
Metal free decarboxylative aminoxylation of carboxylic acids using a biphasic solvent system†
Göran
Schulz
 and
Andreas
Kirschning
*
and
Andreas
Kirschning
*
Institute of Organic Chemistry and Center of Biomolecular Drug Research (BMWZ), Leibniz Universität Hannover, Schneiderberg 1B, 30167 Hannover, Germany. E-mail: andreas.kirschning@oci.uni-hannover.de
First published on 13th October 2020
Abstract
The smooth oxidative radical decarboxylation of carboxylic acids with TEMPO and other derivatives as radical scavengers is reported. The key to success was the use of a two-phase solvent system to avoid otherwise predominant side reactions such as the oxidation of TEMPO by persulfate and enabled the selective formation of synthetically useful alkoxyamines. The method does not require transition metals and was successfully used in a new synthetic approach for the antidepressant indatraline.
Introduction
The Minisci reaction is an oxidative radical decarboxylation reaction of carboxylic acids 1 first reported in the early 1970s (Scheme 1).1 Mechanistically, the reaction is initiated by the oxidation of a silver(I) catalyst using potassium- or ammonium persulfate 8 as an oxidant. In the presence of an Ag(I) salt persulfate dissociates to give two sulfate radicals 9 from where an electron is transferred onto Ag(I). Sulfate 10 is formed as a by-product. This process can be repeated with the second radical 9. The active silver(II) species is next able to abstract the hydrogen atom from the carboxyl group in 1. | ||
| Scheme 1 Proposed mechanism of the classical Minisci protocol for generation of alkyl radicals from alkyl carboxylic acids using silver(II).2 | ||
The resulting carboxyl radical 2 releases CO2 to yield alkyl radical 3, which adds to pyridinium cation 4. The resulting radical cation 5 then rearomatises via radical 6 after deprotonation and oxidation to form the alkylated pyridine 7 (Scheme 1). Thereby, silver(II) acts as oxidant being reduced to silver(I). Alternatively, it was reported that the oxidation of radical 6 can also be promoted by the sulfate radical 9 to furnish sulfate 10.2
Over the past decade, this decarboxylative alkyl radical forming process has been utilised in numerous C–C3 and C–S4 coupling reactions, ring closing cascades,5 radical substitutions6 and azidations.7
Despite extensive research in this field, controlled and selective decarboxylations of this type still have some disadvantages. Often up to stoichiometric amounts of the expensive silver(I) salts are required.3,4,5a In selected rare cases it has been shown that the addition of a base can eliminate the need for transition metal catalysts.8,9 In fact, the sulfate radical 9 with a redox potential of 2.5–3.1 V is a strong single electron oxidant and can therefore form carboxyl radicals 2 from the carboxylates generated in situ.10 Particularly with the metal-free variants, but also with the silver-catalysed methods, the low selectivity is revealed by the fact that carboxylic acid 1 must be used in excess.8,9,11 In addition, the strongly oxidising conditions significantly limit the substrate and reagent range. This obstacle becomes clear, for example, when using a very common method for investigating radical mechanisms. By adding aminoxyl radicals such as 2,2,6,6-tetramethylpiperidin-1-yloxyl (TEMPO, 11) to the reaction mixture, highly reactive radical intermediates can be captured and identified as alkoxyamines.12,13 In the Minisci reaction, this method was used to determine mechanistic details and the resulting alkoxyamines were usually confirmed by mass spectrometry.8,14 One reason why this reaction has not been used preparatively so far is probably that TEMPO can be oxidised by peroxodisulfate salts.15
But alkoxyamines are an important group of reagents and functional groups widely used in organic chemistry (Scheme 2). They serve as initiators for the nitroxide-mediated polymerisation (NMP), a radical polymerisation induced by homolytic cleavage of the C–ON bond, in most cases under thermolytic conditions. The highly reactive alkyl radical and the persistent aminoxyl radical react with alkenes by a migratory insertion into the double bond.16 This reaction type has also been successfully employed in synthetic organic chemistry for ring-closing reactions.17 In 2016, Knowles et al. reported on the mesolytic cleavage of alkoxyamines by photoredox catalysis. The cross-coupling of the resulting carbocations with various nucleophiles led to the formation of new C–C, C–N or C–O bonds.18 Beyond these applications, alkoxyamines can easily be converted into alcohols19 or carbonyl compounds.20 Existing methods to form alkoxyamines start from haloalkanes,21 boronates,22 boranes,23 aldehydes12,13,24 or redox active esters.25
 | ||
| Scheme 2 Selected applications of alkoxyamines in organic chemistry. | ||
In view of these diverse considerations, we initiated a program to develop a method for the formation of alkoxyamines from carboxylic acids by metal-free radical decarboxylation.
Results and discussion
Optimisation of reaction conditions
In literature, single-phase solvent systems have been used for metal-free as well as the common silver-catalysed decarboxylative transformations using peroxodisulfate salts. Usually water is combined with a polar solvent such as acetonitrile,8,19 conditions that are not suitable if aminoxyl radicals like TEMPO 11 are employed due to the presence of peroxodisulfate salts.20 We assumed that this side reaction can be suppressed by using a two-phase system to keep the oxidant in a separate compartment from the aminoxyl radical. Mechanistically, the carboxylic acid 1 is first deprotonated at the phase boundary using potassium carbonate as base. Since the carboxylate and the peroxodisulfate salt 8 are water-soluble, the oxidative decarboxylation should take place in the aqueous phase. As a result a less polar alkyl radical is formed, which in turn is trapped by TEMPO (11) in the organic phase to yield the alkoxyamine 12 (Scheme 3). | ||
| Scheme 3 Proposed phase-transfer concept for the decarboxylative aminoxylation reported here. | ||
Following this hypothesis, we optimised the conditions of decarboxylative aminoxylation using biphenylacetic acid (1a) as model acid. A mixture of 1a, TEMPO 11 (2.0 eq.), potassium persulfate (3.0 eq.) and K2CO3 (1.5 eq.) was stirred for 20 h at rt in 1,2-dichloroethane and water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). We started with an excess of TEMPO 11 because in single-phase solvent systems the yields were greatly reduced due to the oxidation of TEMPO by persulfate. After extraction of the reaction mixture with EtOAc, alkoxyamine 12a was collected in 11% yield. However, we also observed the formation of benzophenone (13) (Table 1, entry 1). This oxidative transformation upon treatment of phenylacetic acid derivatives with K2S2O8 in water has already been reported in 2017 by Bhat et al. They proposed that the benzyl radical could be oxidised to a cation by the sulfate radical. After the nucleophilic addition of water, a second oxidation takes place, which leads to the observed carbonyl product.26
:1). We started with an excess of TEMPO 11 because in single-phase solvent systems the yields were greatly reduced due to the oxidation of TEMPO by persulfate. After extraction of the reaction mixture with EtOAc, alkoxyamine 12a was collected in 11% yield. However, we also observed the formation of benzophenone (13) (Table 1, entry 1). This oxidative transformation upon treatment of phenylacetic acid derivatives with K2S2O8 in water has already been reported in 2017 by Bhat et al. They proposed that the benzyl radical could be oxidised to a cation by the sulfate radical. After the nucleophilic addition of water, a second oxidation takes place, which leads to the observed carbonyl product.26
|
|
|||||
|---|---|---|---|---|---|
| Entry | K2S2O8 [eq.] | Solvent mix. (v:v) |
T [°C] |
12aa [%] |
13a [%] |
|
a 1H NMR-yields using naphthalene as an internal standard; yields in brackets are isolated yields.
b 1.0 eq. of TEMPO.
c Na2S2O8 was used instead of K2S2O8.
d (NH4)2S2O8 was used instead of K2S2O8; n.d. = not detected.
|
|||||
| 1 | 3.0 | DCE/H2O (1:1) |
25 | 11 | 39 |
| 2 | 3.0 | DCE/H2O (2:1) |
80 | 28 | 30 |
| 3 | 1.0 | DCE/H2O (2:1) |
80 | 49 | 13 |
| 4 | 1.0 |
DCE/H
2
O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1) :1)
|
80 | 60 (59) | 11 |
| 5b | 1.0 | DCE/H2O (3:1) |
80 | (56) | n.d. |
| 6c | 1.0 | DCE/H2O (3:1) |
80 | 46 | 22 |
| 7d | 1.0 | DCE/H2O (3:1) |
80 | 27 | 28 |
| 8 | 1.0 | MeCN/H2O (3:1) |
80 | 2 | 36 |
| 9 | 1.0 | DMF/H2O (3:1) |
80 | 3 | 2 |
| 10 | (NH4)2S2O8 (1.0 eq.), AgNO3 (2.5 mol%), CH2Cl2/H2O (1:1), rt., 6 h (ref. 27) |
<1 | 18 | ||

Following this proposal, we assumed that changing the ratio of 1,2-dichloroethane and water in favour to the former should reduce by-product formation. Additionally, we raised the reaction temperature to 80 °C to accelerate the decarboxylation step. With these changes of conditions the yield for alkoxyamine 12a could be raised to 28% (entry 2) with benzophenone (13) still being formed as the main product. As a result, the amount of oxidising agent was reduced to one equivalent. The yield for 12a was now at 49% (entry 3). In contrast, both a higher amount of base and its absence led to a decrease in yield.
A 1,2-dichloroethane–water ratio of 3:1 further improved the yield in favour of 12a to 60% (59% isolated yield) (entry 4). The reduction of the TEMPO amount from two to one equivalent resulted in comparable yield (entry 5). This indicates that only a very small amount of TEMPO is oxidized under the reaction conditions. Principally, the use of one equiv. of TEMPO is also feasible. However, if more polar, water-soluble aminoxyl radicals are used, an excess of the radical would likely be beneficial. The counter ion of the oxidising agent exerts a non-negligible influence on the transformation, as shown by the lower yields when using sodium or ammonium persulfate (entries 6 and 7). Finally, the reaction was repeated using acetonitrile or DMF as co-solvents (entries 8 and 9), with a dramatic decrease in yield, confirming the need for a two-phase system for the reasons given above.
In 2014, Xia et al. reported on the alkylation of purine nucleosides, whereby silver catalysis was also used in a Minisci-type reaction.27 The authors investigated the mechanism of this transformation by adding TEMPO to the reaction mixture. According to their experimental data, the resulting alkoxyamine could be obtained in 87% yield from cyclohexanecarboxylic acid. We repeated the experiment under exactly the conditions described by Xia and colleagues. When cyclohexanecarboxylic acid was used, we observed a lower conversion rate, with only traces of the desired product being formed. For diphenylacetic acid (1a) the yield of 12a was less than 1%, while benzophenone (13) formed as the main product (entry 10).
Substrate scope and limitations
With the present optimised conditions, we extended the usability of this reaction to further carboxylic acids (Scheme 4). If aromatic substituents are present, the positioning of this substituent relative to the carboxyl group has a major influence on the result. Arylacetic acid derivatives provide medium to high yields, precisely because it is assumed that an aryl-stabilised benzylic radical is formed intermediately. With an increasing number of carbon atoms between the carboxyl group and the aryl substituent, the yields decrease (1b–d → 12b–d). Benzyl cation stabilising substituents like the 4-methoxy group, however, favour over-oxidation and formation of the acetophenone derivative, resulting in a reduced yield of 54% for alkoxyamine 12e. The use of the toluene derivative 1g reveals the chemoselectivity of this method, as the methyl substituent remained untouched. Other functional groups noteworthy alcohols (tropic acid 1i → 12i) are tolerated. However, we found that the presence of amino groups as in N-Boc-phenylglycine, p-aminophenylacetic acid and 3-amino-2-phenylpropionic acid resulted in complex mixtures, which in the range of δ = 10 ppm in the 1H-NMR spectrum showed signals indicating the presence of aldehydes and/or imines or, in the case of the aniline derivative, polymeric materials. The decarboxylative aminoxylation presented here also allows easy access to alkoxyamine 12h, an important initiator of the nitroxide-mediated polymerisation (NMP).28 When using α-keto acid 1k, a biologically significant reaction type is achieved, which provides the TEMPO ester 12k. Such oxidative decarboxylations are widespread processes in nature. For example, the oxoglutarate dehydrogenase complex (OGDC), which is found in the citrate cycle, catalyses the oxidative decarboxylation of α-ketoglutarate to succinyl-CoA.29 Finally, we also performed decarboxylations on two more complex acids, namely indometacin and ibuprofen, which provided the TEMPO derivatives 12l and 12m in moderate to good yields. When using acids that lack aryl components, such as cyclohexanecarboxylic acid, 1-adamantanecarboxylic acid and 2-methylpentanoic acid, the formation of the desired alkoxyamines was only detected in trace amounts. Instead, we observed mainly non-decarboxylating C–H activations in different positions within the alkyl backbone.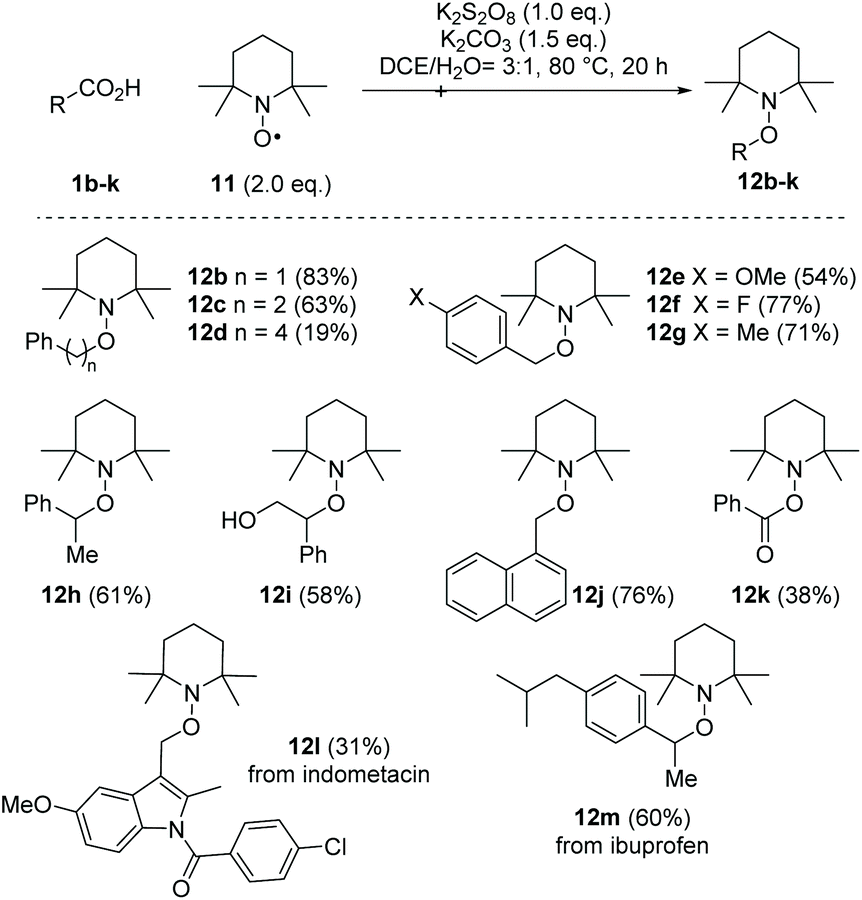 | ||
| Scheme 4 Substrate scope of the radical decarboxylative aminoxylation. | ||
This radical decarboxylation can be conducted with other TEMPO derivatives 14a–f using phenylacetic acid 1b as model substrate (Scheme 5). In applications of alkoxyamines in nitroxide-mediated polymerisations and also in the cyclisation reactions in organic synthesis, the structure of the aminoxyl radicals used plays an important role.16
 | ||
| Scheme 5 Use of other aminoxyl radicals 14a–f in oxidative decarboxylations with phenylacetic acid (1b) (aisolated yields, b15f turned out to be unstable at elevated temperatures). | ||
Both the dissociation and the recombination rate of the C–O bond, whose homolytic cleavage initiates the radical cross-coupling reaction cascade, depends strongly on the structure of the aminoxyl radical. This concerns the ring size, the steric size as well as the polarity of the persistent radical.30
Synthesis of (±)-indatraline
Indatraline (Lu 19-005, 16) is a non-selective inhibitor of the monoamine transporter. It is able to block the reuptake of dopamine, norepinephrine, and serotonin very similar to cocaine. In addition, indatraline is used to block the effects of methamphetamine and MDMA.31 Sertraline (17), a structurally closely related drug, is one of the best-selling antidepressants and, with over 38 million prescriptions in 2017, the 14th most prescribed drug in the United States.32 Here we show the application of our decarboxylative aminoxylation in a new synthetic approach to (±)-indatraline (16). The starting point is the advanced intermediate 18 from the industrial sertraline (17) synthesis (Scheme 6).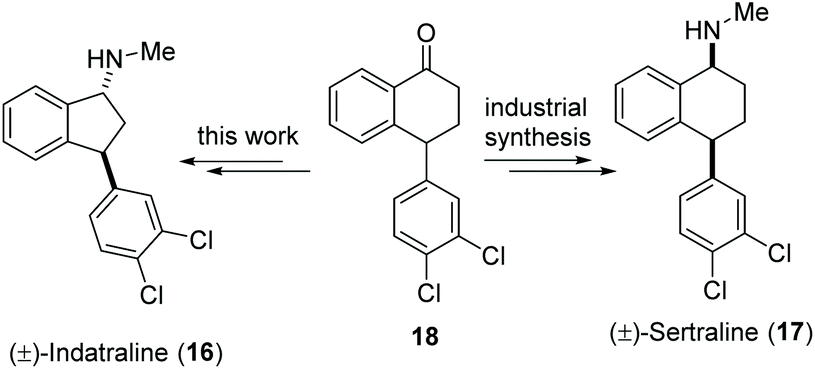 | ||
| Scheme 6 Advanced intermediate 18 of the industrial sertraline (17) synthesis as a starting point for a new total synthesis of indatraline (16). | ||
Our synthesis started with a Favorskii-type ring contraction using a slightly modified two-step protocol originally reported by the Herzon group (Scheme 7).33 First, we treated tetralone 18 with trimethylsilyltrifluoromethanesulfonate and triethylamine to obtain TMS-enol ether 19 almost quantitatively. The subsequent click reaction with nonaflyl azide and the resulting nitrogen-releasing rearrangement led to amide 20 as anti-diastereomer dr (9:1) in 71% yield.
 | ||
| Scheme 7 A new total synthesis approach for (±)-indatraline (16) based on the decarboxylative aminoxylation of carboxylic acid 21. | ||
The saponification of amide 20 proved to be problematic, since methods for the hydrolysis of nonaflyl amides have not yet been reported. Alkaline conditions were unsuccessful, probably because of the strong electron-withdrawing effect of the nonaflyl group, which stabilises a negative charge on the nitrogen atom. However, under acidic conditions, using a mixture of aqueous sulfuric acid and 1,4-dioxane at 100 °C, carboxylic acid 21 was obtained in 87% yield. Next, the key step of the synthesis, the protocol of oxidative decarboxylation presented here, was investigated, and fortunately we collected alkoxyamine 22 in 51% yield. The oxidation of alkoxyamine by mCPBA resulted in indenone 23 in 92% yield. The synthesis was then completed according to the established protocols of Davies34 and Froimowitz.35 This included diastereoselective reduction with K-Selectride®. The resulting syn-indenol 24 was finally converted into (±) indatraline (16) by mesylation and subsequent nucleophilic substitution with methylamine.
Experimental procedures
Procedure for the radical decarboxylative aminoxylation exemplified for phenylacetic acid (1b)
A vial charged with phenylacetic acid (1b) (27 mg, 200 μmol, 1.00 equiv.), TEMPO (11) (64 mg, 400 μmol, 2.0 equiv.), potassium carbonate (41 mg, 300 μmol, 1.50 equiv.) and potassium peroxodisulfate (54 mg, 200 μmol, 1.0 equiv.) was evacuated, purged with argon and 1,2-dichloroethane (1.50 mL) and water (0.50 mL) were added. The biphasic mixture was stirred at 80 °C under an argon atmosphere for 20 h. Then, the reaction mixture was diluted with water (5.0 mL) and extracted with EtOAc (3× 10.0 mL). The combined organic phases were washed with brine (10.0 mL), dried over MgSO4, filtered and concentrated under reduced pressure. The product was purified by flash column chromatography (petroleum ether:EtOAc = 50:1). The alkoxyamine 12b was collected as a pale-yellow oil (41 mg, 166 μmol, 83%). Rf = 0.50 (petroleum ether:EtOAc 50:1); 1H-NMR (CDCl3, 600 MHz): δ [ppm] 7.38–7.28 (m, 5H, ArH), 4.83 (s, 2H, CH2ON), 1.64–1.34 (m, 6H, 3 × CH2), 1.26 (s, 6H, 2 × CH3), 1.16 (s, 6H, 2 × CH3); 13C-NMR (CDCl3, 150 MHz): δ [ppm] 138.5 (q, ArC), 128.4 (t, 2 × ArC), 127.6 (t, 2 × ArC), 127.4 (t, ArC), 78.9 (s, CH2ON), 60.2 (q, 2 × C(CH3)2), 39.9 (s, 2 × CH2), 33.2 (p, 2 × CH3), 20.4 (p, 2 × CH3), 17.3 (s, CH2(CH2)2). The analytical data are consistent with those reported in the literature.36
Conclusions
In summary, we reported on the first general method for the direct synthesis of alkoxyamines from carboxylic acids. By using a biphasic solvent system, we were able to suppress known side reactions and thus perform radical decarboxylation and subsequent capture of the alkyl radical with structurally different aminoxyl radicals with high chemoselectivity. This allowed us to completely avoid the use of transition metal catalysts.37Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded in part by the BMBF (project: SILVIR, grant no. 16GW0202). We thank Judith Synofzik (Leibniz University Hannover) and Kjeld Gerdes (Leibniz University Hannover) for performing some of the experiments.Notes and references
- F. Minisci, R. Bernardi, F. Bertini, R. Galli and M. Perchinummo, Tetrahedron, 1971, 27, 3575 CrossRef CAS.
- R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666 CrossRef CAS.
- W.-M. Zhao, X.-L. Chen, J.-W. Yuan, L.-B. Qu, L.-K. Duan and Y.-F. Zhao, Chem. Commun., 2014, 50, 2018 RSC.
- P.-F. Wang, X.-Q. Wang, J.-J. Dai, Y.-S. Feng and H.-J. Xu, Org. Lett., 2014, 16, 4586 CrossRef CAS.
- (a) K. Yan, D. Yang, W. Wei, F. Wang, Y. Shuai, Q. Li and H. Wang, J. Org. Chem., 2015, 80, 1550 CrossRef CAS; (b) S. Seo, M. Slater and M. F. Greaney, Org. Lett., 2012, 14, 2650 CrossRef CAS.
- L. Cui, H. Chen, C. Liu and C. Li, Org. Lett., 2016, 18, 2188 CrossRef CAS.
- C. Liu, X. Wang, Z. Li, L. Cui and C. Li, J. Am. Chem. Soc., 2015, 137, 9820 CrossRef CAS.
- S. Lu, Y. Gong and D. Zhou, J. Org. Chem., 2015, 80, 9336 CrossRef CAS.
- J. K. Laha, K. V. Patel, K. S. S. Tummalapalli and N. Dayal, Chem. Commun., 2016, 52, 10245 RSC.
- (a) M. Brienza and I. Katsoyiannis, Sustainability, 2017, 9, 1604 CrossRef; (b) L. W. Matzek and K. E. Carter, Chemosphere, 2016, 151, 178 CrossRef CAS.
- S. Mandal, T. Bera, G. Dubey, J. Saha and J. K. Laha, ACS Catal., 2018, 8, 5085 CrossRef CAS.
- A. Kipke, K.-U. Schöning, M. Yusubov and A. Kirschning, Eur. J. Org. Chem., 2017, 6906 CrossRef CAS.
- K.-U. Schöning, W. Fischer, S. Hauck, A. Dichtl and M. Kuepfert, J. Org. Chem., 2009, 74, 1567 CrossRef.
- (a) L. Lv, S. Lu, Y. Chen and Z. Li, Org. Chem. Front., 2017, 4, 2147 RSC; (b) Y. Li, M. Hu and J.-H. Li, ACS Catal., 2017, 7, 6757 CrossRef CAS; (c) G. Hong, J. Yuan, J. Fu, G. Pan, Z. Wang, L. Yang, Y. Xiao, P. Mao and X. Zhang, Org. Chem. Front., 2019, 6, 1173 RSC; (d) J. Li, W.-J. Hao, P. Zhou, Y.-L. Zhu, S.-L. Wang, S.-J. Tu and B. Jiang, RSC Adv., 2017, 7, 9693 RSC.
- P. Gao, J. Wang, Z. Bai, M.-J. Fan, D.-S. Yang and Z.-H. Guan, Org. Lett., 2018, 20, 3627 CrossRef CAS.
- L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 5034 CrossRef CAS.
- (a) A. Studer, Angew. Chem., Int. Ed., 2000, 39, 1108 CrossRef CAS; (b) M. Šimek, K. Bártová, R. Pohl, I. Císařová and U. Jahn, Angew. Chem., 2020, 59, 6160 CrossRef.
- Q. Zhu, E. C. Gentry and R. R. Knowles, Angew. Chem., Int. Ed., 2016, 55, 9969 CrossRef CAS.
- A. R. Howell and G. Pattenden, J. Chem. Soc., Chem. Commun., 1990, 103 RSC.
- T. Inokuchi and H. Kawafuchi, Tetrahedron, 2004, 51, 11969 CrossRef.
- (a) R. Braslau, A. Tsimelzon and J. Gewandter, Org. Lett., 2004, 6, 2233 CrossRef CAS; (b) J.-L. Counturier and O. Guerret, WO 2002012149, 2002; (c) J.-L. Counturier and O. Guerret, WO 2000061544, 2000; (d) T. J. Connolly, M. V. Baldoví, N. Mohtat and J. C. Scaiano, Tetrahedron Lett., 1996, 37, 4919 CrossRef CAS.
- C. Cadot, P. I. Dalko, J. Cossy, C. Ollivier, R. Chuard and P. Renaud, J. Org. Chem., 2002, 67, 7193 CrossRef CAS.
- C. Ollivier, R. Chuard and P. Renaud, Synlett, 1999, 807 CrossRef CAS.
- K.-U. Schöning, W. Fischer, A.-I. Basbas and A. Dichtl, WO 2008003602, 2008.
- C. Zheng, Y. Wang, Y. Xu, Z. Chen, G. Chen and S. H. Liang, Org. Lett., 2018, 20, 4824 CrossRef CAS.
- T. B. Mete, T. M. Khopade and R. G. Bhat, Tetrahedron Lett., 2017, 58, 2822 CrossRef CAS.
- R. Xia, M.-S. Xie, H.-Y. Niu, G.-R. Qu and H.-M. Guo, Org. Lett., 2014, 16, 444 CrossRef CAS.
- T. J. Connolly, M. V. Baldoví, N. Mohtat and J. C. Scaiano, Tetrahedron Lett., 1996, 37, 4919 CrossRef CAS.
- L. A. Moran, H. R. Horton, K. G. Scrimgeour and M. D. Perry, Principles of biochemistry, Pearson Education, 2012, pp. 394–402 Search PubMed.
- G. Audran, P. Brémond and S. R. A. Marque, Chem. Commun., 2014, 50, 7921 RSC.
- R. B. Rothman, J. S. Partilla, M. H. Baumann, C. M. Dersch, F. I. Carroll and K. C. Rice, Synapse, 2000, 35, 222 CrossRef CAS.
- ClinCalc DrugStats Database, “The Top 300 of 2020”, https://clincalc.com/DrugStats/Top300Drugs.aspx, 2020.
- M. J. Mitcheltree, Z. A. Konst and S. B. Herzon, Tetrahedron, 2013, 69, 5634 CrossRef CAS.
- H. M. L. Davies and T. M. Gregg, Tetrahedron Lett., 2002, 43, 4951 CrossRef CAS.
- M. Froimowitz, K. M. Wu, A. Moussa, R. M. Haidar, J. Jurayj, C. George and E. L. Gardner, J. Med. Chem., 2000, 43, 4981 CrossRef CAS.
- Y. Yasu, T. Koike and M. Akita, Adv. Synth. Catal., 2012, 354, 3414 CrossRef CAS.
- For a recent copper-mediated oxidative decarboxylation followed by alkylation of secondary amines see: D. Kong, P. J. Moon, O. Bsharat and R. J. Lundgren, Angew. Chem., 2020, 59, 1313 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob01773f |
| This journal is © The Royal Society of Chemistry 2021 |