
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Identification of organophosphorus simulants for the development of next-generation detection technologies†
Rebecca J.
Ellaby
a,
Ewan R.
Clark
*a,
Nyasha
Allen
b,
Faith R.
Taylor
a,
Kendrick K. L.
Ng
a,
Milan
Dimitrovski
a,
Dominique F.
Chu
c,
Daniel P.
Mulvihill
b and
Jennifer R.
Hiscock
 *a
*a
aSchool of Physical Sciences, University of Kent, Park Wood Road, Canterbury, Kent CT2 7NH, UK. E-mail: J.R.Hiscock@Kent.ac.uk; E.R.Clark@Kent.ac.uk; Tel: +44(0) 1227 816467 Tel: +44(0) 1227 816152
bSchool of Biosciences, University of Kent, Park Wood Road, Canterbury, Kent CT2 7NH, UK
cSchool of Computing, University of Kent, Darwin Road, Canterbury, Kent CT2 7NZ, UK
First published on 15th February 2021
Abstract
Organophosphorus (OP) chemical warfare agents (CWAs) represent an ongoing threat but the understandable widespread prohibition of their use places limitations on the development of technologies to counter the effects of any OP CWA release. Herein, we describe new, accessible methods for the identification of appropriate molecular simulants to mimic the hydrogen bond accepting capacity of the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety, common to every member of this class of CWAs. Using the predictive methodologies developed herein, we have identified OP CWA hydrogen bond acceptor simulants for soman and sarin. It is hoped that the effective use of these physical property specific simulants will aid future countermeasure developments.
O moiety, common to every member of this class of CWAs. Using the predictive methodologies developed herein, we have identified OP CWA hydrogen bond acceptor simulants for soman and sarin. It is hoped that the effective use of these physical property specific simulants will aid future countermeasure developments.
Introduction
Organophosphorus (OP) compounds have been used on a global scale as pesticides and chemical warfare agents (CWAs) for almost a century.1 The uncontrolled release of these highly toxic compounds presents a continuing global risk as evidenced by recent small-scale events within Germany (2020),2 the UK (2018)3 and Malaysia (2017),4 combined with larger-scale events in Syria (2013)5 and Japan (1995).6 The development of novel detection technologies is therefore of high global importance.Recently, supramolecular systems have been effectively targeted to fulfil this need.7 This has included the development of fluorescent8 and luminescent sensors,9–11 and a body of work from Gale and co-workers in which hydrogen bond donating compounds have been shown to: selectively form OP CWA hydrogen bonded complexes;12,13 act as OP CWA hydrolysis organocatalysts;14 produce supramolecular organogels that act both as OP CWA sensory and decontamination materials.15–17 Additionally, Cragg and co-workers have developed a crown-(thio)urea based receptor capable of sensing the presence of sarin and soman hydrolysis breakdown products.18 Here the resultant fluoride ion binds to the thio(urea) moiety, while the phosphate group coordinates to the crown ether, resulting in a colourless to orange colorimetric change. Finally, a body of work by Ward and co-workers has shown that self-assembled cages can bind CWA simulants inducing a fluorescence response,19 and act as catalysts for the hydrolysis of phospho-ester species.20
OP simulants are typically the only available option for developing novel approaches to combat OP CWA release due to the highly toxic nature of, and legal restrictions placed upon, the live agents themselves. However, no single simulant can simultaneously mimic all the chemical properties of an OP CWA without also inheriting undesirable molecular traits. It is therefore vital to consider the appropriate properties of any simulant chosen to aid in the development of novel OP CWA detection, decontamination, or remediation methodologies.21–24 Recently, work undertaken by Snurr and Mendonca, has shown that density functional theory (DFT) can be used to study the mechanism of OP hydrolysis, which has resulted in the development of a quantitative structure activity relationship (QSAR) model that enables the identification of appropriate OP CWA simulants for decontamination purposes.25
However, when considering supramolecular technologies, the moiety target is often selective coordination of the polar PO functionality. This chemical group, common to all OP CWAs, plays a significant role in both molecular surface coordination properties and reactivity. Herein, we extend our prior work, which showed that simple, low level computational modelling may be used to predict hydrogen bond mediated aggregation events26,27 and antimicrobial activity,28 to identify appropriate OP CWA simulants for the development of supramolecular detection technologies, complementing the recent advances made in this area for hydrolysis siumulants.25
Here, we hypothesised that neutral hydrogen bond donating receptors 1–4 (Fig. 1) would form hydrogen bonded complexes with potential OP CWA simulants (5–26) and, due to the structural simplicity of 1–4, the strength of the complex formed would depend on the properties associated with the principal hydrogen bond acceptor (HBA) group. The association constants (Kass) relating to these hydrogen bonded complexation processes should therefore provide information relating to the simulants’ principal HBA site, which corresponds to the moiety mimicking the PO group of a specific OP CWA. These experimental data are then correlated with parameters derived from accessible computational modelling methods, to enable the identification of an appropriate simulant. Compounds 11–15 are commercially available species commonly used to simulate OP CWAs. The structures of simulants 5–10 and 16–26 were designed to fulfil the following criteria: charge neutrality; not readily ionised under experimental conditions; containing a single electrophilic site (S or P); and containing a single HBA centre (either a PO (5–15), or OSO (16–26)).
 | ||
| Fig. 1 Chemical structure of 1–26, sarin and soman. | ||
Synthesis
The syntheses of 1–4, 7–9, 17–21, and 23–26 have previously been reported.29–40 Compounds 5, 6 and 10 were obtained as colourless oils in 65%, 90%, and 85% yields respectively, from the reaction of diethyl chlorophosphate with the respective alcohol in chloroform. Compound 16 was obtained as a white solid in a 59% yield from the reaction of 4-(trifluoromethyl)phenol with p-toluene sulfonylchloride in chloroform. Compound 22 was obtained as a pale-yellow solid in a 91% yield from the reaction of 4-(trifluoromethyl)phenol with methylsulfonyl chloride in chloroform.Results and discussion
Association constant determination
Experimental association constants for receptor![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :simulant complexes were determined in CD3CN using 1H NMR titration techniques, following the downfield change in chemical shift of the thiourea/pyrrole NH resonances upon increasing concentration of the simulant with respect to the appropriate receptor (1–4). In all cases the experimental data were fitted to 1:1, 2:1, and 1:2 host:guest binding isotherms using Bindfit v0.5.41 The errors produced from fitting these data were compared and found to support the formation of 1:1 receptor:simulant complexes. Job plot experiments were also performed but did not give conclusive results.42
:simulant complexes were determined in CD3CN using 1H NMR titration techniques, following the downfield change in chemical shift of the thiourea/pyrrole NH resonances upon increasing concentration of the simulant with respect to the appropriate receptor (1–4). In all cases the experimental data were fitted to 1:1, 2:1, and 1:2 host:guest binding isotherms using Bindfit v0.5.41 The errors produced from fitting these data were compared and found to support the formation of 1:1 receptor:simulant complexes. Job plot experiments were also performed but did not give conclusive results.42
A series of initial association studies were conducted to identify a lead receptor; the results are summarised in Table 1. Here, only the most acidic receptor (1) was found to produce association constants that span the ≥two orders of magnitude necessary to validate our hypothesis. Therefore, further data analysis utilises results obtained with 1 only. These initial results were then expanded to include all simulants (5–26); this final data set is given in Table 2.
:1 host:guest binding isotherm using Bindfit v0.541
:1 host:guest binding isotherm using Bindfit v0.541
| Simulant | K ass | Simulant | K ass |
|---|---|---|---|
| 5 | 36 (±6%) | 16 | 25 (±7%) |
| 6 | 17 (±1%) | 17 | <10 |
| 7 | <10 | 18 | <10 |
| 8 | <10 | 19 | 48 (±11%) |
| 9 | <10 | 20 | 106 (±8%) |
| 10 | <10 | 21 | 41 (±8%) |
| 11 | 11 (±1%) | 22 | 64 (±5%) |
| 12 | 52 (±6%) | 23 | 46 (±9%) |
| 13 | <10 | 24 | 76 (±6%) |
| 14 | <10 | 25 | <10 |
| 15 | 12 (±2%) | 26 | <10 |
A single crystal X-ray structure,‡ obtained for a 1:1 complex of 1 and DMSO (Fig. 2), shows the formation of one hydrogen bond from each of the thiourea NH groups to the SO HBA group of the DMSO solvent molecule. It is therefore reasonable to hypothesise that the analogous hydrogen bonded complexes may be formed between 1 and simulants 5–15, which contain a single oxygen atom acting as the principal HBA (Fig. 3c). However, when considering simulants 16–26, the possibility exists that, because of the two HBA oxygen atoms within the sulfonate group, the formation of hydrogen bonds may be permitted to one (Fig. 3a, O1′) or both of these atoms (Fig. 3b, O2′).
 | ||
| Fig. 2 Single crystal X-ray structure showing 1 binding a single DMSO solvent molecule through the formation of two hydrogen bonds. Disorder associated with the DMSO molecule and CF3 moieties have been omitted for clarity. Grey = carbon; white = hydrogen; green = fluorine; blue = nitrogen; yellow = sulphur. Red dashed line indicates hydrogen bonding. | ||
 | ||
| Fig. 3 Possible binding modes for the formation of 1:1 complexes of 1 with (a) 16–26, (b) 16–26, (c) 5–15 and (d) 5–15. Hydrogen bonding angles are shown in red. | ||
In-silico modelling
As it is feasible that 16–26 could adopt one of two 1:1 binding modes with 1 (Fig. 3b and c), the potential for one of these binding modes prevailing within the solution state was further explored computationally (M06-2X/6-311 g(d,p)), (PCM = acetonitrile, Gaussian16).43–45
The geometries of 6, 9, 10, 11, 16–20, 25, 26, sarin and soman with receptor 1, were optimised individually and as interacting pairs in both O1′ and O2′ configurations.† These calculations confirmed the viability of both O1′ and O2′ binding modes for the sulfonyl species. However, as expected no stable O2′ binding modes were found for the phosphonyl species (Fig. 3d). The energies of binding are significant (−41.2 to −84.6 kJ mol−1) for both sulfonyl and phosphonyl species, with no clear preference for O1′ over O2′ found for the sulfonyls, although significant substituent dependence was observed. This is attributed to the variable changes in steric profile on moving the simulant between O1′ and O2′ bonding and the resultant effect on secondary interactions.
Having confirmed the binding modes, we then sought to develop an accessible predictive model for association strength. DFT (M06-2X/6-311 g(d,p)) is a good computational model for systems such as these as it accounts for non-covalent interactions but is computationally expensive.46 We have previously shown that low-level calculations can supply useful guest-only parameters for predicting trends in association constant without significant computational overhead. Reducing the size of the calculations requires far less computational time; additionally, Spartan ‘16 runs on a conventional desktop making this approach accessible to a wider chemical audience. The range of parameters output by default for PM6 calculations is sparse, and so to expand the scope of searchable parameter space, DFT (B3LYP/6-3G1*) calculations were also performed using Spartan ‘16.47–49 The analogous parameters obtained were consistent with those values produced from PM6 and M06-2X/6-311 g(d,p) calculations.† In total, a list of 19 computationally derived parameters (P), summarised in Table 3, were produced for each potential simulant/OP CWA alone or when in a 1:1 complex with 1. This list of parameters are easily accessible, unfiltered by assumption of their relevance to avoid prejudicing the parametric search. These parameters were then used alongside the experimentally derived association constant data (Table 2) to produce initial predictive association constant models.
| P | P description | SI unit | Derivation method | |
|---|---|---|---|---|
| PM6 | B3LYP/6-31G* | |||
| N/A = non-applicable. ‘×’ identifies the computational modeling method used to generate the values for a particular parameter (P). | ||||
| P 1 | E min | kJ mol−1 | × | |
| P 2 | E max | kJ mol−1 | × | |
| P 3 | Molecular volume | Å3 | × | |
| P 4 | Molecular area | Å2 | × | |
| P 5 | Solvent accessible area | Å2 | × | |
| P 6 | Polar surface area | Å2 | × | |
| P 7 | % Polar surface area | % | × | |
| P 8 | Polarizability | C m−2 | × | |
| P 9 | Steric weighting factor (SWF) | Å2 | × | |
| P 10 | Steric accessibility factor (SAF) | Å2 | × | |
| P 11 | HOMO | eV | × | |
| P 12 | LUMO | eV | × | |
| P 13 | Total energy | kJ | × | |
| P 14 | Electrostatic charge | C | × | |
| P 15 | Additive N⋯O bond length | Å | × | |
| P 16 | logP |
N/A | × | |
| P 17 | Dipole moment | D | × | |
| P 18 | Additive NH⋯O bond length | Å | × | |
| P 19 | H-Bond angle difference from optimal 180° | ° | × | |
Single parameter predictive models
Initial comparison identified correlations between the experimentally derived association constants and host:guest hydrogen bond angle (obtained from B3LYP-6-31G* calculations), Fig. 4. For ease of interpretation, the hydrogen bond angles involved in complex formation are represented as the sum of the difference from the optimal 180° hydrogen bonding angle for those hydrogen bonds formed during the potential simulant:receptor complexation processes,50,51 such as those exemplified in Fig. 3. Here, we identify three trends, one for the phosphoryl species and two for the sulfonyl species. The two trends identified for the sulfonyl species correspond to the two different classes of sulfonate molecules present within the library of simulants studied, those substituted with a toluene group (16–20) and those substituted with a methyl group (21–26). As the limitation of the experimental association constant determination methodology is ≈10 M−1, we suggest that any values incorporating ≤10 M−1 may be considered inappropriate when included into a model such as this and should be treated with caution. We hypothesise that for a complex formed utilising a single HBA atom, increasing hydrogen bonding strength will result in an overall decrease in hydrogen bonding angle away from 180°, as the host and guest move closer together, resulting in a positive correlation as observed for the phosphoryl species (blue and black trends, Fig. 4). However, for complexes formed using two HBA oxygen atoms, the hydrogen bonding angles will move towards 180°, resulting in a negative correlation as observed for both sulfonate species (blue and black trends, Fig. 4). The two different trends observed for the methyl and toluene substituted sulphonyl groups we believe are due to additional R-group specific interactions.
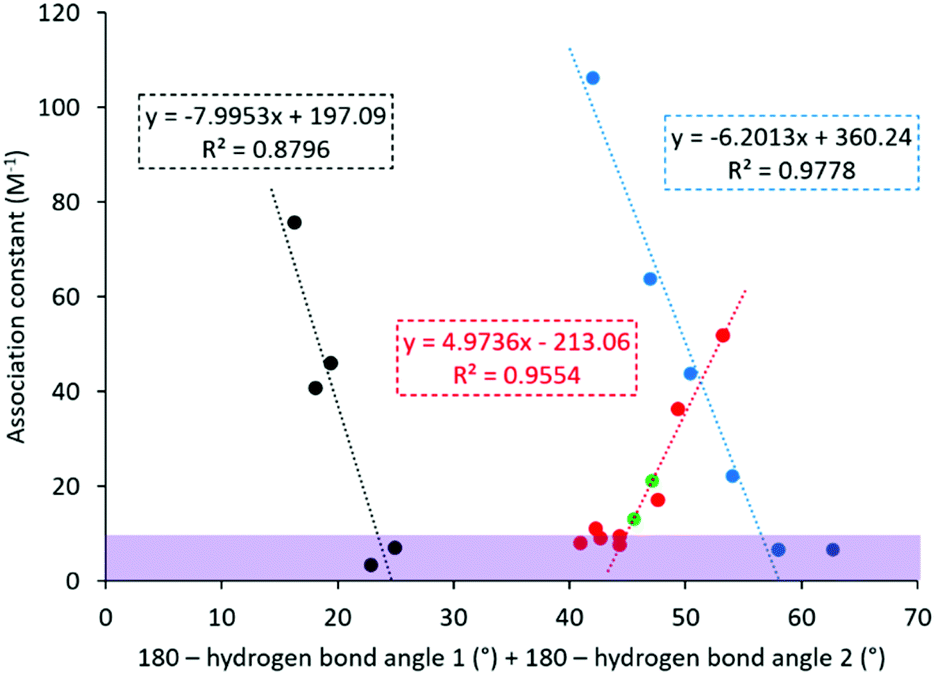 | ||
| Fig. 4 One parameter association constant (Kass) prediction. Red = phosphoryl species; blue = toluene substituted sulfonyl species; black = methyl substituted sulfonyl species; green = predicted OP CWAs; purple shaded section = Kass ≤ 10 M−1. | ||
The OP CWA association constants were then predicted for sarin and soman (Fig. 4, green), using the trend shown for the phosphoryl species (Fig. 4, red), resulting in predicted association constant values of 22 M−1 and 13 M−1 for sarin and soman respectively. Using the three trends shown in Fig. 4, compounds 6 and 16 were identified as the most appropriate simulants for sarin, while 10 and 11 were identified as the most appropriate simulants for soman.
Exhaustive parameter search
The energetics of association do not depend solely on a single molecular property, and we have previously shown that the use of an exhaustive, high throughput parameter search allows the rapid identification of predictive models.28 To enable the elucidation of further association constant predictive models using more than one computational parameter (Table 3), R data analysis software52 was used to complete an exhaustive search of all potential parameter combinations for direct and inverse correlations of up to three parameters (eqn (1) and (2)). These searches included all relevant association constant data (Table 2) where Kass > 10, to identify a single linear correlation. | (1) |
 | (2) |
The form of these equations is suggested by the manner in which factors contribute to the free energy of binding and thus association constant.† The top 10 fits of these data to each of these two equations as identified through R2 analysis are detailed within the supplementary materials. Although this approach did not result in the identification of any viable predictive models in this instance, it does offer an insight into which factors contribute to binding. Examination of the top 10 fits identified through either two parameter or three parameter searches shows that only some parameters exhibit any energetic significance, these results are summarised in Table 3. From this analysis, P10 (steric accessibility factor) and P14 (HBA electrostatic charge) are the two parameters that appear most commonly, with a total of 17 and 9 occurrences respectively; P11 (HOMO energy) is the third most common parameter, with 8 occurrences. The prevalence of P14 is not surprising as the electrostatic charge at the acceptor atoms would be expected to contribute to the electrostatic component of hydrogen bonding. Likewise, the HOMO energies, as associated with lone pairs at oxygen, will be involved in the potential for covalent contribution to the hydrogen bonding. More perplexing is the dominance of P10, which describes the exposed electrophilic site of the simulant and not the HBA oxygen at all. However, the trends identified show an inverse correlation with P10 – the larger the exposed electrophilic site, the smaller the binding constant. This therefore corresponds to the steric bulk about the P or S atom and the extent to which the substituents are folded away from the electrophilic site and towards the HBA oxygens. We can therefore regard P10 as a proxy for steric bulk about the HBA centres. Taken together, these indicate that simple electrostatic models of hydrogen bonding are not sufficient to describe these systems, and that consideration of secondary bonding (and repulsive) interactions will be key in properly mimicking OP CWAs in future work (Table 4).
Potential simulant toxicity
It is imperative that any simulants do not duplicate the toxicity of OP CWAs. The toxicity of these potential simulants against the commonly used model species Schizosaccharomyces pombe (S. pombe) was ascertained to gain some insight as to the potential toxicity of 5–12 and 16–26 towards eukaryotic cells in a biological environment.53,54 The growth of this model yeast system was monitored by optical density measurements at 600 nm (OD600) in the presence and absence of 5–12 and 16–26, which were supplied in a 1:19 EtOH:H2O solution to aid simulant solubility. The results of these studies are summarised in Fig. 5.
 | ||
| Fig. 5 S. pombe toxicity screening % cellular growth (recorded as maximum OD600) reached at stationary phase in the presence of 5–12, 16–26 (3.3 mM) relative to controls after 45 hours, supplied to the microbial broth solution in a 5% ethanol solution. Data shown represents an average of three experiments. Green – cell growth impeded by <16% over the course of 45 hours; Amber – cell growth impeded by 16–50% over the course of 45 hours; Red – cell growth impeded by >50% over the course of 45 hours. Control = 5% aqueous ethanol solution. Error = standard error of the mean. | ||
These preliminary results show that, at 3.3 mM, the simulants 5, 8, 10, 16–20, 22, 25 and 26 have no significant impact on cell growth therefore demonstrating little/no toxic effects, while simulants 6, 7, 9, 11, 12, 21, 23 and 24 show a degree of toxicity that warrants further investigation. The lead simulants identified to mimic complex formation for soman and sarin were shown to reduce the growth of S. pombe by 22.1%, 17.8%, 7.6%, 9.3%, 22.3% and 6.5% for 6, 8–11, and 16 respectively over a period of 45 h relative to control samples. This indicates simulants 8, 10 and 16 pose no significant toxicity risk to eukaryotic organisms, while simulants 6, 9 and 11 would require further investigation. It is also important to note that 17, 19 and 22 showed greater than 100% cell growth and further investigations into this are underway. Nevertheless, full pharmacodynamic and pharmacokinetic studies should be undertaken before these simulants are considered safe to handle without the use of personal protective equipment.
Conclusions
We have produced a first-generation predictive model to aid in OP CWA simulant selection for the development of defensive technologies relating to OP CWA sensing through complexation, combining low level computational modelling and exhaustive parameter searches. The lead simulants currently identified using this model are 6 and 16 (for sarin), and 10 and 11 (for soman). Additionally, we hypothesise that the single parameter relationships identified here maybe developed towards the identification of solution state binding mode.Finally, these results show that when choosing a simulant for the development of novel OP CWA detection and remediation methodologies, the specific physicochemical properties of interest should be considered. It is hoped that the uncovering of structure activity relationships achieved using similar parametric searching methodologies to those detailed herein will enable the evermore effective identification of appropriate simulants to aid in the development of novel technologies for the effective detection of OP CWAs. Our work in this area is ongoing as we seek to apply this highly accessible methodology to enable the identification of suitable simulants not only for CWAs but also for pesticides, a common agricultural pollutant.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. Hiscock and R. Ellaby would like to thank the University of Kent for funding. We would also like to thank K. Howland (School of Biosciences, University of Kent) for his mass spectrometry assistance.Notes and references
- R. T. Delfino, T. S. Ribeiro and J. D. Figueroa-Villar, J. Braz. Chem. Soc., 2009, 20, 407–428 CrossRef CAS.
- German Federal Government, Statement by the Federal Government on the Navalny case, https://www.bundesregierung.de/breg-en/news/statement-by-the-federal-government-on-thenavalny-case-1781882 (accessed 24th September 2020).
- M. Peplow, Chem. Eng. News, 2018, 96, 3–3 Search PubMed.
- T. Nakagawa and A. T. Tu, Forensic Toxicol., 2018, 36, 542–544 CrossRef CAS.
- H. John, M. J. van der Schans, M. Koller, H. E. T. Spruit, F. Worek, H. Thiermann and D. Noort, Forensic Toxicol., 2018, 36, 61–71 CrossRef CAS.
- T. Okumura, N. Takasu, S. Ishimatsu, S. Miyanoki, A. Mitsuhashi, K. Kumada, K. Tanaka and S. Hinohara, Ann. Emerg. Med., 1996, 28, 129–135 CrossRef CAS.
- M. R. Sambrook and S. Notman, Chem. Soc. Rev., 2013, 42, 9251–9267 RSC.
- B. D. de Grenu, D. Moreno, T. Torroba, A. Berg, J. Gunnars, T. Nilsson, R. Nyman, M. Persson, J. Pettersson, I. Eklind and P. Wasterby, J. Am. Chem. Soc., 2014, 136, 4125–4128 CrossRef.
- G. H. Dennison, C. G. Bochet, C. Curty, J. Ducry, D. J. Nielsen, M. R. Sambrook, A. Zaugg and M. R. Johnston, Eur. J. Inorg. Chem., 2016, 9, 1348–1358 CrossRef.
- G. H. Dennison, M. R. Sambrook and M. R. Johnston, RSC Adv., 2014, 4, 55524–55528 RSC.
- G. H. Dennison, M. R. Sambrook and M. R. Johnston, Chem. Commun., 2014, 50, 195–197 RSC.
- M. R. Sambrook, J. R. Hiscock, A. Cook, A. C. Green, I. Holden, J. C. Vincent and P. A. Gale, Chem. Commun., 2012, 48, 5605–5607 RSC.
- J. R. Hiscock, N. J. Wells, J. A. Ede, P. A. Gale and M. R. Sambrook, Org. Biomol. Chem., 2016, 14, 9560–9567 RSC.
- J. R. Hiscock, M. R. Sambrook, P. B. Cranwell, P. Watts, J. C. Vincent, D. J. Xuereb, N. J. Wells, R. Raja and P. A. Gale, Chem. Commun., 2014, 50, 6217–6220 RSC.
- J. R. Hiscock, I. L. Kirby, J. Herniman, G. J. Langley, A. J. Clark and P. A. Gale, RSC Adv., 2014, 4, 45517–45521 RSC.
- J. R. Hiscock, M. R. Sambrook, J. A. Ede, N. J. Wells and P. A. Gale, J. Mater. Chem. A, 2015, 3, 1230–1234 RSC.
- J. R. Hiscock, M. R. Sambrook, N. J. Wells and P. A. Gale, Chem. Sci., 2015, 6, 5680–5684 RSC.
- H. Cave, J. A. Ede, M. R. Sambrook, H. Dodd, F. Fucassi, A. S. Cragg, A. H. Lansley and P. J. Cragg, Supramol. Chem., 2019, 31, 703–712 CrossRef CAS.
- C. G. P. Taylor, J. R. Piper and M. D. Ward, Chem. Commun., 2016, 52, 6225–6228 RSC.
- C. G. P. Taylor, A. J. Metherell, S. P. Argent, F. M. Ashour, N. H. Williams and M. D. Ward, Chem. – Eur. J., 2020, 26, 3065–3073 CrossRef CAS.
- J. A. Ede, P. J. Cragg and M. R. Sambrook, Molecules, 2018, 23(1), 207–219 CrossRef.
- M. R. Sambrook, I. A. Gass and P. J. Cragg, Supramol. Chem., 2018, 30, 206–217 CrossRef CAS.
- M. R. Sambrook, J. C. Vincent, J. A. Ede, I. A. Gass and P. J. Cragg, RSC Adv., 2017, 7, 38069–38076 RSC.
- S. L. Bartelt-Hunt, D. R. U. Knappe and M. A. Barlaz, Crit. Rev. Environ. Sci. Technol., 2008, 38, 112–136 CrossRef CAS.
- M. L. Medonca and R. Q. Snurr, Chem. – Eur. J., 2019, 25, 9217–9229 CrossRef.
- L. J. White, S. N. Tyuleva, B. Wilson, H. J. Shepherd, K. K. L. Ng, S. J. Holder, E. R. Clark and J. R. Hiscock, Chem. – Eur. J., 2018, 24, 7761–7773 CrossRef CAS.
- L. J. White, N. J. Wells, L. R. Blackholly, H. J. Shepherd, B. Wilson, G. P. Bustone, T. J. Runacres and J. R. Hiscock, Chem. Sci., 2017, 8, 7620–7630 RSC.
- N. Allen;, L. J. White, J. E. Boles, G. T. Williams, D. F. Chu, R. J. Ellaby, H. J. Shepherd, K. K. L. Ng, L. R. Blackholly, B. Wilson, D. P. Mulvihill and J. R. Hiscock, ChemMedChem, 2020, 15, 2193–2205 CrossRef.
- N. Busschaert, I. L. Kirby, S. Young, S. J. Coles, P. N. Horton, M. E. Light and P. A. Gale, Angew. Chem., Int. Ed., 2012, 51, 4426–4430 CrossRef CAS.
- C. M. Chau, T. J. Chuan and K. M. Liu, RSC Adv., 2014, 4, 1276–1282 RSC.
- S. M. S. Chauhan, B. Garg and T. Bisht, Molecules, 2007, 12, 2458–2466 CrossRef CAS.
- Y. F. Chen, C. L. Kao, W. K. Lee, P. C. Huang, C. Y. Hsu and C. H. Kuei, J. Chin. Chem. Soc., 2016, 63, 751–757 CrossRef CAS.
- D. S. Panmand, A. D. Tiwari, S. S. Panda, J. C. M. Monbaliu, L. K. Beagle, A. M. Asiri, C. V. Stevens, P. J. Steel, C. D. Hall and A. R. Katritzky, Tetrahedron Lett., 2014, 55, 5898–5901 CrossRef CAS.
- H. Huang, J. Ash and J. Y. Kang, Org. Lett., 2018, 20, 4938–4941 CrossRef CAS.
- S. M. Feng, J. Li and J. F. Wei, New J. Chem., 2017, 41, 4743–4746 RSC.
- D. V. Schoonover and H. W. Gibson, Tetrahedron Lett., 2017, 58, 242–244 CrossRef CAS.
- J. W. W. Chang, E. Y. R. Chia, C. L. L. Chai and J. Seayad, Org. Biomol. Chem., 2012, 10, 2289–2299 RSC.
- T. Noji, H. Fujiwara, K. Okano and H. Tokuyama, Org. Lett., 2013, 15, 1946–1949 CrossRef CAS.
- N. Ortega, A. Feher-Voelger, M. Brovetto, J. I. Padron, V. S. Martin and T. Martin, Adv. Synth. Catal., 2011, 353, 963–972 CrossRef CAS.
- J. J. Wang, Y. Y. Zhao, W. Zhao, P. Wang and J. Li, J. Carbohydr. Chem., 2016, 35, 445–454 CrossRef CAS.
- P. Thordarson, K. Sewell and V. Efremova, supramolecular.org.
- F. Ulatowski, K. Dabrowa, T. Balakier and J. Jurczak, J. Org. Chem., 2016, 81, 1746–1756 CrossRef CAS.
- M. Walker, A. J. A. Harvey, A. Sen and C. E. H. Dessent, J. Phys. Chem. A, 2013, 12590–12600 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- A. F. Rodrigues-Oliveira, F. W. M. Ribeiro, G. Cervi and T. C. Correra, ACS Omega, 2018, 3, 9075–9085 CrossRef CAS.
- Y. Shao, L. F. Molnar, Y. Jung, J. Kussmann, C. Ochsenfeld, S. T. Brown, A. T. B. Gilbert, L. V. Slipchenko, S. V. Levchenko, D. P. O'Neill, R. A. DiStasio Jr., R. C. Lochan, T. Wang, G. J. O. Beran, N. A. Besley, J. M. Herbert, C. Y. Lin, T. Van Voorhis, S. H. Chien, A. Sodt, R. P. Steele, V. A. Rassolov, P. E. Maslen, P. P. Korambath, R. D. Adamson, B. Austin, J. Baker, E. F. C. Byrd, H. Dachsel, R. J. Doerksen, A. Dreuw, B. D. Dunietz, A. D. Dutoi, T. R. Furlani, S. R. Gwaltney, A. Heyden, S. Hirata, C.-P. Hsu, G. Kedziora, R. Z. Khalliulin, P. Klunzinger, A. M. Lee, M. S. Lee, W. Z. Liang, I. Lotan, N. Nair, B. Peters, E. I. Proynov, P. A. Pieniazek, Y. M. Rhee, J. Ritchie, E. Rosta, C. D. Sherrill, A. C. Simmonett, J. E. Subotnik, H. L. Woodcock III, W. Zhang, A. T. Bell, A. K. Chakraborty, D. M. Chipman, F. J. Keil, A. Warshel, W. J. Hehre, H. F. Schaefer, J. Kong, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Phys. Chem. Chem. Phys., 2006, 8, 3172–3191 RSC.
- C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310–5324 CrossRef CAS.
- J. J. P. Stewart, J. Mol. Model., 2007, 13, 1173–1213 CrossRef CAS.
- L. R. Blackholly, H. J. Shepherd and J. R. Hiscock, CrystEngComm, 2016, 18, 7021–7028 RSC.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- R Core Team, R: A language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria, 2018, https://www.R-project.org/ Search PubMed.
- J. Hayles and P. Nurse, Introduction to Fission Yeast as a Model System, Cold Sping Harbor Protocols, 2018 Search PubMed.
- X. H. Gan, J. Yang, J. Li, H. Y. Yu, H. M. Dai, J. Y. Liu and Y. Huang, Biochem. J., 2011, 435, 103–111 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, mass spectrometry, NMR, crystallography‡ and computational modelling data. CCDC 1855728. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ob02523b |
| ‡ A suitable crystal of 1 with DMSO was selected and mounted on a Rigaku Oxford Diffraction Supernova diffractometer. Data were collected using Cu Kα radiation at 100 K. Structures were solved with ShelXT55 structure solution programmes via direct methods and refined with ShelXL56 on least squares minimisation. Olex257 was used as an interface to all ShelX programmes. |
| This journal is © The Royal Society of Chemistry 2021 |