
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterization of bichromophoric 1-deoxyceramides as FRET probes†
Eduardo
Izquierdo
ab,
Mireia
Casasampere
b,
Gemma
Fabriàs
 bc,
José Luís
Abad
b,
Josefina
Casas
*bc and
Antonio
Delgado
*ab
bc,
José Luís
Abad
b,
Josefina
Casas
*bc and
Antonio
Delgado
*ab
aDepartment of Pharmacology, Toxicology and Medicinal Chemistry, Unit of Pharmaceutical Chemistry (Associated Unit to CSIC). Faculty of Pharmacy and Food Sciences. University of Barcelona (UB), Joan XXIII 27-31, 08028 Barcelona, Spain. E-mail: antonio.delgado@ub.edu
bResearch Unit on BioActive Molecules, Department of Biological Chemistry, Institute for Advanced Chemistry of Catalonia (IQAC-CSIC), Jordi Girona 18-26, 08034-Barcelona, Spain. E-mail: fina.casas@iqac.csic.es
cLiver and Digestive Diseases Networking Biomedical Research Centre (CIBEREHD), ISCIII, 28029 Madrid, Spain
First published on 23rd February 2021
Abstract
The suitability as FRET probes of two bichromophoric 1-deoxydihydroceramides containing a labelled spisulosine derivative as a sphingoid base and two differently ω-labelled fluorescent palmitic acids has been evaluated. The ceramide synthase (CerS) catalyzed metabolic incorporation of ω-azido palmitic acid into the above labeled spisulosine to render the corresponding ω-azido 1-deoxyceramide has been studied in several cell lines. In addition, the strain-promoted click reaction between this ω-azido 1-deoxyceramide and suitable fluorophores has been optimized to render the target bichromophoric 1-deoxydihydroceramides. These results pave the way for the development of FRET-based assays as a new tool to study sphingolipid metabolism.
Introduction
Sphingolipids (SLs) are one of the major classes of lipids in eukaryotes. Canonical SLs derive from the sphingoid base dihydrosphingosine (dhSo) or (2R,3S) 2-amino-1,3-octadecanediol (Scheme 1), which is metabolically modified to account for the different families of SLs known to date.1 Among them, ceramides (Cer) occupy a pivotal position in the metabolic pathways and they are considered as a metabolic hub.2 Apart from their fundamental structural role in cell membranes,3 Cer are also important second messengers in several cellular processes. In this regard, Cer have been reported to activate apoptosis in response to a variety of cell stress inducing agents4–6 and, consistent with their role in apoptosis, various types of cancer cells have been shown to reduce their Cer levels as a survival strategy through the overexpression of CDase enzymes.7 Furthermore, Cer also participate in the regulation of autophagy and stimulate cell cycle arrest, cell differentiation,8 and senescence, the last by modifying telomerase activity.9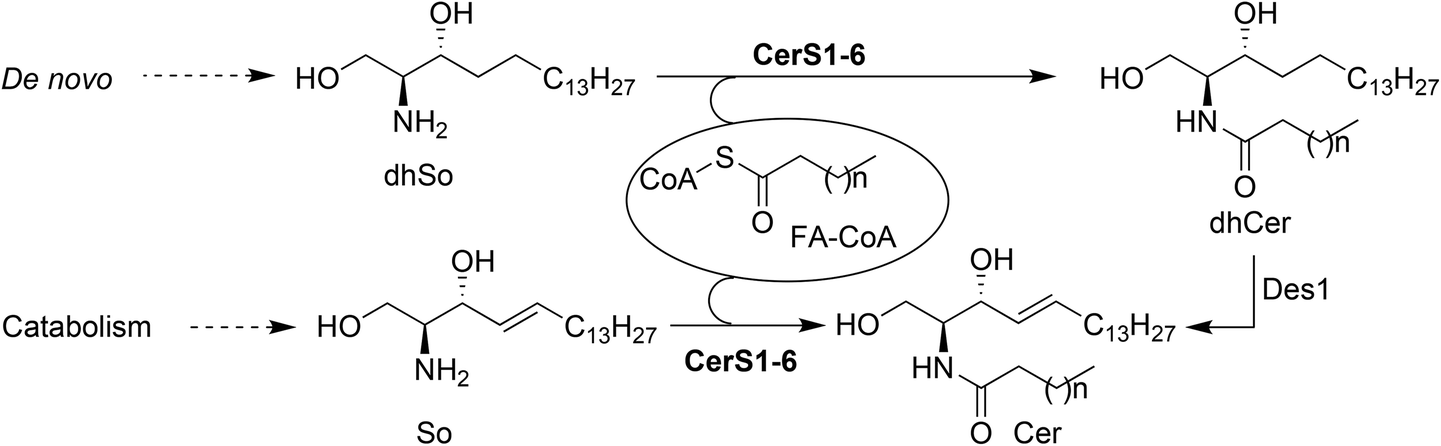 | ||
| Scheme 1 Metabolic routes leading to Cer. | ||
The intracellular levels of Cer are the result of the catabolic processes from higher SLs (sphingomyelin, glycosphingolipids and ceramide-1-phosphate), together with biosynthesis de novo by the N-acylation of dhSo with a variety of fatty acids (FA), prior to their desaturation by a specific desaturase (Des1) that introduces a C4(E) double bond into the sphingoid base. In this context, ceramide synthases (CerS) are a family of enzymes responsible for the N-acylation of dhSo (in the de novo pathway) or So (in the catabolic pathway) to form dhCer and Cer, respectively (Scheme 1).
Six isoforms of CerS have been identified in mammals, each one of them encoded by a unique gene (CerS1-6).10 CerS enzymes are expressed differently in various tissues11 and their levels of expression change during development, suggesting that populations of Cer with particular acyl chain lengths might be generated to meet the specific physiological needs of each tissue.12 Moreover, the nature of the acyl chains is a determinant of the biophysical properties of the resulting Cer and also the signaling pathways in which they participate.13 The development of modern lipidomic techniques14 has allowed determination of the relative abundance of the various Cer types in a range of biological contexts, and has provided some insight into the effect of the acyl chain composition on the physiological role of Cer.15 Given the importance of CerS activity in cell fate, we became interested in the development of new chemical probes16 towards this end. In a previous work, we reported on the use of 1-deoxydihydrosphingosine (doxdhSo, spisulosine or ES285) as a suitable probe for the profiling of CerS activity in intact cells.17 On the basis that 1-deoxysphingolipids (doxSLs) can be virtually considered as “dead-end” metabolites, due to the lack of the C1-OH group, we envisioned that a fluorescent probe derived from spisulosine, together with a suitable FA analogue, could be used to develop a FRET-based assay to monitor CerS activity.18
The affinity towards lipid phases, fluorescent properties and sensitivity to the polarity of the surrounding environment of the fluorophores NBD and Nile red (NR) has promoted the use of these groups in several biological applications related to the study of the cell membrane.19,20 Due to the existing overlap between the emission band of NBD and the absorption band of NR, these two fluorophores have also been incorporated into lipids as a donor–acceptor fluorophore pair to perform FRET experiments.21–24 More recently, 7-methoxycoumarin-3-carboxylate (MCC) has also been used as an alternative fluorophore partner for NBD in FRET experiments.25 In this case, however, NBD played the role of the acceptor fluorophore, whereas MCC was used as the donor. On this basis, we considered the NBD-labelled spisulosine RBM5-155 and the ω-azido palmitic acid (ωN3PA) as suitable CerS substrates for an ideal experiment design. A strained-promoted alkyne–azide cycloaddition (SPAAC) of the resulting doxdhCer RBM5-159 with a BCN-derived fluorescent dye (RBM5-142 (MCC) or RBM5-143 (NR)) should render the bichromophoric doxdhCer RBM5-160 or RBM5-161, respectively, whose FRET emission should correlate with CerS activity (Scheme 2).
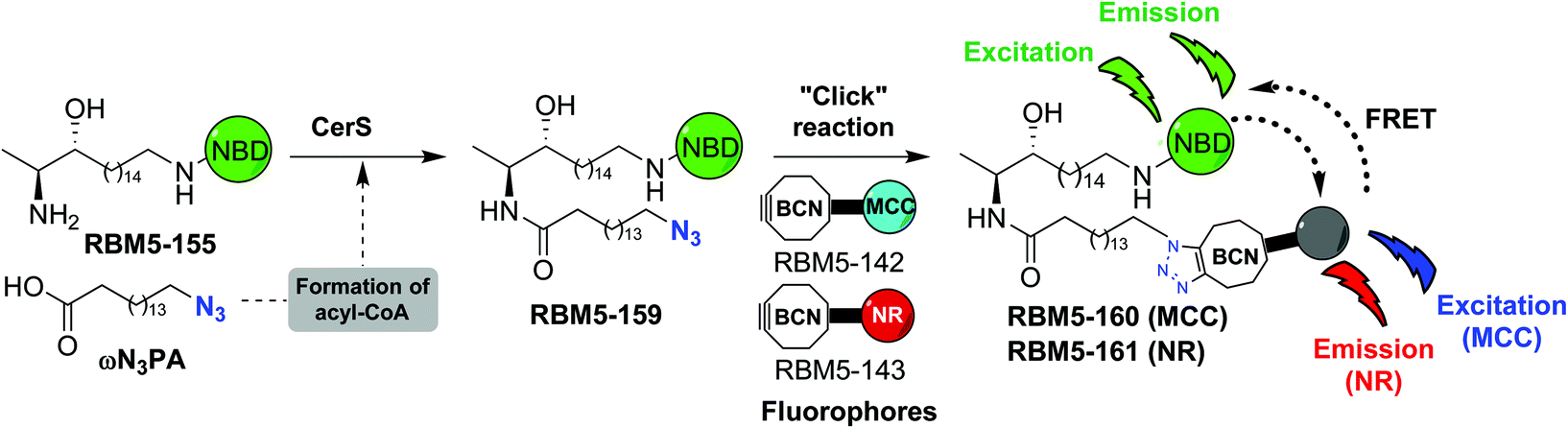 | ||
| Scheme 2 Conceptual design of the required probes for a FRET-based CerS assay. | ||
In this paper, we wish to report on the synthesis and photochemical characterization of the above probes and reagents, their validation as CerS substrates and their suitability as FRET partners.18
Synthesis of the probes
The synthesis of the NBD probe RBM5-155 was carried out as depicted in Scheme 3. Starting from the anti-configured allylic alcohol RBM5-084,26 a cross-metathesis reaction with the bromoalkene RBM5-149, using the second generation Grubbs’ catalyst, afforded a highly E-enriched E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z mixture of RBM5-150, in agreement with the literature.26 This mixture was subjected to catalytic hydrogenation on Rh/Al2O3 to give the corresponding saturated ω-bromo intermediate RBM5-151. Subsequent nucleophilic displacement with sodium azide, followed by the reduction of the azido group with the Pd/C-TES system,27 reaction of the intermediate amine hydrochloride with NBD-Cl,24 and final N-Boc removal afforded RBM5-155, which was isolated and further manipulated as the corresponding hydrochloride, given its apparent sensitivity to the alkaline conditions required for the formation of the free amine.28 Finally, the N-acylation of RBM5-155 with ωN3PA29 afforded the doxdhCer RBM5-159, which was used as the standard for quantitative LC-MS in cell assays (see below).
:Z mixture of RBM5-150, in agreement with the literature.26 This mixture was subjected to catalytic hydrogenation on Rh/Al2O3 to give the corresponding saturated ω-bromo intermediate RBM5-151. Subsequent nucleophilic displacement with sodium azide, followed by the reduction of the azido group with the Pd/C-TES system,27 reaction of the intermediate amine hydrochloride with NBD-Cl,24 and final N-Boc removal afforded RBM5-155, which was isolated and further manipulated as the corresponding hydrochloride, given its apparent sensitivity to the alkaline conditions required for the formation of the free amine.28 Finally, the N-acylation of RBM5-155 with ωN3PA29 afforded the doxdhCer RBM5-159, which was used as the standard for quantitative LC-MS in cell assays (see below).
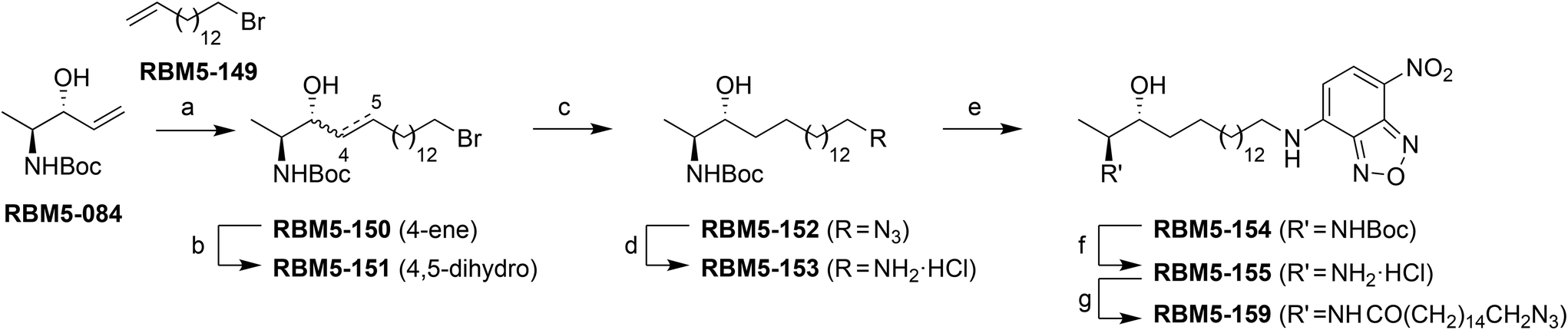 | ||
| Scheme 3 Synthesis of doxSL probes. Reagents and conditions: (a) RBM5-149, Grubbs’ 2nd gen. catalyst, CH2Cl2, reflux, 2 h, 44%, E/Z = 95:5; (b) H2, 5 wt% Rh on Al2O3, MeOH, rt, 3 h, 86%; (c) NaN3, DMF, 80 °C, 3 h, 95%; (d) TES, Pd–C, MeOH:CHCl3 (9:1), rt, 10 min, 85%; (e) NBD-Cl, DIPEA, MeOH, 0 °C to rt, overnight, 84%; (f) AcCl, MeOH, 0 °C to rt, overnight, 80%; (g) ωN3PA, EDC, HOBt, EtN3, CH2Cl2, rt, 2 h, 74%. | ||
The fluorophores required for the SPAAC click reactions were obtained from the modified ω-aminoalkyl MCC and NR derivatives RBM5-135 and RBM5-136, respectively, obtained as shown in Scheme 4.18
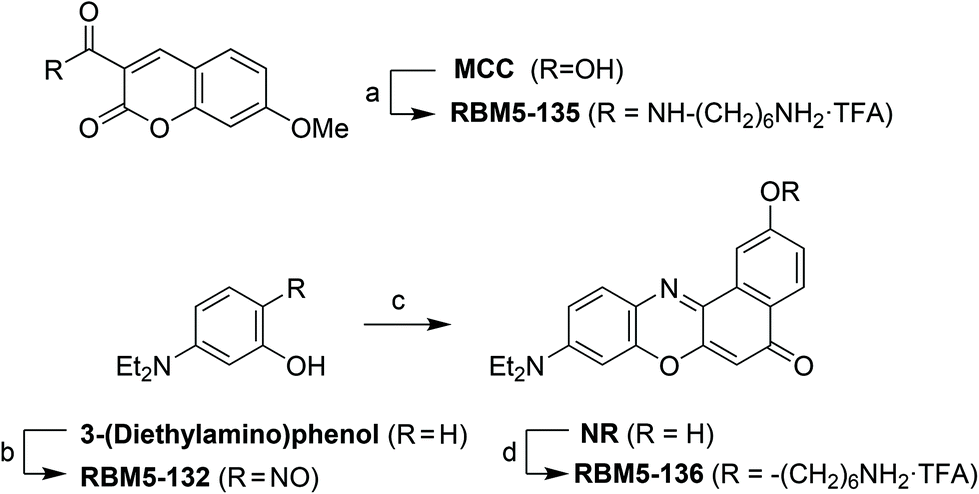 | ||
| Scheme 4 Synthesis of the MCC and NR fluorophores. (a) (1). N-Boc-1,6-hexanediamine, EDC, HOBt, Et3N, CH2Cl2, rt, 2 h, 57%; (2). TFA:CH2Cl2 (1:2), 0 °C to rt, 1 h, quantitative; (b) NaNO2, HCl (aq.), 0 °C, 5 h, 67%; (c) naphthalene-1,6-diol, DMF, 160 °C, 4 h, 19%; (d) (1). N-Boc-6-bromohexanamine, K2CO3, DMF, 85 °C, overnight, 78%; (2). TFA:CH2Cl2 (1:2), 0 °C to rt, 1 h, quantitative. | ||
The clickable reagents RBM5-142 and RBM5-143 were synthesized by the condensation of RBM5-135 and RBM5-136, respectively, with the p-nitrophenyl carbonate RBM5-141, obtained by the reaction of commercial trans-9-hydroxymethyl[6.1.0]bicyclonon-4-yne (BCN-OH) with 4-nitrophenyl chloroformate.30 The corresponding bichromophoric click adducts RBM5-160 and RBM5-161 were prepared for their complete photochemical characterization and as standards for FRET studies (Scheme 5).
 | ||
| Scheme 5 Synthesis of clickable fluorophores. Reagents and conditions: (a) RBM5-135, Et3N, CH2Cl2, rt, overnight, 89%; (b) RBM5-136, Et3N, CH2Cl2, rt, overnight (90%); (c) RBM5-159, CH2Cl2, rt, overnight, 84–93% (for assay-like conditions, see text). | ||
FRET efficiency of the bichromophoric probes RBM5-160 and RBM5-161
The suitability of the selected FRET partners was corroborated by calculation of the spectral overlap integral and Förster radius (R0) (Table S1†) from the spectral data of the monochromophoric compounds RBM5-142, RBM5-143 and RBM5-154 (Table S2†). The normalized absorption and emission spectra of the bichromophoric probes RBM5-160 and RBM5-161 in DMSO, EtOH and PBS buffer presented two bands owing to the presence of the two fluorescent labels (Fig. 1). Compound RBM5-160 presented two maxima at around 350 nm (MCC moiety) and 470 nm (NBD moiety), while the maxima for RBM5-161 were located around 485 nm (NBD moiety) and 550 nm (NR moiety). The fluorescence intensity was strongly affected by the solvent, the highest fluorescence intensities being recorded in EtOH for both compounds. However, their emission in PBS buffer was reduced as a result of their lower aqueous solubility. The position and shape of the emission spectra in the two probes were slightly modified in the different solvents, due to the solvatochromic effects. Furthermore, there were also considerable deviations in the absorption spectra of compounds RBM5-160 and RBM5-161, when compared with the spectra of the related monochromophoric compounds, probably due to the intramolecular attractive interactions between the two fluorophores in each compound.31 The molar extinction coefficients (ε) of compounds RBM5-160 and RBM5-161, calculated at their corresponding two absorption maxima in DMSO and EtOH are listed in Table S3.†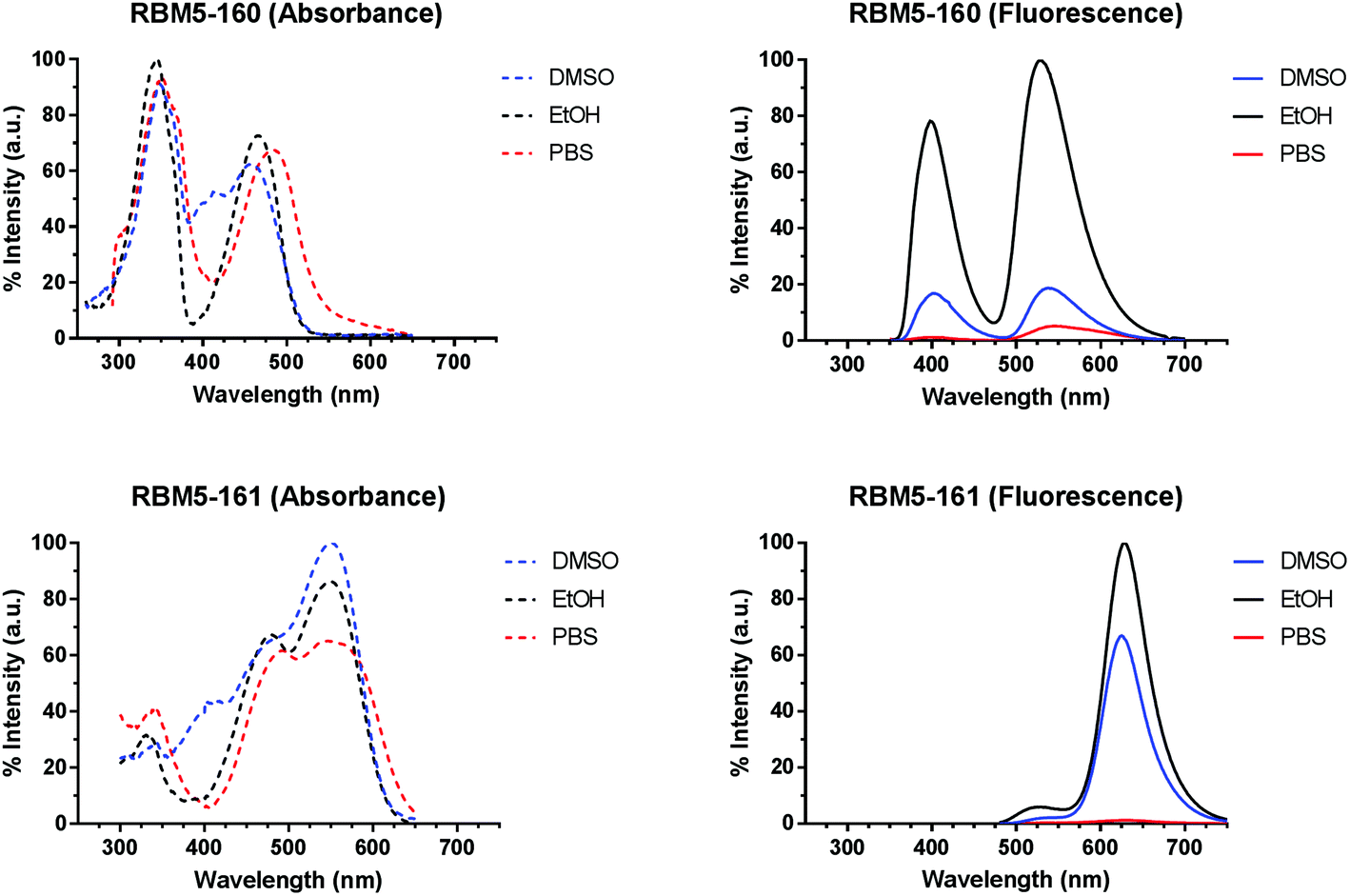 | ||
| Fig. 1 Normalised absorption (left panels) and emission (right panels) spectra for the bichromophoric compounds RBM5-160 (top) and RBM5-161 (bottom) at 5 μM in DMSO (blue), EtOH (black) and PBS (red). Emission spectra upon excitation at 340 nm (MCC in RBM5-160) and 455 nm (NBD in RBM5-161), respectively. The absorption and emission spectra of RBM5-160 in DMSO and PBS were normalised to those of the same compound in EtOH. The absorption spectra of RBM5-161 in EtOH and PBS were normalised to those in DMSO. The emission spectra of RBM5-161 in DMSO and PBS were normalised to those in EtOH. | ||
The intramolecular FRET efficiencies of compounds RBM5-160 and RBM5-161 were estimated in DMSO and EtOH, based on the loss of donor fluorescence in the presence of the acceptor. To this end, we compared the integrated fluorescence intensities (I), within the donor-specific wavelength interval, of the donor (D) compounds (RBM5-142 and RBM5-154) to those of the related donor + acceptor (DA) compounds (RBM5-160 and RBM5-161, respectively). The calculated FRET efficiency of the NBD/NR pair in RBM5-161 (ENBD/NR = 0.88–0.96) was higher than that of the MCC/NBD pair in RBM5-160 (EMCC/NBD = 0.56–0.88). For both fluorophore pairs, the FRET process was more efficient in EtOH than in DMSO and, in the case of the NBD/NR pair in RBM5-161, the two studied excitation wavelengths gave the same E value in EtOH (Table 1).
| Compound | Solvent | λ ex (nm) | E |
|---|---|---|---|
| a FRET efficiencies (E) were calculated from the decrease of the donor emission (see Experimental section). | |||
| RBM5-160 | DMSO | 340 | 0.56 |
| EtOH | 340 | 0.86 | |
| RBM5-161 | DMSO | 470 | 0.90 |
| 455 | 0.88 | ||
| EtOH | 470 | 0.96 | |
| 455 | 0.96 | ||
Optimization of the SPAAC reaction from RBM5-159
The SPAAC reaction between the azide-tagged doxdhCer RBM5-159 and the fluorescent dyes RBM5-142 and RBM5-143 was studied under different conditions in order to determine the best reagent ratios and reaction times at a concentration scale compatible with that expected in the CerS activity assay in intact cells. The reaction progress was first monitored in DMSO by analyzing the changes in the fluorescence emission of the mixture upon irradiation at the donor-specific excitation wavelength. For RBM5-160 (donor: MCC, acceptor: NBD, see Scheme 2), the fluorescence emission at λem = 535 (NBD emission) at two single concentrations of RBM5-159 (5 and 20 μM) and increasing concentrations of the BCN-MCC reagent RBM5-142 (from 5 to 50 μM) was always lower than that of a standard solution of the expected adduct RBM5-160 at 5 and 20 μM (results not shown). However, a similar experiment using the BCN-NR reagent RBM5-143 (from 5 to 50 μM) led to fluorescence intensities at λem = 625 (NR emission) comparable to those of a standard solution of the expected adduct RBM5-161 using a 2.5 to 4 fold excess of the BCN-NR reagent RBM5-143 (Fig. 2).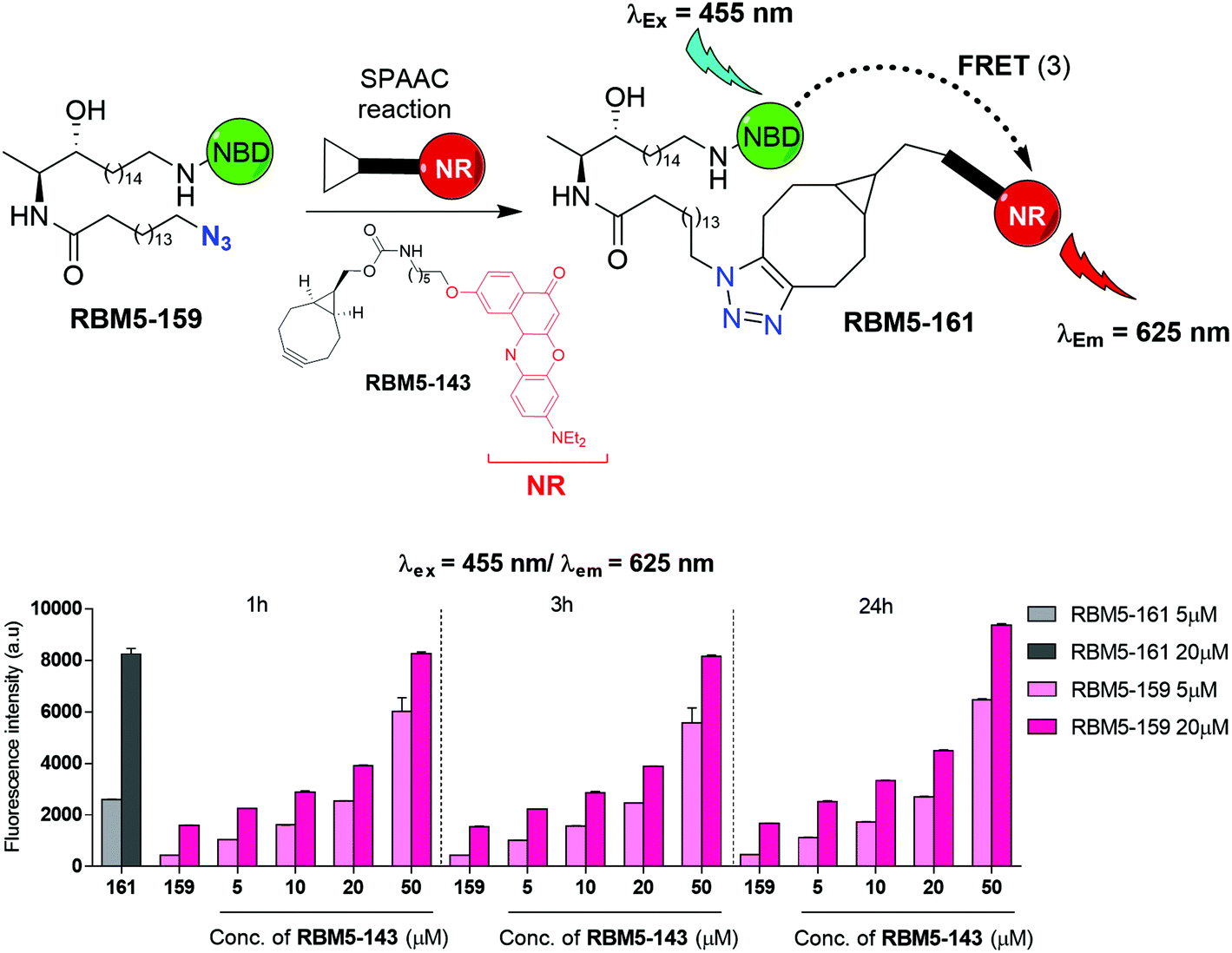 | ||
| Fig. 2 Bar diagram representing the changes in fluorescence emission at 625 nm (bottom), resulting from the excitation at 455 nm, of mixtures containing different ratios of compounds RBM5-159 and RBM5-143 in DMSO at various reaction times. Compound RBM5-159 was used as the negative control, equivalent to 0% conversion, whereas compound RBM5-161 was used as the positive control, equivalent to 100% conversion. The results correspond to the mean ± standard deviation of at least two independent experiments with triplicates. | ||
Experiments in MeOH, EtOH and sodium acetate buffer (NaOAc 250 mM, NaCl 200 mM, 0.1% Triton X-100) also showed a remarkable enhancement of the emission at 625 nm (acceptor emission, λexc = 455 nm) in comparison with RBM5-159, together with an attenuation of the fluorescence emission at 535 nm (donor emission, λexc = 455 nm), in agreement with the formation of the desired cycloadduct RBM5-161 (Fig. S1†). The background emission of RBM5-143 at 625 nm observed at the donor excitation (λexc = 455 nm) is indicative of an excitation cross-talk (or acceptor excitation bleed-through, AEB), a phenomenon related to the overlap of the donor and acceptor absorption spectra. This phenomenon, together with the related emission cross-talk (or donor emission bleed-through, DEB), arising from the overlap of the donor and acceptor emission spectra32 must be carefully considered in the design of ratiometric experiments based on the development of the FRET effect. In contrast, ratiometric experiments based on FRET attenuation21,22,24 may benefit from the disappearance of these interferences. The experimental AEB and DEB calculated for RBM5-161 are listed in Table S4 and Fig. S2, S3.†
CerS catalyzed incorporation of ω-azidopalmitic acid into RBM5-155 in cells
The suitability of the probe RBM5-155 and ωN3PA as CerS substrates was evaluated by examining the extent of their metabolic conversion into the corresponding N-acylated doxdhCer RBM5-159 by LC-MS. Initial experiments were carried out in lung adenocarcinoma A549 cells and mouse embryonic fibroblasts (MEF). Even though the total amount of doxdhCers, arising from acylation of RBM5-155 with endogenous FAs, was very similar in the two cell lines (∼120–130 pmol equiv./106 cells), the profile of the different doxdhCer species differed considerably. In A549 cells, there was a high proportion of elongated C18N3 and C20N3 doxdhCer species (resulting from the action of the endogenous elongases),33 whereas the non-elongated doxdhCer RBM5-159 accounted for less than a third of the total doxdhCers (∼28%). Conversely, in MEF cells, the non-elongated RBM5-159 was the most abundant metabolite (85% of the total doxdhCers), whereas the formation of elongated species was minimal. These results show that the elongation process is unpredictable and highly dependent on the cell line, although it appears to be independent on the overall CerS activity. Attempts to reduce or avoid the acyl chain elongation by using the FA biosynthesis inhibitors TOFA or cerulenin were fruitless.34–36 Moreover, the incorporation of ωN3C13 acid, as well as that of branched αN3PA and (ω-1)N3PA, was also tested. The first two were deemed as poor CerS substrates since only modest amounts of the corresponding doxdhCers were formed under the standard assay. Conversely, the branched (ω-1)N3PA was efficiently incorporated, although the high ratio of the elongated doxdhCer observed made us disregard its use (results not shown). For practical purposes, the overexpression of the CerS under study may improve the signal-to-noise ratio of the assay. As a proof of concept, human embryonic kidney cells (HEK293T) were transfected with the human CerS5 gene using the cationic polymer polyethylenimine (PEI),37 and were subsequently incubated with the doxdhSo probe RBM5-155 and ωN3PA. UPLC-TOF analysis of the lipid extracts showed a higher ratio of the C16-doxdhCer species in the transfected cells, in agreement with the expected preference of the CerS5 isoform for the C16 FAs38 (Fig. 3).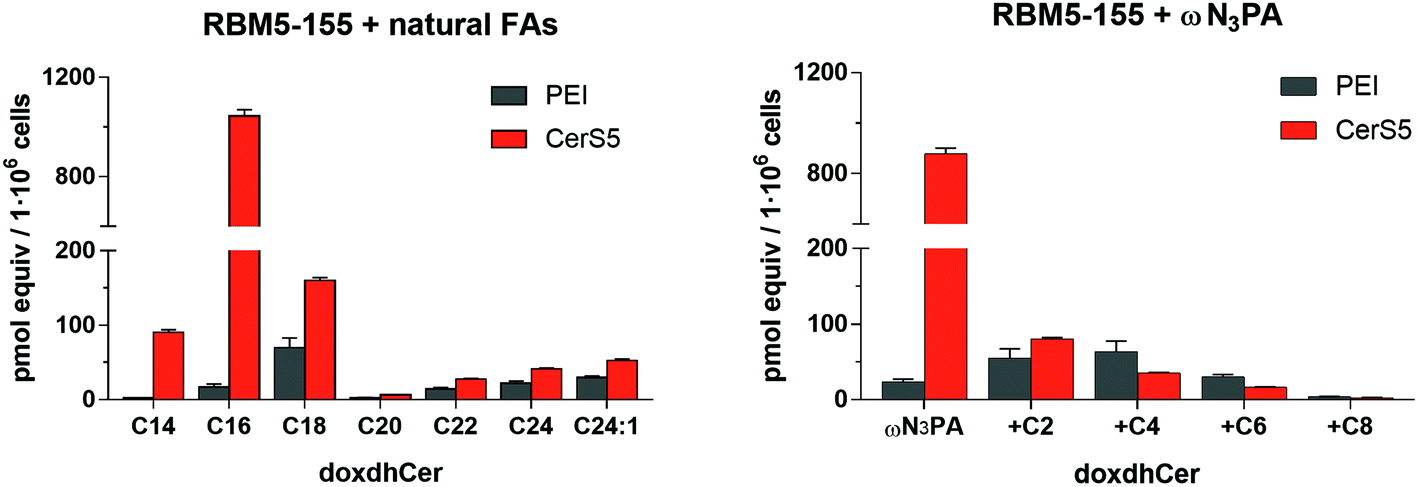 | ||
| Fig. 3 N-Acylation of the doxdhSo probe RBM5-155 with the endogenous natural FAs or ωN3PA. HEK293T cells transfected with the plasmid containing human CerS5 (bars in orange) or simply treated with the transfection reagent PEI (bars in grey) were incubated for 90 min with RBM5-155 (5 μM) in the absence (left) or in the presence (right) of ωN3PA (0.5 mM) complexed in 0.5% acid-free BSA. After lipid extraction, the different doxdhCer species were quantified by UPLC-TOF. Left: Amount of doxdhCers containing endogenous natural FAs; right: amount of doxdhCers containing the administered ωN3PA and the corresponding elongated FAs. The results correspond to the mean ± standard deviation of at least two independent experiments with triplicates. | ||
Conclusions
In summary, the NBD probe RBM5-155 and the modified fatty acid ωN3PA behave as suitable CerS substrates in cells. Despite the degree of incorporation and elongation of the fatty acid being strongly dependent on the particular cell line, studies carried out with the doxdhCer RBM5-159 indicated its suitability as a SPAAC reaction partner with the BCN modified NR fluorophore RBM5-143 to render the bichromophoric doxdhCer RBM5-161, exhibiting a high FRET efficiency in the solvent systems used. These results represent an interesting addition to the available chemical toolbox of labelled sphingolipids for FRET-based studies in cells. Efforts along this line are underway in our lab and will be reported in due course.Experimental section
Synthesis
:30) as the mobile phase. Samples were analyzed using a 10 μL volume injection. The m/z ratios are reported as values in atomic mass units.
((1R,8S,9S)-Bicyclo[6.1.0]non-4-yn-9-yl)methyl (6-(7-methoxy-2-oxo-2H-chromene-3-carboxamido)hexyl)carbamate (RBM5-142). To an ice-cooled solution of N-Boc protected RBM5-135
18 (93 mg, 0.22 mmol) in dry CH2Cl2 (6 mL) was added dropwise neat TFA (1.5 mL). After stirring at rt for 2 h in the dark, the reaction mixture was concentrated to dryness to afford the corresponding crude amine trifluoroacetate (71 mg). This crude was taken up in CH2Cl2 (10 mL), followed by the sequential addition of Et3N (108 μL, 0.78 mmol) and p-nitrophenol activated carbonate ester RBM5-14130 (70 mg, 0.22 mmol). After stirring overnight at rt in the dark, the reaction mixture was evaporated in vacuo and the residue was flash chromatographed (from 0 to 20% EtOAc in CH2Cl2) to give the desired carbamate RBM5-142 (98 mg, 89%) as an off-white solid.
1H NMR (400 MHz, CDCl3) δ 8.83 (s, 1H), 8.76 (br s, 1H), 7.58 (d, J = 8.7 Hz, 1H), 6.93 (dd, J = 8.7, 2.4 Hz, 1H), 6.86 (d, J = 2.4 Hz, 1H), 4.69 (br s, 1H), 4.13 (d, J = 8.1 Hz, 2H), 3.91 (s, 3H), 3.44 (td, J = 7.1, 5.8 Hz, 2H), 3.20–3.12 (m, 2H), 2.34–2.15 (m, 6H), 1.67–1.24 (m, 11H), 0.99–0.86 (m, 2H).
13C NMR (101 MHz, CDCl3) δ 164.9, 162.1, 162.1, 156.9, 156.8, 148.3, 131.0, 115.0, 114.1, 112.6, 100.4, 99.0, 62.7, 56.2, 41.0, 39.7, 30.0, 29.5, 29.2, 26.7, 26.5, 21.6, 20.2, 18.0.
HRMS calcd for C28H35N2O6 ([M + H]+): 495.2490, found: 495.2496.
((1R,8S,9S)-Bicyclo[6.1.0]non-4-yn-9-yl)methyl (6-((9-(diethylamino)-5-oxo-5H-benzo[a]phenoxazin-2-yl)oxy)hexyl)carbamate (RBM5-143). To an ice-cooled solution of N-Boc protected RBM5-136
18 (100 mg, 0.19 mmol) in dry CH2Cl2 (6 mL) was added dropwise neat TFA (1.5 mL). After stirring at rt for 2 h in the dark, the reaction mixture was concentrated to dryness to afford the corresponding crude amine trifluoroacetate (81 mg). This crude was taken up in CH2Cl2 (10 mL), followed by the sequential addition of Et3N (93 μL, 0.67 mmol) and p-nitrophenol activated carbonate ester RBM5-14130 (60 mg, 0.19 mmol). After stirring overnight at rt in the dark, the reaction mixture was evaporated in vacuo and the residue was flash chromatographed (from 0 to 20% EtOAc in CH2Cl2) to give the desired carbamate RBM5-143 (104 mg, 90%) as a shiny dark-red solid.
1H NMR (400 MHz, CDCl3) δ 8.21 (d, J = 8.7 Hz, 1H), 8.03 (d, J = 2.6 Hz, 1H), 7.60 (d, J = 9.0 Hz, 1H), 7.15 (dd, J = 8.7, 2.6 Hz, 1H), 6.65 (dd, J = 9.1, 2.7 Hz, 1H), 6.45 (d, J = 2.7 Hz, 1H), 6.29 (s, 1H), 4.70 (br s, 1H), 4.19–4.11 (m, 4H), 3.46 (q, J = 7.1 Hz, 5H), 3.25–3.15 (m, 2H), 2.34–2.16 (m, 6H), 1.90–1.82 (m, 2H), 1.67–1.30 (m, 10H), 1.26 (t, J = 7.0 Hz, 6H), 0.97–0.87 (m, 2H).
13C NMR (101 MHz, CDCl3) δ 183.4, 161.9, 156.9, 152.2, 150.9, 147.0, 140.3, 134.2, 131.2, 127.9, 125.7, 124.8, 118.4, 109.6, 106.7, 105.5, 99.0, 96.5, 68.3, 62.8, 45.2, 41.1, 30.1, 29.3, 29.2, 26.6, 25.9, 21.6, 20.2, 17.9, 12.8.
HRMS calcd for C37H44N3O5 ([M + H]+): 610.3275, found: 610.3279.
15-Bromopentadec-1-ene (RBM5-149). A stirred solution of 14-pentadecen-1-ol39 (2.77 g, 12.23 mmol) and PPh3 (3.53 g, 13.46 mmol) in anhydrous CH2Cl2 (100 mL) was cooled to 0 °C, and NBS (2.61 g, 14.68 mmol) was added in small portions over 10 min. The resultant dark yellow solution was allowed to warm to rt and stirred for 1 h, after which the solvent was removed by vacuum evaporation. The residue was then diluted with water (50 mL), extracted with hexanes (3 × 50 mL) and worked-up as usual to give a crude, which was purified by flash chromatography on silica gel (isocratic 100% hexanes) to afford RBM5-149 as a colorless oil, (2.62 g, 74%).
1H NMR (400 MHz, CDCl3) δ 5.81 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 4.99 (dq, J = 17.1, 1.9 Hz, 1H), 4.93 (ddt, J = 10.2, 2.4, 1.4 Hz, 1H), 3.41 (t, J = 6.9 Hz, 2H), 2.08–2.00 (m, 2H), 1.85 (dt, J = 14.5, 7.0 Hz, 2H), 1.46–1.33 (m, 3H), 1.32–1.25 (m, 16H).
13C NMR (101 MHz, CDCl3) δ 139.4, 114.2, 34.2, 34.0, 33.0, 29.8, 29.8, 29.7, 29.7, 29.6, 29.3, 29.1, 28.9, 28.3.
(2′S,3′R,4′EZ)-tert-Butyl (18-bromo-3-hydroxyoctadec-4-en-2-yl)carbamate (RBM5-150). To a stirred solution of RBM5-084
26,40 (480 mg, 2.38 mmol) and olefin RBM5-149 (1.59 g, 5.49 mmol) in degassed CH2Cl2 (20 mL), second generation Grubbs’ catalyst (101 mg, 0.12 mmol) was added in one portion at rt. The resulting mixture was refluxed in the dark for 2 h, cooled to rt and concentrated in vacuo to afford a crude, which was purified by flash chromatography on silica gel (from 0 to 5% MTBE in CH2Cl2). Compound RBM5-150 was obtained as an inseparable mixture of E/Z isomers (colourless thick oil, 485 mg, 44%).
1H NMR (400 MHz, CDCl3) (E isomer) δ 5.71 (dt, J = 14.6, 6.7 Hz, 1H), 5.42 (dt, J = 15.0, 7.5 Hz, 1H), 4.76 (br s, 1H), 4.11 (dd, J = 6.4, 3.0 Hz, 1H), 3.73–3.58 (m, 1H), 3.40 (t, J = 6.9 Hz, 2H), 2.09–1.96 (m, 2H), 1.85 (dt, J = 14.5, 6.9 Hz, 2H), 1.44 (s, 9H), 1.43–1.23 (m, 20H), 1.07 (d, J = 6.9 Hz, 3H).
13C NMR (101 MHz, CDCl3) (mixture of E/Z isomers) δ 156.1, 155.7, 134.2, 133.5, 129.4, 128.7, 125.6, 124.9, 79.4, 79.2, 75.4, 73.5, 51.0, 50.0, 37.3, 33.9, 33.4, 32.8, 32.6, 32.3, 29.6, 29.5, 29.5, 29.4, 29.2, 28.7, 28.4, 28.1, 15.2, 14.3.
(2′S,3′R)-tert-Butyl (18-bromo-3-hydroxyoctadecan-2-yl)carbamate (RBM5-151). A solution of RBM5-150 (450 mg, 0.97 mmol) in degassed MeOH (45 mL) was hydrogenated at 1 atm and rt in the presence of Rh/Al2O3 (60 mg, 15% w/w). After stirring for 3 h, the catalyst was removed by filtration through a Celite® pad, and the solid was rinsed with MeOH (3 × 10 mL). The combined filtrates were concentrated in vacuo, and the residue was subjected to flash chromatography on silica gel (from 0 to 1% MeOH in CH2Cl2) to yield RBM5-151 as a white solid (395 mg, 87%).
[α]20D = −3.65 (c 1, CHCl3).
1H NMR (400 MHz, CDCl3) δ 4.74 (br s, 1H), 3.70–3.67 (m, 1H), 3.64 (td, J = 8.0, 7.2, 2.7 Hz, 1H), 3.41 (t, J = 6.9 Hz, 2H), 1.85 (dt, J = 14.5, 7.0 Hz, 2H), 1.71 (br s, 1H), 1.44 (s, 9H), 1.43–1.35 (m, 4H), 1.34–1.23 (m, 22H), 1.08 (d, J = 6.8 Hz, 3H).
13C NMR (101 MHz, CDCl3) δ 156.0, 79.6, 74.6, 50.8, 34.2, 33.6, 33.0, 29.8, 29.7, 29.6, 28.9, 28.6, 28.3, 26.2, 14.5.
HRMS calcd for C23H47BrNO3 ([M + H]+): 464.2734, 466.2713, found: 464.2729, 466.2718.
(2′S,3′R)-tert-Butyl (18-azido-3-hydroxyoctadecan-2-yl)carbamate (RBM5-152). To a stirred solution of RBM5-151 (395 mg, 0.85 mmol) in anhydrous DMF (8 mL) was added NaN3 (166 mg, 2.55 mmol). The mixture was heated to 80 °C and stirred for 3 h under Ar. Water (20 mL) was next added, and the mixture was extracted with Et2O (3 × 20 mL). Usual workup afforded a crude, which was purified by flash chromatography (from 0 to 1% MeOH in CH2Cl2) to give RBM5-152 (off-white wax, 346 mg, 95%).
[α]20D = −4.13 (c 1, CHCl3).
1H NMR (400 MHz, CDCl3) δ 4.75 (s, 1H), 3.71–3.67 (m, 1H), 3.66–3.61 (m, 1H), 3.25 (t, J = 7.0 Hz, 2H), 1.75 (br s, 1H), 1.66–1.54 (m, 2H), 1.44 (s, 9H), 1.40–1.35 (m, 4H), 1.34–1.23 (m, 22H), 1.07 (d, J = 6.8 Hz, 3H).
13C NMR (101 MHz, CDCl3) δ 156.0, 79.6, 74.6, 51.6, 50.7, 33.6, 29.8, 29.8, 29.7, 29.7, 29.7, 29.7, 29.6, 29.3, 29.0, 28.5, 26.8, 26.2, 14.4.
HRMS calcd for C23H47N4O3 ([M + H]+): 427.3643, found: 427.3636.
(2′S,3′R)-tert-Butyl (18-amino-3-hydroxyoctadecan-2-yl)carbamate (RBM5-153). To a solution of RBM5-152 (300 mg, 0.70 mmol) in degassed MeOH (16 mL) containing Pd–C (60 mg, 20% w/w) neat TES (1.1 mL, 7.03 mmol) was added dropwise, and the resultant suspension was stirred at rt under an Ar-filled balloon. After stirring for 1 h, the reaction mixture was filtered through a pad of Celite® and the solid was rinsed with MeOH (3 × 5 mL). The combined filtrates were concentrated in vacuo and the residue was triturated with hexanes (4 × 2 mL) to give the desired amine hydrochloride, without the need of further purification.
1H NMR (400 MHz, CD3OD) δ 3.53–3.47 (m, 1H), 3.47–3.41 (m, 1H), 2.67 (t, J = 7.1 Hz, 2H), 1.53–1.46 (m, 4H), 1.44 (s, 9H), 1.30 (s, 24H), 1.08 (d, J = 6.6 Hz, 3H).
13C NMR (101 MHz, CD3OD) δ 157.8, 79.9, 75.3, 51.8, 42.3, 34.8, 33.0, 30.8, 30.7, 30.6, 28.8, 28.0, 27.1, 15.5.
HRMS calcd for C23H49N2O3 ([M + H]+): 401.3738, found: 401.3742.
(2′S,3′R)-tert-Butyl [3-hydroxy-18-[(7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)amino]octadecan-2-yl]carbamate (RBM5-154). To a stirred solution of 4-chloro-7-nitrobenzo[c][1,2,5]oxadiazole (NBD-Cl) (132 mg, 0.66 mmol) in MeOH (15 mL) containing DIPEA (522 μL, 3.00 mmol) at 0 °C was added dropwise a solution of RBM5-153 (240 mg, 0.60 mmol) in MeOH (10 mL). After the addition was complete, the mixture was allowed to warm to rt and stirred overnight. Then, the volatiles were removed under reduced pressure and the residue was directly subjected to flash chromatography on silica gel (from 0 to 1% MeOH in CH2Cl2) to afford RBM5-154 as a shiny orange wax (278 mg, 82%).
1H NMR (400 MHz, CDCl3) δ 8.49 (d, J = 8.6 Hz, 1H), 6.31 (br s, 1H), 6.17 (d, J = 8.7 Hz, 1H), 4.75 (br s, 1H), 3.73–3.66 (m, 1H), 3.66–3.62 (m, 1H), 3.49 (ap q, J = 6.7 Hz, 2H), 1.86 (br s, 1H), 1.81 (dt, J = 14.9, 7.4 Hz, 2H), 1.53–1.44 (m, 2H), 1.44 (s, 9H), 1.41–1.35 (m, 4H), 1.33–1.23 (m, 20H), 1.07 (d, J = 6.8 Hz, 3H).
13C NMR (101 MHz, CDCl3) δ 156.0, 144.3, 144.2, 144.0, 136.7, 123.7, 98.6, 79.6, 74.6, 50.8, 44.2, 33.6, 29.8, 29.7, 29.7, 29.7, 29.6, 29.6, 29.5, 29.3, 28.6, 28.5, 27.0, 26.1, 14.5.
HRMS calcd for C29H50N5O6 ([M + H]+): 564.3756, found: 564.3761.
(2S,3R)-2-Amino-18-[(7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)amino]octadecan-3-ol hydrochloride (RBM5-155). An ice-cooled solution of RBM5-154 (85 mg, 0.15 mmol) in MeOH (10 mL) was treated with neat AcCl (54 μL, 0.75 mmol) and the resulting mixture was allowed to warm to rt and stirred overnight in the dark. Then, the solvent was evaporated in vacuo and the residue was purified by flash chromatography on silica gel (from 0 to 20% MeOH in CH2Cl2) to afford RBM5-155 as a shiny orange wax (65 mg, 86%).
1H NMR (400 MHz, CD3OD) δ 8.52 (d, J = 8.7 Hz, 1H), 6.35 (d, J = 8.9 Hz, 1H), 3.75–3.65 (m, 1H), 3.53 (br s, 2H), 3.31–3.22 (m, 1H), 1.77 (app p, J = 7.4 Hz, 2H), 1.55–1.37 (m, 8H), 1.36–1.25 (m, 18H), 1.22 (d, J = 6.8 Hz, 3H).
13C NMR (101 MHz, CD3OD) δ 146.7, 145.8, 145.5, 138.6, 122.7, 99.6, 71.7, 52.6, 44.8, 34.0, 30.7, 30.6, 30.3, 29.2, 28.0, 27.0, 12.1.
HRMS calcd for C24H42N5O4 ([M + H]+): 464.3231, found: 464.3228.
(2′S,3′R)-16-Azido-N-[3-hydroxy-18-[(7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)amino]octadecan-2-yl]hexadecanamide (RBM5-159). EDC·HCl (12 mg, 64 μmol) and HOBt (7 mg, 52 μmol) were sequentially added to an ice-cooled solution of ω-azidopalmitic acid29 (13 mg, 44 μmol) in anhydrous CH2Cl2 (6 mL). The resulting mixture was vigorously stirred at rt under Ar for 15 min, and next added dropwise to a solution of RBM5-155 (20 mg, 40 μmol) and Et3N (89 μL, 0.20 mmol) in anhydrous CH2Cl2 (6 mL), and the reaction was stirred at rt for additional 2 h. The mixture was next diluted with CH2Cl2 (50 mL), washed with brine (2 × 25 mL) and worked-up as usual to afford a crude, which was flash chromatographed (from 0 to 14% EtOAc in CH2Cl2) to afford RBM5-159 as a shiny orange solid (22 mg, 74%).
1H NMR (400 MHz, CDCl3) δ 8.48 (d, J = 8.6 Hz, 1H), 6.49 (br s, 1H), 6.17 (d, J = 8.7 Hz, 1H), 5.81 (d, J = 7.9 Hz, 1H), 4.05–3.96 (m, 1H), 3.66–3.59 (m, 1H), 3.53–3.45 (m, 2H), 3.24 (t, J = 7.0 Hz, 2H), 2.51 (br s, 1H), 2.17 (t, J = 7.6 Hz, 2H), 1.80 (app p, J = 7.4 Hz, 2H), 1.69–1.53 (m, 4H), 1.49–1.20 (m, 48H), 1.09 (d, J = 6.8 Hz, 3H).
13C NMR (101 MHz, CDCl3) δ 173.4, 144.4, 144.1, 144.1, 136.7, 123.9, 98.6, 74.5, 51.6, 49.6, 44.2, 37.0, 33.7, 29.7, 29.7, 29.5, 29.5, 29.4, 29.3, 29.0, 28.6, 27.1, 26.8, 26.1, 25.9, 14.3.
HRMS calcd for C40H71N8O5 ([M + H]+): 743.5542, found: 743.5547.
Compound RBM5-160. Compound RBM5-142 (7 mg, 15 μmol) was added to a stirred solution of RBM5-159 (9 mg, 12 μmol) in CH2Cl2 (4 mL). After stirring overnight at rt in the dark, the reaction mixture was concentrated to dryness and the residue was flash chromatographed on silica gel (from 0 to 5% MeOH in CH2Cl2) to afford the desired SPAAC reaction adduct RBM5-160 (14 mg, 93%, inseparable mixture of diastereomers) as a dark-orange solid.
1H NMR (400 MHz, DMSO-d6) (mixture of diastereomers) δ 9.54 (s, 1H), 8.81 (s, 1H), 8.63 (t, J = 5.7 Hz, 1H), 8.50 (d, J = 9.1 Hz, 1H), 7.90 (d, J = 8.7 Hz, 1H), 7.49 (d, J = 8.5 Hz, 1H), 7.14–7.04 (m, 2H), 7.04 (dd, J = 8.7, 2.3 Hz, 1H), 6.41 (d, J = 9.3 Hz, 1H), 4.47 (d, J = 6.1 Hz, 1H), 4.18 (t, J = 7.0 Hz, 2H), 4.07–3.96 (m, 2H), 3.89 (s, 3H), 3.66–3.58 (m, 1H), 3.50–3.39 (m, 2H), 3.34–3.26 (m, 2H), 3.26–3.17 (m, 1H), 2.99–2.88 (m, 4H), 2.77–2.61 (m, 2H), 2.15–2.05 (m, 3H), 2.01 (t, J = 7.3 Hz, 2H), 1.73–1.60 (m, 4H), 1.56–1.05 (m, 60H), 0.97 (d, J = 6.7 Hz, 3H), 0.94–0.82 (m, 2H).
HRMS calcd for C68H105N10O11 ([M + H]+): 1237.7959, found: 1237.7983.
Compound RBM5-161. Compound RBM5-143 (10 mg, 15 μmol) was added to a stirred solution of RBM5-159 (10 mg, 13 μmol) in CH2Cl2 (4 mL). After stirring overnight at rt in the dark, the reaction mixture was concentrated to dryness and the residue was flash chromatographed on silica gel (from 0 to 5% MeOH in CH2Cl2) to afford the desired SPAAC reaction adduct RBM5-161 (16 mg, 88%, inseparable mixture of diastereomers) as a dark-red solid.
1H NMR (400 MHz, DMSO-d6) (mixture of diastereomers) δ 9.53 (br s, 1H), 8.48 (d, J = 8.9 Hz, 1H), 8.03 (d, J = 8.6 Hz, 1H), 7.94 (d, J = 2.5 Hz, 1H), 7.64–7.59 (m, 1H), 7.49 (d, J = 8.6 Hz, 1H), 7.25 (dd, J = 8.7, 2.5 Hz, 1H), 7.10 (t, J = 5.7 Hz, 1H), 6.81 (d, J = 9.2 Hz, 1H), 6.64 (d, J = 2.4 Hz, 1H), 6.38 (d, J = 9.0 Hz, 1H), 6.18 (s, 1H), 4.47 (d, J = 6.1 Hz, 1H), 4.21–4.10 (m, 4H), 4.02 (d, J = 7.8 Hz, 2H), 3.67–3.57 (m, 1H), 3.50 (q, J = 7.0 Hz, 4H), 3.46–3.39 (m, 2H), 3.25–3.18 (m, 1H), 3.15–3.04 (m, 4H), 3.05–2.95 (m, 2H), 2.94–2.86 (m, 2H), 2.78–2.59 (m, 2H), 2.13–1.90 (m, 7H), 1.83–1.73 (m, 2H), 1.70–1.59 (m, 4H), 1.54–1.10 (m, 58H), 0.96 (d, J = 6.7 Hz, 3H), 0.93–0.82 (m, 2H).
HRMS calcd for C77H114N11O10 ([M + H]+): 1352.8745, found: 1352.8760.
Spectroscopic studies
Absorption spectra were recorded on a Jasco V-730 UV-Vis spectrophotometer using a spectral bandwidth of 1 nm, a response time of 0.24 s, a data interval of 1 nm (except for quinine sulphate and compound RBM5-142, in which case the data interval was of 0.2 nm) and a scan rate of 200 nm min−1. Measurements were carried under an inert atmosphere (continuous flow of nitrogen gas) at a constant temperature of 20 °C.Fluorescence emission spectra were recorded on a Photon Technology International (PTI) QuantaMaster fluorometer at room temperature. The excitation and emission monochromators were set at 0.5 mm, giving a spectral bandwidth of 2 nm (except for fluorescein and compounds RBM5-155 and RBM5-159, in which case the monochromators were set at 0.35 mm, giving a spectral bandwidth of 1.4 nm). The data interval was 1 nm and the integration time was 1 s. All measurements were carried out using a Hellma 1.5 mL PTFE-stoppered fluorescence quartz cuvette (4 clear windows) with a 1 cm path length.
Molar extinction coefficients (ε) were calculated according to Lambert–Beer's law from solutions at concentrations ranging between 0.25 and 25 μM in the appropriate solvent system (spectrophotometric grade solvents). The absorbance values at the λAbsmax were then plotted against the corresponding concentrations and adjusted to a linear regression function forced through the origin (i.e. the line was forced to intercept (0,0)) using GraphPad Prism version 7.00 for Windows (GraphPad Software Inc., La Jolla, USA). Only the absorbance values in the range between 0.05 and 1 were used. Since we used a 1 cm path length cuvette, ε equals the slope of the graph.
Fluorescence quantum yields were measured (ΦF) following the comparative method described by Resch-Genger and Rurack41 (IUPAC technical report). The integrated fluorescence intensity (i.e. the area under the curve of the emission spectrum) was plotted against the corresponding Abs value at the λex and adjusted to a linear regression function forced through the origin using GraphPad Prism version 7.00 for Windows (GraphPad Software Inc., La Jolla, CA, USA). Then, ΦF was calculated by using eqn (1), where the subscripts x and Std. denote sample and standard, respectively, Grad equals the slope of the plot of the integrated fluorescence intensity vs. absorbance at the λex and η is the refractive index of the solvent.
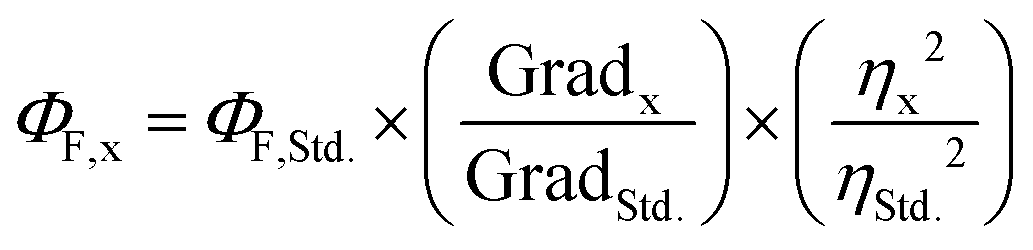 | (1) |
Spectral overlap integrals (J(λ)) of the donor–acceptor pairs were calculated using the specific J(λ) calculator tool from a|e UV-Vis-IR Spectral Software version 2.2 for Windows from FluorTools. To calculate J(λ), a|e UV-Vis-IR Spectral Software uses eqn (2), where FD is the normalised donor emission spectrum, εA is the extinction coefficient spectrum of the acceptor, and λ is the wavelength.
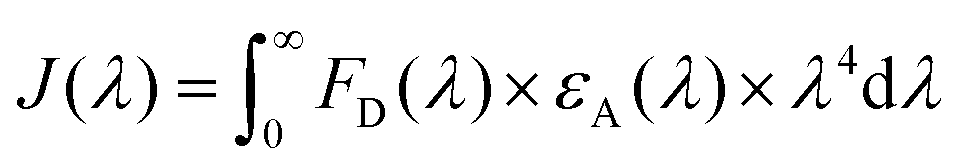 | (2) |
Förster radius (R0) of the two different donor–acceptor pairs were calculated by using eqn (3), where ΦD is the quantum yield of the donor, J(λ) is the spectral overlap integral, η is the refractive index of the solvent and κ is a constant that reflects the relative orientation of the excited donor's electric field and the acceptor's absorption dipole. For molecules in which the rotational diffusion of the dyes is faster than the donor's fluorescence lifetime, κ takes a value of 2/3.42R0 was expressed in Å.
| R0 = 0.211 × [κ2 × ΦD × J(λ) × η−4]1/6 | (3) |
Composite spectra deconvolution
The absorption and emission spectra of RBM5-161 were decomposed into their individual components using the Composite Spectrum Regression App for OriginPro version 2018 (OriginLab Corporation, Northampton, MA, USA). The spectra of RBM5-154 (donor) and RBM5-143 (acceptor) were used as the reference for x1 and x2, respectively, to express the spectra of RBM5-161 as the linear combination of the spectra of their individual components, according to the general eqn (4), where x1 and x2 are the spectra of the donor and the acceptor, respectively, multiplied for an appropriate coefficient (a1 and a2).| y = a1x1 + a2x2 | (4) |
The deconvolution regression was carried out forcing an intercept with the origin, thereby obtaining a fitted curve (xi,yi values), the values for the a1 and a2 coefficients and a fitting score (R-square and SE); the reference spectra of the individual components (x1 and x2) were then multiplied by the a1 and a2 coefficients to obtain the calculated spectra.
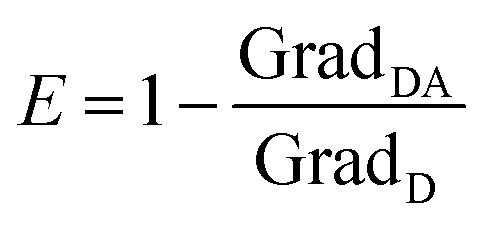 | (5) |
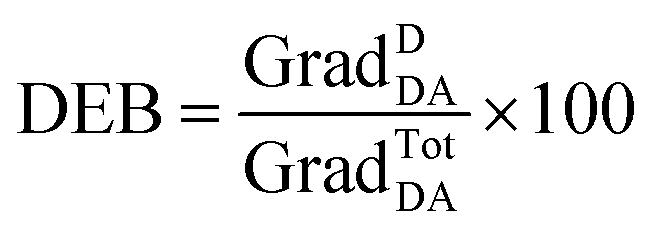 | (6) |
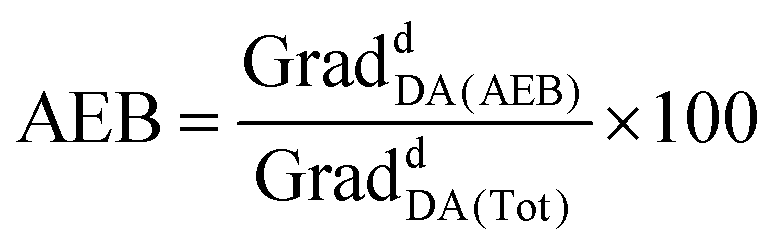 | (7) |
Cell studies
:chloroform, 2:1. Samples were heated at 48 °C overnight and next day, 75 μL of 1 M KOH in methanol was added, followed by 2 h of incubation at 37 °C. Afterwards, the saponification was neutralised with 75 μL of 1 M acetic acid and solvent was removed using a Speed Vac Savant SPD131DDA (Thermo Scientific).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Partial financial support from the Project CTQ2017-85378-R (AEI/FEDER, UE) of the Spanish Ministry of Science, Innovation and Universities is acknowledged. We are grateful to Prof. Anthony H. Futerman (Weizmann Institute of Science, Rehovot, Israel) for providing the CerS5 plasmid and to Prof. Vicente Marchán (UB) for technical support. Experimental contributions from Mr Alexandre García and Mr Pedro Rayo are also acknowledged. About the source of the cells, we purchased from ATCC (https://www.lgcstandards-atcc.org/en.aspx). We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).References
- A. C. Carreira, T. C. Santos, M. A. Lone, E. Zupančič, E. Lloyd-Evans, R. F. M. de Almeida, T. Hornemann and L. C. Silva, Mammalian Sphingoid Bases: Biophysical, Physiological and Pathological Properties, Prog. Lipid Res., 2019, 75, 100988 CrossRef CAS.
- Y. A. Hannun and L. M. Obeid, Principles of Bioactive Lipid Signalling: Lessons from Sphingolipids, Nat. Rev. Mol. Cell Biol., 2008, 9(2), 139–150 CrossRef CAS.
- A. Alonso and F. M. Goñi, The Physical Properties of Ceramides in Membranes, Annu. Rev. Biophys., 2018, 47(1), 633–654 CrossRef CAS.
- Y. A. Hannun, Functions of Ceramide in Coordinating Cellular Responses to Stress, Science, 1996, 274(5294), 1855–1859 CrossRef CAS.
- G. M. Jenkins, A. Richards, T. Wahl, C. Maos, L. Obeid and Y. A. Hannun, Involvement of Yeast Sphingolipids in the Heat Stress Response of Saccharomyces Cerevisiae, J. Biol. Chem., 1997, 272, 32566–32572 CrossRef CAS.
- H. Sietsma, R. J. Veldman and J. W. Kok, The Involvement of Sphingolipids in Multidrug Resistance, J. Membr. Biol., 2001, 181, 153–162 CrossRef CAS.
- S. M. Cho and H. J. Kwon, Acid Ceramidase, an Emerging Target for Anti-Cancer and Anti-Angiogenesis, Arch. Pharmacal Res., 2019, 42(3), 232–243 CrossRef CAS.
- T. Okazaki, R. M. Bell and Y. A. Hannun, Sphingomyelin Turnover Induced by Vitamin D3 in HL-60 Cells. Role in Cell Differentiation, J. Biol. Chem., 1989, 264(32), 19076–19080 CrossRef CAS.
- M. E. Venable, J. Y. Lee, M. J. Smyth, A. Bielawska and L. M. Obeid, Role of Ceramide in Cellular Senescence, J. Biol. Chem., 1995, 270(51), 30701–30708 CrossRef CAS.
- T. D. Mullen, Y. A. Hannun and L. M. Obeid, Ceramide Synthases at the Centre of Sphingolipid Metabolism and Biology, Biochem. J., 2012, 441, 789–802 CrossRef CAS.
- J. Kurz, M. J. Parnham, G. Geisslinger and S. Schiffmann, Ceramides as Novel Disease Biomarkers, Trends Mol. Med., 2018, 25(1), 20–32 CrossRef.
- J. Stiban, R. Tidhar and A. H. Futerman, Ceramide Synthases: Roles in Cell Physiology and Signaling, Adv. Exp. Med. Biol., 2010, 688, 60–71 CrossRef CAS.
- J. W. Park, W. J. Park and A. H. Futerman, Ceramide Synthases as Potential Targets for Therapeutic Intervention in Human Diseases, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2014, 1841(5), 671–681 CrossRef CAS.
- A. H. Merrill Jr., M. C. Sullards, J. C. Allegood, S. Kelly and E. Wang, Sphingolipidomics: High-Throughput, Structure-Specific, and Quantitative Analysis of Sphingolipids by Liquid Chromatography Tandem Mass Spectrometry, Methods, 2005, 36(2), 207–224 CrossRef.
- S. Grösch, S. Schiffmann and G. Geisslinger, Chain Length-Specific Properties of Ceramides, Prog. Lipid Res., 2012, 51(1), 50–62 CrossRef.
- I. Nieves, P. Sanllehí, J.-L. L. Abad, G. Fabriàs, J. Casas and A. Delgado, Chemical Probes of Sphingolipid Metabolizing Enzymes, in Bioactive Sphingolipids in Cancer Biology and Therapy, ed. Y. Hannun, C. Luberto, L. Obeid and C. Mao, Springer International Publishing, Cham, 2015, pp. 437–469 Search PubMed.
- J. L. Abad, I. Nieves, P. Rayo, J. Casas, G. Fabriàs and A. Delgado, Straightforward Access to Spisulosine and 4,5-Dehydrospisulosine Stereoisomers: Probes for Profiling Ceramide Synthase Activities in Intact Cells, J. Org. Chem., 2013, 78(12), 5858–5866 CrossRef CAS.
- An initial version of this work was deposited in ChemRxiv on August 6, 2020, DOI: 10.26434/chemrxiv.127641.
- S. Haldar and A. Chattopadhyay, in Application of NBD-Labeled Lipids in Membrane and Cell Biology, ed. Y. Mély and G. Duportail, Springer Berlin Heidelberg, 2012, pp. 37–50 Search PubMed.
- P. J. G. Coutinho, Photophysics and Biophysical Applications of Benzo[a]Phenoxazine Type Fluorophores, in Reviews in Fluorescence, Springer, New York, NY, 2009, vol. 2007, pp. 335–362 Search PubMed.
- O. Wichmann, M. H. Gelb and C. Schultz, Probing Phospholipase A2 with Fluorescent Phospholipid Substrates, ChemBioChem, 2007, 8(13), 1555–1569 CrossRef CAS.
- O. Wichmann, J. Wittbrodt and C. Schultz, A Small-Molecule FRET Probe To Monitor Phospholipase A2 Activity in Cells and Organisms, Angew. Chem., Int. Ed., 2006, 45(3), 508–512 CrossRef CAS.
- O. Wichmann and C. Schultz, FRET Probes to Monitor Phospholipase A2 Activity, Chem. Commun., 2001, 1(23), 2500–2501 RSC.
- K. P. Bhabak, A. Hauser, S. Redmer, S. Banhart, D. Heuer and C. Arenz, Development of a Novel FRET Probe for the Real-Time Determination of Ceramidase Activity, ChemBioChem, 2013, 14(9), 1049–1052 CrossRef CAS.
- T. Pinkert, D. Furkert, T. Korte, A. Herrmann and C. Arenz, Amplification of a FRET Probe by Lipid–Water Partition for the Detection of Acid Sphingomyelinase in Live Cells, Angew. Chem., Int. Ed., 2017, 56(10), 2790–2794 CrossRef CAS.
- J. G. Mina, J. A. Mosely, H. Z. Ali, P. W. Denny and P. G. Steel, Exploring Leishmania Major Inositol Phosphorylceramide Synthase (LmjIPCS): Insights into the Ceramide Binding Domain, Org. Biomol. Chem., 2011, 9(6), 1823–1830 RSC.
- P. K. Mandal and J. S. McMurray, Pd−C-Induced Catalytic Transfer Hydrogenation with Triethylsilane, J. Org. Chem., 2007, 72(17), 6599–6601 CrossRef CAS.
- In this sense, our early attempts to obtain the free amine by washing the organic extracts with 0.5 M NaOH resulted in the formation of unidentified by-products, as evidenced by the appearance of new UV active spots in the TLC plate, and a poor recovery after column chromatography of the crude.
- T. Walter, J. Schlegel, A. Burgert, A. Kurz, J. Seibel and M. Sauer, Incorporation Studies of Clickable Ceramides in Jurkat Cell Plasma Membranes, Chem. Commun., 2017, 53(51), 6836–6839 RSC.
- J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Readily Accessible Bicyclononynes for Bioorthogonal Labeling and Three-Dimensional Imaging of Living Cells, Angew. Chem., Int. Ed., 2010, 49(49), 9422–9425 CrossRef CAS.
- I. Tosi, Model Systems for Artificial Photosynthesis: Calix[4]Arenes Functionalized with Chromophoric Units for Energy and Charge Transfer, Doctoral thesis, Universita’ degli studi di Parma, 2015 Search PubMed.
- E. A. Bykova and J. Zheng, Spectra FRET: A Fluorescence Resonance Energy Transfer Method in Live Cells, in Reviews in Fluorescence 2007, ed. C. D. Geddes, Springer, New York, NY, 2009, pp. 87–101 Search PubMed.
- S. Smith, A. Witkowski and A. K. Joshi, Structural and Functional Organization of the Animal Fatty Acid Synthase, Prog. Lipid Res., 2003, 42(4), 289–317 CrossRef CAS.
- S. A. McCune and R. A. Harris, Mechanism Responsible for 5-(Tetradecyloxy)-2-Furoic Acid Inhibition of Hepatic Lipogenesis, J. Biol. Chem., 1979, 254(20), 10095–10101 CrossRef CAS.
- D. L. Halvorson and S. A. McCune, Inhibition of Fatty Acid Synthesis in Isolated Adipocytes by 5-(Tetradecyloxy)-2-Furoic Acid, Lipids, 1984, 19(11), 851–856 CrossRef CAS.
- D. Vance, I. Goldberg, O. Mitsuhashi, K. Bloch, S. Ōmura and S. Nomura, Inhibition of Fatty Acid Synthetases by the Antibiotic Cerulenin, Biochem. Biophys. Res. Commun., 1972, 48(3), 649–656 CrossRef CAS.
- S. Lahiri, H. Lee, J. Mesicek, Z. Fuks, A. Haimovitz-Friedman, R. N. Kolesnick and A. H. Futerman, Kinetic Characterization of Mammalian Ceramide Synthases: Determination of Km Values towards Sphinganine, FEBS Lett., 2007, 581(27), 5289–5294 CrossRef CAS.
- S. Lahiri and A. H. Futerman, LASS5 Is a Bona Fide Dihydroceramide Synthase That Selectively Utilizes Palmitoyl-CoA as Acyl Donor, J. Biol. Chem., 2005, 280(40), 33735–33738 CrossRef CAS.
- A. N. Rai and A. Basu, Sphingolipid Synthesis via Olefin Cross Metathesis: Preparation of a Differentially Protected Building Block and Application to the Synthesis of D-Erythro-Ceramide, Org. Lett., 2004, 6(17), 2861–2863 CrossRef CAS.
- T. Ibuka, H. Habashita, A. Otaka, N. Fujii, Y. Oguchi, T. Uyehara and Y. Yamamoto, A Highly Stereoselective Synthesis of (E)-Alkene Dipeptide Isosteres via Organocyanocopper-Lewis Acid Mediation Reaction, J. Org. Chem., 1991, 56(14), 4370–4382 CrossRef CAS.
- U. Resch-Genger and K. Rurack, Determination of the Photoluminescence Quantum Yield of Dilute Dye Solutions (IUPAC Technical Report), Pure Appl. Chem., 2013, 85(10), 2005–2013 CAS.
- S. M. Müller, H. Galliardt, J. Schneider, B. George Barisas and T. Seidel, Quantification of Förster Resonance Energy Transfer by Monitoring Sensitized Emission in Living Plant Cells, Front. Plant Sci., 2013, 413 Search PubMed , Frontiers Research Foundation.
Footnote |
| † Electronic supplementary information (ESI) available: Calculated spectral overlap integrals and Förster critical distances (Table S1), photophysical properties of the monochromophoric compounds (Table S2), photophysical properties of the bichromophoric compounds (Table S3), and the NMR spectra of the synthesized compounds. See DOI: 10.1039/d1ob00113b |
| This journal is © The Royal Society of Chemistry 2021 |