
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Studies on a catalytic version of the Matteson asymmetric homologation reaction†
Keith
Smith
 *a,
Basil A.
Saleh
ab,
Mohammed B.
Alshammari
ac,
Gamal A.
El-Hiti
d and
Mark C.
Elliott
*a
*a,
Basil A.
Saleh
ab,
Mohammed B.
Alshammari
ac,
Gamal A.
El-Hiti
d and
Mark C.
Elliott
*a
aSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK. E-mail: smithk13@cardiff.ac.uk; elliottmc@cardiff.ac.uk
bDepartment of Chemistry, College of Science, University of Basrah, Iraq
cChemistry Department, College of Sciences and Humanities, Prince Sattam bin Abdulaziz University, P.O. Box 83, Al-Kharij 11942, Saudi Arabia
dCornea Research Chair, Department of Optometry, College of Applied Medical Sciences, King Saud University, P.O. Box 10219, Riyadh 11433, Saudi Arabia
First published on 22nd April 2021
Abstract
Studies of a catalytic asymmetric version of the Matteson reaction between dichloromethylboronates and organolithium reagents have been undertaken. From several different chiral catalytic systems studied, only one based on a mannitol derivative has given substantial asymmetric induction close to that previously achieved with a bis(oxazoline) derivative and ytterbium triflate. More detailed study of the latter reaction revealed that fresh ytterbium triflate actually reduced the level of asymmetric induction, while “aged” ytterbium triflate, or a fresh sample that had been treated with water, brought about improved induction. The implications of these findings are discussed.
Introduction
Boronic acid derivatives are important intermediates with potential applications in areas such as medicine and organic synthesis,1 while the use of chiral catalysts has also proven to be efficient for the asymmetric synthesis of various organic compounds.2–6 Boronic esters are generally stable compounds with low to moderate toxicity and have been widely used in the production of either carbon–carbon or carbon-heteroatom bonds.7 A particularly useful reaction of chiral boronic esters (1) involves their reactions as electrophiles with dichloromethyllithium as a nucleophile, followed by rearrangement of the ate complexes produced, to give the corresponding chiral α-haloboronic esters (2) with high levels of asymmetric induction at the newly created stereogenic centre (Matteson asymmetric homologation, Scheme 1).8–11 Further stereospecific reactions of the α-haloboronic esters with organometallic reagents provide access to sec-alkylboronic esters (3), which on oxidation give sec-alcohols. Alternatively, 3 can be subjected to a similar sequence over again to generate a second contiguous stereogenic centre in a new boronic ester (4) and further iterations are also possible to give products with multiple contiguous stereogenic centres. Such reactions can provide routes to many useful targets in which the stereochemistry is controlled.8,12 For example, the elm bark beetle pheromone 5 (R1 = n-Pr, R2 = Me, R3 = Et) was synthesized with high stereoselection from 1 (R = i-Pr, R1 = n-Pr) and the corresponding Grignard reagents.13 The other diastereoisomers can be obtained by varying the stereoisomer of the boronate and/or interchanging the groups R1 and R2. Similar methodology can also be extended in a limited way to the synthesis of tertiary alcohols by use of a more complicated dichloroalkyllithium reagent. For example, (R)-(+)-2-phenyl-2-butanol (84% enantiomeric excess; ee) was synthesized using a reaction like that in Scheme 1 from a chiral phenylboronate (R1 = Ph), but with 1,1-dichloroethy1lithium instead of dichloromethyllithium and then ethylmagnesium bromide (R2 = Et).14 The role played by ZnCl2 as a Lewis acid is vital to improve the yield or stereoisomer predominance (ee or de) of the products in all of these reactions.13,15,16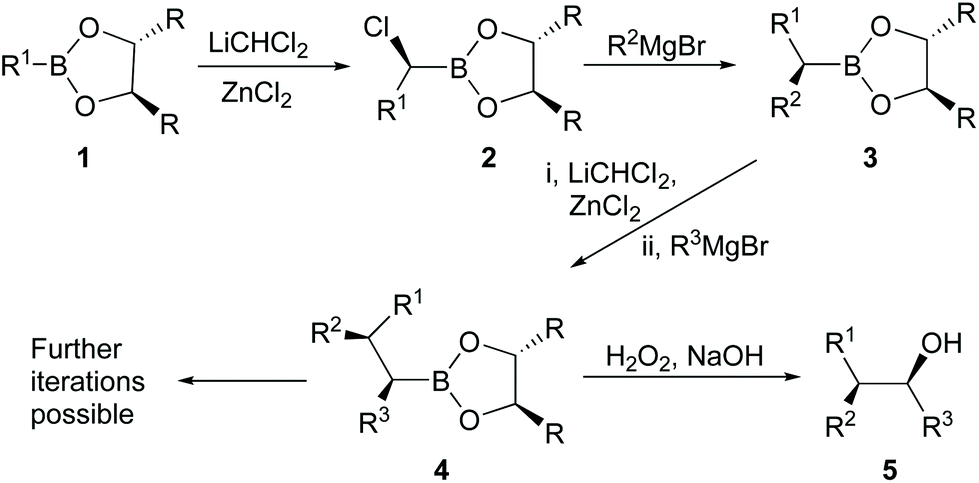 | ||
| Scheme 1 Matteson asymmetric homologation. | ||
In the Matteson reaction the chiral auxiliary is located in the boronate ester (substrate control), but others have adapted the approach to allow use of an achiral boronate with a chiral organolithium reagent (reagent control). For example, Aggarwal has made extensive and elegant use of reactions of chiral lithiated alkylcarbamates with B-alkyl pinacolboronates17–19 and such reactions are more widely applicable to synthesis of tertiary alcohols and even to generation of quaternary stereogenic centres.20 Blakemore's approach has been to generate labile chiral main group metal carbenoids, such as α-chloroalkyllithiums in situ.21–23
Although these approaches are undoubtedly valuable additions to the synthetic toolbox, as with the Matteson approach they require a stoichiometric amount of the chiral auxiliary, and it would be desirable to be able to introduce the chiral control through a catalytic agent. A successful catalytic procedure has been introduced that uses a chiral nickel catalyst, racemic α-chloroalkylboronates and achiral organozinc reagents and it provides good yields and enantioselectivities for a range of different boronates and organozinc reagents with 16 mol% of the chiral ligand,24 but in a stereo-convergent manner (both enantiomers of the α-chloroalkylboronate leading to the same enantiomer of the product), indicating that the mechanism is different from that of the Matteson reaction and potentially not as general. Nevertheless, the nickel-catalysed procedure has been applied successfully in the stereoselective synthesis of cholesterols and other complex substrates.24
To our knowledge, there is only one report of the use of a catalytic procedure for the Matteson type of reaction, involving use of an achiral boronic ester (e.g. 2-(dichloromethyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane), and an achiral organolithium reagent in the presence of a chiral ligand (e.g. a bis-oxazoline), and a metal triflate as a Lewis acid.25 However, the process needed a significant (five-fold) excess of the chiral ligand in order to provide a good level of asymmetric induction in the reaction studied, ostensibly because lithium chloride generated in situ competed for the ligand.25 As part of our interest in the use of organometallic intermediates and in particular boron reagents in organic syntheses,26–32 we decided to investigate this procedure in more detail in the hope that we may be able to overcome the difficulties and develop a procedure that was successful with a much smaller quantity of chiral ligand. We now report our results.
Results and discussion
First, we attempted an alternative way to carry out the Jadhav reaction25 in which reaction of alkylboronate and dichloromethyllithium rather than dichloromethylboronate and alkyllithium was investigated. The synthesis of various alkylboronates should be easier than synthesis of dichloromethylboronate and a range of alkyllithiums. Alternative catalysts 6 and 7 (Fig. 1), which incorporated both the Lewis acid and chiral auxiliary components in a single molecule, were synthesised using published procedures33–35 and used in comparison with the Jadhav catalyst (8, Fig. 1, +Yb(OTf)3) in the reaction of 9 to give 10 followed by reaction with (1S,2S,3R,5S)-2,3-pinanediol [(S)-pinanediol] to produce 11 (Scheme 2), which allowed the level of asymmetric induction in 10 to be monitored (there is no epimerisation α- to boron and a high yield of the pinanediol boronate ensures that the de for this compound reflects the ee in the pinacol boronate precursor; for background information on NMR analysis of diastereoisomer ratios of asymmetric boron-containing products, see ref. 36). Reaction of 9 with n-BuLi (−78 °C, THF) in the presence of chiral reagents 6, 7, and [8 + Yb(OTf)3] gave 10 in 88–98% yields (Scheme 2).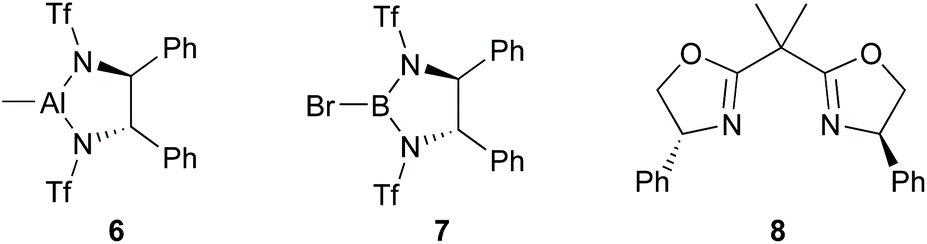 | ||
| Fig. 1 Chiral reagents 6–8.33–35,37 | ||
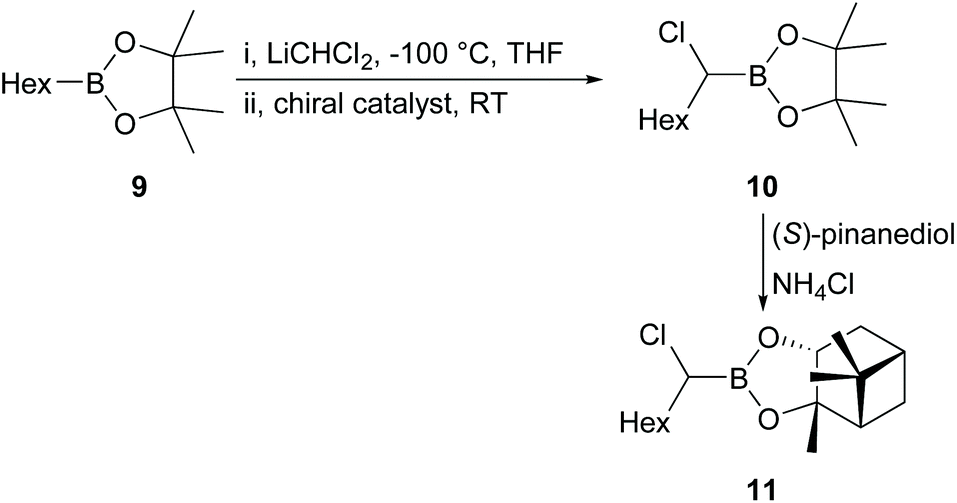 | ||
| Scheme 2 Alternative Jadhav reaction. | ||
Product 11 was produced in high yields (81–90%) after purification, but the results obtained showed that use of reagent 8 (ratio 9/8/Yb(OTf)3 = 1/2.5/0.3) gave only 4% ee in 10 based on the de calculated from the 1H NMR spectrum of 11. Such a low percentage de was surprising, even for a reaction in THF, which had been shown to give lower levels of induction than non-coordinating solvents,25 since the intermediate involved in the reaction of 9 to give 10 was assumed to be very similar to that in the original Jadhav reaction.25 However, catalysts 6 and 7 (ratio 9/6 or 7 = 1/2.5) gave essentially no induction (≤1% de). Therefore, we decided to revert to the original Jadhav reaction to check that everything was in order and attempted reaction of 12 with n-BuLi (−40 °C, hexane) in the presence of chiral ligand 8 and Yb(OTf)3 (Scheme 3; ratio 12/8/Yb(OTf)3 = 1/0.5/0.2). The product 13 was produced in 89% yield after purification. Reaction of 13 with (S)-pinanediol gave 14 in 88% yield, but even under these conditions the product 14 was formed with only 21% de, in contrast to the figure of 55% de reported by Jadhav under similar conditions.25
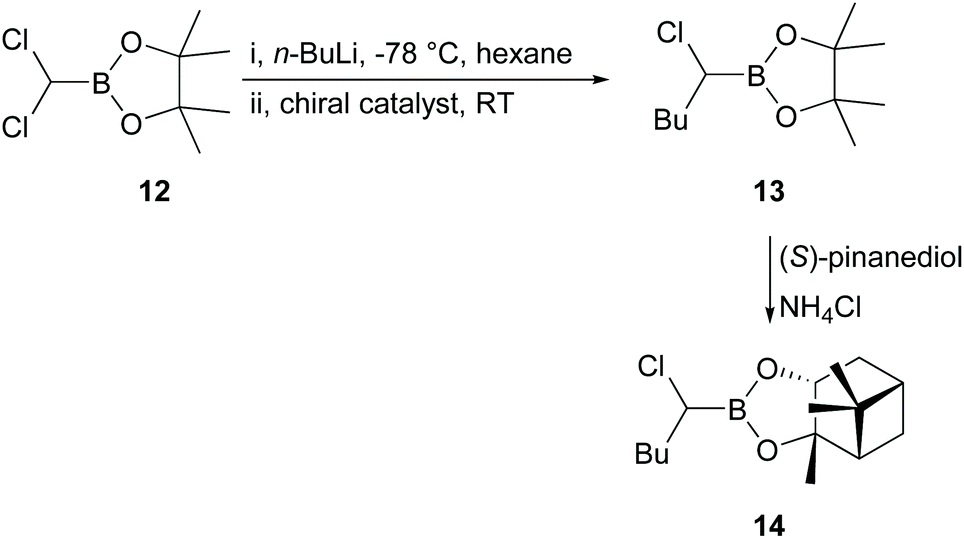 | ||
| Scheme 3 Jadhav reaction with various chiral catalysts. | ||
Nevertheless, we decided to conduct the reaction represented in Scheme 3 in one-pot under conditions similar to the optimal ones reported by Jadhav25 but with chiral catalysts 6 and 7 (Fig. 1), and with chiral ligands 15, 16, 17, 18, and 19 (Fig. 2) in the presence of Yb(OTf)3. The results obtained are shown in Table 1.
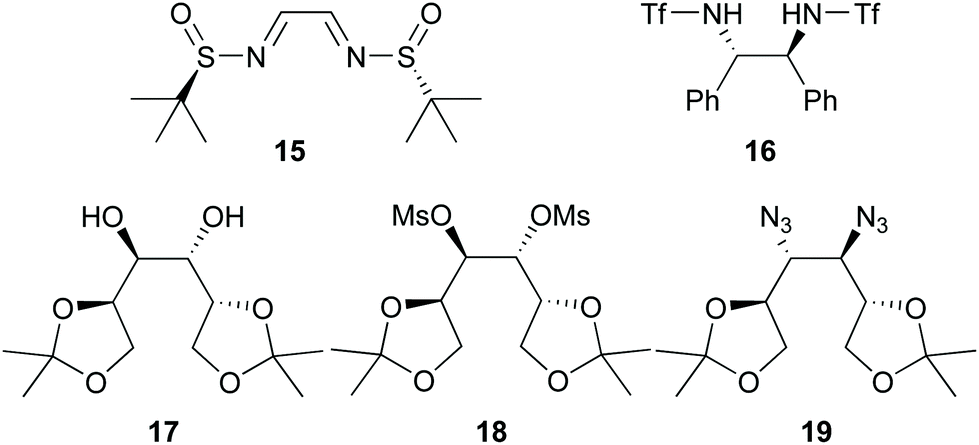 | ||
| Fig. 2 Chiral ligands 15–19.34,38–41 | ||
| Entry | Chiral catalyst | Yield (%) | de (%) |
|---|---|---|---|
| a Ratio 12/6 or 7 = 1/2.5; ratio 12/15, 16, 17, 18 or 19/Yb(OTf)3 = 1/2.5/0.3. | |||
| 1 | 6 | 86 | 11 |
| 2 | 7 | 89 | 2.5 |
| 3 | 15 + Yb(OTf)3 | 84 | 4 |
| 4 | 16 + Yb(OTf)3 | 85 | 16 |
| 5 | 17 + Yb(OTf)3 | 89 | 52 |
| 6 | 18 + Yb(OTf)3 | 84 | 4 |
| 7 | 19 + Yb(OTf)3 | 88 | 2 |
Although all of the chiral catalysts gave some induction, none gave a de as high as that reported by Jadhav (79% with 8 and Yb(OTf)3 under similar conditions).25 The highest induction (52% de) was achieved when chiral ligand 17 (Fig. 2) was used in the presence of Yb(OTf)3 (Table 1; entry 5). In order to gauge the importance of the Lewis acid, we conducted this reaction in the absence of Yb(OTf)3. The level of induction achieved (46% de) was within experimental variation of that achieved when Yb(OTf)3 was present, raising doubts about the role of Yb(OTf)3 in this case. Chiral catalyst 6 and ligand 16 in the presence of Yb(OTf)3 gave modest induction (11 and 16%, respectively; Table 1, entries 1 and 4), but catalyst 7 and ligands 15, 18, and 19 in the presence of Yb(OTf)3 (Table 1; entries 2, 3, 6, 7, respectively), gave very poor induction (only 2–4% de). In view of the poor levels of induction, even with Jadhav's catalyst 8 in our hands under conditions similar to ones reported by Jadhav,25 we decided to investigate in more detail the reaction with the Jadhav catalyst (8).25
It had been speculated by Jadhav that the presence of Li cations within the system resulted in competition with the Yb for complexation with the chiral ligand. This had been suggested as the reason why such a large amount of the chiral ligand was needed in order to provide good induction.25 Such speculation was not entirely consistent with the results reported by Jadhav,25 but nevertheless we decided to investigate the matter further by observing the influence of addition of 12-crown-4, which forms a strong complex with Li cations, on the level of induction in the reaction depicted in Scheme 3 in the presence of 8 (ratio 12/8/Yb(OTf)3 = 1/0.5/0.21). If the speculation be correct, the effective removal of Li from complexation with bis(oxazoline) should allow the ligand to be free to complex with Yb, thereby increasing the likelihood of good asymmetric induction. The addition of small quantities of 12-crown-4 did indeed lead to an increase in the level of induction. For example, the use of 0.6 equivalents of the crown ether led to a maximum of around 50% de (Fig. 3; each point represents the result of a separate experiment). However, larger quantities of the crown ether (up to 1.5 equivalents) caused the level of induction to drop again (Fig. 3).
 | ||
| Fig. 3 Effect of 12-crown-4 in the reaction shown in Scheme 3 under Jadhav conditions with 8 (ratio 12/8/Yb(OTf)3 = 1/0.5/0.21) on the level of asymmetric induction. | ||
In order to interpret the effect of addition of the crown ether, it is important to recognise that Yb also forms a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (ligand:Yb) complex with 12-Crown-4.42 Therefore, the shape of the graph (Fig. 3) could be explained qualitatively if the 12-crown-4 preferentially complexes Li, freeing up some additional chiral ligand for complexation to Yb, resulting in an increase in de, until the amount of crown ether added becomes sufficient for significant competition with the chiral ligand for complexation of Yb to occur, at which point the de begins to decrease again. However, a qualitatively similar shape of graph would result if the Li [bis(oxazoline)] complex is the important one for induction and if the Yb complex with the crown ether is stronger than the Li complex, so that with a small amount of crown ether complexation of the crown ether with Yb would free up chiral ligand to increase induction, but with larger quantities of crown ether the crown ether competes with the chiral ligand for complexation of Li, so that induction diminishes.
:1 (ligand:Yb) complex with 12-Crown-4.42 Therefore, the shape of the graph (Fig. 3) could be explained qualitatively if the 12-crown-4 preferentially complexes Li, freeing up some additional chiral ligand for complexation to Yb, resulting in an increase in de, until the amount of crown ether added becomes sufficient for significant competition with the chiral ligand for complexation of Yb to occur, at which point the de begins to decrease again. However, a qualitatively similar shape of graph would result if the Li [bis(oxazoline)] complex is the important one for induction and if the Yb complex with the crown ether is stronger than the Li complex, so that with a small amount of crown ether complexation of the crown ether with Yb would free up chiral ligand to increase induction, but with larger quantities of crown ether the crown ether competes with the chiral ligand for complexation of Li, so that induction diminishes.
In order to distinguish these two possibilities, an experiment was conducted in which the amount of Yb(OTf)3 was varied while keeping other materials constant (and no crown ether present). If the Yb complex with the chiral ligand is the important one for induction, as the proportion of Yb:Li increases, the level of induction should increase, whereas the induction should decrease if the Li complex with the chiral ligand is the important one. In practice, the level of induction decreased as the proportion of Yb increased (Fig. 4; each point represents the result of a separate experiment). For example, the de was highest (45%) in the absence of Yb(OTf)3 and was lowest (18%) when 0.1 equivalents of Yb(OTf)3 was used. This result suggests that the Li complex with the chiral ligand is more important than the Yb one for bringing about induction. This suggested that to achieve the highest level of induction it would be better to have no Yb present.
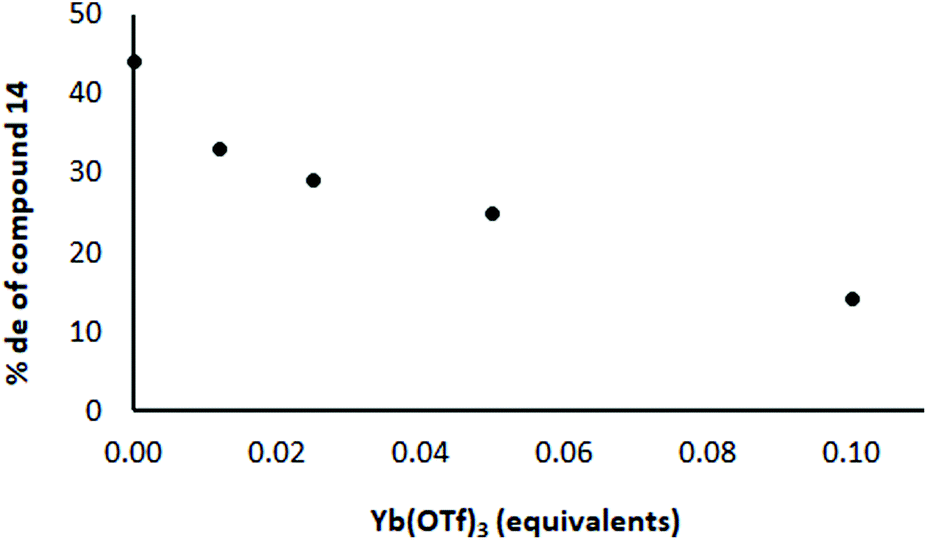 | ||
| Fig. 4 Effect of Yb(OTf)3 on the % de in the reaction shown in Scheme 3 with ratio 12/8/Yb(OTf)3 = 1/0.5/as shown in the diagram. The ligand 8 and the appropriate quantity of Yb(OTf)3 were premixed and added to the reaction mixture as a solution. | ||
Next, we conducted the reaction represented in Scheme 3 in the absence of Yb(OTf)3 using different quantities of the chiral ligand 8 under otherwise identical conditions. It turned out that the level of induction increased almost linearly with the quantity of chiral ligand 8 present until one equivalent of ligand had been introduced and then levelled off, at ca. 60% de (Fig. 5; each point represents the result of a separate experiment). This supports the hypothesis that it is the Li complex that is important for induction and not the Yb complex, but suggested that there was no catalytic turnover. However, the maximum level of induction was still somewhat lower than Jadhav had reported even with Yb present.25
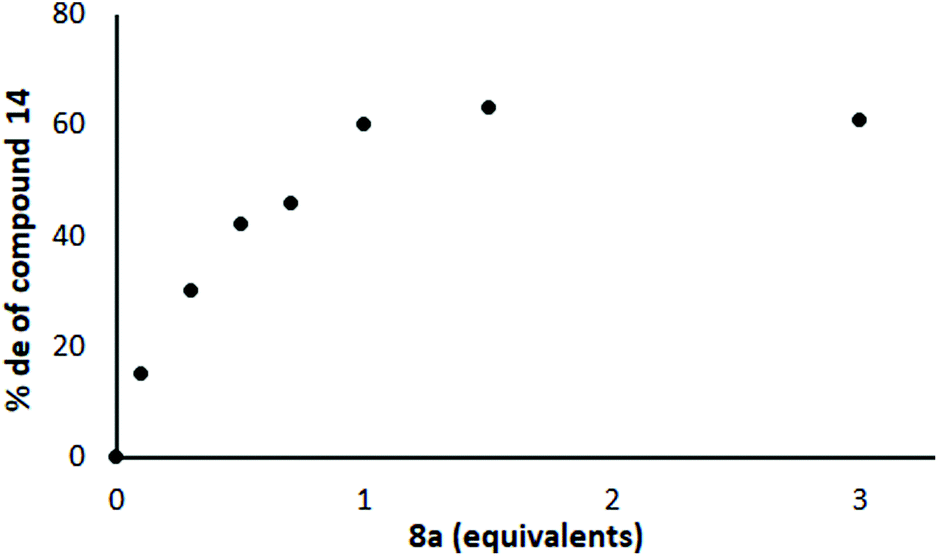 | ||
| Fig. 5 Effect of chiral ligand 8 on the % de in the reaction shown in Scheme 3 (no Yb(OTf)3 present). | ||
In the light of that result, we wondered whether different samples of Yb(OTf)3 might behave differently. Our experiments reported above had all been conducted with samples of Yb(OTf)3 from a new, previously unopened bottle. We therefore tested Yb(OTf)3 from a bottle that had been opened on numerous occasions. With only 0.5 equivalents of the ligand, 70% de was achieved, comparable with Jadhav's result25 under similar conditions and suggesting some level of catalytic turnover. A similar result was achieved using a fresh supply of Yb(OTf)3 and treating it with water for 24 h before use in the reaction. It would seem likely, therefore, that something arising from the reaction of Yb(OTf)3 with water could enhance the level of induction a little and bring about a modicum of catalytic turnover. However, it is also possible that some direct involvement of water in the reaction occurs, as has been observed in some main group organometallic reactions.43–45 Neither varying the reaction temperature (0 to −78 °C) nor the solvent produced any further improvement in the de. Further research is therefore required in order to identify a suitable catalyst system to provide both high levels of asymmetric induction and good catalytic turnover.
Conclusions
In conclusion, therefore, from a range of alternative catalyst systems, only a chiral catalyst derived from mannitol has provided a reasonable level of asymmetric induction and even this catalyst gave a lower level of induction than the bis(oxazoline) compound introduced by Jadhav. Yb(OTf)3, which was proposed by Jadhav to be an important Lewis acid catalyst for the process, when sourced from a new bottle, does not appear to have any beneficial effect on the level of asymmetric induction and better induction is achieved in its absence. However, “aged” Yb(OTf)3, which has been exposed to air/moisture for some time, does appear to give better levels of induction even than the reaction without Yb(OTf)3 present and even allows a modicum of catalytic turnover, but the extent of the turnover is insufficient to render the process attractive. More research is needed in order to identify the nature of the “aged” Yb(OTf)3 and to identify a catalyst system that provides both high levels of asymmetric induction and good catalytic turnover in Matteson-type reactions.Author contributions
Conceptualization and experimental design: K. S. and M. C. E.; methodology: K. S., G. A. E.-H. and M. C. E.; supervision: K. S., G. A. E.-H. and M. C. E.; investigation and formal analysis: B. A. S. and M. B. A.; writing: K. S., G. A. E.-H. and M. C. E. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Cardiff University and the Governments of Saudi Arabia and Iraq for support. G. A. El-Hiti is grateful to the Deanship of Scientific Research, King Saud University, for funding through the Vice Deanship of Scientific Research Chairs. B. A. Saleh is grateful to the University of Basrah for supporting him during his PhD study leave. We are grateful for Dr B. D. Ward (Cardiff University) for assistance with the analysis of NMR spectra of stereoisomers.Notes and references
-
Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. G. Hall, Wiley-VCH, Weinheim, Germany, 2011 Search PubMed
.
-
Lewis Acids in Organic Synthesis, ed. H. Yamamoto, Wiley-VCH, Weinheim, Germany, 2002 Search PubMed
- T. N. Nguyen, P.-A. Chen, K. Setthakarn and J. A. May, Molecules, 2018, 23, 2317 CrossRef PubMed
- S. Namirembe and J. P. Morken, Chem. Soc. Rev., 2019, 48, 3464–3474 RSC
- S. Panda and J. M. Ready, J. Am. Chem. Soc., 2017, 139, 6038–6041 CrossRef CAS PubMed
- L. Zhang, G. J. Lovinger, E. K. Edelstein, A. A. Szymaniak, M. P. Chierchia and J. P. Morken, Science, 2016, 351, 70–74 CrossRef CAS PubMed
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS
- D. S. Matteson, J. Org. Chem., 2013, 78, 10009–10023 CrossRef CAS PubMed
- D. S. Matteson, Tetrahedron, 1989, 45, 1859–1885 CrossRef CAS
- D. S. Matteson, Tetrahedron, 1998, 54, 10555–10607 CrossRef CAS
-
D. S. Matteson, Stereodirected Synthesis with Organoboranes, Springer, Berlin, 1995 Search PubMed
- D. S. Matteson, Chem. Rev., 1989, 89, 1535–1551 CrossRef CAS
- D. S. Matteson, K. M. Sadhu and M. L. Peterson, J. Am. Chem. Soc., 1986, 108, 810–819 CrossRef CAS
- D. S. Matteson and G. D. Hurst, Heteroat. Chem., 1990, 1, 65–74 CrossRef CAS
- D. S. Matteson, P. K. Jesthi and K. M. Sadhu, Organometallics, 1984, 3, 1284–1288 CrossRef CAS
- D. S. Matteson and K. M. Sadhu, J. Am. Chem. Soc., 1983, 105, 2077–2078 CrossRef CAS
- B. S. L. Collins, C. M. Wilson, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2017, 56, 11700–11733 CrossRef CAS PubMed
- C. Sandford and V. K. Aggarwal, Chem. Commun., 2017, 53, 5481–5494 RSC
-
S. G. Aiken, J. M. Bateman and V. K. Aggarwal, Boron “Ate” Complexes for Asymmetric Synthesis, in Science of Synthesis: Advances in Organoboron Chemistry towards Organic Synthesis, 2020, ch. 13, pp. 393–458 Search PubMed
- H. K. Scott and V. K. Aggarwal, Chem. – Eur. J., 2011, 17, 13124–13132 CrossRef CAS PubMed
- P. R. Blakemore, S. P. Marsden and H. D. Vater, Org. Lett., 2006, 8, 773–776 CrossRef CAS PubMed
- P. R. Blakemore and M. S. Burge, J. Am. Chem. Soc., 2007, 129, 3068–3069 CrossRef CAS PubMed
- X. Sun and P. R. Blakemore, Org. Lett., 2013, 15, 4500–4503 CrossRef CAS PubMed
- J. Schmidt, J. Choi, A. T. Liu, M. Slusarczyk and G. C. Fu, Science, 2016, 354, 1265–1269 CrossRef CAS PubMed
- P. K. Jadhav and H.-W. Man, J. Am. Chem. Soc., 1997, 119, 846–847 CrossRef CAS
- K. Smith, M. B. Alshammari and G. A. El-Hiti, Synthesis, 2018, 3634–3652 CrossRef CAS
- G. A. El-Hiti, K. Smith and A. S. Hegazy, Heterocycles, 2015, 91, 479–504 CrossRef CAS
- B. A. Saleh, K. Smith, M. C. Elliott, D. H. Jones, B. M. Kariuki and G. A. El-Hiti, Tetrahedron, 2016, 72, 6914–6928 CrossRef CAS
- D. H. Jones, K. Smith, M. C. Elliott and G. A. El-Hiti, Tetrahedron, 2015, 71, 6285–6289 CrossRef CAS
- M. C. Elliott and K. Smith, Organometallics, 2013, 32, 4878–4881 CrossRef CAS
- M. C. Elliott, K. Smith, D. H. Jones, A. Hussain and B. A. Saleh, J. Org. Chem., 2013, 78, 3057–3064 CrossRef CAS PubMed
- K. Smith, A. A. Balakit, R. T. Pardasani and G. A. El-Hiti, J. Sulfur Chem., 2011, 32, 287–295 CrossRef CAS
- S. Pikul and E. J. Corey, Org. Synth., 1993, 71, 30 CrossRef CAS
- S. Pikul and E. J. Corey, Org. Synth., 1993, 71, 22 CrossRef CAS
- D. C. Braddock, J. M. Redmond, S. A. Hermitage and A. J. P. White, Adv. Synth. Catal., 2006, 348, 911–916 CrossRef CAS
- J. S. Fossey, E. V. Anslyn, W. D. G. Brittain, S. D. Bull, B. M. Chapin, C. S. Le Duff, T. D. James, G. Lees, S. Lim, J. A. C. Lloyd, C. V. Manville, D. T. Payne and K. A. Roper, J. Chem. Educ., 2017, 94, 79–84 CrossRef CAS
- A. Sakakura, R. Kondo and K. Ishihara, Org. Lett., 2005, 7, 1971–1974 CrossRef CAS PubMed
- X. Sun, S. Wang, S. Sun, J. Zhu and J. Deng, Synlett, 2005, 2776–2780 CAS
- E. S. Alvarenga, V. M. T. Carneiro, F. O. Silvério and W. A. Saliba, J. Chil. Chem. Soc., 2006, 51, 986–988 Search PubMed
- A. M. A. Al Majid, H. M. A. Al-Hazimi and M. B. Al-Shammery, Jordan J. Chem., 2010, 5, 221–230 CAS
- S. Hanessian, J.-Y. Gauthier, K. Okamoto, A. L. Beauchamp and T. Theophanides, Can. J. Chem., 1993, 71, 880–885 CrossRef CAS
- C. Qian and L. Wang, Tetrahedron: Asymmetry, 2000, 11, 2347–2357 CrossRef CAS
- S.-C. Kuo, F. Chen, D. Hou, A. Kim-Meade, C. Bernard, J. Liu, S. Levy and G. G. Wu, J. Org. Chem., 2003, 68, 4984–4987 CrossRef CAS PubMed
- J. García–Álvarez, E. Hevia and V. Capriati, Chem. – Eur. J., 2018, 24, 14854–14863 CrossRef PubMed
- K. Smith, Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 1975, 72, 136–149 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data and NMR spectra of compounds. See DOI: 10.1039/d1ob00604e |
| This journal is © The Royal Society of Chemistry 2021 |