
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, DNA-binding and antiproliferative properties of diarylquinolizinium derivatives†
Roberta
Bortolozzi
 a,
Heiko
Ihmels
*b,
Robin
Schulte
b,
Christopher
Stremmel
b and
Giampietro
Viola
*a
a,
Heiko
Ihmels
*b,
Robin
Schulte
b,
Christopher
Stremmel
b and
Giampietro
Viola
*a
aDepartment of Women's and Child's health, Oncohematology laboratory, University of Padova, Via Giustiniani 2, I-35128 Padova, Italy. E-mail: giampietro,viola.1@unipd.it
bDepartment of Chemistry and Biology, University of Siegen, Adolf-Reichwein-Str. 2, 57068 Siegen, Germany. E-mail: ihmels@chemie.uni-siegen.de
First published on 23rd December 2020
Abstract
A series of ten 2,7- and 2,8-diarylquinolizinium derivatives was synthesized and their DNA-binding and cytotoxic properties were investigated. Except for one nitro-substituted derivative all tested diarylquinolizinium ions bind to DNA with sufficient affinity (2 × 104 M−1–2 × 105 M−1). It was shown with photometric, fluorimetric and polarimetric titrations as well as with flow-LD analysis that the ligands bind mainly by intercalation to duplex DNA, however, depending on the ligand–DNA ratio, groove binding and backbone association were also observed with some derivatives. The biological activity was further investigated with tests of cytotoxicity and antiproliferative properties towards non-tumor cells and selected cancer cells, along with cell cycle analysis and an annexin-V assay. Notably, substrates that carry donor-functionalities in the 4-position of the phenyl substituents revealed a strong, and in some cases selective, antiproliferative activity as quantified by the growth inhibition, GI50, at very low micromolar and even submicromolar level both in leukemia and solid tumors.
Introduction
The association of external ligands with DNA is still a key element in anticancer therapy.1 Specifically, DNA-targeted chemotherapy makes use of the occupation of binding sites of DNA by the applied drug and the resulting structural changes of the nucleic acid,2 because both processes can interfere with the regular biological functions of DNA and eventually lead to growth inhibition and apoptosis of cancer cells.3 This principle of chemotherapeutic treatment of cancer with small molecules is an established approach in modern medicine, but it still has some serious drawbacks.4 Along with the general problem to provide sufficient biocompatibility and cell permeability of the exogenous ligands, their low selectivity towards cancer cells often causes adverse side effects in real applications, because the drugs can also bind to DNA in healthy cells. Therefore, an efficient and selective DNA-targeting drug has to keep a delicate balance between chemical, physical–chemical and biological properties that is very difficult to accomplish. As a result, very few DNA-binding drugs are actually available as admitted drugs that fulfil these requirements, despite the fact that a large number of structurally diverse DNA-binding compounds is already available.5 In view of this background, there is still a demand for DNA-binding ligands as novel lead structures for drug development.6In this context, DNA intercalators, i.e. aromatic ligands that bind to DNA by a coplanar association of the π system between two DNA base pairs, are attractive targets because their construction and development is based on reliable design principles.5 To accomplish this binding mode, an intercalator usually consists of an essentially planar (hetero)aromatic system, most often with fused arene units, to enable a large overlap of the π systems. In addition, a cationic charge commonly increases the affinity of an intercalator toward DNA because of thermodynamically favorable charge-transfer and electrostatic interactions, along with the energy gained from the resulting counter ion release from the DNA backbone.5 Representative and traditional examples of cationic DNA intercalators are the acridines,7 phenothiazines,8 phenanthridinium derivatives,9 and cyanine dyes,10 all of which are still employed as useful scaffolds in drug development. Along these lines, we and others have established the class of annelated quinolizinium ions as versatile DNA intercalators with a highly variable substitution and annelation pattern,11 and we have demonstrated in recent years with several examples that annelated quinolizinium derivatives bind efficiently and selectively to several types of nucleic acids.12 The special interest in this class of compounds is supported and motivated by their close structural resemblance to the berberine-type alkaloids, such as berberine, sanguinarine, sempervirine, or flavopereirine, which are well-established drug candidates whose bioactivity is also based on their DNA-binding properties.13 A survey of DNA-binding quinolizinium derivatives, however, showed that except for the parent compound and a few derivatives,14 the main focus has been on the annelated quinolizinium ions, so far, whereas the investigation of non-fused systems has been somewhat neglected. This observation is surprising because according to the minimal DNA-intercalator concept, a bicyclic fused ring system is not only sufficient to establish a DNA-intercalating unit, but often even increases its potential for application in chemotherapy.15 To fill this gap in the field of quinolizinium-based DNA binders, we focussed our attention to aryl-substituted quinolizinium derivatives. These biaryl-type systems still have an extended π system that is comparable to the benzo- or naphtho-annelated quinolizinium, but the former provide significantly more structural flexibility by torsional rotation about the biaryl axis. We proposed that this structural variability facilitates the access of the ligand to the intercalation site and also enables the ligand to fit well in the binding site by adapting the optimal conformation. To examine this proposal, we synthesized the diaryl-substituted quinolizinium derivatives 3a–j. And herein we demonstrate that these compounds represent a class of DNA intercalators with cytotoxic properties that depend significantly on the substitution pattern and which induce efficiently apoptosis in cancer cells.
Results
Synthesis
The 2,7-diarylquinolizinium derivatives 3a–3i were obtained in a Suzuki–Miyaura coupling of 2,7-dibromoquinolizinium bromide16 (1a) with the potassium aryltrifluoroborates172a–2i. The reactions were conducted in water with Pd(OAc)2 as catalyst and K2CO3 as base18 to give the corresponding products 3a–3i as bromide salts in 15–43% yield (Scheme 1). The compounds 3a–3i were identified and fully characterized with NMR spectroscopy (1H, 13C, COSY, HSQC, HMBC), mass spectrometry, and elemental analysis. In particular, the structure of the quinolizinium unit was identified by the characteristic, low field-shifted 1H NMR signals of the 4-H and 6-H protons at ca. 9.4 ppm and 9.7 ppm (cf. Experimental section). | ||
| Scheme 1 Synthesis of the 2,7-diarylquinolizinium bromides 3a–3i. | ||
The 2,8-bis(4-methoxyphenyl)quinolizinium bromide (3j) was synthesized in a multistep synthesis. In the first step, 4-bromo-2-methylpyridine (4) was deprotonated with LDA followed by an acylation with methyl-3-trimethylsilylpropiolate (5) to give the pyridylbutynone 6. The latter is in equilibrium with the corresponding enol form 6enol and could not be isolated. Therefore, the intermediate 6 was directly desilylated by treatment with TBAF, which in turn induced the cyclization to the 8-bromoquinolizin-2-one (7) in 26% yield overall yield (Scheme 2).19 Subsequent bromination in 2-position with PBr3 at 160 °C according to published procedure20 gave the 2,8-dibromoquinolizinium bromide (1b) only in a very low yield of 6%, which has also been observed with the parent hydroxyquinolizinium.20 The yield was only slightly improved to 9% by the addition of DMF as solvent and catalyst. Finally, the 2,8-bis(4-methoxyphenyl)quinolizinium bromide (3j) was obtained in 38% yield in a Suzuki–Miyaura coupling of 1b with the aryltrifluoroborate 2d (Scheme 2). The compounds 7, 1b and 3j were identified and fully characterized with NMR spectroscopy (1H, 13C, COSY, HSQC, HMBC), mass spectrometry, and elemental analysis. The structure and substitution pattern of the quinolizinium unit were identified by the characteristic shifts and multiplicities of the 1H NMR signals at 8.41 (dd, 3-H, 7-H), 8.81 (d, 9-H), and 9.25 (d, 4-H, 6-H) ppm.
 | ||
| Scheme 2 Synthesis of 2,8-bis(4-methoxyphenyl)quinolizinium bromide (3j). | ||
DNA-binding properties
The interactions of 3a–3j with calf thymus (ct) DNA were investigated by photometric and fluorimetric titrations (Fig. 1 and 2). In all cases, the addition of ct DNA led to an initial decrease of the ligand absorption followed by a red shift of the absorption maximum by 8–20 nm (Fig. 1, Table 1; cf. ESI Fig. S1†). The emission of the ligands 3a, 3b, 3c, and 3g is efficiently quenched upon the addition of ct DNA (Fig. 2; cf. ESI, Fig. S2†). Although the emission of 3d, 3f, and 3j also decreased at the beginning of the titration, i.e. at higher ligand–DNA ratios (LDR > 0.5), further addition of DNA eventually led to an increase of the emission intensity with a blue shift (3d, Δλfl = 17 nm) or red shift (3f, Δλfl = 50 nm) of the emission maximum (Fig. 2; Table 1; cf. ESI, Fig. S2†). As the only exception, the ligand 3e showed a relatively strong emission light-up effect from the start of the DNA titration with a light-up factor of 37 at saturation. Ligand 3h has only a very weak intrinsic emission, and upon addition of ct DNA it showed only an initial slight increase of the emission intensity that is eventually followed by a decrease (cf. ESI, Fig. S2†). | ||
| Fig. 1 Photometric titration of 3a (a), 3c (c) and 3d (d) (c = 20 μM) with ct DNA (c = 2.0 mM in base pairs) in BPE buffer (pH = 7.0; 5% v/v DMSO). The arrows indicate the development of the absorption bands during titration. Inset: Plot of absorption Abs. versus cDNA. | ||
 | ||
| Fig. 2 Fluorimetric titration of 3a (a), 3c (c), 3d (d), 3e (e) and 3f (f) (c = 20 μM) with ct DNA (c = 2.0 mM in base pairs) in BPE buffer (pH = 7.0; 5% v/v DMSO). The arrows indicate the development of the emission bands during titration. Inset: Plot of normalized emission intensity I/I0versus cDNA. | ||
| Ligand | K b/104a (M−1) | n | ΔλAbs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (nm) b (nm) |
Δλflb (nm) |
|---|---|---|---|---|
| a Binding constant, Kb, and neighbor exclusion parameter, n, determined from photometric or fluorimetric titrations, cDNA in base pairs. b Shift of absorption and emission maximum upon ligand–DNA complexation. c Could not be determined. | ||||
| 3a | 3.5 | 2.5 | +11 | <1 |
| 3b | 15 | 1.0 | +13 | <1 |
| 3c | 2.3 | 1.0 | +13 | −12 |
| 3d | 5.1 | 1.5 | +14 | −17 |
| 3e | 3.1 | 4.0 | +20 | −23 |
| 3f | 1.6 | 5.5 | +12 | +50 |
| 3g | 2.3 | 3.0 | +10 | <1 |
| 3h | —c | —c | +8 | <1 |
| 3i | 15 | 1.0 | +10 | <1 |
| 3j | 5.2 | 2.0 | +27 | <18 |
The data from the photometric titrations were used for the determination of the binding constants of 3a–3j with ct DNA. For that purpose, the experimental binding isotherms were fitted to the neighbor exclusion model of McGhee and von Hippel21 (cf. ESI, Fig. S3†). The resulting binding constants are in the range from 1.6 × 104 M−1 (3f) to 1.5 × 105 M−1 (3b). In the case of the nitro-substituted derivative 3h, a binding constant could not be obtained by this method.
To gain further information about the DNA-binding modes of 3a–j with ct DNA, circular dichroism (CD) and flow linear dichroism (flow-LD) spectra at different ligand–DNA ratios (LDR) were recorded (Fig. 3 and 4, cf. ESI, Fig. S4 and S5†). In general, the derivatives 3a–j may be divided in two groups with regard to the CD pattern observed on ligand–DNA complex formation (Fig. 3; cf. ESI, Fig. S4†). The first group consists of derivatives 3a, 3c, and 3g that showed only a very weak induced CD (ICD) signal (LDR 1.00) in the absorption range of the respective ligand in the presence of DNA, along with a slight increase of the intensity of the positive DNA band at 260 nm. Likewise, the ligand 3i also exhibited a weak positive ICD signal in the absorption range of the ligand at lower LDR, but at LDR = 1.0 a weak negative band developed in this region. In contrast, the group of ligands 3b, 3d, 3e, 3f, 3h, and 3j gave much stronger positive ICD bands between 380 nm and 420 nm upon addition of DNA, with 3b, 3d, 3e, and 3j showing significantly stronger ICD signals than 3f and 3h. In addition, the ligands 3d and 3e exhibited a positive ICD signal at 355 nm (3d) and 382 nm (3e) at LDR = 1, while 3j had an additional positive band at 340 nm.
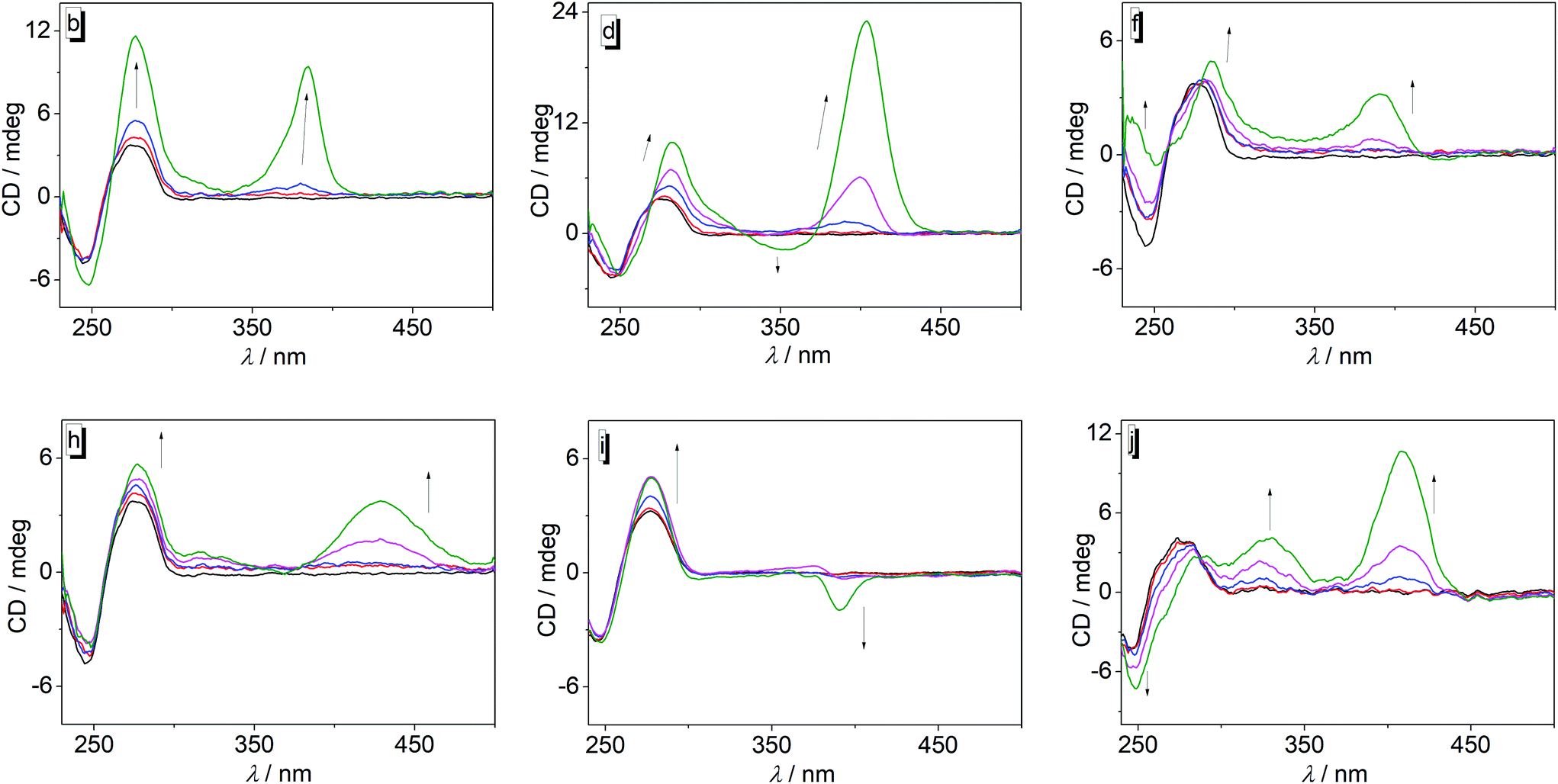 | ||
| Fig. 3 CD spectra of 3b,d,f,h,i, and j in the presence of ct DNA in BPE buffer (pH = 7.00, 5% DMSO) at LDR 0.00 (black), 0.05 (red), 0.20 (blue), 0.50 (magenta), and 1.00 (green); cDNA = 20 μM; T = 20 °C. The arrows indicate the development of the CD bands with increasing LDR. | ||
 | ||
| Fig. 4 LD spectra of 3b,d,e,h,i, and j in the presence of ct DNA in BPE buffer (pH = 7.00, 5% DMSO) at LDR 0.00 (black), 0.05 (red), 0.20 (blue), 0.50 (magenta) and 1.00 (green); cDNA = 20 μM; T = 20 °C. The arrows indicate the development of the LD bands with increasing LDR. | ||
The complementary flow-LD measurements revealed the development of clear negative LD signals for the ligands 3a–j in the absorption range of the corresponding ligands (Fig. 4, cf. ESI, Fig. S5†); however, in the case of 3d and 3i a weak positive LD signal was also formed at ca. 400 nm and 380 nm at an LDR of 1.00, respectively. Notably, compound 3h did not show a significant LD signal in the absorption range of the ligand when bound to DNA (Fig. 4h). The intensity of the DNA band at 260 nm increased slightly during the addition of the ligand 3a, whereas it decreased or fluctuated in the presence of 3b and 3f, or 3c, 3e, 3g, 3i, and 3j, respectively (Fig. 4; cf. ESI Fig. S5†).
Biological activity in cancer cells
With the purpose to analyze the antiproliferative effect of the substrates 3a–j on cells, a representative panel of four human cell lines derived from different cancer types, including cervix carcinoma (HeLa), breast cancer (MDA-MB-468), and B-cell leukemia (SEM and RCH-ACV) was treated with these compounds. The viability of the incubated cells was determined, and the effect of the substrate was quantified as growth inhibition, GI50 (Table 2). As reference compound we included doxorubicin, a well-known DNA intercalator, in the series. As expected, incubation with doxorubicin resulted in GI50 values in the range of 0.004–0.042 μM for MDA-MB468 and leukemia cells and in the micromolar level (1.1 μM) for HeLa cells. Within the tested series of quinolizinium derivatives, the parent compound 3a exhibited a modest activity against the four employed cell lines (GI50 = 23–36 μM). As a general trend, the introduction of an electron donor functionality in the 4-position of the phenyl substituents significantly increased the cytotoxic activity with a GI50 at the low micromolar (HeLa and MDA-MB-468) and even submicromolar level (SEM and RCH-SCV) for the leukemic cell lines (3b, 3d and 3e). Interestingly, the attachment of two additional donor groups, namely methoxy functionalities, in the 3- and 5-position in compound 3f, led to a loss of the antiproliferative activity. At the same time, the introduction of an electron-withdrawing group at the phenyl substituents in derivatives 3g, 3h and 3i essentially abolished the cytotoxicity. A bulky hydrophobic substituent such as naphthalene (3c), on the other hand, maintained good activity. The geometry of the quinolizinium ring substituents also seems to have little influence on the activity, as the 2,8-diarylquinolizinium derivative 3j exhibited the same activity as the 2,7-diarylquinolizinium 3d.| HeLa | MDA-MB468 | SEM | RCH-ACV | PBL | PBL (+PHA) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| GI50/μMa | SIb | GI50/μMa | SIb | GI50/μMa | SIb | GI50/μMa | SIb | GI50/μMa | GI50/μMa | |
| a GI50 = concentration required to inhibit tumor cell proliferation by 50%. Values are the mean ± SE from the dose–response curves of at least three independent experiments carried out in triplicate. b SI = ratio between GI50 calculated on PHA-stimulated PBL and GI50 of cancer cell lines. c Doxo = Doxorubicin. | ||||||||||
| 3a | 36.1 ± 4.8 | 25.6 ± 4.6 | 29.0 ± 0.9 | 23.0 ± 1.4 | ||||||
| 3b | 1.5 ± 0.5 | 0.48 | 1.7 ± 0.3 | 0.42 | 0.013 ± 0.005 | 55.4 | 0.21 ± 0.05 | 3.44 | 3.1 ± 0.2 | 0.7 ± 0.5 |
| 3c | 2.5 ± 0.9 | 0.58 | 2.4 ± 0.7 | 0.61 | 0.28 ± 0.09 | 5.21 | 0.82 ± 0.1 | 1.78 | 4.2 ± 1.2 | 1.5 ± 0.1 |
| 3d | 2.9 ± 0.8 | 0.18 | 1.4 ± 0.2 | 0.37 | 0.31 ± 0.1 | 1.68 | 0.27 ± 0.08 | 1.92 | 2.6 ± 0.1 | 0.5 ± 0.2 |
| 3e | 2.7 ± 0.2 | 0.39 | 2.2 ± 0.3 | 0.47 | 0.53 ± 0.06 | 1.96 | 0.62 ± 0.08 | 1.68 | 8.7 ± 2.1 | 1.04 ± 0.06 |
| 3f | >100 | >100 | >100 | >100 | ||||||
| 3g | >50 | >50 | >50 | >50 | ||||||
| 3h | >100 | 43.5 ± 1.3 | >100 | >100 | ||||||
| 3i | 66.0 ± 8.3 | 72.5 ± 5.7 | >100 | >100 | ||||||
| 3j | 1.6 ± 0.2 | 0.75 | 0.54 ± 0.12 | 2.22 | 0.17 ± 0.05 | 7.05 | 0.46 ± 0.06 | 2.61 | 3.7 ± 0.8 | 1.2 ± 0.1 |
| Doxo | 1.1± 0.06 | 0.32 | 0.042 ± 0.009 | 8.45 | 0.043 ± 0.002 | 8.26 | 0.0037 ± 0.0002 | 95.95 | 2.3 ± 0.3 | 0.36 ± 0.02 |
The most active compounds 3b, 3c, 3d, 3e, 3j, and doxorubicin also showed toxicity in healthy human cells, as tested either in quiescent and in active proliferation phase (PHA-stimulated) peripheral blood lymphocytes PBL, with GI50 values in the range of 0.4–8.7 μM (Table 2). In this context, the selectivity towards cancer cells was evaluated by the selectivity index (SI) (Table 2). Hence, the derivatives 3b, 3c, 3d, 3e, and 3j have a low selectivity index concerning HeLa and MDA-MB468 cancer cells (SI = 0.2–2.2), while they displayed a good selectivity toward leukemia cell lines with values as large as SI = 55 for derivative 3b.
The effect of compounds with the most pronounced antiproliferative activity (3b, 3d, 3e, and 3j) on cell cycle progression was examined by flow cytometry in HeLa cells (Fig. 5). After 24 h of treatment, derivatives 3b, 3d and 3e did not induce significant changes in the cell cycle (Fig. 5, panel a) while compound 3j (Fig. 5, panel b) induced an accumulation of cells in G2/M from 13.1% in the untreated cell to 26.6% at the highest employed concentration accompanied by a progressive decrease of the G1 phase (from 60% in the untreated cells to 44.6% at 10 μM).
 | ||
| Fig. 5 Flow cytometric cell cycle analysis of HeLa cells treated with 3b (panel a), 3d (panel b), 3e (panel c) and 3j (panel d) for 24 h at c = 0 μM, 5 μM, and 10 μM; : G1, : G2/M, : S. Data are represented as mean of two independent experiments ± SEM. | ||
The mode of cell death induced by the most active compounds 3b, 3d, 3e and 3j in HeLa cells was examined with the annexin-V/PI assay.22 Dual staining with dye-labeled annexin-V and propidium iodide (PI) permits discrimination between live cells (annexin-V−/PI−), early apoptotic cells (annexin-V+/PI−), late apoptotic cells (annexin-V+/PI+) and necrotic cells (annexin-V−/PI+) (+: positive indicator signal; −: no indicator signal). The cells that were treated with the test compounds showed a significant accumulation of annexin-V positive cells after a 24 h treatment at 5.0 μM, and apoptosis was even more evident at higher concentration (10 μM) of both compounds (Fig. 6). Derivatives 3b, 3d and 3j all appear to have the same cytotoxic potency to induce apoptosis, which is in good agreement with their antiproliferative activity (Table 2). The percentage of apoptotic cells increased further after 48 h, at which time a marked increase in necrotic cells was also observed, namely 10–25% (3b, 3d and 3j) or even 40–60% (3e) vs. approx. 5% in the untreated cells, clearly indicating that these compounds induced substantial cell death by both apoptosis and necrosis at longer incubation times.
 | ||
| Fig. 6 Flow cytometric analysis of apoptotic cells after treatment of HeLa cells with 3b (a), 3d (b), 3e (c) and 3j (d) after incubation for 24 h or 48 h at c = 5 and 10 μM. The cells were harvested and labeled with annexin-V-FITC (FITC = fluorescein isothiocyante) and propidium iodide (PI) and analyzed by flow cytometry. Data are represented as mean ± SEM of three independent experiments (+: positive indicator signal; −: no indicator signal). | ||
Discussion
The data from the spectrometric titrations revealed the association of the ligands 3a–3j with DNA, as unambiguously indicated by the typical changes of the absorption and emission properties as well as by the formation of ICD bands in the absorption range of the ligands during titration.23 Except for the derivatives 3b and 3i the ligands have a moderate affinity to the DNA as shown by binding constants in the range of 2–5 × 104 M−1 which are comparable to the ones reported for substituted or annelated quinolizinium derivatives.11 In the case of ligands 3b and 3i, the somewhat larger binding constant of 2 × 105 M−1 may be attributed to the “methyl effect”, i.e. increased host–guest affinity based on attractive dispersion interactions of alkyl groups (here methyl and ethyl),24 that has been observed frequently for DNA ligands.14,25 However, it should be noted that the inconsistent developments of absorption and emission bands during each titration with varying LDR indicate a heterogeneous binding mode of the ligands to the DNA, which is further supported by the lack of isosbestic points during the photometric titrations. We propose that at very high LDR, i.e. at the beginning of the photometric and fluorimetric titrations, the excess of ligands relative to the DNA binding sites leads to the aggregation of the ligands along the DNA backbone, as often observed for cationic dyes.26 With decreasing LDR, however, sufficient DNA binding sites become available so that the ligands can also bind by groove binding and intercalation. The LD spectra of most ligand–DNA complexes revealed an intercalative binding mode at lower LDR as manifested by the negative LD bands in the absorption range of the ligands 3a, 3b, 3c, 3f, 3g, and 3j.27 All of these ligands also gave a positive ICD signal when bound to DNA, which is assigned to a binding mode with the transition dipole moment perpendicular to the one of the DNA bases.27 Thus, a binding mode is suggested in which the ligand is aligned with its long molecular axis essentially perpendicular to the long axis of the intercalation site, which is often observed for the resembling annelated quinolizinium derivatives.11 Because the LD spectra did not reveal any sign of groove binding, namely a clear positive band was not detected at any of the tested LDR, it is assumed that at larger LDR these ligands form aggregates along the DNA backbone instead.In the case of derivatives 3d, 3e, and 3i, the binding situation appears to be slightly different than with the ligands discussed above. Specifically, at LDR = 1.0 the LD spectra of complexes of DNA with 3d and 3i, and to very small extent the one with 3e, developed weak red-shifted positive signals along with the major negative bands, which suggests additional groove binding. This switch of binding modes with LDR was also observed, even more clearly, in the CD spectra of these derivatives, where the maxima and phases of the ICD bands change also significantly when going from LDR = 0.05 to LDR = 1.0, which indicates at least two different orientations of the ligands relative to the DNA base pairs.27 Moreover, the CD bands of 3d and 3e are relatively strong as often observed for groove binders. Hence, the combination of CD and LD spectra of the ligands 3d, 3e, and 3i also revealed a heterogeneous binding that depends on the LDR; however, in this case, these ligands have the tendency to groove binding if sufficient space for binding is available, i.e. at low LDR. It should be emphasized, however, that the relative intensities of the CD and LD bands do not reflect the degree of the different contributions of the binding modes, because intercalated ligands give relatively strong LD bands, but weak CD bands, whereas in the case of groove binders weak LD bands and very strong CD bands are observed.27
With the discussion of the heterogeneous binding situation of the diarylquinolizinium derivatives in mind, it should be emphasized that the binding constants (Table 1) represent just an average global binding constant with different and varying contribution of the distinct binding modes, which is also explained by the broad range of the neighbor exclusion parameters n = 1–5.5 that indicate the binding site size.
A special case is provided by the nitro-substituted derivative 3h. Although the photometric titrations clearly point to the association of this compound with DNA, the LD spectra did not show significant bands in the absorption region of the ligand, except for very weak fluctuations. These observations suggest that this compound only binds to DNA by backbone association with no significant contribution by intercalation or groove binding.
Overall, the series of ligands 3a–j does not provide a reasonable trend that allows to deduce any relationship between the substitution pattern and DNA-binding properties. Obviously, all ligands bind to DNA by a combination of the different binding modes whose contribution to the overall binding depends strongly on the ligand–DNA ratio. The actual preference for a particular binding mode apparently depends on a delicate balance between the stereoelectronic and steric influence of the particular aryl substituent.
Except for compound 3e, the emission of all tested diarylquinolizinium derivatives is significantly quenched upon addition of DNA, which shows that the initially formed aggregates along the DNA backbone (see above) are not fluorescent. In the case of ligands without functional groups, 3a, 3b, and 3c, and those with acceptor substituents, 3g and 3i, the emission further decreased steadily in the course of the titration, suggesting that the fluorescence of the intercalated and/or groove-bound ligand is also quenched, presumably by a photoinduced electron transfer (PET) reaction between the DNA bases and the DNA-bound quinolizinium, as shown for other quinolizinium derivatives.28 In contrast, the emission intensity of the methoxy-substituted ligands 3d, 3f, and 3j increased again with continuing titration. Obviously, the emission of these compounds is no longer strongly quenched when they are intercalated, assumedly because the donor substituents reduce the reduction potential of the quinolizinium core such that a PET reaction with the DNA bases is no longer energetically favorable. Moreover, the restricted environment in the respective DNA binding site most likely reduces the conformational flexibility of the ligand, which may also contribute to the enhancement of emission intensity by suppression of the radiationless deactivation of the excited state by torsional relaxation processes. Most notably, the shift of the emission maxima of the DNA-bound ligands 3d and 3f changes significantly as compared to the unbound species. This effect may also be caused by the accommodation in the sterically constrained binding site when the ligand is forced to adapt a different torsion angle between the biaryl units than in solution, which in turn leads to a different donor–acceptor interplay between the methoxy-substituted phenyl rings and the quinolizinium unit and thus to a change of emission energy. With respect to the emission properties, the derivative 3e figures as a special case because upon binding to DNA the emission intensity increases steadily with a light up factor of I/I0 ≈ 37 at saturation. Obviously, the emission of this compound is not quenched at all when bound to the nucleic acid, independent of the binding mode. Instead, it appears as if at least one pathway for a radiationless deactivation of the excited state is significantly suppressed in the ligand–DNA complex, most likely the torsional relaxation (see above).
Considering the pronounced DNA-binding properties of diarylquinolizinum derivatives 3a–j to bind to DNA, we evaluated their antiproliferative activity and their ability to induce apoptosis in human tumor cell lines. Notably, most of the tested derivatives are endowed with a good antiproliferative activity both in leukemia and solid tumors. A direct comparison with the antiproliferative activity of doxorubicin, a well-established cytotoxic DNA intercalator, showed that the cytotoxic activity of some diarylquinolizinium derivatives towards HeLa, and SEM cell lines is essentially in the same range. Nevertheless, most of these quinolizinium derivatives have also a high activity towards healthy cells and – other than doxorubicin – a low selectivity toward solid tumors (HeLa and MDA-MB468). As remarkable exceptions, the derivatives 3b, 3c, and 3j stand out in this series as they have a resembling (3c, 3j) or significantly higher (3b) activity and selectivity towards leukemic SEM cell lines as doxorubicin, which signifies the general potential of this class of compounds as selective cytotoxic antitumor agents.
It should be noted that the antiproliferative activity of the tested diarylquinolizinium derivatives does not correlate well with ability to bind to DNA. These ligands bind to DNA through intercalation or backbone association, but this property does not seem to have the main impact on their biological activity. Indeed, the close structure–activity relationships observed among the different derivatives, namely highest activity of 4-donor-substituted substrates, pointed out that the cytotoxicity is based on more factors than just the interaction with DNA, e.g. cell permeability, accumulation in different cell components, or mode of action that involve different targets along with DNA. In this context, it has been shown that the antiproliferative activity of many DNA-binding agents is directly linked to the inhibition of DNA processing enzymes such as Topoisomerases. Along these lines, future biological studies shall be performed to clarify the mechanism of action.
Conclusions
In summary, we have demonstrated that diaryl-substituted quinolizinium derivatives constitute a promising class of readily available DNA-binding and cytotoxic compounds with a relatively strong antiproliferative effect on selected tumor cells, and even with a significant selectivity in some cases. Within the series of tested derivatives, the donor-substituted substrates show the most pronounced effect, thus pointing to an underlying structure–activity relationship. Based on these fundamental results we conclude that these properties warrant further studies of this class of compounds as novel lead structure for antitumor agents with a focus on the detailed mechanistic investigation of the biological activity and on the assessment of quantitative structure–activity relationships (QSAR) to identify even more potent derivatives with efficient and selective antitumor activity. Moreover, it should be assessed in detail whether these compounds operate as topoisomerase inhibitors or topoisomerase poisons.Experimental section
Materials
Commercially available reagents were used without further purification. Potassium aryltrifluoroborates 2a–2i,17 2,7-dibromoquinolizinium bromide (1a),16 and methyl 3-trimethylsilylpropiolate19 (5) were synthesized according to the published procedures. BPE-buffer [6.0 mM Na2HPO4, 2.0 mM NaH2PO4, 1.0 mM Na2EDTA; pH = 7.0, c(Na+) = 16 mM] was prepared from biochemical grade chemicals in E-Pure water (resistivity ≥ 18 MΩ m) and filtered through a PVDF membrane filter (pore size 0.45 μM). Doxorubicin hydrochloride was purchased by Sigma-Aldrich (Milano, Italy).Equipment
NMR spectra were recorded with a Bruker AV 400 spectrometer [400 MHz (1H); 100 MHz (13C)] at room temperature, or with a Varian VNMR-S600 spectrometer [600 MHz (1H); 150 MHz (13C)] at 35 °C. The NMR spectra were processed with the software ACD/NMR Processor Academic Version 12.01 and referenced to the corresponding solvent peaks [δ(DMSO-d5) = 2.50 (1H) and 39.5 (13C); δ(CHCl3) = 7.26 (1H) and 77.2 (13C)]. Elemental analysis data were determined with a HEKAtech EURO EA combustion analyzer by Mr Rochus Breuer (Universität Siegen, Organic Chemistry I, University of Siegen). Mass spectra (ESI) were recorded on a Finnigan LCQ Deca (U = 6 kV; working gas: argon; auxiliary gas: N2; temperature of the capillary: 200 °C). Absorption spectra were obtained with a Varian Cary 100 bio spectrometer in quartz cells (10 mm) with baseline correction. Emission spectra were recorded in quartz cells (10 mm) with a Cary Eclipse spectrometer at 20 °C. The CD-and LD-spectroscopic measurements were performed on a Chirascan spectrometer (Applied Photophysics). To record the LD Spectra with a shear gradient of 1200 s−1 in a rotating couette, the CD spectrometer was equipped with a High Shear Couette Cell Accessory (Applied Photophysics). The melting points were measured with a Büchi 545 (Büchi, Flawil, CH) and are uncorrected.Methods
The excitation and emission slits were adjusted to 5 nm for the detection of the emission spectra with a detection speed of 120 min−1 at 20 °C and detection voltages between 400 V and 500 V depending on the fluorescence emission intensity. All spectra were smoothed with the implemented “moving-average” function by a factor 5. The relative fluorescence quantum yields Φfl were determined relative to coumarin 120 (Φfl, S = 0.56 in EtOH;293a,b, 3g and 3h), coumarin 1 (Φfl, S = 0.73 in EtOH;303d) and coumarin 102 (Φfl, S = 0.95 in EtOH30) (for 3c and 3e–f).31
For the CD and flow-LD spectra, five solutions of ct DNA and the ligands (LDR 0.00, 0.05, 0.20, 0.50, and 1.00) in BPE buffer/DMSO (cf. ESI Tables 2 and 3†) were recorded after an equilibration time of 30 min.
After extensive washing, cells were resuspended (1.0 × 106 cells per mL) in RPMI-1640 with 10% FBS and incubated overnight. For cytotoxicity evaluations in proliferating PBL cultures, non-adherent cells were resuspended at 5 × 105 cells per mL in growth medium, containing 2.5 μg mL−1 PHA (Irvine Scientific). Different concentrations of the test compounds were added, and viability was determined 72 h later by the MTT test.33 For cytotoxicity evaluations in resting PBL cultures, non-adherent cells were resuspended (5 × 105 cells per mL) and treated for 72 h with the new derivatives, as described above.
Synthesis
:0 to 90:10; Rf = 0.4 (90:10)] to give the ketone 7 as brown solid (17.8 mmol, 4.00 g, 26%). An analytically pure sample was obtained upon protonation with HBr and crystallization from MeOH to give 8-bromo-2-hydroxyquinolizinium bromide 7·HBr as brown powder that was directly submitted to the next synthetic step without further purification; mp 265–268 °C. – 1H NMR (400 MHz, DMSO-d6): δ = 7.52 (d, 4J = 4 Hz, 1 H, 1-H), 7.58 (dd, 3J = 7 Hz, 4J = 3 Hz, 1 H, 3-H), 7.87 (dd, 3J = 7 Hz, 4J = 2 Hz, 1 H, 7-H), 8.61 (d, 4J = 2 Hz, 1 H, 9-H), 8.88 (d, 3J = 7 Hz, 1 H, 6-H), 9.07 (d, 3J = 7 Hz, 1 H, 4-H). – 13C NMR (100 MHz, DMSO-d6): δ = 107.5 (C1), 116.7 (C3), 122.8 (C7), 127.0 (C9), 129.7 (C8), 136.4 (C6), 139.7 (C4), 145.4 (C9a), 165.4 (C2). – MS (ESI+): m/z = 226 (100) [M − Br]+.
:20; Rf = 0.2]. The combined fractions were filtered through a pad of Celite and concentrated by removal of the solvent under reduced pressure. After recrystallization from MeOH/EtOAc the product 1b was obtained as brown needles (170 mg, 463 μmol, 9%); mp >300 °C. – 1H NMR (400 MHz, DMSO-d6): δ = 8.40 (dd, 3J = 7 Hz, 4J = 2 Hz, 2 H, 3-H, 7-H), 8.85 (d, 4J = 2 Hz, 2 H, 1-H, 9-H), 9.27 (d, 3J = 7 Hz, 2 H, 4-H, H-6). – 13C NMR (100 MHz, DMSO-d6): δ = 127.1 (C3, C7), 128.2 (C1, C9), 132.8 (C2, C8), 137.7 (C4, C6), 143.0 (C9a). – MS (ESI+): m/z = 288 (100) [M − Br]+. – El. Anal. for C9H6Br3N (367.87), calcd (%): C 29.39, H 1.64, N 3.81, found (%): C 29.43, H 1.54, N 3.58.
General procedure for the Suzuki–Miyaura coupling of dibromoquinolizinium derivatives with potassium aryltrifluoroborates 3a–3j (GP 1)15
Under an argon gas atmosphere, the corresponding aryltrifluoroborate (480 μmol) was added to an oxygen-free suspension of dibromoquinolizinium bromide 1 or 7 (73.6 mg, 200 μmol), K2CO3 (82.8 mg, 600 μmol) and Pd(OAc)2 (2.4 mg, 8.0 μmol) in water (5.0 mL), and the solution was stirred for 4.5–7.0 h at 70 °C. The reaction mixture was cooled to 0 °C, the precipitated solid was filtered off and washed with THF (5.0 mL). The residue was suspended in methanol (∼100 mL) and the remaining solid (Pd black) was filtered off and washed with methanol (3 × 10 mL). After combination of the MeOH fractions and removal of the solvent the desired product was obtained by crystallization from MeOH/EtOAc.:1); Rf = 0.3] to give product 3a as yellow needles (27.0 mg, 74.5 μmol, 37%). An analytical pure sample was obtained by crystallization from MeOH/EtOAc (5.1 mg, 14 μmol, 14%); mp 254–257 °C (decomp.). – 1H NMR (600 MHz, DMSO-d6): δ = 7.61 (dd, 3J = 7 Hz, 3J = 7 Hz, 1 H, 4′′-H), 7.64–7.70 (m, 5 H, 3′-H, 4′-H, 5′-H, 3′′-H, 5′′-H), 7.98 (d, 3J = 7 Hz, 2 H, 2′′-H, 6′′-H), 8.13 (d, 3J = 7 Hz, 2 H, 2′-H, 6′-H), 8.60 (d, 3J = 8 Hz, 1 H, 3-H), 8.62 (d, 3J = 9 Hz, 1 H, 9-H), 8.77 (d, 3J = 9 Hz, 1 H, 8-H), 9.04 (s, 1 H, 1-H), 9.40 (d, 3J = 7 Hz, 1 H, 4-H), 9.78 (s, 1 H, 6-H). – 13C NMR (150 MHz, DMSO-d6): δ = 121.9 (C3), 122.4 (C1), 127.2 (C9), 127.3 (C2′′, C6′′), 127.6 (C2′, C6′), 129.6 (C3′′, C5′′), 129.7 (C3′, C5′), 130.1 (C4′′), 131.2 (C4′), 133.5 (C6, C1′′), 134.2 (C1′), 134.7 (C7), 135.5 (C8), 137.0 (C4), 141.6 (C9a), 146.3 (C2). – MS (ESI+): m/z = 282 (100) [M − Br]+. – El. Anal for C21H16BrN·1.5H2O (389.30), calcd (%): C 64.79, H 4.92, N 3.60, found (%): C 64.77, H 4.45, N 3.61.
:1); Rf = 0.2] to give product 3i as pale brown needles (25.0 mg, 49.5 μmol, 25%). An analytical pure sample was obtained by twofold crystallization from MeOH/EtOAc (8.0 mg, 16 μmol, 8%); mp >285 °C (decomp.). – 1H NMR (600 MHz, DMSO-d6): δ = 1.37 (t, 3J = 7 Hz, 6 H, 4′-OCH2![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3, 4′′-OCH23), 4.37 (q, 3J = 7 Hz, 4 H, 4′-O2CH3, 4′′-O2CH3), 8.13 (d, 3J = 8 Hz, 2 H, 2′′-H, 6′′-H), 8.20 (d, 3J = 8 Hz, 4 H, 3′-H, 3′′-H, 5′-H, 5′′-H), 8.26 (d, 3J = 8 Hz, 2 H, 2′-H, 6′-H), 8.66–8.68 (m, 2 H, 9-H, 3-H), 8.85 (d, 3J = 9 Hz, 1 H, 8-H), 9.15 (s,1 H, 1-H), 9.48 (d, 3J = 7 Hz, 1 H, 4-H), 9.96 (s, 1 H, 6-H). – 13C NMR (150 MHz, DMSO-d6): δ = 16.2 (4′-OCH23, 4′′-OCH23), 61.2 (4′-O2CH3, 4′′-O2CH3), 122.1 (C3), 123.5 (C1), 127.6 (C9), 127.8 (C2′′, C6′′), 128.1 (C2′, C6′), 130.2 (C3′, C5′, C3′′, C5′′), 131.0 (C4′′), 131.9 (C4′), 133.8 (C7), 134.6 (C6), 135.7 (C8), 137.3 (C4), 137.3 (C1′′), 138.4 (C1′), 141.8 (C9a), 145.2 (C2), 165.1 (4′′-C
3, 4′′-OCH23), 4.37 (q, 3J = 7 Hz, 4 H, 4′-O2CH3, 4′′-O2CH3), 8.13 (d, 3J = 8 Hz, 2 H, 2′′-H, 6′′-H), 8.20 (d, 3J = 8 Hz, 4 H, 3′-H, 3′′-H, 5′-H, 5′′-H), 8.26 (d, 3J = 8 Hz, 2 H, 2′-H, 6′-H), 8.66–8.68 (m, 2 H, 9-H, 3-H), 8.85 (d, 3J = 9 Hz, 1 H, 8-H), 9.15 (s,1 H, 1-H), 9.48 (d, 3J = 7 Hz, 1 H, 4-H), 9.96 (s, 1 H, 6-H). – 13C NMR (150 MHz, DMSO-d6): δ = 16.2 (4′-OCH23, 4′′-OCH23), 61.2 (4′-O2CH3, 4′′-O2CH3), 122.1 (C3), 123.5 (C1), 127.6 (C9), 127.8 (C2′′, C6′′), 128.1 (C2′, C6′), 130.2 (C3′, C5′, C3′′, C5′′), 131.0 (C4′′), 131.9 (C4′), 133.8 (C7), 134.6 (C6), 135.7 (C8), 137.3 (C4), 137.3 (C1′′), 138.4 (C1′), 141.8 (C9a), 145.2 (C2), 165.1 (4′′-C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 165.2 (4′-CO). – MS (ESI+): m/z = 426 [M − Br]+. – El. Anal. for C27H24BrNO4·H2O (524.41), calcd (%): C 61.84, H 5.00, N 2.67; found (%): C 61.53, H 4.84, N 3.07.
O), 165.2 (4′-CO). – MS (ESI+): m/z = 426 [M − Br]+. – El. Anal. for C27H24BrNO4·H2O (524.41), calcd (%): C 61.84, H 5.00, N 2.67; found (%): C 61.53, H 4.84, N 3.07.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Generous support by the Deutsche Forschungsgemeinschaft is gratefully acknowledged. We thank Ms Jennifer Hermann and Ms. Sandra Uebach for technical assistance.Notes and references
- (a) A. Ali and S. Bhattacharya, Bioorg. Med. Chem., 2014, 22, 4506 CrossRef CAS; (b) S. M. V. de Almeida, A. G. Ribeiro, G. C. d. L. Silva, J. E. F. Alves, E. I. C. Beltrão, J. F. de Oliveira, L. B. de Carvalho and M. D. A. de Lima, Biomed. Pharmacother., 2017, 96, 1538 CrossRef CAS; (c) Y. H. Du, J. Huang, X. C. Weng and X. Zhou, Curr. Med. Chem.: Anti-Cancer Agents, 2010, 17, 173 CrossRef CAS; (d) S. Neidle and D. E. Thurston, Nat. Rev. Cancer, 2005, 5, 285 CrossRef CAS.
- G. S. Goldberg and R. Airley, Cancer Chemotherapy: Basic Science to the Clinic, Wiley, Chichester, 2nd edn, 2020 Search PubMed.
- (a) L. Pett, J. A. Hartley and K. Kiakos, Curr. Top. Med. Chem., 2015, 15, 1293 CrossRef CAS; (b) M. Frezza, S. Hindo, D. Chen, A. Davenport, S. Schmitt, D. Tomco and Q. P. Dou, Curr. Pharm. Des., 2010, 16, 1813 CrossRef CAS.
- R. Martinez and L. Chacon-Garcia, Curr. Med. Chem., 2005, 12, 127 CrossRef CAS.
- Selected reviews: (a) A. Rahman, P. O'Sullivan and I. Rozas, Med. Chem. Commun., 2019, 10, 26 RSC; (b) S. Bhaduri, N. Ranjan and D. P. Arya, Beilstein J. Org. Chem., 2018, 14, 1051 CrossRef CAS; (c) S. Boga, D. Bouzada, D. G. Pena, M. V. Lopez and M. E. Vasquez, Eur. J. Org. Chem., 2018, 249 CrossRef CAS; (d) M. Wang, Y. Yu, C. Liang, A. Lu and G. Zhang, Int. J. Mol. Sci., 2016, 17, 779 CrossRef; (e) A. Rescifina, C. Zagni, M. G. Varrica, V. Pistara and A. Corsaro, Eur. J. Med. Chem., 2014, 74, 95 CrossRef CAS; (f) S. Banerjee, E. B. Veale, C. M. Phelan, S. A. Murphy, G. M. Tocci, L. J. Gillespie, D. O. Frimannsson, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2013, 42, 1601 RSC; (g) X. Wang and Z. Guo, Chem. Soc. Rev., 2013, 42, 202 RSC; (h) A. Mukherjee and W. D. Sasikala, Adv. Protein Chem. Struct. Biol., 2013, 92, 1 CAS; (i) A. C. Komor and J. K. Barton, Chem. Commun., 2013, 49, 3617 RSC.
- K. Gurova, Future Oncol., 2009, 5, 1685 CrossRef CAS.
- P. Prasher and M. Sharma, Med. Chem. Commun., 2018, 9, 1589 RSC.
- R. W. Sabnis, Handbook of Biological Dyes and Stains, Wiley, Chichester, 2010 Search PubMed.
- (a) L.-M. Tumir, M. R. Stojković and I. Piantanida, Beilstein J. Org. Chem., 2014, 10, 2930 CrossRef; (b) N. C. Garbett, N. B. Hammond and D. E. Graves, Biophys. J., 2004, 87, 3974 CrossRef CAS.
- (a) M. Levitus and S. Ranjit, Q. Rev. Biophys., 2011, 44, 123 CrossRef CAS; (b) B. Armitage, Top. Curr. Chem., 2005, 253, 55 CrossRef CAS.
- (a) P. Bosch, D. Sucunza, F. Mendicuti, A. Domingo and J. J. Vaquero, Org. Chem. Front., 2018, 5, 1916 RSC; (b) D. Sucunza, A. M. Cuadro, J. Alvarez-Builla and J. J. Vaquero, J. Org. Chem., 2016, 81, 10126 CrossRef CAS; (c) H. Ihmels, K. Faulhaber, D. Vedaldi, F. Dall'Acqua and G. Viola, Photochem. Photobiol., 2005, 81, 1107 CrossRef CAS.
- A. Granzhan and H. Ihmels, Synlett, 2016, 27, 1775 CrossRef CAS.
- (a) N. Singh and B. Sharma, Front. Mol. Biosci., 2018, 5, 21 CrossRef; (b) C. W. Xiao, Q. A. Ji, Q. Wei, Y. Liu and G. L. Bao, BMC Complementary Altern. Med., 2015, 15, 177 CrossRef; (c) A. Croaker, G. J. King, J. H. Pyne, S. Anoopkumar-Dukie, V. Simanek and L. Liu, Mutat. Res., Rev. Genet. Toxicol., 2017, 774, 46 CrossRef CAS.
- (a) S. Kölsch, H. Ihmels, J. Mattay, N. Sewald and B. O. Patrick, Beilstein J. Org. Chem., 2020, 16, 111 CrossRef; (b) X. Xie, M. Zuffo, M.-P. Teulade-Fichou and A. Granzhan, Beilstein J. Org. Chem., 2019, 15, 1872 CrossRef CAS; (c) H. Ihmels, M. Karbasiyoun, K. Löhl and C. Stremmel, Org. Biomol. Chem., 2019, 17, 6404 RSC; (d) A. K. Das, H. Ihmels and S. Kölsch, Photochem. Photobiol. Sci., 2019, 18, 1373 RSC; (e) H. Yao, L. Chang, C. Liu, X. Jiao, S. He, H. Liu, E. Zacharioudakis, T. Cañeque, R. Custodio, S. Müller, A. M. Cuadro, J. J. Vaquero and R. Rodriguez, Bioorg. Med. Chem. Lett., 2017, 27, 203 CrossRef; (f) X. Zeng, J. Fluoresc., 2015, 25, 1637 CrossRef; (g) K. Benner, H. Ihmels, S. Kölsch and P. M. Pithan, Org. Biomol. Chem., 2014, 12, 1725 RSC; (h) L. Chang, C. Liu, S. He, Y. Lu, S. Zhang, L. Zhao and X. Zeng, Sens. Actuators, B, 2014, 202, 483 CrossRef CAS; (i) E. Maçoas, G. Marcelo, S. Pinto, T. Cañeque, A. M. Cuadro, J. J. Vaquero and J. M. G. Martinho, Chem. Commun., 2011, 47, 7374 RSC.
- W. A. Denny, in Small Molecule DNA and RNA Binders, ed. M. Demeunynck, C. Bailly and W. D. Wilson, Wiley–VCH, Weinheim, 2004, p. 482 Search PubMed.
- G. M. Sanders, M. van Dijk and H. van der C. Plas, Heterocycles, 1981, 15, 213 CrossRef CAS.
- A. J. J. Lennox and G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2012, 51, 9385 CrossRef CAS.
- T. Cañeque, A. M. Cuadro, J. Alvarez-Builla and J. J. Vaquero, Tetrahedron Lett., 2009, 50, 1419 CrossRef.
- M. J. James, N. D. Grant, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Org. Lett., 2016, 18, 6256 CrossRef CAS.
- B. J. Margolis, K. A. Long, D. L. T. Laird, J. C. Ruble and S. R. Pulley, J. Org. Chem., 2007, 72, 2232 CrossRef CAS.
- J. D. McGhee and P. H. von Hippel, J. Mol. Biol., 1974, 86, 469 CrossRef CAS.
- I. Vermes, C. Haanen, H. Steffens-Nakken and C. Reutelingsperger, J. Immunol. Methods, 1995, 184, 39 CrossRef CAS.
- M. Aleksić and V. Kapetanović, Acta Chim. Solv., 2014, 61, 555 Search PubMed.
- H. Schönherr and T. Cernak, Angew. Chem., 2013, 125, 12480 CrossRef.
- (a) A. Serobian, C. P. Pracey, D. S. Thomas, W. A. Denny, G. E. Ball and L. P. G. Wakelin, J. Mol. Recognit., 2020, 33, e2843 CrossRef CAS; (b) A. K. F. Martensson and P. Lincoln, Phys. Chem. Chem. Phys., 2018, 20, 11336 RSC; (c) K. Benner, A. Bergen, H. Ihmels and P. M. Pithan, Chem. – Eur. J., 2014, 20, 9883 CrossRef CAS; (d) Y. Sato, S. Nishizawa, K. Yoshimoto, T. Seino, T. Ichihashi, K. Morita and N. Teramae, Nucleic Acids Res., 2009, 37, 1411 CrossRef CAS.
- (a) E. M. Tuite and B. Nordén, J. Phys. Chem. B, 2018, 122, 2891 CrossRef CAS; (b) R. Nandini and B. Vishalakshi, Spectrochim. Acta, Part A, 2009, 74, 1025 CrossRef CAS; (c) W. D. Sasikala and A. Mukhe, Phys. Chem. Chem. Phys., 2016, 18, 10383 RSC; (d) A. Biancardi, T. Biver, A. Burgalassi, M. Mattonai, F. Secco and M. Venturini, Phys. Chem. Chem. Phys., 2014, 16, 20061 RSC; (e) L. H. Fornander, L. Wu, M. Billeter, P. Lincoln and B. Norden, J. Phys. Chem. B, 2013, 117, 5820 CrossRef CAS; (f) A. Chakraborty, M. Ali and A. K. Saha, Spectrochim. Acta, Part A, 2010, 75, 1577 CrossRef; (g) J. L. Seifert, R. E. Connor, S. A. Kushon, M. Wang and B. A. Armitage, J. Am. Chem. Soc., 1999, 121, 2987 CrossRef CAS; (h) L. M. Wilhelmsson, B. Norden, K. Mukherjee, M. T. Dulay and R. N. Zare, Nucleic Acids Res., 2002, 30, 3e CrossRef; (i) D. Hüglin, W. Seiffert and H. W. Zimmermann, Histochemistry, 1986, 86, 71 CrossRef.
- (a) T. Šmidlehner, I. Piantanida and G. Pescitelli, Beilstein J. Org. Chem., 2018, 14, 84 CrossRef; (b) B. Nordén, A. Rodger and T. Dafforn, Linear dichroism and circular dichroism. A textbook on polarized-light spectroscopy, RSC Publishing, Cambridge, 2010 Search PubMed.
- C. Bohne, K. Faulhaber, B. Giese, A. Häfner, A. Hofmann, H. Ihmels, A.-K. Köhler, S. Perä, F. Schneider and M. A. L. Sheepwash, J. Am. Chem. Soc., 2005, 127, 76 CrossRef CAS.
- H. Pal, S. Nad and M. Kumbhakar, J. Chem. Phys., 2003, 119, 443 CrossRef CAS.
- G. Jones, W. R. Jackson, C. Y. Choi and W. R. Bergmark, J. Phys. Chem., 1985, 89, 294 CrossRef CAS.
- (a) B. Valeur and M. N. Berberan-Santos, Molecular fluorescence. Principles and applications, Wiley-VCH, Weinheim, 2nd edn, 2012 CrossRef; (b) G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991 CrossRef CAS.
- R. Bortolozzi, H. Ihmels, L. Thomas, M. Tian and G. Viola, Chem. – Eur. J., 2013, 19, 8736 CrossRef CAS.
- R. Romagnoli, P. G. Baraldi, M. K. Salvador, F. Prencipe, C. Lopez-Cara, S. S. Ortega, A. Brancale, E. Hamel, I. Castagliuolo, S. Mitola, R. Ronca, R. Bortolozzi, E. Porcù, G. Basso and G. Viola, J. Med. Chem., 2015, 58, 3209 CrossRef CAS.
- R. Bortolozzi, A. Luraghi, E. Mattiuzzo, A. Sacchetti, A. Silvani and G. Viola, J. Nat. Prod., 2020, 83, 2434 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, additional spectroscopic data, 1H and 13C NMR spectra. See DOI: 10.1039/d0ob02298e |
| This journal is © The Royal Society of Chemistry 2021 |