
Synthesis of spirocyclic heterocycles from α,β-unsaturated N-acyliminium ions†
Thanphat
Thaima
 a,
Arife
Yazici
a,
Chiramet
Auranwiwat
ab,
Anthony C.
Willis
c,
Uta
Wille
d,
Thunwadee
Limtharakul
b and
Stephen. G.
Pyne
*a
a,
Arife
Yazici
a,
Chiramet
Auranwiwat
ab,
Anthony C.
Willis
c,
Uta
Wille
d,
Thunwadee
Limtharakul
b and
Stephen. G.
Pyne
*a
aSchool of Chemistry and Molecular Bioscience, University of Wollongong, Wollongong, New South Wales 2522, Australia. E-mail: spyne@uow.edu.au
bDepartment of Chemistry, Faculty of Science, and the Research Center on Chemistry for Development of Health Promoting Products from Northern Resources, Chiang Mai University, Chiang Mai 50200, Thailand
cResearch School of Chemistry, Australian National University, Canberra, ACT 2601, Australia
dSchool of Chemistry, Bio21 Institute, University of Melbourne, Parkville, Victoria 3010, Australia
First published on 11th November 2020
Abstract
The reactions of α,β-unsaturated N-acyliminium ions, generated in situ from 4(S)-O-substitutedhydroxy-5-hydroxy-5-vinyl-N-alkylpyrrolidin-2-ones, with allylsilanes and indoles leading to the formation of spirocyclic heterocycles, are reported. Six single crystal X-ray structures and extensive 2D NMR experiments confirmed the structures and stereochemistries of these products. In addition, computational studies provided mechanistic insights and an understanding of the stereochemical outcomes of these reactions.
Introduction
N-Acyliminium ions are important reactive intermediates in C–C and C-heteroatom bond forming reactions.1–7 Intermolecular versions have been extensively developed to provide novel heterocyclic ring structures.1,2,5,6 We recently demonstrated that α,β-unsaturated N-acyliminium ions8,9 and α-cyclopropyl N-acyliminium ions10 are attractive reactive intermediates for the one-pot synthesis of novel spirocyclic and bridged heterocycles (C, Scheme 1(a)). In an earlier communication we reported that the reactions of α,β-unsaturated N-acyliminium ions B, generated in situ from the treatment of 4(S)-benzyloxy-5-hydroxy-5-vinyl-N-alkylpyrrolidin-2-ones A with BF3·Et2O, with latent bis-nucleophiles (Nu1–Nu2), including allylsilanes and aromatic and heteroaromatic compounds, gave rise to spirocycles Cvia sequential conjugate (1,4-addition) and 1,2-addition reactions (Scheme 1(a)).8,9 We report here a full account of this work using allylsilanes and indoles including further examples aimed at increasing the diastereoselectivities and substrate scope of these reactions. Six single crystal X-ray structures and more extensive 2D NMR experiments confirmed the structures and stereochemistries of these products. In addition, density functional theory (DFT) studies provided mechanistic insights and a better understanding of the stereochemical outcomes of these reactions.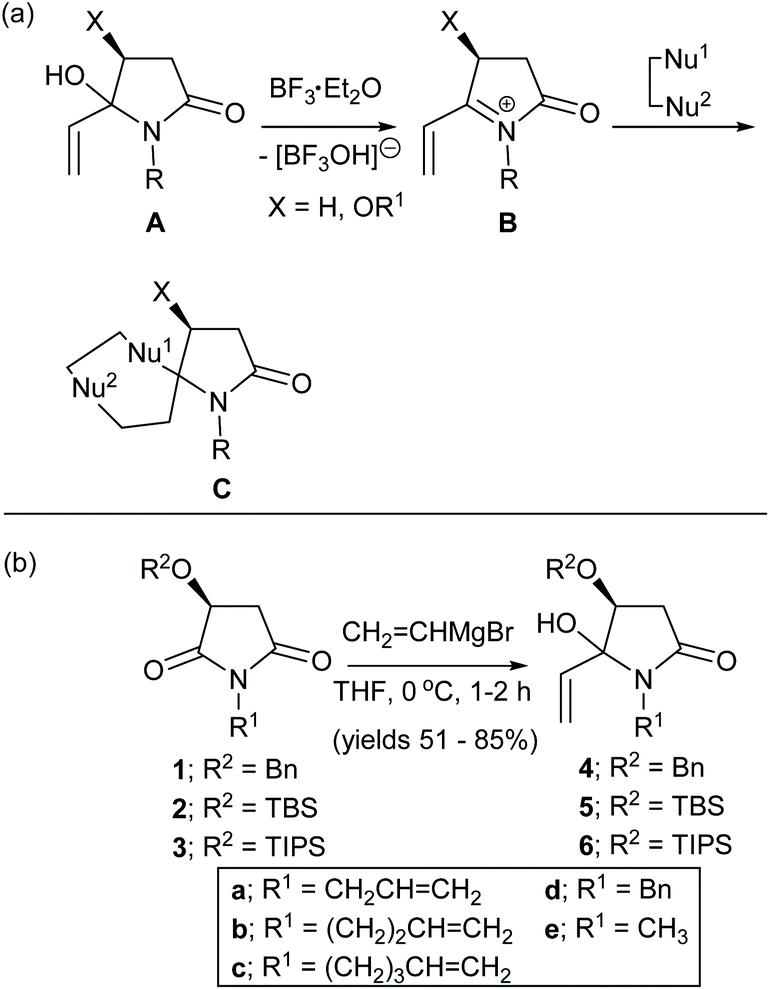 | ||
| Scheme 1 (a) Synthesis of spirocycles C from 5-vinylpyrrolidin-2-ones A and latent bis-nucleophiles (Nu1–Nu2) via α,β-unsaturated N-acyliminium ions B. (b) Synthesis of the α,β-unsaturated N-acyliminium ion precursors 4a-e, 5d,e and 6e; compounds 4a–d were prepared previously.8 | ||
Results and discussion
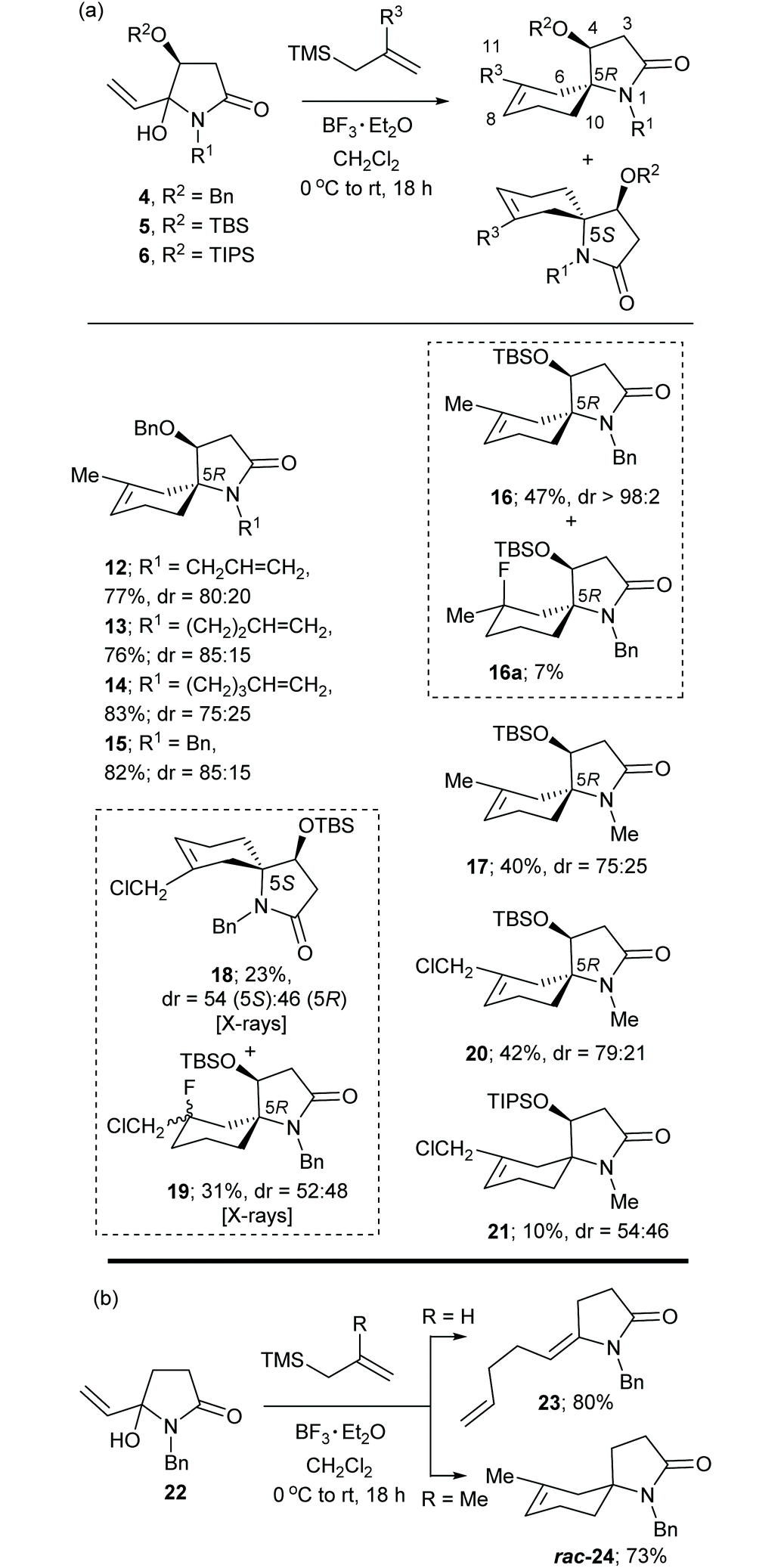
The α,β-unsaturated N-acyliminium ion precursors 4a–e, 5d,e and 6e were readily prepared from the O-protected 3(S)-hydroxy-N-alkylpyrrolidin-2,5-diones 1–3 by treatment with vinylmagnesium bromide in THF at 0 °C for 1–2 h (Scheme 1(b)). Compounds 4a–d were prepared previously using this method.8Compounds 4a–e, 5d,e and 6e were isolated in yields ranging from 51–85% as mixtures of diastereomers, which were used as such in subsequent reactions.
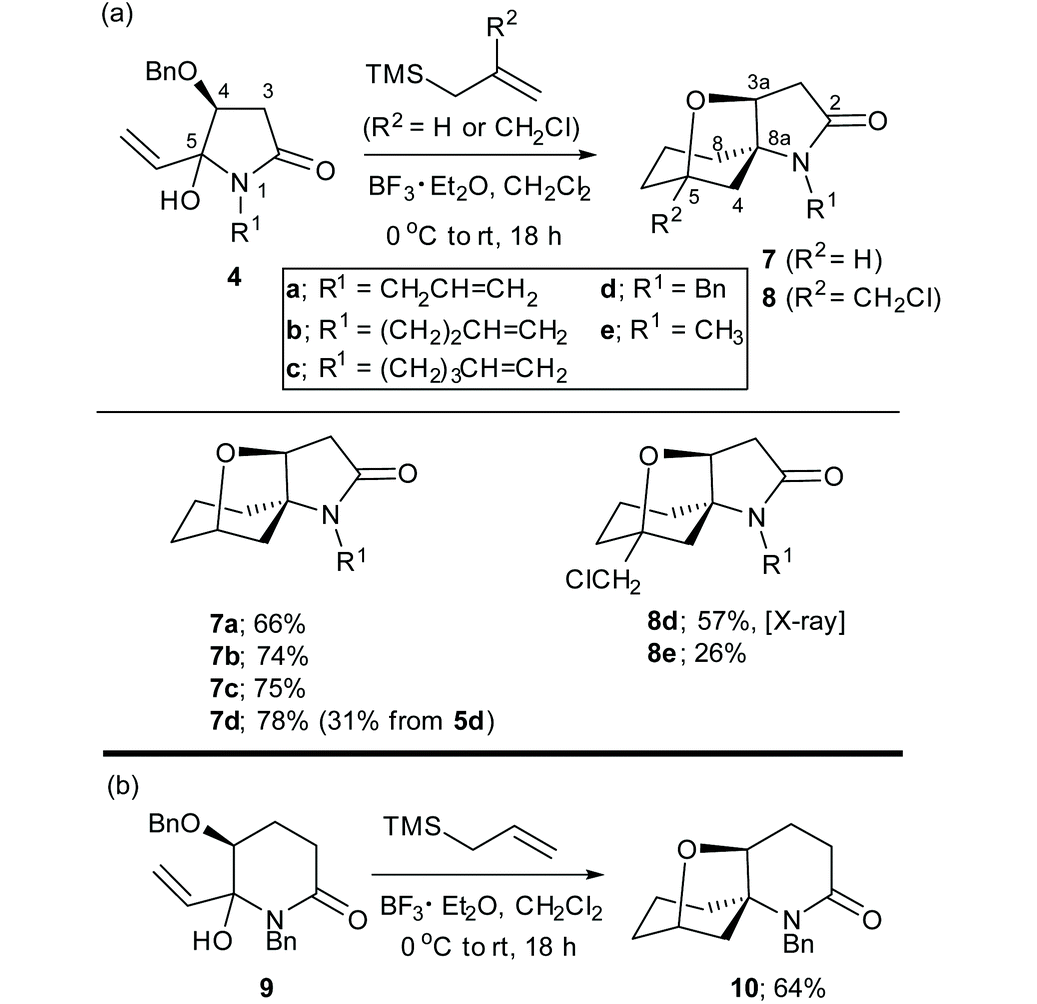
Under optimised reaction conditions,8,9 treatment of a solution of the N-benzyl-4(S)-benzyloxy-5-hydroxy-5-vinylpyrrolidin-2-ones 4a–d and allyltrimethylsilane (1.5 equiv.) at 0 °C with BF3·Et2O (2.0 equiv.) gave the spirocyclic products 7a–d in purified yields ranging from 66–78% as single diastereomers (Table 1(a)). The spirocyclic compound 7d could also be obtained from the OTBS analogue of 4d, (compound 5d) but in a much lower yield of 31%. Treatment of 4d or 4e with 2-chloromethylallyltrimetylsilane in the presence of BF3·Et2O provided access to the more highly functionalized spirocycles 8d and 8e, respectively.
|
The absolute configuration of 8d was established by X-ray diffraction of a single crystal as shown in Fig. 1. Comparisons of the 1H and 13C NMR chemical shifts for compounds 7a–d and 8a,b indicated that they all had the related spirocyclic core motif. In particular, the 1H NMR chemical shifts for H-4 (δHca. 4.4 ppm [J = ca. 8 Hz]) and H2-3 [δHca. 2.7 ppm (dd, J = ca. 17, 9 Hz) and δHca. 2.5 ppm (dd, J = ca. 17, 8 Hz)] and the 13C NMR chemical shifts for C-2 (δCca. 172 ppm), C-3 (δCca. 39 ppm), C-4 (δCca. 80 ppm) and C-5 (δCca. 70 ppm) were all in close agreement (see Table 1 for compound numbering). Thus, the structures that we proposed for compounds 7a–d in our original communication,8 for example compound 7d, was originally assigned as the isomeric structure 7d′ shown in Scheme 2, are incorrect. The original assignments were purely based on NMR experiments, because X-ray diffraction data of 8d were not available at that time, which would have allowed us to directly compare NMR data with earlier synthesised examples.
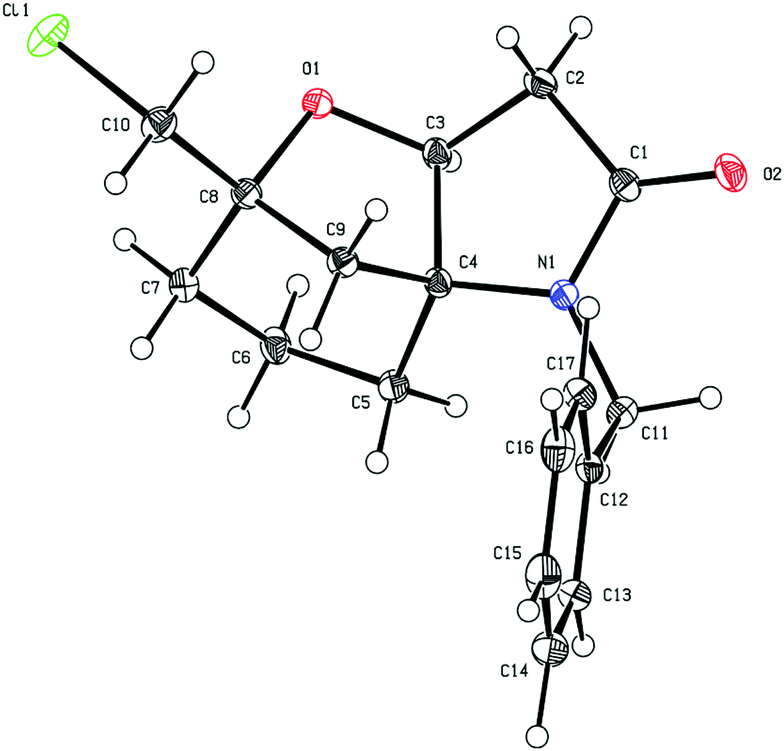 | ||
| Fig. 1 ORTEP plot of 8d, anisotropic displacement ellipsoids display 30% probability levels. Hydrogen atoms are drawn as circles with small radii (CCDC 2010550†). | ||
 | ||
| Scheme 2 Proposed intermediates in the synthesis of the spirocycle 7d. | ||
The reaction of the six-membered ring homologue 9 with allyltrimethylsilane/BF3·Et2O gave the related spirocyclic compound 10 (Table 1(b)). Notably, these reactions resulted in the loss of the O-benzyl protecting group (and the O-TBS group in the reaction of 5d to give 7d) with participation of the C-4 oxygen atom in the cyclization process to give a tetrahydrofuran ring. Related spirocyclic products were reported by Inouye et al. from the cyclization of two 5-(4-alkenyl)-3,4-dibenzyloxy-5-hydroxy-pyrrolidin-2-ones in formic acid.11
A suggested mechanism for the formation of products 7 and 8, as illustrated for the synthesis of 7d from 4d, is provided in Scheme 2. This mechanistic scheme involves formation of the (E)-enamide 11, via 1,4-addition of allyltrimethylsilane to the α,β-unsaturated N-acyliminium ion D, followed by protonation of this moiety to give the iminium ion E which undergoes cyclization with participation of the C-4 oxygen atom of the O-benzyl group to give the observed product 7d. The N-allyl analogue of (E)-enamide 11 (NOE correlation observed between the enamide alkene proton and the N-allyl methylene protons) has been isolated in 66% yield after work up of a reaction after a short reaction time. Subsequent treatment of this N-allyl enamide with BF3·Et2O under the standard reaction conditions also gave 7a.8
There are two possible modes of cyclization of E as shown in Scheme 2(b). The observed products (7/8) indicate that they arise from cyclization of the alkene moiety syn to the O-benzyl group of the N-acyliminium ion intermediate E (via reactive conformation E1) with participation by the O-benzyl group (either in a concerted or stepwise fashion) giving rise to a more stable cis-fused tetrahyrofuran ring product. Anti-attack to the O-benzyl group (via reactive conformation E2) would give the product 7d′ which was not observed.
The preference for syn over anti cyclisation has been evaluated using DFT calculations at the M062X/6-31+G* level of theory in dichloromethane for different substituents R1–R3 in E3 (Table 2). Exemplary single point calculations for one reaction system at the M062X/6-311++G**//M062X/6-31+G* level of theory revealed a very minor impact of the basis set size on the energy (entry 5), which demonstates that the smaller basis set predicts the energies associated with the competing pathways with sufficient precision.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | TS1 | ΔGsyn-spiro | TS2 | ΔGanti-cycl | ΔGanti-spiro |
| a M062X/6-31+G* free energies in kJ mol−1 relative to E3, in dichloromethane, free energies in italics from single point calculations using M062X/6-311++G** on M062X/6-31+G* optimised geometries. b Elongated newly formed C–C bond (1.63–1.66 Å). c “—” not determined. | ||||||||
| 1 | Me | Me | H | 47.0 | −47.9 | 57.4 | 56.7b | 24.7 |
| 2 | Me | TMS | H | 43.0 | −57.1 | 63.4 | 61.3b | —c |
| 3 | Me | Me | Me | 25.0 | −65.1 | 34.1 | 18.7 | 16.8 |
| 4 | Bn | Me | Me | 19.4 | −71.4 | 32.4 | 9.8 | — |
| 5 | Me | TMS | Me | 35.0 | −51.6 | 31.8 | 17.0 | 19.2 |
| 31.1 | −53.4 | 31.3 | 19.1 | 18.3 | ||||
| 6 | Me | TBS | Me | 33.9 | −36.5 | 32.8 | 17.4 | — |
| 7 | Me | Me | CH2Cl | 47.6 | −34.7 | 62.5 | 59.2b | — |
| 8 | Me | TMS | CH2Cl | 50.7 | −33.3 | 58.1 | 57.5b | — |

Animation of the vibration associated with the transition state of the syn cyclisation, TS1, shows formation of the C–C bond, which should lead to the (5S) configured spiro intermediate syn-cycl. However, it was not possible to locate a ground state structure of syn-cycl. Instead the tetrahydrofuran adduct syn-spiro was obtained, suggesting that the O–C bond forms after passing TS1. Formation of syn-spiro is exothermic for all R. As would be expected for an oxonium ion, the O–C bond distance in syn-spiro (1.563 Å) is longer than a typical O–C bond (1.43 Å).12 Subsequent loss of R2 to form the uncharged products of type 7/8 requires assistance by a nucleophile, such as F− (calculations not shown). The geometry of TS1 confirms a sequential process with a C–C distance of 1.996 Å (with H for R3 and Me for R1 and R2), which indicates an early transition state, and a distance between O and the cationic C of 2.658 Å (Fig. 2(a)). TS1 is strongly influenced by the nature of R3, where substituents that do not provide stabilisation to the developing positive charge on C-7, such as H (entries 1, 2) or CH2Cl (entries 7 and 8), raise the barrier for cyclisation by at least 10 kJ mol−1, compared with R3 = Me (entries 3–6). No significant influence of R1 on the energy profile can be found, which is due to the considerable conformational flexibility in these intermediates allowing the large N-benzyl group to rotate ‘out of the way’ (entry 3 vs. 4).
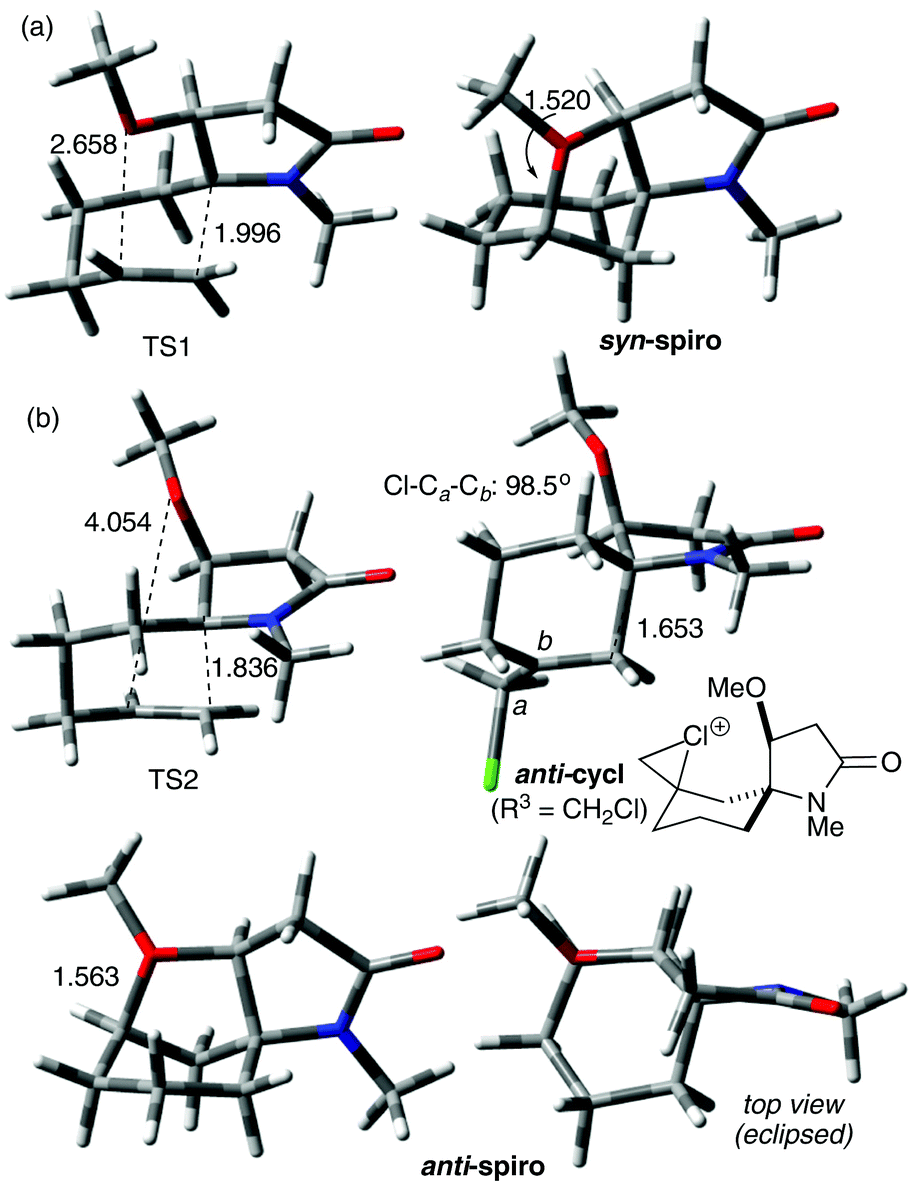 | ||
| Fig. 2 Optimised geometries for selected stationary points of the (a) syn- and (b) anti-cyclisation of N-acyliminium ion E calculated with M062X/6-31+G* in dichloromethane. Distances in Å. R1, R2 = Me, R3 = H, unless stated otherwise. | ||
In contrast, anti-cyclisation does not lead to the corresponding anti-spiro intermediate but gives the bicyclic anti-cycl, which possesses the (5R) configuration at the spiro centre, in an endothermic reaction. The different outcome can be rationalised by the fact that in the syn pathway the OR2 group is ideally positioned to close the tetrahydrofuran ring after passing TS1, whereas a similar cyclisation in anti-cycl requires significant conformational changes, since the OR2 group points away from the carbocation (the transition state for this process was not calculated). Because of the insufficient stabilisation of the carbocation, all isomeric anti-cycl shown in Table 2 feature an elongated C–C bond (ca. 1.63–1.66 Å), and stabilisation through rapid subsequent reactions, for example through deprotonation and formation of an alkene or trapping by a nucleophile, such as F− (see below) should occur. On the other hand, formation of anti-spiro is endothermic by >15 kJ mol−1 relative to E3, which is likely due to an energetically unfavourable eclipsed arrangement of the tetrahydrofuran and cyclohexyl ring that includes the R2 and R3 substituents (Fig. 2(b)). Such a constrained arrangement is absent in the syn-spiro intermediate (geometry not shown).
Comparison of TS1 and TS2 reveals that the latter is considerably higher in energy by 8–20 kJ mol−1 in those cases where the R3 substituent does not stabilise the developing carbocation very well (i.e., R3 = H, CH2Cl, entries 1, 2, 7 and 8). When R3 = Me, TS2 drops to about 33 ± 1 kJ mol−1 which is independent of the size of R2, whereas TS1 ranges from 20–35 kJ mol−1 where the higher energies are found for the bulkier TMS and TBS substituents (entries 3–6). These data suggest that the ether O could provide some stabilisation to the developing positive charge on C-7 in TS1 through electrostatic interactions, which become less important with increasing steric bulk of R2. In fact, analysis of the O–C distance in TS1 revealed an increase with increasing size of R2 from 2.788 Å (R2 = Me) < 2.850 Å (R2 = TMS) < 2.914 Å (R2 = TBS). In contrast, the O–C distance in TS2 is >4 Å (see Fig. 2(b)) which is too large to assist in the C–C bond forming process by stabilising the developing carbocation. Thus, in the case of silyl ethers, the barriers for the syn and anti cyclisation of E3 become essentially similar, with the anti pathway being potentially even slightly faster, and the reaction outcome depends on the stability of the resulting products and their subsequent reactions.
Interestingly, when R3 = H or CH2Cl, the corresponding anti-cycl is only about 2 kJ mol−1 lower in energy than TS2 (entries 1, 2, 7 and 8) which indicates a highly reversible cyclisation that could compete with other processes, for example alkene formation or trapping by a nucleophile. Fig. 2(b) shows exemplary the geometry of the anti-cycl intermediate of entry 7, which is characterised by a considerably compressed tetrahedral angle at the CH2Cl group (Cl–Ca–Cb 98.5°). While these data suggest that the chlorine atom stabilises, at least to some extent, the positive charge through formation of a chloronium ion, the high barrier TS2 clearly indicates that the electron-withdrawing effect of the chlorine atom outweighs the resonance effect.
To further probe the scope of these reactions substrates 4–6 having a Me or Bn group at R1, and a Bn, TBS or TIPS at R2 were studied to confirm the computational prediction that changing the Bn group at R2 for the more sterically demanding TBS or TIPS groups might prevent participation of the C-4 oxygen in the cyclization process giving rise to bicyclic products and perhaps with enhanced diastereoselectivities. Trimethylallylsilanes having a 2-Me or 2-CH2Cl substituent were also studied with the view of disfavouring participation by the C-4 oxygen substituent.
The results of this study using substrates 4–6 and 2-methyl- and 2-chloromethyl-allyltrimethylsilane are shown in Table 3(a). Substrates 4a–d reacted with 2-methylallyltrimethylsilane to give the bicyclic spirocyclic products 12–15, respectively in yields ranging from 76–83% and diastereoselectivities ranging from 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 to 85:15. In each case the major diastereomer could be isolated as a pure entity, while in the case of 15 both diastereomers could be separated to allow their individual characterization. The position of the double bond in both these isomers was indicated from the HMBC correlations between H-6 and H-10 and the spirocyclic carbon C-5. Our more recent X-ray crystal structures of the individual (5S) and (5R) diastereomers of 18 (Fig. 3 and 4, respectively) and the unexpected fluorinated products 19 (Fig. 5), both having the (5R)-configuration, further assisted with these structural assignments. Fluorinated products of type 19 obviously resulted from trapping of a cationic intermediate by fluoride ions, which are present in the reaction system through the Lewis acid BF3·Et2O. The crystal structure of (5S)-18 indicated the cyclohexene ring adopted a half-chair conformation in which the C–N bond of the pyrrolidinone ring was in a pseudo-axial position, while in (5R)-18 the corresponding C–N bond was in a pseudo-equatorial position of the half-chair cyclohexene ring. In the original communication8 the position of the double bond of the major isomer of compounds 12–15 was incorrectly assigned at C-6/C-7 based on the broad singlet multiplicity of the alkene proton resonance. The 1H and 13C NMR chemical shifts of the protons and carbons of the bicyclic ring core structure (except for those associated with positions 7, 8 and 11) of (5R)-15 (major diastereomer) were similar to those of (5R)-18 and those of (5S)-15 (minor diastereomer) were similar to those of (5S)-18. Of significance, were the similar and more shielded 13C NMR chemical shifts of C-6 and the similar and more deshielded ones for C-10 for (5R)-15 and (5R)-18 [(5R)-15: δC-6 26.2 and δC-10 37.4; (5R)-18: δC-6 25.6 and δC-10 32.6]. While for the related two (5S) compounds the C-6 resonance is significantly more deshielded and the C-10 resonance shielded relative to those in their 5R diastereomers [(5S)-15: δC-6 36.1 and δC-10 30.3; (5S)-18: δC-6 29.4 and δC-10 30.6]. Similar patterns in chemical shifts were observed in the 13C NMR spectra of compounds 12–15. Furthermore, NOESY NMR experiments on (5R)-15 showed correlations between H-3α (δH 2.68, dd, J = 17.3, 4.9 Hz) and H-6α (δH 1.63, d, J = 17.4 Hz); the same two correlations were also observed in the NOESY spectrum of (5R)-18. The distance between these protons in the single crystal structure of (5R)-18 was 2.37 Å, consistent with the observed NOEs (Fig. 5). In contrast, the NOESY NMR spectrum of (5S)-18 showed correlations between H-4 and H-6α and between H-3α and H-10β and no correlation between H-3α and H-6α, consistent with the observed inter-hydrogen distances measured from the X-ray structure (see Fig. 4 for specific details). The assignments of H-3α and H-3β in the aforementioned compounds were consistent with the above NOE correlations and their 1H NMR multiplicities and coupling constants [H-3α (dd, J = ca. 17, 5 Hz) and H-3β (d, J = ca. 17 Hz)] and the measured torsional angles (ϕ) between these protons and H-4 in the crystal structures of (5S)-18 [ϕ(H − 3α, H − 4) = −28.8o, ϕ(H − 3β, H − 4) = 90.8o] and (5R)-18 [ϕ(H − 3α, H − 4) = −34.9o, ϕ(H − 3β, H − 4) = 89.8o].
:25 to 85:15. In each case the major diastereomer could be isolated as a pure entity, while in the case of 15 both diastereomers could be separated to allow their individual characterization. The position of the double bond in both these isomers was indicated from the HMBC correlations between H-6 and H-10 and the spirocyclic carbon C-5. Our more recent X-ray crystal structures of the individual (5S) and (5R) diastereomers of 18 (Fig. 3 and 4, respectively) and the unexpected fluorinated products 19 (Fig. 5), both having the (5R)-configuration, further assisted with these structural assignments. Fluorinated products of type 19 obviously resulted from trapping of a cationic intermediate by fluoride ions, which are present in the reaction system through the Lewis acid BF3·Et2O. The crystal structure of (5S)-18 indicated the cyclohexene ring adopted a half-chair conformation in which the C–N bond of the pyrrolidinone ring was in a pseudo-axial position, while in (5R)-18 the corresponding C–N bond was in a pseudo-equatorial position of the half-chair cyclohexene ring. In the original communication8 the position of the double bond of the major isomer of compounds 12–15 was incorrectly assigned at C-6/C-7 based on the broad singlet multiplicity of the alkene proton resonance. The 1H and 13C NMR chemical shifts of the protons and carbons of the bicyclic ring core structure (except for those associated with positions 7, 8 and 11) of (5R)-15 (major diastereomer) were similar to those of (5R)-18 and those of (5S)-15 (minor diastereomer) were similar to those of (5S)-18. Of significance, were the similar and more shielded 13C NMR chemical shifts of C-6 and the similar and more deshielded ones for C-10 for (5R)-15 and (5R)-18 [(5R)-15: δC-6 26.2 and δC-10 37.4; (5R)-18: δC-6 25.6 and δC-10 32.6]. While for the related two (5S) compounds the C-6 resonance is significantly more deshielded and the C-10 resonance shielded relative to those in their 5R diastereomers [(5S)-15: δC-6 36.1 and δC-10 30.3; (5S)-18: δC-6 29.4 and δC-10 30.6]. Similar patterns in chemical shifts were observed in the 13C NMR spectra of compounds 12–15. Furthermore, NOESY NMR experiments on (5R)-15 showed correlations between H-3α (δH 2.68, dd, J = 17.3, 4.9 Hz) and H-6α (δH 1.63, d, J = 17.4 Hz); the same two correlations were also observed in the NOESY spectrum of (5R)-18. The distance between these protons in the single crystal structure of (5R)-18 was 2.37 Å, consistent with the observed NOEs (Fig. 5). In contrast, the NOESY NMR spectrum of (5S)-18 showed correlations between H-4 and H-6α and between H-3α and H-10β and no correlation between H-3α and H-6α, consistent with the observed inter-hydrogen distances measured from the X-ray structure (see Fig. 4 for specific details). The assignments of H-3α and H-3β in the aforementioned compounds were consistent with the above NOE correlations and their 1H NMR multiplicities and coupling constants [H-3α (dd, J = ca. 17, 5 Hz) and H-3β (d, J = ca. 17 Hz)] and the measured torsional angles (ϕ) between these protons and H-4 in the crystal structures of (5S)-18 [ϕ(H − 3α, H − 4) = −28.8o, ϕ(H − 3β, H − 4) = 90.8o] and (5R)-18 [ϕ(H − 3α, H − 4) = −34.9o, ϕ(H − 3β, H − 4) = 89.8o].
 | ||
| Fig. 3 X-ray crystal structure of (5S)-18, displayed in Mercury. Double-headed arrows show NOSEY correlation in solution (CCDC 2010551†). | ||
 | ||
| Fig. 4 X-ray crystal structure of (5R)-18, displayed in Mercury. Double-headed arrows show NOSEY correlation in solution (CCDC 2010552†). | ||
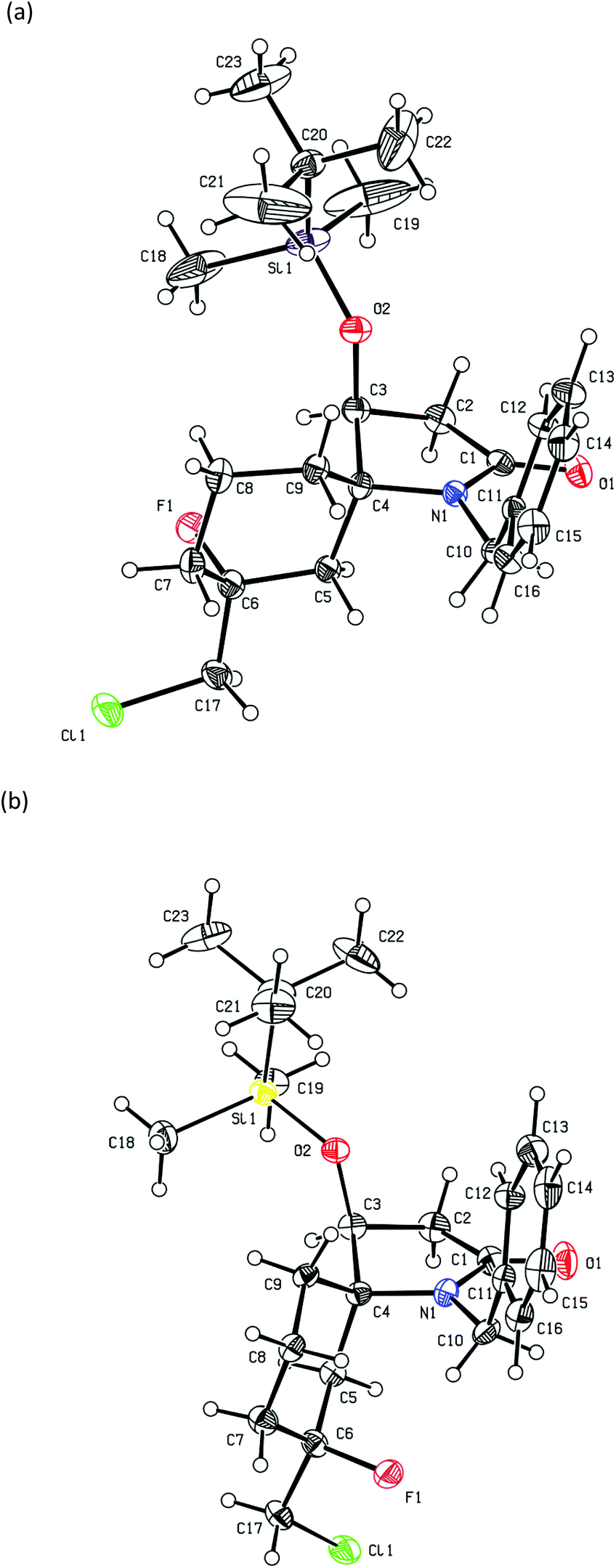 | ||
| Fig. 5 ORTEP plot of (a) (5R,7R)-19 (CCDC 2010552†) and (b) (5R,7S-19) (CCDC 2010553†), anisotropic displacement ellipsoids display 30% probability levels. Hydrogen atoms are drawn as circles with small radii. | ||
|
The OTBS analogue of 4d (5d) reacted with 2-methylallyltrimethylsilane under the standard reaction conditions to give (5R)-16 essentially as a single diastereomer indicating high facial control in the cyclization step due to the much bulkier TBS group. The 13C NMR chemical shifts for C-6 and C-10 for 16 (δC-6 25.9 and δC-10 36.7) were in close correlation with those of (5R)-12–15 (major) and (5R)-18. The NOESY spectrum of this compound showed correlations between H-3α (δH 2.75, dd, J 17.0, 5.1 Hz) and H-6α (δH 1.66, d, J = 17.6 Hz) and H-4 (δH 4.06, dd, J 5.1, 1.6 Hz) and H-6α which further supported the (5R)-configuration. While the yield of (5R)-16 was low, the presence of its (5S)-diastereomer could not be observed in the 1H NMR spectrum of the crude reaction product mixture or upon isolation by column chromatography. This reaction also produced the fluorinated compound 16a in 7% yield. This compound had the (5R) configuration from the NOESY correlation of H-4 with H-6. The N-Me analogue 5e was less diasteroselective giving 17 with a dr of 75:25. The major diastereomer was assigned as having the (5R)-configuration based on the 13C NMR chemical shifts of C-6 and C-10 (δC-6 25.9 and δC-10 36.7).
In contrast, the reactions of 5d (R1 = Bn) with 2-chloromethylallyl-trimetylsilane and BF3·Et2O gave some unexpected and contrasting results. This reaction gave a 54:46 mixture of (5S)-18 and (5R)-18, respectively in 23% yield along with a mixture of the fluorinated products 19 (dr = 52:48) in 31% yield. Compounds (5S)-18 and (5R)-18 could be separated by further purification by column chromatography and their single crystal structures are shown in Fig. 3 and 4, respectively. In contrast, the diastereomers of compound 19 could not be separated by column chromatography however they formed unique crystal types that could be separated manually allowing for their crystal structures to be determined and their individual spectroscopic data to be obtained. Their single crystal X-ray structures showed that both had the (5R)-configuration and were epimeric at C-7 (Fig. 5). In the solid state, the cyclohexane rings of these compounds adopted a chair-like conformation in which the C-7 fluorine substituent was axial (Me equatorial), with the C-5 N substituent adopting an equatorial and axial like position in (7R)-19 and (7S)-19, respectively on the cyclohexene ring. These products likely arise from trapping of the carbocation intermediate F (Scheme 3) by F−. Based on the isolated yields of 18 and 19 and the dr of 18 the estimated ratio of (5R)-F to (5S)-G (Scheme 3), formed in the initial cyclization reaction is 77:33. This stereochemical preference for the (5R) isomer is consistent with that found in products 12–15. Because of the sterically more hindered environment of the alkene group in the crystal structure of (5S)-18 (Fig. 3), over that of (5R)-18 (Fig. 4) the lack of fluorinated products from the former diastereomer might be expected and the carbocation G instead stabilises through alkene formation.
 | ||
| Scheme 3 Proposed intermediates in the synthesis of the spirocycles 12–18, 20 and 21. | ||
The major diastereomer of 20 (Table 3(a)) prepared from the reaction of 5e with 2-chloromethylallyltrimetylsilane and BF3·Et2O was secured after purification by column chromatography. Its (5R)-configuration was apparent from the 1D NOE correlation between H-4 (δH 4.07) and H10β (δH 1.46); this inter-hydrogen distance was 2.31 Å in the crystal structure of (5R)-18 (Fig. 4) and thus a NOE enhancement between these protons would be expected. In contrast, the reaction of 6e with 2-chloromethylallyltrimetylsilane and BF3·Et2O showed little preference for either the (5R) or (5S) diastereomer of the product 21 which was isolated as a mixture (dr 54:46) in 10% yield.
The racemic N-acyliminium ion precursor 22, lacking the C-4 oxygen substituent found in related compounds 4–6, underwent BF3·Et2O promoted reactions with allyltrimethyl- and 2-methylallylsilane to give the diene 23 and the spirocyclic product 24, respectively in good yields (Table 3(b)). The former result indicates that spirocyclization is not favoured when R = H due to the lack of carbocation stabilization in the spirocyclization step.
With the view of preparing spirocyclic scaffolds bearing two or more functional groups that could potentially allow the synthesis of chemical libraries for future drug discovery we examined the reactions of the chloromethylene compounds 8d and 20 with sodium azide (Scheme 4). We had anticipated that the corresponding azides 25 and 26 could be converted via to their respective amino derivatives which would be useful handles for further derivitization. Not surprisingly, treatment of the neopentyl-like chloride 8d with sodium azide in DMF at 60 °C for 96 h resulted in only recovered starting material. In contrast, the allylic chloride 20 (dr = 79:21) reacted with sodium azide in DMF at 80 °C over 5 h to give azide 26 (dr = 79:21) in 82% yield. This was converted to the primary amine 27 (dr = 79:21) in 50% yield via a Staudinger reaction12,13 as shown in Scheme 4.
 | ||
| Scheme 4 Synthesis of 26 and 27. | ||
In our earlier communication we reported the synthesis of a single spirocyclicindole derivative (28 in Table 4) in 45% yield (dr = 3:1) from the reaction of 4a and indole.8 We have now found that the analogous reaction of 4d, the N-Bn analogue of 4a, gave the spiroindole derivative 29 (dr = 2:1). The major diastereomer of this compound could be separated by column chromatography and gave suitable crystals for X-ray diffraction analysis, which established the (3R) configuration at the spirocarbon of 29 (Fig. 6). A related reaction between 4a and N-methylindole was also poorly diastreoselective (dr = 2:1), however the major diastereomer could be separated by column chromatography; its NOESY NMR spectrum showed a correlation between H-3′ and the N-Me group indicating the (3R) configuration (Table 4). In contrast, the analogous reaction between indole and the OTBS derivative 5d gave the spirocycle 31 in a highly diasteroselective fashion (dr = 9:1). Unfortunately NOESY or ROESY NMR experiments did not allow us to establish the relative configuration of 31. However, based on the similar 13C NMR chemical shifts for C-1 (δC 23.0 for 29 and δC 22.8 for 31) and C-2 (δC 33.9 for 29 and δC 33.4 for 31) in this compound and 29 we tentatively assign the (3R) configuration to compound 31.
 | ||
| Fig. 6 ORTEP plot of 29, anisotropic displacement ellipsoids display 30% probability levels. Hydrogen atoms are drawn as circles with small radii (CCDC 2010550†). | ||

|
Conclusions
The reactions of α,β-unsaturated N-acyliminium ions generated in situ from 4(S)-O-substitutedhydroxy-5-hydroxy-5-vinyl-N-alkylpyrrolidin-2-ones with allylsilanes and indoles led to the formation of spirocyclic heterocycles, often in a diastereoselective fashion. Six single crystal X-ray structures and extensive 2D NMR experiments helped confirm the structures and stereochemistries of these products. The spirocycle 20, incorporating a chloromethyl substituent, was shown to have potential as a scaffold for chemical library synthesis. In addition, computational studies provided mechanistic insights and helped an understanding of the stereochemical outcomes of these reactions.Experimental
Synthetic procedures and general methods
General methods were as described previously.8–10 The synthesis of compounds 1a–d, 4a–d, 7a–7d, 9, 10, 12–15 and 23 and rac-24 was described in detail earlier, including copies of their 1H and 13C NMR spectra.8(S)-3-((tert-Butyldimethylsilyl)oxy)-1-methylpyrrolidine-2,5-dione (2e)14 this is a known compound however no details of its preparation or spectroscopic data were given
To a solution of commercially sourced (S)-3-hydroxy-1-methylpyrrolidine-2,5-dione (1.291 g, 10.0 mmol, 1.0 equiv.) in CH2Cl2 (10 mL) was added imidazole (0.681 g, 15.0 mmol, 1.5 equiv.) followed by TBSCl (1.507 g, 15.0 mmol, 1.5 equiv.) and the reaction mixture was stirred at 50 °C for 24 h. Water (30 mL) was added and the mixture was extracted with EtOAc (2 × 50 mL). The organic layer was washed with brine, dried (MgSO4), filtered and concentrated in vacuo. The crude product was purified by column chromatography on silica gel eluting with 10:90 EtOAc/hexanes to afford the desired product as a white solid (2.127 g, 87%). [α]22D −55.7 (c 0.46, CHCl3). IR (neat): νmax 2964, 2893, 1699, 1378, 1247, 1034, 840775 cm−1. 1H NMR (500 MHz, CDCl3) δ 4.59 (dd, J = 8.2, 4.4 Hz, 1H, H-3), 3.01 (dd, J = 17.9, 8.2 Hz, 1H, H-4a), 3.00 (s, 3H, NCH3), 2.60 (dd, J = 17.9, 4.4 Hz, 1H, H-4b), 0.92 (s, 9H, (CH3)3CSi), 0.19 (s, 3H, CH3Si), 0.18 (s, 3H, CH3Si). 13C NMR (125 MHz, CDCl3) δ 177.0 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 174.6 (CO), 68.2 (C-3), 39.1 (C-4), 25.9 ((CH3)3CSi), 25.0 (CH3), 18.5 ((CH3)3CSi), −4.4 (CH3Si), −5.0 (CH3Si). HRMS (ESI): m/z calcd for C11H21NO3SiNa [M + Na]+: 266.1182; found: 266.1183.
O), 174.6 (CO), 68.2 (C-3), 39.1 (C-4), 25.9 ((CH3)3CSi), 25.0 (CH3), 18.5 ((CH3)3CSi), −4.4 (CH3Si), −5.0 (CH3Si). HRMS (ESI): m/z calcd for C11H21NO3SiNa [M + Na]+: 266.1182; found: 266.1183.
(S)-1-Methyl-3-((triisopropylsilyl)oxy)pyrrolidine-2,5-dione (3e)
To a solution of commercially sourced (S)-3-hydroxy-1-methylpyrrolidine-2,5-dione (0.571 g, 4.4 mmol, 1 equiv.) in CH2Cl2 (9 mL) was added DMAP (0.591 g, 4.8 mmol, 1.1 equiv.) and TIPSCl (1.272 g, 1.4 mL, 6.6 mmol, 1.5 equiv.) and the reaction mixture was stirred at 50 °C for 5 h. Water (10 mL) was added and the mixture was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with brine, dried (MgSO4), filtered and concentrated in vacuo. The crude product was purified by column chromatography on silica gel eluting with 10:90 EtOAc/hexanes to afford the desired product as a colourless oil (0.993 g, 79%). [α]22D −40.7 (c 0.91, CHCl3). IR (neat): νmax 2944, 2867, 1788, 1706, 1435, 1280, 1114, 881, 683 cm−1. 1H NMR (500 MHz, CDCl3) δ 4.71 (dd, J = 8.0, 4.4 Hz, 1H, H-4), 3.04 (dd, J = 17.9, 8.1 Hz, 1H, H-3), 3.00 (s, 3H, NCH3), 2.63 (dd, J = 17.8, 4.5 Hz, 1H, H-3), 1.22–1.05 (m, 21H, (CH3)2CHSi). 13C NMR (125 MHz, CDCl3) δ 176.6 (CO), 174.2 (CO), 68.1 (C-4), 39.4 (C-3), 24.7 (NCH3), 17.8 (3 × CH3), 17.7 (3 × CH3), 12.0 (3 × CH). HRMS (ESI): m/z calcd for C14H27NO3SiNa [M + Na]+: 308.1658; found: 308.1659.
General procedure for synthesis of compounds 4–6 (Scheme 1(b))
A solution of pyrrolidinedione 1, 2 or 3 (1 equiv.) in anhydrous THF (0.1 M) was cooled to 0 °C, and then vinylmagnesium bromide (1.0 M in THF, 1.5 equiv.) was added dropwise. After stirring for 1–2 h at 0 °C, the reaction mixture was quenched with sat. aqueous NH4Cl and the aqueous layer was extracted with EtOAc (three times). The combined organic layer was dried over MgSO4, filtered and concentrated in vacuo. The crude product was purified by column chromatography.(4S,5S)-4-(Benzyloxy)-5-hydroxy-1-methyl-5-vinylpyrrolidin-2-one and (4S,5R)-4-(benzyloxy)-5-hydroxy-1-methyl-5-vinylpyrrolidin-2-one (4e)
The title compound was prepared following the general procedure using 1e15 (0.877 g, 4.0 mmol, 1.0 equiv.), vinylmagnesium bromide (6.0 mL, 1.2 equiv.) and THF (40 mL). Purification by column chromatography on silica gel eluting with 50:50 EtOAc/hexanes gave the desired product as a pale yellow oil (0.745 g, 75%) and as a mixture of diastereomers (4.3:1). A small amount of the major isomer was obtained as a colourless oil in pure form by further separation using column chromatography. [α]22D +19.2 (c 0.26, CHCl3). IR (neat): νmax 3311, 3031, 2926, 2871, 1669, 1392, 1348, 1074, 984, 936, 736, 697 cm−1. 1H NMR (500 MHz, CDCl3) δ 7.37–7.27 (m, 6H, ArH), 6.02 (dd, J = 17.4, 10.8 Hz, 1H, H-6), 5.54 (d, J = 17.4 Hz, 1H, H-7a), 5.51 (d, J = 10.8 Hz, 1H, H-7b), 4.64 (d, J = 11.8 Hz, 1H, H-8a), 4.56 (d, J = 11.8 Hz, 1H, H-8b), 3.99 (dd, J = 7.3, 5.6 Hz, 1H, H-4, 2.76 (dd, J = 17.0, 7.3 Hz, 1H, H-3a), 2.71 (s, 3H, NCH3), 2.56 (s, 1H, OH), 2.44 (dd, J = 17.0, 5.6 Hz, 1H, H-3b). 13C NMR (125 MHz, CDCl3) δ 171.6 (CO), 137.4 (ArC), 134.6 (ArC), 128.7 (ArCH), 128.5 (ArCH), 128.0 (ArCH), 127.9 (ArCH), 127.7 (ArCH), 118.2 (C-7), 92.8 (C-5), 81.8 (C-4), 72.2 (PhCH2O), 36.5 (C-3), 24.3 (NCH3). HRMS (ESI): m/z calcd for C14H17NO3Na [M + Na]+: 270.1106; found: 270.1098.
(4S,5R)-1-Benzyl-4-((tert-butyldimethylsilyl)oxy)-5-hydroxy-5-vinylpyrrolidin-2-one (5d)
Prepared following the general procedure using 2d16 (2.23 g, 0.531 mmol), vinyl magnesium bromide (0.996 mL, 0.797 mmol) and THF (5.3 mL). Purification by column chromatography (3:7, EtOAc/hexanes) gave the desired product as a colourless viscous oil (93.7 mg, 48%) and as a mixture of diastereomers (3:1). A small amount of the each diastereomer was obtained in pure from by further column chromatography. Major isomer: 1H NMR (500 MHz, CDCl3) δ 7.30–7.20 (m, 5H, ArH), 5.64 (dd, J = 17.1, 10.3 Hz, 1H, CH), 5.46 (dd, J = 17.1, 1.3 Hz, 1H, CH2), 5.31 (dd, J = 10.3, 1.3 Hz, 1H, CH2), 4.56 (d, J = 15.3 Hz, 1H, NCH2), 4.29 (d, J = 15.3 Hz, 1H, NCH2), 4.09 (dd, J = 6.2, 3.8 Hz, 1H, H-4), 3.69 (s, 1H, OH), 2.69 (dd, 16.8, 6.2 Hz, 1H, H-3), 2.42 (dd, J = 16.8, 3.8 Hz, 1H, H-3), 0.88 (s, 9H CH3), 0.11 (s, 3H, CH3), 0.08 (s, 3H, CH3). 13C NMR (125 MHz, CDCl3) δ 172.0 (CO), 138.6 (ArC), 136.8 (CH), 128.3 (ArCH), 127.9 (ArCH), 127.0 (ArCH), 118.5 (CH2), 91.0 (C), 71.9 (CH), 43.2 (CH2), 38.6 (CH2), 25.7 (CH3), 18.2 (C), −4.6 (CH3), −4.9 (CH3). HRMS (ESI): m/z calcd for C19H29NO3SiNa [M + Na]+: 370.1814; found: 370.1808. Minor isomer: 1H NMR (500 MHz, CDCl3) δ 7.30–7.23 (m, 5H, ArH), 5.99 (dd, J = 17.5, 10.8 Hz, 1H, CH), 5.47 (dd, J = 17.5, 1.1 Hz, 1H, CH2), 5.41 (dd, J = 10.7, 1.1 Hz, 1H, CH2), 4.70 (d, J = 15.2 Hz, 1H, NCH2), 4.17 (d, J = 15.2 Hz, 1H, NCH2), 4.07 (dd, J = 5.7, 2.2 Hz, 1H, H-4), 2.90 (dd, J = 16.8, 5.7 Hz, 1H, H-3), 2.31 (ddd, J = 16.8, 2.2, 0.8 Hz, 1H, H-3), 0.87 (s, 9H, CH3), 0.05 (s, 3H, CH3), 0.02 (s, 3H, CH3). 13C NMR (125 MHz, CDCl3) δ 174.0 (CO), 138.3 (ArC), 136.5 (CH), 128.9 (ArCH), 128.1 (ArCH), 127.6 (ArCH), 118.5 (CH2), 94.1 (C), 75.7 (CH), 42.9 (CH2), 39.7 (CH2), 25.8 (CH3), 18.1 (C), −4.6 (CH3), −4.8 (CH3).
(4S,5S)-4-((tert-Butyldimethylsilyl)oxy)-5-hydroxy-1-methyl-5-vinylpyrrolidin-2-one and (4S,5R)-4-((tert-butyldimethylsilyl)oxy)-5-hydroxy-1-methyl-5-vinylpyrrolidin-2-one (5e)
The title compound was prepared following the general procedure using 5e (0.243 g, 1.0 mmol, 1.0 equiv.), vinylmagnesium bromide (2.63 mL, 1.5 equiv.) and THF (10 mL). Purification by column chromatography on silica gel eluting with 30:70 EtOAc/hexanes gave the desired product as a colourless oil (0.714 g, 85%) and as a mixture of diastereomers (1.5:1). A small amount of each isomer was obtained in pure form by further column chromatography. Major isomer: [α]22D −59.0 (c 0.05, CHCl3). IR (neat): νmax 3355, 2929, 2856, 2679, 1453, 1357, 1250, 1072, 997, 828, 777 cm−1. 1H NMR (500 MHz, CDCl3) δ 5.70 (dd, J = 17.1, 10.5 Hz, 1H, H-6), 5.42 (d, J = 17.1 Hz, 1H, H-7a), 5.36 (d, J = 10.5 Hz, 1H, H-7b), 4.09 (dd, J = 6.5, 3.4 Hz, 1H, H-4), 3.86 (s, 1H, OH), 2.75 (s, 3H, NCH3), 2.65 (dd, J = 17.1, 6.5 Hz, 1H, H-3a), 2.34 (dd, J = 17.1, 3.4 Hz, 1H, H-3b), 0.91 (s, 9H, (CH3)3CSi), 0.12 (s, 6H, CH3Si). 13C NMR (125 MHz, CDCl3) δ 171.7 (CO), 136.7 (C-6), 118.5 (C-7), 90.3 (C-5), 71.7 (C-4), 38.7 (C-3), 25.8 ((CH3)3CSi), 25.1 (CH3), 18.2 ((CH3)3CSi), −4.6 (CH3Si), −4.9 (CH3Si). HRMS (ESI): m/z calcd for C13H26NO3Si [M + H]+: 272.1682; found: 272.1687. Minor isomer: [α]22D +29.9 (c 1.41, CHCl3). IR (neat): νmax 3354, 2928, 2856, 1677, 1463, 1391, 1253, 1084, 833, 777 cm−1. 1H NMR (500 MHz, CDCl3) δ 5.70 (dd, J = 17.2, 10.6 Hz, 1H, H-6), 5.53 (d, J = 17.2 Hz, 1H, H-7a), 5.36 (d, J = 10.6 Hz, 1H, H-7b), 4.26 (dd, J = 6.0, 2.8 Hz, 1H, H-4), 3.04 (s, 1H, OH), 2.75 (s, 3H, NCH3), 2.32 (dd, J = 13.7, 6.0 Hz, 1H, H-3a), 2.02 (dd, J = 13.7, 2.8 Hz, 1H, H-3b), 0.91 (s, 9H, (CH3)3CSi), 0.18 (s, 6H, CH3Si). 13C NMR (125 MHz, CDCl3) δ 172.8 (CO), 137.0 (C-6), 117.8 (C-7), 89.4 (C-5), 70.6 (C-4), 44.8 (C-3), 25.7 ((CH3)3CSi), 24.7 (NCH3), 18.1 ((CH3)3CSi), −4.6 (CH3Si), −5.2 (CH3Si).
(4S,5S)-5-Hydroxy-1-methyl-4-((triisopropylsilyl)oxy)-5-vinylpyrrolidin-2-one and (4S,5R)-5-hydroxy-1-methyl-4-((triisopropylsilyl)oxy)-5-vinylpyrrolidin-2-one (6e)
The title compound was prepared following the general procedure using 3e (0.428 g, 1.5 mmol, 1.0 equiv.), vinylmagnesium bromide (1.8 mL, 1.2 equiv.) and THF (15 mL). Purification by column chromatography on silica gel eluting with 20:80 EtOAc/hexane gave the desired product as a pale yellow oil (0.240 g, 51%) and as a mixture of diastereomers (2.1:1). IR (neat): νmax 3359, 2942, 2866, 1681, 1463, 1388, 1068, 992, 881, 681 cm−1. 1H NMR (500 MHz, CDCl3, major diastereomer) δ 5.71 (dd, J = 17.2, 10.5 Hz, 1H, H-6), 5.45 (dd, J = 17.2, 1.1 Hz, 1H, H-7a), 5.37 (dd, J = 10.5, 1.1 Hz, 1H, H-7b), 4.23 (dd, J = 6.6, 3.3 Hz, 1H, H-4), 2.76 (s, 3H, NCH3), 2.70 (dd, J = 17.0, 6.6 Hz, 1H, H-3a), 2.42 (dd, J = 17.0, 3.3 Hz, 1H, H-3b), 1.22–1.03 (m, 21H, (CH3)3CHSi). 13C NMR (125 MHz, CDCl3, major diastereomer) δ 171.4 (CO), 136.7 (C-6), 118.5 (C-7), 90.3 (C-5), 71.6 (C-4), 39.1 (C-3), 24.9 (NCH3), 17.8 (6 × CH3), 12.0 (3 × CH). HRMS (ESI): m/z calcd for C16H31NO3SiNa [M + Na]+: 336.1971; found: 336.1969.
General procedure for synthesis of spiro compounds 7–19 (Tables 1 and 2)
To an ice-bath cooled solution of N-acyliminium ion precursor (4–6) (1 equiv.) in anhydrous CH2Cl2 (0.1 M) was added 2-(chloromethyl)allyl-trimethylsilane (1.5 equiv.). To this mixture was added BF3·Et2O (2.0 equiv.) dropwise and the reaction mixture was stirred at room temperature for 4 h. The reaction mixture was quenched with sat. aqueous NaHCO3 and the aqueous layer was extracted with CH2Cl2 (three times). The combined organic layer was dried over MgSO4, filtered and concentrated in vacuo. The crude product was purified by column chromatography.(3aS,5R,8aR)-1-Benzyl-5-(chloromethyl)hexahydro-5,8a-methanooxepino[3,2-b]pyrrol-2(1H)-one (8d)
The title compound was prepared following the general procedure using a diastereomeric mixture of mixture of 4d (0.065 g, 0.2 mmol, 1.0 equiv.), 2-(chloromethyl)allyl-trimethylsilane (0.049 g, 34.3 μL, 0.3 mmol, 1.5 equiv.), BF3·Et2O (0.057 g, 49.3 μL, 0.4 mmol, 2.0 equiv.) and CH2Cl2 (2 mL). Purification by column chromatography on silica gel eluting with 50:50 EtOAc/hexanes gave 8d as a white solid (0.035 g, 57%) and as a single diastereomer. [α]22D +145.7 (c 0.12, CHCl3). IR (neat): νmax 3029, 2926, 2860, 1684, 1398, 1095, 1046, 932, 744, 702 cm−1. 1H NMR (500 MHz, CDCl3) δ 7.38–7.13 (m, 5H, ArH), 4.80 (d, J = 15.0 Hz, 1H, PhCHAHBN), 4.46 (t, J = 8.1 Hz, 1H, H-3a), 4.16 (d, J = 15.0 Hz, 1H, PhCHAHBN), 3.50 (d, J = 11.7 Hz, 1H, CHAHBCl), 3.42 (d, J = 11.7 Hz, 1H, CHAHBCl), 2.78 (dd, J = 17.3, 8.6 Hz, 1H, H-3), 2.52 (dd, J = 17.3, 7.7 Hz, 1H, H-3), 1.93–1.83 (m, 3H, H-6 and H-7), 1.77–1.69 (m, 2H, H-4 and H-8), 1.64–1.59 (m, 1H, H-6), 1.36–1.22 (m, 2H, H-4 and H-8). 13C NMR (125 MHz, CDCl3) δ 172.0 (CO), 137.6 (ArC), 128.7 (ArCH), 128.5 (ArCH), 127.8 (ArCH), 87.6 (C-5), 79.6 (C-3a), 70.1 (C-8a), 49.1 (CH2Cl), 44.6 (C-4), 43.8 (PhCH2N), 39.0 (C-3), 33.3 (C-8), 32.7 (C-6), 19.1 (C-7). HRMS (ESI): m/z calcd for C17H21NO235Cl [M + H]+: 306.1255; found: 306.1254.
(3aS,5R,8aR)-5-(Chloromethyl)-1-methylhexahydro-5,8a-methanooxepino[3,2-b]pyrrol-2(1H)-one (8e)
The title compound was prepared following the general procedure for the spiro-cyclization reaction of N-acyliminium ion precursor, using a mixture of (4S,5S)-4-(benzyl oxy)-5-hydroxy-1-methyl-5-vinylpyrrolidin-2-one and (4S,5R)-4-(benzyloxy)-5-hydroxy-1-methyl-5-vinylpyrrolidin-2-one (0.099 g, 0.4 mmol, 1.0 equiv.), 2-(chloromethyl)allyl-trimethylsilane (0.101 g, 111.9 μL, 0.6 mmol, 1.5 equiv.), BF3·Et2O (0.113 g, 98.3 μL, 0.8 mmol, 2.0 equiv.) and CH2Cl2 (4 mL). Purification by column chromatography on silica gel eluting with 2:98 MeOH/CH2Cl2 gave the spirotricyclic product as a pale yellow oil (0.0204 g, 26%) and as a single diastereomer. [α]22D +33.9 (c 0.10, CHCl3). IR (neat): νmax 2926, 2875, 1674, 1390, 1336, 1044, 744, 698 cm−1. 1H NMR (500 MHz, CDCl3) δ 4.45 (dd, J = 8.6, 7.5 Hz, 1H, H-3a), 3.61 (d, J = 11.7 Hz, 1H, CHAHBCl), 3.54 (d, J = 11.6 Hz, 1H, CHAHBCl), 2.80 (s, 3H, CH3N), 2.71 (dd, J = 17.5, 8.6 Hz, 1H, H-3), 2.45 (dd, J = 17.5, 7.5 Hz, 1H, H-3), 1.99–1.92 (m, 4H, H-4, H-7 and H-8), 1.81 (d, J = 10.7 Hz, 1H, H-4), 1.81–1.77 (m, 1H, H-6), 1.71–1.64 (m, 1H, H-8), 1.52–1.43 (m, 1H, H-6). 13C NMR (125 MHz, CDCl3) δ 171.7 (CO), 87.3 (C-5), 79.1 (C-3a), 70.1 (C-8a), 49.0 (CH2Cl), 43.3 (C-4), 39.0 (C-3), 33.3 (C-6), 32.1 (C-8), 25.9 (CH3N), 18.9 (C-7). HRMS (ESI): m/z calcd for C11H17NO235Cl [M + H]+: 230.0984; found: 230.0952.
(4S,5R)-1-Benzyl-4-((tert-butyldimethylsilyl)oxy)-7-methyl-1-azaspiro[4.5]dec-7-en-2-one (16) and (4S,5R,7R)-1-Benzyl-4-((tert-butyldimethylsilyl)oxy)-7-fluoro-7-methyl-1-azaspiro[4.5]decan-2-one (16a)
The title compound was prepared following the general procedure using a diastereomeric mixture of 5d (37.7 mg, 0.108 mmol), methylallyltrimethylsilane (28.6 μL, 0.163 mmol), BF3·Et2O (26.7 μL, 0.216 mmol) and CH2Cl2 (1.1 mL). Purification by column chromatography on silica gel eluting with 30:70 EtOAc/hexanes gave the desired product as a pale yellow oil (19.6 mg, 47%) as a white solid and the fluorocompound 16a (2.9 mg, 7%) as a colourless oil. 16: [α]22D +22.5 (c 0.4, CHCl3); IR (neat):νmax 2929, 2855, 1690, 1407, 1359, 1252, 1084, 830, 776, 702; 1H NMR (500 MHz, CDCl3) δ 7.29–7.20 (m, 5H, ArH), 5.41 (br s, 1H, H-8), 4.75 (d, J = 15.9 Hz, 1H, NCH2), 4.16 (d, J = 15.9 Hz, 1H, NCH2), 4.06 (dd, J = 5.1, 1.6 Hz, 1H, H-4), 2.75 (dd, J = 17.0, 5.1 Hz, 1H, H-3), 2.38 (dd, J = 17.0, 1.6, 1H, H-3), 2.14–2.03 (m, 2H, H-6 and H-9), 1.96 (m, 1H, H-6), 1.84 (m, 1H, H-10), 1.66 (d, J = 17.6 Hz, 1H, H-6), 1.61 (br s, 3H, CH3-7), 1.44 (m, 1H, H-10), 0.89 (s, 9H, CH3), 0.12 (s, 3H, CH3), 0.05 (s, 3H, CH3). 13C NMR (125 MHz, CDCl3) 173.4 (CO), 138.9 (ArC), 131.1 (CH), 128.4 (ArCH), 127.0 (ArCH), 126.9 (ArCH), 121.6 (CH2), 71.0 (C-4), 67.5 (C-5), 42.7 (NCH2), 39.9 (C-3), 36.7 (C-6), 25.9 (C-10), 25.8 ((CH3)3CSi), 23.4 (CH3), 22.8 (C-9), 18.0 ((CH3)3CSi), −3.6 (CH3Si), −5.2 (CH3Si). HRESI-MS m/z 386.2533 (calcd for 386.2515, C23H36NO2Si, [M + H]+).
16a: 1H NMR (500 MHz, CDCl3) δ 7.27–7.20 (m, 5H, ArH), 4.85 (d, J = 16.2 Hz, 1H, NCH2), 4.69 (dd, J = 4.7, 1.8 Hz, 1H, H-4), 3.98 (d, J = 16.2 Hz, 1H, NCH2), 2.79 (dd, J = 17.2, 4.7 Hz, 1H, H-3), 2.34 (d, J = 17.2 Hz, 1H, H-3), 2.07 (br d, J = 12.8 Hz, 1H, H-6), 1.88 (t, J = 12.7 Hz, 1H, H-8), 1.72 (ddt, J = 15.1, 10.3, 2.5 Hz, 1H, H-10), 1.58 (m, 1H, H-7), 1.47 (m, 1H, H-7), 1.51 (d, J = 15.0 Hz, 1H, H-10), 1.43 (d, J = 15.0 Hz, 1H, H-10), 1.30 (d, J = 2.1 Hz, 3H, CH3), 1.24 (m, 1H, H-8), 1.09 (td, J = 13.2, 3.8 Hz, 1H, H-6), 0.89 (s, 9H, CH3), 0.13 (s, 3H, CH3), 0.08 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3) δ 174.4 (CO), 138.9 (ArC), 128.6 (ArCH), 127.3 (ArCH), 127.0 (ArCH), 96.1 (d, 1JC,F = 167.3 Hz, C-7), 69.6 (d, 4JC,F = 9.8 Hz, C-4), 69.1 (d, 3JC,F = 2.5 Hz, C-5), 42.5 (NCH2), 41.7 (d, 2JC,F = 21.6 Hz, C-6), 40.0 (C-3), 36.4 (d, 2JC,F = 22.8 Hz, C-8), 28.9 (d, 4JC,F = 7.5 Hz, C-6), 28.8 (d, 2JC,F = 25.4 Hz, CH3), 26.1 (CH3), 18.3 (d, 3JC,F = 17.0 Hz, C-9), 18.2 (CH3), −3.6 (CH3), −4.9 (CH3); HRESI-MS m/z 428.2407 (calcd for 428.2397, C23H36NO2FNaSi) [M + Na]+.
(4S,5R)-4-((tert-Butyldimethylsilyl)oxy)-1,7-dimethyl-1-azaspiro[4.5]dec-7-en-2-one (17)
The title compound was prepared following the general procedure using a diastereomeric mixture of 5e (0.054 g, 0.2 mmol, 1.0 equiv.), methallyltrimethylsilane (0.057 g, 38.5 μL, 0.3 mmol 1.5 equiv.), BF3·Et2O (0.057 g, 49.4 μL, 0.4 mmol, 2.0 equiv.) and CH2Cl2 (2 mL). Purification by column chromatography on silica gel eluting with 20:80 EtOAc/hexanes gave the desired product as a pale yellow oil (0.0250 g, 40%) and as a mixture of diastereomers (75:25). A small amount of the major isomer was obtained in pure form by further separation using column chromatography. [α]22D +38.2 (c 0.25, CHCl3). IR (neat): νmax 3039, 2927, 2855, 1692, 1397, 1252, 1080, 929, 830, 775, 668 cm−1. 1H NMR (400 MHz, CDCl3) δ 5.46 (s, 1H, H-8), 3.99 (dd, J = 5.8, 2.8 Hz, 1H, H-4), 2.76 (s, 3H, NCH3), 2.61 (dd, J = 16.9, 5.8 Hz, 1H, H-3), 2.28 (dd, J = 16.9, 2.8 Hz, 1H, H-3), 2.24–2.17 (m, 1H, H-9), 2.13–2.03 (m, 2H, H-6 and H-9), 2.00–1.90 (m, 1H, H-10), 1.67 (s, 3H, CH3), 1.65–1.57 (m, 2H, H-6 and H-10), 0.88 (s, 9H, (CH3)3CSi), 0.07 (s, 3H, CH3Si), 0.06 (s, 3H, CH3Si). 13C NMR (100 MHz, CDCl3) δ 172.5 (CO), 131.1 (C-7), 121.8 (C-8), 71.6 (C-4), 66.1 (C-5), 39.7 (C-3), 35.9 (C-6), 25.8 ((CH3)3CSi), 25.3 (C-10), 25.2 (CH3), 23.5 (NCH3), 23.0 (C-9), 18.1 ((CH3)3CSi), −3.8 (CH3Si), −5.1 (CH3Si). HRMS (ESI): m/z calcd for C17H32NO2Si [M + H]+: 310.2202; found: 310.2203.
(4S,5S)-1-Benzyl-4-((tert-butyldimethylsilyl)oxy)-7-(chloromethyl)-1-azaspiro[4.5]dec-7-en-2-one (18)
The title compound was prepared following the general procedure using a mixture of 5d (0.062 g, 0.18 mmol, 1.0 equiv.), 2-(chloromethyl)allyl-trimethylsilane (0.044 g, 48.4 μL, 0.27 mmol, 1.5 equiv.), BF3·Et2O (0.051 g, 43.9 μL, 0.36 mmol, 2.0 equiv.) and CH2Cl2 (1.7 mL). Purification by column chromatography on silica gel eluting with 40:60 EtOAc/hexanes gave the desired product as a white solid (0.0173 g, 23%) and as a mixture of diastereomers (54:46). A small amount of each isomer was obtained in pure form by further recrystallization. Major 5S-isomer: [α]22D −83.9 (c 0.13, CHCl3). IR (neat): νmax 3033, 2924, 2851, 1678, 1407, 1257, 1075, 943, 828, 775, 744, 710 cm−1. 1H NMR (500 MHz, CDCl3) δ 7.32–7.12 (m, 5H, ArH), 5.77 (s, 1H, H-8), 4.59 (d, J = 16.0 Hz, 1H, PhCHAHBN), 4.39 (d, J = 16.0 Hz, 1H, PhCHAHBN), 4.10 (dd, J = 6.0, 3.7 Hz, 1H, H-4), 3.87 (d, J = 11.3 Hz, 1H, CHAHBCl), 3.84 (d, J = 11.3 Hz, 1H, CHAHBCl), 2.79 (dd, J = 16.8, 6.0 Hz, 1H, H-3), 2.41 (dd, J = 16.8, 3.7 Hz, 1H, H-3), 2.31 (d, J = 17.4 Hz, 1H, H-6), 2.23–2.12 (m, 3H, H-6 and H-9), 1.80–1.73 (m, 1H, H-10), 1.47–1.40 (m, 1H, H-10), 0.87 (s, 9H, (CH3)3CSi), 0.08 (s, 3H, CH3Si), 0.00 (s, 3H, CH3Si). 13C NMR (125 MHz, CDCl3) δ 172.7 (CO), 138.6 (ArC), 133.0 (C-7), 128.4 (ArCH), 127.0 (ArCH), 126.9 (ArCH), 125.4 (C-8), 72.5 (C-4), 66.3 (C-5), 49.5 (CH2Cl), 43.1 (PhCH2N), 39.7 (C-3), 30.6 (C-10), 29.4 (C-6), 25.7 ((CH3)3CSi), 23.2 (C-9), 17.8 ((CH3)3CSi), −4.3 (CH3Si), −5.2 (CH3Si). HRMS (ESI): m/z calcd for C23H35NO2Si35Cl [M + H]+: 420.2126; found: 420.2121. Minor 5R-isomer: [α]22D–282.6 (c 0.04, CHCl3). IR (neat): νmax 3030, 2926, 2853, 1688, 1408, 1261, 1075, 932, 828, 774, 703 cm−1.1H NMR (500 MHz, CDCl3) δ 7.30–7.19 (m, 5H, ArH), 5.83 (s, 1H, H-8), 4.78 (d, J = 16.0 Hz, 1H, PhCHAHBN), 4.16 (d, J = 16.0 Hz, 1H, PhCHAHBN), 4.06 (dd, J = 5.1, 1.3 Hz, 1H, H-4), 3.97 (d, J = 11.1 Hz, 1H, CHAHBCl), 3.91 (d, J = 11.1 Hz, 1H, CHAHBCl), 2.78 (dd, J = 17.1, 5.1 Hz, 1H, H-3), 2.40 (dd, J = 17.1, 1.3 Hz, 1H, H-3), 2.20 (d, J = 17.3 Hz, 1H, H-6), 2.16 (s, 1H, H-9), 2.06 (d, J = 18.7 Hz, 1H, H-9), 1.94 (d, J = 17.3 Hz, 1H, H-6), 1.91–1.86 (m, 1H, H-10), 1.52–1.43 (m, 1H, H-10), 0.88 (s, 9H, (CH3)3CSi), 0.13 (s, 3H, CH3Si), 0.04 (s, 3H, CH3Si). 13C NMR (125 MHz, CDCl3) δ 173.4 (CO), 138.8 (ArC), 131.8 (C-7), 128.5 (ArCH), 127.8 (ArCH), 127.1 (C-8), 127.0 (ArCH), 70.7 (C-4), 67.1 (C-5), 49.5 (CH2Cl), 42.7 (PhCH2N), 39.8 (C-3), 32.6 (C-10), 25.8 ((CH3)3CSi), 25.6 (C-6), 23.1 (C-9), 18.0 ((CH3)3CSi), −3.6 (CH3Si), −5.2 (CH3Si). HRMS (ESI): m/z calcd for C23H35NO2Si35Cl [M + H]+: 420.2126; found: 420.2140.
(4S,5R,7S)-1-Benzyl-4-((tert-butyldimethylsilyl)oxy)-7-(chloromethyl)-7-fluoro-1-azaspiro[4.5]decan-2-one and (4S,5R,7R)-1-benzyl-4-((tert-butyldimethylsilyl)oxy)-7-(chloromethyl)-7-fluoro-1-azaspiro[4.5]decan-2-one (19)
These compounds were obtained as a by-product from the above spiro-cyclization reaction of a mixture of 5d, 2-(chloromethyl)allyltrimethylsilane and BF3·Et2O as a white solid (0.024 g, 31%) and as a mixture of diastereomers (52:48) A small amount of each isomer was obtained in pure form by further recrystallization and separation of the different crystal types by hand. Major isomer: [α]22D +250.5 (c 0.02, CHCl3). IR (neat): νmax 2927, 2855, 1671, 1428, 1353, 1074, 920, 834, 773, 698 cm−1. 1H NMR (500 MHz, CDCl3) δ 7.26 (m, 5H, ArH), 4.83 (d, J = 16.2 Hz, 1H, PhCHAHBN), 4.64 (d, J = 4.8 Hz, 1H, H-4), 4.03 (d, J = 16.2 Hz, 1H, PhCHAHBN), 3.54–3.41 (m, 2H, CH2Cl), 2.80 (dd, J = 17.2, 4.8 Hz, 1H, H-3), 2.36 (d, J = 17.3 Hz, 1H, H-3), 2.11 (d, J = 13.0 Hz, 1H, H-10), 1.94 (t, J = 12.8 Hz, 1H, H-8), 1.85–1.77 (m, 1H, H-6), 1.66–1.49 (m, 3H, H-6 and H-9), 1.43 (td, J = 13.7, 4.8 Hz, 0.5H, H-8), 1.35 (td, J = 13.7, 4.9 Hz, 0.5H, H-8), 1.15 (td, J = 13.0, 4.2 Hz, 1H, H-10), 0.89 (s, 9H, (CH3)3CSi), 0.13 (s, 3H, CH3Si), 0.09 (s, 3H, CH3Si). 13C NMR (125 MHz, CDCl3) δ 173.7 (CO), 138.3 (ArC), 128.4 (ArCH), 127.0 (ArCH), 126.7 (ArCH), 95.7 (d, 1JC,F = 173 Hz, C-7), 69.1 (C-4), 68.3 (C-5), 50.4 (d, 2JC,F = 30 Hz, CH2Cl), 42.1 (PhCH2N), 39.4 (C-3), 37.1 (d, 2JC,F = 21 Hz, C-6), 32.1 (d, 2JC,F = 25 Hz, C-8), 28.6 (C-10), 25.7 ((CH3)3CSi), 17.9 ((CH3)3CSi), 17.3 (C-9), −4.0 (CH3Si), −5.3 (CH3Si). HRMS (ESI): m/z calcd for C23H36NO2SiClF [M + H]+: 440.2188; found: 440.2201. Minor isomer:[α]22D −242.4 (c 0.05, CHCl3). IR (neat): νmax 2929, 2856, 1667, 1404, 1360, 1248, 1191, 1057, 933, 840, 779, 710 cm−1. 1H NMR (500 MHz, CDCl3) δ 7.33–7.16 (m, 5H, ArH), 4.95 (d, J = 16.4 Hz, 1H, PhCHAHBN), 4.24 (d, J = 16.4 Hz, 1H, PhCHAHBN), 4.03 (dd, J = 5.2, 2.5 Hz, 1H, H-4), 3.74–3.54 (m, 2H, CH2Cl), 2.81 (dd, J = 16.8, 5.2 Hz, 1H, H-3), 2.41 (dd, J = 16.8, 2.5 Hz, 1H, H-3), 2.04 (dd, J = 19.6, 15.0 Hz, 1H, H-6), 1.87 (t, J = 15.0 Hz, 1H, H-6), 1.83–1.65 (m, 3H, H-8, H-9 and H-10), 1.50–1.41 (m, 1H, H-10), 1.36–1.26 (m, 1H, H-9), 0.90 (s, 9H, (CH3)3CSi), 0.14 (s, 3H, CH3Si), 0.08 (s, 3H, CH3Si). 13C NMR (125 MHz, CDCl3) δ 173.0 (CO), 138.5 (ArC), 128.4 (ArCH), 126.8 (ArCH), 126.6 (ArCH), 94.2 (d, 1JC,F = 177 Hz, C-7), 72.8 (C-4), 67.8 (C-5), 50.1 (d, 2JC,F = 29 Hz, CH2Cl), 42.8 (PhCH2N), 39.3 (C-3), 38.9 (d, 2JC,F = 22 Hz, C-6), 32.8 (d, 2JC,F = 21 Hz, C-8), 28.1 (C-10), 25.6 ((CH3)3CSi), 18.3 (d, 3JC,F = 6 Hz, C-9), 17.9 ((CH3)3CSi), −4.0 (CH3Si), −5.1 (CH3Si). HRMS (ESI): m/z calcd for C23H36NO2Si35ClF [M + H]+: 440.2188; found: 440.2206.
(4S,5S)-4-((tert-Butyldimethylsilyl)oxy)-7-(chloromethyl)-1-methyl-1-azaspiro[4.5]dec-7-en-2-one (20)
The title compound was prepared following the general procedure using a diastereomeric mixture of 5e (0.054 g, 0.2 mmol, 1.0 equiv.), 2-(chloromethyl)allyltrimethylsilane (0.049 g, 54.3 μL, 0.3 mmol, 1.5 equiv.), BF3·Et2O (0.057 g, 49.4 μL, 0.4 mmol, 2.0 equiv.) and CH2Cl2 (2 mL). Purification by column chromatography on silica gel eluting with 30:70 EtOAc/hexanes gave the desired product as a pale yellow oil (0.0290 g, 42%) and as a mixture of diastereomers (79:21). A small amount of the major isomer was obtained in pure form by further separation using column chromatography. Major: [α]22D +35.0 (c 0.60, CHCl3). IR (neat): νmax 3050, 2926, 2854, 1695, 1396, 1258, 1122, 1079, 932, 836, 777, 667 cm−1. 1H NMR (500 MHz, CDCl3) δ 5.80 (s, 1H, H-8), 4.07 (t, J = 6.7, 4.7 Hz, 1H, H-4), 4.04 (d, J = 11.5 Hz, 1H, CHAHBCl), 4.00 (d, J = 11.5 Hz, 1H, CHAHBCl), 2.79 (s, 3H, NCH3), 2.67 (dd, J = 16.8, 6.7 Hz, 1H, H-3), 2.36 (d, J = 17.9 Hz, 1H, H-6), 2.33–2.23 (m, 3H, H-3, H-6 and H-9), 1.88 (apparent dt, J = 13.0, 7.9 Hz, 1H, H-10), 1.46 (apparent dt, J = 13.0, 5.4 Hz, 1H, H-10), 0.85 (s, 9H, (CH3)3CSi), 0.05 (s, 3H, CH3Si), 0.01 (s, 3H, CH3Si). 13C NMR (125 MHz, CDCl3) δ 171.9 (CO), 133.1 (C-7), 125.7 (C-8), 72.5 (C-4), 65.2 (C-5), 49.8 (CH2Cl), 39.8 (C-3), 29.9 (C-10), 28.4 (C-6), 25.7 ((CH3)3CSi), 25.3 (NCH3), 23.1 (C-9), 17.8 ((CH3)3CSi), −4.32 (CH3Si), −5.19 (CH3Si). HRMS (ESI): m/z calcd for C17H31NO235ClSi [M + H]+: 344.1813; found: 344.1826.
(4S,5R) and (4S,5S)-7-(Chloromethyl)-1-methyl-4-((triisopropylsilyl)oxy)-1-azaspiro[4.5]dec-7-en-2-one (21)
The title compound was prepared following the general procedure using a diastereomeric mixture of 6e (0.063 g, 0.2 mmol, 1.0 equiv.), 2-(chloromethyl)allyltrimethylsilane (0.049 g, 54.3 μL, 0.3 mmol, 1.5 equiv.), BF3·Et2O (0.057 g, 49.4 μL, 0.4 mmol, 2.0 equiv.) and CH2Cl2 (2 mL). Purification by column chromatography on silica gel eluting with 20:80 EtOAc/hexanes gave the desired product as a pale yellow oil (0.0079 g, 10%) and as a mixture of diastereomers (54:46). IR (neat): νmax 2941, 2865, 1692, 1462, 1391, 1180, 1088, 882, 679 cm−1. 1H NMR (500 MHz, CDCl3) δ 5.84 (s, 0.54H, H-8 (minor)), 5.73 (s, 0.46H, H-8 (major)), 4.24 (dd, J = 6.3, 4.3 Hz, 0.46H, H-4 (minor)), 4.21 (dd, J = 5.8, 2.0 Hz, 0.56H, H-8 (major)), 4.09–3.99 (m, 2H, CH2Cl), 2.81 (s, 1.38H, NCH3 (minor)), 2.77–2.69 (m, 1H, H-3), 2.70 (s, 1.62H, NCH3 (major)), 2.51 (d, J = 17.5 Hz, 0.54H, H-6 (major)), 2.40 (dd, J = 16.7, 4.3 Hz, 0.46H, H-3 (minor)), 2.37 (dd, J = 17.1, 2.0 Hz, 0.54H, H-3 (major)), 2.33–2.26 (m, 1.46H, H-6 (major) and H-9 (minor)), 2.20–2.12 (m, 0.92H, H-6 (minor)), 1.91–1.81 (m, 1.54H, H-9 (major) and H-10), 1.76–1.68 (m, 0.54H, H-9 (major)), 1.56–1.47 (m, 1H, H-10), 1.07–1.01 (m, 21H, (CH3)3CHSi). 13C NMR (125 MHz, CDCl3) δ 172.4 (C-2 major), 172.0 (C-2 minor), 139.9 (C-7 major), 133.1 (C-7 minor), 127.4 (C-8 major), 126.0 (C-8 minor), 74.1 (C-4 major), 73.7 (C-4 minor), 67.3 (C-5 major), 65.5 (C-5 minor), 49.8 (CH2Cl minor), 49.0 (CH2Cl major), 40.4 (C-3 major), 39.8 (C-3 minor), 30.1 (C-10 minor), 29.4 (C-10 major), 28.6 (C-6 major), 25.7 (C-6 minor), 25.4 (NCH3), 23.2 (C-9 minor), 19.9 (C-9 major), 18.1 ((CH3)3CHSi minor), 18.0 ((CH3)3CHSi major), 12.7 ((CH3)3CHSi minor), 12.2 ((CH3)3CHSi major). HRMS (ESI): m/z calcd for C20H37NO235ClSi [M + H]+: 386.2282; found: 386.2285.
(4S,5S)-7-(Azidomethyl)-4-((tert-butyldimethylsilyl)oxy)-1-methyl-1-azaspiro[4.5]dec-7-en-2-one (26)
To a solution of a diastereomeric mixture of 20 (12 mg, 0.035 mmol, 1.0 equiv.) in anhydrous DMF (0.17 mL) was added sodium azide (11.3 mg, 0.174 mmol, 5.0 equiv.) and TBAI (1.1 mg, 0.0035 mmol, 0.1 equiv.) and the reaction mixture was stirred at 80 °C for 5 h. EtOAc (5 mL) was added and the solution was washed with water (3 × 5 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo. The crude product was purified by column chromatography on silica gel eluting with 40:60 EtOAc/hexanes to afford the desired product as a pale yellow oil (10 mg, 82%) and as a mixture of diastereomers (79:21). A small amount of the major isomer was obtained in pure form by further separation using column chromatography. Major isomer: [α]22D −8.0 (c 0.05, CHCl3). IR (neat): νmax 3050, 2928, 2856, 2095, 1689, 1396, 1252, 1122, 1077, 832, 775 cm−1. 1H NMR (500 MHz, CDCl3) δ 5.75–5.67 (m, 1H, H-8), 4.08 (dd, J = 6.7, 4.4 Hz, 1H, H-4), 3.81 (d, J = 13.5 Hz, 1H, CHAHBN3), 3.64 (d, J = 13.5 Hz, 1H, CHAHBN3), 2.78 (s, 3H, NCH3), 2.67 (dd, J = 16.9, 6.7 Hz, 1H, H-3), 2.34–2.24 (m, 4H, H-3, H-6 and H-9), 2.16 (d, J = 17.4 Hz, 1H, H-6), 1.89 (ddd, J = 13.1, 9.2, 6.9 Hz, 1H, H-10), 1.47 (ddd, J = 13.1, 5.6, 4.0 Hz, 1H, H-10), 0.85 (s, 9H, (CH3)3CSi), 0.05 (s, 3H, CH3Si), 0.02 (s, 3H, CH3Si). 13C NMR (125 MHz, CDCl3) δ 171.9 (C-2), 131.5 (C-7), 124.6 (C-8), 72.2 (C-4), 65.2 (C-5), 57.3 (CH2N3), 39.8 (C-10), 29.9 (C-10), 28.8 (C-6), 25.6 ((CH3)3CSi), 25.2 (NCH3), 23.0 (C-9), 17.9 ((CH3)3CSi), −4.3 (CH3Si), −5.2 (CH3Si). HRMS (ESI): m/z calcd for C17H31N4O2Si [M + H]+: 351.2216; found: 351.2213.
(4S,5S)-7-(Aminomethyl)-4-((tert-butyldimethylsilyl)oxy)-1-methyl-1-azaspiro[4.5]dec-7-en-2-one (27)
To a solution of a diastereomeric mixture (79:21) of 26 (7.1 mg, 0.020 mmol, 1.0 equiv.) in THF (75 μL) and water (5 μL) was added triphenylphosphine (10.6 mg, 0.04 mmol, 2.0 equiv.) and the reaction mixture was stirred at room temperature for 5 h. The mixture was acidified to pH 1 with 1 M HCl. The aqueous layer was basified to pH 12 with 1 M NaOH and extracted with EtOAc (3 × 2 mL). The combined organic layer was dried (MgSO4), filtered and concentrated in vacuo. The crude product was purified by column chromatography on silica gel eluting with 5:95 MeOH/CH2Cl2 to afford the desired product as a pale yellow oil (3.2 mg, 50%) and as a mixture of diastereomers (79:21) including a small amount (5%) of triphenylphosphine oxide. 1H NMR (500 MHz, CDCl3) δ 5.70 (s, 1H, H-8), 4.08 (dd, J = 6.8, 4.8 Hz, 1H, H-4), 3.44–3.24 (m, 2H, CH2NH2), 2.79 (s, 3H, NCH3), 2.69–2.62 (m, 1H, H-3), 2.35–2.21 (m, 5H, H-3, H-6 and H-9), 1.93–1.83 (m, 1H, H-10), 1.53–1.42 (m, 1H, H-10), 0.84 (s, 9H, (CH3)3CSi), 0.05 (s, 3H, CH3Si), 0.01 (s, 3H, CH3Si). 13C NMR (125 MHz, CDCl3) δ 171.9 (C-2), 122.4 (C-8), 72.2 (C-4), 65.3 (C-5), 46.6 (CH2NH2), 39.8 (C-3), 29.8 (C-10), 28.9 (C-6), 25.7 ((CH3)3CSi), 25.2 (NCH3), 23.0 (C-9), 17.8 ((CH3)3CSi), −4.3 (CH3Si), −5.2 (CH3Si). (The quaternary carbons C-7 was not detected.) HRMS (ESI): m/z calcd for C17H33N2O2Si [M + H]+: 325.2311; found: 325.2325.
(3R,3′S)-1′-Benzyl-3′-(benzyloxy)-1,4-dihydro-2H-spiro[cyclopenta[b]indole-3,2′-pyrrolidin]-5′-one (29)
To solution of 4d (36.9 mg, 0.113 mmol) and indole (19.9 mg, 0.17 mmol) in dry CH2Cl2 (1.1 mL) under N2 atmosphere at 0 °C was added dropwise BF3·Et2O (28 μL, 0.227 mmol) and the mixture was stirred for 4 h. The sat. NaHCO3 was added to the mixture and extracted with CH2Cl2 (3 × 2 mL). The mixture was concentrated in vacuo and purified by column chromatography (5:95, EtOAc/CH2Cl2) to afford the desire product (20.3 mg, 43%) as a brown viscous and as a mixture of diastereomers (2:1). A small amount of the major diastereomer could be obtained in pure from further separation by column chromatography and was crystalized using CH2Cl2:hexanes (5:1). Major isomer: [α]D25 +85.9 (c 0.6, CHCl3); IR (neat) νmax 3266, 2927, 2924, 2857, 1673, 1411, 1109, 740, 701; 1H NMR (500 MHz, CDCl3) δ 7.51 (d, J = 6.5 Hz, 1H, ArH), 7.23–7.11 (m, 8H, ArH), 7.09 (t, J = 7.7 Hz, 1H, ArH), 7.03 (d, J = 6.2 Hz, 2H, ArH), 6.97 (d, J = 7.2 Hz, 2H, ArH), 4.41 (d, J = 14.8 Hz, 1H, NCH2), 4.33 (dd, J = 9.2, 7.6 Hz, 1H, H-3′), 4.28 (apparent d, J = 3.7 Hz, 2H, OCH2), 4.14 (d, J = 14.8 Hz, 1H, NCH2), 3.26 (m, 1H, H-2), 2.83 (t, J = 6.5 Hz, 2H, H-1), 2.77 (dd, J = 16.4, 7.6 Hz, 1H, H-4′), 2.64 (dd, J = 16.4, 9.2 Hz, 1H, H-4′), 2.24 (m, 1H, H-2). 13C NMR (125 MHz, CDCl3) 171.5 (CO), 142.4 (ArC), 140.4 (ArC), 138.3 (ArC), 137.5 (ArC), 128.5 (ArCH), 128.4 (ArCH), 128.3(ArCH), 127.9 (ArCH), 127.4 (ArCH), 124.2 (ArC), 124.1 (ArC), 122.6 (ArCH), 119.9 (ArCH), 119.6 (ArCH), 112.4 (ArCH), 78.3 (C-3′), 72.9 (C-3), 72.1 (CH2OBn), 43.6 (NCH2), 36.8 (C-4′), 33.9 (C-2), 23.0 (C-1). HRESI-MS m/z 423.2088 (calcd for 423.2073, C28H27N2O2) [M + H]+. Minor isomer: 1H NMR (500 MHz, CDCl3, in part from analysis of the mixture) δ 4.46 (d, J = 11.6 Hz, 1H, NCH2), 4.28 (d, J = 15.0 Hz, 1H, NCH2), 4.24 (d, J = 11.6 Hz, 1H, NCH2), 4.16 (d, J = 15.0 Hz, 1H, NCH2), 4.05 (d, J = 4.4 Hz, 1H, H-3′).
(3R,3′S)-1′-Benzyl-3′-(benzyloxy)-4-methyl-1,4-dihydro-2H-spiro[cyclopenta[b]indole-3,2′-pyrrolidin]-5′-one (30)
To solution of 4d (55.1 mg, 0.17 mmol) and 1-methylindole (31.9 μL, 0.256 mmol) in dry CH2Cl2 (2.1 mL) under N2 atmosphere at 0 °C was added dropwise BF3·Et2O (42.1 μL, 0.341 mmol) and the mixture was stirred for 4 h. The sat. NaHCO3 was added to the mixture and extracted with CH2Cl2 (3 × 2 mL). The mixture was concentrated in vacco and purified by column chromatography (3:7, EtOAc/hexanes) to afford the desire product (23.3 mg, 32%) as a brown viscous and as a mixture of diastereomers (2:1) A small amount of the major diastereomer could be obtained in pure from further separation by column chromatography. Major isomer: [α]26D +73.3 (c 0.2, CHCl3); IR (neat) νmax 2925, 2856, 1680, 1468, 1402, 1353, 1098, 740, 701; 1H NMR (500 MHz, CDCl3) δ 7.55 (dt, J = 7.8, 1.0 Hz, 1H, ArH), 7.25 (m, 1H, ArH), 7.17–7.12 (m, 2H, ArH), 7.09 (t, J = 7.4 Hz, 4H, ArH), 7.03 (t, J = 7.6 Hz, 2H, ArH), 6.90 (d, J = 7.5 Hz, 2H, ArH), 6.83 (d, J = 7.0 Hz, 2H, ArH), 4.51 (d, J = 14.3 Hz, 1H, NCH2), 4.40 (dd, J = 10.1, 7.9 Hz, 1H, H-3′), 4.32 (d, J = 12.2 Hz, 1H, OCH2), 4.24 (d, J = 12.2 Hz, 1H, OCH2), 4.03 (d, J = 14.3 Hz, 1H, NCH2), 3.37 (m, 1H, H-2), 2.90 (m, 2H, H-1), 2.78 (dd, J = 16.4, 7.9 Hz, 1H, H-4′), 2.73 (s, 3H, NCH3), 2.59 (dd, J = 16.4, 10.1 Hz, 1H, H-4′), 2.21 (m, 1H, H-2). 13C NMR (125 MHz, CDCl3) δ 171.1 (CO), 142.8 (ArC), 140.0 (ArC), 137.7 (ArC), 137.2 (ArC), 129.3 (ArCH), 128.4 (ArCH), 128.3 (ArCH), 128.0 (ArCH), 127.6 (ArCH), 127.5 (ArCH), 123.5 (ArC), 123.1 (ArC), 122.2 (ArCH), 119.7 (ArCH), 119.5 (ArCH), 76.3 (C-3′), 72.8 (C-3), 71.9 (OCH2Bn), 44.2 (NCH2), 36.7 (C-4′), 34.2 (C-2), 29.3 (NCH3), 22.6 (C-1). HRESI-MS m/z 437.2241 (calcd for 437.2229, C29H29N2O2) [M + H]+. Minor isomer: δH (500 MHz, CDCl3, in part from analysis of the mixture) 4.50 (1H, d, J = 14.7 Hz, NCH2), 4.23 (1H, d, J = 11.7 Hz, OCH2), 4.17 (1H, d, J = 14.7 Hz, NCH2), 4.09 (1H, dd, J = 6.8, 2.1 Hz, H-4), 4.04 (1H, d, J = 11.7 Hz, OCH2).
(3S,3′S)-1′-Benzyl-3′-((tert-butyldimethylsilyl)oxy)-1,4-dihydro-2H-spiro[cyclopenta[b]indole-3,2′-pyrrolidin]-5′-one (31)
To solution of 5d (57.9 mg, 0.167 mmol) and indole (23.9 mg, 0.25 mmol) in dry CH2Cl2 (1.7 mL) under N2 atmosphere at 0 °C was added dropwise BF3·Et2O (41.0 μL, 0.333 mmol) and the mixture was stirred for 4 h. The sat. NaHCO3 was added to the mixture and extracted with CH2Cl2 (3 × 2 mL). The crude product was concentrated in vacco and purified by column chromatography (3:7, EtOAc/hexanes) yields the title compound 31 (31.7 mg, 42%, dr = 9:1) as a yellow oil. [α]25D +11.1 (c 1.8, CHCl3); IR (neat) νmax 3266, 2928, 2857, 1673, 1410, 1251, 1146, 1123, 1088, 836, 776, 740, 703; 1H NMR (500 MHz, CDCl3) δ 7.45 (m, 1H, ArH), 7.23–7.16 (m, 4H, ArH), 7.13–7.00 (m, 4H, ArH), 4.54 (d, J = 15.0 Hz, 1H, NCH2), 4.46 (dd, J = 8.7, 7.3 Hz, 1H, H-3′), 4.09 (d, J = 15.0 Hz, 1H, NCH2), 3.15 (m, 1H, H-2), 2.86–2.67 (m, 3H, H-1 and H-4′), 2.55 (dd, J = 16.3, 8.7 Hz, 1H, H-4′), 2.21 (m, 1H, H-2), 0.76 (s, 9H, CH3), −0.18 (s, 3H, CH3), −0.38 (s, 3H, CH3). 13C NMR (125 MHz, CDCl3) δ 173.4 (CO), 142.4 (ArC), 139.7 (ArCH), 138.6 (ArCH), 128.6 (ArCH), 128.1 (ArCH), 127.4 (ArCH), 124.5 (ArC), 124.2 (ArC), 122.4 (ArCH), 119.8 (ArCH), 119.4 (ArCH), 112.2 (ArCH), 74.3 (C-3), 73.2 (C-3′), 43.7 (NCH2), 39.5 (C-4′), 33.4 (C-2), 25.7 ((CH3)3CSi), 22.8 (C-1), 17.9 ((CH3)3CSi), −5.20 (CH3Si), −5.33 (CH3Si). HRESI-MS m/z 447.2467 (calcd for 447.2468, C27H35N2O2Si) [M + H]+.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Australian Research Council (DP130101968) and AnalytiCon Discovery GmbH, Germany, and the Universities of Wollongong (SMAH Research Partnership Grant) and Melbourne (High Performance Computing service) for their support. We thank Dr Oliver Kunz (AnalytiCon) for useful discussions. C. A. and T. L. thanks Chiang Mai University for partial support.Notes and references
- W. N. Speckamp and H. Hiemstra, Tetrahedron, 1985, 41, 4367 CrossRef CAS.
- W. N. Speckamp and M. J. Moolenaar, Tetrahedron, 2000, 56, 3817 CrossRef CAS.
- B. E. Maryanoff, H. Zhang, J. H. Cohen, I. J. Turchi and C. A. Maryanoff, Chem. Rev., 2004, 104, 1431 CrossRef CAS.
- S. N. Gaskell, L. J. Duffy and S. M. Allin, Nat. Prod. Commun., 2008, 3, 1825 CrossRef CAS.
- A. Yazici and S. G. Pyne, Synthesis, 2009, 339 CAS.
- A. Yazici and S. G. Pyne, Synthesis, 2009, 513 CAS.
- U. Martinez-Estibalez, A. Gomez-SanJuan, A. O. Garcia-Calvo, E. Aranzamendi, E. Lete and N. Sotomayor, Eur. J. Org. Chem., 2011, 3610 CrossRef CAS.
- A. Yazici and S. G. Pyne, Org. Lett., 2013, 15, 5878 CrossRef CAS.
- A. Yazici, U. Wille and S. G. Pyne, J. Org. Chem., 2016, 81, 1434 CrossRef CAS and references cited therein.
- G. M. Ryder, U. Wille, A. C. Willis and S. G. Pyne, Org. Biomol. Chem., 2019, 17, 7025 RSC.
- Y. Inouye, N. T. Noriko, A. Watanabe, Y. Aoki and H. Kakisawa, Bull. Chem. Soc. Jpn., 1992, 65, 2866 CrossRef CAS.
- G. Gunbas, N. Hafezi, W. L. Sheppard, M. M. Olmstead, I. V. Stoyanova, F. S. Tham, M. P. Meyer and M. Mascal, Nat. Chem., 2012, 4, 1018 CrossRef CAS.
- F. Yokokawa, T. Asano and T. Shioiri, Org. Lett., 2000, 2, 4169 CrossRef CAS.
- D. J. Hart, L.-Q. Sun and A. P. Kozikowski, Tetrahedron Lett., 1995, 36, 7787 CrossRef CAS.
- P.-Q. Huang, S.-L. Wang, J.-L. Ye, Y.-P. Ruan, Y.-Q. Huang, H. Zheng and J.-X. Gao, Tetrahedron, 1998, 54, 12547 CrossRef CAS.
- B.-Y. He, T.-J. Wu, X.-Y. Yu and P.-Q. Huang, Tetrahedron: Asymmetry, 2003, 14, 2101 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed; L. Goerigk, A. Hansen, C. Bauer, S. Ehrlich, A. Najibi and S. Grimme, Phys. Chem. Chem. Phys., 2017, 19, 32184 RSC; L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670 RSC.
- Y. Takano and K. N. Houk, J. Chem. Theory Comput., 2005, 1, 70 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental and spectroscopic details and copies of 1H and 13C NMR spectra; Gaussian archive entries free energies and imaginary frequencies (transition states) for the calculations in Table 2 and Fig. 2. CCDC 2010550–2010555. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ob02075c |
| This journal is © The Royal Society of Chemistry 2021 |