
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiocatalytic enantioselective hydroaminations enabling synthesis of N-arylalkyl-substituted L-aspartic acids†
Mohammad Z.
Abidin
 ab,
Thangavelu
Saravanan
*ac,
Laura
Bothof
a,
Pieter G.
Tepper
a,
Andy-Mark W. H.
Thunnissen
d and
Gerrit J.
Poelarends
*a
ab,
Thangavelu
Saravanan
*ac,
Laura
Bothof
a,
Pieter G.
Tepper
a,
Andy-Mark W. H.
Thunnissen
d and
Gerrit J.
Poelarends
*a
aDepartment of Chemical and Pharmaceutical Biology, Groningen Research Institute of Pharmacy, University of Groningen, Antonius Deusinglaan 1, 9713 AV Groningen, The Netherlands. E-mail: g.j.poelarends.rug.nl; Fax: +31-50-3633000; Tel: +31-50-3633354
bDepartment of Animal Product Technology, Faculty of Animal Science, Gadjah Mada University, Bulaksumur, Yogyakarta, 55281, Indonesia
cSchool of Chemistry, University of Hyderabad, P.O. Central University, Hyderabad-500046, India. E-mail: tsaravanan@uohyd.ac.in
dMolecular Enzymology Group, Groningen Institute of Biomolecular Sciences and Biotechnology, University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands
First published on 30th June 2021
Abstract
N-Substituted L-aspartic acids are important chiral building blocks for pharmaceuticals and food additives. Here we report the asymmetric synthesis of various N-arylalkyl-substituted L-aspartic acids using ethylenediamine-N,N′-disuccinic acid lyase (EDDS lyase) as a biocatalyst. This C–N lyase shows a broad non–natural amine substrate scope and outstanding enantioselectivity, allowing the efficient addition of structurally diverse arylalkylamines to fumarate to afford the corresponding N-arylalkyl-substituted L-aspartic acids in good isolated yield (up to 79%) and with excellent enantiopurity (>99% ee). These results further demonstrate that C–N lyases working in reverse constitute an extremely powerful synthetic tool to prepare difficult noncanonical amino acids.
Introduction
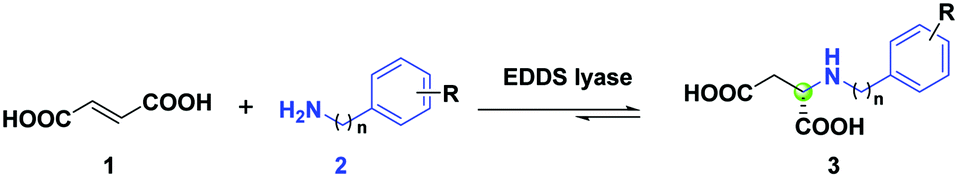
N-Substituted L-aspartic acids and their derivatives are key intermediates in developing promising pharmaceuticals and nutraceuticals with potent biological activities.1–5 Therefore, there is a need to prepare these L-aspartic acid derivatives expediently. The conventional chemical synthesis of N-substituted L-aspartic acids usually relies on reductive amination or asymmetric hydroamination strategies, which often involves multiple steps and harsh reaction conditions.6–9 In this respect, alternative methods need to be pursued to achieve environmentally friendlier and more-step economic synthesis of optically pure N-substituted L-aspartic acids. Hence, an enantioselective biocatalytic hydroamination approach would be an interesting option.Two well-studied carbon–nitrogen (C–N) lyases, aspartate ammonia lyase (DAL)10–12 and 3-methylaspartate ammonia lyase (MAL),10,11,13 have been employed as biocatalysts in the enantioselective hydroamination of fumaric acid to afford N-substituted L-aspartic acids. In addition, we started to investigate another interesting C–N lyase, ethylenediamine-N,N′-disuccinic acid (EDDS) lyase from Chelativorans sp. BNC1, which utilizes a characteristic serine residue (Ser-280) as the catalytic base to facilitate the deamination of (S,S)-EDDS to give ethylenediamine and two fumarate molecules.14 We showed that EDDS lyase accepts various non-native amine substrates in the hydroamination of fumaric acid, including various linear mono- and diamines, homo- and heterocycloalkyl amines, arylamines, and arylhydrazines, enabling the enantioselective synthesis of a wide variety of N-substituted L-aspartic acid derivatives.14–16 The broad amine scope of EDDS lyase also supported the chemoenzymatic synthesis of the fungal natural products aspergillomarasmine A, aspergillomarasmine B, and toxin A, as well as related aminocarboxylic acids.17 EDDS lyase was recently optimized by structure-inspired engineering for the enantioselective synthesis of several challenging N-substituted aspartic acids, which are important chiral precursors to artificial dipeptide sweeteners such as neotame and advantame.18
The absence of cofactors and its capacity to catalyze enantioselective hydroaminations with a variety of amines, make EDDS lyase an attractive enzyme for biocatalytic applications. In this study, we further explored the non-native amine substrate scope of EDDS lyase using a panel of structurally diverse arylalkylamines in the hydroamination of fumaric acid to yield various enantiopure N-arylalkyl-substituted L-aspartic acids.
Results and discussion
We previously reported that both aniline (2x, n = 0) and benzylamine (2y, n = 1) are accepted as non-native substrates in the hydroamination of fumaric acid (1) by EDDS lyase (Scheme 1).16,18 This observation prompted us to test a wide variety of arylalkylamines (2a–2u) with different aliphatic linkers between the amino and aryl groups, as well as having different substituents, as potential substrates in the hydroamination reaction catalyzed by EDDS lyase. The progress of the enzymatic hydroaminations was monitored by 1H NMR spectroscopy (Fig. S1–S21†). Pleasingly, amines 2a–2o, 2r, 2t and 2u were accepted as non-native substrates by EDDS lyase (up to 91% conversion), while amines 2p, 2q and 2s were not processed (Scheme 1).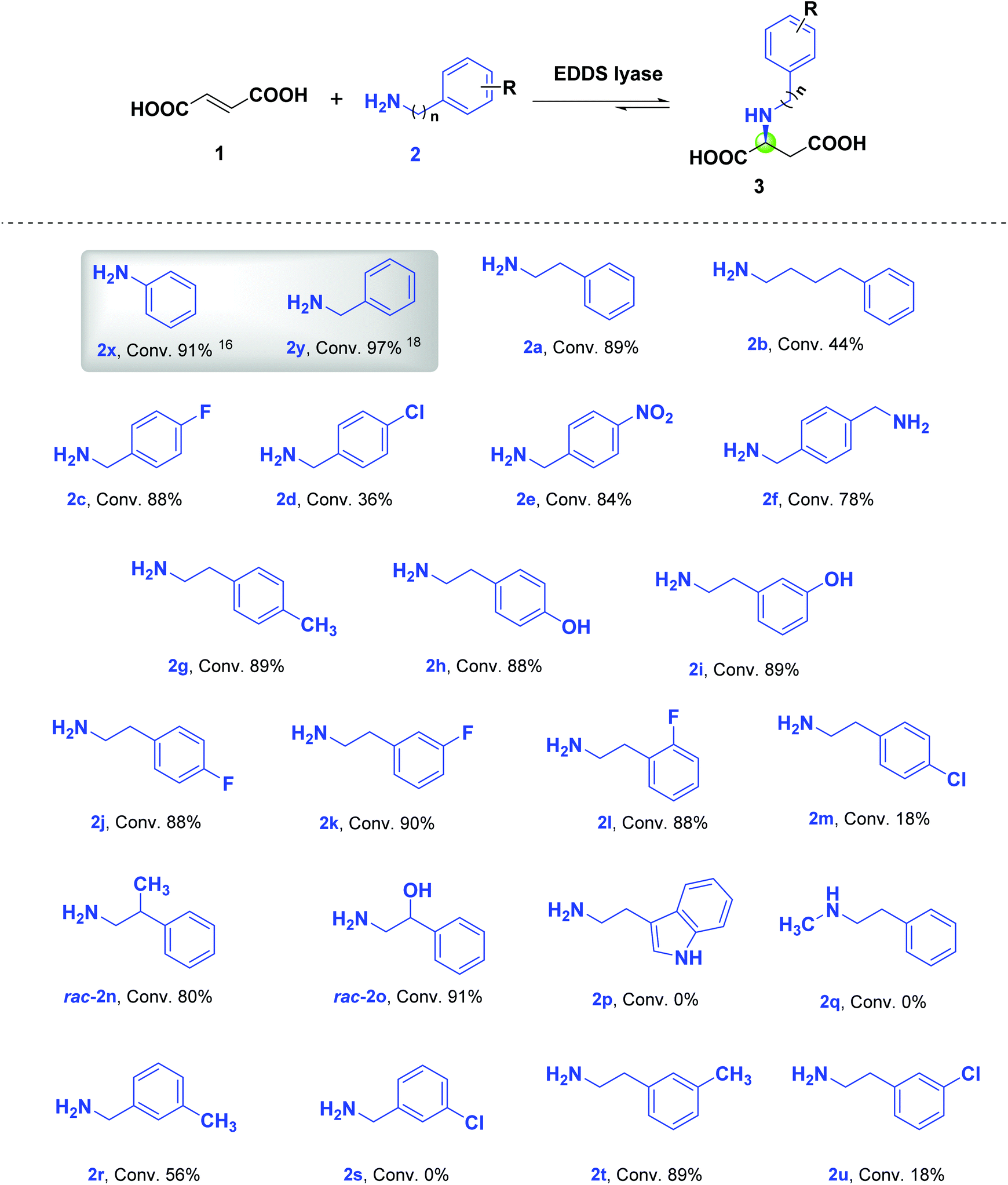 | ||
| Scheme 1 The broad arylalkylamine substrate scope of EDDS lyase enables the asymmetric synthesis of difficult N-substituted L-aspartic acids. Reaction mixtures contained EDDS lyase (15 μM), 1 (10 mM) and 2a–2u (50 mM) in 3 mL NaH2PO4 buffer (20 mM, pH 8.5). Reaction mixtures were incubated at room temperature for 24 h; except for those containing 2c, 2e, 2f, and 2n, which were incubated for 48 h. Conversion was determined by 1H NMR spectroscopy by comparing signals of the amine substrates and corresponding products. | ||
The EDDS lyase catalyzed hydroamination of 1 with 2-phenylethylamine (2a) and 4-phenylbutylamine (2b) resulted in 89% and 44% conversions, respectively (Scheme 1, Fig. S1 and S2†). The lower conversion observed with 2b compared to 2a (as well as 2x and 2y) clearly shows that EDDS lyase prefers amine substrates with short (≤2) aliphatic linkers between the amino and aryl groups. Interestingly, the enzyme accepts distinct para-substituted benzylamines (2c–2e) with reasonable to good conversions (Fig. S3–S5†). Under the conditions used, the EDDS lyase catalyzed hydroamination reaction between diamine 2f (1,4-phenylenedimethanamine) and 1 resulted in the single-addition product with 78% conversion (Fig. S6†).
To further examine the versatility of EDDS lyase, a variety of phenethylamines with different substitutions on the aryl ring such as p-methyl-phenethylamine (2g), p- and m-hydroxy-phenethylamine (2h and 2i), p-, m- and o-fluoro-phenethylamine (2j–2l) and p-chloro-phenethylamine (2m) were tested as non-native amine substrates. EDDS lyase accepted these phenethylamines as substrates in the hydroamination of 1, giving excellent conversion (88–90%) for all amines with the exception of 2m (18%) (Fig. S7–S13†). Overall, the electronic nature and position of the substituents on the aryl ring did not show a significant effect on conversion. However, the p-chloro substituent on phenethylamine (2m) had a negative influence on conversion, which resembles the negative consequence of the p-chloro substituent on the transformation of benzylamine (2d). The crystal structure of EDDS lyase does not provide an immediate explanation for the difficulties in accommodating the p-chloro-phenyl group.14
Notably, prolonged incubation of EDDS lyase with amines 2d and 2m resulted in limited enzyme precipitation, which may also at least partly explain their lower conversion. This effect was not observed upon incubation of the enzyme with the other amines. Further investigation indeed shows that amines 2r and 2t (carrying a m-methyl group) are well converted, whereas the corresponding amines 2s and 2u (carrying a m-chlorine group) are not or poorly processed (Fig. S18–S21†), showing limited enzyme precipitation upon extended incubation.
We further tested racemic 2-substituted phenethylamines (rac-2n and rac-2o) as non-native amine substrates to study the enantiopreference of EDDS lyase. The enzyme accepted both enantiomers of rac-2n and rac-2o, resulting in 80% and 91% conversion, respectively (Fig. S14 and S15†). Hence, the relatively small methyl and hydroxyl substituents are not able to induce enantioselective enzymatic transformation. Lastly, tryptamine (2p) and N-methyl-phenethylamine (2q) were tested as potential amine substrates for EDDS lyase. These two compounds were not accepted by EDDS lyase (Fig. S16 and S17†), indicating that the enzyme does not accept very bulky amines nor secondary amines as substrates for hydroamination of 1.
Despite the use of non-native amine substrates, respectable specific activities are observed, illustrated by the addition of 2a (140 mU mg−1), 2c (21 mU mg−1) and 2j (119 mU mg−1) to 1 (Fig. S71†). For comparison, EDDS lyase exhibits a specific activity of 543 mU mg−1 for the addition of ethylenediamine to 1. To further demonstrate the synthetic usefulness of the EDDS lyase catalyzed hydroamination reactions, we performed semi-preparative scale synthesis of a few selected N-substituted aspartic acids (3a, 3c, 3d, and 3g–3j). High conversions, excellent enantiocontrol (>99% ee), and good isolated product yields (up to 76%) were achieved (Table 1, Fig. S22–S28 and S43–S49†). Conveniently, EDDS lyase exclusively produced the desired S enantiomer of the N-substituted-aspartic acids (Fig. S64–S70†), consistent with the previously reported enantiopreference of this enzyme, and in accordance with the experimentally determined binding mode of (S,S)-EDDS in the crystal structure of EDDS lyase (Fig. S72†).14–18 With fumarate being firmly bound at the back of the active site pocket, the amino group of the second substrate can only perform a pro-S attack (re-face of Cα) because of steric reasons (si-face is not accessible) and because of a favourable hydrogen bond formed with the side chain of Asn113. Thus, despite the use of structurally diverse amines, ranging from small alkylamines to large amino acids, the stereochemical outcome of the EDDS lyase catalyzed hydroamination reaction is not affected.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Amine | Product | Reaction time (h) | Yieldb (%) (mg) | eec (%) | Abs. conf.d |
| a Reaction mixtures contained 1 (10 mM, 0.15 mmol), 2a, 2c, 2d or 2g–j (50 mM), and EDDS lyase (15 μM) in 15 ml NaH2PO4 buffer (20 mM, pH 8.5, room temperature). b Isolated product yield after ion-exchange chromatography. c The ee was determined by high-performance liquid chromatography (HPLC) using a chiral stationary phase and chemically synthesized authentic standards. d Determined by HPLC using a chiral stationary phase and chemically synthesized authentic standards. | ||||||
| 1 | 2a |
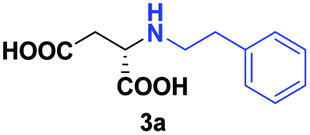
|
24 | 70 (25) | >99 | S |
| 2 | 2c |
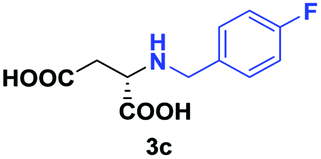
|
48 | 75 (27) | >99 | S |
| 3 | 2d |
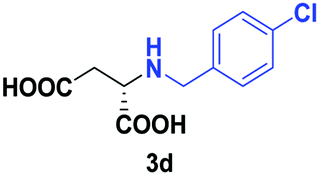
|
24 | 28 (11) | >99 | S |
| 4 | 2g |
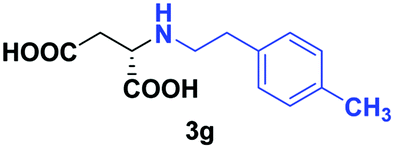
|
24 | 66 (25) | >99 | S |
| 5 | 2h |
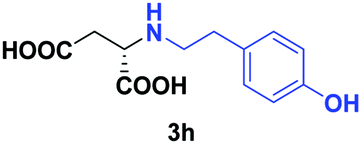
|
24 | 61 (23) | >99 | S |
| 6 | 2i |
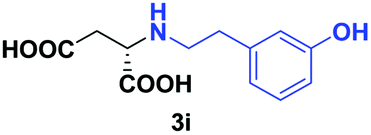
|
24 | 63 (24) | >99 | S |
| 7 | 2j |
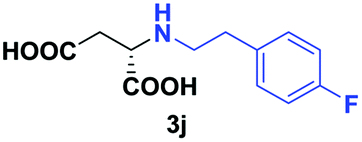
|
24 | 76 (29) | >99 | S |
Next, we investigated whether the stereochemical outcome of the enzymatic hydroamination reaction could be altered by using maleic acid (i.e., cis-butenedioic acid) as electrophile instead of fumarate (i.e., trans-butenedioic acid). However, the enzyme was found to be stereospecific for fumarate, with maleic acid not accepted as alternative electrophile (Fig. S73†). Given that EDDS lyase can also not accept fumaric acid monomethyl ester, crotonic acid, mesaconic acid, itaconic acid, 2-pentenoic acid, and glutaconic acid as alternative substrates,14 the redesign of EDDS lyase for hydroamination of alternative electrophiles remains an important challenge.
In summary, we have shown that EDDS lyase accepts a wide variety of arylalkylamines, with different aliphatic linkers between the amino and aryl groups, as well as carrying different substituents, in the asymmetric hydroamination of fumarate yielding the desired N-substituted L-aspartic acids with high optical purity and in good isolated yield. Together with previously reported work from our laboratory,14–18 this study demonstrates that EDDS lyase has an exceptionally broad amine scope, making it a very attractive biocatalyst for enantioselective production of various hard to synthesize L-aspartate derivatives. Examples of important L-aspartate derivatives include biodegradable metal chelators, metallo-beta-lactamase and glutamate transporter inhibitors, photoswitchable and photocaged drug-like molecules, as well as various N-heterocycles and other chiral synthons that are widely used in pharmaceutical synthesis.19–21 We have recently initiated structure-based and computer-aided protein engineering studies with the aim to enlarge the electrophile substrate scope of EDDS lyase to further increase its synthetic usefulness.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Netherlands Organization of Scientific Research (VICI grant 724.016.002) and from the European Research Council (PoC grant 713483). M. Z. A. acknowledges funding from the Indonesia Endowment Fund for Education (LPDP).References
- M. A. T. Blaskovich, J. Med. Chem., 2016, 59, 10807–10836 CrossRef CAS PubMed.
- M. Maiti, M. Maiti, J. Rozenski, S. De Jonghe and P. Herdewijn, Org. Biomol. Chem., 2015, 13, 5158–5174 RSC.
- A. M. King, S. A. Reid-Yu, W. Wang, D. T. King, G. De Pascale, N. C. Strynadka, T. R. Walsh, B. K. Coombes and G. D. Wright, Nature, 2014, 510, 503 CrossRef CAS PubMed.
- B. C. Mcllwain, R. J. Vandenberg and R. M. Ryan, Biochemistry, 2016, 55, 6801–6810 CrossRef.
- S. Chattopadhyay, U. Raychaudhuri and R. Chakraborty, J. Food Sci. Technol., 2014, 51, 611–621 CrossRef CAS PubMed.
- A. Zilkha and M. D. Bachi, J. Org. Chem., 1959, 24, 1096–1098 CrossRef CAS.
- D. M. Roundhill, Chem. Rev., 1992, 92, 1–27 CrossRef CAS.
- P. S. Piispanen and P. M. Pihko, Tetrahedron Lett., 2005, 46, 2751–2755 CrossRef CAS.
- M. Boros, J. Kökösi, J. Vámos, I. Kövesdi and B. Noszál, Amino Acids, 2007, 33, 709–717 CrossRef CAS PubMed.
- M. de Villiers, V. Puthan Veetil, H. Raj, J. de Villiers and G. J. Poelarends, ACS Chem. Biol., 2012, 7, 1618–1628 CrossRef CAS PubMed.
- F. Parmeggiani, N. J. Weise, S. T. Ahmed and N. J. Turner, Chem. Rev., 2018, 118, 73–118 CrossRef CAS PubMed.
- R. Li, H. J. Wijma, L. Song, Y. Cui, M. Otzen, Y. Tian, J. Du, T. Li, D. Niu, Y. Chen, J. Feng, J. Han, H. Chen, Y. Tao, D. B. Janssen and B. Wu, Nat. Chem. Biol., 2018, 14, 664–670 CrossRef CAS PubMed.
- H. Raj, W. Szymański, J. De Villiers, H. J. Rozeboom, V. Puthan Veetil, C. R. Reis, M. De Villiers, F. J. Dekker, S. De Wildeman, W. J. Quax, A.-M. W. H. Thunnissen, B. L. Feringa, D. B. Janssen and G. J. Poelarends, Nat. Chem., 2012, 4, 478–484 CrossRef CAS PubMed.
- H. Poddar, J. de Villiers, J. Zhang, V. Puthan Veetil, H. Raj, A.-M. W. H. Thunnissen and G. J. Poelarends, Biochemistry, 2018, 57, 3752–3763 CrossRef CAS PubMed.
- J. Zhang, H. Fu, P. G. Tepper and G. J. Poelarends, Adv. Synth. Catal., 2019, 361, 2433–2437 CrossRef CAS.
- H. Fu, A. Prats Luján, L. Bothof, J. Zhang, P. G. Tepper and G. J. Poelarends, ACS Catal., 2019, 9, 7292–7299 CrossRef CAS.
- H. Fu, J. Zhang, M. Saifuddin, G. Cruiming, P. G. Tepper and G. J. Poelarends, Nat. Catal., 2018, 1, 186–191 CrossRef CAS.
- J. Zhang, E. Grandi, H. Fu, T. Saravanan, L. Bothof, P. G. Tepper, A.-M. W. H. Thunnissen and G. J. Poelarends, Angew. Chem., 2020, 59, 429–435 CrossRef CAS PubMed.
- K. H. M. E. Tehrani, H. Fu, N. C. Brüchle, V. Mashayekhi, A. Prats Luján, M. J. van Haren, G. J. Poelarends and N. I. Martin, Chem. Commun., 2020, 56, 3047–3049 RSC.
- J. Zhang, M. Z. Abidin, T. Saravanan and G. J. Poelarends, ChemBioChem, 2020, 21, 2733–2742 CrossRef CAS PubMed.
- V. Arkhipova, H. Fu, M. W. H. Hoorens, G. Trinco, L. N. Lameijer, E. Marin, B. L. Feringa, G. J. Poelarends, W. Szymanski, D. J. Slotboom and A. Guskov, J. Am. Chem. Soc., 2021, 143, 1513–1520 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob00748c |
| This journal is © The Royal Society of Chemistry 2021 |