Thiol–ene enabled preparation of S-lipidated anti-HBV peptides†
Received
30th September 2020
, Accepted 1st November 2020
First published on 6th November 2020
Abstract
Despite significant efforts made towards treatments for Hepatitis B virus (HBV), a long-term curative treatment has thus far eluded scientists. Recently, the Sodium Taurocholate Co-Transporting Polypeptide (NTCP) receptor has been identified as the entry pathway of HBV into hepatocytes. Myrcludex B, an N-terminally myristoylated 47-mer peptide mimic of the preS1 domain of the Hepatitis B virion, was identified as a potent protein–protein interaction (PPI) inhibitor blocking HBV fusion (IC50 = 140 pM). Herein we report an optimised chemical synthesis of Myrcludex B and a series of novel analogues. Employing a small modification to the Cysteine Lipidation of a Peptide or Amino acid (CLipPA) thiol–ene reaction, a library of S-lipidated Myrcludex B and truncated (21-mer) analogues were prepared, providing novel chemical space to probe for the discovery of novel anti-HBV peptides. The S-lipidated analogues showed an equivalent or a slight decrease (∼2-fold) in binding effectiveness to NTCP expressing hepatocytes compared to Myrcludex B. Three S-lipidated analogues were highly potent HBV inhibitors (IC50 0.97–3.32 nM). These results demonstrate that incorporation of heteroatoms into the lipid ‘anchor’ is tolerated by this antiviral scaffold and to the best of our knowledge constitutes the first report of potent S-lipidated antiviral peptides. Interestingly, despite only moderate reductions in binding effectiveness, truncated analogues possessed dramatically reduced inhibitory activity thus providing new insights into the structure activity relationship of these hitherto unreported antiviral S-lipopeptides.
Introduction
Hepatitis B virus (HBV) is responsible for chronic infection in millions of people worldwide and is a major factor in the development of liver cirrhosis and hepatocellular carcinoma.1
Currently, HBV infection is treated with either nucleoside analogues, non-/PEGylated interferon-α or a combination of the two.2,3 Although these current HBV treatments exist, their cost often limits availability in the countries of highest need. Furthermore, while treatment is effective in suppression of viral replication to low or undetectable levels, it is rarely curative, requiring lifelong therapy to maintain a virus-free status in patients. Thus, there is a need to develop new or novel combinatorial treatments that can eliminate persistent viral infection in chronic HBV sufferers.3,4
HBV infects hepatocytes via interaction of the preS1 region of the HBV large surface protein with the virus entry receptor, the Sodium (Na) Taurocholate Co-transporting Polypeptide (NTCP).5 Identification of NTCP as an HBV receptor elucidates the fusion process as an attractive target for drug development. While searching for new methods of treatment for chronic HBV, Gripon et al. developed a 47-mer N-lipidated peptide analogue of the N-terminal sequence of the preS1 domain with the ability to inhibit HBV infection.6,7 Subsequent studies involving variation of the peptide sequence and the nature of the lipid established Bulervitide, coined ‘Mycludex B’, as the lead analogue for clinical trials, with IC50 potency of 140 pM towards HBV.8,9 Myrcludex B is a “first-in-class” drug that acts as a competitive protein–protein interaction (PPI) inhibitor of HBV and the NTCP receptor.10 Urban et al. determined that lipidation was essential for the inhibitory activity of Myrcludex B, and proposed it acts as a membrane ‘anchor’ increasing local concentration through membrane affinity.5 Further investigations by Schulze et al. determined that a myristoylated and truncated sequence of Myrcludex B (21-mer) retained activity, albeit with reduced potency (IC50 = 3 nM).11 A similar approach using lipopeptides as fusion inhibitors has recently been investigated for coronaviruses and HIV.12,13
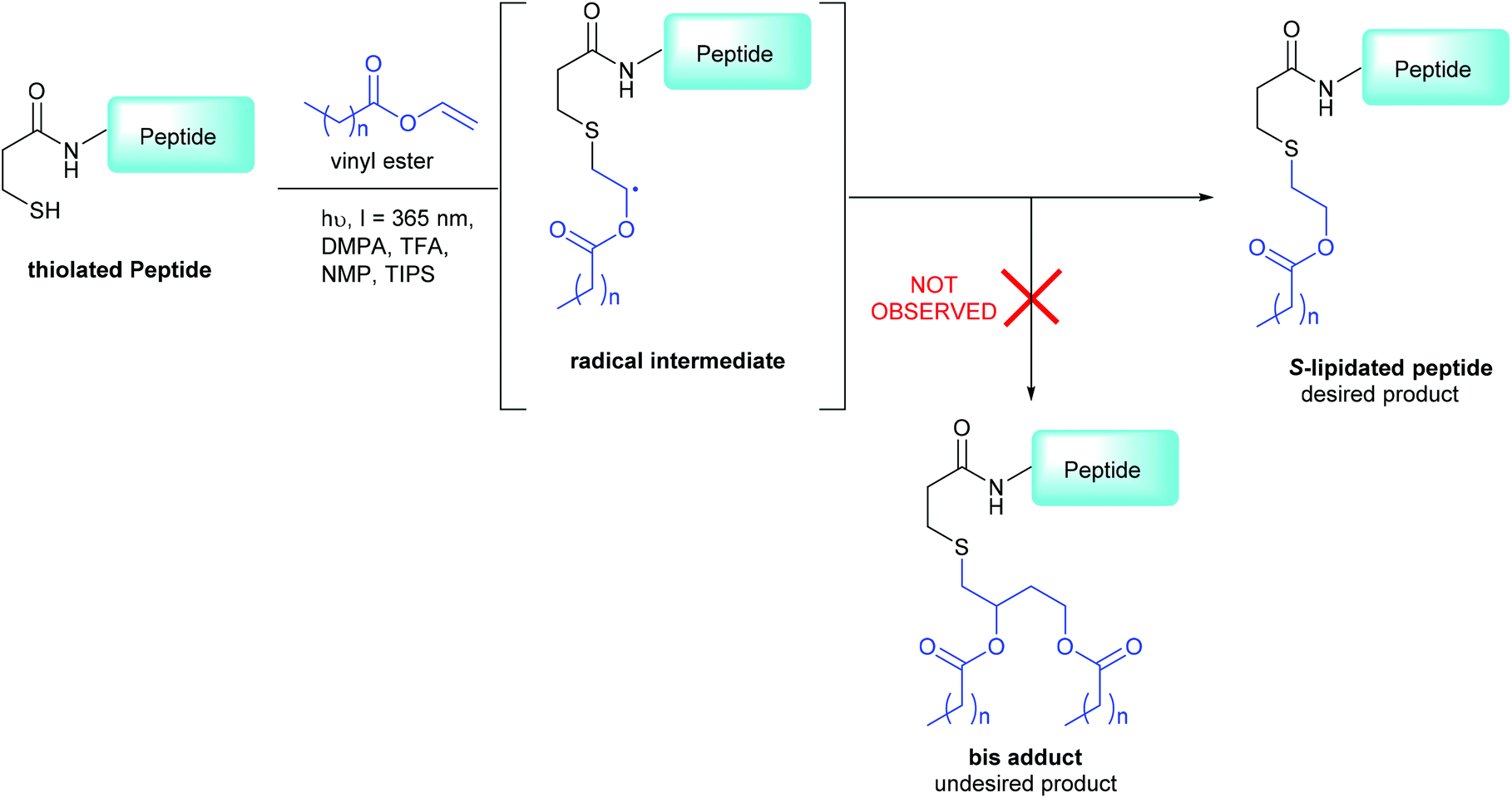
We recently reported a novel lipidation technology, based on thiol–ene chemistry, termed “CLipPA” – Cysteine Lipidation on a Peptide or Amino Acid, that enables a lipid to be installed selectively onto a peptide upon UV irradiation (Scheme 1).14–17 This strategy has previously been applied to the preparation of S-lipidated peptides with a range of therapeutic properties, including antibacterial lipopeptides, self-adjuvating vaccines and calcitonin gene-related peptide (CGRP) receptor antagonists.18–23
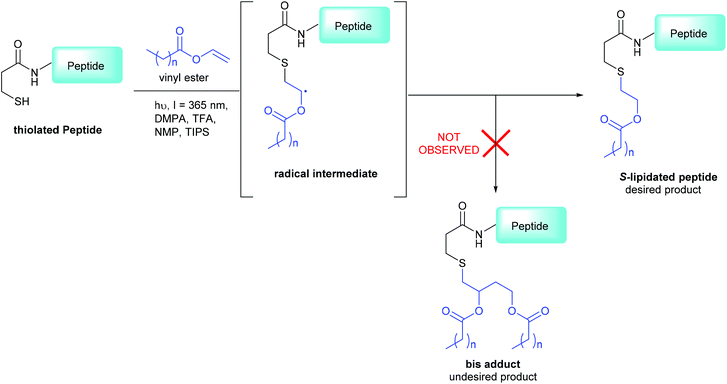 |
| | Scheme 1 3-MPA modified CLipPA lipidation of a model peptide (blue) via a radical intermediate leading to the desired S-lipidated product; the undesired bis-adduct is not observed under our optimized conditions.15 | |
The CLipPA reaction is carried out by irradiation (365 nm) of an NMP solution containing a peptide bearing a thiol handle, a vinyl ester containing the lipid of a desired chain length and a photoinitiator, 2,2-dimethoxy-2-pheynlacetophenone (DMPA). The sulfur radical species thus generated quickly reacts with the unsaturated vinyl group of the ester to give a stabilised radical intermediate. Hydride transfer by tert-nonylthiol (tNonSH) and triisopropylsilane (TIPS) then affords the desired S-lipidated product (Scheme 1). In the absence of a hydride reagent, an undesired bis-adduct forms together with the desired product. The apparent pH of the reaction is lowered using trifluoroacetic acid (TFA) to protonate electron rich side-chain residues, thus preventing propagation of radical species by single-electron transfer. The reaction conditions are mild, generally high-yielding, atom economical, and chemo-selective. Thus, fully deprotected peptides are amenable to CLipPA.
We envisaged the CLipPA lipidation strategy could be used for the facile preparation of a library of S-lipidated Myrcludex B analogues from a common precursor, in which the lipid portion was varied to encompass structural diversity, including aromatic moieties. We herein report a library of potent S-lipidated Myrcludex B and truncated Myrcludex B analogues bearing distinctive lipid moieties prepared from a modified Myrcludex B scaffold using the CLipPA thiol–ene reaction, by incorporation of the thiol handle, 3-mercaptopropionic acid (3-MPA). The analogues were additionally prepared as fluorescently labelled variants for the determination of binding efficiency and ultimately for correlation with HBV inhibitory potency. To the best of our knowledge this is the first report of potent S-lipidated antiviral peptides and the first study attempting to correlate NTCP binding with inhibitory activity.
Results/discussion
Library design
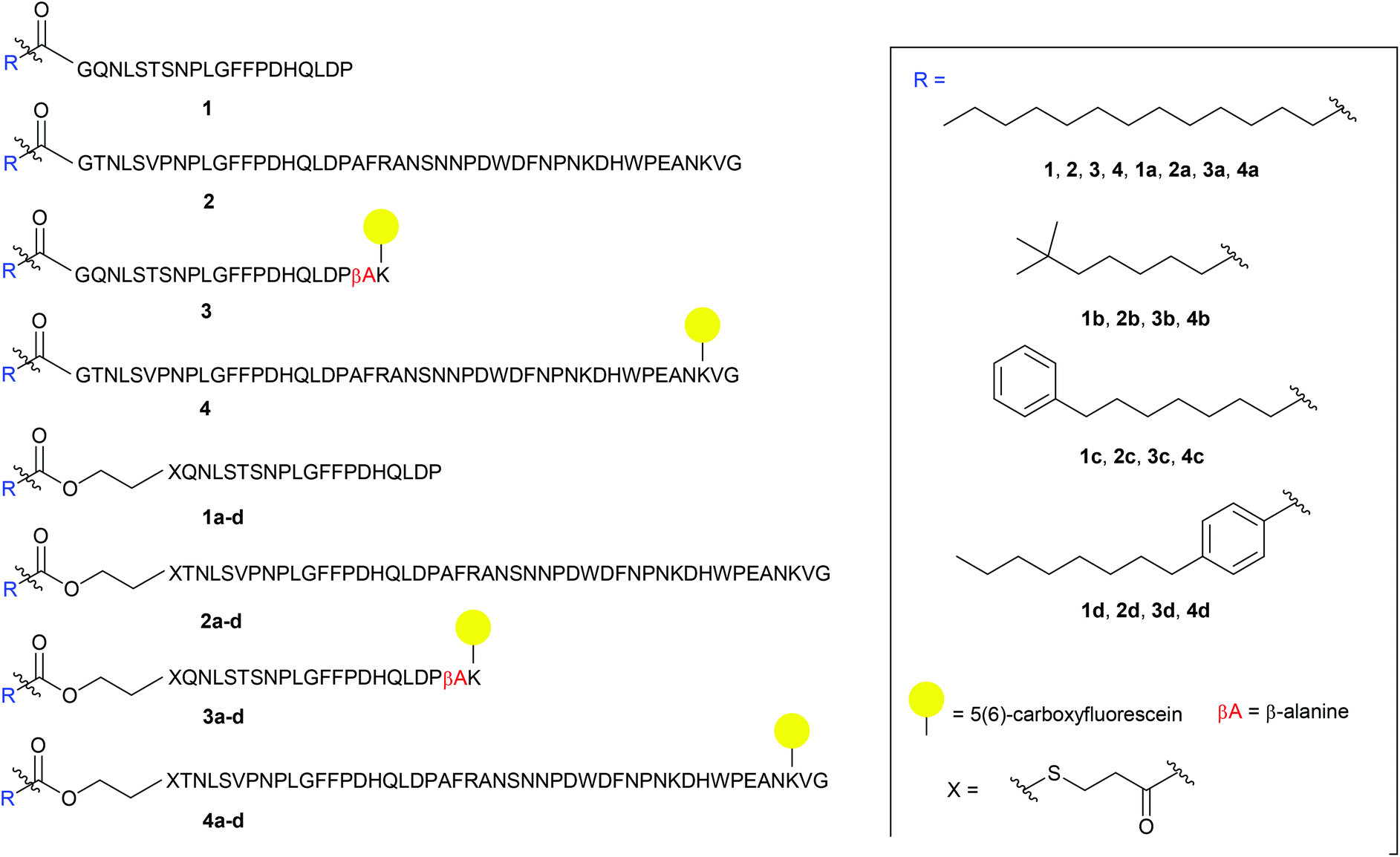
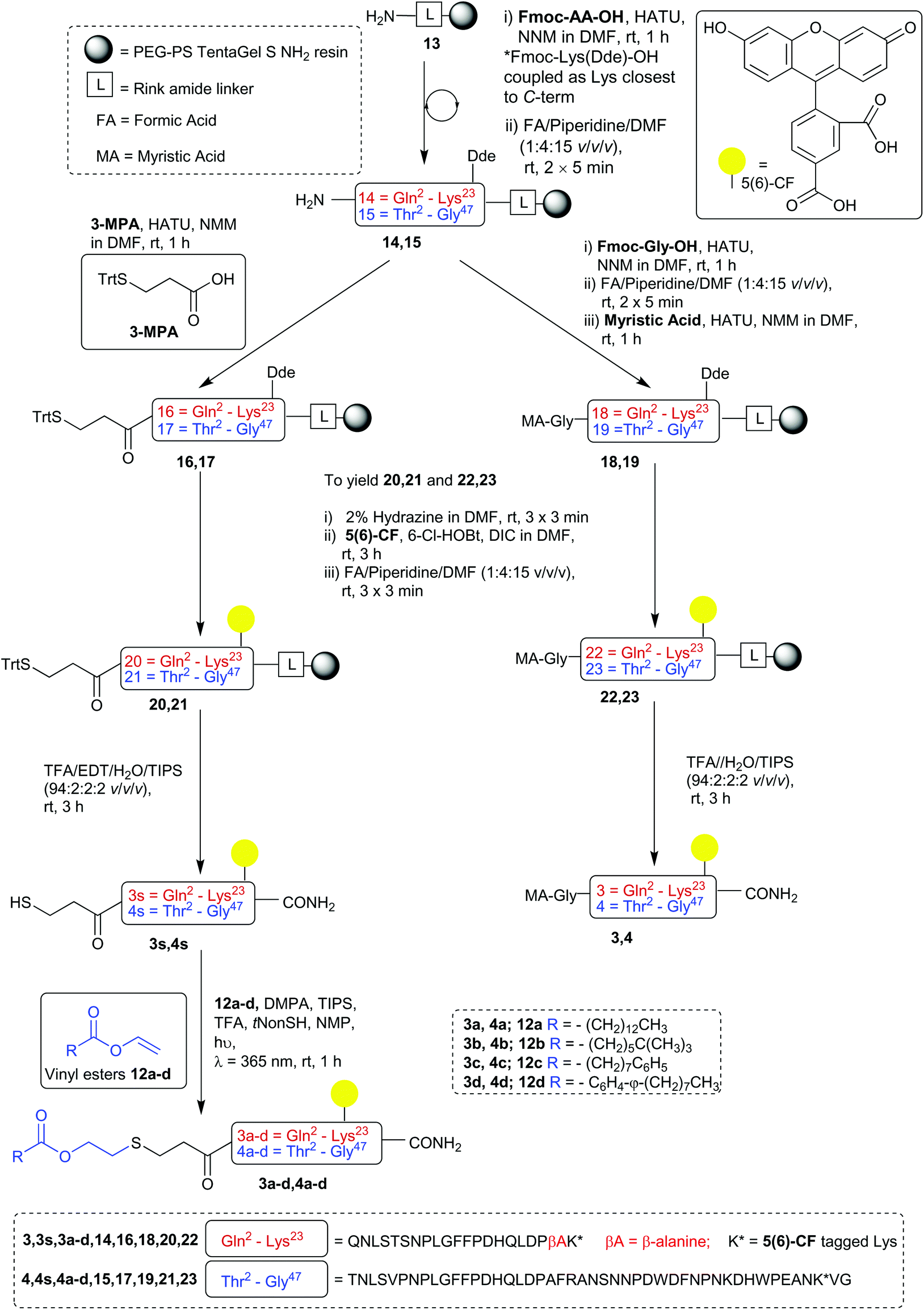
The S-lipidated analogue series was designed to incorporate two unique peptide sequences, one mimicking the truncated sequence reported by Schulze et al. (residues 2–21 of Myrcludex B) and one equivalent to Myrcludex B (47 residues) (Fig. 1).9,11 By replacing Gly1 at the N-terminus with 3-mercaptopropionic acid (3-MPA), these sequences were thus functionalised with the necessary thiol handle required for implementation of the desired chemo-selective S-lipidation strategy. Towards this end, 3-MPA was chosen rather than cysteine to avoid the addition of a positively charged amino group (at physiological pH) to the hydrophobic region at the N-terminus of the peptides. A series of four lipids (Fig. 1) were selected to encompass a variety of different functionalities, such as aromatic groups, branched lipids and long chain alkyl lipids similar to the lipid in the parent sequence. This would enable a direct comparison of the effects imparted by the heteroatoms present in the CLipPA bridging unit.
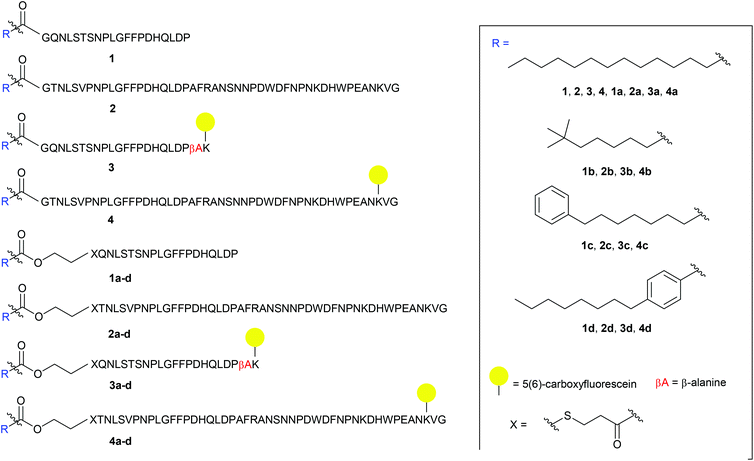 |
| | Fig. 1 Library of envisioned analogues for truncated/full length/labelled and non-labelled peptides. Lipidation highlighted in blue. Amino acids represented as single letter code; X = 3-mercaptopropanoyl. | |
A series of 5(6)-carboxyfluorescein (5(6)-CF) labelled analogues were additionally prepared for binding assays with NTCP expressing hepatocytes. As the literature in the past has not been definitive as to the site of fluorescent labelling, we determined the position of our tag from previous SAR studies. Given that the C-terminus is reported to be less essential for activity, the side chain of Lys45 near the C-terminus of the full-length peptides was labelled. As the truncated sequence possessed no Lys in close proximity to the C-terminus, a β-Ala spacer followed by a Lys residue were inserted at the C-terminus to enable fluorescent labelling (3 and 3a–d, Fig. 1).
The truncated sequence (1), the parent compound (Myrcludex B, 2) and their 5(6)-CF labelled derivatives (3 and 4) were prepared using the native myristic acid as the lipid component to serve as controls during biological evaluations. In addition, a non-lipidated fluorescently labelled 47-mer peptide (5) (see ESI: S5.20†) was prepared as a further control for the binding assays.
Peptide synthesis
Controls and CLipPA precursors.
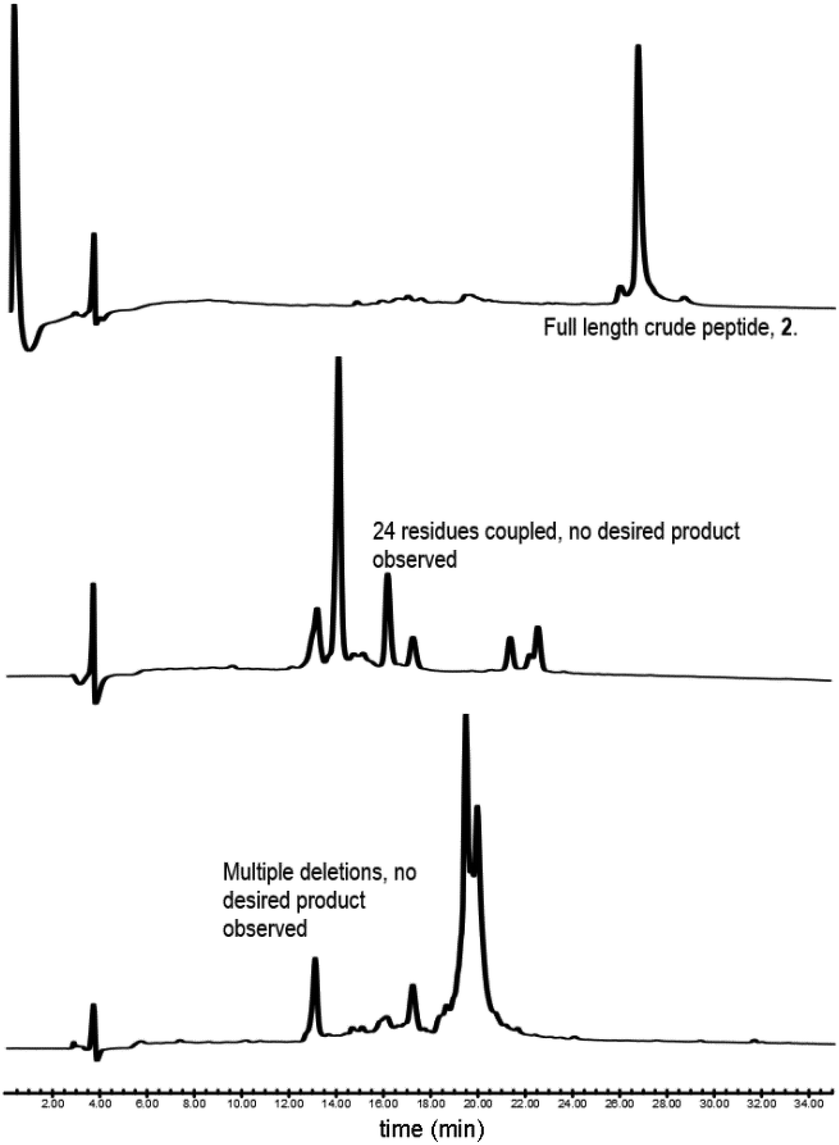
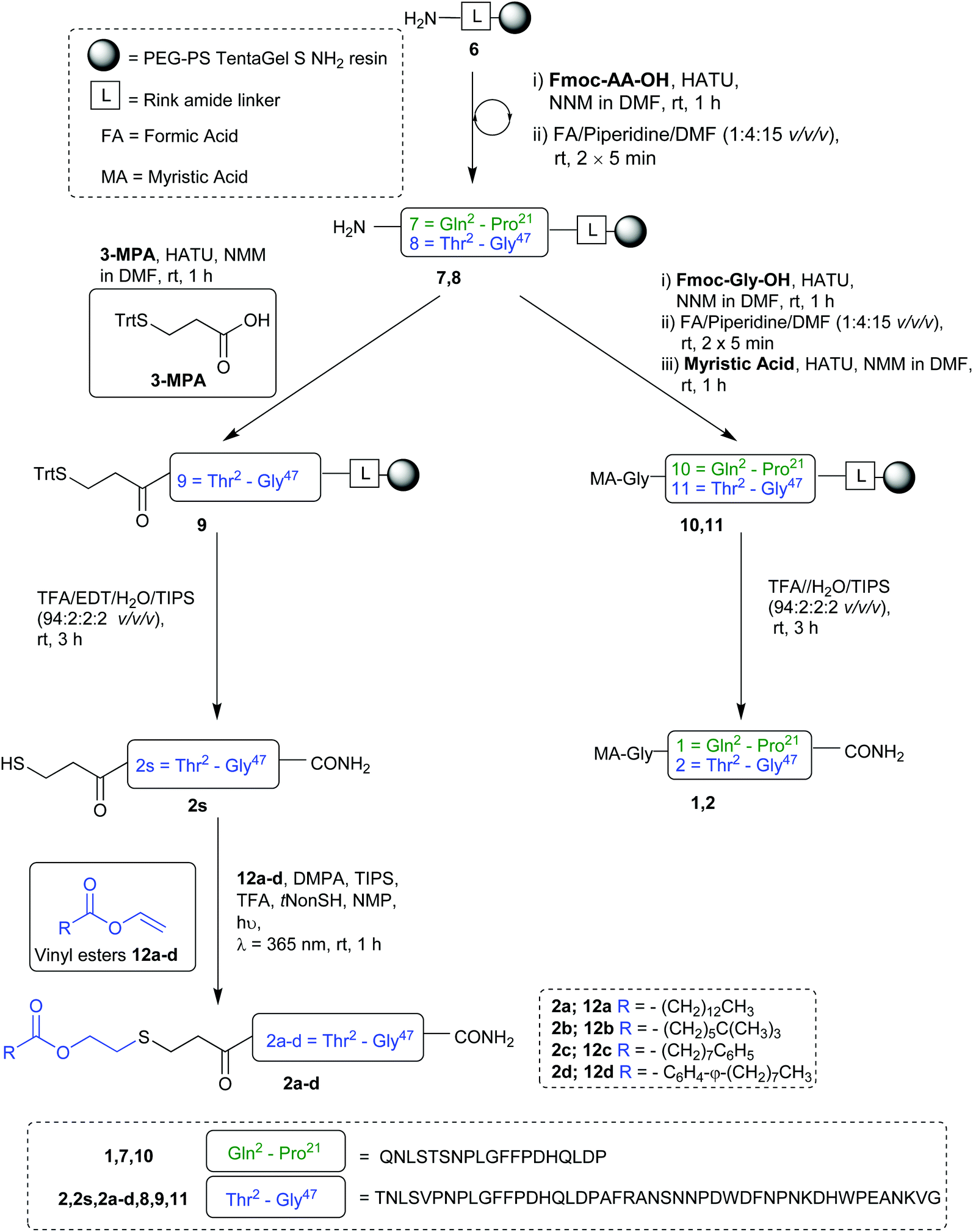
Our synthesis began with the reference peptide Myrcludex B (2) employing 9-fluorenylmethoxycarbonyl solid phase peptide synthesis (Fmoc-SPPS), where couplings were performed with 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,-tertramethyluronium hexafluorophosphate (HATU) and N,N-diisopropylethylamine (DIPEA) (1 h, rt). Fmoc removal was performed with piperidine/dimethylformamide (DMF) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 v/v) (2 × 5 min, rt). During the attempted synthesis, upon mini-cleavage of the partially elongated peptidyl resin (residues 27 to 47), ESI-MS revealed masses of +66 m/z and +132 m/z (relative to the desired mass), attributed to piperidide adduct(s) formed on aspartic acid residue(s) during iterative Fmoc removals with piperidine/DMF (1:4 v/v) (2 × 5 min, rt). Michels et al. reported that the addition of formic acid (5% v/v) to the Fmoc deprotection solution was effective in suppressing the formation of these difficult to separate adducts.24,25 While implementing this modification to our synthesis proved successful in eliminating the formation of piperidide adducts, the synthesis ultimately remained unsuccessful, with a mini cleavage of the peptide at residue 23 revealing multiple deletions and complete absence of the desired peptide mass (Fig. 2, lower). Upon switching from a polystyrene based resin (0.94 mmol g−1) to an aminomethyl ChemMatrix® resin (0.62 mmol g−1), little improvement was achieved (Fig. 2, middle), however, the use of the lower loading (0.27 mmol g−1) TentaGel® S NH2 resin resulted in a highly successful synthesis of Myrcludex B (2) (Fig. 2, upper). Implementing this modified synthesis, crude 47-mer lipopeptide (2) was yielded in ∼85% purity (Fig. 2, upper) after resin cleavage and global deprotection using a cocktail of TFA/EDT/H2O/TIPS (94:2:2:2 v/v/v/v) for 3 h at rt. Having optimised an effective synthetic procedure, the amended protocol was adapted for use with an automated synthesiser, in which the base was swapped from DIPEA to N-methylmorpholine (NMM) (Scheme 2) for improved miscibility in DMF, with little to no effect on the success of the synthesis. Following this procedure, the truncated analogue (1) and CLipPA precursor peptide (2s) in which the N-terminal glycine was replaced with 3-mercaptopropionic acid were successfully prepared with crude purities >75%. The crude peptides were then purified to >95% by RP-HPLC to afford the pure peptides (1) and (2s) in 21% and 30% yield respectively, for either direct use in biological assays or CLipPA S-lipidation as appropriate. While not necessary for successful CLipPA thiol–ene lipidation, the thiol bearing precursor peptides were first purified to facilitate reaction monitoring and comparison of the conversion rates for the four structurally diverse lipids.
:4 v/v) (2 × 5 min, rt). During the attempted synthesis, upon mini-cleavage of the partially elongated peptidyl resin (residues 27 to 47), ESI-MS revealed masses of +66 m/z and +132 m/z (relative to the desired mass), attributed to piperidide adduct(s) formed on aspartic acid residue(s) during iterative Fmoc removals with piperidine/DMF (1:4 v/v) (2 × 5 min, rt). Michels et al. reported that the addition of formic acid (5% v/v) to the Fmoc deprotection solution was effective in suppressing the formation of these difficult to separate adducts.24,25 While implementing this modification to our synthesis proved successful in eliminating the formation of piperidide adducts, the synthesis ultimately remained unsuccessful, with a mini cleavage of the peptide at residue 23 revealing multiple deletions and complete absence of the desired peptide mass (Fig. 2, lower). Upon switching from a polystyrene based resin (0.94 mmol g−1) to an aminomethyl ChemMatrix® resin (0.62 mmol g−1), little improvement was achieved (Fig. 2, middle), however, the use of the lower loading (0.27 mmol g−1) TentaGel® S NH2 resin resulted in a highly successful synthesis of Myrcludex B (2) (Fig. 2, upper). Implementing this modified synthesis, crude 47-mer lipopeptide (2) was yielded in ∼85% purity (Fig. 2, upper) after resin cleavage and global deprotection using a cocktail of TFA/EDT/H2O/TIPS (94:2:2:2 v/v/v/v) for 3 h at rt. Having optimised an effective synthetic procedure, the amended protocol was adapted for use with an automated synthesiser, in which the base was swapped from DIPEA to N-methylmorpholine (NMM) (Scheme 2) for improved miscibility in DMF, with little to no effect on the success of the synthesis. Following this procedure, the truncated analogue (1) and CLipPA precursor peptide (2s) in which the N-terminal glycine was replaced with 3-mercaptopropionic acid were successfully prepared with crude purities >75%. The crude peptides were then purified to >95% by RP-HPLC to afford the pure peptides (1) and (2s) in 21% and 30% yield respectively, for either direct use in biological assays or CLipPA S-lipidation as appropriate. While not necessary for successful CLipPA thiol–ene lipidation, the thiol bearing precursor peptides were first purified to facilitate reaction monitoring and comparison of the conversion rates for the four structurally diverse lipids.
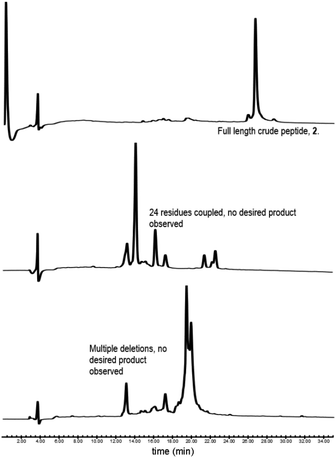 |
| | Fig. 2 RP-HPLC traces of initial attempts to synthesise Myrcludex B (2). (lower) Polystyrene based resin, (middle) aminomethyl ChemMatrix® resin and (upper) TentaGel® S NH2 resin. Desired final product only observed for TentaGel® S NH2. Other syntheses were aborted mid sequence due to the presence of many unidentified by-products. | |
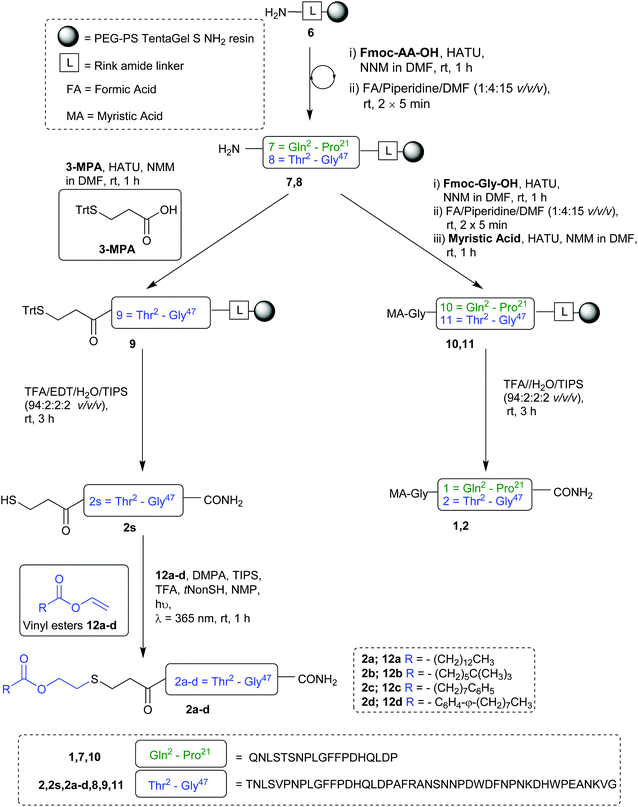 |
| | Scheme 2 Preparation of Myrcludex B analogues. | |
5(6)-Carboxyfluorescein labelled peptides.
By incorporating a Lys residue with the fully orthogonal (1-(4,4′-dimethyl-2,6-dioxocyclohexylidene)-3-ethyl) Dde as a side chain protecting group in the aforementioned positions of the sequence, fluorescently labelled analogues of each biological control peptides (3 and 4) and CLipPA precursor peptides (3s and 4s) were prepared (Scheme 3) alongside the non-lipidated biological control peptide (5, shown in ESI, S5.20†).26 After complete sequence elongation, the Dde protecting group of each peptidyl-resin was then selectively removed by treatment with a solution of hydrazine in DMF (2% v/v) (3 × 3 min, rt). Having liberated the lysine ε-amino group, 5(6)-CF was coupled under base free conditions using 6-chloro-1-hydroxylbenzotriazole (6-Cl-HOBt) and N,N′-diisopropylcarbodiimide (DIC) (3 h, rt), followed by three treatments with the standard Fmoc removal solution to effect aminolysis of the 5(6)-CF groups polymerised onto the peptidyl resin via the 5(6)-CF phenol (as observed by RP-HPLC and ESI-MS upon resin mini-cleavage, in accordance with the literature).27 Upon resin cleavage with TFA/EDT/H2O/TIPS (94:2:2:2 v/v/v/v) (3 h, rt) the crude native myristoylated peptides (3 and 4) and thiol bearing CLipPA precursors (3s and 4s) were afforded in >70% purity. These fluorescently labelled peptides were purified via semi-preparative RP-HPLC to afford the desired peptides in >95% purity with yields ranging from 12%–30%.
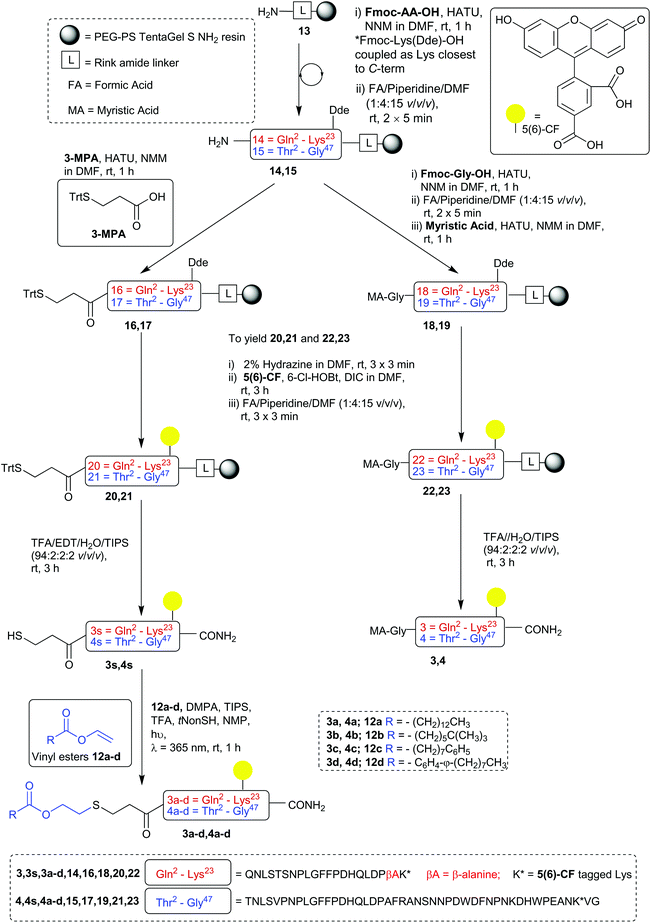 |
| | Scheme 3 Preparation of 5(6)-carboxyfluorecein labelled Myrcludex B analogues. | |
Peptide S-lipidation
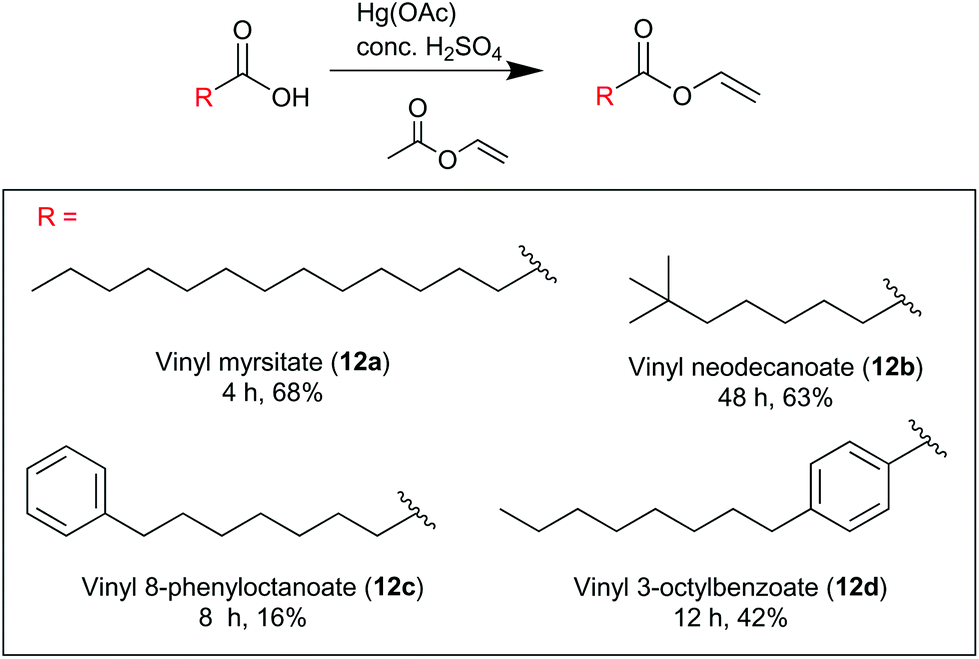
The desired lipid moieties were functionalised as vinyl esters (Scheme 4), following the procedures reported by Magrone et al.28 Purified peptides (2s/3s and 4s) were subjected to the CLipPA thiol–ene reaction with each of the vinyl esters (12a–12d). Each peptide, the photoinitiator (DMPA) and the CLipPA additives were dissolved in a solution of NMP with the desired vinyl ester and the reaction mixture irradiated under UV (365 nm) for one hour to effect CLipPA S-lipidation.16 After irradiation, the reaction mixture containing the crude CLipPA product was triturated with diethyl ether prior to purification, thereby removing non-peptidyl by-products (Fig. 3).
 |
| | Scheme 4 Synthesis of vinyl esters for CLipPA chemistry. Reagents and conditions: (Hg(OAc)2 (0.025 equiv.), conc. H2SO4 (cat., 15 μL), 74 °C; reactions were conducted on gram scale with vinyl acetate (10 equiv.) as solvent. Reaction time and % yield stated below. 12b structure indicative only (mixture of vinyl neodecanoate isomers). | |
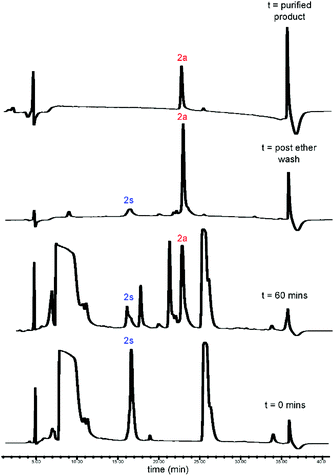 |
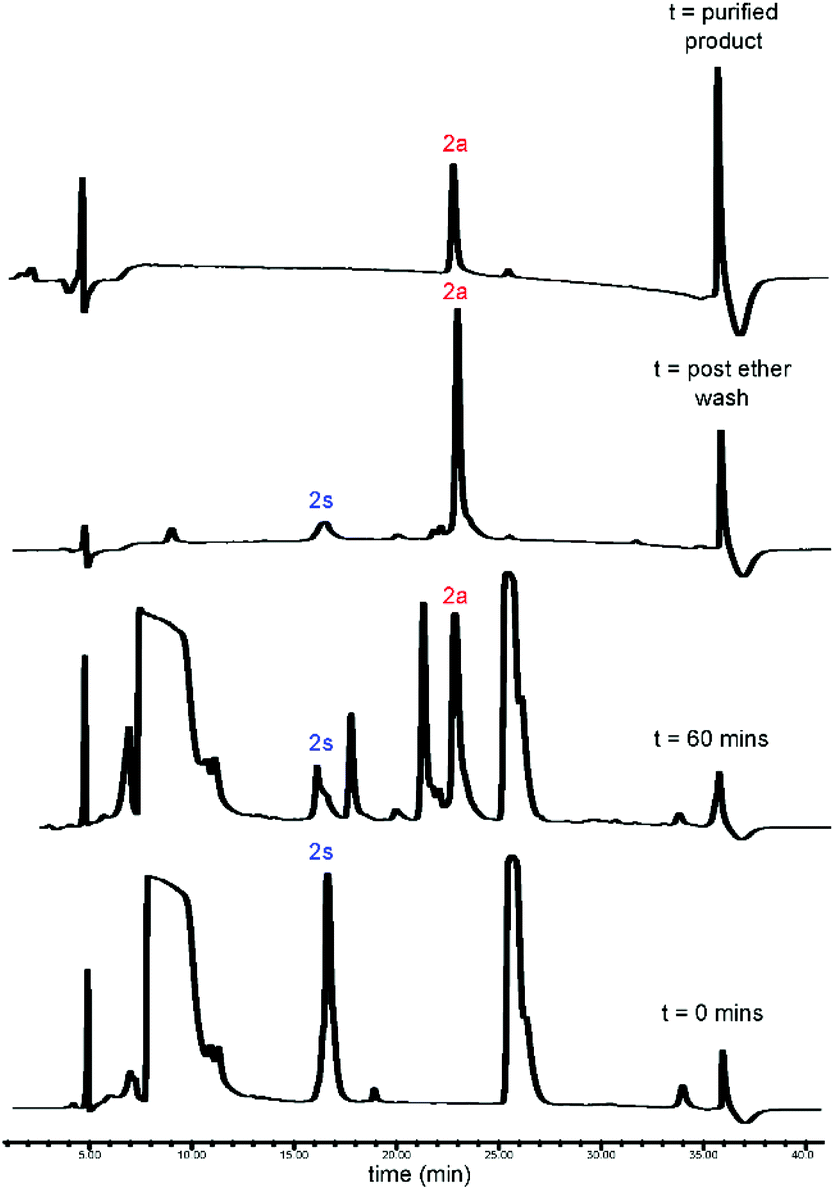
| | Fig. 3 RP-HPLC traces for CLipPA addition of vinyl myristate (12a) to precursor peptide (2s), yielding S-lipidated peptide 2a. | |
CLipPA conjugation of vinyl esters (12a–d) with peptide precursors 2s and 4s afforded S-lipidated 47-mer Myrcludex B analogues 2a–d and 4a–d, respectively, while conjugation with peptide precursors 3s afforded truncated analogues (3a–d) (Schemes 2 and 3) with conversions ranging 60%–90% and isolated yields ranging 6%–37%. Conversion rates of the S-lipidation were moderately dependent on the precursor peptide, vinyl ester and reaction scale. The 47-mer thiol functionalised peptides 2s and 4s and the straight chain vinyl ester (vinyl myristate, 12a) provided the greatest conversion rates. The S-lipidated peptide analogues were then purified via semi-preparative RP-HPLC to >95% purity.
Lipopeptide binding to HepG2-NTCP cells
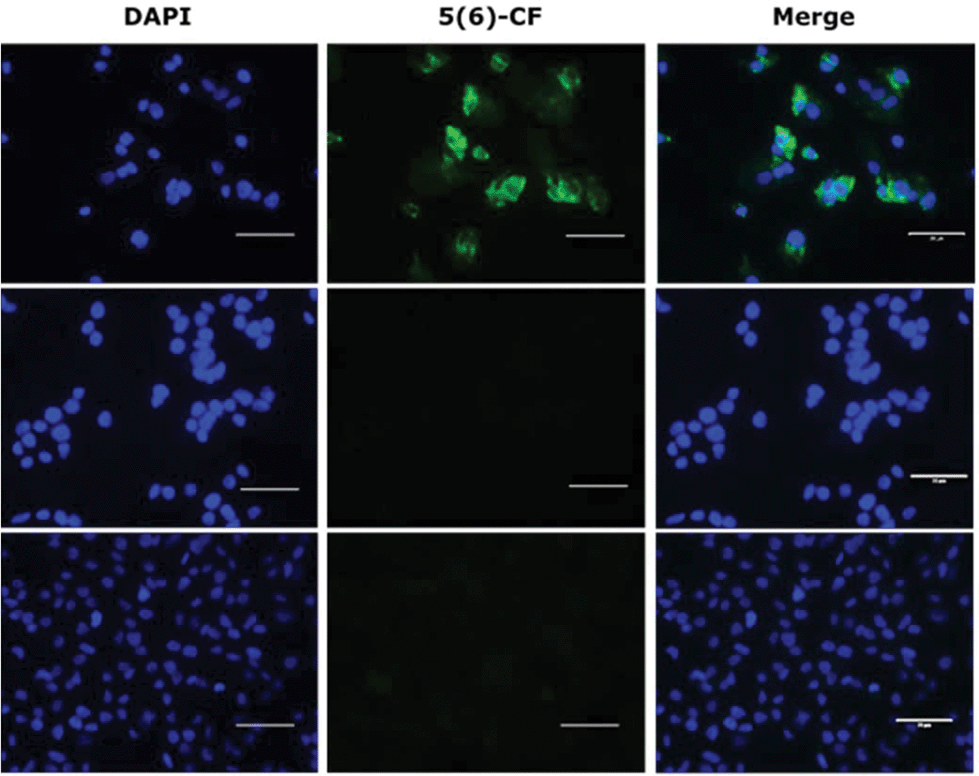
The binding of the lipopeptides to hepatocytes expressing NTCP was assessed rather than the isolated receptor, as the lipid is thought to play an important role as an ‘anchor’ in the cell membrane.5,29 All ten fluorescently labelled peptides (3, 3a–d and 4, 4a–d) were titrated (800 nM–5 nM) in triplicate experiments against HepG2 NTCP cells and their relative receptor binding affinity measured by flow cytometry, employing Myrcludex B (4) as a positive control. Lipopeptide 4 bound exclusively to NTCP expressing HepG2 cells and not to the parental HepG2 cell line that lacks expression of the HBV receptor. The non-lipidated 47-mer peptide (5) was incubated at 400 nM with both HepG2 and HepG2 NTCP cell lines, showing no binding with either cell line, confirming N-terminal lipidation as a prerequisite for peptide–receptor binding. Observation by fluorescent microscopy confirmed these findings (Fig. 4).
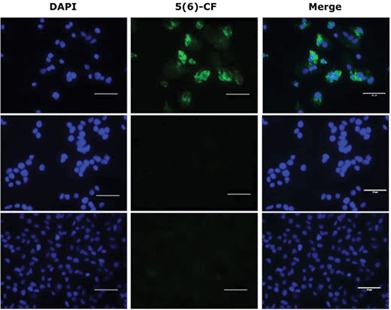 |
| | Fig. 4 Fluorescent microscopy of 5(6)-CF labelled peptides; (lower) HepG2 cells treated with 400 nM of Myrcludex B (4); (middle) HepG2 NTCP cells treated with 400 nM non-lipidated peptide (5); (upper) HepG2 NTCP cells treated with 400 nM Myrcludex B (4). Nuclei in blue and 5(6)-CF labelled peptides in green. Bar = 50 μm. | |
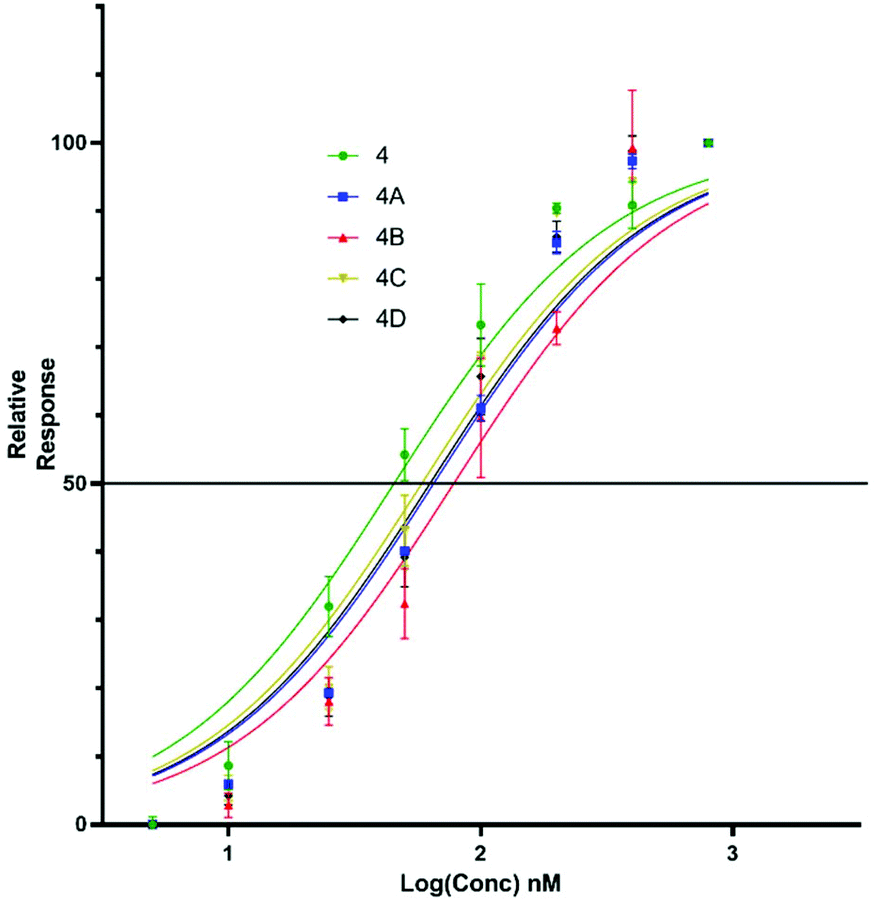
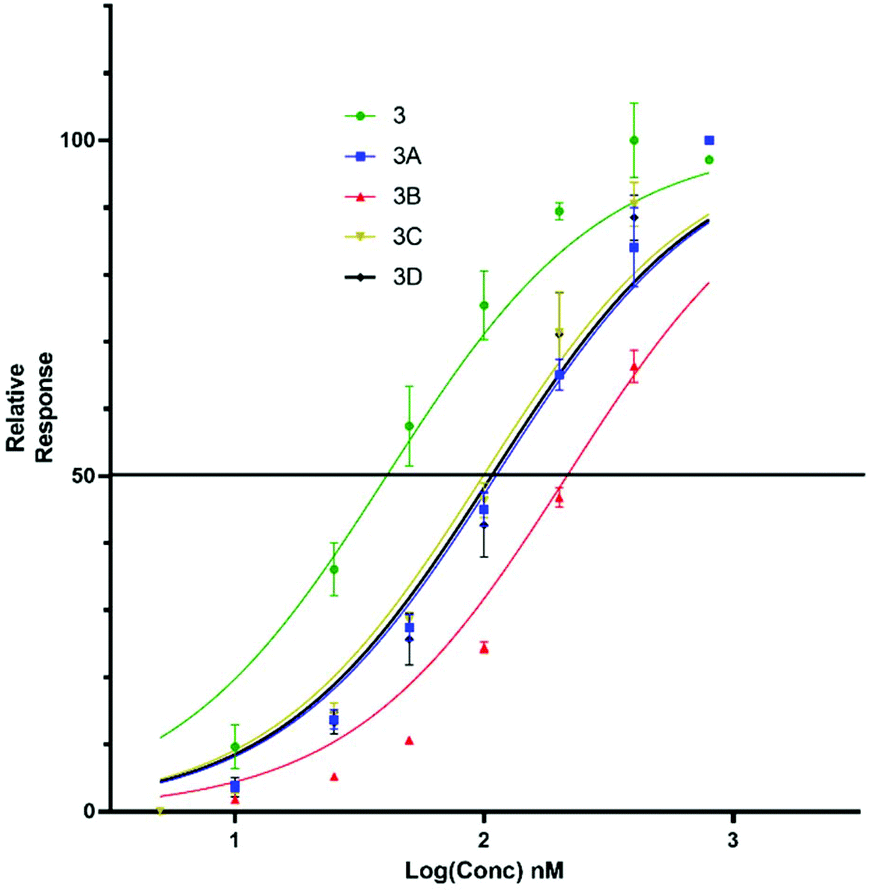
Upon titration of the S-lipidated analogues, a concentration dependant relationship was observed. Increasing peptide concentrations produced greater mean fluorescence, with the saturation at ∼200 nM for the majority of lipopeptides. No further increase in mean fluorescence was observed upon increasing peptide concentrations to 800 nM. These experiments were used to derive EC50 values to compare the binding effectiveness of each peptide for NTCP expressing HepG2 cells. The S-lipidated analogues demonstrated comparable binding (Table 1) to the native myristoylated sequences (Tables 1 and 2 and Fig. 5 and 6). The 47-mer lipopeptides (Table 1, Fig. 5) demonstrated EC50 values ranging 45.69 nM to 79.85 nM and in general bound more effectively than their equivalently S-lipidated truncated analogues [EC50 values ranging 40.55 nM to 213.1 nM] (Table 2 and Fig. 6).
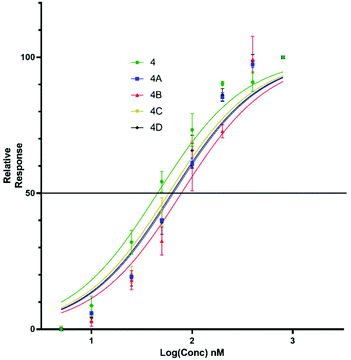 |
| | Fig. 5 EC50 plots for 47-mer of 5(6)-CF labelled lipopeptides (4 and 4a–d) binding HepG2 NTCP cells. Error bars correspond to ±1 SE. | |
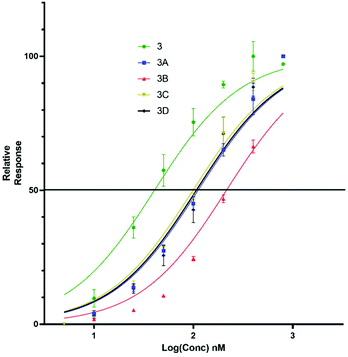 |
| | Fig. 6 EC50 plots for truncated 5(6)-CF labelled lipopeptides (3, 3a–d) binding HepG2 NTCP cells. Error bars correspond to ±1 SE. | |
Table 1 EC50 values for binding of Myrcludex B (4) and S-lipidated analogues, 4a–d to HepG2 NTCP cells
| Peptide |
4
|
4a
|
4b
|
4c
|
4d
|
| EC50 (nM) |
45.69 |
66.99 |
79.85 |
59.72 |
64.86 |
Table 2 EC50 values for binding of truncated Myrcludex B (3) and S-lipidated analogues, 3a–d to HepG2 NTCP cells
| Peptide |
3
|
3a
|
3b
|
3c
|
3d
|
| EC50 (nM) |
40.55 |
112.4 |
213.1 |
101.0 |
107.1 |
For both the full length 47 residue and truncated analogue series, the S-lipidated 8-phenyloctanoate analogue showed the best binding [3c, EC50 = 101.0 nM and 4c, EC50 = 59.72 nM], suggesting aromatic moieties can be well-tolerated by the lipid ‘anchor’ during NTCP binding. Interestingly, the truncated analogue bearing native myristoylation (3) appeared to bind most strongly of the lipopeptides. This was surprising given previous literature reported the viral inhibitory effect of the truncated analogue to be ∼20-fold less than that of the full-length 47-mer.11 While a possible explanation could be that incorporation of the relatively large fluorescent tag favoured interaction with the NTCP receptor, this observation did not hold true for our S-lipidated analogues (vide supra), which bound less effectively than their corresponding 47-mers. Overall, the binding experiments were encouraging and the S-lipidated compound series were progressed for evaluation of inhibitory activity in order to examine the correlation between binding and inhibitory potency.
Antiviral activity of lipopeptides
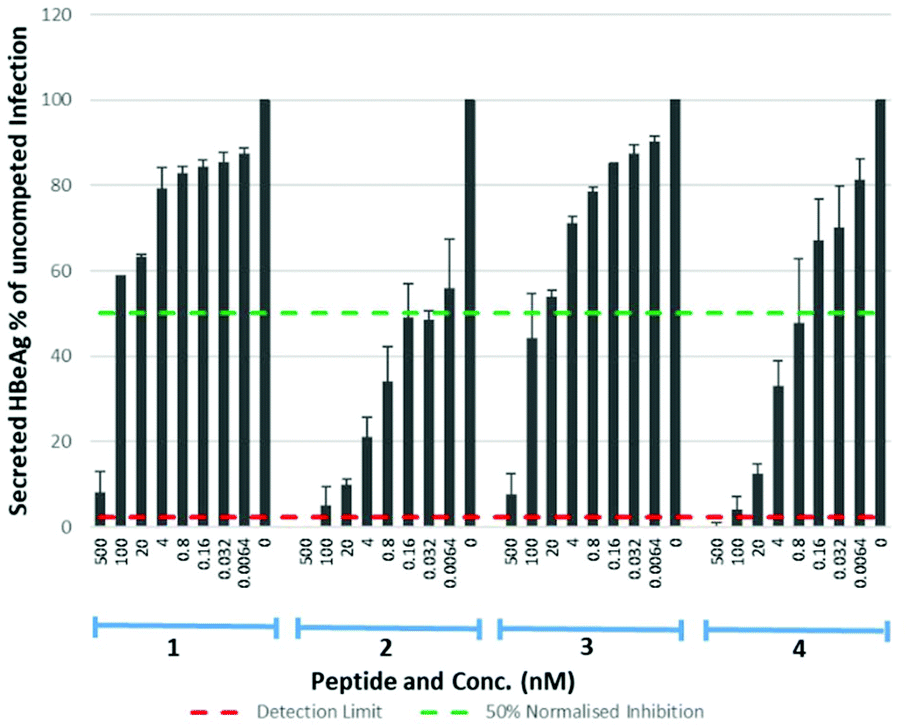
Eight lipopeptides were investigated for their HBV inhibitory effect. Inhibition was determined by measurement of HBeAg secreted into the cell culture medium 9 days post-infection, by enzyme-linked immunosorbent assay (ELISA), and data normalised to a control infection in the absence of an inhibitor. The truncated lipopeptide (1) and its 5(6)-CF labelled counterpart (3) were first evaluated for inhibitory activity (Table 3, Fig. 7), to investigate if the fluorescent tag may have contributed to the unexpectedly strong binding of 3. Their inhibitory effect was also compared against the native myristoylated 47-mer (2, Myrcludex B), which demonstrated an IC50 of 0.056 nM, in accordance with previous work from Schulze et al.11
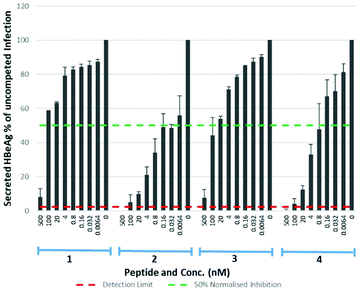 |
| | Fig. 7 HBV replication inhibitory potency of native (myristoylated), lipopeptides. Error bars correspond to ±1 SE. | |
Table 3 IC50 values for native (myristoylated) lipopeptides 1–4 on HBV replication
| Peptide |
1
|
2
|
3
|
4
|
| IC50 (nM) |
47.11 |
0.056 |
30.37 |
0.645 |
Both the truncated lipopeptides 1 and 3 exhibited reduced inhibitory activity (∼800-fold and ∼500-fold respectively) compared to the corresponding 47-mer (2). Their inhibitory effect therefore did not correlate with the strong binding of 3 and the 5(6)-CF label appeared to have little effect upon inhibitory potency. The drastic reduction in potency upon truncation is particularly interesting given it produced only a small (approx. two-fold) reduction in their EC50 for binding to NTCP expressing HepG2 cells. Somewhat surprisingly, for the 47-mer peptide, incorporation of the C-terminal 5(6)-CF label (4) reduced inhibitory potency ∼10-fold, despite this region being reported less crucial for inhibition.11 Given the poorer inhibitory potency of truncated lipopeptides 1 and 3, their S-lipidated analogues (1a–d) were not further investigated.
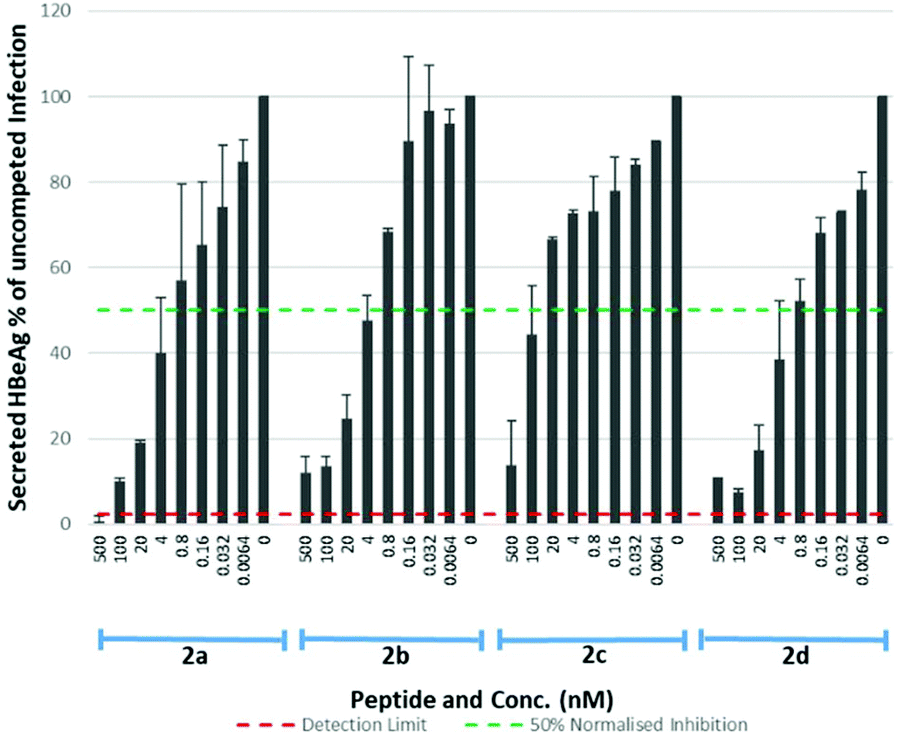
The S-lipidated 47-mer analogues (2a–d) were however, assayed for their inhibitory activity (Table 4, Fig. 8), exhibiting IC50 values in the nM range. Analogues 2a, 2b and 2d exhibited IC50 values of 1.23 nM, 3.33 nM, and 0.97 nM respectively, within ∼17–60-fold the potency of Myrcludex B (2) (IC50 0.056 nM). The CLipPA S-lipidation approach is therefore tolerated moderately well for these antiviral lipopeptides, and the novel thioether analogues retain remarkable potency despite the presence of the additional heteroatoms in their lipid ‘anchor’. The 47-mer analogue (2c) bearing the S-linked 8-phenyloctanoic lipid (12c), exhibited significantly reduced potency [IC50 50.32], in the same range as the truncated analogues 1 and 3, despite being the most effective binder of NTCP expressing HepG2 cells. Importantly, the results from this aromatic S-lipidated analogue further corroborate our findings from truncated lipopeptides 1 and 3, that the anti-HBV potency is poorly correlated to binding of NTCP expressing hepatocytes.
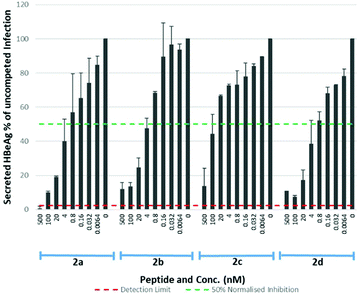 |
| | Fig. 8 HBV replication inhibitory potency of S-lipidated 47-mer analogues (2a–d). Error bars correspond to ±1 SE. | |
Table 4 IC50 values for 47-mer S-lipidated analogues 2a–d on HBV replication
| Peptide |
2a
|
2b
|
2c
|
2d
|
| IC50 (nM) |
1.230 |
3.320 |
50.32 |
0.970 |
Lipids have previously been identified as membrane ‘anchors’ that support peptides in maintaining a high local concentration at the membrane and thereby facilitate interaction with the relevant receptor.29 A similar role has been proposed for the lipid of Myrcludex B (2).5 Despite numerous analogues (4a–d) binding to NTCP expressing hepatocytes with similar effectiveness to 5(6)-CF labelled Myrcludex B (4), this doesn't correlate with their inhibitory activity (2, 2a–d). This phenomenon was exemplified by the S-linked 8-phenyloctanoate analogue (4c), which demonstrated the most effective binding of all 47-mer lipopeptides whereas the equivalent lipopeptide (2c) exhibited by far the lowest inhibitory potency, reduced by 15–50-fold compared to the other S-lipidated analogues (2a,b,d) and 900-fold compared to Myrcludex B (2). This discrepancy between binding and inhibitory potency suggests the lipid may play an additional role in blocking the NTCP–HBV interaction, beyond simply acting as a membrane anchor.
Conclusions
We report the first SPPS-based synthesis of Myrcludex B that includes a detailed and optimised synthetic procedure. Using the 3-MPA modified CLipPA thiol–ene approach, a series of S-lipidated peptides bearing structurally diverse lipids were prepared chemoselectively from a common precursor, in moderate to excellent conversions. These PPI inhibitors appear to constitute the first report of potent S-lipidated antiviral agents, with three analogues demonstrating IC50 values ranging 0.97–3.32 nM. With the potency retained by these analogues, the incorporation of heteroatoms within the lipid ‘anchor’ warrants further investigation into the pharmacological properties of these S-lipidated peptides. In particular, the presence of both an ester and thioether within the lipid portion of these compounds favours solubility and may also provide a potentially beneficial metabolic route, allowing the lipid to dissociate from the peptide through ester hydrolysis. Interestingly, the effectiveness with which these lipopeptides bind NTCP expressing hepatocytes was found to correlate very poorly with their inhibitory potency and, to the best of our knowledge, also represents the first investigation of this inter-relationship. This apparent discrepancy highlights how the exact role of the proposed lipid ‘anchor’ remains unclear and warrants further investigation.
Experimental section
General procedures
Analytical reverse phase high-performance liquid chromatography (RP-HPLC) was performed on either a Waters Alliance analytical HPLC or Dionex Ultimater 3000 equipped with a Phenomenex Luna C18 column (100 A, 5 μm, 4.6 mm × 250 mm) operated at rt, with chromatograms recorded at 210/214 nm and 254 nm. Semi-preparative RP-HPLC was performed on a Waters 1525 Binary HPLC pump equipped with a Waters 2489 UV/visible detector using either a Waters XTerra Prep MS C18 OBD column (125 A, 10 μm, 300 × 19 mm) or Waters XTerra Prep MS C18 OBD column (125 A, 10 μm, 250 × 10 mm). For both analytical and semi-preparative RP-HPLC, solvents used were as follows: solvent A = 0.1% TFA in water (MQ H2O) and solvent B = 0.1% TFA in MeCN. For analytical RP-HPLC the gradient employed was 5–95% of solvent B over 30 minutes at flow rate 1 ml min−1, unless specified otherwise. For semi-preparative RP-HPLC gradients were adjusted as indicated in the experimental procedures, according to elution times and peak profiles obtained during analytical analysis. Analytical thin-layer chromatography (TLC) was performed on Kieselgel F254 200 μm silica plates. The compounds were then visualised by ultraviolet fluorescence or by staining with potassium permanganate or vanillin followed by heating of the plate. Column chromatography was performed using Grace Davison Discovery Sciences LC60A 40-63 Micron Chromatographic Silica Media with the indicated eluent. Low-resolution mass spectrometry was performed on a Waters Quattro micro API Mass Spectrometer in ESI positive mode. Ultraviolet irradiation was performed using a handheld Spectroline UV lamp at a peak wavelength of 365 nm. Nuclear magnetic resonance (NMR) experiments were recorded as shown on a Bruker AVANCE 400 spectrometer operating at 400 MHz for 1H nuclei and 100 MHz for 13C nuclei. Chemical shifts were reported in parts per million (ppm) on the δ scale from tetramethylsilane (TMS), at 0.00 ppm. Shifts were referenced to residual solvent peaks of CDCl3 at δ = 7.26 ppm for 1H NMR, and CDCl3δ = 77.16 ppm for 13C NMR or DMSO-d6 at δ = 2.50 ppm for 1H NMR and δ = 39.52 ppm for 13C NMR. The 1H NMR shift values are reported as chemical shift δ, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublets, td = triplet of doublets, qd = quartet of doublets), coupling constant (J in Hz), relative integral and assignments.
The purity of all final tested compounds was confirmed to be 95% or greater by analytical HPLC using a 5 μm Phenomenex Luna reverse phase C18 column (4.6 mm × 250 mm) (ESI Fig. S5.1–S5.20†).
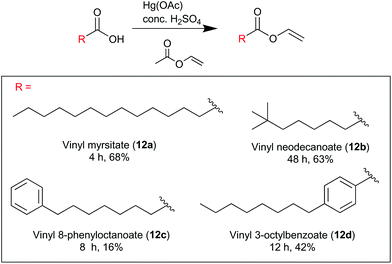
Vinyl ester synthesis
Vinyl myristate (12a).
Myristic acid (2.00 g, 8.76 mmol) was added to an excess of vinyl acetate (8.07 ml, 87.6 mmol), mercury acetate (60 mg, 0.219 mmol) and fuming sulfuric acid (H2SO4, 15.0 μL). The reaction proceeded under reflux (74 °C) under a nitrogen atmosphere for 4 hours until completion, as judged by TLC monitoring of the reaction mixture. Upon completion of the reaction the reaction mixture was cooled, the sulfuric acid quenched portion-wise with NaOAc·3H2O (3 × 25.0 mg) and filtered through Celite. Excess vinyl acetate was evaporated and the crude reaction mixture purified by flash column chromatography on silica gel with an eluent of ethyl acetate:petroleum ether (1:99), to yield vinyl myristate (12a) a colourless oil (1.52 g, 68%). δH (400 MHz, CDCl3) 7.25–7.32 (1H, m), 4.85 (1H, dd, J = 4.4, 2.0), 4.55 (1H, dd, J = 6.4, 2.0), 2.38 (2H, t, J = 10), 1.65 (2H, t, J = 5.6), 1.26 (20H, s), 0.88 (3H, t, J = 8.4). δC (100 MHz, CDCl3) 171.0, 141.4, 97.5, 34.1, 32.1, 29.8, 29.8, 29.7, 29.6, 29.5, 29.4, 29.2, 24.8, 22.8, 14.3. Spectroscopic data was consistent with that reported in literature.30
Vinyl neodecanoate (12b).
Neodecanoic acid (98%, mixture of isomers) (2.17 ml, 11.6 mmol) was added to an excess of vinyl acetate (10.7 ml, 116 mmol), mercury acetate (92 mg, 0.290 mmol) and fuming sulfuric acid (H2SO4, 15.0 μL). The reaction proceeded under reflux (74 °C) under a nitrogen atmosphere for 4 hours until completion, as judged by TLC monitoring of the reaction mixture. Upon completion of the reaction the reaction mixture was cooled, the sulfuric acid quenched portion-wise with NaOAc·3H2O (3 × 25.0 mg) and filtered through Celite. Excess vinyl acetate was evaporated and the crude reaction mixture purified by flash column chromatography on silica gel with an eluent of ethyl acetate:petroleum ether (1:99), to yield vinyl neodecanoate (12b) a colourless oil (1.44 g, 63%). νmax/cm−1: 2962, 2935, 2875, 1745, 1645, 1464, 1212, 1138. δH (400 MHz, DMSO-d6) 7.25–7.17 (1H, m), 4.92–4.87 (1H, m), 4.67–4.65 (1H, m). Other signals unable to be clearly assigned due to overlaps from a mixture of neodecanoic acid isomers that originate from the commercial starting material (see Fig. S4.5†). δC (100 MHz, DMSO-d6) 174.3, 141.4, 98.2. Other signals unable to be clearly assigned due to overlaps from a mixture of neodecanoic acid isomers that originate from the commercial starting material. ESI-HRMS: m/z [M + Na]+ calculated for C12H22NaO2: 221.1514, observed: 221.1514.
Vinyl 8-phenyloctanoate (12c).
8-Phenyloctanoic acid (1.58 mg, 7.72 mmol) was added to an excess of vinyl acetate (7.22 ml, 77.2 mmol), mercury acetate (55 mg, 0.193 mmol) and fuming sulfuric acid (H2SO4, 15.0 μL). The reaction proceeded under reflux (74 °C) under a nitrogen atmosphere for 8 hours until completion, as judged by TLC monitoring of the reaction mixture. Upon completion of the reaction the reaction mixture was cooled, the sulfuric acid quenched portion-wise with NaOAc·3H2O (3 × 25.0 mg) and filtered through Celite. Excess vinyl acetate was evaporated and the crude reaction mixture purified by flash column chromatography on silica gel with an eluent of ethyl acetate:petroleum ether (1:99), to yield vinyl 8-phenyloctanoate (12c) a colourless oil (0.300 g, 16%). νmax/cm−1: 3027, 2929, 2856, 1753, 1646, 1294, 1266, 1138. δH (400 MHz, CDCl3) 7.25–7.31 (5H, m), 7.15–7.18 (1H, m), 4.87 (1H, dd, J = 12.8, 1.6), 4.55 (1H, dd, J = 5.2, 1.6), 2.60 (2H, t, J = 8.0), 2.37 (2H, t, J = 7.6), 1.55–1.67 (4H, m), 1.34 (6H, s). δC (100 MHz, CDCl3) 142.8, 170.9, 141.2, 128.4, 128.3, 125.6, 97.5, 35.9, 33.9, 31.4, 29.1, 28.9, 24.6. ESI-HRMS:m/z [M + Na]+ calculated for C16H22NaO2: 269.1507, observed 269.1512.
Vinyl 4-octylbenzoate (12d).
4-Octylbenzoic acid (1.55 mg, 6.60 mmol) was added to an excess of vinyl acetate (6.09 ml, 66.0 mmol), mercury acetate (53 mg, 0.165 mmol) and fuming sulfuric acid (H2SO4, 15.0 μL). The reaction proceeded under reflux (74 °C) under a nitrogen atmosphere for 8 hours until completion, as judged by TLC monitoring of the reaction mixture. Upon completion of the reaction the reaction mixture was cooled, the sulfuric acid quenched portion-wise with NaOAc·3H2O (3 × 25.0 mg) and filtered through Celite. Excess vinyl acetate was evaporated and the crude reaction mixture purified by flash column chromatography on silica gel with an eluent of ethyl acetate:petroleum ether (1:99), to yield vinyl 4-octylbenzoate(12d) a colourless oil (0.710 g, 42%). νmax/cm−1: 2925, 2856, 1730, 1645, 1611, 1294, 1260, 1178, 1138, 1087. δH (400 MHz, CDCl3) 8.01 (2H, d, J = 8), 7.51 (1H, dd, J = 8, 6.0), 7.28 (2H, d, J = 8.4), 5.05 (1H, dd, J = 12.0, 1.6), 4.68 (1H, dd, J = 4.4, 1.6), 2.67 (t, J = 8), 1.65 (2H, t, J = 7.2), 1.26–1.31 (10H, m), 0.88 (3H, s). δC (100 MHz, CDCl3) 163.8, 149.4, 141.5, 130.1, 128.6, 126.4, 97.9, 36.1, 31.9, 31.1, 29.4, 29.3, 29.2, 22.7, 14.1. ESI-HRMS: m/z [M + Na]+ calculated for C17H24NaO2: 283.1662, observed 283.1669.
Peptide synthesis
General procedures.
Linear peptides were synthesised by 9-fluorenylmethoxycarbonyl (Fmoc) solid phase peptide synthesis (SPPS) at rt on a 0.1 mmol scale either manually using a fritted glass reaction vessel or using a PS3 automated peptide synthesiser. TentaGel® S NH2 Resin (0.370 g, 0.100 mmol, 0.270 mmol g−1 loading) was swollen in CH2Cl2 (3 ml) for 5 min followed by DMF (3 ml) for 5 min. Fmoc-Rink amide linker (216 mg, 0.4 mmol, 4 equiv.) was used as a linker to the solid TentaGel support and was coupled to the resin using the same method as the subsequent amino acids. Using the PS3 Peptide Synthesiser, all amino acids were coupled in single, hour long cycles at rt. Protected amino-acids were coupled using a mixture of Fmoc-AAx-OH (0.4 mmol, 4 equiv.), HATU (144 mg, 0.38 mmol, 3.8 equiv.) and NMM (1 mmol, 10 equiv.) in DMF (3 ml). The Fmoc protecting group was removed with a solution of 20% piperidine in DMF (v/v) containing 5% formic acid (v/v) (3 ml, 2 × 5 min). Between couplings and following Fmoc removal, the resin was washed with DMF (6 × 5 ml).
Method for coupling of 3-(tritylthio)propionic acid (3-MPA) to the N-terminus.
A mixture of 3-(tritylthio)propionic acid (0.139 mg, 0.4 mmol, 4 equiv.) HATU (144 mg, 0.38 mmol, 3.8 equiv.) and DIPEA (175 μL, 1 mmol, 10 equiv.) in DMF (3 ml) was coupled for 2 hours at rt to the deprotected N-terminus of the peptidyl resin.
Method for coupling of myristic acid (MA).
A mixture of myristic acid (90 mg, 0.4 mmol, 4 equiv.) HATU (144 mg, 0.38 mmol, 3.8 equiv.) and DIPEA (175 μL, 1 mmol, 10 equiv.) in DMF (3 ml) was coupled for 1 hour at rt to the deprotected N-terminus of the peptidyl resin.
Method of removal of Dde protecting group from amino acid side chain.
The resin-bound peptide was treated with 2% hydrazine monohydrate in DMF v/v (3 × 3 min) at rt and vigorously agitated.
Method for coupling of 5(6)-carboxyfluorescein (5(6)-CF).
After removal of the sidechain Dde group, the resin-bound peptide was treated with a mixture of 5(6)-CF (151 mg, 0.4 mmol, 3 equiv.) 6-Cl HOBT (50.8 mg, 0.400 mmol, 3 equiv.) and DIC (46.0 μL, 0.4 mmol, 3 equiv.) in DMF and vigorously agitated for 4 hours at rt. The resin was then washed with 20% piperidine in DMF (v/v) containing 5% formic acid (v/v) (3 ml, 3 × 3 min).
Method for cleavage of peptide from resin.
After washing the resin with CH2Cl2 (3 × 5 ml) a cleavage mixture of TFA/TIS/EDT/H2O (10 ml, v/v/v/v; 94/2/2/2) was added to the on-resin peptide and agitated for 2 hours at rt. The cleavage mixture was drained and chilled diethyl ether (45 ml) was used to precipitate the peptide. The peptide was isolated by centrifugation and the pellet washed with diethyl ether (45 ml), dissolved in MeCN:MQ H2O (1:1) and lyophilised.
Cell culture
HepG2 (HB8065) cells were obtained from ATCC. HepG2-NTCP cells were kindly provided by Stephan Urban, University Hospital Heidelberg. Both cell lines were maintained in DMEM supplemented media with 10% v/v FCS and 0.1% v/v P/S with additional supplementation of 4 μg ml−1 G418 for HepG2-NTCP. Cell were cultivated at 37 °C and 5.0% CO2 and subcultured every 3–5 days at a ratio from 1:3 to 1:6. HepAD38 cells were kindly provided by Stephen Locarnini, Victorian Infectious Diseases Reference Laboratory and grown in DMEM containing 5% heat inactivated FCS and 0.3 μg ml−1 of doxycycline.31
HBV production
HepAD38 cells were grown to 80% confluence whereupon the media was replaced with doxycycline-free DMEM containing 5% heat inactivated FCS (day 0). Cells continued to produce virus for 15 days with media being changed every 3–4 days. Every 3–4 days from day 15 the supernatant media was harvested. Every second harvest, virus was concentrated and stored at −80 °C. To concentrate the supernatant, it was filtered through a sterile 0.45 μm filter and a solution of 40% polyethylene glycol (PEG) added to result in a final solution with 6% PEG v/v. After inversion this solution was stored overnight at 4 °C. Remaining at 4 °C the PEG solution was centrifuged at a fixed angle for 1 hour (10000 rcf). Post centrifugation the supernatant was removed, and the pellet resuspended in 1/50 of the volume in a solution of PBS/FCS (90%/10% v/v). The viral suspension in PBS/FCS was rotated and inverted overnight at 4 °C. To remove insoluble precipitate the viral suspension was centrifuged twice at 4 °C for 10 minutes (3000 rcf). The final viral suspension was aliquoted, and the supernatant stored at −80 °C after quantification by quantitative Polymerase Chain Reaction (qPCR).
Lipopeptide binding assays
24-Well microtitre plates were seeded with HepG2 NTCP or HepG2 cells at a density of 2 × 105 per well in a growth media of DMEM containing 10% heat inactivated FCS and 0.1% P/S (v/v) and incubated overnight at 37 °C in a 5% CO2 atmosphere until cells were adhered and confluent. The cells were washed twice with PBS. Peptide solutions were prepared in identical growth media. A dilution series (800 nM–5 nM) was made across the plate from standard solutions prepared from the target peptide in DMSO stock, with the final DMSO concentration being less than 0.1% v/v. For each peptide one additional well was treated with a control peptide at fixed concentration as an internal standard, and one well treated with media devoid of peptide. The plates were then incubated for 30 minutes once again at 37 °C in a 5% CO2 atmosphere. Cells were then twice washed with PBS and trypsinised. Cells were centrifuged at 500 rcf in growth media initially and twice in PBS to be finally suspended in a solution of 1% FCS in PBS (FACS buffer). The final cell suspension solutions were then each analysed using a BD Accuri Flow Cytometer and the raw data gated and analysed with FlowJo® to acquire mean fluorescent values. All peptides were analysed in triplicate. The data was normalised relative to a standard of Myrcludex B at a concentration (400 nM) exceeding the saturation point of binding. Using Prism 8.0.2 the mean fluorescent values of each titration were used to create EC50 curves and EC50 values for each peptide. All fluorescence assays were performed in duplicate and repeated at least twice independently.
HBV inhibition assay
Peptides (500 nM–0.0064 nM final concentration) were diluted in a 96 well plate in DMEM containing 2% heat inactivated FCS, 0.1% P/S, 2% DMSO, 1% NEAA and 4% PEG v/v and 1000 genome equivalent units of HBV. HepG2 NTCP cells (5 × 104 per well) were added and infection initiated by spinoculation as described.32 Cells were incubated for up to 12 days with media replaced at days 1,3,6 and 9 with the peptide dilution series maintained during the 12-day incubation period. Peptide inhibition of HBV replication was determined by HBeAg chemiluminescence immunoassay of secreted HBeAg, 50 μL of culture media collected at day 9 were applied to 96-well plates of a chemiluminescence immunoassay (CLIA) according to the manufacturer's instructions (Autobio Diagnostics Co., Zhengzhou, China). Results were normalised to peptide-free controls. All competition assays were performed in duplicate and repeated at least twice independently.
Abbreviations
| 3-MPA | 3-Mercaptopropionic acid |
| 5(6)-CF | 5(6)-Carboxyfluorescein |
| 6-Cl-HOBt | 6-Chloro-1-hydroxylbenzotriazole |
| approx. | Approximately |
| CLIA | Chemiluminescence immunoassay |
| CLipPA | Cysteine lipidation on a peptide or amino acid |
| Dde | 1-(4,4′-Dimethyl-2,6-dioxocyclohexylidene)-3-ethyl |
| DIC |
N,N′-diisopropylcarbodiimide |
| DIPEA |
N,N-Diisopropylethylamine |
| DMEM | Dulbecco's Modified Eagle's Medium |
| EDT | 1,2-Ethanedithiol |
| Et2O | Diethylether |
| Et(OAc)2 | Ethylacetate |
| FA | Formic acid |
| HATU | 2-(7-Aza-1H-benzotriazole-1-yl)-1,1,3,-tertramethyluronium hexafluorophosphate |
| Fmoc | 9-Fluorenylmethoxycarbonyl |
| FACS | Fluorescence activate cell sorting |
| HBeAg | HBV E antigen |
| Hg(OAc)2 | Mercury acetate |
| MeCN | Acetonitrile |
| MQ | Milli-q water |
| NEAA | Non-essential amino acids |
| NMM |
N-Methylmorpholine |
| NTCP | Sodium (Na) taurocholate co-transporting polypeptide |
| RP-HPLC | Reverse-phase high performance liquid chromatography |
| SE | Standard error |
| SPPS | Solid-phase peptide synthesis |
|
tNonSH |
tert-Nonylthiol. |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the Maurice Wilkins Centre for Molecular Biodiscovery for financial support.
References
- Hepatitis B, https://www.who.int/news-room/fact-sheets/detail/hepatitis-b, (accessed 18 January 2019).
- A. S. F. Lok and B. J. McMahon, Hepatology, 2007, 45, 507–539 CrossRef CAS.
- A. S. Lok, Gastroenterology, 2007, 132, 1586–1594 CrossRef CAS.
- P. Martin, D. T.-Y. Lau, M. H. Nguyen, H. L. A. Janssen, D. T. Dieterich, M. G. Peters and I. M. Jacobson, Clin. Gastroenterol. Hepatol., 2015, 13, 2071–2087 CrossRef.
- Y. Ni, F. A. Lempp, S. Mehrle, S. Nkongolo, C. Kaufman, M. Fälth, J. Stindt, C. Königer, M. Nassal, R. Kubitz, H. Sültmann and S. Urban, Gastroenterology, 2014, 146, 1070–1083 CrossRef CAS.
- D. Glebe, S. Urban, E. V. Knoop, N. Çaǧ, P. Krass, S. Grün, A. Bulavaite, K. Sasnauskas and W. H. Gerlich, Gastroenterology, 2005, 129, 234–245 CrossRef CAS.
- P. Gripon, I. Cannie and S. Urban, J. Virol., 2005, 79, 1613–1622 CrossRef CAS.
- H. Wedemeyer, P. Bogomolov, A. Blank, L. Allweiss, M. Dandri, B. Bremer, N. Voronkova, K. Schöneweis, A. Pathil, J. Burhenne, M. Haag, M. Schwab, W. Haefeli, J. S. Z. Wiesch, A. Alexandrov and S. Urban, J. Hepatol., 2018, 68, S3 CrossRef.
- S. Urban, R. Bartenschlager, R. Kubitz and F. Zoulim, Gastroenterology, 2014, 147, 48–64 CrossRef CAS.
- Y. Ni and S. Urban, Methods Mol. Biol., 2017, 1540, 15–25 CrossRef CAS.
- A. Schulze, A. Schieck, Y. Ni, W. Mier and S. Urban, J. Virol., 2010, 84, 1989–2000 CrossRef CAS.
- J.-E. Park and T. Gallagher, Virology, 2017, 511, 9–18 CrossRef CAS.
- P. Ingallinella, E. Bianchi, N. A. Ladwa, Y.-J. Wang, R. Hrin, M. Veneziano, F. Bonelli, T. J. Ketas, J. P. Moore, M. D. Miller and A. Pessi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5801–5806 CrossRef CAS.
- T. H. Wright, A. E. S. Brooks, A. J. Didsbury, G. M. Williams, P. W. R. Harris, P. R. Dunbar and M. A. Brimble, Angew. Chem., Int. Ed., 2013, 52, 10616–10619 CrossRef CAS.
- S.-H. Yang, P. W. R. Harris, G. M. Williams and M. A. Brimble, Eur. J. Org. Chem., 2016, 2608–2616 CrossRef CAS.
- S.-H. Yang, Y. O. J. Hermant, P. W. R. Harris and M. A. Brimble, Eur. J. Org. Chem., 2020, 944–947 CrossRef CAS.
-
World Intellectual Property Organization, WO2014207708A2, 2014.
- V. Yim, P. Harris, M. A. Brimble, J. R. Allison, A. Chakraborty, S. Swift, A. Cameron, S. A. Ferguson, M. K. Knottenbelt, I. Kavianinia and G. Cook, Chem. Sci., 2020, 11, 5759–5765 RSC.
- V. V. Yim, I. Kavianinia, A. J. Cameron, P. W. R. Harris and M. A. Brimble, Org. Biomol. Chem., 2020, 18, 2838–2844 RSC.
- T. H. Wright, A. E. S. Brooks, A. J. Didsbury, G. M. Williams, P. W. R. Harris, P. R. Dunbar and M. A. Brimble, Angew. Chem., Int. Ed., 2013, 52, 10616–10619 CrossRef CAS.
- E. T. Williams, P. W. R. Harris, M. A. Jamaluddin, K. M. Loomes, D. L. Hay and M. A. Brimble, Angew. Chem., Int. Ed., 2018, 57, 11640–11643 CrossRef CAS.
- J. T. W. Tong, I. Kavianinia, S. A. Ferguson, G. M. Cook, P. W. R. Harris and M. A. Brimble, Org. Biomol. Chem., 2020, 18, 4381–4385 RSC.
- B. L. Lu, G. M. Williams, D. J. Verdon, P. R. Dunbar and M. A. Brimble, J. Med. Chem., 2020, 63, 2282–2291 CrossRef CAS.
- M. Mergler, F. Dick, B. Sax, P. Weiler and T. Vorherr, J. Pept. Sci., 2003, 9, 36–46 CrossRef CAS.
- T. Michels, R. Dölling, U. Haberkorn and W. Mier, Org. Lett., 2012, 14, 5218–5221 CrossRef CAS.
- J. J. Díaz-Mochón, L. Bialy and M. Bradley, Org. Lett., 2004, 6, 1127–1129 CrossRef.
- R. Fischer, O. Mader, G. Jung and R. Brock, Bioconjugate Chem., 2003, 14, 653–660 CrossRef CAS.
- P. Magrone, F. Cavallo, W. Panzeri, D. Passarella and S. Riva, Org. Biomol. Chem., 2010, 8, 5583–5590 RSC.
-
R. Kowalczyk, P. W. R. Harris, G. M. Williams, S.-H. Yang and M. A. Brimble, in Peptides and Peptide-based Biomaterials and their Biomedical Applications, ed. A. Sunna, A. Care and P. L. Bergquist, Springer International Publishing, Cham, 2017, pp. 185–227 Search PubMed.
-
AIST, Spectral Database for Organic Compounds,SDBS, https://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_disp.cgi?sdbsno=7737, (accessed 26 June 2019) Search PubMed.
- S. K. Ladner, M. J. Otto, C. S. Barker, K. Zaifert, G. H. Wang, J. T. Guo, C. Seeger and R. W. King, Antimicrob. Agents Chemother., 1997, 41, 1715–1720 CrossRef CAS.
- K. Okuyama-Dobashi, H. Kasai, T. Tanaka, A. Yamashita, J. Yasumoto, W. Chen, T. Okamoto, S. Maekawa, K. Watashi, T. Wakita, A. Ryo, T. Suzuki, Y. Matsuura, N. Enomoto and K. Moriishi, Sci. Rep., 2015, 5, 17047 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation of intermediate and final compounds (1H, 13C spectra, RP-HPLC traces, and high and low-res MS) along with biological data. See DOI: 10.1039/d0ob01997f |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *abc,
Carol J.
Wang
b,
Paul W. R.
Harris
*abc,
Carol J.
Wang
b,
Paul W. R.
Harris