
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in the synthesis of α-amino ketones
Lewis A. T.
Allen
 a,
Robert-Cristian
Raclea
b,
Philipp
Natho
a and
Philip J.
Parsons
*a
a,
Robert-Cristian
Raclea
b,
Philipp
Natho
a and
Philip J.
Parsons
*a
aDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, W12 0BZ, London, UK. E-mail: p.parsons@imperial.ac.uk
bMassachusetts Institute of Technology, Department of Chemistry, 77, Massachusetts Avenue, Cambridge, MA 02139-4307, USA
First published on 8th December 2020
Abstract
Due to the importance of the amino ketone motif in synthetic and medicinal chemistry, the number of protocols developed in recent years has considerably increased. This review serves to collate and critically evaluate novel methodologies published since 2011 towards this high value synthon. The chapters are divided by the requisite functionality in the starting material, and an emphasis is placed on discussing functional group compatibility and resultant product substitution patterns. Throughout, applications to medicinal targets are highlighted and mechanistic details are presented, and we further provide a short outlook for future development and emerging potential within this area.
 Lewis A. T. Allen | Lewis Allen is a postdoctoral research scientist based at Imperial College London. Lewis received his PhD from Imperial College London in 2015 after completing studies on the total synthesis of aphidicolin under the guidance of Prof. Philip Parsons. Lewis was then employed as a senior research scientist at Pareon Chemicals Ltd before becoming an associate principal scientist. Lewis returned to Imperial College London to investigate the synthesis of novel analgesics and cognitive enhancers based on truncated natural products. |
 Robert-Cristian Raclea | Robert-Cristian Raclea is a PhD student at the Massachusetts Institute of Technology. He received his MSci degree from Imperial College London in 2020 after completing his final year research project with Prof. Philip Parsons on the oxidative deconstruction of azetidinols. Robert has also previously conducted synthetic and medicinal chemistry research in the academic groups of Prof. Movassaghi, Prof. Hoelder, Dr Cordier, and Prof. Dinica. |
 Philipp Natho | Philipp Natho is a PhD student at Imperial College London working under the supervision of Prof. Philip Parsons. He received his M.Sci in Chemistry from Imperial College London where he investigated the total synthesis of morphine under the supervision of Prof. Don Craig. He also worked on the synthesis of highly substituted pyridines under the supervision of Prof. Rick Danheiser at the Massachusetts Institute of Technology. Philipp's current work in the Parsons group is focused on the ring expansion of cyclobutanols to tetralones under transition-metal-free conditions and the application of this technology to natural product synthesis. |
 Philip J. Parsons | Prof. Philip J. Parsons started his independent research career at the University of Southampton in 1979, after completing postdoctoral studies with Prof. Gilbert Stork at Columbia University. While at the University of Southampton, Prof. Parsons cofounded Cookson Chemicals Ltd, now Tocris Bioscience a brand of Bio-Techne. In 1990, he was appointed Professor and Head of Organic Chemistry at the University of Reading. He later moved to the University of Sussex in 1995 where he was appointed Dean of the School of Chemistry, Physics, and Environmental Science. Recently, he was appointed Senior Research Investigator and now Professor at Imperial College London. |
1. Introduction
The versatility and importance of α-amino ketones for widespread applications across synthetic and medicinal chemistry have long been appreciated (Scheme 1).1–6 Indeed, the large range of modulatory activities that α-amino ketones exhibit on important biological receptors has not only led to the common recreational use of naturally occurring examples (e.g. (−)-cathinone in Khat leaves),7 but has also enabled the discovery of vital pharmaceutical compounds, such as bupropion (antidepressant drug),8 amfepramone (appetite suppressant drug),9 keto-ACE (ACE-inhibitor for the treatment of hypertension)10,11 or Effient (antiplatelet drug) through careful structure–activity relationship studies. Moreover, these bifunctional substrates serve as indispensable building blocks for the synthesis of common heterocycles,12–15 such as pyrazines16,17 and pyrroles,18,19 and the construction of chiral 1,2-amino alcohols,20–22 which are extensively used in synthetic chemistry as ligands and chiral auxiliaries.23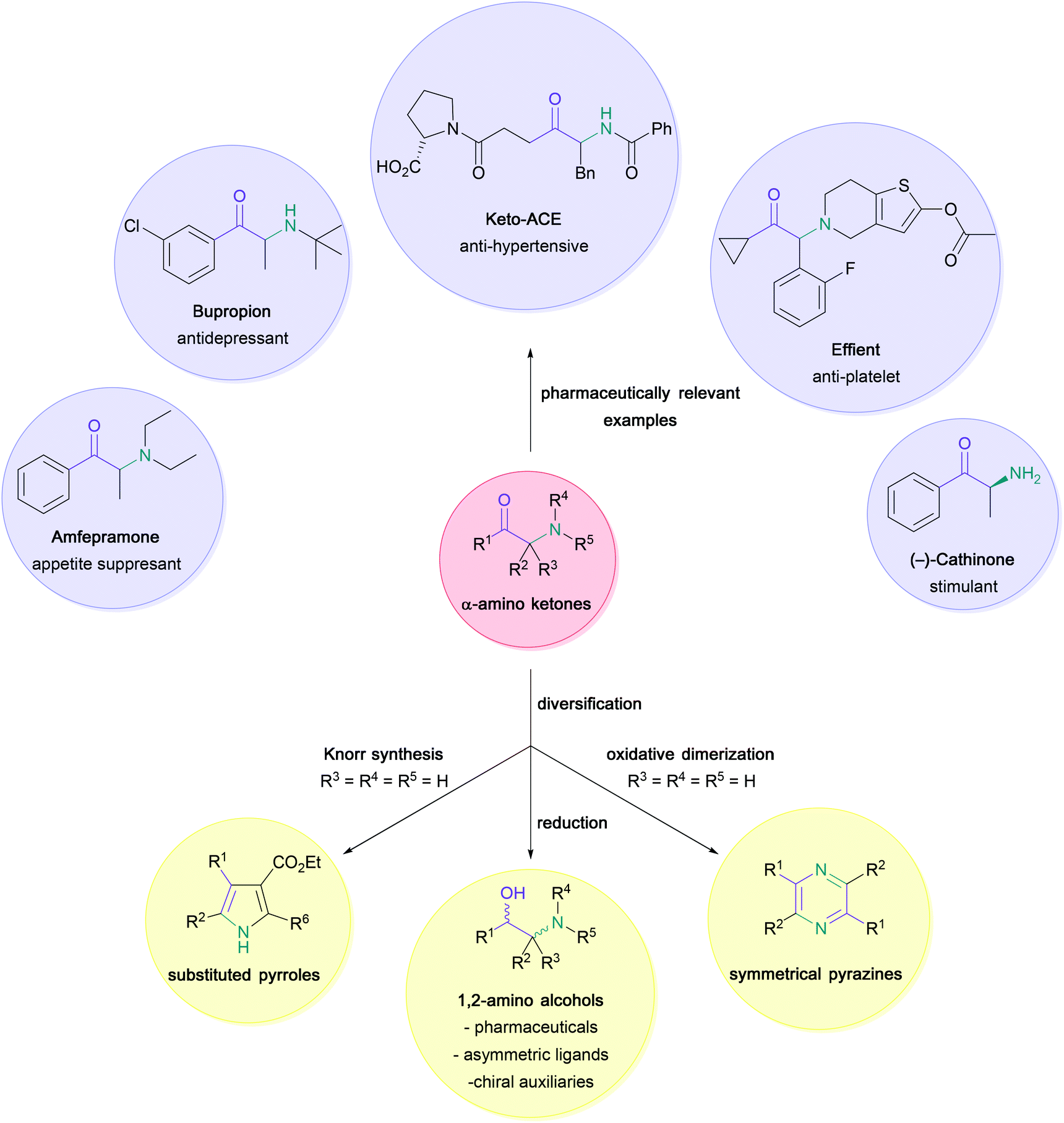 | ||
| Scheme 1 Importance of α-amino ketones in synthetic and medicinal chemistry. | ||
In view of their remarkable impact on several areas of organic chemistry, a large number of synthetic studies have focused on the construction of these high-value synthons. Classically, these processes rely on the nucleophilic substitution of an α-halogenated ketone (1) with an amine (2) or azide (4) (Scheme 2, 1),24 or the electrophilic amination of enolates (6) with suitable N-based electrophiles, such as haloamines (7), hydroxylamines (8) or azo compounds (10) (Scheme 2, 2).25–31 However, these methods often face several challenges, including the scarce availability of nitrogen electrophiles, the obtainment of racemic products, or the requirement for additional steps either through prefunctionalisation of starting materials (e.g. regioselective halogenation of ketones), or subsequent chemoselective reduction of nitrogen-containing precursors (e.g. azides, hydrazinyls, or hydroxylamines) to amines, often under harsh reaction conditions.
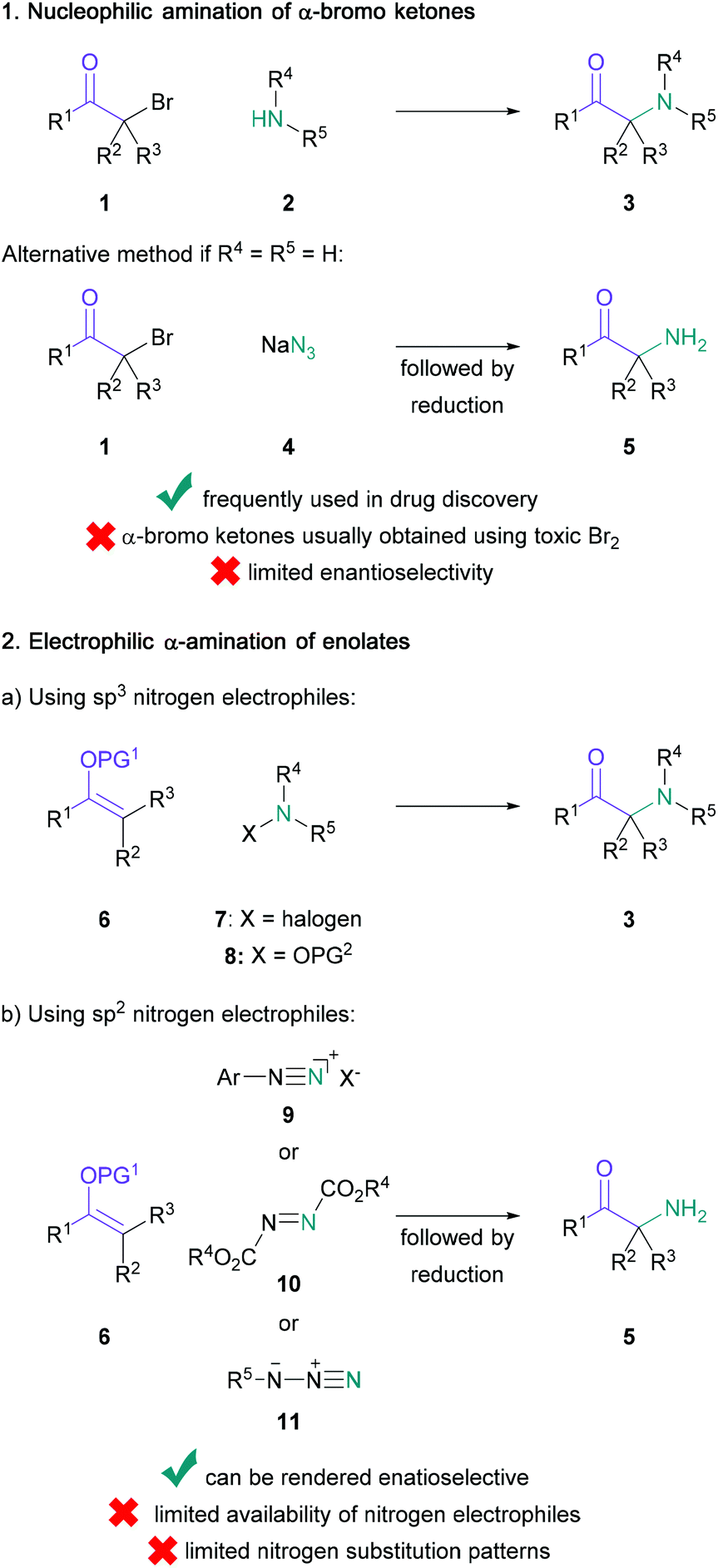 | ||
| Scheme 2 1) Synthesis of α-amino ketones by nucleophilic substitution at the carbonyl α-center; (2) synthesis of α-amino ketones by electrophilic amination of enolates. | ||
A key factor that has influenced the recent surge in the development of new protocols for the synthesis of α-amino ketones has been the recognition that improved atom economy, product enantioselectivity, functional group compatibility, and step count can facilitate the exploration of compounds with benefits to society.32–34 The aim of this review is to collate and critically discuss newly developed methodologies for the synthesis of α-amino ketones starting from 2011. Significant and relevant protocols that construct these scaffolds without the requirement for further functional group manipulation will be introduced. Categorised by the key functional groups present in the starting material, this review has been structured into eight sections, as follows:
When discussing these methodologies, an emphasis is placed on the synthetic utility, novel bond disconnections and the extent to which some of the previously mentioned drawbacks are addressed, providing the reader with valuable insights into the current state of the ‘art’ and varied approaches towards of α-amino ketones.
2. Discussion
2.1. Ketones
Classical syntheses of α-amino ketones have principally relied on the nucleophilic substitution of α-haloketones. As the generation of these starting materials has significant drawbacks, such as increased step count and the handling of toxic and corrosive bromine, it is desirable to achieve direct amination of cheap and readily available ketones within a single synthetic operation. Examples of direct aminations were independently developed by Bella35 and Liang36 in 2012. Bella used electrophilic N-chloropyrrolidine and N-chloropiperidine to obtain the amino ketones in 22–63% yield, with the pyrrolidine or piperidine unit incorporated in the final product. Liang demonstrated that N-bromosuccinimide (NBS) could serve as both the electrophilic brominating agent and the nucleophilic nitrogen source. In Liang's net transformation, the succinimide moiety was added to aromatic ketones (12) in up to 91% yield (Scheme 3). Kumar later developed a related NBS-mediated synthesis with cyclic amines, allowing the efficient synthesis of α-arylated amino ketones.37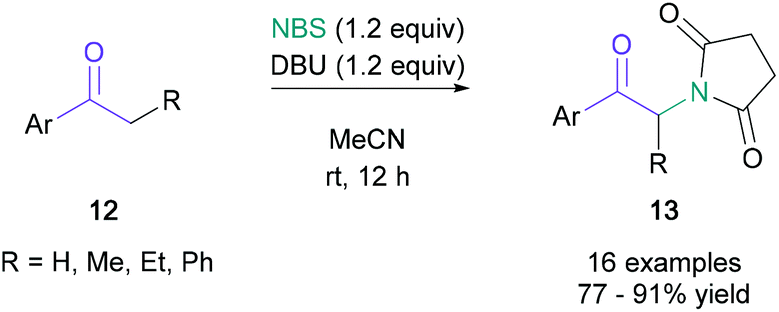 | ||
| Scheme 3 Liang's α-amino ketone synthesis through a dual role of NBS. | ||
The quantity of the halogen source may be reduced by using a stoichiometric peroxide-based oxidant. In 2012, Lamani reported that aryl/heteroaryl-ketones (14) with aliphatic or aromatic substituents on the α-position were successfully aminated with cyclic secondary amines (15) in up to 80% yield when treated with N-iodosuccinimide (30 mol%) and tert-butyl hydroperoxide (Scheme 4).38 The use of the methodology was demonstrated by the synthesis of an ifenprodil derivative (17) in 50% yield from commercially available starting materials.
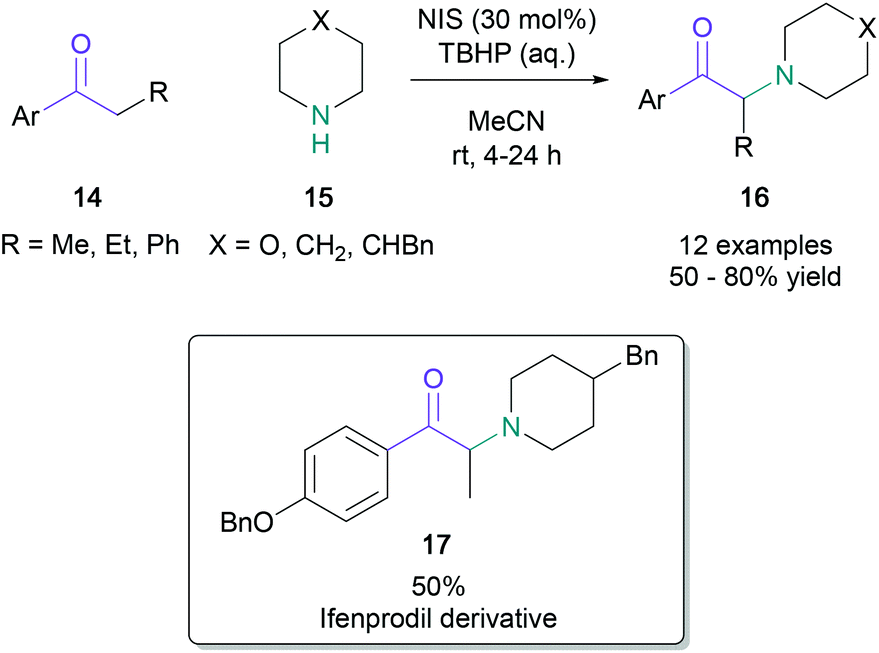 | ||
| Scheme 4 Lamani's α-amino ketone synthesis utilising a stoichiometric co-oxidant. | ||
In a conceptually related contribution, Guo showed that the substrate scope could be extended to include primary and acyclic secondary amines (19) when employing ammonium iodide (15 mol%) and sodium percarbonate (Scheme 5).39 Notably, amines containing reactive functional groups, such as allyl (23) or aniline (24), were tolerated in this methodology. The utility of the procedure was demonstrated by the synthesis of amfepramone in 72% yield. Upon investigating the mechanism, the authors indicated that an α-iodo intermediate was unlikely; with the support of a series of control experiments, the intermediacy of an α-radical (26) was suggested, followed by oxidation to the α-cation (27) (Scheme 6). This mechanistic suggestion contrasts with earlier halogen-mediated pathways. In the same year, Zhang and co-workers employed a combination of tert-butyl ammonium iodide and tert-butyl hydrogen peroxide to aminate ketones with imides, such as phthalimide, saccharin, and succinimide.40
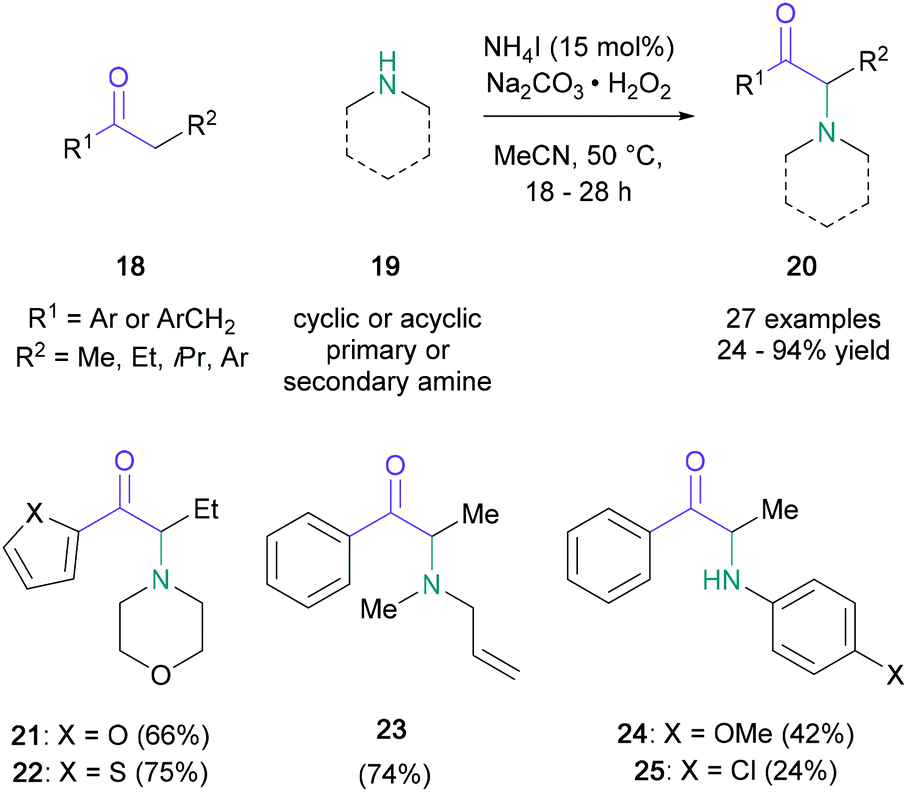 | ||
| Scheme 5 Guo's α-amino ketone synthesis with ammonium iodide and sodium percarbonate. | ||
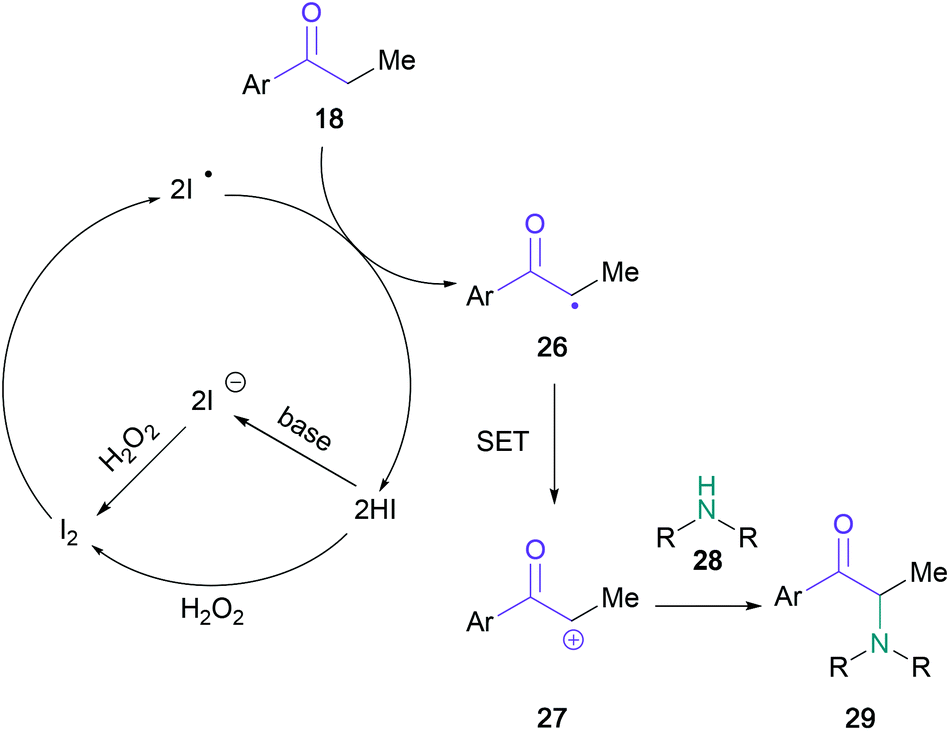 | ||
| Scheme 6 Proposed mechanism for the ammonium iodide-mediated α-amino ketone synthesis by Guo. | ||
With the foresight to recognise the potential of a direct amination amenable to a broad substrate scope, MacMillan disclosed a general procedure utilising copper(II) bromide (0.1 equiv.) as the brominating agent and air as the oxidant (Scheme 7).41 Although the initial studies focused on the reaction of propiophenone and morpholine, the direct amination was found to be suitable for aliphatic ketones (30) and several cyclic and acyclic secondary amines (31). Further modifications of the standard conditions, e.g. elevated reaction temperatures for substrates containing electron-rich π-systems (34), or the addition of Lewis acids for aliphatic ketones to facilitate the enolization event (36), enabled the successful conversion of otherwise challenging substrates. The utility and generality of the procedure was demonstrated with substrates usually susceptible to 1,2-elimination events (35), and the one-step synthesis of the appetite suppressant amfepramone in 80% yield, which is comparable Guo's peroxide-mediated synthesis (72%). In 2014, Huang and co-workers showed that the use of a non-commercial dimethyl-Pybox ligand with MacMillan's conditions marginally increased the yields for comparable substrates.42 And in 2017, Truong and Nguyen reported a novel copper-based metal–organic framework that functioned as an efficient heterogeneous catalyst for the oxidative amination of ketones and esters.43
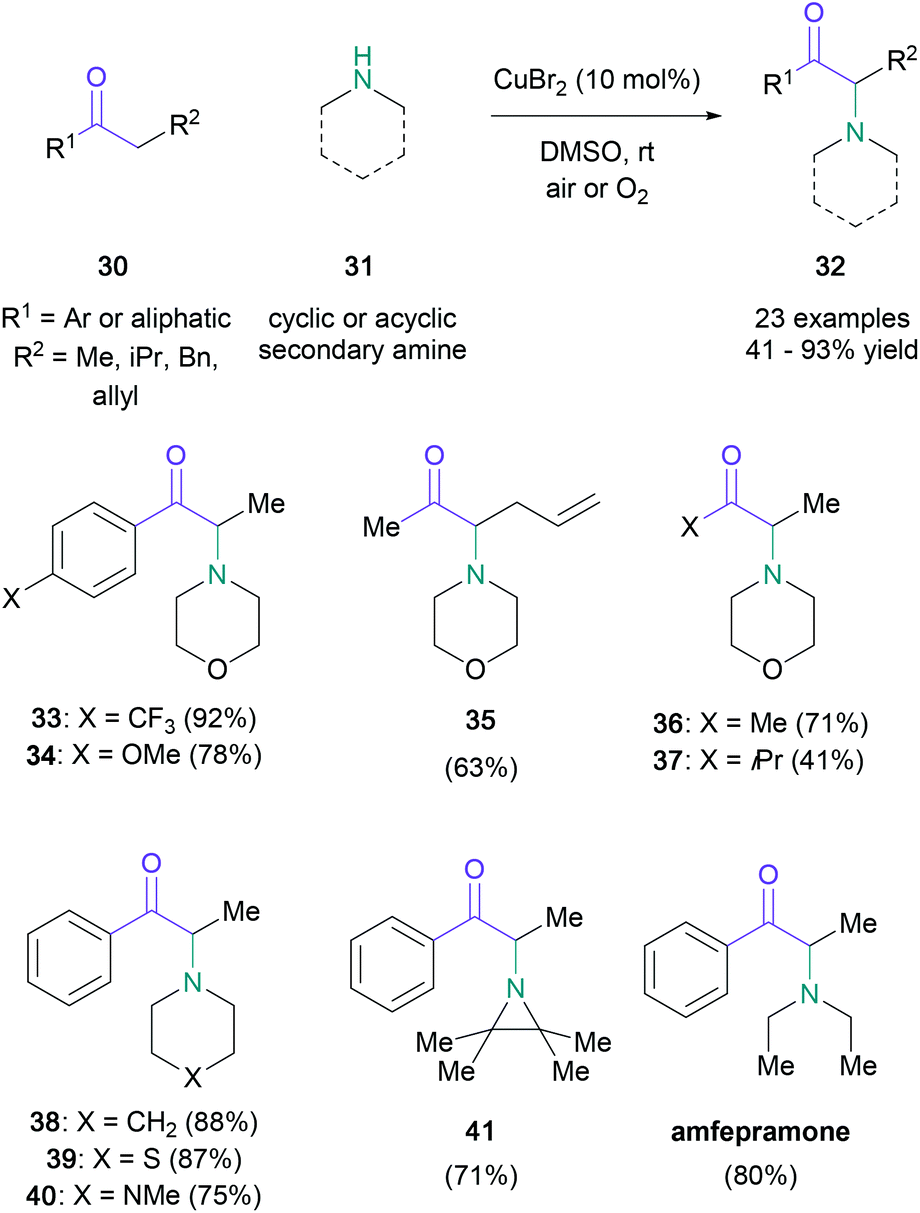 | ||
| Scheme 7 MacMillan's α-amino ketone synthesis with substoichiometric quantities of copper(II) bromide. | ||
In 2016, electrochemical α-halogenation was applied to amino ketone synthesis to remove the requirement of stoichiometric oxidants or metals. The groups of Sun and Zeng demonstrated that molecular iodine could be generated from ammonium iodide (0.5 equiv.) at the cell's graphite anode in an undivided cell, allowing for α-iodination of several mono/disubstituted-ketones (42) (Scheme 8).44 The transient α-iodocarbonyl was substituted in situ with a variety of secondary amines (43) to obtain the desired α-amino ketones (44) in up to 75% yield, with cyclic amines performing on average better than acyclic substrates. The success of this direct electrochemical amination hinges on α-centres with low steric hindrance and generally produced lower yields than those obtained by MacMillan.41
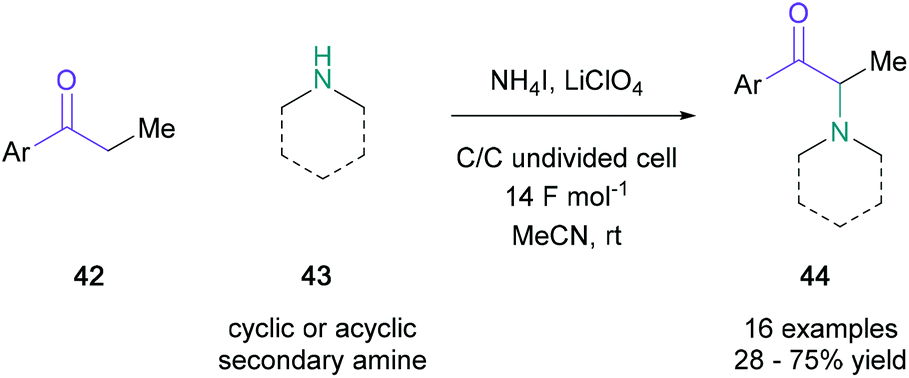 | ||
| Scheme 8 Sun and Zeng's electrochemical approach to α-amino ketones. | ||
The amination of pre-functionalised α-centres has also been developed recently.45,46 A significant contribution to this area is Zhou's highly enantioselective synthesis of α-amino ketones (47) from α-diazo ketones (45), co-catalysed by [Rh2(TFA)4] and chiral spiro phosphoric acid 46 (Scheme 9).46 This N–H Insertion procedure featured exceedingly short reaction durations (1 minute) and excellent enantioselectivities (up to 98% ee). Although the amine fragment was limited to tert-butyl carbamate, the ketone fragment could be substituted by both electron-rich and electron-poor aromatic rings, as well as aliphatic groups. The yield was found to be consistently high (≈80%), unless reactive side groups were present, such as alkenes or methoxy groups, as they enabled cyclopropanation or CH-insertion side reactions, respectively.
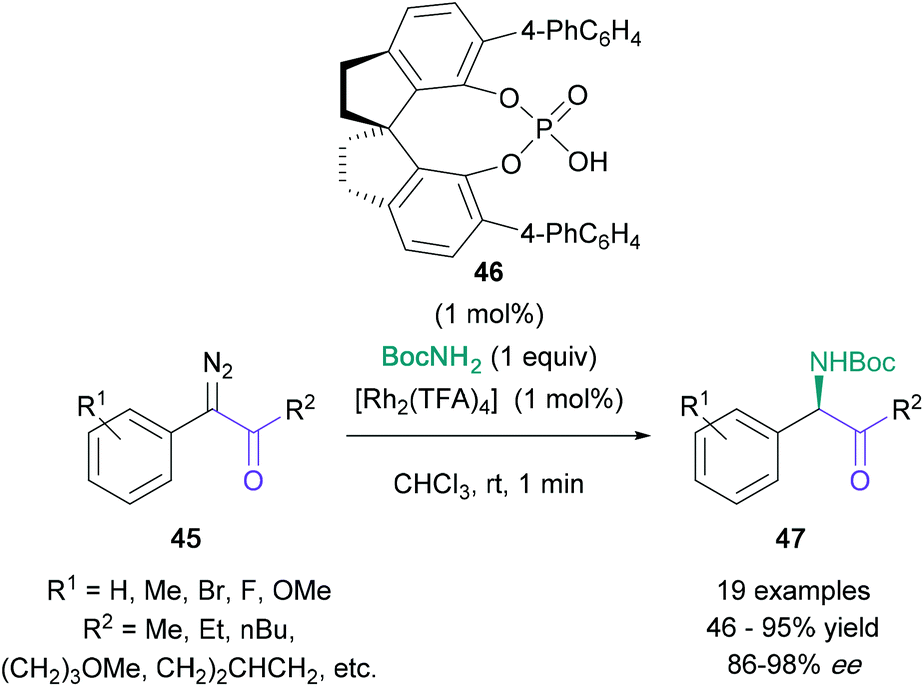 | ||
| Scheme 9 Zhou's enantioselective synthesis of α-amino ketones from α-diazo ketones. | ||
A popular pre-functionalised starting motif for α-amino ketone synthesis is the α-hydroxy ketone, as it can readily undergo Heyns rearrangement. Mechanistically, this rearrangement is thought to be initiated by the formation of an α-hydroxy imine 50, which proceeds to rearrange to the α-amino ketone 52via the hydroxy enamine intermediate 51 in an intramolecular redox reaction (Scheme 10).47 Notably, in the net transformation, a migration of the carbonyl group to the neighbouring carbon is achieved.
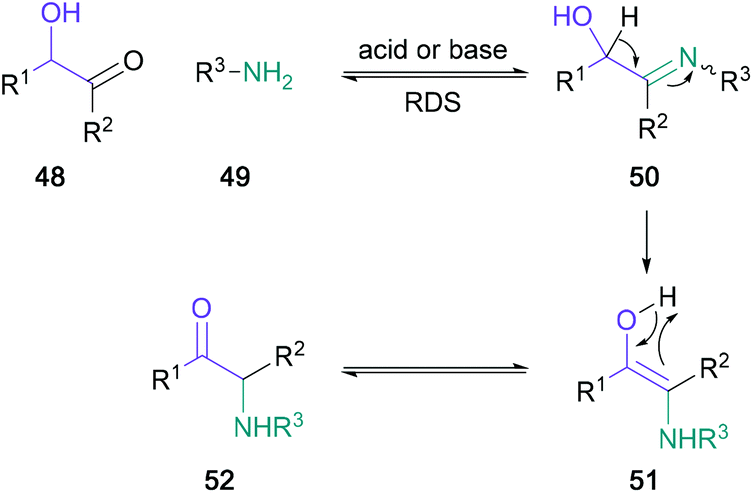 | ||
| Scheme 10 General mechanism for the Heyns rearrangement. | ||
In 2013, the first asymmetric Heyns rearrangement of racemic α-hydroxy ketones was demonstrated by Frongia (Scheme 11).48 In this study, the organocatalyst β-isocupreidine 54 was identified for the enantioselective reaction between acetoin and p-anisidine, producing the rearrangement product in 95% yield and 71% ee. A further 17 examples were produced using these optimised conditions to investigate the impact of steric and electronic factors of aniline (55) on the outcome of this protocol. Optimal results were obtained with para- & meta-substituted anilines with up to 82% yield and up to 81% ee. The challenge associated with a chiral Heyns rearrangement is reflected by the detrimental effects on yield and enantioselectivity observed with electronically or sterically challenging anilines. For example, the use of ortho-substituted anilines significantly reduced yield and enantiomeric excess (26% yield, 11% ee) due to steric reasons, and para-cyano substituted aniline failed to participate in the reaction due to its lower basicity. In a related contribution, Frongia and co-workers further modified this procedure to enable the enantioselective synthesis of α-arylamino cyclobutanones from racemic α-hydroxy cyclobutanones.49
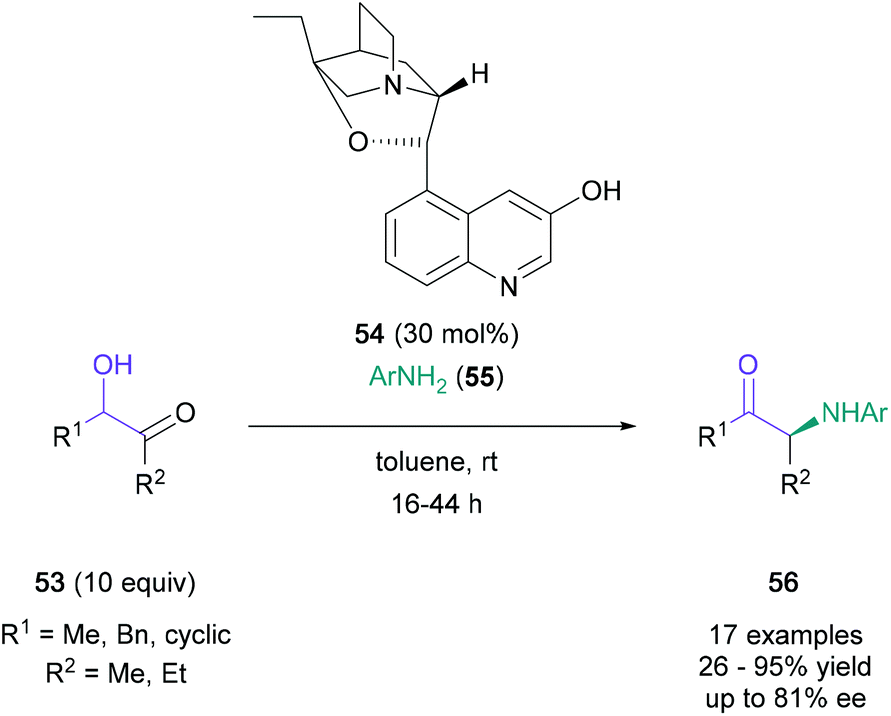 | ||
| Scheme 11 Frongia's enantioselective Heyns rearrangement. | ||
Subsequently, several other racemic variants targeting a similar substrate scope under silica tungstic acid- or tin(II) chloride catalysis have been developed, which dramatically accelerated substrate conversion.50,51 Notably, Tang and co-workers demonstrated that cyclic and acyclic secondary amines (58) can also undergo the Heyns rearrangement by para-toluenesulfonic acid catalysis, affording tertiary amines (59) without the apparent migration of the carbonyl group in up to 83% yield (Scheme 12).52
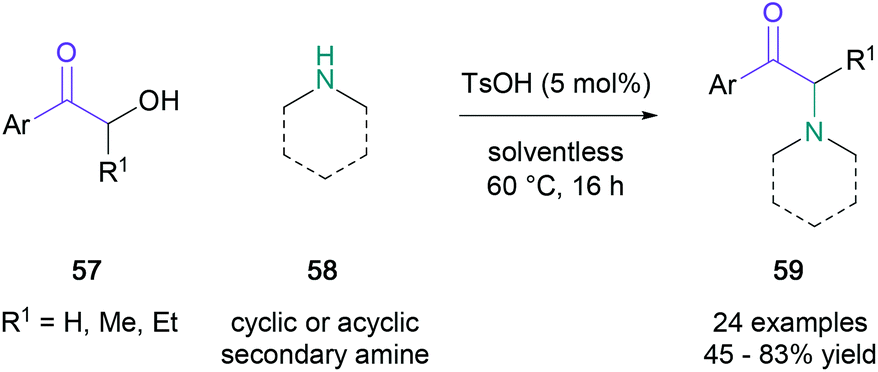 | ||
| Scheme 12 Tang's Heyns rearrangement without migration of the carbonyl group. | ||
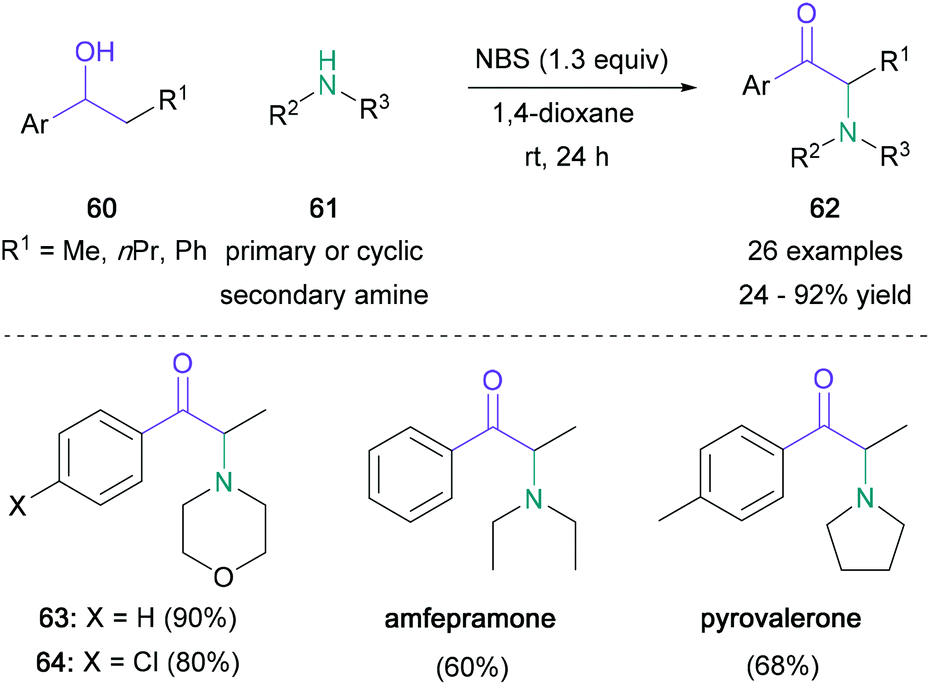
Benzylic alcohols may also serve as convenient synthetic analogues to carbonyl derivatives if their oxidation can be achieved in situ under mild conditions.53 In 2015, the Sekar group unveiled a one-pot NBS-mediated open-flask synthesis of α-amino ketones which uses benzylic alcohols (60) and nitrogen nucleophiles (61) (Scheme 13).54 This operationally simple transformation led to the desired products (62) in up to 94% yield, and the best results were obtained with secondary cyclic amines. Although the yields were not influenced by the electronic properties of the aromatic substituent, a significant decrease was observed when using sterically hindered starting materials. Medicinally relevant amfepramone and pyrovalerone were obtained in 60% and 68% respectively in one step from commercially available starting materials. Later, Sekar reported that α-imido ketones could also be prepared by using a modification of this procedure.55
 | ||
| Scheme 13 Sekar's one-pot oxidation and α-amination of benzylic alcohols. | ||
In 2016, Park and Rhee demonstrated that the α-hydroxy imine intermediate present in a Heyns rearrangement could be also formed from 1,2-azidoacetates by ruthenium catalysis.56 The authors reported an improved yield for 1,2-azidoacetates, and this approach was subsequently applied to the synthesis of a wide range of α-amido ketones, forming the products in 53–94% yield.
2.2. Aldehydes
The use of aldehydes in the synthesis of α-amino ketones has been mainly reserved to the aza-benzoin condensation. Rovis published an enantioselective version of this transformation in 2012, utilising aliphatic aldehydes (65) and aryl-substituted boc-imines (66) (Scheme 14).57 Aldehydes featuring oxygen, sulfur and nitrogen-containing linear tethers were well-tolerated, and the corresponding α-amino ketones (68) were obtained in 33–93% yields with satisfactory enantioselectivity. The enantiopure triazolium salt 67, which generates the active NHC catalyst in situ, was shown to facilitate the slow release of imine 66 from an aza-Breslow intermediate, thus counteracting racemisation issues present in previous modifications of this reaction.58 The following year, Ye reported an additional protocol which proved to be compatible with α,β-unsaturated aldehydes and fully substituted electron-poor imines, allowing for the direct generation of quaternary stereocentres.59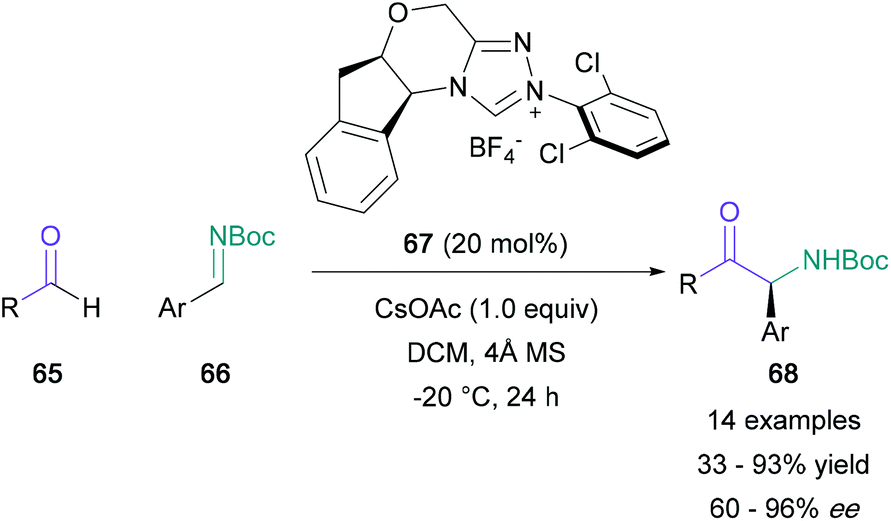 | ||
| Scheme 14 Rovis’ enantioselective aza-benzoin condensation. | ||
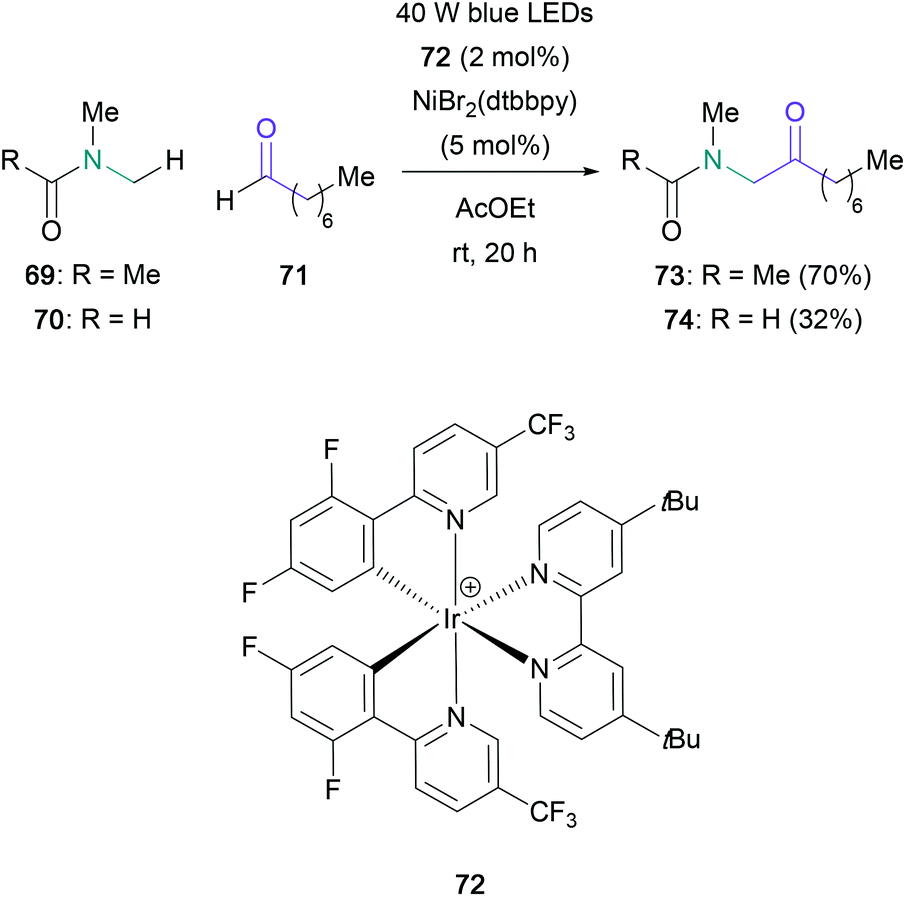
An alternative to employing the umpolung reactivity of aldehydes in the aza-benzoin reaction is to instead identify methods for the direct C–H functionalisation of this moiety. Recently, Murakami reported a photoredox dual metal-catalysed dehydrogenative coupling of aldehydes and toluene derivatives, with two examples using substituted amides instead (Scheme 15).60 The coupling of octanal (71) with DMA (69) or DMF (70), which were present in a 100-fold excess, led to the corresponding α-amino ketone (73 and 74) in 70% and 32% yields respectively. Despite the large excess of amide required, this protocol represents a promising avenue for the derivatisation of aldehydes.
 | ||
| Scheme 15 Murakami's photoredox dual metal-catalysed dehydrogenative coupling. | ||
2.3. Carboxylic acids and derivatives
The direct transformation of α-amino acids to methyl ketones was originally reported by Dakin and West almost a century ago. In 2016, Schreiner reported the first example of an asymmetric Dakin–West reaction by exploiting modern enantioselective decarboxylative protonation chemistry.61 To accomplish this transformation, Schreiner used a methylimidazole-containing oligiopeptide (76) (10 mol%) in combination with DCC and Ac2O to convert several amino acid derivatives (75) to the corresponding α-acetoamido ketone products (77) in 52–98% yield (Scheme 16). Although the yields were found to be broadly acceptable, the enantioselectivity of the transformation varied significantly with the substitution of the starting material. Notably, leucine and cyclohexylalanine derivatives gave products in 54–58% ee, whereas substrates with small substituents, such as alanine and methionine derivatives, converted to the expected products with poor selectivity (11–25% ee).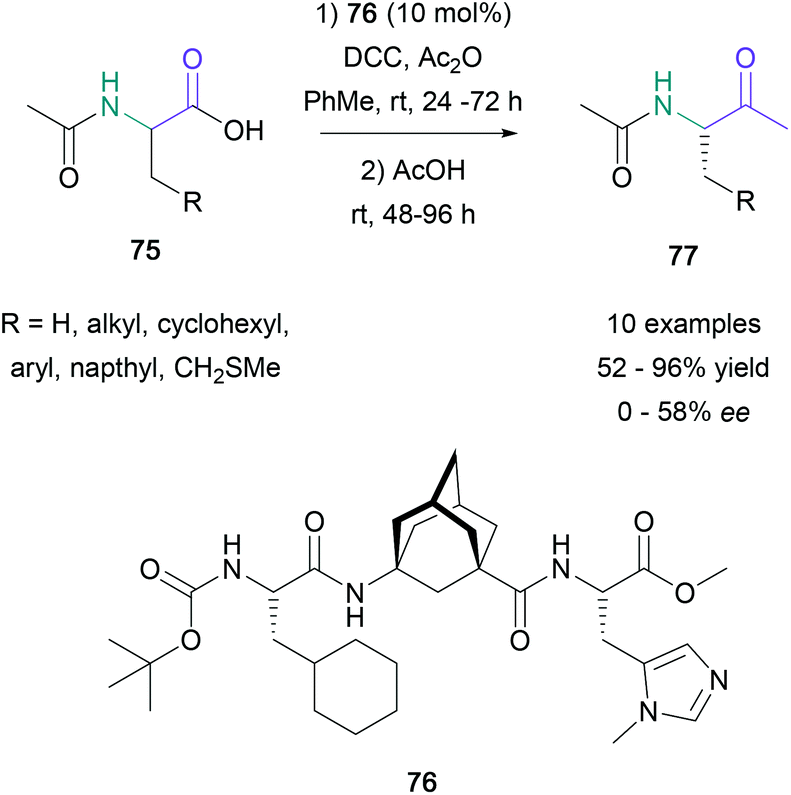 | ||
| Scheme 16 Schreiner's enantioselective Dakin–West reaction. | ||
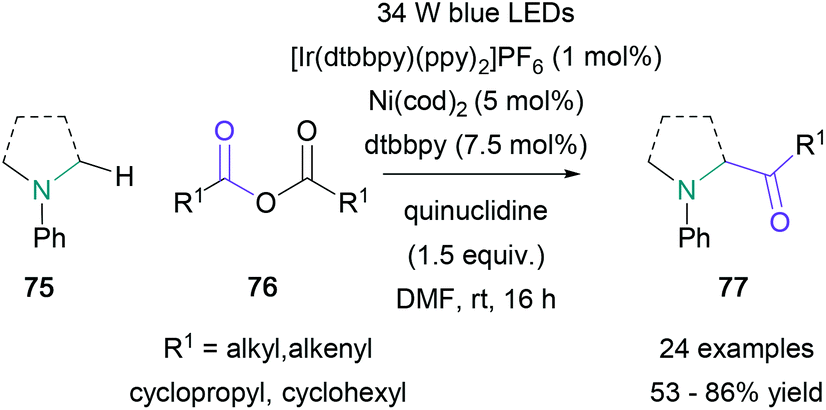
In the same year, Doyle employed carboxylic acid derivatives for the direct α-acylation of N-aryl amines using photoredox and nickel catalysis.62 Model studies for this transformation focused on the acylation of N-phenylpyrrolidine with propionic anhydride, providing the expected product in 83% yield. With optimised conditions in hand, a wide range of cyclic and acyclic amines (78) were successfully acylated with propionic anhydride in 60–86% yield, and N-phenylpyrrolidine was acylated with several symmetric anhydrides (79) in 57–74% yield (Scheme 17). To expand the scope of the process beyond symmetric anhydrides, Doyle also noted that thioesters may be successfully employed in this transformation, providing the expected products with no appreciable loss in yield (65–77%). This modification allowed for the incorporation of biotin and steroidal residues on to N-phenylpyrrolidine in up to 76% yield.
 | ||
| Scheme 17 Doyle's direct acylation of N-aryl amines. | ||
2.4. Enol ethers
The use of enol ethers in α-amination processes presents a direct advantage over unprotected ketones by removing potential regioselectivity issues. Largely speaking, protocols used for the generation of α-amino ketones from enol ethers can be divided into two camps: (1) electrophilic nitrogen sources; (2) nucleophilic nitrogen with umpoled enolates. A contemporary alternative involves photocatalytically-generated nitrogen-centered radicals using 1-aminopyridinium salts.63In 2017, Baidya published a convenient α-amination protocol involving the in situ generation of nitroso arenes from anilines (82) in the presence of super-stoichiometric amounts of Oxone® (Scheme 18).64 These intermediates underwent a nitroso aldol reaction with silyl enol ethers (81), and the addition products (84) were easily converted to the desired α-amino ketones (83) in up to 82% yield by assistance of the disilyl protecting group. A related transformation developed by Hashimoto uses an imidoiodinane as the nitrogen electrophile in the presence of a polymer-supported dirhodium(II) catalyst.65
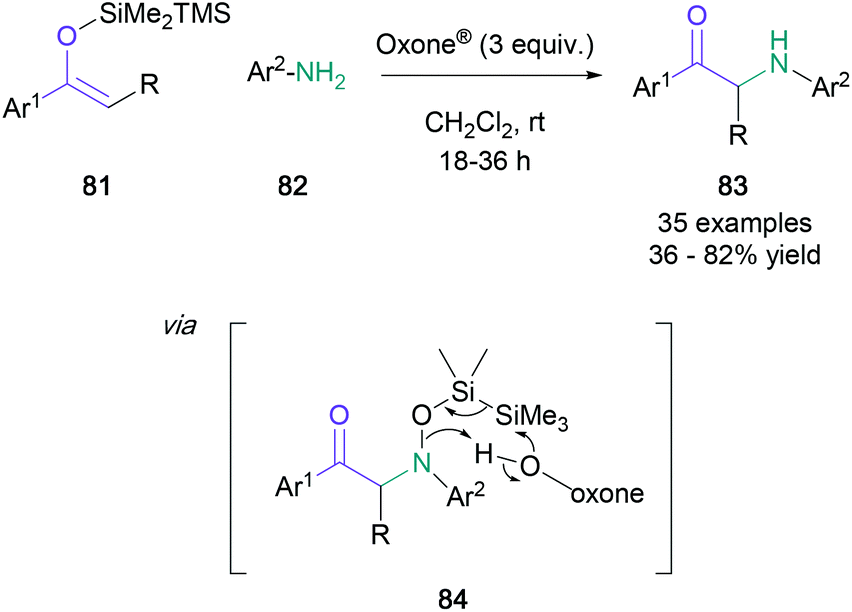 | ||
| Scheme 18 Baidya's Oxone®-mediated α-amino ketone synthesis between silyl enol ethers and anilines. | ||
Electrophilic nitrogen was also used by Kürti in an aza-derivative of the Rubottom oxidation (Scheme 19).66 Treatment of tetrasubstituted silyl enol ethers (85) with protected hydroxylamine 86 in HFIP generated amino ketones featuring a quaternary centre (87) in up to 82% yield. Even though most of the reported examples involved the synthesis of aromatic α,α-dimethyl substituted ketones, the methodology was also found to be compatible with unsaturated substituents (88) and alicyclic ketones (89). Electron-deficient substrates did not react under these conditions, although their desired transformation was instead achieved under transition metal catalysis. An enantioselective version of this novel protocol was investigated by the Corey group in the same year.67
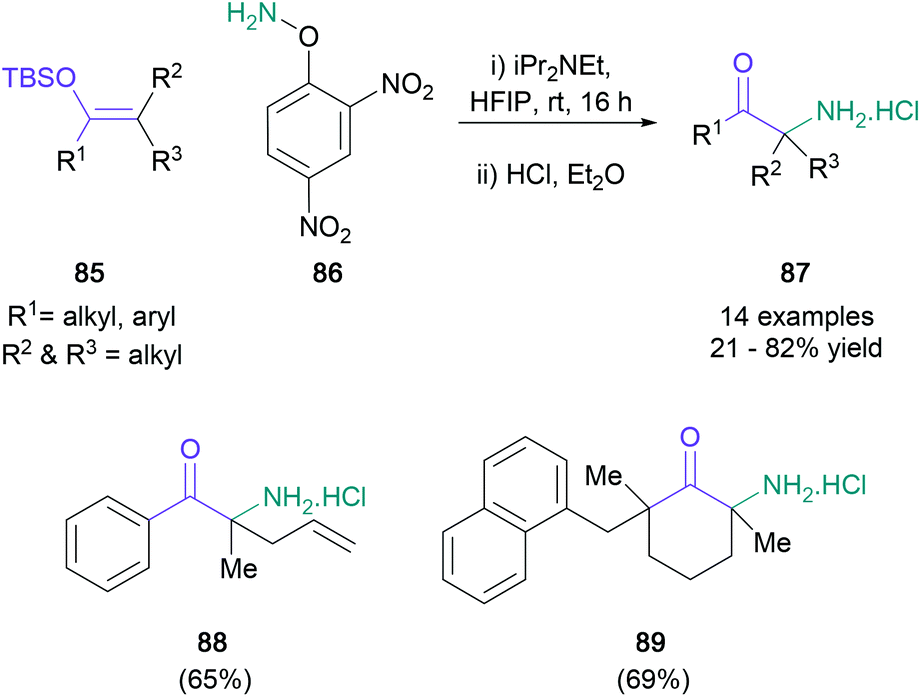 | ||
| Scheme 19 Kürti's aza-Rubottom oxidation to α-amino ketones. | ||
In the category of nucleophilic aminations with umpoled enolates, Wirth's 2014 report on an iodine(III)-mediated intramolecular transformation stands out as particularly useful (Scheme 20).68 Using this procedure, enol ethers bearing an amino-functionalised silyl protecting group (90) were converted to the corresponding α-amino ketones (93) in up to 94% yield by employing PIDA. When the chiral hypervalent iodine reagent 91 was used instead, the resulting products (92) were produced in up to 94% ee. This protocol is compatible with numerous aromatic, bicyclic, and aliphatic ketones, and it has been used to efficiently functionalise two steroid-derived enol ethers. Another valuable umpolung methodology was presented by Xu in 2018, which involved the use of N-alkenoxypyridinium salts.69
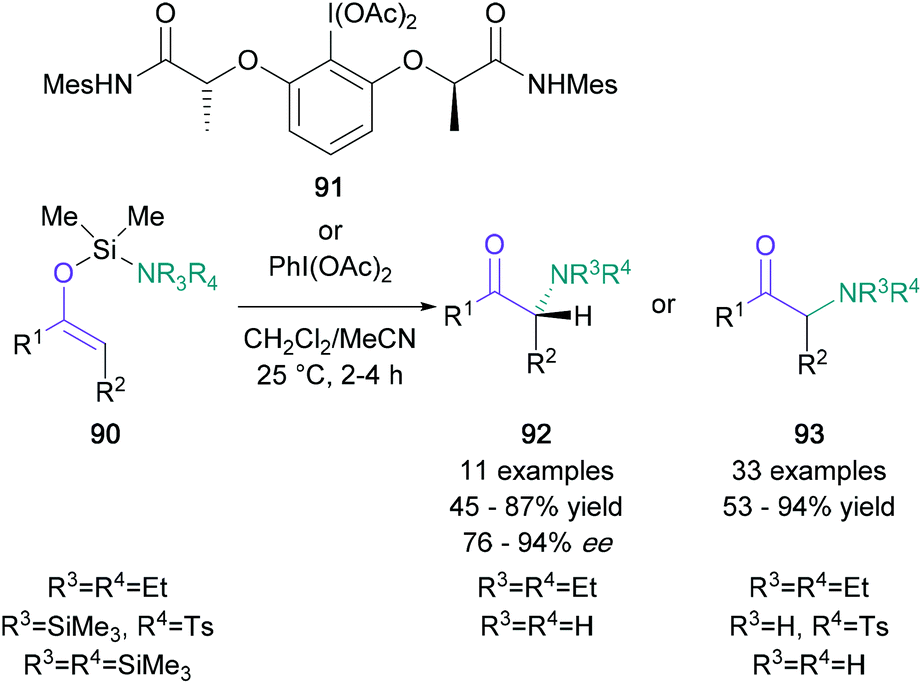 | ||
| Scheme 20 Wirth's hypervalent iodine-mediated α-amino ketone synthesis. | ||
Finally, a creative α-amination strategy published by Hartwig in 2018 made use of silyl-protected conjugated dienolates bearing a γ-carbonato group (Scheme 21).70 These starting materials (94) can be subjected to an iridium-catalysed SN2′ process with substituted amines (96) in the presence of asymmetric phosphonamidite ligand 95, resulting in enantioenriched protected α-amino ketones (97) in up to 96% yield with excellent regio- and stereoselectivity. Additionally, a wide range of aliphatic (98) and aromatic amines (99) were efficiently employed in this reaction, including peptide derivatives (100).
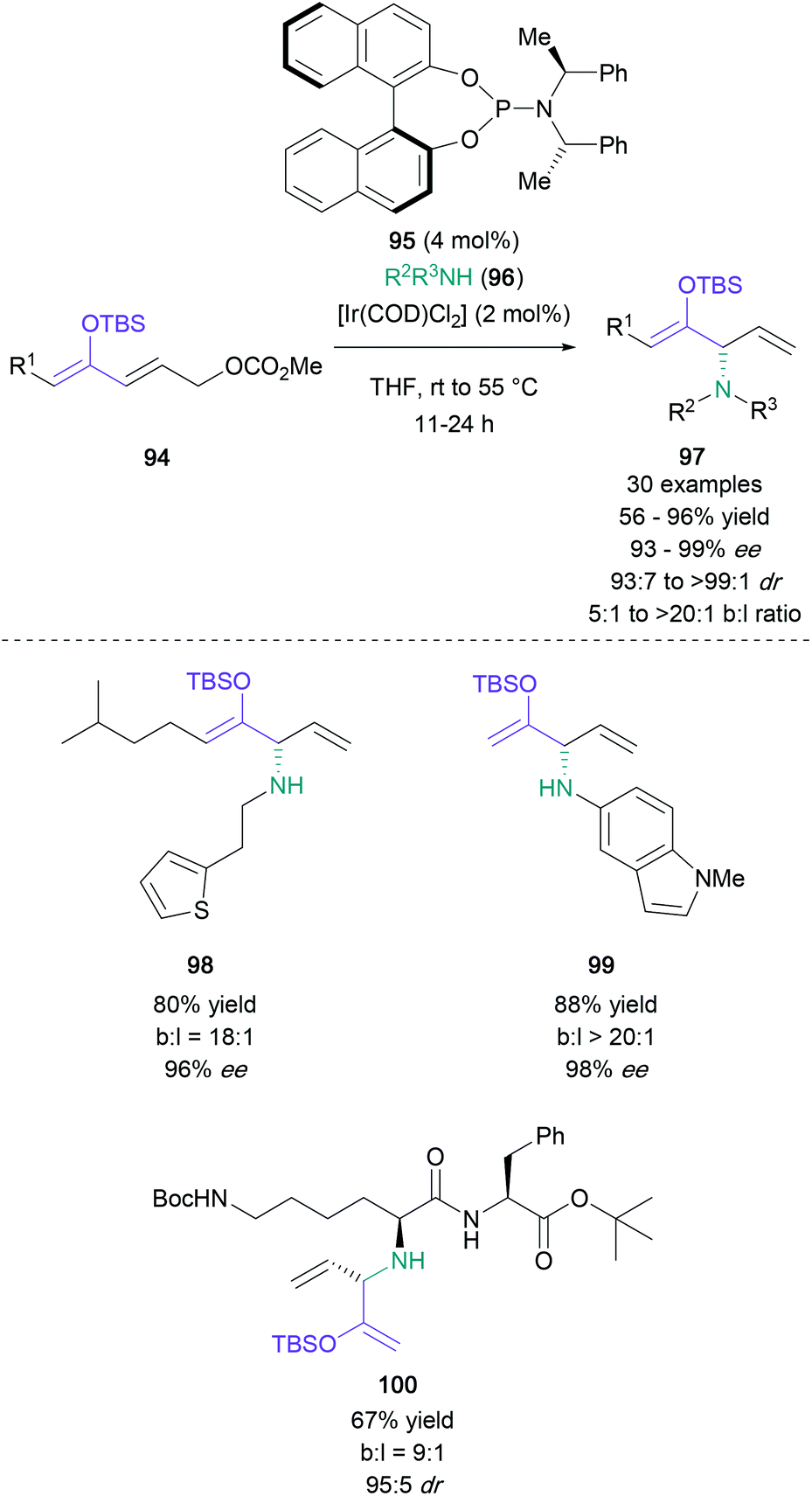 | ||
| Scheme 21 Hartwig's iridium-catalysed SN2′ reaction of silyl-protected conjugated dienolates. | ||
2.5. Enones
A common method for the generation of enantioenriched α-amino ketones is the catalytic hydrogenation of α-amino enones, which can in turn be prepared from the corresponding diketones. Zhang achieved this transformation with excellent enantioselectivity using a rhodium(I) diphosphine complex,71 and they subsequently expanded the substrate scope to include an increased range of aliphatic substituents in 2016.72 Dimeric cobalt phosphine complexes are also suitable for hydrogenation of α-amino enones, as shown by Cabrera in 2013.73An elegant two-step sequence to obtain highly functionalised α-amino ketones from enones without an α-amino group was reported by Lee in 2014 (Scheme 22).74 Upon treating cyclic enones (101) with lithium trimethylsilyldiazomethane (102), a 1,4-addition to obtain pyrazolines (103) was observed to occur in up to 92% yield. These heterocycles or their N-alkylated analogues (105) were then ring-opened under acidic conditions to generate β-cyano-α-amino ketones (104 and 106) in up to 95% yield. This methodology was compatible even with exocyclic and endocyclic tetrasubstituted enones, with the resulting products (107 and 108) obtained in 66–68% yield over 2–3 steps. Additionally, ketamine analogues (109) were successfully synthesised, providing a novel avenue for exploration of this pharmacologically relevant chemical space.
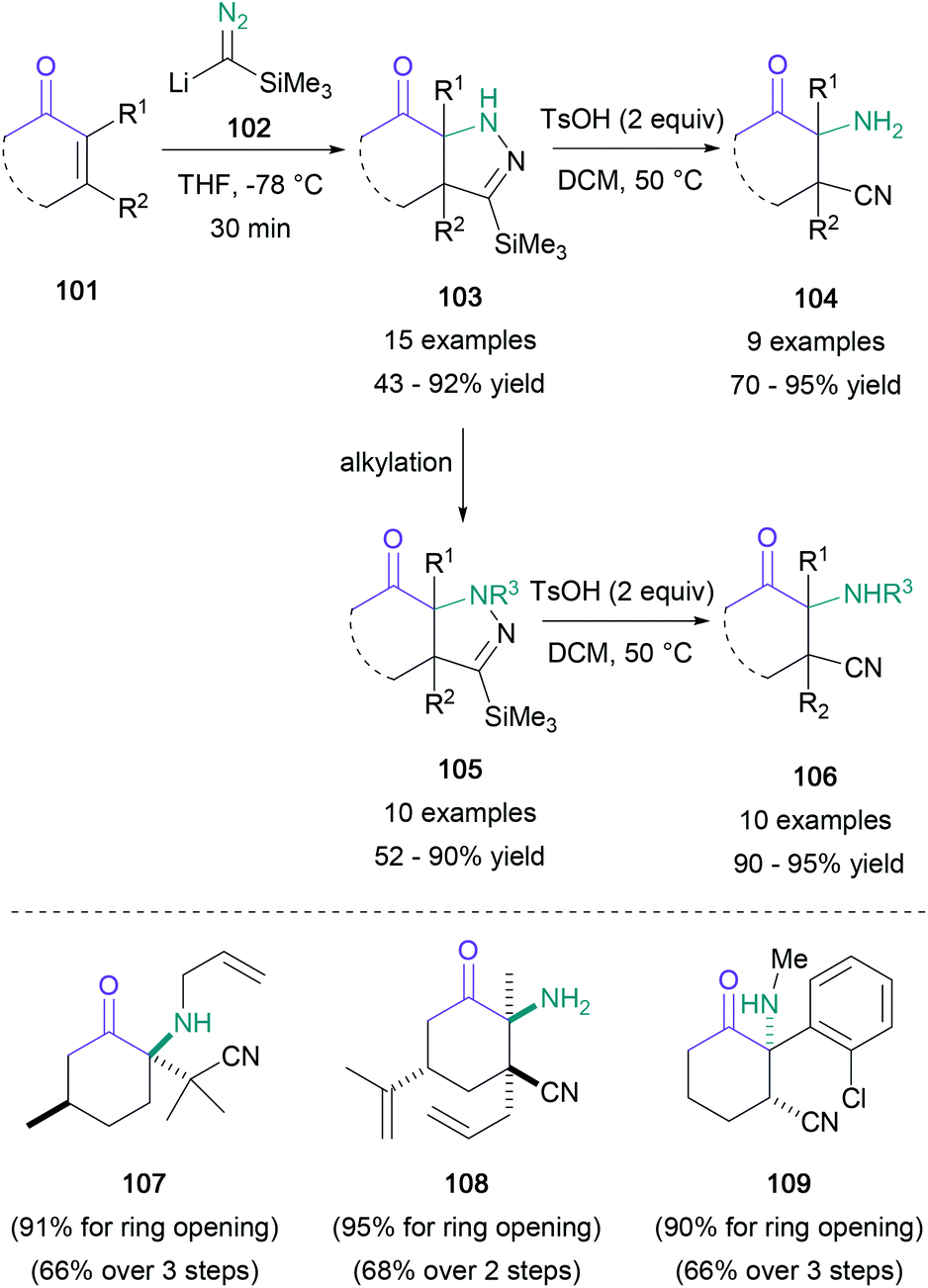 | ||
| Scheme 22 Lee's two-step synthesis of α-amino ketones via pyrazolines. | ||
2.6. Imines
Imines are a versatile platform for the preparation of substituted amino ketones. In 2012, Kimpe reported a three-step approach for the synthesis of α-amino ketones from chiral N-tert-butanesulfinyl imines, derived from the corresponding α-halo ketones (Scheme 23).75 The imines (110) were then treated with sodium methoxide to give the N-tert-butanesulfinyl 2-amino acetals (111), which were deprotected in two sequential steps. First, treatment with TMSOTf gave the N-tert-butanesulfinyl α-amino ketones (112), and second, methanolic 3 M HCl provided the final products. The synthetic sequence was found to be suitable for aromatic and non-aromatic substituted α-halo ketones, as well as the synthesis of (S)-cathinone hydrochloride, formed in a combined 15% yield over three steps (88% ee). Yus also explored the use of N-tert-butylsulfinyl imines in the synthesis of amino ketones and amino acids.76 In these experiments, imines were treated with nitroethane to give β-nitroamine adducts, which were then oxidised to give amino ketones.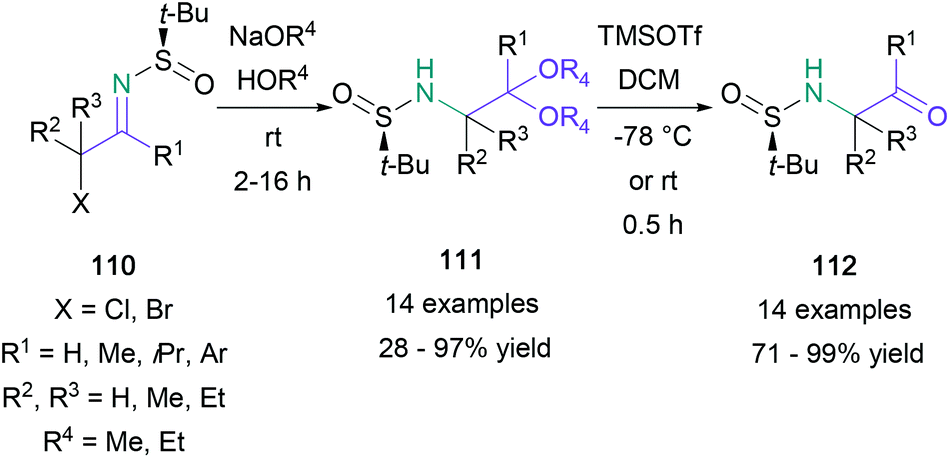 | ||
| Scheme 23 Kimpe's synthesis of α-amino ketones from chiral N-tert-butanesulfinyl imines. | ||
Another asymmetric synthesis of amino ketones from imines was accomplished by Guo using a transfer hydrogenation approach with chiral Brønsted acid catalysis (Scheme 24).77 The symmetrical diaryl α-keto ketimines (113) produced the expected products (116) in 95–97% yield and 90–96% ee. Unsymmetrical aryl- and alkyl-substituted diketones were also employed in a regioselective one-pot procedure, furnishing amino ketones with only a marginal reduction in yield.
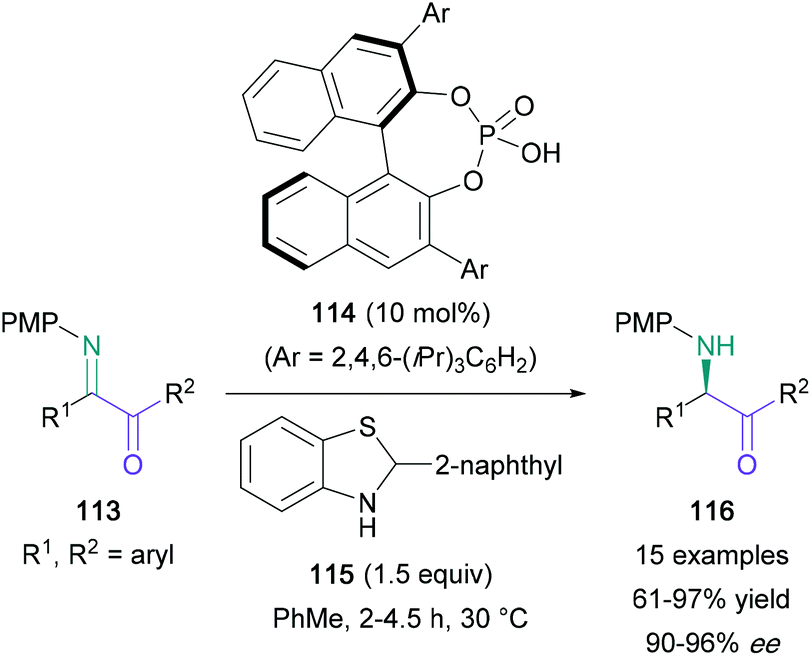 | ||
| Scheme 24 Guo's Brønsted acid-catalysed enantioselective transfer hydrogenation of imines. | ||
In 2014, Wulff reported a novel asymmetric α-iminol rearrangement.78 In this report, hydroxyimines (117) were treated with a VANOL Zr catalyst (118) at 80 °C in toluene to give α-amino ketones (119) in 96% yield and 95% ee (Scheme 25). This successful procedure was subsequently applied to a broad range of electron poor/rich aryl-substituted imines, as well as non-aromatic substituted imines, providing the corresponding amino ketones in 88–99% yield and up to 99% ee. Wulff also later reported an operationally-straightforward high-yielding achiral α-iminol rearrangement using silica or montmorillonite K10.79
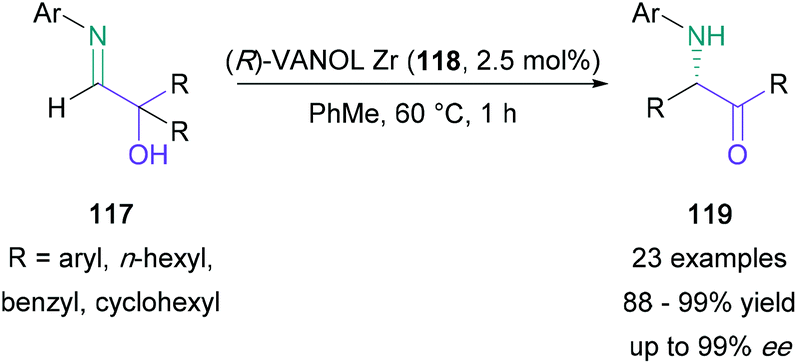 | ||
| Scheme 25 Wulff's (R)-VANOL Zr-catalysed enantioselective iminol rearrangement. | ||
Although the classical acyloin condensation is known to generate α-hydroxy ketones suitable for a Heyns rearrangement, a related titanium(III)-catalysed reductive coupling between imines (120) and nitriles (121), developed by Streuff, enabled direct access to α-amino ketones (122), where the competing pinacol-type homodimerisation was suppressed (Scheme 26). Remarkably, when ketimines were used as precursors, this methodology allowed for the synthesis of sterically challenging fully substituted α-carbonyl centres in up to 77% yield, which were otherwise difficult to prepare. The authors noted that aromatic and benzylic nitriles underwent this reductive coupling, and even acyclic aliphatic nitriles were tolerated, albeit the obtained yield is reduced to 47% in this case.80 Amino ketones were also formed by a reductive cross-coupling reaction between imines and nitriles in the presence of titanium(IV), proceeding via a 2,5-diazatitanacyclopentene intermediate.81
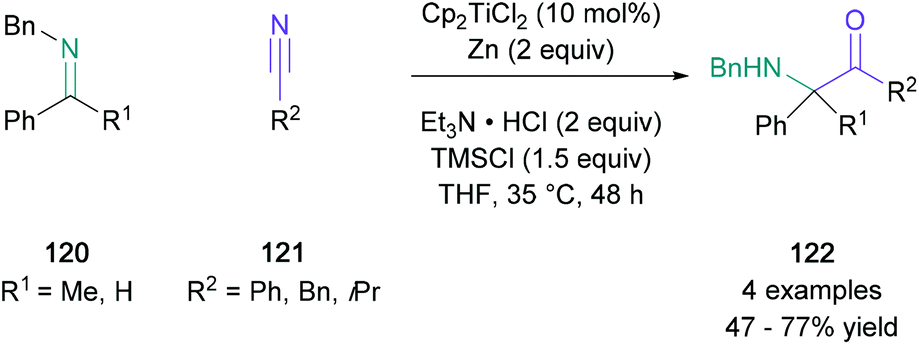 | ||
| Scheme 26 Streuff's titanium(III)-catalysed reductive coupling between imines and nitriles. | ||
2.7. Enamines
There have been a limited number of synthetic methodologies that employ enamine derivatives in the synthesis of α-amino ketones. Toste's recent work made use of the azoalkene moiety, obtained via mild oxidation of hydrazones (Scheme 27). In this protocol, treatment of azoalkenes (123) with chiral phosphoric acid 125 promoted a nucleophilic amination using anilines (124), providing highly enantioenriched α-amino hydrazones (126) in up to 93% yield.82 Three of the resulting hydrazone products were then converted to the corresponding α-amino ketones (127) under mild conditions in 72–84% yield, with no racemisation of the newly formed stereocenter.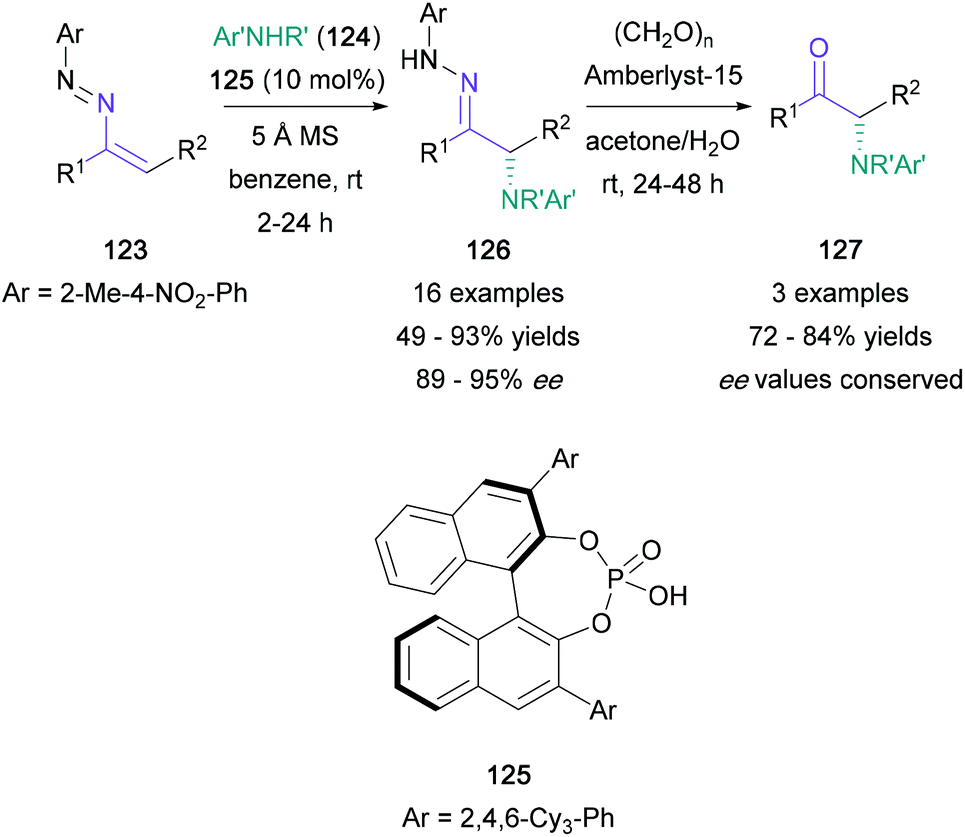 | ||
| Scheme 27 Toste's chiral nucleophilic amination of azoalkenes. | ||
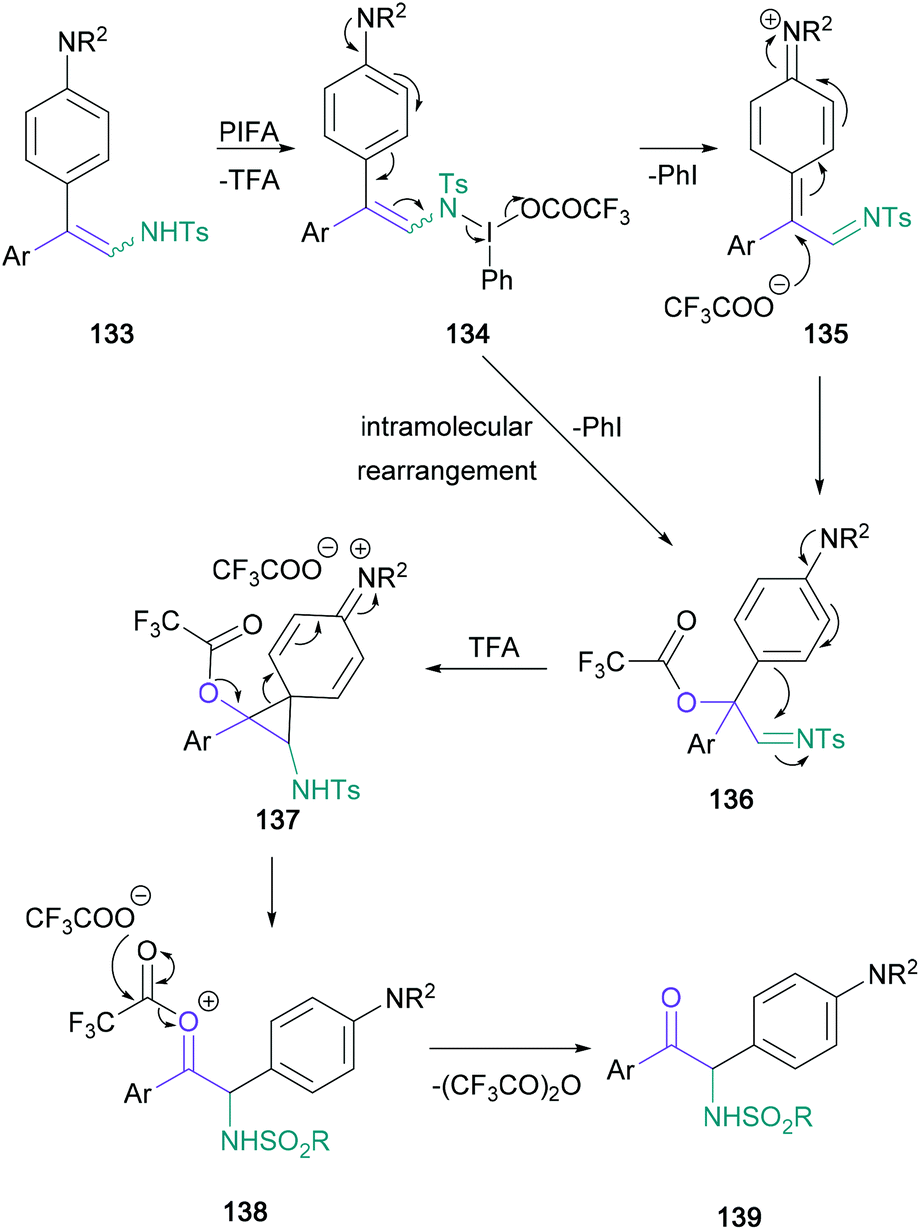
In contrast with Toste's approach, Anbarasan introduced the required C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond by an oxidative rearrangement, while conserving the nitrogen functionality of the starting material.83 In this transformation, diaryl arylsulfonyl-protected enamines (128) are converted to α-amino ketones (129) in up to 81% yield, with a concomitant migration of the more electron-rich aromatic substituent (Scheme 28). Although the migrating aromatic ring usually featured a disubstituted amine substituent (130 and 131), para-methoxy aryl groups were also shown to migrate effectively (132). The authors proposed a mechanism starting with substitution of the PIFA triflate, followed by an intramolecular rearrangement to form imine 136 and subsequent semi-pinacol rearrangement to give the product (139) (Scheme 29).
O bond by an oxidative rearrangement, while conserving the nitrogen functionality of the starting material.83 In this transformation, diaryl arylsulfonyl-protected enamines (128) are converted to α-amino ketones (129) in up to 81% yield, with a concomitant migration of the more electron-rich aromatic substituent (Scheme 28). Although the migrating aromatic ring usually featured a disubstituted amine substituent (130 and 131), para-methoxy aryl groups were also shown to migrate effectively (132). The authors proposed a mechanism starting with substitution of the PIFA triflate, followed by an intramolecular rearrangement to form imine 136 and subsequent semi-pinacol rearrangement to give the product (139) (Scheme 29).
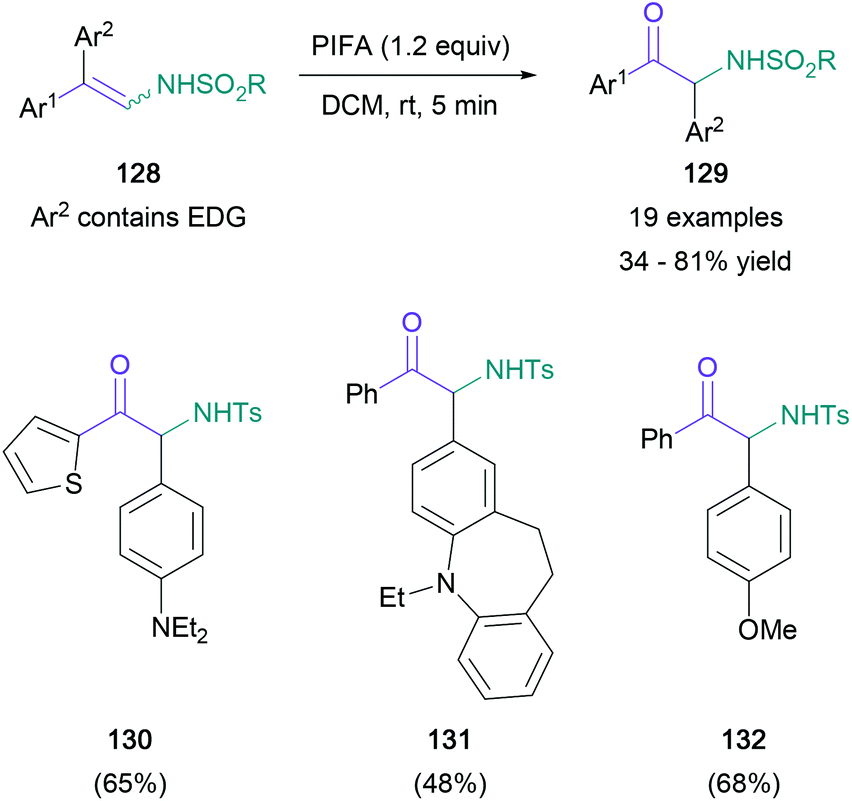 | ||
| Scheme 28 Anbarasan's oxidative rearrangement of diaryl arylsulfonyl-protected enamines. | ||
 | ||
| Scheme 29 Underlying mechanism for Anbarasan's oxidative rearrangement of diaryl arylsulfonyl-protected enamines. | ||
2.8. Unsaturated hydrocarbons
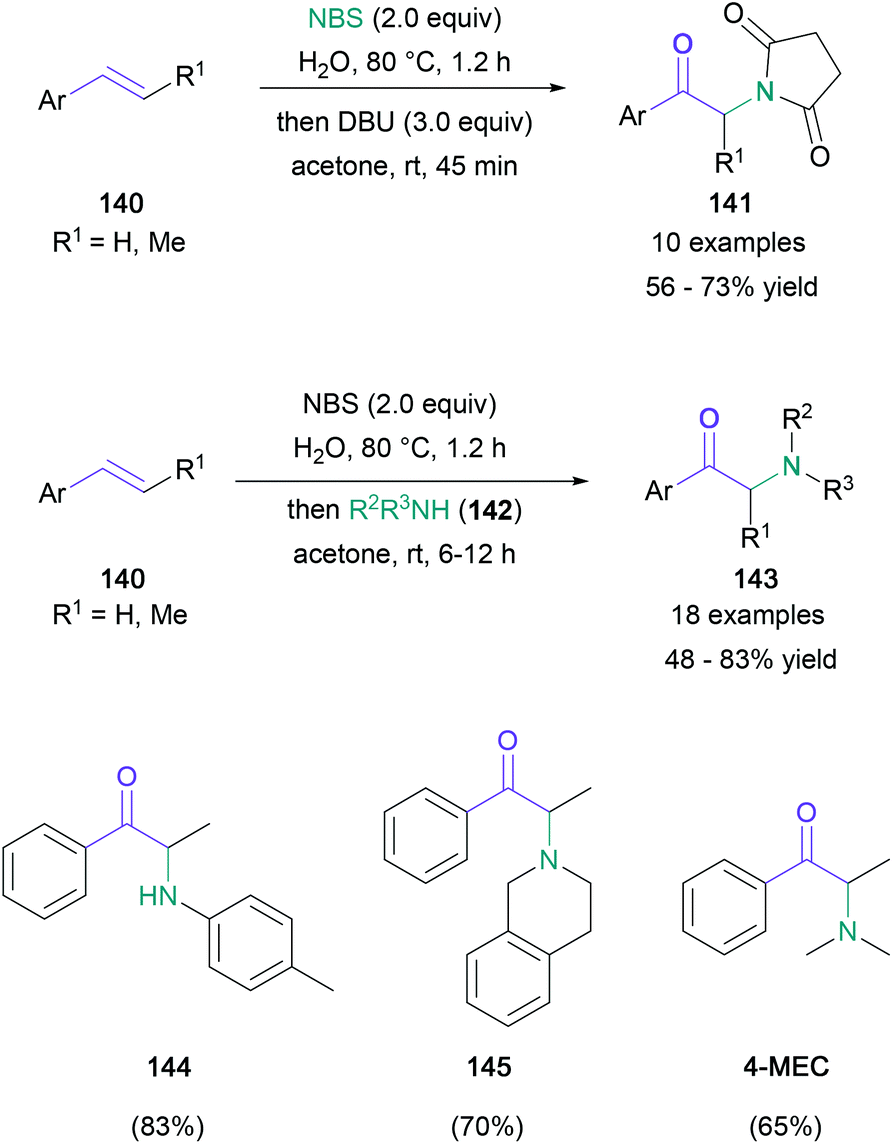
Unsaturated hydrocarbons are particularly desirable starting materials due to their commercial availability and lower cost. There are two commonly explored methods of introducing the desired α-amino ketone motif to these building blocks: the first is the formation of halohydrin intermediates, which undergo oxidation and nucleophilic substitution; the second involves a radical or nucleophilic addition of nitrogen species to the unsaturated C–C bond, followed by the oxidative or solvolytic generation of the carbonyl functionality.An early example in the halohydrin category was reported by Kshirsagar in 2016.84 In this work, the treatment of mono- or disubstituted styrenes (140) with NBS in water followed by the addition of either DBU or a nucleophilic amine (142) with acetone led to the formation of α-imido (141) or α-amino ketones (143) respectively (Scheme 30). Although monoalkylated anilines were used predominately as a source of nucleophilic amine (144), secondary amines (145) were also successfully implemented, leading to the synthesis of known stimulants, including 4-methylethcathinone (4-MEC) and amfepramone. Notably, NBS plays the dual role of a bromonium ion source and an oxidant for the bromohydrin. In 2016, Zhang also achieved the base-switchable functionalisation of styrenes, with 1,3-dibromo-5,5-dimethylhydantoin (DBH) functioning as the brominating agent and oxidant,85 and in the same year, the Sudalai group presented an anhydrous version of Kshirsagar's protocol, employing DMSO as both the solvent and oxidant.86
 | ||
| Scheme 30 Kshirsagar's halohydrin-mediated α-amino ketone synthesis from styrenes. | ||
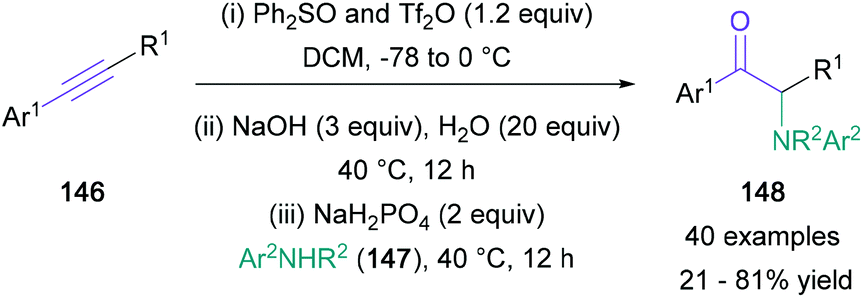
When alkynes were used as starting materials in NBS-mediated functionalisations, solvolysis of the intermediary bromonium ion directly generates an α-halo ketone that is readily aminated, as demonstrated by Liang in 2016 and by Zeng in 2019.87,88 An alternative approach to alkyne functionalisation employing an activated sulfoxide was presented by Xu in 2019 (Scheme 31).89
 | ||
| Scheme 31 Xu's sulfoxide-mediated functionalisation of alkynes. | ||
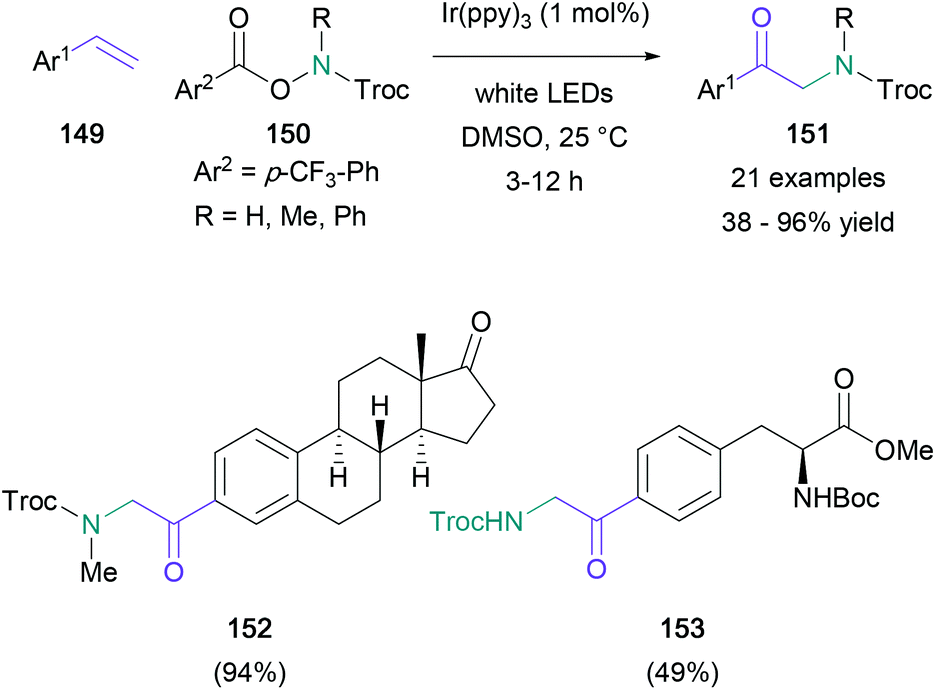
A useful example of a radical functionalisation of styrene derivatives was published by the Yu group in 2017.90 This functionalisation (Scheme 32) involved the attack of a terminal alkene (149) by a nitrogen-centered radical, generated from a protected hydroxylamine (150), and further conversion of the resultant radical to a cation that reacted with DMSO to give the α-amino carbonyl (151). The reaction proceeded in up to 96% yield with a wide range of substituted aromatics, and the functional group tolerance of this methodology was proven through the synthesis of steroid and amino acid-derived aminocarbonyls 152 and 153 respectively. This work followed Zhang's 2015 publication on a copper(II)-mediated radical addition of NFSI and its analogues to alkynes, yielding 1,2-diarylated α-amino ketones.91
 | ||
| Scheme 32 Yu's photoredox-mediated generation of nitrogen-centred radicals for the functionalisation of styrenes. | ||
2.9. Nitrogen heterocycles
The copper(I)-catalysed azide alkyne cycloaddition (CuAAC) provides efficient access to 1,4-disubstituted triazole building blocks, convenient starting materials for Murakami's work on the synthesis of amino ketones. Murakami converted 4-phenylsulfonyl-1,2,3-triazole to an α-imino rhodium-(II) carbenoid that reacted with water to give a phenyl-substituted α-amino ketone in 91% yield.92 After this initial success, a wide range of aromatic-, heteroaromatic-, alkyl-, alicyclic-substituted triazoles (154) were subjected to the optimal reaction conditions, providing the expected products (155) in up to 99% yield (Scheme 33). Further investigations demonstrated that the process could be conveniently performed in one-pot with the click reaction required to form the tetrazole starting material. Additionally, Murakami found that water may be substituted for tert-butyl alcohol to give an α-amino enol ether product, a significant result developed subsequently by the group.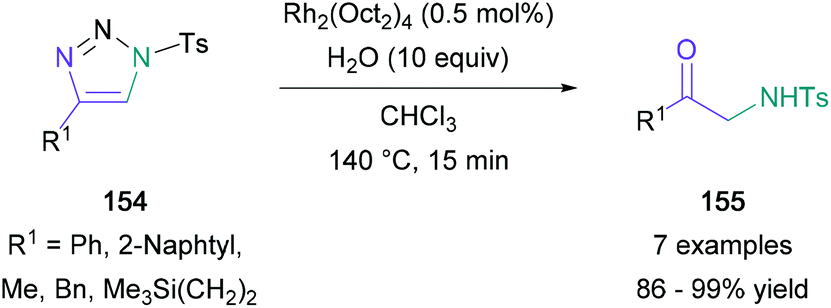 | ||
| Scheme 33 Murakami's rhodium-catalysed α-amino ketone synthesis from 4-substituted sulfonyl-1,2,3-triazoles. | ||
Following this initial publication, a domino reaction involving a regioselective 1,2-aminohydroxlation of terminal alkynes in tandem with a [3,3]-sigmatropic rearrangement was published by Murakami and Miura, providing access to α-substituted α-amino ketones in one-pot from substituted triazoles (156) (Scheme 34).93 In this later investigation, water in the reaction medium was replaced by substituted allylic/propargylic alcohols. Notably, α-substituted allylic alcohols provided the E-alkene product exclusively, supporting the authors’ proposed transition state with an α-substituent in the pseudo-equatorial position for a six-membered chair (165) (Scheme 35). In addition, (S)-1-methyl-2-propen-1-ol (159) (97% ee) was used providing the expected product (160) in 93% yield with good chirality transfer (91% ee); this approach was later applied to the enantioselective synthesis of (−)-α-conhydrine.94 Lee also exploited this chemistry to form α-substituted α-amino ketones with a view to forming γ-oxo-β-amino acids and δ-oxo-γ-amino acids.95,96
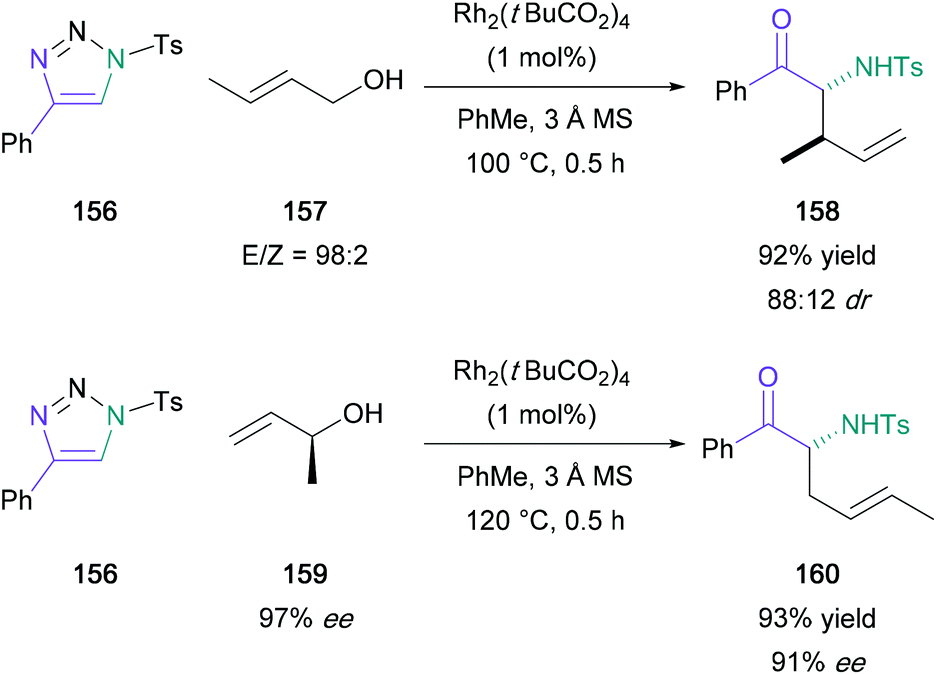 | ||
| Scheme 34 Murakami's rhodium-catalysed domino 1,2-aminohydroxlation/[3,3]-sigmatropic rearrangement. | ||
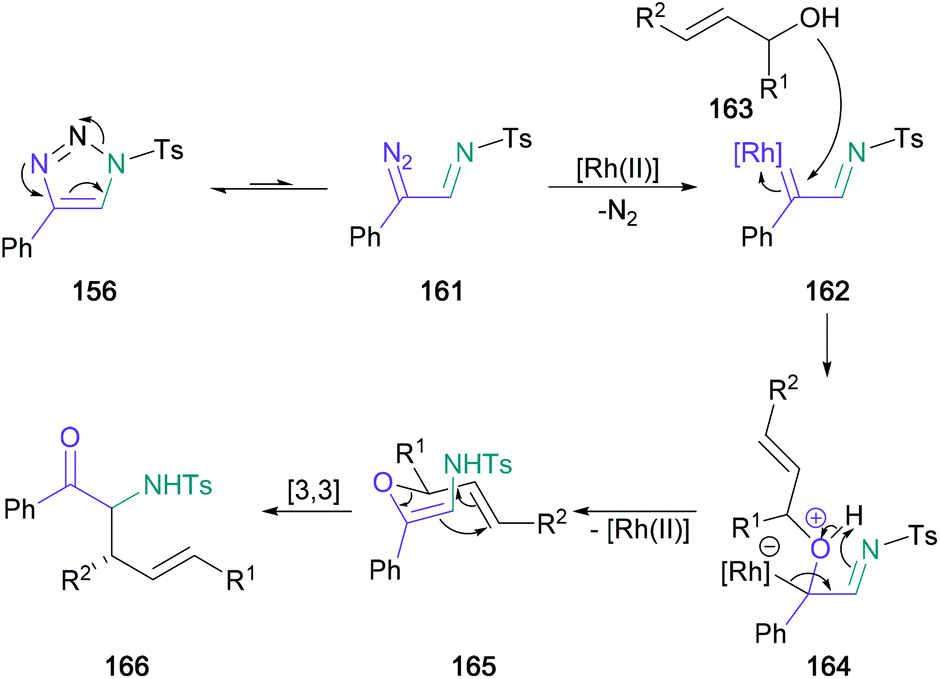 | ||
| Scheme 35 Mechanistic proposal for Murakami's rhodium-catalysed domino 1,2-aminohydroxlation/[3,3]-sigmatropic rearrangement. | ||
Further building on this approach, Bi investigated the 1,2-aminohydroxylation with a concomitant [1,3]-sigmatropic shift.97 This formal [1,3]-shift provided access to α-substituted α-amino ketones without a dependence on unsaturated alcohol starting materials, a requirement in Murakami's work. After a successful initial study with 2-phenyl-2-propanol and a range of substituted triazoles, Bi found that primary, secondary, and tertiary alcohols were also suitable for this transformation, producing the expected products in up to 91% yield. This domino reaction was developed further by Dong and Feng, who reported an asymmetric [1,3]-rearrangement of the amino enol ether intermediate (Scheme 36).98 Phenyl-substituted triazoles (167) were successfully reacted with a range of substituted-benzyl alcohols (168) in the presence of both rhodium and a chiral indium catalyst, and the corresponding α-substituted α-amino ketone products (170) were isolated in 72–93% yield and 82–96% ee for phenyl-substituted derivatives. Based on mechanistic studies, the authors proposed that the chiral N,N′-dioxide-indium(III) species coordinates to the enol ether oxygen, weakening the bond and blocking the Si face of the enolate.
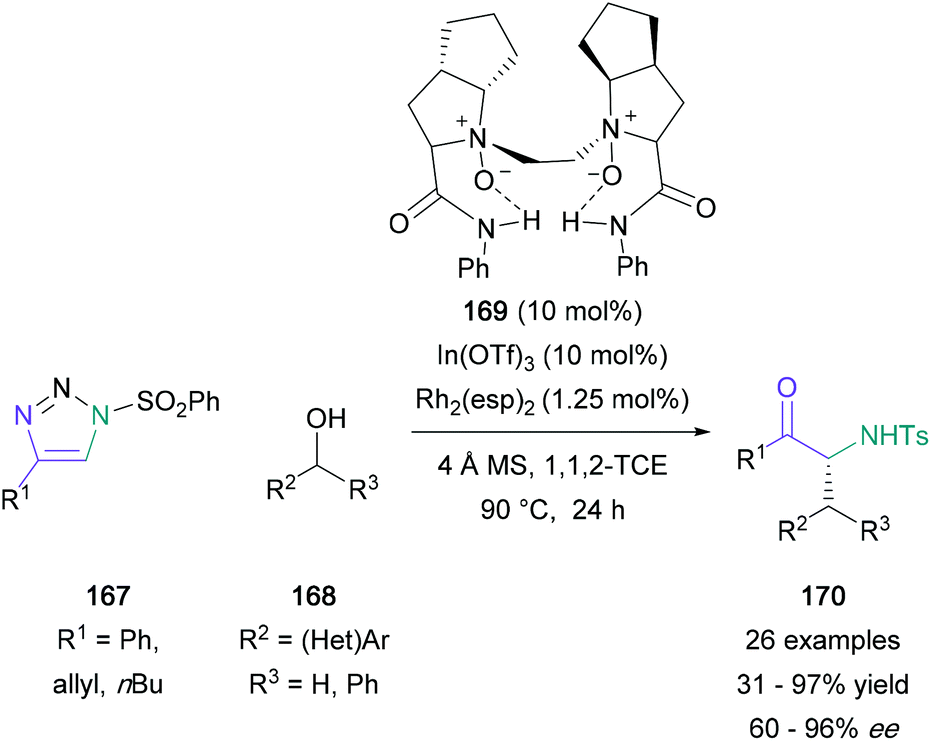 | ||
| Scheme 36 Dong and Feng's dual indium/rhodium-catalysed domino 1,2-aminohydroxlation/[1,3]-sigmatropic rearrangement. | ||
While the chemistry of triazoles has been heavily exploited in the synthesis of α-amino ketones, other nitrogen-containing heterocycles have not received the same level of attention. Azetidin-3-ones and their corresponding alcohol derivatives, for instance, represent an interesting category of starting materials, as they contain the required 1,2-relationship between the oxygen and nitrogen functionalities. As such, the development of ring opening procedures for these easily accessible heterocycles can lend itself to novel approaches towards α-amino ketones.
The first protocol in this class involved ring opening of 2-alkyl-azetidin-3-ones (171) with titanium(IV) to generate a diverse range of α-amino ketone derivatives (Scheme 37).99 Treatment with titanium(IV) iodide leads predominantly to a reductive ring opening at the less hindered position (173), whereas the addition of an equal amount of titanium(IV) chloride/bromide reverses this regioselectivity (172). Furthermore, the sole use of titanium(IV) bromide leads to α-amino-α′-bromo ketones (174), whereas using similar conditions on O-alkyl oxime analogues (175) leads to products with the opposite regioselectivity (176). The high versatility of this methodology is provided by the various mechanistic pathways encountered therein.
 | ||
| Scheme 37 Titanium(IV)-mediated ring opening of 2-alkyl-azetidin-3-ones. | ||
In contrast to these reductive or redox-neutral conditions, Parsons disclosed an oxidative deconstruction process that converted tertiary aryl-/heteroaryl-substituted azetidin-3-ols (177) to the corresponding primary α-amino ketones (178) with the loss of one carbon atom (Scheme 38).100 In the presence of the silver(I)/persulfate oxidant pair, the desired products were obtained in up to 80% yield. This reaction was compatible with a variety of aromatic substitution patterns (179 and 180) and heterocyclic scaffolds (181) and offers a new retrosynthetic disconnection for pharmaceutically relevant aryl-α-amino ketones.
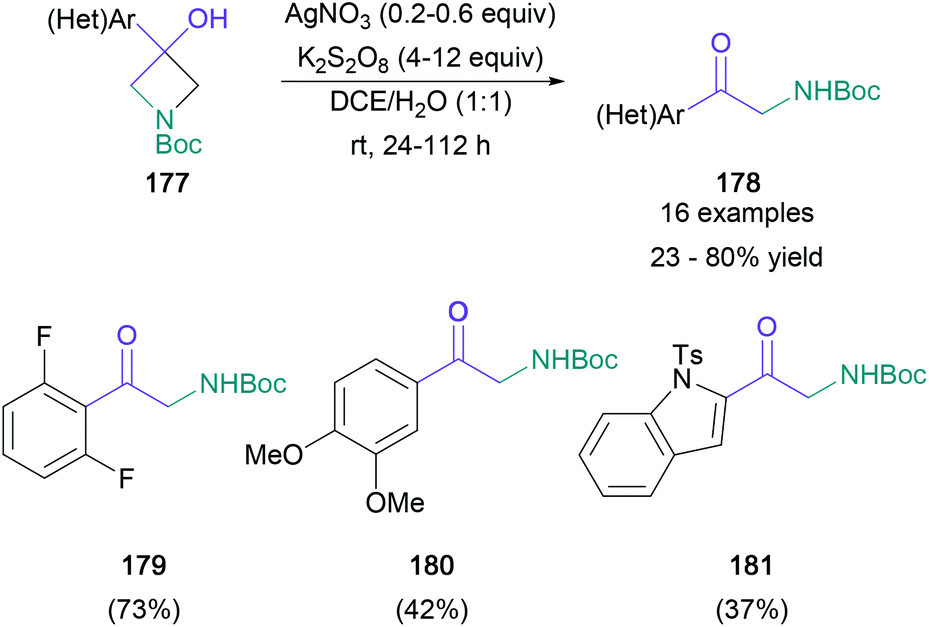 | ||
| Scheme 38 Parsons’ oxidative deconstruction of azetidinols. | ||
3. Conclusions and future outlook
This review collates and compares novel procedures for the synthesis of amino ketones published in recent years (2011–2020). In the surveyed period, there are two notable themes. The first theme is a shift away from traditional bond disconnections (Scheme 2), as alternative functional groups, including unsaturated hydrocarbons and nitrogen-heterocycles, are clearly shown to be feasible precursors. We anticipate that the expanded repertoire of procedures will allow for the practical introduction of the desired amino ketone motif into a diverse range of complex scaffolds. The second theme is a significant improvement of green metrics for the synthesis of this high-value synthon, which is in line with the global anticipated paradigm shift towards reaction design based on the Twelve Principles of Green Chemistry.32–34 The need for subsequent functional group modification steps was substituted with protocols achieving direct access to the desired motif, and the requirement for stoichiometric quantities of toxic or hazardous reagents was replaced with more economical and environmentally benign transition-metal-free or catalytic protocols. Remarkably, the application of alternative synthetic techniques such as electrochemical, photochemical, or biocatalytic protocols101,102 addressed some of the aforementioned shortcomings and with their potential to access otherwise challenging reactivity, and further improve on environmental factors, we expect further advances within these areas in the near future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The award of an EPSRC President's Scholarship (to P. N.) is gratefully acknowledged. Additional funding from Dr Isabel Bader and her late husband Dr Alfred Bader is also gratefully recognised. This review is dedicated to the late philanthropist Dr Alfred Bader and also in memory of the late Oxana Bennett.References
- J. Concellon and H. Rodriguez-Solla, Curr. Org. Chem., 2008, 12, 524–543 CrossRef CAS.
- B. E. Blough, A. Landavazo, J. S. Partilla, M. H. Baumann, A. M. Decker, K. M. Page and R. B. Rothman, ACS Med. Chem. Lett., 2014, 5, 623–627 CrossRef CAS PubMed.
- P. C. Meltzer, D. Butler, J. R. Deschamps and B. K. Madras, J. Med. Chem., 2006, 49, 1420–1432 CrossRef CAS PubMed.
- M. C. Myers, J. Wang, J. A. Iera, J. Bang, T. Hara, S. Saito, G. P. Zambetti and D. H. Appella, J. Am. Chem. Soc., 2005, 127, 6152–6153 CrossRef CAS PubMed.
- K. F. Foley and N. V. Cozzi, Drug Dev. Res., 2003, 60, 252–260 CrossRef CAS.
- R. Kolanos, J. S. Partilla, M. H. Baumann, B. A. Hutsell, M. L. Banks, S. S. Negus and R. A. Glennon, ACS Chem. Neurosci., 2015, 6, 771–777 CrossRef CAS PubMed.
- S. W. Toennes, S. Harder, M. Schramm, C. Niess and G. F. Kauert, Br. J. Clin. Pharmacol., 2003, 56, 125–130 CrossRef CAS PubMed.
- D. M. Perrine, J. T. Ross, S. J. Nervi and R. H. Zimmerman, J. Chem. Educ., 2000, 77, 1479 CrossRef CAS.
- T. Silverstone, Drugs, 1992, 43, 820–836 CrossRef CAS PubMed.
- A. T. Nchinda, K. Chibale, P. Redelinghuys and E. D. Sturrock, Bioorg. Med. Chem. Lett., 2006, 16, 4612–4615 CrossRef CAS PubMed.
- R. G. Almquist, W.-R. Chao, M. E. Ellis and H. L. Johnson, J. Med. Chem., 1980, 23, 1392–1398 CrossRef CAS PubMed.
- P. Langer and A. Bodtke, Tetrahedron Lett., 2003, 44, 5965–5967 CrossRef CAS.
- T. N. Sorrell and W. E. Allen, J. Org. Chem., 1994, 59, 1589–1590 CrossRef CAS.
- D. E. Frantz, L. Morency, A. Soheili, J. A. Murry, E. J. J. Grabowski and R. D. Tillyer, Org. Lett., 2004, 6, 843–846 CrossRef CAS PubMed.
- J. Huang, L. Luo, N. Xing, L. Gu, C. Li, Q. Han, S. Zheng and L. He, RSC Adv., 2020, 10, 13815–13819 RSC.
- H. Iida, K. Hayashida, M. Yamada, K. Takahashi and K. Yamada, Synth. Commun., 1973, 3, 225–230 CrossRef CAS.
- T. Chiba, H. Sakagami, M. Murata and M. Okimoto, J. Org. Chem., 1995, 60, 6764–6770 CrossRef CAS.
- L. Knorr, Ber. Dtsch. Chem. Ges., 1884, 17, 1635–1642 CrossRef.
- L. Knorr, Justus Liebigs Ann. Chem., 1886, 236, 290–332 CrossRef.
- F. D. Klingler, Acc. Chem. Res., 2007, 40, 1367–1376 CrossRef CAS PubMed.
- S. K. Gediya, G. J. Clarkson and M. Wills, J. Org. Chem., 2020, 85, 11309–11330 CrossRef CAS PubMed.
- W. Wu, C. You, C. Yin, Y. Liu, X. Q. Dong and X. Zhang, Org. Lett., 2017, 19, 2548–2551 CrossRef CAS PubMed.
- D. J. Ager, I. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835–876 CrossRef CAS PubMed.
- L. E. Fisher and J. M. Muchowski, Org. Prep. Proced. Int., 1990, 22, 399–484 CrossRef CAS.
- E. Erdik, Tetrahedron, 2004, 60, 8747–8782 CrossRef CAS.
- C. Greck, B. Drouillat and C. Thomassigny, Eur. J. Org. Chem., 2004, 2004, 1377–1385 CrossRef.
- J. M. Janey, Angew. Chem., Int. Ed., 2005, 44, 4292–4300 CrossRef CAS PubMed.
- P. Selig, Angew. Chem., Int. Ed., 2013, 52, 7080–7082 CrossRef CAS PubMed.
- A. M. R. Smith and K. K. (Mimi) Hii, Chem. Rev., 2011, 111, 1637–1656 CrossRef CAS PubMed.
- T. Vilaivan and W. Bhanthumnavin, Molecules, 2010, 15, 917–958 CrossRef CAS PubMed.
- M. Shang, X. Wang, S. M. Koo, J. Youn, J. Z. Chan, W. Yao, B. T. Hastings and M. Wasa, J. Am. Chem. Soc., 2017, 139, 95–98 CrossRef CAS PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- I. T. Horváth and P. T. Anastas, Chem. Rev., 2007, 107, 2169–2173 CrossRef PubMed.
- C. J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197–13202 CrossRef CAS PubMed.
- D. M. Scarpino Schietroma, M. R. Monaco, V. Bucalossi, P. E. Walter, P. Gentili and M. Bella, Org. Biomol. Chem., 2012, 10, 4692–4695 RSC.
- Y. Wei, S. Lin and F. Liang, Org. Lett., 2012, 14, 4202–4205 CrossRef CAS PubMed.
- Y. Kumar, Y. Jaiswal, R. Thakur and A. Kumar, ChemistrySelect, 2018, 3, 5614–5619 CrossRef CAS.
- M. Lamani and K. R. Prabhu, Chem. – Eur. J., 2012, 18, 14638–14642 CrossRef CAS PubMed.
- Q. Jiang, B. Xu, A. Zhao, J. Jia, T. Liu and C. Guo, J. Org. Chem., 2014, 79, 8750–8756 CrossRef CAS PubMed.
- Y. Lv, Y. Li, T. Xiong, Y. Lu, Q. Liu and Q. Zhang, Chem. Commun., 2014, 50, 2367–2369 RSC.
- R. W. Evans, J. R. Zbieg, S. Zhu, W. Li and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 16074–16077 CrossRef CAS PubMed.
- W.-G. Jia, D.-D. Li, Y.-C. Dai, H. Zhang, L.-Q. Yan, E.-H. Sheng, Y. Wei, X.-L. Mu and K.-W. Huang, Org. Biomol. Chem., 2014, 12, 5509–5516 RSC.
- T. V. Tran, H. T. N. Le, H. Q. Ha, X. N. T. Duong, L. H. T. Nguyen, T. L. H. Doan, H. L. Nguyen and T. Truong, Catal. Sci. Technol., 2017, 7, 3453–3458 RSC.
- S. Liang, C.-C. Zeng, H.-Y. Tian, B.-G. Sun, X.-G. Luo and F. Ren, J. Org. Chem., 2016, 81, 11565–11573 CrossRef CAS PubMed.
- M. N. Vander Wal, A. K. Dilger and D. W. C. MacMillan, Chem. Sci., 2013, 4, 3075–3079 RSC.
- B. Xu, S.-F. Zhu, X.-D. Zuo, Z.-C. Zhang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2014, 53, 3913–3916 CrossRef CAS PubMed.
- Z. Wang, Compr. Org. Name React. Reagents, 2010, 1403–1407 Search PubMed.
- A. Frongia, F. Secci, F. Capitta, P. P. Piras and M. L. Sanna, Chem. Commun., 2013, 49, 8812–8814 RSC.
- D. J. Aitken, P. Caboni, H. Eijsberg, A. Frongia, R. Guillot, J. Ollivier, P. P. Piras and F. Secci, Adv. Synth. Catal., 2014, 356, 941–945 CrossRef CAS.
- M. Farahi, F. Tamaddon, B. Karami and S. Pasdar, Tetrahedron Lett., 2015, 56, 1887–1890 CrossRef CAS.
- F. Tamaddon and A. Dehghani Tafti, Synlett, 2016, 2217–2220 CrossRef CAS.
- L. Li, Q. Zeng, G. Li and Z. Tang, Synlett, 2019, 694–698 CAS.
- C. Liu, Z. Fang, Z. Yang, Q. Li, S. Guo and K. Guo, RSC Adv., 2016, 6, 25167–25172 RSC.
- S. Guha, V. Rajeshkumar, S. S. Kotha and G. Sekar, Org. Lett., 2015, 17, 406–409 CrossRef CAS PubMed.
- M. Muneeswara, A. Muthukumar and G. Sekar, ChemistrySelect, 2018, 3, 12524–12529 CrossRef CAS.
- Y. Kim, H. K. Pak, Y. H. Rhee and J. Park, Chem. Commun., 2016, 52, 6549–6552 RSC.
- D. A. Dirocco and T. Rovis, Angew. Chem., Int. Ed., 2012, 51, 5904–5906 CrossRef CAS PubMed.
- S. M. Mennen, J. D. Gipson, Y. R. Kim and S. J. Miller, J. Am. Chem. Soc., 2005, 127, 1654–1655 CrossRef CAS PubMed.
- L. H. Sun, Z. Q. Liang, W. Q. Jia and S. Ye, Angew. Chem., Int. Ed., 2013, 52, 5803–5806 CrossRef CAS PubMed.
- T. Kawasaki, N. Ishida and M. Murakami, J. Am. Chem. Soc., 2020, 142, 3366–3370 CrossRef CAS PubMed.
- R. C. Wende, A. Seitz, D. Niedek, S. M. M. Schuler, C. Hofmann, J. Becker and P. R. Schreiner, Angew. Chem., Int. Ed., 2016, 55, 2719–2723 CrossRef CAS PubMed.
- C. L. Joe and A. G. Doyle, Angew. Chem., Int. Ed., 2016, 55, 4040–4043 CrossRef CAS PubMed.
- L. Wang, P. Cheng, X. Wang, W. Wang, J. Zeng, Y. Liang and O. Reiser, Org. Chem. Front., 2019, 6, 3771–3775 RSC.
- I. Ramakrishna, V. Bhajammanavar, S. Mallik and M. Baidya, Org. Lett., 2017, 19, 516–519 CrossRef CAS PubMed.
- T. Oohara, H. Nambu, M. Anada, K. Takeda and S. Hashimoto, Adv. Synth. Catal., 2012, 354, 2331–2338 CrossRef CAS.
- Z. Zhou, Q.-Q. Cheng and L. Kürti, J. Am. Chem. Soc., 2019, 141, 2242–2246 CrossRef CAS PubMed.
- Y. Han and E. J. Corey, Org. Lett., 2019, 21, 283–286 CrossRef CAS PubMed.
- P. Mizar and T. Wirth, Angew. Chem., Int. Ed., 2014, 53, 5993–5997 CrossRef CAS PubMed.
- X. Xia, B. Chen, X. Zeng and B. Xu, Org. Biomol. Chem., 2018, 16, 6918–6922 RSC.
- Z. T. He and J. F. Hartwig, Nat. Chem., 2019, 11, 177–183 CrossRef CAS PubMed.
- T. Sun, G. Hou, M. Ma and X. Zhang, Adv. Synth. Catal., 2011, 353, 253–256 CrossRef CAS.
- W. Gao, Q. Wang, Y. Xie, H. Lv and X. Zhang, Chem. – Asian J., 2016, 11, 231–233 CrossRef CAS PubMed.
- M. Amézquita-Valencia, R. Ramírez-Garavito, R. A. Toscano and A. Cabrera, Catal. Commun., 2013, 33, 29–33 CrossRef.
- C. Sun, M. J. O'Connor, D. Lee, D. J. Wink and R. D. Milligan, Angew. Chem., Int. Ed., 2014, 53, 3197–3200 CrossRef CAS PubMed.
- F. Colpaert, S. Mangelinckx, B. Denolf and N. De Kimpe, J. Org. Chem., 2012, 77, 6023–6032 CrossRef CAS PubMed.
- M. J. García-Muñoz, H. K. Dema, F. Foubelo and M. Yus, Tetrahedron: Asymmetry, 2014, 25, 362–372 CrossRef.
- W. Wen, Y. Zeng, L. Y. Peng, L. N. Fu and Q. X. Guo, Org. Lett., 2015, 17, 3922–3925 CrossRef CAS PubMed.
- X. Zhang, R. J. Staples, A. L. Rheingold and W. D. Wulff, J. Am. Chem. Soc., 2014, 136, 13971–13974 CrossRef CAS.
- X. Zhang, Y. Dai and W. D. Wulff, Synlett, 2018, 2015–2018 CAS.
- M. Feurer, G. Frey, H.-T. Luu, D. Kratzert and J. Streuff, Chem. Commun., 2014, 50, 5370–5372 RSC.
- H. Chen, G. Fan, S. Li, K. Mao and Y. Liu, Tetrahedron Lett., 2014, 55, 1593–1596 CrossRef CAS.
- D. H. Miles, J. Guasch and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 7632–7635 CrossRef CAS PubMed.
- D. Yadagiri and P. Anbarasan, Chem. Commun., 2015, 51, 14203–14206 RSC.
- M. H. Shinde and U. A. Kshirsagar, Org. Biomol. Chem., 2016, 14, 858–861 RSC.
- S. Xu, P. Wu and W. Zhang, Org. Biomol. Chem., 2016, 14, 11389–11395 RSC.
- P. K. Prasad, R. N. Reddi and A. Sudalai, Org. Lett., 2016, 18, 500–503 CrossRef CAS PubMed.
- M. Li, L. Zhang, B. Zhao and F. Liang, RSC Adv., 2016, 6, 93325–93329 RSC.
- T. Wei, Y. Zeng, W. He, L. Geng and L. Hong, Chin. Chem. Lett., 2019, 30, 383–385 CrossRef CAS.
- Z. Zhang, Y. Luo, H. Du, J. Xu and P. Li, Chem. Sci., 2019, 10, 5156–5161 RSC.
- Q. Qin, Y. Y. Han, Y. Y. Jiao, Y. He and S. Yu, Org. Lett., 2017, 19, 2909–2912 CrossRef CAS PubMed.
- G. Zheng, Y. Li, J. Han, T. Xiong and Q. Zhang, Nat. Commun., 2015, 6, 1–9 Search PubMed.
- T. Miura, T. Biyajima, T. Fujii and M. Murakami, J. Am. Chem. Soc., 2012, 134, 194–196 CrossRef CAS.
- T. Miura, T. Tanaka, T. Biyajima, A. Yada and M. Murakami, Angew. Chem., Int. Ed., 2013, 52, 3883–3886 CrossRef CAS PubMed.
- T. Miura, T. Tanaka, Q. Zhao, S. G. Stewart and M. Murakami, Helv. Chim. Acta, 2017, 100, e1600320 CrossRef.
- H. J. Jeon, M. S. Kwak, D. J. Jung, J. Bouffard and S. G. Lee, Org. Biomol. Chem., 2016, 14, 11238–11243 RSC.
- D. J. Jung, H. J. Jeon, J. H. Lee and S. G. Lee, Org. Lett., 2015, 17, 3498–3501 CrossRef CAS PubMed.
- P. Mi, R. Kiran Kumar, P. Liao and X. Bi, Org. Lett., 2016, 18, 4998–5001 CrossRef CAS PubMed.
- Y. Chen, Y. Liu, Z. Li, S. Dong, X. Liu and X. Feng, Angew. Chem., 2020, 59, 8052–8056 CrossRef CAS PubMed.
- S. Ariga, S. Hata, D. Fukuda, T. Nishi, I. Hachiya and M. Shimizu, Heterocycles, 2012, 86, 1187–1210 CrossRef CAS.
- R.-C. Raclea, P. Natho, L. A. T. Allen, A. J. P. White and P. J. Parsons, J. Org. Chem., 2020, 85, 9375–9385 CrossRef CAS PubMed.
- S. W. Chun, M. E. Hinze, M. A. Skiba and A. R. H. Narayan, J. Am. Chem. Soc., 2018, 140, 2430–2433 CrossRef CAS PubMed.
- S. W. Chun and A. R. H. Narayan, Synlett, 2019, 1269–1274 CAS.
| This journal is © The Royal Society of Chemistry 2021 |