
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne pot synthesis of thio-glycosides via aziridine opening reactions†
Nives
Hribernik
 ,
Alice
Tamburrini
,
Ermelinda
Falletta
and
Anna
Bernardi
*
,
Alice
Tamburrini
,
Ermelinda
Falletta
and
Anna
Bernardi
*
Università degli Studi di Milano, Dipartimento di Chimica, via Golgi 19, 20133 Milano, Italy. E-mail: anna.bernardi@unimi.it
First published on 7th December 2020
Abstract
A one-pot aziridine opening reaction by glycosyl thiols generated in situ from the corresponding anomeric thio-acetates affords thio-glycosides with a pseudo-disaccharide structure and an N-linked tether. The scope of the one-pot aziridine opening reaction was explored on a series of mono- and disaccharides, creating a class of pseudo-glycosidic compounds with potential for further functionalization. Unexpected anomerization of glycosyl thiols was observed under the reaction conditions and the influence of temperature, base and solvent on the isomerization was investigated. Single isomers were obtained in good to acceptable yields for mannose, rhamnose and sialic acid derivatives. The class of thio-glycomimetics synthesized can potentially be recognized by various lectins, while presenting hydrolytic and enzymatic stability. The nitrogen functionality incorporated in the glycomimetics can be exploited for further functionalization, including tethering to linkers, scaffolds or peptide residues.
Introduction
It is well known that interactions between carbohydrates and proteins play an important role in physiological and pathological processes of major relevance and yet the therapeutic use of carbohydrates is limited by their difficult synthesis, their low intrinsic activity and their chemical nature, which does not fit expectations for drug-like molecules.1 To overcome at least some of these drawbacks, chemically modified analogues of natural carbohydrates, so called glycomimetics, have been proposed.2 The development of functional carbohydrate mimics has been tackled either by structure-based design,1,3–13 or by discovery campaigns, often planned taking advantage of microarray technology.14–16 The latter approach has been frustrated by the cumbersome synthesis of oligosaccharides, that severely limits the diversity and the number of compounds that can be rapidly generated.We have recently reported that the one-pot opening reaction of epoxide 2 by the glycosyl thiol generated in situ from 2,3,4,6-tetra-O-acetyl-1-S-acetyl-α-D-mannopyranose 1 provides a facile access to the 1,2-dimannoside mimic 3, which is produced as a single isomer resulting from exclusive trans-diaxial opening of the epoxide (Scheme 1).17 The pseudo-thio-1,2-dimannoside 3 binds to the dendritic cell receptor DC-SIGN with an affinity comparable to that of the natural disaccharide Manα(1,2)Man, but is significantly more stable to enzymatic hydrolysis18 and arguably simpler to synthesize.
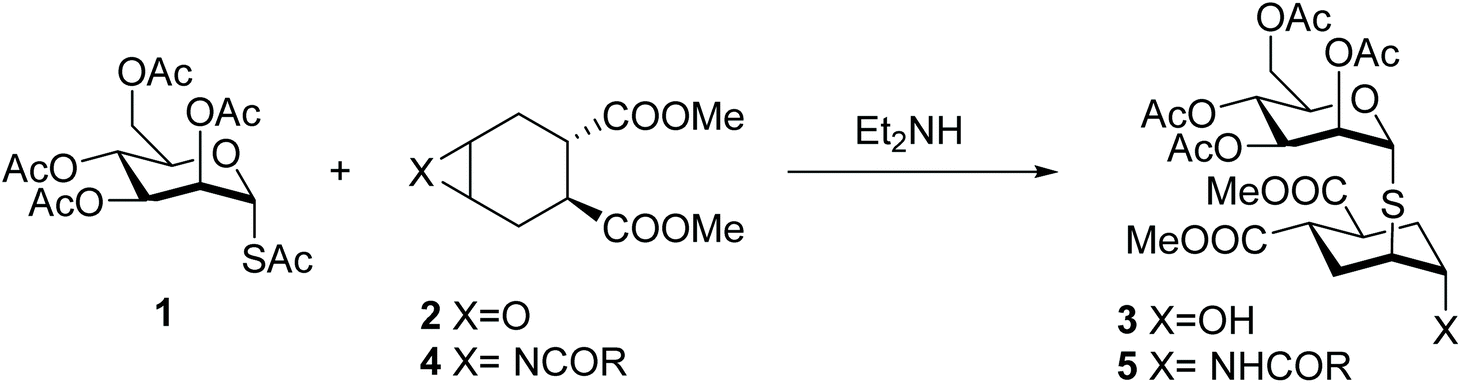 | ||
| Scheme 1 One-pot synthesis of the pseudo-thio-1,2-dimannoside 3 by epoxide opening (from ref. 17) and the planned approach to the aziridine-opening process (this article). | ||
Inspired by the potential of this system, we aimed at developing a one-pot procedure for opening the corresponding aziridine 4. This approach would afford N-linked-pseudo-thio-disaccharides such as 5. Natural N-linked glycans, which represent one of the most common covalent modification of proteins,19 are generally connected to an Asn side-chain through a β-GlcNAc residue. Thus, here we are not trying to reproduce or mimic the structure of these glycans, but are using a totally artificial structure (an α-Man mimic) to allow the rapid generation of hydrolytically stable pseudo-glycoconjugates that can be tested in a variety of fashions against relevant carbohydrate binding proteins in drug discovery programs. Initially, we developed the approach using the mannosyl thio-acetate 1 as the model compound. We then further explored the scope of the one-pot aziridine opening reaction on various mono- and disaccharides, to create a class of glycomimetics (N-linked-pseudo-thio-disaccharides) with improved stability and the potential of being recognized by various lectins. While looking into the reaction scope, we observed interesting phenomena connected to the configurational stability of anomeric thiols, some of which unexpectedly underwent anomeric isomerization under the aziridine opening conditions. Our results are reported below.
Results
Synthesis of peracetylated glycosyl thio-acetates
Various methods for the synthesis of peracetylated glycosyl thio-acetates have been reported. To simplify the synthesis and at the same time achieve satisfying yields, we selected one-pot procedures with conditions tailored for each sugar individually. The conditions used are summarized in Scheme 2 and described in detail in the ESI.† Briefly, the 1-S-acetyl derivatives of mannose (Man), rhamnose (Rha), glucose (Glc), galactose (Gal) and lactose (Lac) (Scheme 2, 1 and 6–9) were obtained by acid catalysed acetylation of the free sugar, followed by reaction with thioacetic acid (AcSH) under TMSOTf or BF3 catalysis (two step, one pot).20,21 The 1-S-acetyl derivatives of sialic acid (Neu5Ac) and N-acetylglucosamine (GlcNAc)22 (Scheme 2, 10–11) were synthesized by reaction of AcSK with the corresponding peracetylated anomeric chloride, generated in situ by reaction of the free sugar with AcCl. In most cases, the peracetylated glycosyl thio-acetates were obtained as single isomers and, when not, the two isomers were chromatographically separated (see ESI†).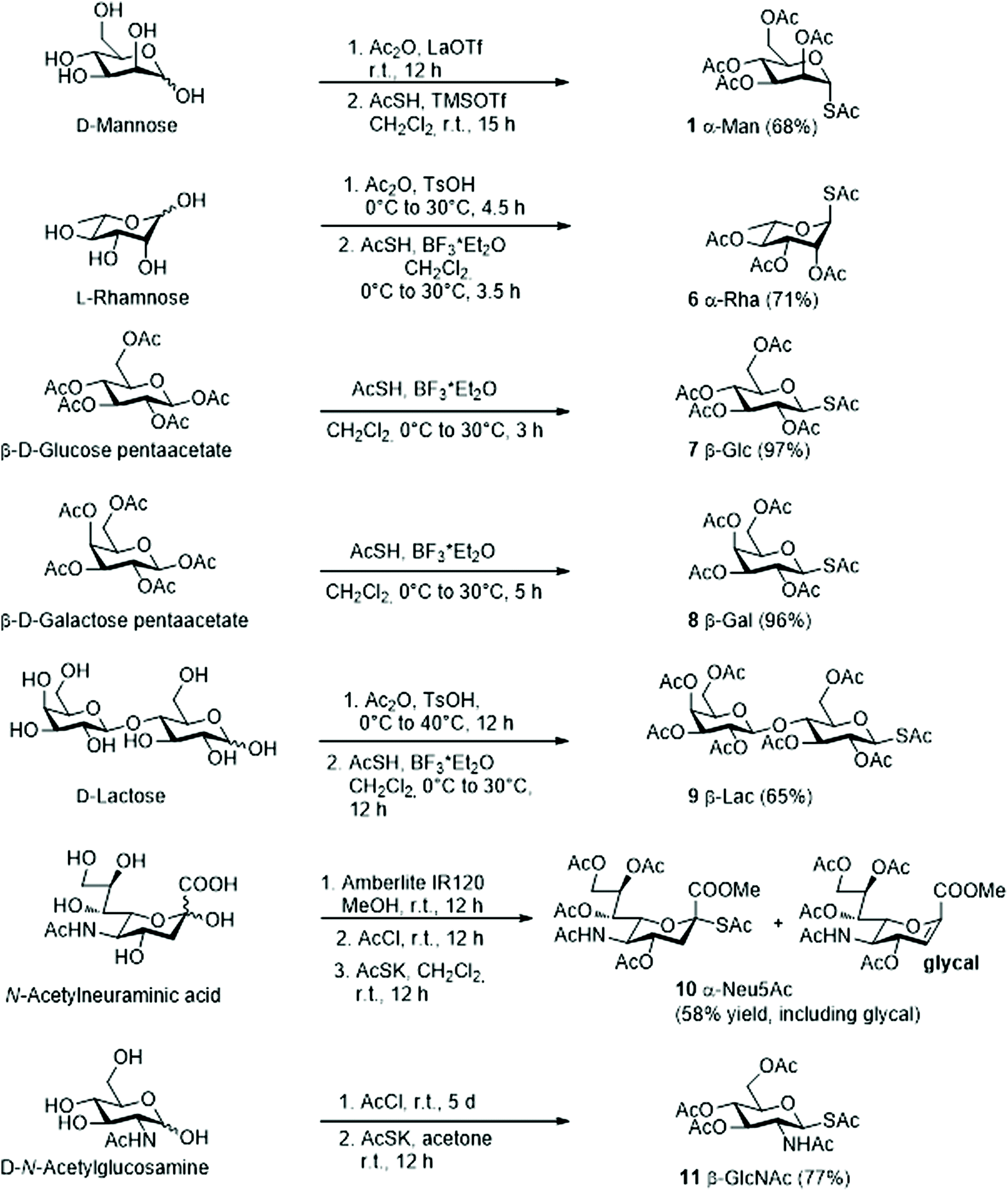 | ||
| Scheme 2 Synthesis of peracetylated glycosyl thio-acetates 1 and 6–11. | ||
Synthesis of aziridine 4
The required aziridine 4 was prepared as a single enantiomer by N–H aziridination of (1S,2S)-1,2-dicarbomethoxy-4-cyclohexene 1223 using the method recently introduced by Falck and co-workers.24 The resulting N–H aziridine 13 can then be transformed in a variety of amides and carbamates of general formula 4 (Scheme 3). Falck's N–H aziridination occurs via homogenous Rhodium catalysis (Rh2(esp)2, Du Bois’ catalyst 14) and uses O-(2,4-dinitrophenyl)hydroxylamine (DPH 15) as the aminating agent, with no external oxidants. The reaction is operationally simple, fast and scalable, but careful optimization of the reaction conditions was required to avoid dimerization of 13, a process which occurs rapidly at room temperature to afford 16 (Scheme 3). Under optimal conditions (6 h at 0 °C, addition of 10% catalyst and DPH after 3 h) on a 1.0 mmol scale no dimer was observed and the reaction yield was 90%, after Boc-protection of 13 to afford 4a (Scheme 3). Boc protection was performed using di-t-butyl-dicarbonate and adapting the conditions reported by Mordini and co-workers for a similar substrate.25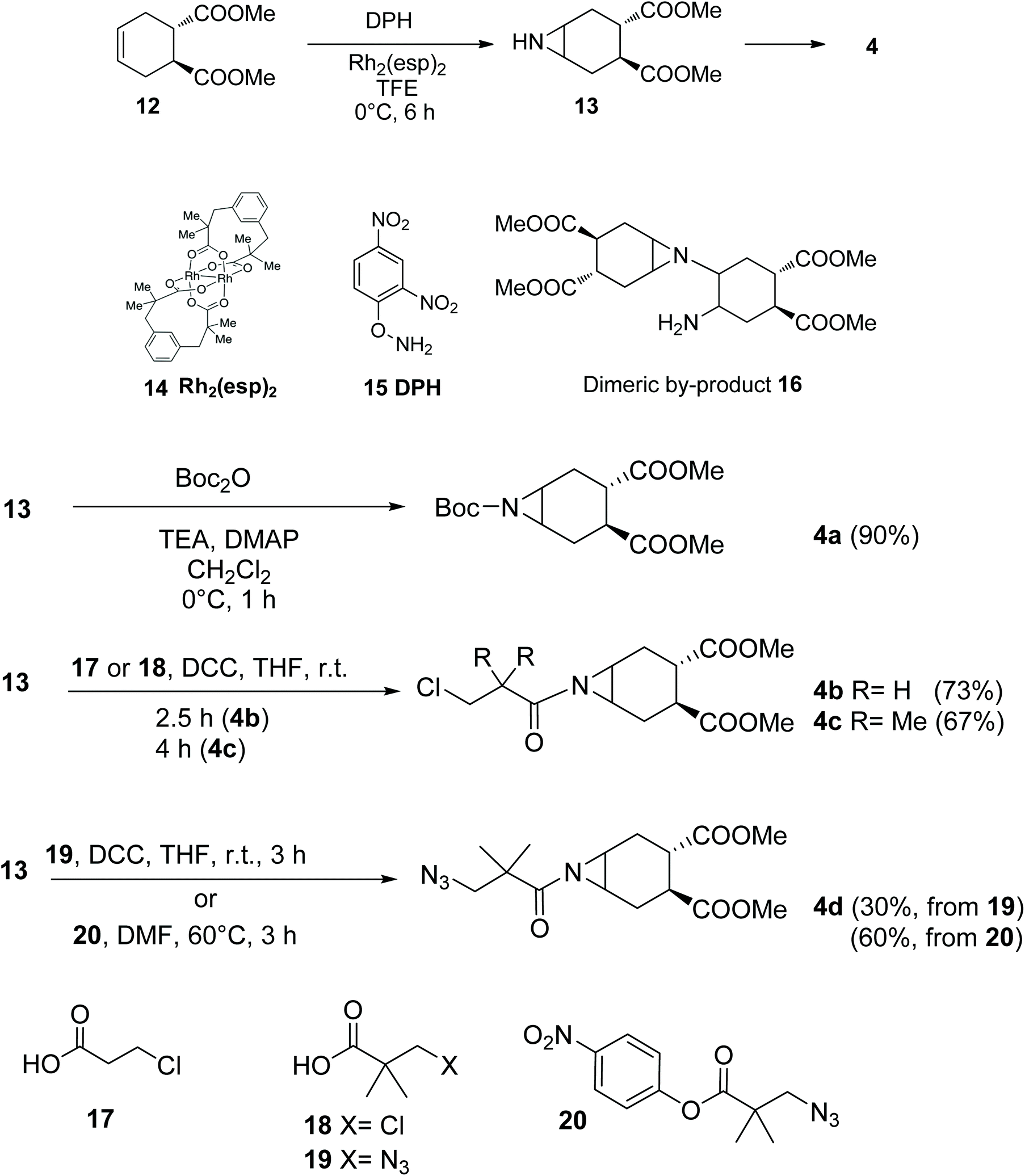 | ||
| Scheme 3 Synthesis and acylation of the N–H aziridine 13, to afford the N-acyl aziridines 4a–d. | ||
Acylation of the aziridine nitrogen protects the molecule against polymerization, while preserving its reactivity as an electrophile in a nucleophilic substitution reaction. The nitrogen protecting group can serve multiple purposes in the design of the glycomimetic structures of our interest, thus a series of different acylating agents and conditions were explored (Scheme 3). Reaction of 13 with acyl chlorides and pyridine did not afford the expected amide. Rather, products resulting from nucleophilic opening of the aziridine by chloride ions were identified by MS analysis of the crude reaction mixtures.26–28 Clean amide formation could be obtained either by carbodiimide-promoted coupling, or using activated esters. The reaction occurred smoothly, including with functionalized and hindered acyl donors 17–20 (Scheme 3), that could be used to install appropriate linkers for further (pseudo)-glycoconjugation of the fully-formed mimics.
One-pot aziridine opening
The aglycone moiety 4 used in this study was carefully selected for its symmetry properties and high conformational stability, both imparted by the 1,2-trans-dicarboxy substituents.29 Additionally, aziridines 4 lack potential chelating groups that have been shown to impinge on the regio- and stereocontrol of nucleophilic substitutions of both cyclitol epoxides and aziridines.30,31 Thus we expected a single reaction product to be formed by trans-diaxial opening of the aziridine, in keeping with the results of the analogous reaction of epoxide 2 with 1-S-acetyl-α-D-mannopyranose 1.17Indeed, reaction of 1 with 4a (Scheme 4) proceeded smoothly under the conditions established for epoxide 2 (1.9 mol equiv. of Et2NH in DMF at room temperature). Product 5a was obtained in 82% yield as a single isomer from a completely selective trans-diaxial opening process, preserving, as expected, the α-configuration of mannose. The product configuration was fully confirmed by coupling constant analysis and NOESY, as previously described for 3.17 To further explore the role of the nitrogen protecting group in this reaction, substrates 4b–d were also examined (Scheme 4). Reaction of 1 with 4b failed to afford the desired aziridine opening product (5b), rather the thiolate anion generated in situ from 1 displaced the terminal chloride of the linker, to give 21.32 Steric hindrance of the primary chloride, as in 4c, abolished this side reaction, and restored the aziridine opening pathway giving 5c in almost quantitative yields. Reaction of the corresponding azide-bearing compound 4d was more sluggish, but microwave irradiation of the reaction mixture at 60 °C for 2 h afforded 5d in 68% yield. The same product was obtained in 71% yield from 5a by Boc removal (TFA, quant) followed by acylation of crude 22 with the p-nitrophenyl ester 20, thus establishing 5a as an appropriate intermediate for later stage elaboration of the scaffold linker (Scheme 4).
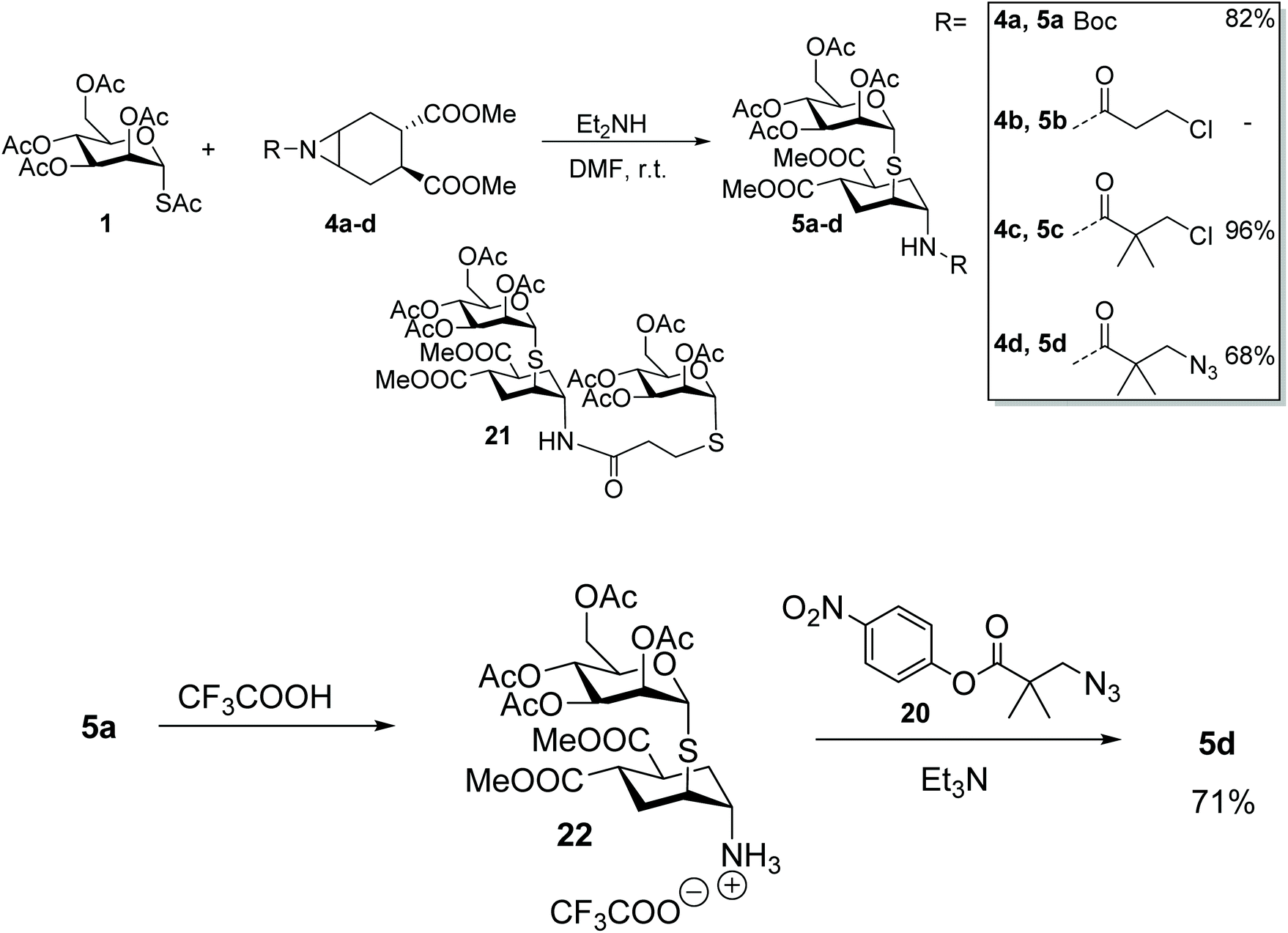 | ||
| Scheme 4 Opening reaction of aziridines 4a–d with 1-S-acetyl-α-D-mannopyranose 1. The azide-bearing product 5d can also be obtained upon acylation of 22 deriving from the N-Boc opening product 5a. | ||
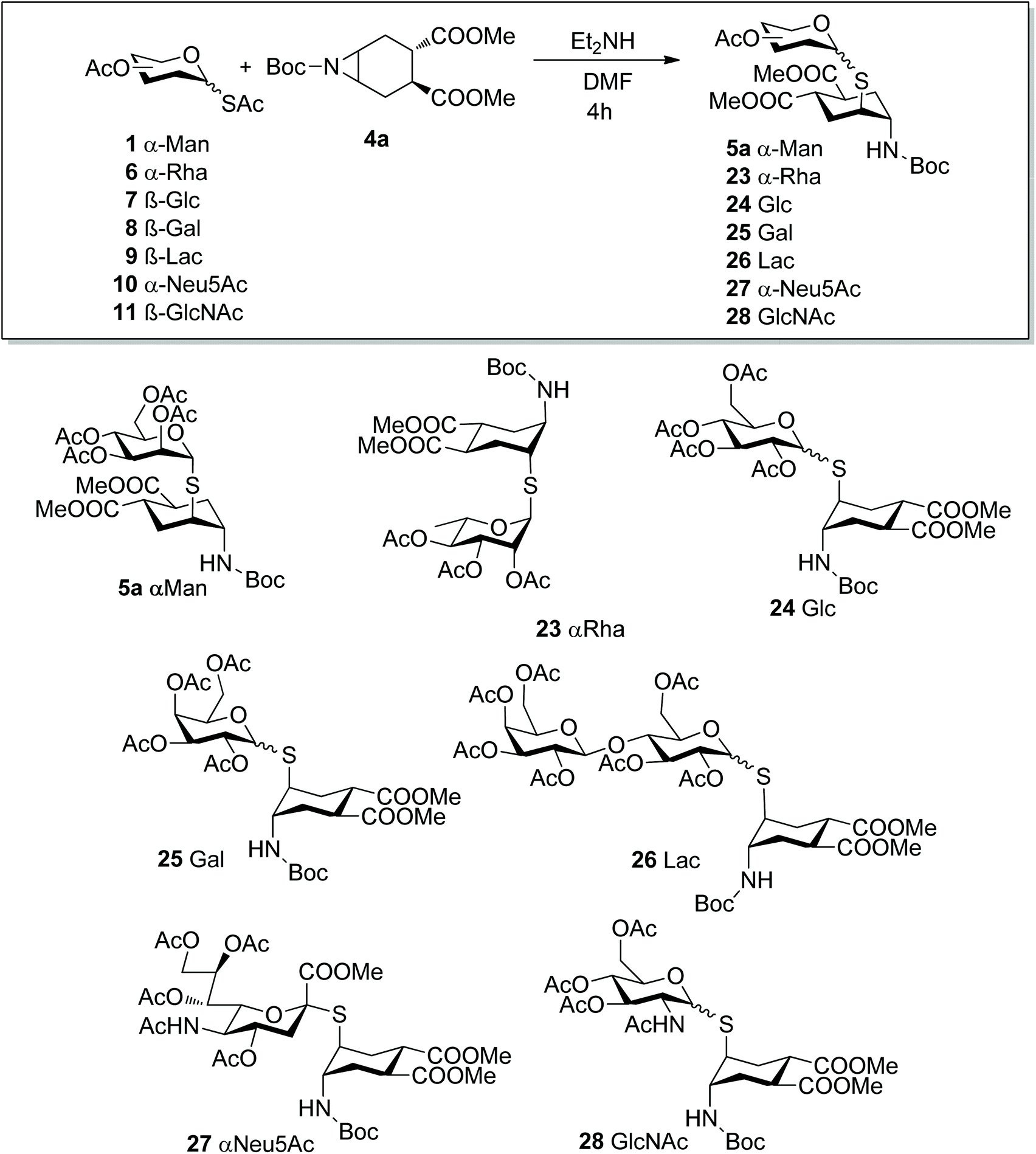
We then moved on to investigate the scope of the aziridine opening approach using the mono- and disaccharide anomeric thio-acetates 6–11 and the N-Boc-aziridine 4a as the model substrate (Scheme 5). The reactions were performed under the conditions established with mannose and their course was followed by LC-MS and/or 1H NMR. LC-MS revealed that the expected products were formed in most cases within 1 h at room temperature. The main by-product observed was the glycosyl disulphide, as exemplified by 29 for the gluco series (Scheme 6). Reaction of 4a with the α-Rha derivative 6 (Scheme 5) under the conditions established for mannose afforded 23 with 92% isolated yield (0.3 mmol scale, Table 2, entry 2) as a single α isomer, as established by NOESY experiments (see ESI†).
 | ||
| Scheme 5 Scope of the aziridine opening reaction. | ||
 | ||
| Scheme 6 Mechanism of the aziridine opening reaction. | ||
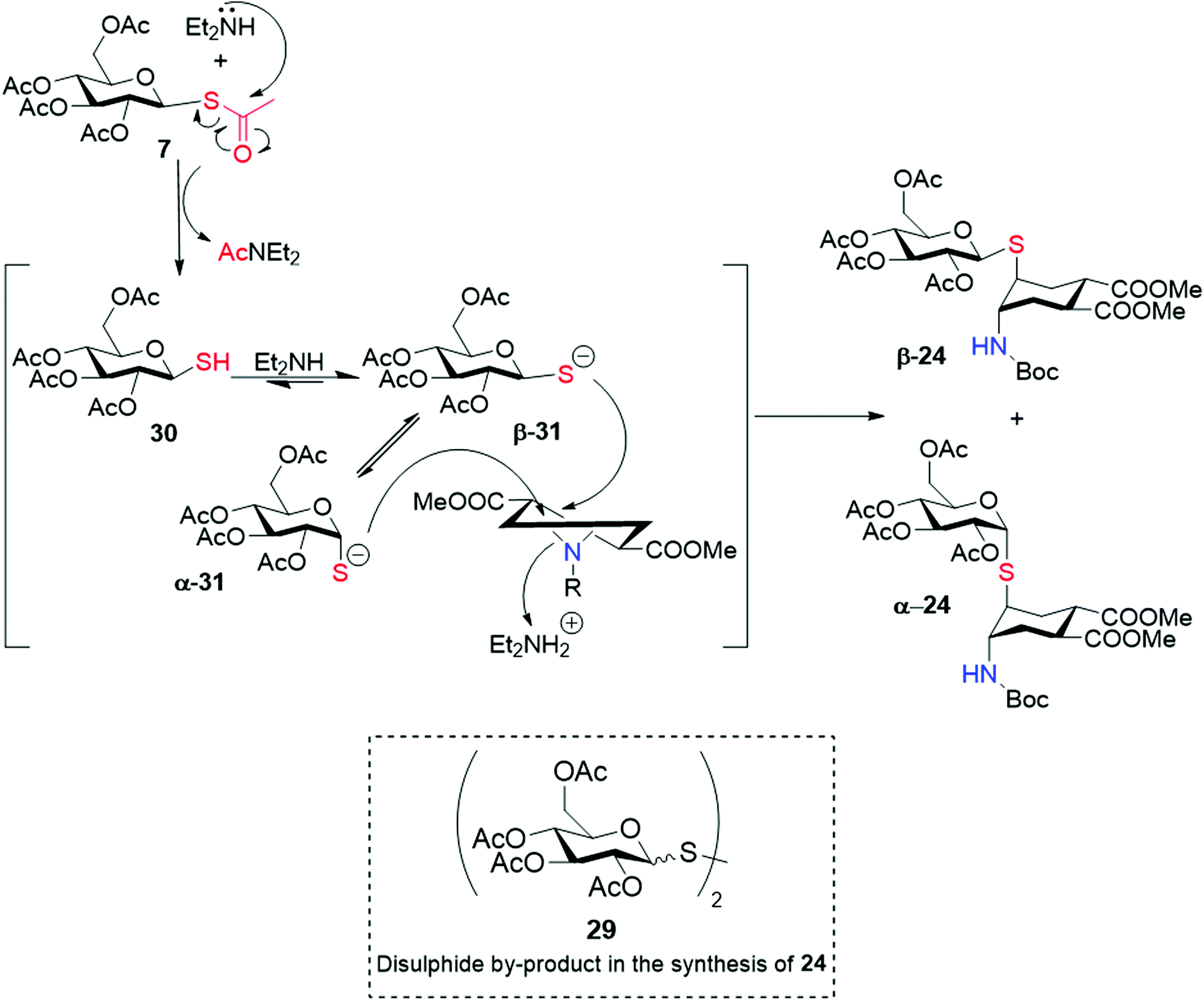
In the same conditions, however, reaction of the β-Glc thioacetate 7 afforded both the β- and α-isomers β-24 and α-24 (Scheme 6) in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, as estimated by integration of the anomeric proton signals at 4.72 and 5.73 ppm, respectively. The two isomers were separated chromatographically (iPr2O:EtOAc eluent), and analysis of coupling constants supported by NOESY-NMR and MS data undoubtedly confirmed their structure and anomeric configuration. Similarly, the disulphide by-product 29 was obtained as an anomeric mixture (see ESI†). This was rather unexpected, because most literature claims configurational stability of glycosyl thiols, particularly under basic conditions.33–35
:1 ratio, as estimated by integration of the anomeric proton signals at 4.72 and 5.73 ppm, respectively. The two isomers were separated chromatographically (iPr2O:EtOAc eluent), and analysis of coupling constants supported by NOESY-NMR and MS data undoubtedly confirmed their structure and anomeric configuration. Similarly, the disulphide by-product 29 was obtained as an anomeric mixture (see ESI†). This was rather unexpected, because most literature claims configurational stability of glycosyl thiols, particularly under basic conditions.33–35
Further experimentation with 7 was based on an analysis of the putative reaction mechanism depicted in Scheme 6: nucleophilic attack of Et2NH on the thioester generates Et2NAc and the free thiol 30, in equilibrium with the thiolate β-31 under the basic reaction conditions. The thiolate can either attack the aziridine, in an SN2-like process, or equilibrate, presumably via ring-opening (mutarotation conditions) to α-31. Additionally, both β and α thiol/thiolate can dimerize to the disulphide, which was found as an anomeric mixture in the reaction crude. Thus, we examined the role of reaction temperature, substrate concentration, base concentration and solvent on the relative rate of these reactions. The amount of anomeric isomerization strongly depended on temperature: at 0 °C (Table 1, entry 3) the ratio changed to 10:1 in favour of the β product β-24, but the overall yield of 24 decreased. Reducing the amount of base and/or the substrate concentration did not have an effect on the β:α ratio, but also slowed down the SN2 reaction process, thus increasing the amount of disulphide by-product (e.g. compare entries 1 and 2, 3 and 4, 3 and 5). Changing the solvent from DMF to CH2Cl2 (entry 6) gave low yields (16%) of almost pure β-product, but dimerization of the glycosyl thiol was the major result. Using both acetonitrile or DMF/CH2Cl2 mixtures of various composition (from 7/3 to 9/1) the reaction was slow and low yields of β:α = 3:1 were obtained (not shown in the table). Thus, it appears that low temperatures (0 °C) favour aziridine opening over anomerization. A high concentration of substrate is also beneficial, by increasing the rate of the nucleophilic substitution. A solvent of lower polarity as CH2Cl2 appears to reduce anomeric isomerization, but it also slows down the SN2 reaction, thus resulting in low yields and extensive dimerization of the thiol. Reducing the amount of base has no influence on the selectivity, but again favors dimerization over the formation of the aziridine opening product 24.
a
| Entry | T (°C) | Et2NH (mol equiv.) |
β-24:α-24b |
Yieldc (%) | 29 (%) |
|---|---|---|---|---|---|
| a Unless otherwise noted, all reactions were performed on a 0.06 mmol scale, with a 0.65 M concentration of 4a in DMF and 1.3 mol equiv. of 7 for 4 h with the amount of base and at the temperature indicated. b As judged by 1H NMR of the crude. c Isolated, combined yields of the two anomeric products 24. d 0.3 M concentration of 4a. e Reaction performed in CH2Cl2. | |||||
| 1 | 20 | 1.9 | 2:1 |
61 | <5 |
| 2 | 20 | 1.4 | 2:1 |
43 | 37 |
| 3 | 0 | 1.9 | 10:1 |
34 | 41 |
| 4 | 0 | 1.4 | 10:1 |
28 | 61 |
| 5d | 0 | 1.9 | 10:1 |
24 | 63 |
| 6e | 20 | 1.9 | 20:1 |
16 | 71 |
As an alternative, the one-pot aziridine procedure could be split in two consecutive reactions. Thus, 7 was selectively deacetylated at the anomeric position (DTT35) and then the free thiol 30 was used in the reaction with 4a (Scheme 7). When using the free thiol, a nucleophilic base is no longer needed and a sub-stoichiometric amount (0.3 mol equiv.) of the bulky non-nucleophilic base iPr2NEt (DIPEA) was used. Both the β- and α-isomers were still formed in 3:1 ratio (34% yield). In the absence of base, no product was formed and the β-configuration of the starting thiol was preserved, which indicates that formation of the thiolate is required for both the aziridine opening reaction and the isomerization.
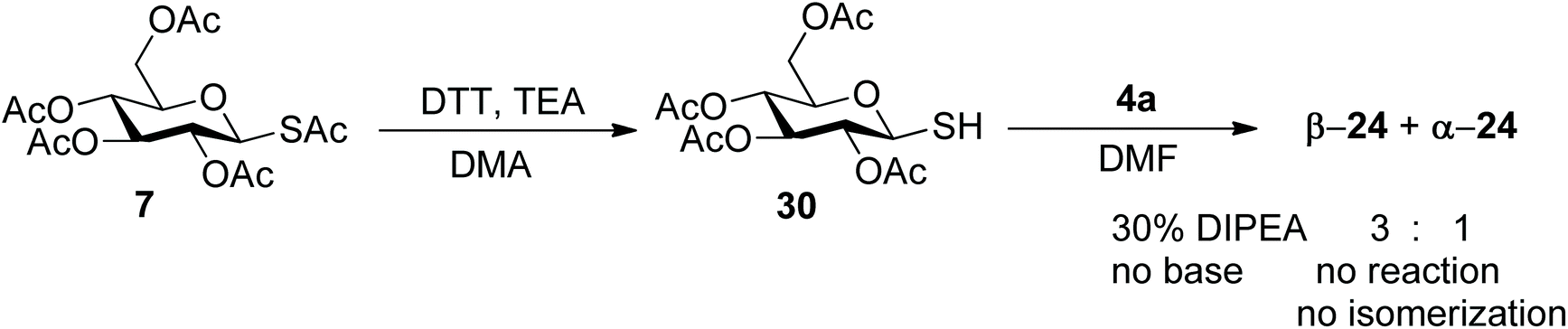 | ||
| Scheme 7 Model studies with glucose derivative 7. Two step conditions: synthesis of the free thiol 30 and reaction with 4a. | ||
Upon suggestion of a referee, we also examined whether the presence of aziridine 4 has an influence on the anomerization process. To this end, β-Glc thioacetate 7 was treated with Et2NH (1.5 mol equiv.) in DMF overnight and, upon quenching, a mixture of 2 steroisomeric disulphides was identified in the crude reaction product. This result is indeed expected, based on the anomeric isomerizarition mechanism proposed in Scheme 6. However, when the thiol 30 was dissolved in degassed DMF and treated with 23% DIPEA for 5 h, it was recovered unchanged. Upon addition of the aziridine 4 to the reaction mixture, formation of both β-24 and α-24 was observed in the reaction crude. Thus, the anomerization mechanism of the thiol/thiolate appears to be somewhat more complex than suggested in Scheme 6 and strongly dependent on the reaction conditions. This aspect will surely deserve further investigations, that we will report in due course.
In conclusion, the highest yields of aziridine opening product 24 were obtained using the reaction conditions developed for mannose and operating at room temperature. Under these conditions, a high isomerization rate was observed. Performing the reaction at 0 °C, improved the β-24:α-24 ratio to synthetically useful levels (10:1), but reduced the yield to 34%, due to extensive formation of the disulphide dimer. For all the other thio-glycosides, thus, we stuck to the conditions adopted for mannose, adjusting the reaction temperature as required by the reactivity of the individual substrate (see Table 2).
| Entry | Sugar | T (°C) | Product | β/α ratiob (yield %c) |
|---|---|---|---|---|
| a All reactions were performed with a 0.65 M concentration of aziridine 4a and 1.9 mol equiv. of Et2NH in DMF, at the indicated temperature. b Evaluated by 1H NMR. c Isolated, both isomers. d Contains 17% glycal. | ||||
| 1 | Man 1 | 20 | 5a | α only (82) |
| 2 | Rha 6 | 20 | 23 | α only (92) |
| 3 | Glc 7 | 0 | 24 | 10:1 (34) |
| 4 | Glc 7 | 20 | 24 | 2:1 (61) |
| 5 | Gal 8 | 0 | 25 | 4.5:1 (36) |
| 6 | Gal 8 | 20 | 25 | 3:1 (44) |
| 7 | Lac 9 | 0 | 26 | 5:1 (42) |
| 8 | Neu5Ac 10 | 20 | 27 | α only (56)d |
| 9 | GlcNAc 11 | 20 | 28 | 2:1 (19) |
Further exploring the scope of the reaction, the GlcNAc derivative 11 (Scheme 5) gave ca. 19% of a 2:1 β:α mixture at room temperature (Table 2, entry 9). At 0 °C, the β:α ratio improved marginally (3:1) and the yields of the reaction became so low that it was not worth optimizing further. Surprisingly, the anomeric isomerization of β-galactose 8 and β-lactose 9 showed lower dependence on temperature in the interval examined (0 °C to room temperature) and 20%–30% of the α-isomer was formed in all cases (Table 2, entries 5 and 6). The reaction of the N-acetylneuraminic acid derivative 10 was complicated by the heterogeneity of the starting material. Thioacetylation of Neu5Ac is known to yield a basically inseparable mixture of products which contains the expected α-thioacetate 10 (Scheme 5), together with the glycal and acetyl disulphide products.36 Using this mixture in the aziridine opening reaction afforded a crude which, upon chromatographic purification, yielded 27 (56%), together with ca. 17% glycal (Table 2, entry 8). The mixture was purified by HPLC for analytical purposes and NMR analysis confirmed that 27 was obtained as a single α-anomer, as shown by the signal of the H3eq proton, which appears at 2.70 ppm as a doublet of doublet (Jgem = 12.8 Hz, J3eq-4 = 4.5 Hz)37 and by the signal of the C1 carbon in a proton not decoupled 13C NMR spectra, which appears at 168.4 ppm as a doublet of quartets with a coupling constant JC1-H3ax = 3.9 Hz (see ESI†).38 Thus, as for mannose and rhamnose, the Neu5Ac thiol is configurationally stable under the reaction conditions. The results obtained in the screening (yields, anomeric ratios and optimal reaction conditions) for the glycosyl thio-acetates 1 and 6–11 are summarized in Table 2.
Finally, the thio-rhamno conjugate 23 was successfully used in the preparation of an N-linked pseudo-glycosylaminoacid scaffold, a viable building block for solid phase pseudo-glycopeptide synthesis (Scheme 8). After removal of the Boc protecting group (TFA, quant), the resulting amine was coupled in solution with the side chain carboxy group of an appropriately protected glutamic acid 32, using HATU as the coupling agent. The pseudo-glycosylaminoacid 33 was isolated in 92% yield after chromatography.
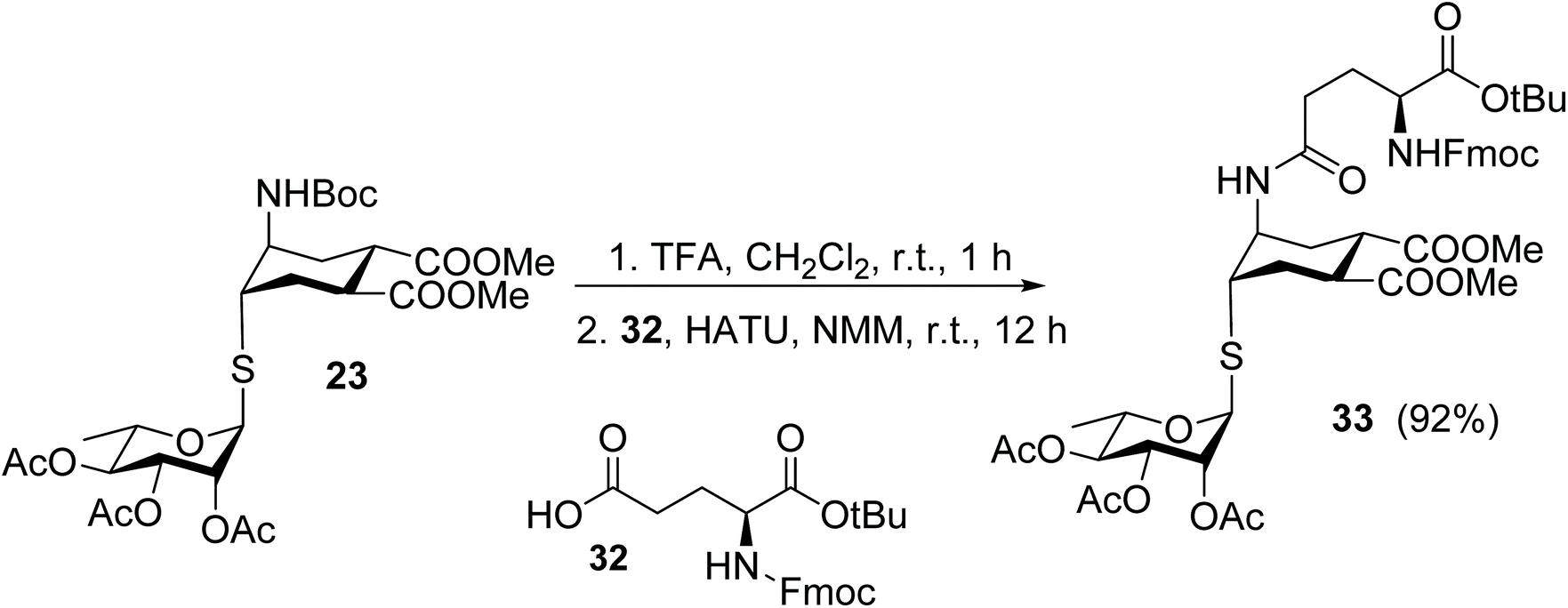 | ||
| Scheme 8 Synthesis of 33, a building block for solid phase pseudo-glycopeptide synthesis. | ||
Discussion and conclusions
A new synthetic approach was devised for the synthesis of thio-glycosides with the structure of N-linked pseudo-disaccharides through aziridine opening. In the developed procedure, formation of the thiol and opening of the aziridine are combined in a one-pot reaction, providing an efficient and operationally simple alternative to classical glycosylation methods. Under the conditions of the aziridine opening reaction, glycosyl thiols of glucose, N-acetylglucosamine, galactose and lactose showed anomeric isomerization. Particularly for glucose, the anomerization proved to be temperature, base and solvent dependant. As one of the main advantages of glycosyl thiols is that – once formed – they are believed to retain their anomeric configuration, the finding is of high importance for future studies on thio-derivatives of carbohydrates.In principle, there are two major pathways through which anomeric isomerization of glycosyl thiols can occur, mutarotation or Lewis acids promoted epimerization. While the mutarotation process of reducing sugars (1-hydroxyaldoses) has been extensively explored, the mutarotation of glycosyl thiols still remains relatively unclear. The configurational stability of glycosyl thiols could be partially explained by poor orbital overlap between the anomeric carbon and the sulphur atom, which does not favor opening and subsequent mutarotation of the pyranose ring.39 However, it was shown that in aqueous media mutarotation of free glycosyl thiols occurs at lower and neutral pH, while it is almost completely blocked under basic conditions. An exception is 1-thio-D-mannopyranose that was observed to mutarotate under both acidic and basic conditions.40 The pH dependence of mutarotation is a result of steric, electronic (anomeric) and solvation effects. Accordingly, there are a few reports of mutarotation during 1-S-glycosylation and similar reactions.39,41,42 Anomerization of glycosyl thiols was also noticed in reactions with Lewis acids such as TiCl4 and SnCl4. In uronic acids such anomerization is particularly fast, presumably favoured by coordination of the C-1 heteroatom and C-6 carbonyl group to the Lewis acid. However, conditions have been found to exploit protecting groups to achieve similar results with many other monosaccharides.43,44 To the best of our knowledge, anomeric epimerization under (slightly) basic conditions such as the ones employed in our system has never been observed. Yet our data clearly show that it occurs for all the β-glycosyl thiols that we have examined, at least in DMF. The configurational stability observed for α-mannosyl and α-rhamnosyl thiols may be related to the axial configuration of the C2 hydroxy group, an element which is known to affect anomerization equilibria.45 The mechanism and the origin of stereoselectivity in this reaction will deserve further attention.
Despite this unforeseen hurdle, the one-pot aziridine opening reaction by glycosyl thio-acetates could be used in a number of cases, including mannose, rhamnose and Neu5Ac, to synthesize in good-to-acceptable yields mimics of thio-disaccharides outfitted with a N-linked tether. These molecules are equipped for easy conjugation and, at the same time, characterized by hydrolytic stability both of the thio-glycosidic linkage and of the conjugation handle. As an example, we described here the thio-mannosyl derivatives 5b–d, which are fully hydrolytically stable analogues of Manα(1,2)Man ready for conjugation. The of Manα(1,2)Man disaccharide is a well-known binding epitope of immune system C-type lectins, including DC-SIGN.17,23 We also described the thio-rhamnosylated glutamic acid derivative 33, which can be used in the synthesis of pseudo-rhamnopeptides. Rhamnosyl glycoconjugates have been recently described as important tools for the development of novel immunotherapeutics.46–51
Carbohydrate-binding proteins (lectins) have been difficult to target selectively, since they often share broad selectivity for individual monosaccharides. One of the effective strategies described so far uses a monosaccharide, acting as the lectin anchor and connected to an aglycone. The aglycone is designed to host additional functionalities and orient them to secondary interaction sites in the lectin binding region, which are often more easily differentiated between different protein targets.52 We believe that the synthetic approach described in this paper, which exploits the expeditious glycosylation of aziridine-containing scaffolds, will be instrumental for the fast synthesis of (pseudo)-glycosylated libraries of compounds that could be used for the identification of selective lectin ligands.
Experimental section
General
Chemicals were purchased from commercial sources and used without further purification, unless otherwise indicated. When anhydrous conditions were required, the reactions were performed under nitrogen or argon atmosphere. Anhydrous solvents were purchased from Sigma-Aldrich® with a content of water ≤0.005%. Triethylamine (TEA), methanol and dichloromethane were dried over calcium hydride, THF was dried over sodium/benzophenone and freshly distilled. N,N-Dimethylformamide (DMF) was dried over 4 Å molecular sieves. Reactions were monitored by analytical thin-layer chromatography (TLC) performed on Silica Gel 60 F254 plates (Merck), and TLC Silica gel 60 RP-18 F254s (Merck) with UV detection (254 nm and 365 nm) and/or staining with ammonium molybdate acid solution, potassium permanganate alkaline solution or ninhydrin. Flash column chromatography was performed using silica gel 60 (40–63 μm, Merck). Automated flash chromatography was performed with Biotage Isolera Prime system and SNAP ULTRA cartridges were employed. Microwave irradiation was performed by a Biotage Initiator+ system. NMR experiments were recorded on a Bruker AVANCE-400 MHz instrument at 298 K. Chemical shifts (δ) are reported in ppm. The 1H and 13C NMR resonances of compounds were assigned with the assistance of COSY, HSQC and in some cases NOESY experiments. Multiplicities are assigned as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), mult. (for multiplets encompassing more than one proton).Mass spectra were recorded on Apex II ICR FTMS (ESI ionization-HRMS), Waters Micromass Q-TOF (ESI ionization-HRMS) or Thermo Fischer LCQ apparatus (ESI ionization). The mass spectrometer was operated with electrospray ionization in the positive ion mode. Full-scan mass spectra were recorded in the mass/charge (m/z) range of 50–2000. Liquid chromatography-mass spectrometry (LC-MS) analyses were carried out on a Thermo Fisher LCQ Fleet ion trap mass spectrometer equipped with a UPLC UltiMate™ 3000 system containing UV detector. A Zorbax RX-C18 (2.1 × 150 mm–5 μm) was used as column. The column oven was maintained at 30 °C. 5 μL of each sample solution were eluted using a binary gradient elution consisting of 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B) as follows: from 2% to 95% B in 25 min, 95% B kept for 10 min, then the eluent composition was brought at 2% B in 5 min. The flow rate was 0.25 mL min−1. The mass spectrometer was operated with electrospray ionization in the positive ion mode. Full-scan mass spectra were recorded in the mass/charge (m/z) range of 50–2000. Specific optical rotation values were measured using a Perkin-Elmer 241, at 589 nm in a 1 dm cell. The following abbreviations are used: DCC (N,N′-dicyclohexylcarbodiimide), DMF (N,N′-dimethylformamide), DMAP (4-dimethylaminopyridine), TFA (trifluoroacetic acid), DPH (2,4-dinitrophenylhydroxylamine 15), Rh2(esp)2 (Du Bois' catalyst 14), Hex (hexane). The olefin 12 was synthesized as previously described.23 The peracetylated glycosyl thio-acetates 1–9 were obtained as described in ref.20,21 The Neu5Ac and GlcNAc derivatives 10 and 11 were synthesized according to ref. 22. Experimental details and characterization of these known compounds are reported as ESI.†
Synthesis of aziridine 13
The reaction was performed in previously well-dried glassware flushed with Ar. Olefin 12 (200 mg, 1.01 mmol) was dissolved in CF3CH2OH (6.7 ml, 0.15 M) and transferred into the flask under Ar. The solution was cooled to 0 °C and then Rh2(esp)2 (7.7 mg, 0.010 mmol) and DPH (241 mg, 1.21 mmol) were added. The reaction mixture was stirred at 0 °C (ice bath) under Ar atmosphere for 6 h. After 3 h additional portions of Rh2(esp)2 (7.7 mg, 0.010 mmol) and DPH (40 mg, 0.20 mmol) were added. After 6 h the reaction mixture was diluted with CH2Cl2 (10 ml) and washed with sat. NaHCO3 (10 ml), then H2O was added (10 ml) to dissolve any forming salts. The aqueous phase was washed twice with CH2Cl2. Combined organic phases were washed with brine and dried over Na2SO4, filtered and concentrated under vacuum. The crude product was used directly and without further purification in the N-protection reaction to avoid decomposition and dimerization that occur during chromatographic purification. For analytical purposes, chromatographic purification (95:5 CH2Cl2:MeOH) was performed on a batch obtaining aziridine 13 in 68% (110 mg, 0.52 mmol) yield starting from 150 mg of olefin 12. Rf = 0.2 (CH2Cl2/MeOH 95:5); 1H NMR (400 MHz, CDCl3): δ 3.66 (s, 3H, OMe), 3.64 (s, 3H, OMe), 2.78 (td, J1-6ax = J1-2 = 11.4 Hz, J1-6eq = 4.6 Hz, 1H, H1), 2.54 (td, J2-1 = J2-3ax = 11.4 Hz, J2-3eq = 6.5 Hz, 1H, H2), 2.39–2.33 (mult., 4H, H3eq, H6eq, H4, H5), 1.86–1.72 (mult., 2H, H3ax, H6ax); 13C NMR (100 MHz, CDCl3): δ 175.9 (CO), 175.0 (CO), 51.9 (OMe), 51.9 (OMe), 41.3 (C2), 38.2 (C1), 29.7 (C4), 28.0 (C6), 27.5 (C3), 27.2 (C5); MS (ESI) calcd for C10H15NO4 [M + H]+m/z: 214.11; found m/z: 214.11.
The dimer 16 was also isolated by chromatography. Characterization of the dimer 16: Rf = 0.39 (CH2Cl2/MeOH 9:1); 1H NMR (400 MHz, CDCl3): δ 3.71–3.64 (mult., 12H, OMe), 3.21 (td, J1-6ax = J1-2 = 9.9 Hz, J1-6eq = 4.4 Hz, 1H, H1), 3.10–3.01 (mult., 2H, H2, H4), 2.80 (td, J10-9ax = J10-11 = 10.3 Hz, J10-9eq = 5.0 Hz, 1H, H10), 2.52 (td, J11-12ax = J11-10 = 10.3 Hz, J11-12eq = 6.3 Hz, 1H, H11), 2.34–2.14 (mult., 3H, H12eq, H9eq, H3eq), 1.96–1.66 (mult., 5H, H12ax, H6eq, H6ax, H9ax, H3ax), 1.66–1.60 (m, 1H, H7), 1.37–1.31 (m, 1H, H5); 13C NMR (100 MHz, CDCl3): δ 175.9 (CO), 175.70 (CO), 175.3 (CO), 174.9 (CO), 69.4 (C5), 52.2 (2 × OMe), 52.1 (2 × OMe), 50.0 (C4), 41.1 (C1), 40.1 (C10), 39.5 (C2), 39.0 (C11), 38.6 (C7), 34.5 (C8), 31.7 (C6), 28.4 (C3), 27.9 (C9), 27.2 (C12).; MS (ESI) calcd for C20H30N2O8 [M + H]+m/z: 427.21; found m/z: 427.59.
Synthesis of the N-Boc-aziridine 4a
To a solution of the free aziridine 13 (crude from the previous step, 1.01 mmol) dissolved in freshly distilled CH2Cl2 under N2 atmosphere, DMAP (a few crystals) were added. The mixture was cooled to 0 °C and Et3N (freshly distilled, 0.84 ml, 6.06 mmol) and Boc2O (1.2 ml, 5.05 mmol) were added. After 1 h under stirring at 0 °C, the reaction was allowed to reach room temperature, then quenched with H2O. The resulting mixture was diluted with a saturated solution of NH4Cl; the aqueous phase extracted twice with EtOAc. The organic layers were washed with saturated solutions of KHSO4, Na2CO3 and NaCl in this order and finally dried over Na2SO4, filtered and concentrated in vacuum. Purification by flash chromatography (7:3 Hex:EtOAc) afforded the t-butylcarbamate 4a as a yellow waxy solid in 90% yield (0.29 g, 0.91 mmol). Rf = 0.33 (Hex/EtOAc 7:3); [α]26D (CHCl3, c 1.05): +34; 1H NMR (400 MHz, CDCl3): δ 3.66 (s, 3H, OMe), 3.65 (s, 3H, OMe), 2.82 (td, J1-6ax = J1-2 = 11.5 Hz, J1-6eq = 4.5 Hz, 1H, H1), 2.72–2.67 (m, 1H, H5), 2.62 (td, J4-3eq = J4-5 = 6.4 Hz, J4-3ax = 1.0 Hz 1H, H4), 2.54 (td, J2-1 = J2-3ax = 11.5 Hz, J2-3eq = 6.4 Hz, 1H, H2), 2.45 (ddd, J6eq-6ax = 14.2 Hz, J6eq-1 = 4.6 Hz, J6eq-5 = 1.6 Hz, 1H, H6eq), 2.35 (dt, J3eq-3ax = 14.9 Hz, J3eq-4 = J3eq-2 = 6.4 Hz, 1H, H3eq), 1.95 (ddd, J3ax-3eq = 14.9 Hz, J3ax-2 = 11.8 Hz, J3ax-4 = 1.0 Hz, 1H, H3ax), 1.74 (ddd, J6ax-6eq = 14.5 Hz, J6ax-1 = 11.4 Hz, J6ax-5 = 3.0 Hz, 1H, H6ax), 1.43 (s, 9H, tBu); 13C NMR (100 MHz, CDCl3): δ 175.4 (CO), 174.5 (CO), 162.3 (CO, carbamate), 81.3 (CIV Boc), 52.0 (2 × OMe), 40.8 (C2), 38.3 (C1), 37.0 (C5), 34.5 (C4), 28.0 (tBu 3 × Me), 27.5 (C6), 26.3 (C3); MS (ESI) calcd for C15H23NO6 [M + Na]+m/z: 336.14; found m/z: 336.18.
Synthesis of N-acyl-aziridine 4b
To a solution of the free aziridine 13 (32 mg, 0.15 mmol) in dry THF (750 μl, 0.2 M), 3-chloropropionic acid 17 (19.5 mg, 0.18 mmol) and DCC (37 mg, 0.18 mmol) were added at room temperature. After 2.5 h, the white precipitate was filtered over celite, washing with CH2Cl2 and finally the solvents were removed under reduced pressure. Amide 4b was isolated through a chromatographic column (1:1 Hex:EtOAc) in 73% yield (34 mg, 0.11 mmol). Rf = 0.3 (Hex/EtOAc 1:1); 1H NMR (400 MHz, CDCl3): δ 3.77 (t, J8-7 = 6.4 Hz, 2H, CH2Cl), 3.68 (s, 3H, OMe), 3.67 (s, 3H, OMe), 2.91–2.78 (mult., 5H, H4, H5, H1, CH2-linker), 2.64 (ddd, J2-3ax = J2-1 = 11.1 Hz, J2-3eq = 6.6 Hz, 1H, H2), 2.44 (ddd, J6eq-6ax = 14.3 Hz, J6eq-1 = 4.2 Hz, J6eq-5 = 1.3 Hz, 1H, H6eq), 2.36 (dt, J3eq-3ax = 14.8 Hz, J3eq-2 = J3eq-4 = 6.4 Hz, 1H, H3eq), 2.03 (ddd, J3ax-3eq = 15.1 Hz, J3ax-2 = 10.8 Hz, J3ax-4 = 0.7 Hz, 1H, H3ax), 1.88 (ddd, J6ax-6eq = 14 Hz, J6ax-1 = 10.2 Hz, J6ax-5 = 0.9 Hz, 1H, H6ax); MS (ESI) calcd for C13H18ClNO5 [M + Na]+m/z: 326.09 (100%), 328.07 (32%); found m/z: 326.38, 328.37.
Synthesis of N-acyl-aziridine 4c
The free aziridine 13 (28 mg, 0.13 mmol) was dissolved in freshly distilled THF (650 μl). 3-Chloro-2,2-dimethylpropionic acid 18 (21 mg, 0.16 mmol) and DCC (32 mg, 0.16 mmol) were added in this order at room temperature and under nitrogen. After 4 h under stirring, the mixture was filtered over celite and washed with CH2Cl2. Flash chromatography (1:1 Hex:EtOAc) afforded the amide 4c in 67% yield (29 mg, 0.087 mmol). Rf = 0.4 (Hex/EtOAc 1:1); 1H NMR (400 MHz, CDCl3): δ 3.69 (s, 3H, OMe), 3.67 (s, 3H, OMe), 3.61 (AB system, Japp = 14.7 Hz, 2H, CH2Cl), 2.93–2.82 (mult., 3H, H4, H5, H1), 2.67 (td, J2-3ax = J2-1 = 10.1 Hz, J2-3eq = 6.6 Hz, 1H, H2), 2.44 (ddd, J6eq-6ax = 14.5 Hz, J6eq-1 = 4.8 Hz, J6eq-5 = 1.3 Hz, 1H, H6eq), 2.48–2.31 (m, 1H, H3eq), 2.02 (ddd, J3ax-3eq = 14.5 Hz, J3ax-2 = 10.4 Hz, J3ax-4 = 1 Hz, 1H, H3ax), 1.9 (ddd, J6ax-6eq = 14 Hz, J6ax-1 = 11 Hz, J6ax-5 = 3.3 Hz, 1H, H6ax), 1.34 (s, 6H, 2 × CH3 linker); 13C NMR (100 MHz, CDCl3): δ 188.7 (CO amide), 174.9 (CO), 174.3 (CO), 52.0 (CH2Cl), 52.0 (2 × OMe), 40.4 (C2), 38.1 (C1), 36.7 (C5), 34.0 (C4), 29.6 (CMe2), 26.4 (C6), 25.5 (C3), 24.1, 23.9 (2 × CH3 linker); MS (ESI) calcd for C15H22ClNO5 [M + Na]+m/z: 354.12 (100%), 356.11 (33%); found m/z: 354.43, 356.41.
Synthesis of the activated ester 20
2,2-Dimethyl-3-chloro-propanoic acid 18 (400 mg, 2.97 mmol) was dissolved in dry DMF (14.6 ml) and NaN3 (990 mg, 15.20 mmol) was added. The mixture was stirred at 50 °C under nitrogen atmosphere. After 3 days, the reaction was cooled to room temperature, diluted with CH2Cl2 and filtered over a celite pad to remove unreacted NaN3 and the generated salts (NaCl). The solvent was evaporated at reduced pressure giving 3-azido-2,2-dimethylpropanoic acid 19 in quantitative yield (425 mg). 1H NMR (400 MHz, CDCl3): δ 3.42 (s, 2H, CH2), 1.25 (s, 6H, 2 × CH3).To a solution of crude acid 19 (311 mg, 2.17 mmol) in dry DMF (7.2 ml, 0.3 M) p-nitrophenyl trifluoacetate (766 mg, 3.25 mmol) and dry pyridine (350 μl) were added. The mixture was warmed to 60 °C and stirred overnight. The following day, the reaction was cooled to room temperature and solvent and pyridine were removed under vacuum. Compound 20 was isolated in 73% (419 mg, 1.58 mmol) yield through a chromatographic column (8:2 Hex:EtOAc). Rf = 0.4 (Hex/EtOAc 8:2); 1H NMR (400 MHz, CDCl3): δ 8.28 (d, J = 8.9 Hz, 2H, Har), 7.27 (d, J = 9.2 Hz, 2H, Har), 3.57 (s, 2H, CH2N3), 1.4 (s, 6H, 2 × CH3); 13C NMR (100 MHz, CDCl3): δ 173.5 (CO), 155.5 (Car), 145.6 (Car), 125.3 (2 × CHar), 122.5 (2 × CHar), 59.59 (CH2), 44.4 (C(CH3)2), 23.03 (2 × CH3).
Synthesis of N-acyl-aziridine 4d
Using DCC: To a solution of 13 (38 mg, 0.18 mmol) in dry THF (890 μl, 0.2 M), carboxylic acid 19 (31 mg, 0.21 mmol) was added and then DCC (44 mg, 0.21 mmol). After 3 h under stirring at room temperature, the mixture was filtered over celite and washed with CH2Cl2. The chromatographic column afforded compound 4d in 30% yield (18 mg, 0.053 mmol). Using the activated ester20: To a solution of aziridine 13 (39 mg, 0.18 mmol) in dry DMF (720 μl, 0.25 M) and pyridine (18 μl) the activated ester 20 (63 mg, 0.24 mmol) was added. The reaction proceeded at 60 °C under stirring for 3 h. The mixture was cooled to room temperature and then solvent and pyridine were removed under reduced pressure. Flash chromatography (1:1 Hex:EtOAc) allowed to isolate compound 4d in 60% yield (36 mg, 0.11 mmol). Rf = 0.38 (Hex/EtOAc 1:1); 1H NMR (400 MHz, CDCl3): δ 3.69 (s, 3H, OMe), 3.67 (s, 3H, OMe), 3.43 (AB system, Japp = 14.7 Hz, 2H, CH2N3), 2.90–2.79 (mult., 3H, H1, H5, H4), 2.66 (td, J2-3ax = J2-1 = 10.6 Hz, J2-3eq = 6.7 Hz, 1H, H2), 2.44 (ddd, J6eq-6ax = 14.3 Hz, J6eq-1 = 4.8 Hz, J6eq-5 = 1.3 Hz, 1H, H6eq), 2.36 (ddd, J3eq-3ax = 15 Hz, J3eq-2 = 6.9 Hz, J3eq-4 = 6.4 Hz, 1H, H3eq), 2.01 (ddd, J3ax-3eq = 14.9 Hz, J3ax-2 = 10.7 Hz, J3ax-4 = 0.6 Hz, 1H, H3ax), 1.26 (s, 6H, 2 CH3 linker); 13C NMR (100 MHz, CDCl3): δ 189.5 (CO amide), 175.1 (CO), 174.4 (CO), 60.3 (CH2N3), 52.2 (OMe), 52.1 (OMe), 40.5 (C2), 38.3 (C1), 36.9 (C5), 34.2 (C4), 29.8 (C(CH3)2 linker), 26.6 (C6), 25.7 (C3), 23.9 (CH3 linker), 23.7 (CH3 linker); MS (ESI) calcd for C15H22N4O5 [M + Na]+m/z: 361.15; found m/z: 361.61.
General procedure for the opening reaction of aziridine 4a
Peracetylated thio-glycoside (1.3 eq.) and aziridine (1 eq.) were dissolved in dry DMF (0.65 M) under N2 atmosphere at the temperature indicated in Table 2 and Et2NH (1.9 eq.) was added. The reaction mixture was stirred for 4 h at the temperature indicated in Table 2, then the reaction mixture was diluted with EtOAc and washed with 1 M HCl. The organic phase was washed three times with H2O and the aqueous phases were additionally extracted with EtOAc. The combined organic phases were dried over Na2SO4, filtered and concentrated under vacuum.General method for deacetylation of the sugar moiety
The substrates were dissolved in a MeNH2/EtOH solution (4 M) at room temperature in order to have a final solution 0.05 M of the substrate. The reactions were monitored by reverse-phase TLC (eluents based on different ratios of H2O/MeOH) and, after completion, the solvent and the excess MeNH2 were removed under reduced pressure. The products were separated from the N-methylacetamide by-product using automated flash chromatography (Ultra HP-Sphere C18 cartridges) in reverse phase (isocratic elution, 1:1 H2O:MeOH).
Synthesis of the pseudo-thio-dimannoside 5a
Prepared from peracetylated thio-mannose 1 (37 mg, 0.091 mmol) and aziridine 4a (22 mg, 0.070 mmol) according to the general procedure and purified by flash chromatography (1:1 Hex:EtOAc) to give 5a in 82% yield (39 mg, 0.058 mmol). Rf = 0.33 (Hex/EtOAc 1:1); [α]19D (CHCl3, c 1.95): +59; 1H NMR (400 MHz, CDCl3): δ 5.40–5.34 (mult., 2H, H1, H2), 5.30 (dd, J4-5 = 10.2 Hz, J4-3 = 9.8 Hz, 1H, H4), 5.21 (dd, J3-4 = 9.8 Hz, J3-2 = 2.9 Hz, 1H, H3), 4.80–4.70 (m, 1H, NH), 4.44–4.29 (mult., 2H, H5, H6a), 4.17–4.07 (m, 1H, H6b), 3.95–3.83 (m, 1H, H4′), 3.71 (s, 3H, OMe), 3.70, (s, 3H, OMe), 3.31–3.21 (m, 1H, H5′), 3.09–3.00 (m, 1H, H1′), 2.88–2.77 (m, 1H, H2′), 2.21–1.95 (mult., 3H, H6′eq, H3′eq, H6′ax), 2.16 (s, 3H, OAc), 2.08 (s, 3H, OAc), 2.04 (s, 3H, OAc), 1.98 (s, 3H, OAc), 1.91–1.80 (m, 1H, H3′ax), 1.43 (s, 9H, tBu); 13C NMR (100 MHz, CDCl3): δ 173.7 (CO), 173.7 (CO), 170.6 (CO), 169.9 (CO), 169.9 (CO), 169.7 (CO), 169.6 (CO, carbammate), 82.1 (C1), 80.0 (CIV Boc), 70.9 (C2), 69.4 (C3), 69.3 (C5), 66.1 (C4), 62.5 (C6), 52.3 (OMe), 52.2 (OMe), 49.0 (C4′), 44.5 (C5′), 39.9 (C1′, C2′), 29.7 (C6′), 29.1 (C3′), 28.3 (tBu, 3 × Me), 20.9 (OAc), 20.7 (OAc), 20.7 (OAc), 20.6 (OAc); MS (ESI) calcd for C29H43NO15S [M + Na]+m/z: 700.24; found m/z: 701.01.
Product 5a (19 mg, 0.028 mmol) was deacetylated following the general procedure for deacetylation (reaction time 4 h). The deacetylated product was obtained after purification in 70% yield (10 mg, 0.020 mmol). Rf = 0.24 (H2O/MeOH 1:1) [α]18D (MeOH, c 0.5): +113; 1H NMR (400 MHz, CD3OD): δ 5.34 (d, J1-2 = 1 Hz, 1H, H1), 3.96–3.81 (mult., 4H, H2, H5, H4′, H6a), 3.78–3.71 (dd, 1H, J6a-6b = 12.5 Hz, J6b-5 = 5.8 Hz, H6b), 3.68 (s, 3H, OMe), 3.68 (s, 3H, OMe), 3.66–3.59 (mult., 2H, H4, H3), 3.24–3.17 (m, 1H, H5′), 3.06–2.94 (mult., 2H, H1′, H2′), 2.25–2.11 (m, 1H, H6′eq), 2.11–1.97 (mult., 2H, H6′ax, H3′eq), 1.9–1.8 (m, 1H, H3′ax), 1.45 (s, 9H, tBu); 13C NMR (100 MHz, CD3OD): δ 176.1 (CO), 176.0 (CO), 157.2 (CO, carbammate), 86.4 (C1), 80.4 (CIV Boc),75.4 (C5), 73.8 (C2), 73.2 (C3), 68.8 (C4), 62.9 (C6), 52.5 (2 × OMe), 50.7 (C4′), 46.0 (C5′), 41.5 (C1′), 41.0 (C2′), 30.3 (C6′), 30.3 (C3′), 28.8 (tBu, 3 × Me). MS (ESI) calcd for C21H35NO11S [M + Na] + m/z: 532.18; found: 532.51; HR-MS (ESI) calcd for C21H35NO11S [M + Na] + m/z: 532.1829; found 532.1835.
Synthesis of the pseudo thio-disaccharide 23
Prepared from peracetylated thio-rhamnose 6 (126 mg, 0.36 mmol) and aziridine 4a (87 mg, 0.28 mmol) according to the general procedure and purified by automated flash chromatography (Biotage Isolera, SNAP Cartridge, gradient 30–50% EtOAc/Hex) to give 23 as a single α isomer in 92% yield (158 mg, 0.26 mmol) as colourless oil. Rf = 0.34 (Hex/EtOAc 1:1); [α]21D (CHCl3, c 1.00): −71; 1H NMR (400 MHz, CDCl3) δ 5.37–5.31 (m, 1H, H2), 5.31 (s, 1H, H1), 5.19–5.11 (m, 1H, H3), 5.13–5.03 (m, 1H, H4), 4.79 (s, 1H, NH), 4.23 (dt, J5-4 = 12.3 Hz, J5-Me = 6.3 Hz, 1H, H5), 3.90–3.83 (m, 1H, H4′), 3.70 (s, 3H, OMe), 3.68 (s, 3H, OMe), 3.35–3.28 (m, 1H, H5′), 3.05–2.99 (m, 1H, H1′), 2.84–2.73 (m, 1H, H2′), 2.14 (s, 3H, OAc), 2.06 (s, 3H, OAc), 2.17–1.93 (mult., 3H, H3′eq, H6′ax, H6′eq), 1.97 (s, 3H, OAc), 1.96–1.85 (m, 1H, H3′ax), 1.43 (s, 9H, tBu), 1.24 (d, JMe-5 = 6.3 Hz, 3H, Me); 13C NMR (101 MHz, CDCl3) δ 175.0 (CO), 174.4 (CO), 170.4 (CO), 170.3 (CO), 170.3 (CO), 83.8 (C1), 71.7 (C2), 71.6 (C4), 69.9 (C3), 67.9 (C5), 52.7 (OMe), 52.6 (OMe), 49.8 (C4′), 45.7 (C5′), 40.3 (C2′), 40.1 (C1′), 30.1 (C6′), 29.3 (C3′), 28.7 (tBu-3 × Me), 21.3 (OAc), 21.2 (OAc), 21.1 (OAc), 17.6 (Me); JH1–C1 = 170.3 (HSQC without 13C decoupling); LC-MS (Rt = 20.41 min) calcd for C27H41NO13S [M + Na]+m/z: 642.23; found m/z: 641.78. MS (HRMS): calcd for C27H41NO13S [M + Na]+m/z: 642.2196; found m/z: 642.2195.
Synthesis of the pseudo thio-disaccharide 24
Prepared from peracetylated thio-glucose 7 (34 mg, 0.083 mmol) and aziridine 4a (20 mg, 0.064 mmol) according to the general procedure and purified by flash chromatography (1:1 Hex:EtOAc) to give 24 in 61% yield (26 mg, 0.039 mmol) as colourless oil (entry 4 in Table 2). The β and α isomers could be separated by flash chromatography (8:1 iPr2O:EtOAc). β-24: Rf = 0.22 (iPr2O/EtOAc 8:1); 1H NMR (400 MHz, CDCl3) δ 5.23 (dd, J3-4 = J3-2 = 10 Hz, 1H, H3), 5.13 (dd, J4-3 = J4-5 = 10 Hz, 1H, H4), 5.04 (dd, J2-1 = J2-3 = 10 Hz, 1H, H2), 4.87 (m, 1H, NH), 4.72 (d, J1-2 = 10 Hz, 1H, H1), 4.27 (dd, J6a-6b = 12.4 Hz, J6a-5 = 4.5 Hz, 1H, H6a), 4.15 (dd, J6b-6a = 12.4 Hz, J6b-5 = 2.4 Hz, 1H, H6b), 3.93–3.80 (m, 1H, H4′), 3.76 (ddd, J5-4 = 10.0 Hz, J5-6a = 4.5 Hz, J5-6b = 2.4 Hz, 1H, H5), 3.69 (s, 6H, 2 × OMe), 3.42–3.26 (m, 1H, H5′), 2.99–2.86 (m, 1H, H1′), 2.81–2.70 (m, 1H, H2′), 2.24–2.08 (mult., 3H, H3′eq, H6′eq, H6′ax), 2.07 (s, 3H, OAc), 2.05 (s, 3H, OAc), 2.02 (s, 3H, OAc), 2.00 (s, 3H, OAc), 1.91–1.81 (m, 1H, H3′ax), 1.45 (s, 9H, tBu); 13C NMR (101 MHz, CDCl3) δ 174.0 (CO), 171.8 (CO), 171.7 (CO), 170.8 (CO), 170.3 (CO), 169.5 (CO), 83.7 (C1), 76.0 (C5), 74.1 (C3), 70.2 (C2), 68.3 (C4), 62.0 (C6), 52.4 (OMe), 52.3 (OMe), 48.2* (C4′), 42.1* (C5′), 40.1 (C1′, C2′), 29.8 (C3′, C6′), 28.5 (tBu-3 × Me), 20.9 (OAc), 20.9 (OAc), 20.8 (OAc), 20.8 (OAc). * These signals are better visible in the HSQC spectrum; LC-MS (Rt = 19.64 min) calcd for C29H43NO15S [M + Na]+m/z: 700.24; found m/z: 699.81; MS (HRMS): calcd for C29H43NO15S [M + Na]+m/z: 700.2251; found m/z: 700.2249. α-24: 1H NMR (400 MHz, CDCl3) δ 5.73 (d, J2-1 = 5.7 Hz, 1H, H1), 5.35 (dd, J3-2 = J3-4 = 9.8 Hz, 1H, H3), 5.14–4.99 (mult., 2H, H4, H2), 4.75 (s, 1H, NH), 4.48–4.39 (m, 1H, H5), 4.34 (dd, J6a-6b = 12.5 Hz, J6a-5 = 4.6 Hz, 1H, H6a), 4.19–4.09 (m, 1H, H6b), 3.92–3.83 (m, 1H, H4′), 3.71 (s, 3H, OMe), 3.70 (s, 3H, OMe), 3.24–3.15 (m, 1H, H5′), 3.11–2.98 (m, 1H, H1′), 2.88–2.73 (m, 1H, H2′), 2.09 (s, 3H, OAc), 2.08 (s, 3H, OAc), 2.10–2.00 (mult., 3H, H3′eq, H6′eq, H6′ax), 2.03 (s, 3H, OAc), 2.01 (s, 3H, OAc), 1.92–1.84 (m, 1H, H3′ax), 1.44 (s, 9H, tBu); 13C NMR (HSQC) (101 MHz, CDCl3) δ 81.2 (C1), 70.2 (C3), 67.8 (C5, C2, C4), 61.6 (C6), 51.9 (2 × OMe), 49.2 (C4′), 42.5 (C5′), 39.7 (C1′, C2′), 29.0 (C3′, C6′), 29.0(tBu-3 × Me), 20.6 (4 × OAc); LC-MS (Rt = 19.87 min) calcd for C29H43NO15S [M + Na]+m/z: 700.24; found m/z: 699.81.
Synthesis of the pseudo thio-disaccharide 25
Prepared from peracetylated thio-galactose 8 (51 mg, 0.13 mmol) and aziridine 4a (30 mg, 0.096 mmol) according to the general procedure and purified by flash chromatography (1:1 Hex:EtOAc) to give 25 as a mixture of α and β isomers in 44% (29 mg, 0.042 mmol) yield as colourless oil. The β and α isomers were separated by a second flash chromatography (8:1 iPr2O:EtOAc) to give pure β-25 in 34% yield (22 mg, 0.033 mmol). β-25: Rf = 0.33 (iPr2O/EtOAc 8:2); 1H NMR (400 MHz, CDCl3) δ 5.45 (dd, J4-3 = J4-5 = 3.4 Hz, 1H, H4), 5.24 (dd, J2-1 = J2-3 = 10.0 Hz, 1H, H2), 5.05 (dd, J3-2 = 10.0 Hz, J3-4 = 3.4 Hz, 1H, H3), 4.86 (m, 1H, NH), 4.69 (d, J1-2 = 10.0 Hz, 1H, H1), 4.13 (mult., 2H, H6a, H6b), 3.98 (m, 1H, H5), 3.87–3.77 (m, 1H, H4′), 3.70 (s, 3H, OMe), 3.69 (s, 3H, OMe), 3.39–3.28 (m, 1H, H5′), 3.02–2.93 (m, 1H, H1′), 2.81–2.69 (m, 1H, H2′), 2.17 (s, 3H, OAc), 2.14–2.09 (mult., 3H, H3′eq, H6′ax, H6′eq), 2.04 (s, 3H, OAc), 2.03 (s, 3H, OAc), 1.98 (s, 3H, OAc), 1.89–1.82 (m, 1H, H3′ax), 1.45 (s, 9H, tBu); 13C NMR (101 MHz, CDCl3) δ 174.0 (CO), 170.4 (CO), 170.4 (CO), 170.2 (CO), 170.1 (CO), 169.7 (CO), 84.4 (C1), 74.5 (C5), 72.0 (C3), 67.5 (C4), 67.3 (C2), 61.3 (C6), 52.3 (OMe), 52.2 (OMe), 49.7 (C4′), 43.4 (C5′), 40.0 (C2′, C1′), 29.8 (C3′, C6′), 28.5 (tBu-3 × Me), 20.9 (OAc), 20.8 (OAc), 20.7 (OAc), 20.7 (OAc); LC-MS (Rt = 19.51 min) calcd for C29H43NO15S [M + Na]+m/z: 700.24; found m/z: 699.74; MS (HRMS): calcd for C29H43NO15S [M + Na]+m/z: 700.2251; found m/z: 700.2259.
Synthesis of the pseudo thio-disaccharide 26
Prepared from peracetylated thio-lactose 9 (56 mg, 0.080 mmol) and aziridine 4a (19 mg, 0.062 mmol) according to the general procedure and purified by flash chromatography (3:2 Hex:EtOAc) to give 26 as a 5:1 β:α anomeric mixture in 42% yield (25 mg, 0.026 mmol) as colourless oil. Rf = 0.14 (Hex/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) β-26 5.35–5.33 (m, 1H, H10), 5.21 (dd, J3-4 = J3-2 = 9.2 Hz, 1H, H3), 5.10 (dd, J8-9 = 10.4 Hz, J8-7 = 7.9 Hz, 1H, H8), 5.02–4.97 (m, 1H, H2), 4.95 (dd, J9-8 = 10.4 Hz, J9-10 = 3.2 Hz, 1H, H9), 4.86 (d, JNH-4′ = 6.9 Hz, 1H, NH), 4.67 (d, J1β-2β = 10.1 Hz, 1H, H1β), 4.49 (mult., 2H, H7, H12a), 4.15–4.01 (mult., 3H, H12b, H6a, H6b), 3.91–3.81 (mult., 3H, H11, H4, H5), 3.70 (s, 1H, H4′), 3.68 (2× s, 6H, 2 × OMe), 3.37–3.30 (m, 1H, H5′), 2.96–2.87 (m, 1H, H1′), 2.82–2.70 (m, 1H, H2′), 2.15 (s, 3H, OAc), 2.09 (s, 3H, OAc), 2.12–2.05 (mult., 3H, H3′eq, H6′ax, H6′eq), 2.06 (s, 6H, 2 × OAc), 2.04 (s, 9H, 3 × OAc), 1.96 (s, 3H, OAc), 1.89–1.81 (m, 1H, H3′ax), 1.44 (s, 9H, tBu), α-26δ 5.62 (d, J1α-2α = 5.7 Hz, 1H, H1α); β-2613C NMR (101 MHz, CDCl3) δ 174.7 (CO), 174.0 (CO), 170.5 (CO), 170.4 (CO), 170.3 (CO), 170.2 (CO), 169.8 (CO), 169.8 (CO), 169.2 (CO), 83.5 (C1), 76.8 (C5), 76.1 (C11), 74.0 (C3), 71.2 (C4), 70.8 (C9), 70.5 (C2), 69.2 (C8), 66.7 (C10), 62.1 (C12), 60.9 (C6), 52.3 (OMe), 52.2 (OMe), 49.7 (C4′), 43.1 (C5′), 40.1 (C1′, C2′), 32.1 (C6′), 29.8 (C3′), 28.5 (tBu-3 × Me), 21.0 (OAc), 20.9 (OAc), 20.9 (OAc), 20.9 (OAc), 20.8 (OAc), 20.8 (OAc), 20.6 (OAc); LC-MS (Rt = 20.56 min (β), 20.66 min (α)) calcd for C41H59NO23S [M + Na]+m/z: 988.32; found m/z: 987.62; MS (HRMS): calcd for C41H59NO23S [M + Na]+m/z: 988.3096; found m/z: 988.3090.
Synthesis of the pseudo thio-disaccharide 27
Prepared from peracetylated thio-sialic acid 1036 (68 mg, 0.12 mmol) and aziridine 4a (30 mg, 0.096 mmol) according to the general procedure and purified by flash chromatography (3:1 CH2Cl2:acetone) to give 27 as a single isomer in 56% yield (containing 17% glycal as estimated by NMR) as yellow waxy solid. Rf = 0.33 (CH2Cl2:acetone = 3:1); 1H NMR (400 MHz, CDCl3) δ 5.51–5.44 (m, 1H, H7), 5.43–5.36 (mult., 2H, H8, NH), 5.06 (d, J = 11.0 Hz, 1H, NH), 4.84 (td, J4-5 = J4-3ax = 11.0 Hz, J4-3eq = 4.5 Hz, 1H, H4), 4.28–4.18 (m, 1H, H9), 4.28–4.13 (mult., 2H, H9a, H9b), 4.02 (ddd, J5-NH = J5-4 = J5-6 = 11.0 Hz, 1H, H5), 3.91–3.86 (m, 1H, H6), 3.85 (s, 3H, COOMe), 3.73 (s, 3H, COOMe), 3.71 (s, 3H, COOMe), 3.60–3.51 (m, 1H, H4′), 3.13–3.07 (m, 1H, H5′), 3.07–3.01 (m, 1H, H1′), 2.91–2.84 (m, 1H, H2′), 2.70 (dd, J3eq-3ax = 12.8 Hz, J3eq-4 = 4.5 Hz, 1H, H3eq), 2.20 (s, 3H, OAc), 2.14 (s, 3H, OAc), 2.06 (s, 3H, OAc), 2.18–2.04 (mult., 4H, H3′eq, H3′ax, H6′eq, H6′ax), 2.03 (s, 3H, OAc), 2.00–1.89 (m, 1H, H3ax), 1.87 (s, 3H, NHAc), 1.47 (s, 9H, tBu); 13C NMR (HSQC) (101 MHz, CDCl3) δ 72.3 (C6), 68.2 (C4), 66.2 (C7), 66.0 (C8), 60.2 (C9), 51.3 (OMe), 50.6 (OMe), 50.5 (OMe), 47.4 (C5), 41.3 (C2′), 38.6 (C1′, C5′), 36.6 (C3), 29.6 (C3′, C6′), 27.1 (tBu-3 × Me), 21.8 (NHAc), 19.8 (Ac), 19.4 (Ac), 19.3 (2 × Ac); LC-MS (Rt = 17.99 min) calcd for C35H52N2O18S [M + Na]+m/z: 843.29; found m/z: 842.71; MS (HRMS) calcd for C35H52N2O18S [M + Na]+m/z: 843.2834; found m/z: 843.2820.
Synthesis of the pseudo thio-disaccharide 28
Prepared from peracetylated thio-N-acetylglucosamine 11 (34 mg, 0.083 mmol) and aziridine 4a (20 mg, 0.064 mmol) according to the general procedure and purified by flash chromatography (9:1 CH2Cl2:MeOH) to give 28 as an inseparable anomeric mixture in 19% yield (8 mg, 0.012 mmol) as yellow waxy solid. Rf = 0.08 (Hex/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) β-28δ 5.63 (d, JNH-2 = 9.3 Hz, 1H, NH), 5.18–5.09 (mult., 2H, H3, H4), 4.92–4.84 (m, 1H, NH), 4.75 (d, J1β-2β = 10.4 Hz, 1H, H1β), 4.40–4.34 (m, 1H, H2), 4.26 (dd, J6a-6b = 12.1 Hz, J6a-5 = 4.3 Hz, 1H, H6a), 4.18–4.13 (m, 1H, H6b), 3.92–3.79 (m, 1H, H4′), 3.76–3.74 (m, 1H, H5), 3.71 (s, 3H, COOMe), 3.68 (s, 3H, COOMe), 3.35–3.30 (m, 1H, H5′), 2.97–2.90 (m, 1H, H1′), 2.75–2.68 (m, 1H, H2′), 2.13–2.01 (mult., 3H, H3′eq, H6′ax, H6′eq), 2.09 (s, 3H, OAc), 2.04 (s, 3H, OAc), 2.03 (s, 3H, OAc), 2.02 (s, 3H, OAc), 1.98 (s, 3H, OAc), 1.88–1.83 (m, 1H, H3′ax) 1.45 (s, 9H, Boc); α-28δ 5.52 (d, J1α-2α = 5.2 Hz, 1H, H1α); 13C NMR (HSQC) (101 MHz, CDCl3) δ 67.9 (C3), 73.7 (C4), 85.2 (C1), 68.0 (C2), 61.3 (C6), 52.1 (OMe), 52.0 (OMe), 49.0 (C4′), 41.0 (C5′, C2′, C1′), 29.1 (C6′), 28.6 (C3′), 28.4 (tBu-3 × Me), 23.1 (NHAc), 20.8 (OAc), 20.6 (OAc), 20.7 (OAc); LC-MS (Rt = 16.67 min (β), 17.33 min (α)) calcd for C29H44N2O14S [M + Na]+m/z: 699.25; found m/z: 698.75; MS (HRMS) calcd for C29H44N2O14S [M + Na]+m/z: 699.2411; found m/z: 699.2413.
Synthesis of the pseudo-thio-dimannoside 5c
Prepared from peracetylated thio-mannose 1 (46 mg, 0.11 mmol) and aziridine 4c (30 mg, 0.090 mmol) according to the general procedure described for the synthesis of 5a and purified by flash chromatography (1:1 Hex:EtOAc) to give 5c in 96% yield (60 mg, 0.086 mmol). Rf = 0.2 (Hex/EtOAc 1:1); [α]19D (CHCl3, c 1.22): +55; 1H NMR (400 MHz, CDCl3): δ 5.96–5.89 (m, 1H, NH), 5.41 (d, J1-2 = 1.5 Hz, 1H, H1), 5.34 (dd, J2-3 = 3.5 Hz, J2-1 = 1.5 Hz, 1H, H2), 5.29 (t, J4-3 = J4-5 = 9.7 Hz, 1H, H4), 5.20 (dd, J3-4 = 9.7 Hz, J3-2 = 3.5, 1H, H3), 4.42–4.30 (mult., 2H, H5, H6a), 4.21–4.07 (mult., 2H, H4′, H6b), 3.72 (s, 3H, OMe), 3.70, (s, 3H, OMe), 3.58 (AB system, Japp = 10.7 Hz, 2H, CH2Cl), 3.33–3.27 (m, 1H, H5′), 3.08 (ddd, J1′-2′ = 13.5 Hz, J1′-6′ax = 8.8 Hz, J1′-6′eq = 5.34 Hz, 1H, H1′), 2.87–2.78 (m, 1H, H2′), 2.23 (ddd, J3′eq-3′ax = 14.1 Hz, J3′eq-2′ = 10 Hz, J3′eq-4′ = 4.4 Hz, 1H, H3′eq), 2.18–1.94 (mult., 2H, H6′eq, H6′ax), 2.15 (s, 3H, OAc), 2.07 (s, 3H, OAc), 2.04 (s, 3H, OAc), 1.97 (s, 3H, OAc), 1.92–1.82 (m, 1H, H3′ax), 1.28 (s, 3H, CH3 linker), 1.27 (s, 3H, CH3 linker); 13C NMR (100 MHz, CDCl3): δ 174.34 (CO), 174.22 (CO), 173.56 (CO), 170.76 (CO), 169.94 (CO), 169.83 (CO), 169.72 (CO), 81.80 (C1), 70.95 (C2), 69.52 (C3), 69.44 (C5), 66.26 (C4), 62.59 (C6), 60.4 (C(CH3)2 linker), 52.82 (CH2 linker), 52.42 (OMe), 52.29 (OMe), 48.16 (C4′), 43.67 (C5′), 40.21 (C1′), 40.03 (C2′), 29.35 (C6′), 28.76 (C3′), 23.63 (CH3 linker), 23.37 (CH3 linker), 20.96 (OAc), 20.77 (OAc), 20.73 (OAc), 20.66 (OAc); MS (ESI) calcd for C29H42ClNO14S [M + Na]+m/z: 718.19; found m/z: 718.6.
Product 5c (22 mg, 0.032 mmol) was deacetylated according to the general procedure for acetylation (reaction time 3 h). The deacetylated product was obtained in 62% (10.5 mg, 0.020 mmol) yield after purification. Rf = 0.31 (H2O/MeOH 1:1) [α]20D (MeOH, c 0.47): +110; 1H NMR (400 MHz, CD3OD): δ 5.34 (d, J1-2 = 1.2 Hz, 1H, H1), 4.1 (ddd, J4′-3′ax = 8.2 Hz, J4′-3′eq = 7.3 Hz, J4′-5′ = 4.2 Hz, 1H, H4′), 3.93–3.82 (mult., 3H, H5, H2, H6a), 3.77–3.56 (mult., 5H, H6b, H4, H3, CH2Cl), 3.72 (s, 3H, OMe), 3.71 (s, 3H, OMe), 3.22–3.15 (mult., 2H, H5′, H2′), 3.15–3.09 (m, 1H, H1′), 2.34 (ddd, J6′eq-6′ax = 14.4 Hz, J6′eq-1′ = 7.7 Hz, J6′eq-5′ = 3.6 Hz, 1H, H6′eq), 2.19–2.10 (m, 1H, H3′eq), 2.02 (ddd, J6′ax-6′eq = 14.2 Hz, J6′ax-5′ = 8.1 Hz, J6′ax-1′ = 4.3 Hz, 1H, H6′ax), 1.88 (ddd, J3′ax-3′eq = 14.2 Hz, J3′ax-4′ = 7.7 Hz, J3′ax-2′ = 4.7 Hz, 1H, H3′ax), 1.28 (s, 6H, 2 × CH3 linker); 13C NMR (100 MHz, CD3OD): δ 177.2 (CO), 175.7 (CO), 175.6 (CO), 85.3 (C1), 75.5 (C5), 73.9 (C2), 73.3 (C3), 68.9 (C4), 63.0 (C6), 53.5 (CH2Cl), 52.8 (OMe), 52.7 (OMe), 49.9 (C4′), 45.1 (C5′), 42.0 (C1′), 41.6 (C2′), 31.4 (C6′), 30.5 (C3′), 24.0 (CH3 linker), 23.8 (CH3 linker); HR-MS (ESI) calcd for C21H34ClNO10S [M + Na] + m/z: 550.1490; found: 550.1495.
Synthesis of the pseudo-thio-dimannoside 5d
Thioacetate 1 (78 mg, 0.19 mmol) and aziridine 4d (50 mg, 0.15 mmol) were dissolved in dry DMF (250 μl, 0.6 M) into a MW-reactor. After addition of Et2NH (30μl, 0.28 mmol), the reaction was stirred at 60 °C under MW irradiation for 1 h. After that, the mixture was diluted with EtOAc and washed with 1 M HCl. The aqueous phase was extracted twice with EtOAc and finally the organic layers collected were dried over Na2SO4, filtered and the solvents removed under vacuum. Compound 5d was isolated through flash chromatography (1:1 Hex:EtOAc) in 68% yield (72 mg, 0.10 mmol). Rf = 0.27 (Hex/EtOAc 1:1); [α]18D (CHCl3, c 1.55): +69; 1H NMR (400 MHz, CDCl3): δ 6.09–5.99 (m, 1H, NH), 5.41 (d, J1-2 = 1.1 Hz, 1H, H1), 5.33 (dd, J2-3 = 3.3 Hz, J2-1 = 1.1 Hz, 1H, H2), 5.29 (t, J4-3 = J4-5 = 10.0 Hz, 1H, H4), 5.20 (dd, J3-4 = 10.0 Hz, J3-2 = 3.3 Hz, 1H, H3), 4.42–4.29 (mult., 2H, H5, H6a), 4.18–4.08 (mult., 2H, H4′, H6b), 3.72 (s, 3H, OMe), 3.71 (s, 3H, OMe), 3.40 (AB system, Japp = 15.0 Hz, 2H, CH2N3), 3.29–3.23 (m, 1H, H5′), 3.08 (ddd, J1′-2′ = 12.6 Hz, J1′-6′ax = 8.8 Hz, J1′-6′eq = 4.2 Hz, 1H, H1′), 2.90–2.80 (m, 1H, H2′), 2.28–2.18 (m, H3′eq), 2.18–2.00 (mult., 2H, H6′eq, H6′ax), 2.15 (s, 3H, OAc), 2.07 (s, 3H, OAc), 2.04 (s, 3H, OAc), 1.97 (s, 3H, OAc), 1.92–1.80 (m, 1H, H3′ax), 1.20 (2× s, 6H, 2 × CH3 linker); 13C NMR (100 MHz, CDCl3): δ 175.0 (CO), 174.2 (CO), 173.5 (CO), 170.7 (CO), 169.9 (CO), 169.8 (CO), 169.7 (CO), 81.8 (C1), 71.0 (C2), 69.5 (C3), 69.5 (C5), 66.3 (C4), 62.6 (C6), 60.0 (CH2N3), 52.4 (OMe), 52.3 (OMe), 48.0 (C4′), 43.9 (C5′), 40.2 (C1′), 40.1 (C2′), 29.5 (C6′), 28.9 (C3′), 23.4 (CH3 linker), 23.4 (CH3 linker), 20.1 (OAc), 20.7 (OAc), 20.7 (OAc), 20.7 (OAc); MS (ESI) calcd for C29H42N4O14 [M + Na]+m/z: 725.23; found m/z: 725.66.
Product 5d (31 mg, 0.044 mmol) was deacetylated according to the general procedure for deacetylation (reaction time 3 h). Deacetylated product was obtained in 68% (16 mg, 0.030 mmol) yield after purification. Rf = 0.21 (CH2Cl2/MeOH 9:1); [α]16D (MeOH, c 0.55): +105; 1H NMR (400 MHz, CD3OD): δ 5.34 (d, J1-2 = 1.2 Hz, 1H, H1), 4.1 (ddd, J4′-3′ax = 8.1 Hz, J4′-3′eq = 7.7 Hz, J4′-5′ = 4.1 Hz, 1H, H4′), 3.93–3.82 (mult., 3H, H5, H2, H6a), 3.78–3.67 (m, 1H, H6b), 3.72 (s, 3H, OMe), 3.72 (s, 3H, OMe), 3.67–3.57 (mult., 2H, H4, H3), 3.44 (AB system, Japp = 15.7 Hz, 2H, CH2N3), 3.22–3.08 (mult., 3H, H5′, H1′, H2′), 2.34 (ddd, J6′eq- 6′ax = 14.3 Hz, J6′eq-1′ = 7.7 Hz, J6′eq-5′ = 3.8 Hz, 1H, H6′eq), 2.15 (ddd, J3′eq-3′ax = 14 Hz, J3′eq-2′ = 7 Hz, J3′eq-4′ = 4 Hz, 1H, H3′eq), 2.02 (ddd, J6′ax-6′eq = 14.3 Hz, J6′ax-5′ = 7.9 Hz, J6′ax-1′ = 4.4 Hz, 1H, H6′ax), 1.88 (ddd, J3′ax-3′eq = 14.1 Hz, J3′ax-4′ = 7.8 Hz, J3′ax-2′ = 4.6 Hz, 1H, H3′ax), 1.21 (s, 6H, 2× CH3 linker); 13C NMR (100 MHz, CD3OD): δ 177.9 (CO), 175.6 (CO), 175.6 (CO), 85.3 (C1), 75.5 (C5), 73.9 (C2), 73.3 (C3), 68.9 (C4), 63.0 (C6), 61.1 (CH2N3), 52.7 (2 × OMe), 49.5 (C4′), 45.2 (C5′), 42.0 (C1′), 41.6 (C2′), 31.4 (C6′), 30.5 (C3′), 23.8 (CH3 linker), 23.5 (CH3 linker); HR-MS (ESI) calcd for C21H34N4O10S [M + Na] + m/z: 557.1893; found: 557.1899.
Synthesis of the rhamnosyl aminoacid 33
Compound 23 (58 mg, 0.093 mmol) was dissolved in CH2Cl2 (1.5 ml) and then TFA (0.4 ml) was added. Reaction mixture was kept stirring at RT for 1 h and then concentrated under vacuum. Residue was co-evaporated with toluene 3×. The crude was dissolved in CH3CN (1.9 ml) and N-methylmorpholine (0.010 ml, 0.093 mmol), amino acid 32 (from Fluorochem, 40 mg, 0.093 mmol) and HATU (35 mg, 0.093 mmol) were added. Reaction mixture was kept stirring overnight. Then the reaction mixture was concentrated, dissolved in CHCl3 and washed with water, sat. KHSO4, water, sat. NaHCO3 and water. Combined aqueous phases were additionally washed with CHCl3 and combined organic phases were dried over Na2SO4 and concentrated under reduced pressure. The crude was further purified by flash chromatography (1:1 Hex:EtOAc) to give 33 in 92% yield (79 mg, 0.086 mmol) as colourless oil. Rf = 0.25 (Hex/EtOAc 1:1); [α]23D (CHCl3, c 1.00): −37; 1H NMR (400 MHz, CDCl3) δ 7.76 (d, J = 7.4 Hz, 2H, CH-Ar-Fmoc), 7.62 (d, J = 7.5 Hz, 2H, CH-Ar-Fmoc), 7.45–7.35 (m, 2H, CH-Ar-Fmoc), 7.36–7.26 (m, 2H, CH-Ar-Fmoc), 6.97 (d, J = 6.7 Hz, 1H, NH), 5.64 (d, J = 8.0 Hz, 1H, NH), 5.35 (s, 2H, H1, H2), 5.16 (dd, J3-4 = 10 Hz, J3-2 = 2.9 Hz, 1H, H3), 5.09 (dd, J4-3 = J4-5 = 10 Hz, 1H, H4), 4.50–4.40 (mult., 2H, CH2-Fmoc), 4.28–4.14 (mult., 4H, CH-Fmoc, H5, CH-Glu, H4′), 3.65 (s, 3H, COOMe), 3.63 (s, 3H, COOMe), 3.37–3.32 (m, 1H, C5′), 3.07–2.96 (mult., 2H, C2′, C1′), 2.30–1.82 (mult., 8H, C3′, C6′, CH2-Glu, CH2-Glu), 2.12 (s, 3H, OAc), 2.06 (s, 3H, OAc), 1.98 (s, 3H, OAc), 1.47 (s, 9H, tBu), 1.23 (d, JCH3-5 = 6.2 Hz, 3H, CH3-Rha); 13C NMR (101 MHz, CDCl3) δ 174.7 (CO), 174.3 (CO), 171.9 (CO), 171.2 (CO), 170.2 (CO), 170.1 (CO), 128.0 (CH-Ar-Fmoc), 127.3 (CH-Ar-Fmoc), 125.3 (CH-Ar-Fmoc), 120.2 (CH-Ar-Fmoc), 83.5 (C1), 71.6 (C2), 71.3 (C4), 69.6 (C3), 67.7 (C5), 67.4 (CH2-Fmoc), 53.9 (CH-Glu), 52.3 (COOMe), 52.2 (COOMe), 48.7 (C4′), 47.4 (CH-Fmoc), 45.1 (C5′), 40.1 (C1′), 39.8 (C2′), 33.0 (CH2-Glu), 32.1 (CH2-Glu), 29.9 (C3′, C6′), 28.2 (tBu-3 × Me), 21.1 (OAc), 21.0 (OAc), 20.8 (OAc), 17.3 (CH3-Rha); MS (HRMS) calcd for C46H58N2O16S [M + Na]+m/z: 949.3405; found m/z: 949.3400.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 765581 (PhD4GlycoDrug).Notes and references
- B. Ernst and J. L. Magnani, Nat. Rev. Drug Discovery, 2009, 8, 661 CrossRef CAS.
- A. Tamburrini, C. Colombo and A. Bernardi, Med. Res. Rev., 2020, 40, 495–531 CrossRef CAS.
- M. von Itzstein, Nat. Rev. Drug Discovery, 2007, 6, 967–974 CrossRef CAS.
- H. C. Kolb and B. Ernst, Chem. – Eur. J., 1997, 3, 1571–1578 CrossRef CAS.
- J. Egger, C. Weckerle, B. Cutting, O. Schwardt, S. Rabbani, K. Lemme and B. Ernst, J. Am. Chem. Soc., 2013, 135, 9820–9828 CrossRef CAS.
- A. Bernardi, A. Checchia, P. Brocca, S. Sonnino and F. Zuccotto, J. Am. Chem. Soc., 1999, 121, 2032–2036 CrossRef CAS.
- V. Porkolab, E. Chabrol, N. Varga, S. Ordanini, I. Sutkevičiūtė, M. Thépaut, M. J. García-Jiménez, E. Girard, P. M. Nieto, A. Bernardi and F. Fieschi, ACS Chem. Biol., 2018, 13, 600–608 CrossRef CAS.
- L. Medve, S. Achilli, J. Guzman-Caldentey, M. Thépaut, L. Senaldi, A. Le Roy, S. Sattin, C. Ebel, C. Vivès, S. Martin-Santamaria, A. Bernardi and F. Fieschi, Chem. – Eur. J., 2019, 25, 14659–14668 CrossRef CAS.
- D. Hauck, I. Joachim, B. Frommeyer, A. Varrot, B. Philipp, H. M. Möller, A. Imberty, T. E. Exner and A. Titz, ACS Chem. Biol., 2013, 8, 1775–1784 CrossRef CAS.
- R. Sommer, S. Wagner, K. Rox, A. Varrot, D. Hauck, E.-C. Wamhoff, J. Schreiber, T. Ryckmans, T. Brunner, C. Rademacher, R. W. Hartmann, M. Brönstrup, A. Imberty and A. Titz, J. Am. Chem. Soc., 2018, 140, 2537–2545 CrossRef CAS.
- E.-C. Wamhoff, J. Schulze, L. Bellmann, M. Rentzsch, G. Bachem, F. F. Fuchsberger, J. Rademacher, M. Hermann, B. Del Frari, R. van Dalen, D. Hartmann, N. M. van Sorge, O. Seitz, P. Stoitzner and C. Rademacher, ACS Cent. Sci., 2019, 5, 808–820 CAS.
- S. V. Shelke, B. Cutting, X. Jiang, H. Koliwer-Brandl, D. S. Strasser, O. Schwardt, S. Kelm and B. Ernst, Angew. Chem., Int. Ed., 2010, 49, 5721–5725 CrossRef CAS.
- S. Ng, N. J. Bennett, J. Schulze, N. Gao, C. Rademacher and R. Derda, Bioorg. Med. Chem., 2018, 26, 5368–5377 CrossRef CAS.
- L. Medve, S. Achilli, S. Serna, F. Zuccotto, N. Varga, M. Thépaut, M. Civera, C. Vivès, F. Fieschi, N. Reichardt and A. Bernardi, Chem. – Eur. J., 2018, 24, 14448–14460 CrossRef CAS.
- J. Schulze, H. Baukmann, R. Wawrzinek, F. F. Fuchsberger, E. Specker, J. Aretz, M. Nazaré and C. Rademacher, ACS Chem. Biol., 2018, 13, 3229–3235 CrossRef CAS.
- A. Brinkø, C. Risinger, A. Lambert, O. Blixt, C. Grandjean and H. H. Jensen, Org. Lett., 2019, 21, 7544–7548 CrossRef.
- A. Tamburrini, S. Achilli, F. Vasile, S. Sattin, C. Vivès, C. Colombo, F. Fieschi and A. Bernardi, Bioorg. Med. Chem., 2017, 25, 5142–5147 CrossRef CAS.
- A. Tamburrini, PhD Thesis, Università degli studi di Milano, 2019.
- A. Helenius and M. Aebi, Annu. Rev. Biochem., 2004, 73, 1019–1049 CrossRef CAS.
- C.-S. Chao, M.-C. Chen, S.-C. Lin and K.-K. T. Mong, Carbohydr. Res., 2008, 343, 957–964 CrossRef CAS.
- S.-J. Zhu, H.-Z. Ying, Y. Wu, N. Qiu, T. Liu, B. Yang, X.-W. Dong and Y.-Z. Hu, RSC Adv., 2015, 5, 103172–103183 RSC.
- G. Yang, R. W. Franck, R. Bittman, P. Samadder and G. Arthur, Org. Lett., 2001, 3, 197–200 CrossRef CAS.
- S. Mari, H. Posteri, G. Marcou, D. Potenza, F. Micheli, F. J. Cañada, J. Jimenez-Barbero and A. Bernardi, Eur. J. Org. Chem., 2004, 5119–5225 CrossRef CAS.
- J. L. Jat, M. P. Paudyal, H. Gao, Q.-L. Xu, M. Yousufuddin, D. Devarajan, D. H. Ess, L. Kürti and J. R. Falck, Science, 2014, 343, 61–65 CrossRef CAS.
- A. Mordini, F. Russo, M. Valacchi, L. Zani, A. Degl'Innocenti and G. Reginato, Tetrahedron, 2002, 58, 7153–7163 CrossRef CAS.
- M. Punk, C. Merkley, K. Kennedy and J. B. Morgan, ACS Catal., 2016, 6, 4694–4698 CrossRef CAS.
- T. Mita and E. N. Jacobsen, Synlett, 2009, 1680–1684 CAS.
- M. Senatore, A. Lattanzi, S. Santoro, C. Santi and G. D. Sala, Org. Biomol. Chem., 2011, 9, 6205–6207 RSC.
- A. Bernardi, D. Arosio, L. Manzoni, F. Micheli, A. Pasquarello and P. Seneci, J. Org. Chem., 2001, 66, 6209–6216 CrossRef CAS.
- J. Jiang, M. Artola, T. J. M. Beenakker, S. P. Schröder, R. Petracca, C. de Boer, J. M. F. G. Aerts, G. A. van der Marel, J. D. C. Codée and H. S. Overkleeft, Eur. J. Org. Chem., 2016, 3671–3678 CrossRef CAS.
- P. Serrano, A. Llebaria, J. Vázquez, J. de Pablo, J. M. Anglada and A. Delgado, Chem. – Eur. J., 2005, 11, 4465–4472 CrossRef CAS.
- T. Belz and S. J. Williams, Carbohydr. Res., 2016, 429, 38–47 CrossRef CAS.
- A. Ané, S. Josse, S. Naud, V. Lacône, S. Vidot, A. Fournial, A. Kar, M. Pipelier and D. Dubreuil, Tetrahedron, 2006, 62, 4784–4794 CrossRef.
- H. Hashimoto, K. Shimada and S. Horito, Tetrahedron: Asymmetry, 1994, 5, 2351–2366 CrossRef CAS.
- P. Shu, J. Zeng, J. Tao, Y. Zhao, G. Yao and Q. Wan, Green Chem., 2015, 17, 2545–2551 RSC.
- G. R. Morais, I. F. Oliveira, A. J. Humphrey and R. A. Falconer, Carbohydr. Res., 2010, 345, 160–162 CrossRef CAS.
- U. Dabrowski, H. Friebolin, R. Brossmer and M. Supp, Tetrahedron Lett., 1979, 20, 4637–4640 CrossRef.
- H. Hori, T. Nakajima, Y. Nishida, H. Ohrui and H. Meguro, Tetrahedron Lett., 1988, 29, 6317–6320 CrossRef CAS.
- L. Deng, X. Wang, S. Uppalapati, O. Norberg, H. Dong, A. Joliton, M. Yan and O. Ramström, Pure Appl. Chem., 2013, 85, 1789–1801 CAS.
- R. Caraballo, L. Deng, L. Amorim, T. Brinck and O. Ramström, J. Org. Chem., 2010, 75, 6115–6121 CrossRef CAS.
- S. Josse, J. Le Gal, M. Pipelier, J. Cléophax, A. Olesker, J.-P. Pradère and D. Dubreuil, Tetrahedron Lett., 2002, 43, 237–239 CrossRef CAS.
- M. Adinolfi, D. Capasso, S. Di Gaetano, A. Iadonisi, L. Leone and A. Pastore, Org. Biomol. Chem., 2011, 9, 6278–6283 RSC.
- L. M. Doyle, S. O'Sullivan, C. Di Salvo, M. McKinney, P. McArdle and P. V. Murphy, Org. Lett., 2017, 19, 5802–5805 CrossRef CAS.
- W. Pilgrim and P. V. Murphy, J. Org. Chem., 2010, 75, 6747–6755 CrossRef CAS.
- E. Juaristi and G. Cuevas, Tetrahedron, 1992, 48, 5019–5087 CrossRef CAS.
- R. T. C. Sheridan, J. Hudon, J. A. Hank, P. M. Sondel and L. L. Kiessling, ChemBioChem, 2014, 15, 1393–1398 CrossRef CAS.
- P. Karmakar, K. Lee, S. Sarkar, K. A. Wall and S. J. Sucheck, Bioconjugate Chem., 2016, 27, 110–120 CrossRef CAS.
- M. Herczeg, E. Mező, N. Molnár, S.-K. Ng, Y.-C. Lee, M. Dah-Tsyr Chang and A. Borbás, Chem. – Asian J., 2016, 11, 3398–3413 CrossRef CAS.
- M. K. Hossain, A. Vartak, P. Karmakar, S. J. Sucheck and K. A. Wall, ACS Chem. Biol., 2018, 13, 2130–2142 CrossRef CAS.
- B. Liet, E. Laigre, D. Goyard, B. Todaro, C. Tiertant, D. Boturyn, N. Berthet and O. Renaudet, Chem. – Eur. J., 2019, 25, 15508–15515 CrossRef CAS.
- J. Alsarraf, P. Gormand, J. Legault, M. Mihoub and A. Pichette, Tetrahedron Lett., 2020, 61, 151706 CrossRef CAS.
- A. Bernardi and S. Sattin, Eur. J. Org. Chem., 2020, 4652–4663 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob01956a |
| This journal is © The Royal Society of Chemistry 2021 |