ethyl acetate (20 mL) and washed with water (4 × 30 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo to give N-propyl-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9c) (0.0560 g, 82%) as a red solid. Mp 83–86 °C; νmax/cm−1 (neat) 2967 (CH), 1573 (CC), 1471, 1446, 1329, 1313, 1185, 1160, 1111, 946, 766; δH (400 MHz, CDCl3) 1.04 (3H, t, J 7.4 Hz, 3′-H3), 2.01 (2H, sext, J 7.4 Hz, 2′-H2), 4.23 (2H, t, J 7.4 Hz, 1′-H2), 7.77 (1H, td, J 7.8, 1.1 Hz, 6-H), 7.88 (1H, td, J 7.8, 1.4 Hz, 7-H), 8.02 (1H, dd, J 7.8, 1.4 Hz, 5-H), 8.04 (1H, dd, J 7.8, 1.1 Hz, 8-H); δC (101 MHz, CDCl3) 11.2 (CH3), 23.7 (CH2), 50.0 (CH2), 120.5 (CH), 125.7 (C), 129.3 (CH), 132.6 (CH), 134.1 (CH), 141.8 (C); m/z (ESI) 248.0468 (MNa+. C9H11N3NaO2S requires 248.0464).
N-(tert-Butyl)-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9d)16.
To a stirred solution of 2-amino-N-(tert-butyl)benzenesulfonamide (8d) (0.0260 g, 0.114 mmol) in methanol (1.1 mL) was added polymer-supported nitrite (0.0980 g, containing 0.342 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.0650 g, 0.342 mmol). The reaction mixture was stirred at 0 °C for 1 h, warmed to room temperature and stirred for a further 1 h. The reaction mixture was diluted with ethyl acetate (10 mL) and washed with water (4 × 10 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography, eluting with 20% ethyl acetate in hexane gave N-(tert-butyl)-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9d) (0.0160 g, 60%) as an orange solid. Mp 78–82 °C (lit.16 78–82 °C); δH (400 MHz, CDCl3) 1.82 (9H, s, NC(CH3)3), 7.74 (1H, td, J 7.8, 1.2 Hz, 7-H), 7.85 (1H, td, J 7.8, 1.4 Hz, 6-H), 7.96–8.03 (2H, m, 5-H and 8-H); δC (101 MHz, CDCl3) 30.5 (3 × CH3), 67.9 (C), 120.4 (CH), 126.3 (C), 128.6 (CH), 132.3 (CH), 134.0 (CH), 141.2 (C); m/z (ESI) 262 (MNa+. 100%).
N-Benzyl-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9e)36.
To a stirred solution of 2-amino-N-benzylbenzenesulfonamide (8e) (0.130 g, 0.496 mmol) in methanol (5 mL) was added polymer-supported nitrite (0.426 g, containing 1.49 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.283 g, 1.49 mmol). The reaction mixture was stirred at 0 °C for 1 h, warmed to room temperature and stirred for a further 1 h. The reaction mixture was diluted with ethyl acetate (20 mL) and washed with water (4 × 20 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo to give N-benzyl-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9e) (0.104 g, 76%) as an orange solid. Spectroscopic data were consistent with the literature.36 Mp 99–102 °C; δH (400 MHz, CDCl3) 5.42 (2H, s, PhCH2), 7.30–7.38 (3H, m, 3′-H, 4′-H and 5′-H), 7.46–7.51 (2H, m, 2′-H and 6′-H), 7.77 (1H, td, J 7.7, 1.2 Hz, 6-H), 7.87 (1H, td, J 7.7, 1.4 Hz, 7-H), 8.01 (1H, dd, J 7.7, 1.4 Hz, 5-H), 8.04 (1H, dd, J 7.7, 1.2 Hz, 8-H); δC (101 MHz, CDCl3) 51.3 (CH2), 120.6 (CH), 126.1 (C), 128.6 (CH), 128.9 (2 × CH), 129.0 (2 × CH), 129.5 (CH), 132.8 (CH), 134.2 (CH), 135.4 (C), 141.8 (C); m/z (ESI) 296 (MNa+. 100%).
N-(4′-Methoxybenzyl)-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9f)36.
To a stirred solution of 2-amino-N-(4′-methoxybenzyl)benzenesulfonamide (8f) (0.0700 g, 0.293 mmol) in methanol (2 mL) was added polymer-supported nitrite (0.206 g, containing 0.718 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.137 g, 0.718 mmol). The reaction mixture was stirred at 0 °C for 1 h, warmed to room temperature and stirred for a further 3.5 h. The reaction mixture was diluted with ethyl acetate (20 mL) and washed with water (4 × 20 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo to give N-(4′-methoxybenzyl)-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9f) (0.0610 g, 85%) as a red solid. Spectroscopic data were consistent with the literature.36 Mp 92–95 °C; δH (400 MHz, CDCl3) 3.79 (3H, s, 4′-OCH3), 5.36 (2H, s, NCH2), 6.85–6.91 (2H, m, 3′-H and 5′-H), 7.40–7.46 (2H, m, 2′-H and 6′-H), 7.76 (1H, td, J 7.8, 1.2 Hz, 7-H), 7.86 (1H, td, J 7.8, 1.4 Hz, 6-H), 8.01 (1H, dd, J 7.8, 1.2 Hz, 5-H), 8.03 (1H, dd, J 7.8, 1.4 Hz, 8-H); δC (101 MHz, CDCl3) 51.0 (CH2), 55.4 (CH3), 114.3 (2 × CH), 120.6 (CH), 126.1 (C), 127.5 (C), 129.5 (CH), 130.6 (2 × CH), 132.7 (CH), 134.2 (CH), 141.8 (C), 159.9 (C); m/z (ESI) 326 (MNa+. 100%).
N-Phenyl-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9g)37.
To a stirred solution of 2-amino-N-(phenyl)benzenesulfonamide (8g) (0.0390 g, 0.157 mmol) in methanol (1.5 mL) was added polymer-supported nitrite (0.135 g, containing 0.471 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.0900 g, 0.471 mmol). The reaction mixture was stirred at 0 °C for 2 h. The reaction mixture was diluted with ethyl acetate (20 mL) and washed with water (4 × 20 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography, eluting with 50% diethyl ether in hexane gave N-phenyl-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9g) (0.0290 g, 71%) as an orange solid. Mp 111–115 °C (lit.37 111 °C); δH (400 MHz, CDCl3) 7.51–7.60 (3H, m, 3′-H, 4′-H and 5′-H), 7.62–7.69 (2H, m, 2′-H and 6′-H), 7.84 (1H, td, J 7.7, 1.1 Hz, 7-H), 7.94 (1H, td, J 7.7, 1.4 Hz, 6-H), 8.10–8.15 (2H, m, 5-H and 8-H); δC (101 MHz, CDCl3) 121.1 (CH), 126.6 (C), 128.1 (2 × CH), 129.7 (2 × CH), 129.8 (CH), 130.1 (CH), 133.2 (CH), 134.4 (CH), 135.0 (C), 141.4 (C); m/z (ESI) 282 (MNa+. 100%).
N-(4′-Methylphenyl)-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9h)36.
To a stirred solution of 2-amino-N-(4′-methylphenyl)lbenzenesulfonamide (8h) (0.0650 g, 0.248 mmol) in methanol (2.5 mL) was added polymer-supported nitrite (0.213 g, containing 0.743 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.141 g, 0.743 mmol). The reaction mixture was stirred at 0 °C for 1 h, warmed to room temperature and stirred for a further 2.5 h. The reaction mixture was diluted with ethyl acetate (20 mL) and washed with water (4 × 20 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography, eluting with 20% ethyl acetate in hexane gave N-(4′-methylphenyl)-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9h) (0.0440 g, 65%) as an orange solid. Spectroscopic data were consistent with the literature.36 Mp 100–102 °C; δH (400 MHz, CDCl3) 2.45 (3H, s, 4′-CH3), 7.35 (2H, br d, J 8.0 Hz, 3′-H and 5′-H), 7.50–7.55 (2H, m, 2′-H and 6′-H), 7.83 (1H, td, J 7.6, 1.2 Hz, 6-H), 7.93 (1H, td, J 7.6, 1.4 Hz, 7-H), 8.08–8.14 (2H, m, 5-H and 8-H); δC (101 MHz, CDCl3) 21.4 (CH3), 121.1 (CH), 126.7 (C), 128.1 (2 × CH), 129.7 (CH), 130.3 (2 × CH), 132.3 (C), 133.1 (CH), 134.4 (CH), 140.5 (C), 141.5 (C); m/z (ESI) 296 (MNa+. 100%).
N-(4′-Methoxyphenyl)-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9i)38.
To a stirred solution of 2-amino-N-(4′-methoxyphenyl)benzenesulfonamide (8i) (0.0550 g, 0.197 mmol) in methanol (2 mL) was added polymer-supported nitrite (0.170 g, containing 0.592 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.113 g, 0.592 mmol). The reaction mixture was stirred at 0 °C for 1 h, warmed to room temperature and stirred for a further 1.3 h. The reaction mixture was diluted with ethyl acetate (20 mL) and washed with water (4 × 20 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography, eluting with 50% diethyl ether in hexane gave N-(4′-methoxyphenyl)-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9i) (0.0390 g, 68%) as a yellow solid. Mp 77–82 °C (lit.38 83–84 °C); δH (400 MHz, CDCl3) 3.88 (3H, s, 4′-OCH3), 7.05 (2H, br d, J 8.8 Hz, 3′-H and 5′-H), 7.56 (2H, br d, J 8.8 Hz, 2′-H and 6′-H), 7.80–7.87 (1H, m, 6-H), 7.90–7.97 (1H, m, 7-H), 8.08–8.17 (2H, m, 5-H and 8-H); δC (101 MHz, CDCl3) 55.8 (CH3), 114.9 (2 × CH), 121.1 (CH), 126.5 (C), 127.1 (C), 129.7 (CH), 130.0 (2 × CH), 133.1 (CH), 134.4 (CH), 141.4 (C), 161.1 (C); m/z (ESI) 284 (MNa+ − N2. 100%).
N-(4′-Bromophenyl)-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9j).
To a stirred solution of 2-amino-N-(4′-bromoophenyl)benzenesulfonamide (8j) (0.100 g, 0.306 mmol) in methanol (3.1 mL) was added polymer-supported nitrite (0.263 g, containing 0.917 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.174 g, 0.917 mmol). The reaction mixture was stirred at 0 °C for 2 h. The reaction mixture was diluted with ethyl acetate (20 mL) and washed with water (4 × 20 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography, eluting with 40% diethyl ether in hexane gave N-(4′-bromophenyl)-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9j) (0.0700 g, 68%) as an orange solid. Mp 107–112 °C; νmax/cm−1 (neat) 2981 (CH), 1572 (CC), 1471, 1448, 1337, 1168, 904, 759; δH (400 MHz, CDCl3) 7.50–7.56 (2H, m, 2′-H and 6′-H), 7.65–7.71 (2H, m, 3′-H and 5′-H), 7.85 (1H, td, J 7.7, 1.2 Hz, 6-H), 7.95 (1H, td, J 7.7, 1.3 Hz, 7-H), 8.12 (1H, dd, J 7.7, 1.3 Hz, 5-H), 8.13 (1H, dd, J 7.7, 1.2 Hz, 8-H); δC (101 MHz, CDCl3) 121.1 (CH), 124.4 (C), 126.6 (C), 129.5 (2 × CH), 129.9 (CH), 132.9 (2 × CH), 133.4 (CH), 134.1 (C), 134.6 (CH), 141.3 (C); m/z (ESI) 359.9408 (MNa+. C12H879BrN3NaO2S requires 359.9413).
N-(4′-Iodophenyl)-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9k).
To a stirred solution of 2-amino-N-(4′-iodophenyl)benzenesulfonamide (8k) (0.119 g, 0.318 mmol) in methanol (3.2 mL) was added polymer-supported nitrite (0.273 g, containing 0.954 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.182 g, 0.954 mmol). The reaction mixture was stirred at 0 °C for 2 h. The reaction mixture was diluted with ethyl acetate (30 mL) and washed with water (4 × 30 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography, eluting with 50% diethyl ether in hexane gave N-(4′-iodophenyl)-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (9k) (0.0720 g, 59%) as an orange solid. Mp 115–118 °C; νmax/cm−1 (neat) 1570 (CC), 1472, 1454, 1338, 1182, 1159, 1128, 808, 762; δH (400 MHz, CDCl3) 7.37–7.41 (2H, m, 2′-H and 6′-H), 7.83–7.91 (3H, m, 6-H, 3′-H and 5′-H), 7.96 (1H, td, J 7.8, 1.4 Hz, 7-H), 8.12 (1H, dd, J 7.8, 1.4 Hz, 5-H), 8.14 (1H, dd, J 7.8, 1.2 Hz, 8-H); δC (101 MHz, CDCl3) 96.0 (C), 121.1 (CH), 126.6 (C), 129.6 (2 × CH), 130.0 (CH), 133.4 (CH), 134.6 (CH), 134.8 (C), 138.9 (2 × CH), 141.3 (C); m/z (ESI) 407.9274 (MNa+. C12H8IN3NaO2S requires 407.9274).
N-Naphthyl-1,2,3-benzotriazin-4(3H)-one (10)26.
To a stirred solution of 1,2,3-benzotriazin-4(3H)-one (7a) (0.100 g, 0.680 mmol), 2-naphthaleneboronic acid (0.175 g, 1.02 mmol) and copper acetate (0.123 g, 0.680 mmol) in dichloroethane (7 mL) was added triethylamine (188 μL, 1.36 mmol). The reaction was stirred at room temperature for 3 h and then heated to 45 °C for a further 2.5 h. The reaction mixture was cooled to room temperature and filtered through a pad of Celite®. The reaction mixture was concentrated in vacuo. Purification by flash column chromatography, eluting with 60% diethyl ether in hexane gave N-naphthyl-1,2,3-benzotriazin-4(3H)-one (10) (0.118 g, 64%) as a yellow solid. Mp 169–173 °C (lit.26 173–175 °C); δH (400 MHz, CDCl3) 7.53–7.61 (2H, m, 4′-H and 5′-H), 7.76 (1H, dd, J 8.8, 2.1 Hz, 8′-H), 7.85–7.90 (1H, m, 6-H), 7.91–7.97 (2H, m, 3′-H and 6′-H), 7.98–8.04 (2H, m, 7-H and 7′-H), 8.18 (1H, d, J 2.1 Hz, 2′-H), 8.26 (1H, br d, J 8.1 Hz 8-H), 8.48 (1H, dd, J 8.0, 1.5 Hz, 5-H); δC (101 MHz, CDCl3) 120.6 (C), 123.8 (CH), 125.1 (CH), 125.8 (CH), 126.9 (CH), 127.2 (CH), 127.9 (CH), 128.6 (CH), 128.7 (CH), 129.1 (CH), 133.0 (CH), 133.2 (C), 133.3 (C), 135.3 (CH), 136.4 (C), 143.9 (C), 155.6 (C); m/z (ESI) 296 (MNa+. 100%).
N-(Ethoxycarbonylmethyl)-1,2,3-benzotriazin-4(3H)-one (11)2c.
To a stirred solution of 1,2,3-benzotriazin-4(3H)-one (7a) (0.150 g, 1.02 mmol) in acetonitrile (10 ml) was added ethyl bromoacetate (0.113 mL, 1.02 mmol) and potassium carbonate (0.141 g, 1.02 mmol). The reaction mixture was heated under reflux for 18 h. The reaction mixture was cooled to room temperature, diluted in chloroform (30 mL) and washed with water (3 × 30 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography, eluting with 25% ethyl acetate in hexane gave N-(ethoxycarbonylmethyl)-1,2,3-benzotriazin-4(3H)-one (11) (0.140 g, 59%) as an off-white solid. Mp 107–111 °C (lit.2c 114–116 °C); δH (400 MHz, CDCl3) 1.31 (3H, t, J 7.1 Hz, OCH2CH3), 4.28 (2H, q, J 7.1 Hz, OCH2CH3), 5.19 (2H, s, CH2CO2Et), 7.83 (1H, ddd, J 8.8, 7.9, 1.2 Hz, 6-H), 7.98 (1H, ddd, J 8.8, 8.2, 1.5 Hz, 7-H), 8.19 (1H, ddd, J 8.2, 1.2, 0.6 Hz, 8-H), 8.37 (1H, ddd, J 7.9, 1.5, 0.6 Hz, 5-H); δC (101 MHz, CDCl3) 14.3 (CH3), 51.0 (CH2), 62.2 (CH2), 119.9 (C), 125.4 (CH), 128.7 (CH), 132.8 (CH), 135.3 (CH), 144.5 (C), 155.7 (C), 167.2 (C); m/z (ESI) 256 (MNa+. 100%).
N-(2′-Diethylaminoethyl)acetamide-1,2,3-benzotriazin-4(3H)-one (3)2c.
To a stirred solution of N-(ethoxycarbonylmethyl)-1,2,3-benzotriazin-4(3H)-one (11) (0.100 g, 0.430 mmol) in anhydrous methanol (4 mL) was added dropwise N,N-diethylethylenediamine (0.0610 mL, 0.430 mmol). The reaction mixture was heated under reflux for 18 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. Purification by flash column chromatography, eluting with 10% methanol in dichloromethane gave N-(2′-diethylaminoethyl)acetamide-1,2,3-benzotriazin-4(3H)-one (3) (0.0940 g, 72%) as a white solid. Mp 147–150 °C (lit.2c 155–156 °C); δH (400 MHz, CDCl3) 0.97 (6H, t, J 7.1 Hz, 2 × NCH2CH3), 2.55 (4H, q, J 7.1 Hz, 2 × NCH2CH3), 2.61 (2H, t, J 5.7 Hz, Et2NCH2), 3.39 (2H, q, J 5.7 Hz, CONHCH2), 5.13 (2H, s, CH2CONH), 6.85 (1H, br s, NH), 7.79–7.85 (1H, m, 6-H), 7.94–8.00 (1H, m, 7-H), 8.19 (1H, dd, J 8.2, 0.5 Hz, 8-H), 8.36 (1H, dd, J 7.9, 1.5 Hz, 5-H); δC (101 MHz, CDCl3) 11.4 (2 × CH3), 36.9 (CH2), 46.9 (2 × CH2), 51.5 (CH2), 52.8 (CH2), 120.0 (C), 125.3 (CH), 128.7 (CH), 132.7 (CH), 135.2 (CH), 144.5 (C), 155.8 (C), 166.1 (C); m/z (ESI) 304 (MNa+. 100%).
N-[2′-(4′′-Benzoylpiperazin-1-yl)ethyl]-2-nitrobenzenesulfonamide (13).
2-Nitrobenzenesulfonyl chloride (0.0560 g, 0.253 mmol) was dissolved in dichloromethane (0.2 mL) and added dropwise to a stirred solution of 1-benzoyl-4-[2′-aminoethyl]piperazine (12) (0.0550 g, 0.236 mmol) and triethylamine (0.0340 mL, 0.248 mmol) in dichloromethane (0.4 mL) at 0 °C. The reaction mixture was warmed to room temperature and stirred for 2 h. The reaction mixture was concentrated in vacuo. Purification by flash column chromatography, eluting with 1% methanol in dichloromethane gave N-[2′-(4′′-benzoylpiperazin-1-yl)ethyl]-2-nitrobenzenesulfonamide (13) (0.0880 g, 83%) as a colourless oil. νmax/cm−1 (neat) 3335 (NH), 2941 (CH), 1627 (CO), 1576 (CC), 1539, 1437, 1364, 1166, 1012, 759; δH (500 MHz, CD3OD) 2.39 (2H, br s, 2′′-H2), 2.49 (2H, br s, 6′′-H2), 2.54 (2H, br t, J 6.1 Hz, 2′-H2), 3.21 (2H, br t, J 6.1 Hz, 1′-H2), 3.37 (2H, br s, 3′′-H2), 3.68 (2H, br s, 5′′-H2), 7.35–7.51 (5H, m, Ph), 7.79–7.86 (2H, m, 4-H and 6-H), 7.90 (1H, br t, J 5.3 Hz, 5-H), 8.10–8.15 (1H, m, 3-H); δC (126 MHz, CD3OD) 41.1 (CH2), 43.0 (CH2), 53.4 (CH2), 54.0 (CH2), 57.5 (2 × CH2), 126.1 (CH), 128.0 (2 × CH), 129.7 (2 × CH), 131.2 (CH), 131.7 (CH), 133.7 (CH), 134.7 (C), 135.1 (CH), 136.6 (C), 149.6 (C), 172.4 (C); m/z (ESI) 441.1211 (MNa+. C19H22N4NaO5S requires 441.1203).
N-[2′-(4′′-Benzoylpiperazin-1-yl)ethyl]-2-aminobenzenesulfonamide (14).
To a stirred solution of N-[2′-(4′′-benzoylpiperazin-1-yl)ethyl]-2-nitrobenzenesulfonamide (13) (0.0830 g, 0.198 mmol) in methanol (2 mL) was added zinc powder (0.129 g, 1.98 mmol) and acetic acid (0.113 mL, 1.98 mmol). The reaction was stirred at room temperature for 5 h. The reaction mixture was filtered through a pad of Celite® and washed with methanol (50 mL). The reaction mixture was concentrated in vacuo. Purification by flash column chromatography, eluting with 5% methanol in dichloromethane gave N-[2′-(4′′-benzoylpiperazin-1-yl)ethyl]-2-aminobenzenesulfonamide (14) (0.0650 g, 84%) as a colourless solid. Mp 65–70 °C; νmax/cm−1 (neat) 3373 (NH), 2944 (CH), 1617 (CO), 1575 (CC), 1454, 1305, 1146, 1012, 711; δH (500 MHz, CD3OD) 2.29 (2H, br s, 2′′-H2), 2.35–2.47 (4H, m, 2′-H2 and 6′′-H2), 2.96 (2H, br t, J 6.4 Hz, 1′-H2), 3.39 (2H, br s, 3′′-H2), 3.71 (2H, br s, 5′′-H2), 6.70 (1H, br t, J 8.1 Hz, 5-H), 6.84 (1H, br d, J 8.1 Hz, 3-H), 7.29 (1H, td, J 8.1, 1.2 Hz, 4-H), 7.35–7.51 (5H, m, Ph), 7.62 (1H, dd, J 8.1, 1.2 Hz, 6-H); δC (126 MHz, CD3OD) 40.6 (CH2), 43.1 (CH2), 53.4 (CH2), 53.9 (CH2), 57.3 (2 × CH2), 117.2 (CH), 118.3 (CH), 121.7 (C), 128.0 (2 × CH), 129.7 (2 × CH), 130.6 (CH), 131.1 (CH), 135.0 (CH), 136.7 (C), 147.7 (C), 172.3 (C); m/z (ESI) 411.1463 (MNa+. C19H24N4NaO3S requires 411.1461).
N-[2′-(4′′-Benzoylpiperazin-1-yl)ethyl]-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (5).
To a stirred solution of N-[2′-(4′′-benzoylpiperazin-1-yl)ethyl]-2-aminobenzenesulfonamide (14) (0.0760 g, 0.196 mmol) in methanol (2 mL) was added polymer-supported nitrite (0.164 g, containing 0.587 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.112 g, 0.587 mmol). The reaction mixture was stirred at 0 °C for 1 h, warmed to room temperature and stirred for a further 1 h. The reaction mixture was filtered and the resulting resin was washed with methanol (10 mL). The reaction mixture was concentrated in vacuo. Purification by flash column chromatography, eluting with 60% ethyl acetate in hexane (with 1% triethylamine) gave N-[2′-(4′′-benzoylpiperazin-1-yl)ethyl]-1,2,3,4-benzothiatriazin-1,1(2H)-dioxide (5) (0.0560 g, 72%) as a yellow solid. Mp 60–65 °C; νmax/cm−1 (neat) 2939 (CH), 1630 (CO), 1575 (CC), 1433, 1337, 1188, 1013, 761; δH (500 MHz, CD3OD) 2.53 (2H, br s, 2′′-H2), 2.66 (2H, br s, 6′′-H2), 2.93 (2H, t, J 6.5 Hz, 2′-H2), 3.41 (2H, br s, 3′′-H2), 3.74 (2H, br s, 5′′-H2), 4.41 (2H, t, J 6.5 Hz, 1′-H2), 7.36–7.50 (5H, m, Ph), 7.91 (1H, td, J 7.7, 1.4 Hz, 7-H), 8.01 (1H, td, J 7.7, 1.2 Hz, 6-H), 8.06–8.11 (2H, m, 5-H and 8-H); δC (126 MHz, CD3OD) 43.2 (CH2), 46.2 (CH2), 54.0 (CH2), 54.2 (CH2), 58.0 (2 × CH2), 121.2 (CH), 127.3 (C), 128.0 (2 × CH), 129.7 (2 × CH), 130.3 (CH), 131.1 (CH), 134.4 (CH), 135.6 (CH), 136.8 (C), 142.9 (C), 172.4 (C); m/z (ESI) 422.1253 (MNa+. C19H21N5NaO3S requires 422.1257).
Methyl (2S)-2-(tert-butoxycarbonylamino)-3-(2′-aminobenzoyl)aminopropanoate (16).
To a stirred solution of methyl (2S)-2-(tert-butoxycarbonylamino)-3-aminopropanoate (15) (0.0900 g, 0.412 mmol) in ethyl acetate (1.5 mL) was added isatoic anhydride (0.0740 g, 0.454 mmol). The reaction was heated to 90 °C and stirred for 18 h. After cooling to room temperature, the reaction mixture was concentrated in vacuo. Purification by flash column chromatography, eluting with 40% ethyl acetate in hexane with 1% triethylamine gave methyl (2S)-2-(tert-butoxycarbonylamino)-3-(2′-aminobenzoyl)aminopropanoate (16) (0.0664 g, 48%) as a white solid. Mp 120–124 °C; νmax/cm−1 (neat) 3360 (NH), 2976 (CH), 2928 (CH), 1741 (CO), 1705 (CO), 1522, 1368, 1256, 1161; [α]19D +25.6 (c 0.3, CHCl3); δH (400 MHz, CDCl3) 1.43 (9H, s, 3 × CH3), 3.67–3.85 (5H, m, OCH3 and 3-H2), 4.45–4.55 (1H, m, 2-H), 5.51 (2H, br s, 2′-NH2), 5.60 (1H, br d, J 4.8 Hz, 2-NH), 6.58–6.69 (2H, m, 3′-H and 5′-H), 6.78 (1H, br s, 3-NH), 7.19 (1H, br t, J 7.8 Hz, 4′-H), 7.33 (1H, br d, J 7.8 Hz, 6′-H); δC (101 MHz, CDCl3) 28.4 (3 × CH3), 42.5 (CH2), 52.9 (CH), 53.9 (CH3), 80.6 (C), 115.5 (C), 116.8 (CH), 117.4 (CH), 127.5 (CH), 132.7 (CH), 148.8 (C), 156.1 (C), 169.9 (C), 171.3 (C); m/z (ESI) 360.1532 (MNa+. C16H23N3NaO5 requires 360.1530).
Methyl (2S)-2-(tert-butoxycarbonylamino)-3-[1′,2′,3′-benzotriazin-4′(3H)-one]propanoate (17).
To a stirred solution of methyl (2S)-2-(tert-butoxycarbonylamino)-3-(2′-aminobenzoyl)aminopropanoate (16) (0.100 g, 0.297 mmol) in methanol (3 mL) at 0 °C was added polymer-supported nitrite (0.256 g, containing 0.890 mmol of NO2) and p-toluenesulfonic acid monohydrate (0.169 g, 0.890 mmol). The reaction mixture was stirred at 0 °C for 1 h and then 2 h at room temperature. The reaction mixture was filtered and the resulting resin was washed with methanol (10 mL). The reaction mixture was concentrated in vacuo. The reaction mixture was diluted in ethyl acetate (20 mL) and washed with water (4 × 20 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography, eluting with 60% diethyl ether in hexane gave methyl (2S)-2-(tert-butoxycarbonylamino)-3-[1′,2′,3′-benzotriazin-4′(3H)-one]propanoate (20) (0.0900 g, 87%) as a colourless oil. νmax/cm−1 (neat) 3333 (NH), 2976 (CH), 2928 (CH), 1746 (CO), 1713 (CO), 1688 (CO), 1508, 1368, 1302, 1163; [α]18D −18.2 (c 0.2, CHCl3); δH (400 MHz, CDCl3) 1.32 (9H, s, 3 × CH3), 3.80 (3H, s, OCH3), 4.72 (1H, dd, J 12.8, 6.8 Hz, 3-HH), 4.85–5.00 (2H, m, 2-H and 3-HH), 5.43 (1H, br d, J 6.4 Hz, 2-NH), 7.81 (1H, br t, J 8.0 Hz, 6′-H), 7.95 (1H, td, J 8.0, 0.9 Hz, 7′-H), 8.14 (1H, br d, J 8.0 Hz, 8′-H), 8.35 (1H, dd, J 8.0, 0.9 Hz, 5′-H); δC (101 MHz, CDCl3) 28.3 (3 × CH3), 50.7 (CH2), 52.9 (CH), 53.0 (CH3), 80.4 (C), 119.8 (C), 125.4 (CH), 128.5 (CH), 132.7 (CH), 135.1 (CH), 144.2 (C), 155.2 (C), 156.2 (C), 170.5 (C); m/z (ESI) 371.1323 (MNa+. C16H20N4NaO5 requires 371.1326).
(2S)-2-(tert-Butoxycarbonylamino)-3-[1′,2′,3′-benzotriazin-4′(3H)-one]propanoic acid.
To a stirred solution of methyl (2S)-2-(tert-butoxycarbonylamino)-3-[1′,2′,3′-benzotriazin-4′(3H)-one]propanoate (17) (0.0700 g, 0.201 mmol) in methanol (3.5 mL), dioxane (1.75 mL) and water (1.75 mL) was added caesium carbonate (0.0850 g, 0.261 mmol). The reaction mixture was stirred at room temperature for 18 h. The reaction mixture was concentrated in vacuo, diluted in water (30 mL) and acidified to pH 1 using 1 M aqueous hydrochloric acid. The reaction mixture was extracted with dichloromethane (3 × 30 mL). The organic layers were combined, dried (MgSO4), filtered and concentrated in vacuo to give (2S)-2-(tert-butoxycarbonylamino)-3-[1′,2′,3′-benzotriazin-4′(3H)-one]propanoic acid (0.0660 g, 99%) as a colourless oil. νmax/cm−1 (neat) 3351 (NH), 2973 (CH), 1689 (CO), 1394, 1367, 1301, 1164, 779; [α]19D −52.5 (c 0.2, MeOH); δH (400 MHz, CD3OD) 1.23 (9H, s, 3 × CH3), 4.62 (1H, dd, J 13.2, 9.6 Hz, 3-HH), 4.79 (1H, dd, J 9.6, 4.4 Hz, 2-H), 5.02 (1H, dd, J 13.2, 4.4 Hz, 3-HH), 7.89 (1H, br t, J 7.9 Hz, 6′-H), 8.04 (1H, br t, J 7.9 Hz, 7′-H), 8.13 (1H, br d, J 7.9 Hz, 8′-H), 8.33 (1H, br d, J 7.9 Hz, 5′-H); δC (101 MHz, CD3OD) 28.5 (3 × CH3), 52.3 (CH2), 54.8 (CH), 80.6 (C), 120.8 (C), 125.9 (CH), 129.1 (CH), 133.8 (CH), 136.4 (CH), 145.5 (C), 157.5 (C), 157.6 (C), 173.0 (C); m/z (ESI) 357.1164 (MNa+. C15H18N4NaO5 requires 357.1169).
(2S)-2-Amino-3-[1′,2′,3′-benzotriazin-4′(3H)-one]propanoic acid hydrochloride (18).
A solution of (2S)-2-(tert-butoxycarbonylamino)-3-[1′,2′,3′-benzotriazin-4′(3H)-one]propanoic acid (0.040 g, 0.12 mmol) in 2 M aqueous hydrochloric acid (2 mL) was stirred at 50 °C for 3 h. The reaction mixture was cooled to room temperature and concentrated in vacuo to give (2S)-2-amino-3-[1′,2′,3′-benzotriazin-4′(3H)-one]propanoic acid hydrochloride (18) (0.026 g, 78%) as a white solid. Mp 175–180 °C; νmax/cm−1 (neat) 3372 (NH), 3220 (CH), 1679 (CO), 1651, 1122, 784; [α]19D −5.4 (c 0.2, MeOH); δH (400 MHz, CD3OD) 4.63 (1H, dd, J 7.2, 4.4 Hz, 2-H), 4.94 (1H, dd, J 14.6, 7.2 Hz, 3-HH), 5.11 (1H, dd, J 14.6, 4.4 Hz, 3-HH), 7.95 (1H, td, J 8.1, 1.2 Hz, 6′-H), 8.10 (1H, td, J 8.1, 1.2 Hz, 7′-H), 8.21 (1H, br d, J 8.1 Hz, 8′-H), 8.35 (1H, dd, J 8.1, 1.2 Hz, 5′-H); δC (101 MHz, CD3OD) 49.7 (CH2), 53.3 (CH), 120.9 (C), 126.0 (CH), 129.6 (CH), 134.4 (CH), 136.8 (CH), 145.4 (C), 157.8 (C), 169.1 (C); m/z (ESI) 257.0643 ([MNa − HCl]+. C10H10N4NaO3 requires 257.0645).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from the University of Glasgow (studentships to R. M. and R. J. F.) is gratefully acknowledged.
Notes and references
- Z. Khalid, H. A. Ahmad, M. A. Munawar, M. A. Khan and S. Gul, Heterocycles, 2017, 94, 3 CrossRef CAS.
- For example, see:
(a) S. M. Gadekar and J. L. Frederick, J. Org. Chem., 1962, 27, 1383 CrossRef CAS;
(b) A. S. Clark, B. Deans, M. F. G. Stevens, M. J. Tisdale, R. T. Wheelhouse, B. J. Denny and J. A. Hartley, J. Med. Chem., 1995, 38, 1493 CrossRef CAS PubMed;
(c) G. Caliendo, F. Fiorino, P. Grieco, E. Perissutti, V. Santagada, R. Meli, G. M. Raso, A. Zanesco and G. De Nucci, Eur. J. Med. Chem., 1999, 34, 1043 CrossRef CAS;
(d)
F. Fiorino, B. Severino, F. De Angelis, E. Perissutti, F. Frecentese, P. Massarelli, G. Bruni, E. Collavoli, V. Santagada and G. Calliendo, Arch. Pharm. Chem. Life Sci, 2008, 341, 20 Search PubMed;
(e)
X. Xu, Z. Li, X. Chen, W. Li, H. Jia, X. Cao, X. Shao and Z. Xu, CN108276352, 2018.
-
(a) T. Miura, M. Yamauchi and M. Murakami, Org. Lett., 2008, 10, 3085 CrossRef CAS PubMed;
(b) M. Yamauchi, M. Morimoto, T. Miura and M. Murakami, J. Am. Chem. Soc., 2010, 132, 54 CrossRef CAS PubMed;
(c) T. Miura, M. Morimoto, M. Yamauchi and M. Murakami, J. Org. Chem., 2010, 75, 5359 CrossRef CAS PubMed.
-
(a) Z.-J. Fang, S.-C. Zheng, Z. Guo, J.-Y. Guo, B. Tan and X.-Y. Liu, Angew. Chem., Int. Ed., 2015, 54, 9528 CrossRef CAS PubMed;
(b) N. Wang, S.-C. Zheng, L.-L. Zhang, Z. Guo and X.-Y. Liu, ACS Catal., 2016, 6, 3496 CrossRef CAS.
- V. H. Thorat, N. S. Upadhyay, M. Murakami and C.-H. Cheng, Adv. Synth. Catal., 2018, 360, 284 CrossRef CAS.
-
(a) T. Miura, Y. Nishida, M. Morimoto, M. Yamauchi and M. Murakami, Org. Lett., 2011, 13, 1429 CrossRef CAS PubMed;
(b) M. H. Balakrishnan and S. Mannathan, Org. Lett., 2020, 22, 542 CrossRef PubMed.
- M. H. Balakrishnan, K. Sathriyan and S. Mannathan, Org. Lett., 2018, 20, 3815 CrossRef PubMed.
- H. Wang and S. Yu, Org. Lett., 2015, 17, 4272 CrossRef CAS PubMed.
- M. S. Ao and E. M. Burgess, J. Am. Chem. Soc., 1971, 93, 5298 CrossRef CAS.
-
(a) G. Ege and E. Beisiegel, Liebigs Ann. Chem., 1972, 763, 46 CrossRef CAS;
(b) Y.-Y. Han, H. Wang and S. Yu, Org. Chem. Front., 2016, 3, 953 RSC.
- Y. Xie, G. Gong, Y. Liu, S. Deng, A. Rinderspacher, L. Branden and D. W. Landry, Tetrahedron Lett., 2008, 49, 2320 CrossRef CAS.
-
(a) E. Van Heyningen, J. Am. Chem. Soc., 1955, 77, 6562 CrossRef CAS;
(b) J. P. Colomer and E. L. Moyano, Tetrahedron Lett., 2011, 52, 1561 CrossRef CAS.
- For example, see:
(a) Y. Yan, H. Li, B. Niu, C. Zhu, T. Chen and Y. Liu, Tetrahedron Lett., 2016, 57, 4170 CrossRef CAS;
(b) N. G. Khaligh, M. R. Johan and J. J. Ching, Aust. J. Chem., 2018, 71, 186 CrossRef CAS;
(c) D. S. Barak, S. Mukhopadhyay, D. J. Dahatonde and S. Batra, Tetrahedron Lett., 2019, 60, 248 CrossRef CAS;
(d) Y.-M. Cai, X. Zhang, C. An, Y.-F. Yang, W. Liu, W.-X. Gao, X.-B. Huang, Y.-B. Zhou, M.-C. Liu and H.-Y. Wu, Org. Chem. Front., 2019, 6, 1481 RSC.
- Y. Yan, B. Niu, K. Xu, J. Yu, H. Zhi and Y. Liu, Adv. Synth. Catal., 2016, 358, 212 CrossRef CAS.
- A. Chandrasekhar and S. Sankararaman, J. Org. Chem., 2017, 82, 11487 CrossRef CAS PubMed.
- Z. Lai, C. Wang, J. Li and S. Cui, Org. Lett., 2020, 22, 2017 CrossRef CAS PubMed.
- Y. Zhou, Y. Wang, Y. Lou and Q. Song, Org. Lett., 2018, 20, 6494 CrossRef CAS PubMed.
-
(a) M. Caldarelli, I. R. Baxendale and S. V. Ley, Green Chem., 2000, 2, 43 RSC;
(b) J. Merrington, M. James and M. Bradley, Chem. Commun., 2002, 140 RSC;
(c) V. D. Filimonov, M. Trusova, P. Postnikov, E. A. Krasnokutskaya, Y. M. Lee, H. Y. Hwang, H. Kim and K.-W. Chi, Org. Lett., 2008, 10, 3961 CrossRef CAS PubMed;
(d) M. E. Trusova, E. A. Krasnokutskaya, P. S. Postnikov, Y. Choi, K.-W. Chi and V. D. Filimonov, Synthesis, 2011, 2154 CAS.
-
(a) N. L. Sloan, S. K. Luthra, G. McRobbie, S. L. Pimlott and A. Sutherland, Chem. Commun., 2017, 53, 11008 RSC;
(b) N. L. Sloan, S. K. Luthra, G. McRobbie, S. L. Pimlott and A. Sutherland, RSC Adv., 2017, 7, 54881 RSC.
- R. J. Faggyas, M. Grace, L. Williams and A. Sutherland, J. Org. Chem., 2018, 83, 12595 CrossRef CAS PubMed.
-
(a) R. J. Faggyas, N. L. Sloan, N. Buijs and A. Sutherland, Eur. J. Org. Chem., 2019, 5344 CrossRef CAS;
(b) J. D. Bell, T. E. F. Morgan, N. Buijs, A. H. Harkiss, C. R. Wellaway and A. Sutherland, J. Org. Chem., 2019, 84, 10436 CrossRef CAS PubMed.
-
(a) J. R. Feldman and E. C. Wagner, J. Org. Chem., 1942, 7, 31 CrossRef CAS;
(b) M. S. Manhas, S. G. Amin and V. V. Rao, Synthesis, 1977, 309 CrossRef CAS.
- See ESI† document for the synthesis of 2-aminobenzamides, N-aryl 2-aminobenzamides, N-substituted 2-aminobenzenesulfonamides and 1-benzoyl-4-[2′-aminoethyl]piperazine (12).
-
F. A. V. Sullivan, US2798055, 1957.
-
W. M. Thomas, N. Heights and F. A. V. Sullivan, US2834764, 1958.
- K. S. Kumar, R. Adepu, S. Sandra, D. Rambabu, G. R. Krishna, C. M. Reddy, P. Misra and M. Pal, Bioorg. Med. Chem. Lett., 2012, 22, 1146 CrossRef PubMed.
- For example, see:
(a) C. M. Reid, K. N. Fanning, L. S. Fowler and A. Sutherland, Tetrahedron, 2015, 71, 245 CrossRef CAS;
(b) L. Gilfillan, R. Artschwager, A. H. Harkiss, R. M. J. Liskamp and A. Sutherland, Org. Biomol. Chem., 2015, 13, 4514 RSC;
(c) A. H. Harkiss, J. D. Bell, A. Knuhtsen, A. G. Jamieson and A. Sutherland, J. Org. Chem., 2019, 84, 2879 CrossRef CAS PubMed;
(d) J. D. Bell, A. H. Harkiss, D. Nobis, E. Malcolm, A. Knuhtsen, C. R. Wellaway, A. G. Jamieson, S. W. Magennis and A. Sutherland, Chem. Commun., 2020, 56, 1887 RSC.
- M. R. Stojkovic, P. Piotrowski, C. Schmuck and I. Piantanida, Org. Biomol. Chem., 2015, 13, 1629 RSC.
- This type of transformation cannot tolerate substrates with strongly nucleophilic substituents such as other amines or hydroxyl groups but as demonstrated by compounds 14 and 16, protected versions of these are acceptable.
- Y. Chang, J. Zhang, X. Chen, Z. Li and X. Xu, Bioorg. Med. Chem. Lett., 2017, 27, 2641 CrossRef CAS PubMed.
-
F. G. Kathawalas, US3678166, 1972.
- J. A. Zablocki, E. Elzein, X. Li, D. O. Koltun, E. Q. Parkhill, T. Kobayashi, R. Martinez, B. Corkey, H. Jiang, T. Perry, R. Kalla, G. T. Nottee, O. Saunders, M. Graupe, Y. Lu, C. Venkataramani, J. Guerrero, J. Perry, M. Osier, R. Strickley, G. Liu, W.-Q. Wang, L. Hu, X.-J. Li, N. El-Bizri, R. Hirakawa, K. Kahlig, C. Xie, C. H. Li, A. K. Dhalla, S. Rajamani, N. Mollova, D. Soohoo, E.-I. Lepist, B. Murray, G. Rhodes, L. Belardinelli and M. C. Desai, J. Med. Chem., 2016, 59, 9005 CrossRef CAS PubMed.
- J. G. Archer, A. J. Barker and R. K. Smalley, J. Chem. Soc., Perkin Trans. 1, 1973, 1169 RSC.
- A. Rosowsky, R. A. Forsch and R. G. Moran, J. Med. Chem., 1992, 35, 2626 CrossRef CAS PubMed.
- R. J. LeBlanc and K. Vaughan, Can. J. Chem., 1972, 50, 2544 CrossRef CAS.
- T. Miura, M. Yamauchi, A. Kosaka and M. Murakami, Angew. Chem., Int. Ed., 2010, 49, 4955 CrossRef CAS PubMed.
- F. Ullmann and C. Groß, Ber. Dtsch. Chem. Ges., 1910, 43, 2694 CrossRef.
- A. Saeed and N. H. Rama, J. Chem. Soc. Pak., 1994, 16, 31 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and data for starting materials, 1H and 13C NMR spectra for all compounds. See DOI: 10.1039/d1ob00968k |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*