
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilver-catalyzed tandem cycloisomerization/hydroarylation reactions and mechanistic investigations for an efficient access to 1,2-dihydroisoquinolines†‡
Maxime
De Abreu
 a,
Yue
Tang
b,
Etienne
Brachet
*a,
Mohamed
Selkti
a,
Véronique
Michelet
b and
Philippe
Belmont
*a
a,
Yue
Tang
b,
Etienne
Brachet
*a,
Mohamed
Selkti
a,
Véronique
Michelet
b and
Philippe
Belmont
*a
aUniversité de Paris, CiTCoM, UMR CNRS 8038, F-75006 Paris, France. E-mail: etienne.brachet@u-paris.fr; philippe.belmont@u-paris.fr
bUniversité Côte d'Azur, Institut de Chimie de Nice, UMR CNRS 7272, Parc Valrose, Faculté des Sciences, 06100 Nice, France
First published on 18th December 2020
Abstract
An efficient silver-catalyzed tandem reaction for the formation of 1,2-dihydroisoquinoline derivatives is herein reported. Highly functionalized multiheterocyclic scaffolds are accessible in a straightforward manner using readily accessible starting materials under mild conditions. This methodology offers an attractive route for the synthesis and development of a biologically relevant new heterocyclic pharmacophore, merging the biological activities of isoquinolines with those of various nitrogen-containing heterocycles (indoles, pyrroles) incorporated during the tandem reaction. Mechanistic investigations were also conducted along with a large scope and limitation study, modifying various sites of this pharmacophore.
Introduction
One of the most interesting subclasses of nitrogen-containing heterocycles is certainly the isoquinoline core and its derivatives due to its prevalence in many biologically active compounds, including marketed drugs. Indeed, isoquinolines have various therapeutic applications such as antipruritic (quinisocaine)1 and antispasmodic (papaverine).2 Recently, some 1,2-dihydroisoquinoline derivatives exhibited an interesting activity towards cancer cell lines (I and II, Scheme 1).3,4 Traditionally, isoquinolines were synthesized using the Bischler–Napieralski,5 the Pomeranz–Fritsch6,7 and the Pictet–Spengler8 processes, developed in the late 19th/early 20th century. However, harsh conditions were often required to perform such reactions (high operating temperatures, use of highly toxic and corrosive PCl5 or POCl3). Thereby, many new synthetic methodologies were developed in the last decades, relying on transition-metal catalysis. Larock's team reported the first example of a Pd-catalyzed synthesis of isoquinolines,9 followed by other examples derived from their methodology.10–12 Wu's team pioneered the development of numerous examples of 1-substituted isoquinolines via Ag-catalyzed 6-endo-dig processes using silver as the catalyst, in the presence of many pro-nucleophiles, such as α,β-unsaturated ketones,13 phosphonates,14 imidazoles,15 ketones,16 indoles17 and even trifluoromethyl groups.18 These methods allow rapid access to highly functionalized scaffolds in very few steps. Moreover, the addition of heterocyclic nitrogen-containing pro-nucleophiles (indoles/pyrroles) at position 1 of isoquinolines gives rise to new potential pharmacophores since indole19 or pyrrole20 derivatives also exhibit key biological activities.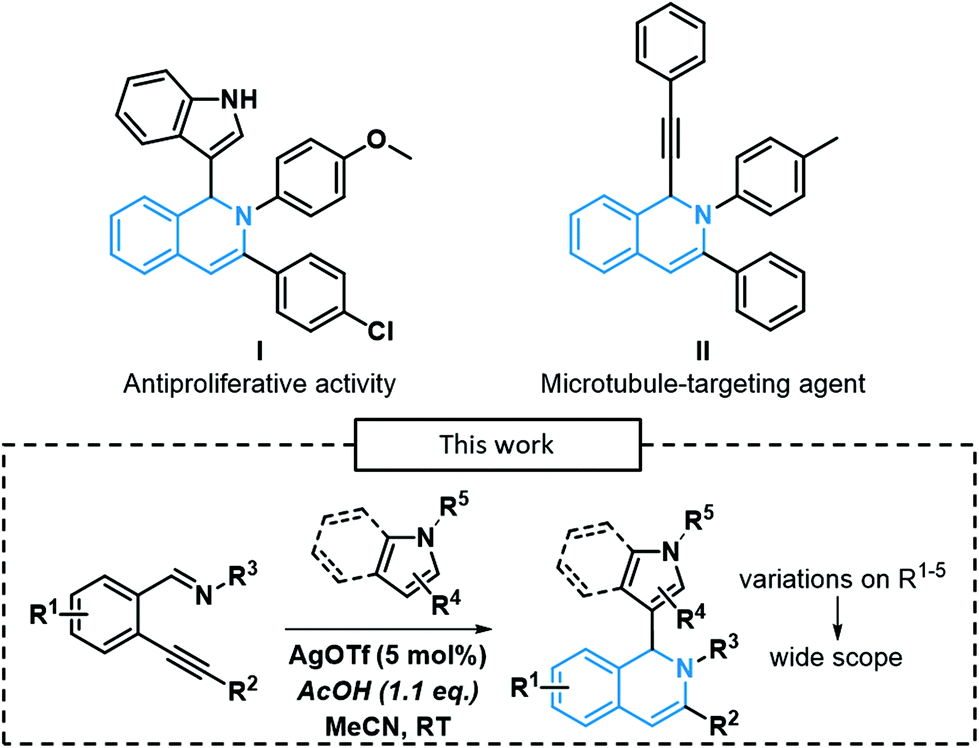 | ||
| Scheme 1 Our approach for the synthesis of 1,2-dihydroisoquinoline derivatives. | ||
Tandem reactions affording these complex scaffolds have been reported in the literature using several modes of activation and a myriad of catalysts, Lewis acids, and electrophiles.3,21 More recently, Han and Lu's team reported a Pd-catalyzed reaction for the synthesis of 1,2-dihydroisoquinolines with the concomitant insertion at position 1 of 3-indoles.22
Such scaffolds can also be obtained using cobalt catalysts, as reported by Tandon et al.23 There is only one report with few examples described in the presence of a silver catalyst.17 Following our interest in Ag-catalyzed cycloisomerization reactions to form furoquinoline/pyranoquinoline,24,25 isochromene26 or pyranoquinoline27 derivatives as well as isobenzofurane or isoindoline derivatives,28 we envisioned to establish efficient reaction conditions for the formation of a wider variety of isoquinoline derivatives compared to previous literature reports.17 We propose herein to study the cyclization of ortho-alkynylarylaldimine derivatives using Ag catalysts (Scheme 1), with the concomitant addition of several types of heterocyclic nucleophiles in order to access highly functionalized 1,2-dihydroisoquinolines in a 3-step pathway.
Results and discussion
To test our hypothesis, we began with the synthesis of the starting materials (Scheme 2) using a Sonogashira cross-coupling reaction performed on 2-bromoarylaldehyde derivatives reacting with terminal alkynes.26 The resulting desired ortho-alkynylarylaldehydes were then combined with aniline derivatives to form the imino starting materials (Scheme 2). A library of ortho-alkynyl(hetero)arylaldimines was obtained bearing various R1, R2 and R3 substituents.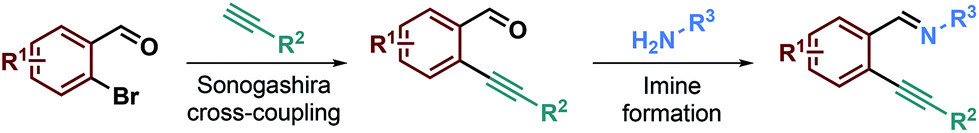 | ||
| Scheme 2 Two-step preparation of the starting materials. | ||
These starting materials were then used for the optimization of the reaction conditions. Based on the conditions previously reported by our team,26,27 we investigated a plausible 6-endo-dig29 ring-closing reaction on model substrate 1a (entries 1 and 2, Table 1). Unfortunately, these conditions were not efficient on that kind of starting material and led only to the slow hydrolysis of the imino group. Since imine hydrolysis can be promoted by traces of water, we added 3 Å M.S. in the reaction medium in order to avoid such side reactions; regrettably, no improvement was observed even after two days of reaction (entry 3). A report from Zhang, Wu et al.30 described the formation of 1-(trifluoromethyl)-1,2-dihydroisoquinolines by incorporating a trifluoromethyl group on 2-alkynylaryl aldimine derivatives in the presence of acetic acid. They concluded that acetic acid, as an additive, was necessary to reach high product yields since it acts as a Brønsted acid for the activation of the imino group. Therefore, inspired by their results, we decided to add 1.1 eq. of acetic acid to the reaction medium using MeCN as the solvent and were pleased to obtain the desired product in a good 79% yield (entry 4). A control experiment, without acetic acid (entry 5), confirmed that the reaction could not proceed without this additive and also that switching the solvent from DCE to CH3CN has no impact on the reaction course. In the same way, no product formation was observed without the silver catalyst, confirming the crucial role of both acetic acid and AgOTf in the formation of the desired product (entries 5 and 6). Several silver catalysts have been tested such as AgSbF6 but no better yields were obtained (2a 53%, entry 7). Solvent change, at the same concentration (0.08 M), to DCE led to the formation of the desired product in a good 78% yield (entry 8), while switching to DMF afforded only traces of isoquinoline 2a (entry 9). We also tried to perform the reaction with a catalytic amount of acetic acid since it should be regenerated during the reaction (see the mechanistic proposal). However, after using 0.1 eq. of acetic acid, we were surprised to obtain a lower 57% yield of 2a after 2.5 days (entry 10), indicating that acetic acid present in slight excess allows faster formation of the desired isoquinoline, preventing the side product formation or hydrolysis reaction.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | Additive | T (°C) | Time (h) | Yielda2a/3 |
| a Isolated yields. b Standard condition: Reactions were performed with 1a (0.16 mmol), AgOTf (5 mol%), N-methylindole (5 eq.) and acetic acid (1.1 eq.) in 2 mL of solvent and the mixture was agitated at room temperature for 16 h. c 0.5 M. d 0.2 M. e 0.08 M. f 1.1 eq. | ||||||
| 1 | AgOTf (10) | DMFc | — | 80 | 16 | — |
| 2 | AgOTf (10) | DCEd | — | 80 | 16 | — |
| 3 | AgOTf (10) | DCEd | 3 Å M.S. | 80 | 48 | — |
| 4 | AgOTf (5) | CH3CNe | AcOHf | rt | 16 | 79/0b |
| 5 | AgOTf (5) | CH3CNe | — | rt | 16 | — |
| 6 | — | CH3CNe | AcOHf | rt | 16 | — |
| 7 | AgSbF6 (5) | CH3CNe | AcOHf | rt | 16 | 53/0 |
| 8 | AgOTf (5) | DCEe | AcOHf | rt | 16 | 78/0 |
| 9 | AgOTf (5) | DMFe | AcOHf | rt | 16 | Traces |
| 10 | AgOTf (5) | CH3CNe | AcOHf | rt | 48 | 57/0 |
| 11 | AgOTf (5) | CH3CNe | PTSAf | rt | 16 | 0/43 |
| 12 | AgOTf (5) | CH3CNe | CSAf | rt | 16 | 0/46 |
| 13 | AgOTf (5) | CH3CNe | TFAf | rt | 16 | 0/56 |
| 14 | AgOTf (1 eq.) | CH3CNe | — | rt | 16 | — |
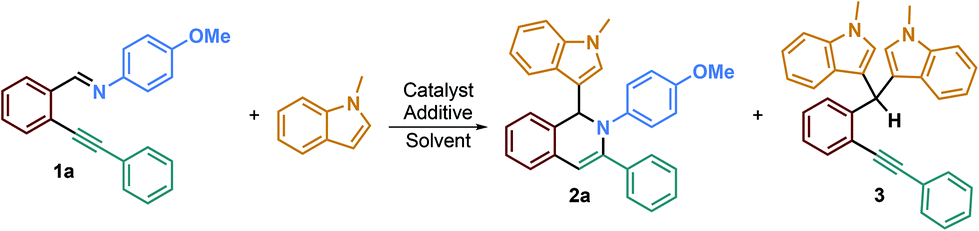
We then investigated the influence of the nature of the acid additive on the reaction by using other common acids. para-Toluenesulfonic acid (PTSA), camphorsulfonic acid (CSA) and trifluoroacetic acid (TFA) did not allow the formation of the desired product; however, when using these acids, we were able to isolate product 3, resulting in double addition of the nucleophile onto the starting material, in yields of 43%, 46% and 56%, respectively (entries 11, 12 and 13).31 The use of stronger acids than acetic acid will be discussed later on, but we may already propose that the iminyl group will be protonated, thereby promoting its hydrolysis and consequently the formation of side product 3 instead of the intramolecular addition of the free iminyl group on the alkynyl bond. Finally, we ran the reaction using 1 eq. of AgOTf in order to study whether the reaction could proceed, in this case, without acetic acid (entry 14). Unfortunately, no product was formed, indicating that both the silver catalyst and acetic acid additive were required to obtain the desired product 2a. Thus, we decided that our reaction conditions are as follows: AgOTf (5 mol%), imino derivative (0.16 mmol), nucleophile (5 eq.), acetic acid (1.1 eq.) in MeCN (0.08 M) at rt for 16 h.
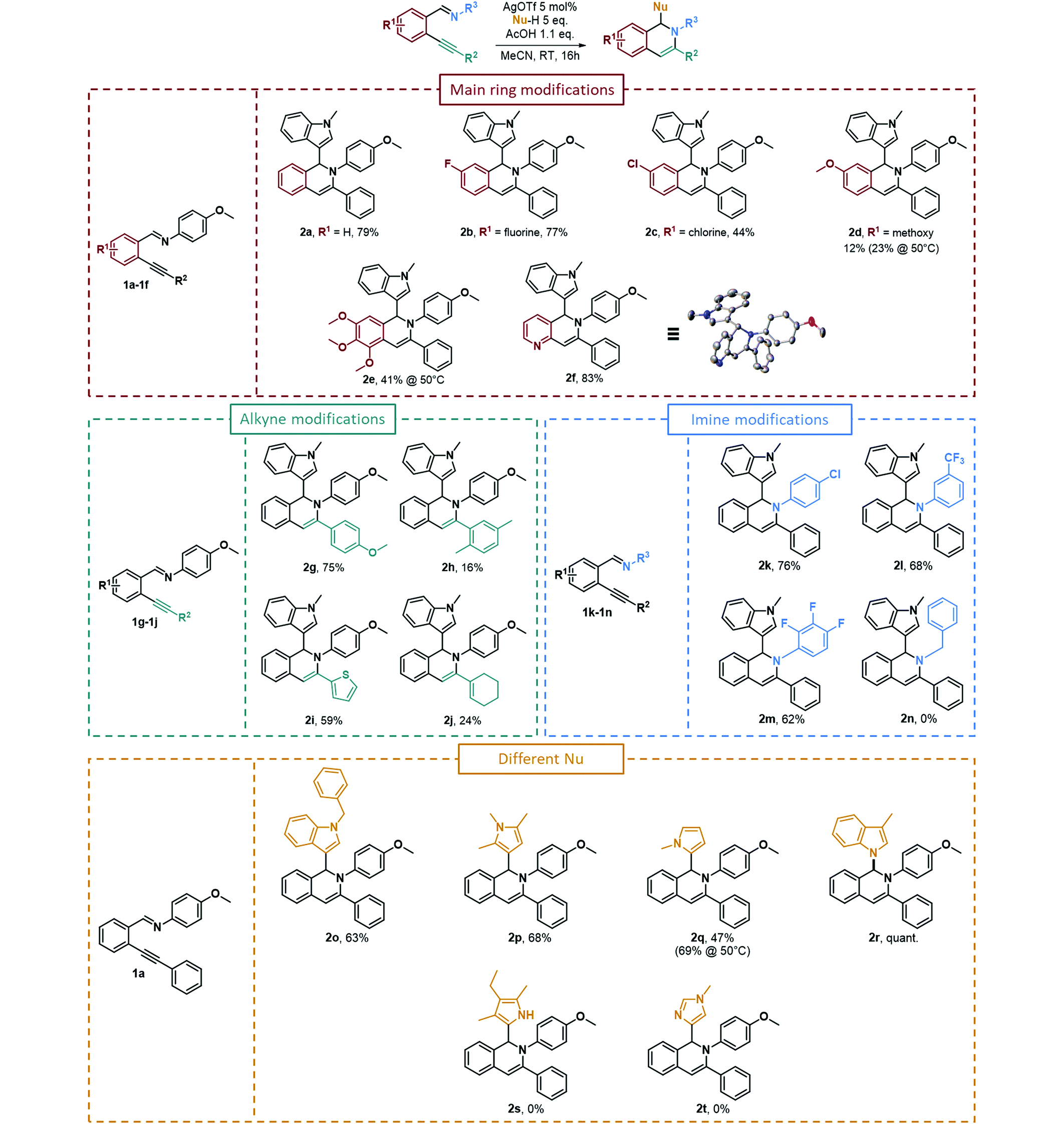
Using these optimized reaction conditions, we then investigated the functional group compatibility of the reaction, modifying the substituents present on various parts of imino derivatives 1. As presented in Table 2, the tandem cycloisomerization/hydroarylation reaction of the model starting material 1a led to the formation of 79% of the corresponding isoquinoline (2a). We then modified the substitutents on the main ring, on the alkynyl part, on the imine substitutent and finally the nucleophile. A fluorine substitutent on the main ring (R1) was not detrimental to the reaction, as the product was isolated in a good 77% yield (2b). However, when another halogen substitutent such as chlorine (2c) was present, the isolated yield decreases to 44%. The yield dropped much further when a methoxy substitutent was present on the same carbon (2d), leading to a low 12% isolated yield that could be slightly increased to 23% upon heating the reaction up to 50 °C. A trimethoxy substitutent afforded the desired isoquinoline (2e) in a higher 41% yield upon heating at 50 °C. It is important to note that the reaction was compatible with the presence of a heterocycle on the main ring such as a pyridine (2f), leading to a high 83% yield. It appeared that the electron-donating groups on the main ring were detrimental to the reaction, while the electron-withdrawing groups led to better isolated yields. As for the alkyne modifications (R2, Table 2), 4-methoxyphenyl substitution on the alkyne of the starting material was well tolerated and led to a good 75% yield (2g), whereas a 2,5-dimethylphenyl group led to a low 16% yield (2h). This result could be explained by the steric hindrance on the ortho position of the alkyne that can interfere with the attack of the imino group on the alkynyl unit. A heterocycle, 2-thiophenyl, substitutent led to a fair 59% yield (2i). Finally, an alkenyl-substituted alkyne (1-cyclohexenyl substitution) could also react and afforded the desired isoquinoline (2j) but in a low 24% yield. We then changed the nature of the substituents on the aniline (R3, Table 2), leading to the formation of the imine starting materials. The reaction could proceed normally when electron-withdrawing substituents were present on the arylimine unit such as 4-chlorophenyl (2k), 3-trifluoromethylphenyl (2l) and 2,3,4-trifluorophenyl (2m) substitutents, which all led to satisfactory yields of 76%, 68% and 62%, respectively. However, an aliphatic imine formed using benzylamine could not afford the corresponding product 2n. Finally, we investigated the reactivity of various nucleophiles towards the starting material 1a and their ability to perform a hydroarylation reaction. We used various nitrogen-containing heterocycles such as a N-methylindole and N-benzylindole that gave the desired products in good yields of 79% (2a) and 63% (2o), respectively. We also used differently substituted pyrroles such as 1,2,5-trimethylpyrrole that led to a good 68% yield (2p) (with an attack on position 3) and a N-methylpyrrole that afforded the desired product 2q in a moderate 47% yield, increasing up to 69% upon heating at 50 °C (with an attack on position 2). The use of 3-methylindole led to the quantitative formation of the hydroamination product 2r. We purposely used this indole derivative to prevent any attack on position 3 and instead to favor the attack on indole's position 2. But, we did not obtain any attack from position 2, which may be because of the steric hindrance of the methyl substitution at position 3 and the only attack observed was from the nitrogen atom. Other nucleophiles such as 3-ethyl-2,4-dimethylpyrrole or 1-methylimidazole could not react, and no traces of the corresponding products 2s and 2t were detected.
|
It is important to note that imines formed using acetophenone or benzophenone derivatives instead of aldehyde derivatives could not lead to the formation of the expected dihydroisoquinoline products. Moreover, we employed numerous heterocyclic nucleophiles in our methodology, from nitrogen to oxygen and sulfur-containing ones. Unfortunately, we did not observe any attack from the latter two (2-methylfuran and 2-methylthiophene for example). This lack of reactivity could be explained by their nucleophilicity. Indeed, using the work of Mayr's team on the establishment of a reactivity scale for various molecules (both for their electrophilicity and nucleophilicity), we can clearly notice a strong difference in their nucleophilicity parameter (N),32–36 with, for instance, values of N for 2-methylfuran and 2-methylthiophene being respectively 3.61 and 1.35. For a comparison, the “weakest” nucleophile that led to a product in this methodology was N-methylindole with a N parameter of 5.75. Another point that should be noted is that the opposite reasoning is not necessarily applicable. Some tested nucleophiles having a high N value did not lead to the desired final compound, such as 3-ethyl-2,4-dimethylpyrrole or 1-methylimidazole, which present N values of 11.63 and 9.91, respectively. Such results might be due to the impossibility to perform a reaction with the carbon atom (C-nucleophile) due to steric hindrance around the nucleophilic atom or even the presence of an unprotected N–H bond. Moreover, during the optimization work (Table 1), we tested acid additives having pKa values much lower than that of acetic acid (4.76). Indeed, PTSA has a pKa value of −6.5, CSA has a pKa value of 1.2 and TFA has a pKa value of 0.52 (in aqueous medium).37 On the other hand, for our iminyl starting material, the pKa prediction is around a value of 2.57.38 Therefore, the acetic acid additive is crucial for the protodemetalation step (Scheme 5), leading to dihydroquinoline formation. In contrast, stronger acids may facilitate iminyl group hydrolysis of the aldimine starting materials, thereby leading to the parent carbonyl derivatives that may undergo the well-known double indole addition, leading to compound 3.39
Finally, in order to gain more insight into the reaction mechanism, we performed some mechanistic experiments, starting with deuteration experiments with N-methylindole (deutared at position 3) or AcOD. Indeed, we wondered if the hydrogen atom incorporated at position 4 of the isoquinoline scaffold (e.g. compound 2k, Scheme 4) originated from the N-methylindole or from the acid additive. We never observed any deuteration after workup at position 4 of 2k. We believe that the absence of deuteration is due to the very reactive and labile H at position 4 (Scheme 4). An NMR experiment confirmed this hypothesis since the proton on position 4 instantly disappeared after the addition of an excess of AcOD, leading to the isolated deuterated isoquinoline 2k–D (see ESI‡).
Moreover, in order to deepen our understanding of the reaction mechanism, we tried to isolate a reaction intermediate.40 Precipitation and recrystallization of the isoquinolinium intermediate 4 were possible by running the reaction in dichloromethane with 1 eq. of AgOTf, without adding a nucleophile, followed by the addition of triflic acid. Thanks to these conditions, we were able to obtain a single crystal of 4 suitable for an X-ray diffraction (Scheme 3).
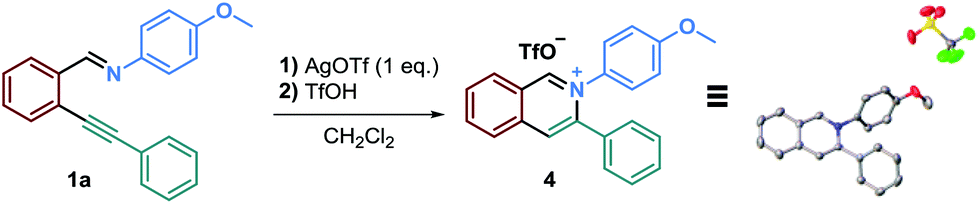 | ||
| Scheme 3 Isolation of the isoquinolinium salt and XRD analysis. | ||
We then used this isoquinolinium salt in the presence of N-methylindole, with and without AcOH, in acetonitrile. To our surprise, no product formation was observed. These results questioned our understanding of this reaction's mechanism since we were expecting a straightforward reaction between the nucleophile and the isoquinolinium ion. We therefore concluded that this isolated salt was probably not a reaction intermediate and the addition of the nucleophile might occur onto the isoquinolinium that still bears the alkenylsilver unit.
To test this hypothesis, we performed new experiments, in a one-pot two-step fashion (Scheme 4). The first step was the reaction of the imine derivative 1k with 1 eq. of AgOTf in order to form the hypothetical isoquinolinium alkenylsilver specie 5, and then, the second step was the addition of 1.1 eq. of AcOH to 5 eq. of N-methylindole. After 10 days of reaction, we were able to isolate the desired isoquinoline 2k in a low 21% yield, which may indicate a plausible alkenylsilver intermediate.
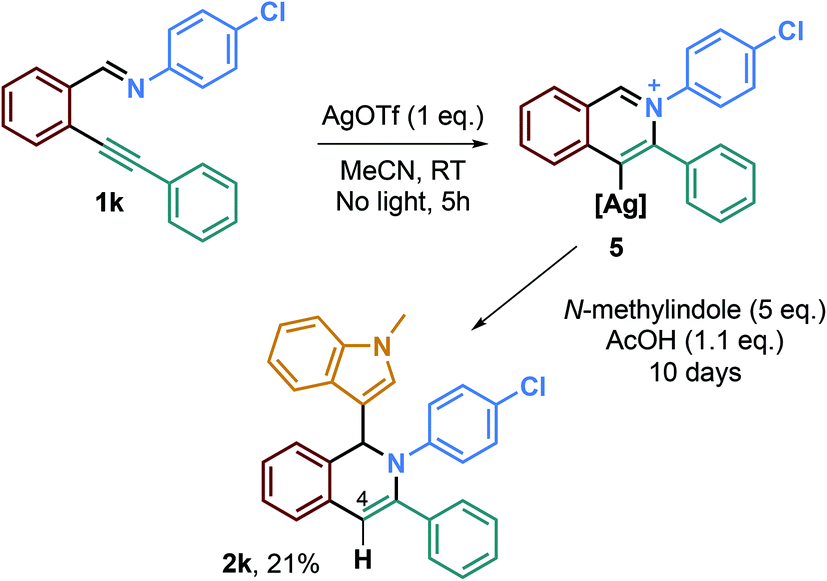 | ||
| Scheme 4 Sequential formation and reaction of the hypothetical alkenyl silver intermediate 5. | ||
Thanks to all these data, we may propose the following reaction mechanism (Scheme 5). The first step of the reaction is expected to be the coordination between the silver catalyst and the alkynyl group of the starting material A, leading to the activated alkynyl compound B. The imine group of B would attack intramolecularly the electron-deficient silver-activated alkynyl unit, leading to the formation of an alkenylsilver intermediate C. We believe that this intermediate would then be attacked by the external nucleophile (N-methylindole here), yielding the intermediate D. Then, after a protodesilveration step, intermediate E would be obtained and would yield the final desired product F upon rearomatization of the indole core.
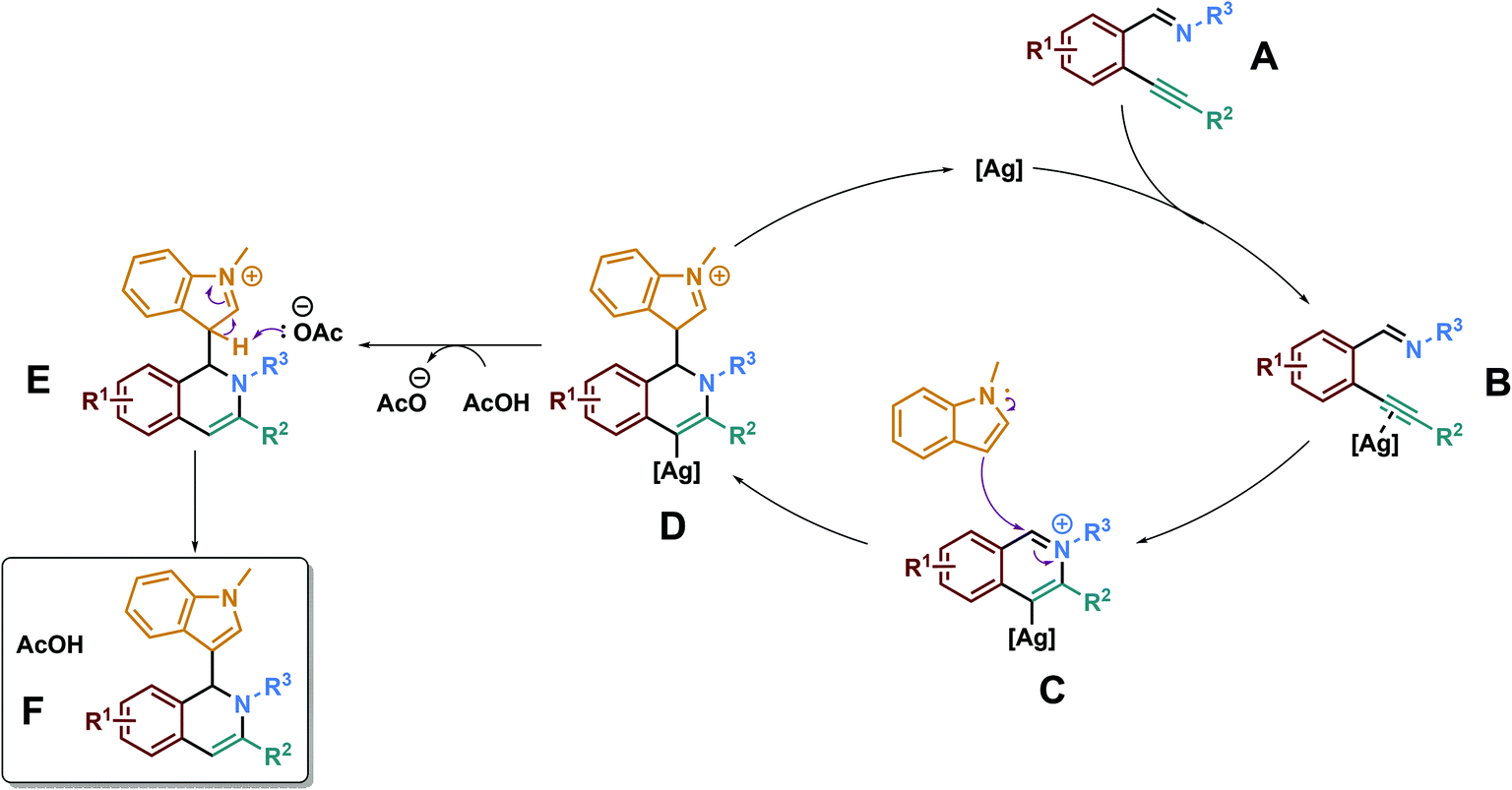 | ||
| Scheme 5 Proposed mechanism. | ||
Conclusions
We herein reported an efficient tandem cycloisomerization/hydroarylation silver-catalyzed reaction leading to the formation of a broad range of substituted isoquinoline structures under mild conditions. Thanks to the synergistic association of AgOTf as a catalyst with acetic acid, the access to a wide library of heterocyclic derivatives, bearing various substitutions, was possible, thanks to modifications of the starting material's main ring, alkynyl and imino parts, as well as the choice of different types of nitrogen-containing heterocycles as nucleophiles. Biological evaluation of these new scaffolds and the study of the central chirality obtained at the end of the tandem reaction are both currently underway.Experimental section
General informations
1H NMR, 13C NMR, 31P NMR and 19F NMR spectra were recorded on a Bruker® Avance™ 300, 400 or 600 MHz spectrometer in CDCl3, DMSO-d6, acetone-d6, acetonitrile-d3 or methanol-d4 solution with the internal solvent signal as a reference. NMR data are reported as follows: chemical shift (ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, dd = doublet of doublets, ddd = doublet of doublet of doublets, td = triplet of doublets, qd = quartet of doublets, m = multiplet, br. s. = broad singlet), coupling constants (Hz) and the number of protons (for 1H NMR). All reactions were monitored by thin-layer chromatography using Merck silica gel plates 60 F254. All the TLC plates used in order to check the completion of the imines formation or cycloisomerization reactions were previously neutralized in a solution of cyclohexane containing 1% triethylamine. Visualization was accomplished with short-wavelength UV light (254 and 365 nm) and/or staining with appropriate stains (anisaldehyde, ortho-phosphomolybdic acid). Purification was performed either via recrystallization, preparative TLC, standard flash chromatography (using silica gel of particle size 40–63 μm) or via a flash purification system (Biotage Isolera One) unless specified otherwise. IR spectra were recorded on a PerkinElmer Spectrum 65 FT-IR spectrometer with the absorption frequencies reported in wavenumber (cm−1). Melting points were measured using capillaries in a melting point apparatus. Silver catalysts were purchased from Strem, Aldrich or Alfa Aesar. All other commercially available reagents and solvents were used without further purification. High-resolution mass spectrometry (HRMS) measurements were recorder either on a Bruker MicrOTOF-Q II™ or Q-TOF maXis™ spectrometer using electrospray ionization (ESI) or atmospheric-pressure chemical ionization (APCI).General procedure for the formation of 1,2-dihydroisoquinolines
An oven-dried re-sealable tube was charged with a mixture of ortho-alkynylbenzaldimine (1 eq.), nucleophile (5 eq.), AgOTf (5 mol%) and acetic acid (1.1 eq.), in acetonitrile (0.08 M). The tube was capped with a rubber septum, evacuated and backfilled with nitrogen. The reaction mixture was stirred at room temperature overnight. After completion of the reaction checked by TLC, the crude reaction mixture was immediately filtered on a short Celite column to eliminate the catalyst. The filtrate was concentrated, and purification of the residue by silica gel column chromatography eluting with cyclohexane/ethyl acetate mixture gave the desired product.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EA, 9:1); ochre solid; 1H NMR (300 MHz, DMSO-d6) δ 8.01 (d, J = 7.3 Hz, 1H), 7.47 (d, J = 7.1 Hz, 2H), 7.36 (d, J = 7.6 Hz, 2H), 7.30–7.07 (m, 8H), 7.09 (d, J = 7.2 Hz, 2H), 6.74 (d, J = 7.2 Hz, 2H), 6.72 (s, 1H), 6.61 (s, 1H), 6.34 (s, 1H), 3.62 (s, 6H); 13C NMR (101 MHz, DMSO-d6) δ 154.4, 141.5, 140.6, 137.6, 136.9, 131.8, 131.6, 128.3, 127.8, 127.7, 127.2, 126.4, 126.0, 125.7, 124.4, 123.1, 121.2, 119.4, 118.9, 116.5, 114.5, 114.2, 111.8, 109.9, 61.3, 55.1, 32.3; IR (neat): 3048, 2922, 2853, 1712, 1607, 1558, 1505, 1488, 1463, 1452, 1372, 1330, 1291, 1242 cm−1; mp: 159–161 °C; HR-MS (ESI): m/z calculated for C31H26N2ONa 465.1943 obtained 465.1931.
:EA, 9:1); ochre solid; 1H NMR (400 MHz, chloroform-d) δ 8.16–8.08 (m, 1H), 7.54–7.47 (m, 2H), 7.25 (h, J = 4.7, 3.8 Hz, 4H), 7.16 (dd, J = 9.8, 7.0 Hz, 3H), 7.09–7.01 (m, 2H), 7.00–6.86 (m, 2H), 6.73–6.64 (m, 2H), 6.61 (d, J = 8.3 Hz, 2H), 6.27 (s, 1H), 3.67 (s, 3H), 3.58 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 161.7 (d, J = 245.4 Hz), 155.1, 141.6 (d, J = 2.3 Hz), 141.1, 138.0, 137.3, 133.9 (d, J = 6.4 Hz), 128.5 (d, J = 2.8 Hz), 128.4, 127.9, 127.9, 127.8, 126.1, 125.8 (d, J = 7.9 Hz), 123.8, 121.8, 119.5 (d, J = 11.6 Hz), 116.8, 114.3, 114.2, 114.1, 113.0 (d, J = 21.8 Hz), 110.4, 109.7, 61.9, 55.5, 32.8; 19F NMR (376 MHz, chloroform-d) δ −115.62; IR (neat): 3052, 2925, 2953, 1712, 1660, 1607, 1566, 1539, 1506, 1492, 1464, 1447, 1423, 1371, 1331, 1273, 1242 cm−1; mp: 158–160 °C; HR-MS (ESI): m/z calculated for C31H25N2OFNa 483.1849 obtained 483.1850.
:EA, 9:1); ochre solid; 1H NMR (300 MHz, chloroform-d) δ 8.19–8.10 (m, 1H), 7.57–7.48 (m, 2H), 7.35–7.16 (m, 9H), 7.07 (d, J = 8.2 Hz, 2H), 6.72 (d, J = 8.2 Hz, 2H), 6.66 (s, 1H), 6.61 (s, 1H), 6.30 (s, 1H), 3.73 (s, 3H), 3.66 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 154.8, 141.8, 140.5, 137.4, 137.0, 133.4, 130.9, 130.3, 128.5, 128.1, 127.9, 127.4, 127.2, 126.1, 125.9, 125.7, 123.4, 121.5, 119.5, 119.2, 116.1, 114.3, 110.8, 110.1, 60.8, 55.2, 32.5; IR (neat): 3059, 2925, 2853, 1720, 1609, 1552, 1506, 1479, 1464, 1446, 1371, 1292, 1242 cm−1; mp: 163–165 °C; HR-MS (APCI): m/z calculated for C31H26ClN2O 477.1728 obtained 477.1718.
:EA, 7:3); yellow solid; 1H NMR (300 MHz, chloroform-d) δ 8.17 (s, 1H), 7.51 (s, 2H), 7.31–7.12 (m, 7H), 7.07 (d, J = 8.4 Hz, 2H), 6.84 (d, J = 9.0 Hz, 1H), 6.78–6.66 (m, 3H), 6.63 (s, 1H), 6.58 (s, 1H), 6.26 (s, 1H), 3.77 (s, 3H), 3.69 (s, 3H), 3.60 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 158.7, 154.8, 141.5, 140.1, 138.4, 137.3, 133.8, 128.3, 128.1, 127.6, 127.5, 126.4, 125.7, 125.6, 123.5, 121.6, 119.5, 119.4, 117.1, 114.2, 113.2, 111.6, 111.5, 109.6, 62.2, 55.5, 55.4, 32.8; IR (neat): 3054, 2925, 2852, 1712, 1607, 1559, 1506, 1463, 1446, 1423, 1371, 1319, 1285, 1243 cm−1; mp: 164–166 °C; HR-MS (ESI): m/z calculated for C32H28N2O2Na 495.2048 obtained 495.2028.
:EA, 7:3); ochre solid; 1H NMR (300 MHz, chloroform-d) δ 8.13 (d, J = 5.4 Hz, 1H), 7.51 (d, J = 7.3 Hz, 2H), 7.26 (s, 3H), 7.16 (q, J = 8.0, 7.3 Hz, 3H), 7.06 (d, J = 8.5 Hz, 2H), 6.83 (s, 1H), 6.70 (d, J = 8.5 Hz, 2H), 6.62 (s, 1H), 6.54 (s, 1H), 6.21 (s, 1H), 4.04 (s, 3H), 3.91 (s, 3H), 3.80 (s, 3H), 3.69 (s, 3H), 3.62 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 154.8, 152.4, 148.5, 141.4, 141.4, 141.1, 140.1, 138.4, 137.3, 128.3, 128.2, 128.1, 127.6, 127.5, 126.3, 123.4, 121.6, 119.4, 119.4, 117.1, 114.2, 109.7, 106.6, 105.5, 61.8, 61.6, 61.1, 56.1, 55.5, 32.9; IR (neat): 3053, 2926, 2853, 1729, 1712, 1595, 1561, 1505, 1486, 1459, 1409, 1370, 1328, 1299 cm−1; mp: 68–70 °C; HR-MS (APCI): m/z calculated for C34H33N2O4 533.2435 obtained 533.2430.
:EA, 7:3); yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 8.39 (d, J = 4.0 Hz, 1H), 7.99 (d, J = 7.8 Hz, 1H), 7.66 (d, J = 7.3 Hz, 1H), 7.49 (d, J = 6.6 Hz, 2H), 7.39 (d, J = 8.1 Hz, 1H), 7.29–7.07 (m, 6H), 6.97 (d, J = 8.8 Hz, 2H), 6.87 (s, 1H), 6.74 (d, J = 8.8 Hz, 2H), 6.64 (s, 1H), 6.44 (s, 1H), 3.65 (s, 3H), 3.62 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 154.9, 150.6, 148.1, 145.8, 140.2, 137.1, 136.9, 133.3, 128.4, 127.7, 127.4, 126.2, 125.3, 123.9, 121.4, 121.1, 119.3, 119.1, 116.3, 114.1, 110.9, 110.0, 79.2, 61.2, 55.1, 32.4; IR (neat): 3055, 2925, 2852, 1711, 1602, 1575, 1557, 1507, 1461, 1437, 1424, 1372, 1243 cm−1; mp: 168–170 °C; HR-MS (APCI): m/z calculated for C30H26N3O 444.2070 obtained 444.2072.
:EA, 9:1); ochre solid; 1H NMR (300 MHz, acetone-d6) δ 8.18–8.12 (m, 1H), 7.47 (d, J = 8.8 Hz, 2H), 7.35–7.32 (m, 1H), 7.31 (t, J = 1.4 Hz, 1H), 7.25 (dq, J = 5.9, 1.5 Hz, 2H), 7.19–7.13 (m, 3H), 7.08 (d, J = 9.0 Hz, 2H), 6.72 (dd, J = 8.9, 4.4 Hz, 4H), 6.66 (s, 1H), 6.61 (d, J = 0.9 Hz, 1H), 6.35 (s, 1H), 3.70 (s, 3H), 3.66 (s, 3H), 3.63 (s, 3H); 13C NMR (75 MHz, acetone-d6) δ 160.4, 155.9, 142.6, 142.2, 138.3, 133.6, 132.7, 131.4, 129.7, 128.8, 128.0, 127.3, 126.8, 126.7, 125.0, 124.5, 122.1, 120.4, 119.8, 117.9, 114.8, 114.4, 111.3, 110.5, 63.0, 55.5, 55.4, 32.7; IR (neat): 3050, 2954, 2925, 2854, 1714, 1606, 1577, 1559, 1505, 1483, 1463, 1451, 1441, 1424, 1371, 1328, 1289, 1242 cm−1; mp: 44–46 °C; HR-MS (APCI): m/z calculated for C32H29N2O2 473.2224 obtained 473.2214.
:EA, 9:1); ochre solid; 1H NMR (300 MHz, chloroform-d) δ 8.01 (d, J = 7.8 Hz, 1H), 7.67 (s, 2H), 7.41–7.19 (m, 4H), 7.13 (s, 1H), 7.05 (s, 1H), 6.98 (d, J = 8.8 Hz, 2H), 6.91 (s, 1H), 6.85 (s, 2H), 6.62 (d, J = 8.9 Hz, 2H), 6.26 (s, 1H), 6.14 (s, 1H), 3.67 (s, 3H), 3.65 (s, 3H), 2.15 (s, 6H). We observed a rapid degradation of this compound so no 13C NMR could be undertaken.
:EA, 9:1); yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 8.03 (d, J = 7.8 Hz, 1H), 7.35 (d, J = 7.8 Hz, 2H), 7.31 (d, J = 5.2 Hz, 1H), 7.29–7.24 (m, 2H), 7.20 (d, J = 7.1 Hz, 1H), 7.18–7.12 (m, 2H), 7.07 (d, J = 9.1 Hz, 3H), 6.89–6.86 (m, 1H), 6.79 (d, J = 7.2 Hz, 3H), 6.47 (s, 1H), 6.24 (s, 1H), 3.65 (s, 3H), 3.59 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 154.8, 142.2, 140.6, 137.0, 135.5, 131.7, 131.3, 127.8, 127.4, 127.3, 126.4, 126.2, 126.2, 125.9, 125.4, 124.4, 123.2, 121.2, 119.7, 118.7, 115.9, 114.3, 111.3, 109.8, 61.9, 55.1, 32.3; IR (neat): 3065, 2926, 2854, 1713, 1604, 1560, 1530, 1505, 1482, 1464, 1440, 1425, 1372, 1351, 1331, 1289, 1243 cm−1; mp: 39–41 °C; HR-MS (ESI): m/z calculated for C29H24N2OSNa 471.1507 obtained 471.1500.
:EA, 9:1); ochre solid; 1H NMR (400 MHz, chloroform-d) δ 8.19 (d, J = 7.9 Hz, 1H), 7.42–7.29 (m, 5H), 7.25 (m, 4H), 6.93 (d, J = 8.9 Hz, 2H), 6.64 (s, 1H), 6.49 (s, 1H), 6.32 (s, 1H), 6.28 (brs, 1H), 3.91 (s, 3H), 3.75 (s, 3H), 2.35–2.25 (m, 1H), 2.23–2.07 (m, 3H), 1.71–1.55 (m, 4H); 13C NMR (101 MHz, chloroform-d) δ 154.8, 144.3, 142.3, 137.4, 134.1, 132.6, 131.7, 129.5, 128.1, 127.1, 126.5, 126.1, 125.8, 124.3, 123.0, 121.5, 119.9, 119.1, 117.4, 114.0, 109.5, 109.1, 62.2, 55.6, 32.8, 26.7, 25.9, 22.8, 22.2; IR (neat): 2928, 2832, 1711, 1601, 1557, 1505, 1483, 1464, 1371, 1241 cm−1; mp: 91–93 °C; HR-MS (APCI): m/z calculated for C31H31N2O 447.2431 obtained 447.2425.
:EA, 9:1); ochre solid; 1H NMR (300 MHz, chloroform-d) δ 8.14–8.07 (m, 1H), 7.50 (dd, J = 7.7, 2.0 Hz, 2H), 7.36–7.18 (m, 10H), 7.13–7.03 (m, 4H), 6.70 (s, 1H), 6.59 (s, 1H), 6.40 (s, 1H), 3.59 (s, 3H); 13C NMR (75 MHz, chloroform-d) δ 146.0, 141.1, 137.7, 137.3, 132.6, 131.9, 128.8, 128.5, 128.1, 128.0, 127.6, 127.5, 126.8, 126.7, 126.1, 126.0, 124.8, 123.0, 121.8, 119.5, 119.3, 116.7, 113.1, 109.7, 61.5, 32.8; IR (neat): 3057, 2923, 2852, 1660, 1625, 1613, 1557, 1532, 1488, 1453, 1419, 1392, 1371, 1341, 1318 cm−1; mp: 65–67 °C; HR-MS (ESI): m/z calculated for C30H23N2ClNa 469.1447 obtained 469.1429.
:EA, 9:1); brown solid; 1H NMR (300 MHz, chloroform-d) δ 8.11–8.05 (m, 1H), 7.50–7.43 (m, 2H), 7.38–7.32 (m, 2H), 7.29–7.25 (m, 4H), 7.24–7.17 (m, 7H), 7.07 (d, J = 6.2 Hz, 1H), 6.72 (s, 1H), 6.59 (s, 1H), 6.45 (s, 1H), 3.61 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 147.7, 140.8, 137.5, 137.3, 132.9, 131.7, 131.1 (q, J = 32.0 Hz), 129.2, 128.6, 128.2, 128.1, 127.6, 127.5, 126.9, 126.1, 126.0, 125.0, 124.5, 124.1 (q, J = 272.0 Hz), 121.9, 119.6, 119.2, 118.1 (q, J = 3.8 Hz), 117.9 (q, J = 3.7 Hz), 116.5, 113.8, 109.8, 61.1, 32.9; 19F NMR (376 MHz, chloroform-d) δ −62.77; IR (neat): 2953, 2923, 2854, 1735, 1654, 1607, 1492, 1453, 1372, 1329, 1248 cm−1; mp: 184–186 °C; HR-MS (APCI): m/z calculated for C31H24F3N2 481.1886 obtained 481.1889.
:EA, 9:1); ochre solid; 1H NMR (400 MHz, chloroform-d) δ 8.08–8.01 (m, 1H), 7.61–7.53 (m, 2H), 7.38–7.28 (m, 2H), 7.28–7.15 (m, 8H), 6.80 (s, 1H), 6.68 (s, 1H), 6.64–6.56 (m, 2H), 6.16 (s, 1H), 3.61 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 147.64 (ddd, J = 247.0, 10.5, 2.3 Hz), 146.06 (ddd, J = 251.0, 10.9, 3.6 Hz), 141.76, 139.42 (dd, J = 16.0, 14.5 Hz), 137.14, 136.96, 132.74 (dd, J = 7.7, 3.4 Hz), 132.53, 131.98, 128.52, 128.42, 128.05, 127.60, 127.49, 127.10, 126.38, 126.14, 125.04, 121.60, 119.97, 119.91, 119.38, 116.25, 113.42, 110.97 (dd, J = 17.7, 3.9 Hz), 109.44, 61.33, 32.80; 19F NMR (282 MHz, chloroform-d) δ −139.96 (dd, J = 21.0, 3.6 Hz), −141.36 (dd, J = 20.2, 3.7 Hz), −158.82 (t, J = 20.6 Hz); IR (neat): 3057, 2923, 2853, 1708, 1606, 1562, 1504, 1491, 1422, 1361, 1264, 1220 cm−1; m.p.: 65–67 °C; HR-MS (APCI): m/z calculated for C30H22F3N2 467.1730 obtained 467.1710.
:EA, 9:1); dark oil; 1H NMR (400 MHz, chloroform-d) δ 8.14–8.06 (m, 1H), 7.53–7.40 (m, 2H), 7.32–7.09 (m, 13H), 7.08–6.98 (m, 2H), 6.95 (dd, J = 6.6, 2.9 Hz, 2H), 6.78 (s, 1H), 6.71–6.62 (m, 2H), 6.58 (s, 1H), 6.32 (s, 1H), 5.15 (s, 2H), 3.68 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 154.9, 142.1, 141.2, 138.3, 137.6, 136.8, 132.2, 131.9, 128.7, 128.3, 127.8, 127.8, 127.6, 127.5, 127.3, 126.6, 126.5, 126.4, 126.1, 124.5, 123.8, 121.8, 119.7, 119.6, 118.1, 114.1, 111.4, 110.2, 62.1, 55.5, 50.1; IR (neat): 3058, 2923, 2853, 1726, 1671, 1623, 1606, 1559, 1506, 1464, 1453, 1384, 1357, 1336, 1301, 1244 cm−1; HR-MS (APCI): m/z calculated for C37H31N2O 519.2431 obtained 519.2428.
:EA, 9:1); brown solid; 1H NMR (400 MHz, chloroform-d) δ 7.50 (d, J = 6.6 Hz, 2H), 7.18 (m, 5H), 7.07 (t, J = 7.9 Hz, 1H), 6.99 (d, J = 7.4 Hz, 1H), 6.86 (d, J = 8.9 Hz, 2H), 6.61 (d, J = 8.9 Hz, 2H), 6.42 (s, 1H), 5.85 (s, 1H), 5.81 (s, 1H), 3.66 (s, 3H), 3.35 (s, 3H), 2.41 (s, 3H), 2.05 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 154.7, 142.6, 141.9, 138.7, 133.6, 131.8, 128.2, 128.0, 127.5, 127.3, 126.8, 126.3, 125.6, 124.2, 123.8, 123.0, 121.4, 113.8, 110.0, 104.6, 62.3, 55.5, 30.2, 12.7, 11.0; IR (neat): 3060, 2923, 2854, 1710, 1652, 1600, 1558, 1506, 1492, 1452, 1398, 1361, 1243 cm−1; mp: 78–80 °C; HR-MS (APCI): m/z calculated for C29H29N2O 421.2274 obtained 421.2267.
:EA, 9:1); ochre solid; 1H NMR (400 MHz, chloroform-d) δ 7.52 (d, J = 7.9 Hz, 2H), 7.19 (m, 6H), 7.05 (d, J = 7.4 Hz, 1H), 6.97 (d, J = 8.9 Hz, 2H), 6.70–6.62 (m, 3H), 6.61–6.56 (m, 1H), 5.98 (s, 1H), 5.93–5.87 (m, 1H), 5.65–5.57 (m, 1H), 3.99 (s, 3H), 3.67 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 155.5, 142.1, 141.2, 137.9, 133.4, 132.8, 130.4, 128.4, 127.9, 127.6, 127.4, 126.6, 126.4, 124.6, 124.3, 123.4, 114.2, 112.0, 110.0, 106.5, 62.4, 55.4, 35.3; IR (neat): 3059, 2927, 1710, 1609, 1560, 1507, 1491, 1452, 1405, 1366, 1301, 1244 cm−1; mp: 62–64 °C; HR-MS (APCI): m/z calculated for C27H25N2O 393.1961 obtained 393.1956.
:EA, 9:1); brown solid; 1H NMR (400 MHz, chloroform-d) δ 7.78 (d, J = 8.2 Hz, 1H), 7.61 (d, J = 7.8 Hz, 1H), 7.44–7.29 (m, 5H), 7.20 (td, J = 8.0, 6.8, 3.5 Hz, 6H), 7.11 (td, J = 7.4, 1.5 Hz, 1H), 6.98–6.94 (m, 1H), 6.87–6.81 (m, 2H), 6.62 (d, J = 8.9 Hz, 2H), 6.48 (s, 1H), 3.67 (s, 3H), 2.28–2.17 (m, 3H); 13C NMR (101 MHz, chloroform-d) δ 156.2, 142.6, 140.5, 137.8, 134.5, 131.4, 129.1, 128.6, 128.4, 128.2, 128, 127.9, 126.6, 126.3, 125.8, 124.8, 123.3, 123.3, 122.1, 119.4, 119.2, 114.1, 112.6, 109.5, 107.4, 72.9, 55.4, 9.9; IR (neat): 3055, 2924, 2854, 1709, 1682, 1605, 1561, 1508, 1492, 1454, 1387, 1347, 1296, 1247 cm−1; mp: 52–54 °C; HR-MS (APCI): m/z calculated for C31H27N2O 443.2118 obtained 443.2104.
:EA, 9:1); white solid; 1H NMR (400 MHz, chloroform-d) δ 7.63–7.57 (m, 1H), 7.48 (d, J = 7.9 Hz, 2H), 7.34–7.28 (m, 3H), 7.26–7.18 (m, 9H), 7.01 (t, J = 8.0, 6.9, 1.1 Hz, 2H), 6.61–6.54 (m, 3H), 3.68 (s, 6H).; 13C NMR (101 MHz, chloroform-d) δ 146.5, 137.6, 132.3, 131.7, 128.7, 128.5, 128.4, 128.3, 128.1, 127.8, 126.1, 123.5, 122.8, 121.5, 120.2, 118.8, 117.9, 109.1, 94.0, 88.5, 37.9, 32.8.; mp: 177–179 °C; HR-MS (ESI+): m/z calculated for C33H26N2Na 473.1994 obtained 473.2004.
:EA, 9:1); ochre solid; 1H NMR (300 MHz, DMSO-d6) δ 8.01 (d, J = 7.3 Hz, 1H), 7.47 (d, J = 7.1 Hz, 2H), 7.36 (d, J = 7.6 Hz, 2H), 7.30–7.07 (m, 8H), 7.09 (d, J = 7.2 Hz, 2H), 6.74 (d, J = 7.2 Hz, 2H), 6.72 (s, 1H), 6.61 (s, 1H), 6.34 (s, 1H), 3.62 (s, 6H); 13C NMR (101 MHz, DMSO-d6) δ 154.4, 141.5, 140.6, 137.6, 136.9, 131.8, 131.6, 128.3, 127.8, 127.7, 127.2, 126.4, 126.0, 125.7, 124.4, 123.1, 121.2, 119.4, 118.9, 116.5, 114.5, 114.2, 111.8, 109.9, 61.3, 55.1, 32.3; IR (neat): 3048, 2922, 2853, 1712, 1607, 1558, 1505, 1488, 1463, 1452, 1372, 1330, 1291, 1242 cm−1; mp: 159–161 °C; HR-MS (ESI): m/z calculated for C31H26N2ONa 465.1943 obtained 465.1931.
:EA, 9:1); ochre solid; 1H NMR (400 MHz, chloroform-d) δ 8.16–8.08 (m, 1H), 7.54–7.47 (m, 2H), 7.25 (h, J = 4.7, 3.8 Hz, 4H), 7.16 (dd, J = 9.8, 7.0 Hz, 3H), 7.09–7.01 (m, 2H), 7.00–6.86 (m, 2H), 6.73–6.64 (m, 2H), 6.61 (d, J = 8.3 Hz, 2H), 6.27 (s, 1H), 3.67 (s, 3H), 3.58 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 161.7 (d, J = 245.4 Hz), 155.1, 141.6 (d, J = 2.3 Hz), 141.1, 138.0, 137.3, 133.9 (d, J = 6.4 Hz), 128.5 (d, J = 2.8 Hz), 128.4, 127.9, 127.9, 127.8, 126.1, 125.8 (d, J = 7.9 Hz), 123.8, 121.8, 119.5 (d, J = 11.6 Hz), 116.8, 114.3, 114.2, 114.1, 113.0 (d, J = 21.8 Hz), 110.4, 109.7, 61.9, 55.5, 32.8; 19F NMR (376 MHz, chloroform-d) δ −115.62; IR (neat): 3052, 2925, 2953, 1712, 1660, 1607, 1566, 1539, 1506, 1492, 1464, 1447, 1423, 1371, 1331, 1273, 1242 cm−1; mp: 158–160 °C; HR-MS (ESI): m/z calculated for C31H25N2OFNa 483.1849 obtained 483.1850.
:EA, 9:1); ochre solid; 1H NMR (300 MHz, chloroform-d) δ 8.19–8.10 (m, 1H), 7.57–7.48 (m, 2H), 7.35–7.16 (m, 9H), 7.07 (d, J = 8.2 Hz, 2H), 6.72 (d, J = 8.2 Hz, 2H), 6.66 (s, 1H), 6.61 (s, 1H), 6.30 (s, 1H), 3.73 (s, 3H), 3.66 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 154.8, 141.8, 140.5, 137.4, 137.0, 133.4, 130.9, 130.3, 128.5, 128.1, 127.9, 127.4, 127.2, 126.1, 125.9, 125.7, 123.4, 121.5, 119.5, 119.2, 116.1, 114.3, 110.8, 110.1, 60.8, 55.2, 32.5; IR (neat): 3059, 2925, 2853, 1720, 1609, 1552, 1506, 1479, 1464, 1446, 1371, 1292, 1242 cm−1; mp: 163–165 °C; HR-MS (APCI): m/z calculated for C31H26ClN2O 477.1728 obtained 477.1718.
:EA, 7:3); yellow solid; 1H NMR (300 MHz, chloroform-d) δ 8.17 (s, 1H), 7.51 (s, 2H), 7.31–7.12 (m, 7H), 7.07 (d, J = 8.4 Hz, 2H), 6.84 (d, J = 9.0 Hz, 1H), 6.78–6.66 (m, 3H), 6.63 (s, 1H), 6.58 (s, 1H), 6.26 (s, 1H), 3.77 (s, 3H), 3.69 (s, 3H), 3.60 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 158.7, 154.8, 141.5, 140.1, 138.4, 137.3, 133.8, 128.3, 128.1, 127.6, 127.5, 126.4, 125.7, 125.6, 123.5, 121.6, 119.5, 119.4, 117.1, 114.2, 113.2, 111.6, 111.5, 109.6, 62.2, 55.5, 55.4, 32.8; IR (neat): 3054, 2925, 2852, 1712, 1607, 1559, 1506, 1463, 1446, 1423, 1371, 1319, 1285, 1243 cm−1; mp: 164–166 °C; HR-MS (ESI): m/z calculated for C32H28N2O2Na 495.2048 obtained 495.2028.
:EA, 7:3); ochre solid; 1H NMR (300 MHz, chloroform-d) δ 8.13 (d, J = 5.4 Hz, 1H), 7.51 (d, J = 7.3 Hz, 2H), 7.26 (s, 3H), 7.16 (q, J = 8.0, 7.3 Hz, 3H), 7.06 (d, J = 8.5 Hz, 2H), 6.83 (s, 1H), 6.70 (d, J = 8.5 Hz, 2H), 6.62 (s, 1H), 6.54 (s, 1H), 6.21 (s, 1H), 4.04 (s, 3H), 3.91 (s, 3H), 3.80 (s, 3H), 3.69 (s, 3H), 3.62 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 154.8, 152.4, 148.5, 141.4, 141.4, 141.1, 140.1, 138.4, 137.3, 128.3, 128.2, 128.1, 127.6, 127.5, 126.3, 123.4, 121.6, 119.4, 119.4, 117.1, 114.2, 109.7, 106.6, 105.5, 61.8, 61.6, 61.1, 56.1, 55.5, 32.9; IR (neat): 3053, 2926, 2853, 1729, 1712, 1595, 1561, 1505, 1486, 1459, 1409, 1370, 1328, 1299 cm−1; mp: 68–70 °C; HR-MS (APCI): m/z calculated for C34H33N2O4 533.2435 obtained 533.2430.
:EA, 7:3); yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 8.39 (d, J = 4.0 Hz, 1H), 7.99 (d, J = 7.8 Hz, 1H), 7.66 (d, J = 7.3 Hz, 1H), 7.49 (d, J = 6.6 Hz, 2H), 7.39 (d, J = 8.1 Hz, 1H), 7.29–7.07 (m, 6H), 6.97 (d, J = 8.8 Hz, 2H), 6.87 (s, 1H), 6.74 (d, J = 8.8 Hz, 2H), 6.64 (s, 1H), 6.44 (s, 1H), 3.65 (s, 3H), 3.62 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 154.9, 150.6, 148.1, 145.8, 140.2, 137.1, 136.9, 133.3, 128.4, 127.7, 127.4, 126.2, 125.3, 123.9, 121.4, 121.1, 119.3, 119.1, 116.3, 114.1, 110.9, 110.0, 79.2, 61.2, 55.1, 32.4; IR (neat): 3055, 2925, 2852, 1711, 1602, 1575, 1557, 1507, 1461, 1437, 1424, 1372, 1243 cm−1; mp: 168–170 °C; HR-MS (APCI): m/z calculated for C30H26N3O 444.2070 obtained 444.2072.
:EA, 9:1); ochre solid; 1H NMR (300 MHz, acetone-d6) δ 8.18–8.12 (m, 1H), 7.47 (d, J = 8.8 Hz, 2H), 7.35–7.32 (m, 1H), 7.31 (t, J = 1.4 Hz, 1H), 7.25 (dq, J = 5.9, 1.5 Hz, 2H), 7.19–7.13 (m, 3H), 7.08 (d, J = 9.0 Hz, 2H), 6.72 (dd, J = 8.9, 4.4 Hz, 4H), 6.66 (s, 1H), 6.61 (d, J = 0.9 Hz, 1H), 6.35 (s, 1H), 3.70 (s, 3H), 3.66 (s, 3H), 3.63 (s, 3H); 13C NMR (75 MHz, acetone-d6) δ 160.4, 155.9, 142.6, 142.2, 138.3, 133.6, 132.7, 131.4, 129.7, 128.8, 128.0, 127.3, 126.8, 126.7, 125.0, 124.5, 122.1, 120.4, 119.8, 117.9, 114.8, 114.4, 111.3, 110.5, 63.0, 55.5, 55.4, 32.7; IR (neat): 3050, 2954, 2925, 2854, 1714, 1606, 1577, 1559, 1505, 1483, 1463, 1451, 1441, 1424, 1371, 1328, 1289, 1242 cm−1; mp: 44–46 °C; HR-MS (APCI): m/z calculated for C32H29N2O2 473.2224 obtained 473.2214.
:EA, 9:1); ochre solid; 1H NMR (300 MHz, chloroform-d) δ 8.01 (d, J = 7.8 Hz, 1H), 7.67 (s, 2H), 7.41–7.19 (m, 4H), 7.13 (s, 1H), 7.05 (s, 1H), 6.98 (d, J = 8.8 Hz, 2H), 6.91 (s, 1H), 6.85 (s, 2H), 6.62 (d, J = 8.9 Hz, 2H), 6.26 (s, 1H), 6.14 (s, 1H), 3.67 (s, 3H), 3.65 (s, 3H), 2.15 (s, 6H). We observed a rapid degradation of this compound so no 13C NMR could be undertaken.
:EA, 9:1); yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 8.03 (d, J = 7.8 Hz, 1H), 7.35 (d, J = 7.8 Hz, 2H), 7.31 (d, J = 5.2 Hz, 1H), 7.29–7.24 (m, 2H), 7.20 (d, J = 7.1 Hz, 1H), 7.18–7.12 (m, 2H), 7.07 (d, J = 9.1 Hz, 3H), 6.89–6.86 (m, 1H), 6.79 (d, J = 7.2 Hz, 3H), 6.47 (s, 1H), 6.24 (s, 1H), 3.65 (s, 3H), 3.59 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 154.8, 142.2, 140.6, 137.0, 135.5, 131.7, 131.3, 127.8, 127.4, 127.3, 126.4, 126.2, 126.2, 125.9, 125.4, 124.4, 123.2, 121.2, 119.7, 118.7, 115.9, 114.3, 111.3, 109.8, 61.9, 55.1, 32.3; IR (neat): 3065, 2926, 2854, 1713, 1604, 1560, 1530, 1505, 1482, 1464, 1440, 1425, 1372, 1351, 1331, 1289, 1243 cm−1; mp: 39–41 °C; HR-MS (ESI): m/z calculated for C29H24N2OSNa 471.1507 obtained 471.1500.
:EA, 9:1); ochre solid; 1H NMR (400 MHz, chloroform-d) δ 8.19 (d, J = 7.9 Hz, 1H), 7.42–7.29 (m, 5H), 7.25 (m, 4H), 6.93 (d, J = 8.9 Hz, 2H), 6.64 (s, 1H), 6.49 (s, 1H), 6.32 (s, 1H), 6.28 (brs, 1H), 3.91 (s, 3H), 3.75 (s, 3H), 2.35–2.25 (m, 1H), 2.23–2.07 (m, 3H), 1.71–1.55 (m, 4H); 13C NMR (101 MHz, chloroform-d) δ 154.8, 144.3, 142.3, 137.4, 134.1, 132.6, 131.7, 129.5, 128.1, 127.1, 126.5, 126.1, 125.8, 124.3, 123.0, 121.5, 119.9, 119.1, 117.4, 114.0, 109.5, 109.1, 62.2, 55.6, 32.8, 26.7, 25.9, 22.8, 22.2; IR (neat): 2928, 2832, 1711, 1601, 1557, 1505, 1483, 1464, 1371, 1241 cm−1; mp: 91–93 °C; HR-MS (APCI): m/z calculated for C31H31N2O 447.2431 obtained 447.2425.
:EA, 9:1); ochre solid; 1H NMR (300 MHz, chloroform-d) δ 8.14–8.07 (m, 1H), 7.50 (dd, J = 7.7, 2.0 Hz, 2H), 7.36–7.18 (m, 10H), 7.13–7.03 (m, 4H), 6.70 (s, 1H), 6.59 (s, 1H), 6.40 (s, 1H), 3.59 (s, 3H); 13C NMR (75 MHz, chloroform-d) δ 146.0, 141.1, 137.7, 137.3, 132.6, 131.9, 128.8, 128.5, 128.1, 128.0, 127.6, 127.5, 126.8, 126.7, 126.1, 126.0, 124.8, 123.0, 121.8, 119.5, 119.3, 116.7, 113.1, 109.7, 61.5, 32.8; IR (neat): 3057, 2923, 2852, 1660, 1625, 1613, 1557, 1532, 1488, 1453, 1419, 1392, 1371, 1341, 1318 cm−1; mp: 65–67 °C; HR-MS (ESI): m/z calculated for C30H23N2ClNa 469.1447 obtained 469.1429.
:EA, 9:1); brown solid; 1H NMR (300 MHz, chloroform-d) δ 8.11–8.05 (m, 1H), 7.50–7.43 (m, 2H), 7.38–7.32 (m, 2H), 7.29–7.25 (m, 4H), 7.24–7.17 (m, 7H), 7.07 (d, J = 6.2 Hz, 1H), 6.72 (s, 1H), 6.59 (s, 1H), 6.45 (s, 1H), 3.61 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 147.7, 140.8, 137.5, 137.3, 132.9, 131.7, 131.1 (q, J = 32.0 Hz), 129.2, 128.6, 128.2, 128.1, 127.6, 127.5, 126.9, 126.1, 126.0, 125.0, 124.5, 124.1 (q, J = 272.0 Hz), 121.9, 119.6, 119.2, 118.1 (q, J = 3.8 Hz), 117.9 (q, J = 3.7 Hz), 116.5, 113.8, 109.8, 61.1, 32.9; 19F NMR (376 MHz, chloroform-d) δ −62.77; IR (neat): 2953, 2923, 2854, 1735, 1654, 1607, 1492, 1453, 1372, 1329, 1248 cm−1; mp: 184–186 °C; HR-MS (APCI): m/z calculated for C31H24F3N2 481.1886 obtained 481.1889.
:EA, 9:1); ochre solid; 1H NMR (400 MHz, chloroform-d) δ 8.08–8.01 (m, 1H), 7.61–7.53 (m, 2H), 7.38–7.28 (m, 2H), 7.28–7.15 (m, 8H), 6.80 (s, 1H), 6.68 (s, 1H), 6.64–6.56 (m, 2H), 6.16 (s, 1H), 3.61 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 147.64 (ddd, J = 247.0, 10.5, 2.3 Hz), 146.06 (ddd, J = 251.0, 10.9, 3.6 Hz), 141.76, 139.42 (dd, J = 16.0, 14.5 Hz), 137.14, 136.96, 132.74 (dd, J = 7.7, 3.4 Hz), 132.53, 131.98, 128.52, 128.42, 128.05, 127.60, 127.49, 127.10, 126.38, 126.14, 125.04, 121.60, 119.97, 119.91, 119.38, 116.25, 113.42, 110.97 (dd, J = 17.7, 3.9 Hz), 109.44, 61.33, 32.80; 19F NMR (282 MHz, chloroform-d) δ −139.96 (dd, J = 21.0, 3.6 Hz), −141.36 (dd, J = 20.2, 3.7 Hz), −158.82 (t, J = 20.6 Hz); IR (neat): 3057, 2923, 2853, 1708, 1606, 1562, 1504, 1491, 1422, 1361, 1264, 1220 cm−1; m.p.: 65–67 °C; HR-MS (APCI): m/z calculated for C30H22F3N2 467.1730 obtained 467.1710.
:EA, 9:1); dark oil; 1H NMR (400 MHz, chloroform-d) δ 8.14–8.06 (m, 1H), 7.53–7.40 (m, 2H), 7.32–7.09 (m, 13H), 7.08–6.98 (m, 2H), 6.95 (dd, J = 6.6, 2.9 Hz, 2H), 6.78 (s, 1H), 6.71–6.62 (m, 2H), 6.58 (s, 1H), 6.32 (s, 1H), 5.15 (s, 2H), 3.68 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 154.9, 142.1, 141.2, 138.3, 137.6, 136.8, 132.2, 131.9, 128.7, 128.3, 127.8, 127.8, 127.6, 127.5, 127.3, 126.6, 126.5, 126.4, 126.1, 124.5, 123.8, 121.8, 119.7, 119.6, 118.1, 114.1, 111.4, 110.2, 62.1, 55.5, 50.1; IR (neat): 3058, 2923, 2853, 1726, 1671, 1623, 1606, 1559, 1506, 1464, 1453, 1384, 1357, 1336, 1301, 1244 cm−1; HR-MS (APCI): m/z calculated for C37H31N2O 519.2431 obtained 519.2428.
:EA, 9:1); brown solid; 1H NMR (400 MHz, chloroform-d) δ 7.50 (d, J = 6.6 Hz, 2H), 7.18 (m, 5H), 7.07 (t, J = 7.9 Hz, 1H), 6.99 (d, J = 7.4 Hz, 1H), 6.86 (d, J = 8.9 Hz, 2H), 6.61 (d, J = 8.9 Hz, 2H), 6.42 (s, 1H), 5.85 (s, 1H), 5.81 (s, 1H), 3.66 (s, 3H), 3.35 (s, 3H), 2.41 (s, 3H), 2.05 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 154.7, 142.6, 141.9, 138.7, 133.6, 131.8, 128.2, 128.0, 127.5, 127.3, 126.8, 126.3, 125.6, 124.2, 123.8, 123.0, 121.4, 113.8, 110.0, 104.6, 62.3, 55.5, 30.2, 12.7, 11.0; IR (neat): 3060, 2923, 2854, 1710, 1652, 1600, 1558, 1506, 1492, 1452, 1398, 1361, 1243 cm−1; mp: 78–80 °C; HR-MS (APCI): m/z calculated for C29H29N2O 421.2274 obtained 421.2267.
:EA, 9:1); ochre solid; 1H NMR (400 MHz, chloroform-d) δ 7.52 (d, J = 7.9 Hz, 2H), 7.19 (m, 6H), 7.05 (d, J = 7.4 Hz, 1H), 6.97 (d, J = 8.9 Hz, 2H), 6.70–6.62 (m, 3H), 6.61–6.56 (m, 1H), 5.98 (s, 1H), 5.93–5.87 (m, 1H), 5.65–5.57 (m, 1H), 3.99 (s, 3H), 3.67 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 155.5, 142.1, 141.2, 137.9, 133.4, 132.8, 130.4, 128.4, 127.9, 127.6, 127.4, 126.6, 126.4, 124.6, 124.3, 123.4, 114.2, 112.0, 110.0, 106.5, 62.4, 55.4, 35.3; IR (neat): 3059, 2927, 1710, 1609, 1560, 1507, 1491, 1452, 1405, 1366, 1301, 1244 cm−1; mp: 62–64 °C; HR-MS (APCI): m/z calculated for C27H25N2O 393.1961 obtained 393.1956.
:EA, 9:1); brown solid; 1H NMR (400 MHz, chloroform-d) δ 7.78 (d, J = 8.2 Hz, 1H), 7.61 (d, J = 7.8 Hz, 1H), 7.44–7.29 (m, 5H), 7.20 (td, J = 8.0, 6.8, 3.5 Hz, 6H), 7.11 (td, J = 7.4, 1.5 Hz, 1H), 6.98–6.94 (m, 1H), 6.87–6.81 (m, 2H), 6.62 (d, J = 8.9 Hz, 2H), 6.48 (s, 1H), 3.67 (s, 3H), 2.28–2.17 (m, 3H); 13C NMR (101 MHz, chloroform-d) δ 156.2, 142.6, 140.5, 137.8, 134.5, 131.4, 129.1, 128.6, 128.4, 128.2, 128, 127.9, 126.6, 126.3, 125.8, 124.8, 123.3, 123.3, 122.1, 119.4, 119.2, 114.1, 112.6, 109.5, 107.4, 72.9, 55.4, 9.9; IR (neat): 3055, 2924, 2854, 1709, 1682, 1605, 1561, 1508, 1492, 1454, 1387, 1347, 1296, 1247 cm−1; mp: 52–54 °C; HR-MS (APCI): m/z calculated for C31H27N2O 443.2118 obtained 443.2104.
:EA, 9:1); white solid; 1H NMR (400 MHz, chloroform-d) δ 7.63–7.57 (m, 1H), 7.48 (d, J = 7.9 Hz, 2H), 7.34–7.28 (m, 3H), 7.26–7.18 (m, 9H), 7.01 (t, J = 8.0, 6.9, 1.1 Hz, 2H), 6.61–6.54 (m, 3H), 3.68 (s, 6H).; 13C NMR (101 MHz, chloroform-d) δ 146.5, 137.6, 132.3, 131.7, 128.7, 128.5, 128.4, 128.3, 128.1, 127.8, 126.1, 123.5, 122.8, 121.5, 120.2, 118.8, 117.9, 109.1, 94.0, 88.5, 37.9, 32.8.; mp: 177–179 °C; HR-MS (ESI+): m/z calculated for C33H26N2Na 473.1994 obtained 473.2004.
General procedure for the formation of 2k–D
To a solution of 2k in CDCl3 was added an excess of AcOD. An aliquot of this mixture was immediately taken for NMR analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a joint Centre National de la Recherche Scientifique (CNRS) and INCa fellowship (ATIP) to P. B. and the University Côte d'Azur. Y. Tang is grateful to Agence Nationale de Recherche (ANR-16-CE07-0006) for a PhD grant. M. De Abreu thanks MESR for funding. We would like to thank Dr R. Guillot from single-crystal X-ray diffraction service ICMMO-UMR 8182 Orsay. Special thanks are extended to the team P.N.A.S. (UMR-CNRS 8038).References
- E. Bangha, H. I. Maibach and P. Elsner, Skin Pharmacol. Physiol., 1996, 9, 376–380 CrossRef CAS.
- N. F. Kassell, G. Helm, N. Simmons, C. D. Phillips and W. S. Cail, J. Neurosurg., 1992, 77, 848–852 CAS.
- Y.-H. Zhao, Y. Yu, D. Hu, L. Zhao, W. Xie and Z. Zhou, Asian J. Org. Chem., 2020, 9, 953–960 CrossRef CAS.
- Y.-M. Ma, Y.-B. Zhou, C.-M. Xie, D.-M. Chen and J. Li, Acta Pharmacol. Sin., 2012, 33, 407–417 CrossRef CAS.
- A. Bischler and B. Napieralski, Ber. Dtsch. Chem. Ges., 1893, 26, 1903–1908 CrossRef.
- C. Pomeranz, Monatsh. Chem. Verw. Teile Anderer Wiss., 1893, 14, 116–119 CrossRef.
- P. Fritsch, Ber. Dtsch. Chem. Ges., 1893, 26, 419–422 CrossRef.
- A. Pictet and T. Spengler, Ber. Dtsch. Chem. Ges., 1911, 44, 2030–2036 CrossRef CAS.
- K. R. Roesch and R. C. Larock, J. Org. Chem., 1998, 63, 5306–5307 CrossRef CAS.
- G. Dai and R. C. Larock, Org. Lett., 2001, 3, 4035–4038 CrossRef CAS.
- G. Dai and R. C. Larock, Org. Lett., 2002, 4, 193–196 CrossRef CAS.
- Q. Huang and R. C. Larock, Tetrahedron Lett., 2002, 43, 3557–3560 CrossRef CAS.
- S. Ye and J. Wu, Tetrahedron Lett., 2009, 50, 6273–6275 CrossRef CAS.
- W. Sun, Q. Ding, X. Sun, R. Fan and J. Wu, J. Comb. Chem., 2007, 9, 690–694 CrossRef CAS.
- H. Lou, S. Ye, J. Zhang and J. Wu, Tetrahedron, 2011, 67, 2060–2065 CrossRef CAS.
- Q. Ding and J. Wu, Org. Lett., 2007, 9, 4959–4962 CrossRef CAS.
- X. Yu and J. Wu, J. Comb. Chem., 2010, 12, 238–244 CrossRef CAS.
- X. Wang, G. Qiu, L. Zhang and J. Wu, Tetrahedron Lett., 2014, 55, 962–964 CrossRef CAS.
- T. P. Singh and O. M. Singh, Mini-Rev. Med. Chem., 2018, 18, 9–25 CrossRef CAS.
- V. Bhardwaj, D. Gumber, V. Abbot, S. Dhiman and P. Sharma, RSC Adv., 2015, 5, 15233–15266 RSC.
- Q. Huang, J. A. Hunter and R. C. Larock, J. Org. Chem., 2002, 67, 3437–3444 CrossRef CAS.
- H. Wang, X. Han and X. Lu, Chin. J. Chem., 2011, 29, 2611–2618 CrossRef CAS.
- U. Urvashi, G. K. Rastogi, S. K. Ginotra, A. Agarwal and V. Tandon, Org. Biomol. Chem., 2015, 13, 1000–1007 RSC.
- T. Godet, C. Vaxelaire, C. Michel, A. Milet and P. Belmont, Chem. – Eur. J., 2007, 13, 5632–5641 CrossRef CAS.
- E. Parker, N. Leconte, T. Godet and P. Belmont, Chem. Commun., 2011, 47, 343–345 RSC.
- G. Mariaule, G. Newsome, P. Y. Toullec, P. Belmont and V. Michelet, Org. Lett., 2014, 16, 4570–4573 CrossRef CAS.
- A. Bontemps, G. Mariaule, S. Desbène-Finck, P. Helissey, S. Giorgi-Renault, V. Michelet and P. Belmont, Synthesis, 2016, 2178–2190 CAS.
- X. Bantreil, A. Bourderioux, P. Mateo, C. E. Hagerman, M. Selkti, E. Brachet and P. Belmont, Org. Lett., 2016, 18, 4814–4817 CrossRef CAS.
- J. E. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 18, 734–736 RSC.
- X. Wang, G. Qiu, L. Zhang and J. Wu, Tetrahedron Lett., 2014, 55, 962–964 CrossRef CAS.
- For recent selected examples: (a) I. Alonso, J. Esquivias, R. Gomez-Arrayas and J. C. Carretero, J. Org. Chem., 2008, 73, 6400–6404 Search PubMed; (b) H. Lin and X.-W. Sun, Tetrahedron Lett., 2008, 49, 5343–5346 CrossRef CAS; (c) D. Enders, A. Narine, F. Toulgoat and T. Bisschops, Angew. Chem., Int. Ed., 2008, 47, 5661–5665 CrossRef CAS.
- H. Mayr and A. R. Ofial, Tetrahedron Lett., 1997, 38, 3503–3506 CrossRef CAS.
- H. Mayr, T. Bug, M. F. Gotta, N. Hering, B. Irrgang, B. Janker, B. Kempf, R. Loos, A. R. Ofial, G. Remennikov and H. Schimmel, J. Am. Chem. Soc., 2001, 123, 9500–9512 CrossRef CAS.
- S. Lakhdar, M. Westermaier, F. Terrier, R. Goumont, T. Boubaker, A. R. Ofial and H. Mayr, J. Org. Chem., 2006, 71, 9088–9095 CrossRef CAS.
- T. A. Nigst, M. Westermaier, A. R. Ofial and H. Mayr, Eur. J. Org. Chem., 2008, 2369–2374 CrossRef CAS.
- M. Baidya, F. Brotzel and H. Mayr, Org. Biomol. Chem., 2010, 8, 1929 RSC.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, 96th edn, CRC Press, 2015 Search PubMed.
- pKa Plugin, Marvin 20.11.0, 2020, ChemAxon ( https://chemaxon.com).
- P. Hazarika, S. Das Sharma and D. Konwar, Synth. Commun., 2008, 38, 2870–2880 CrossRef CAS.
- N. Asao, S. S. Yudha, T. Nogami and Y. Yamamoto, Angew. Chem., Int. Ed., 2005, 44, 5526–5528 CrossRef CAS; N. Asao, S. S. Yudha, T. Nogami and Y. Yamamoto, Heterocycles, 2008, 76, 471–483 CrossRef.
Footnotes |
| † This paper is dedicated to professor Jean Lhomme. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2040640 and 2042160. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ob02197k |
| This journal is © The Royal Society of Chemistry 2021 |