
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Designed membrane protein heterodimers and control of their affinity by binding domain and membrane linker properties†
Chenyang
Lan
abc,
Anja
Stulz
d,
Nicolas P. F.
Barthes
 d,
Susan
Lauw
ef,
Pavel
Salavei
ef,
Manfred
Jung
cd,
Heiko
Heerklotz
cdeg and
Maximilian H.
Ulbrich
*eh
d,
Susan
Lauw
ef,
Pavel
Salavei
ef,
Manfred
Jung
cd,
Heiko
Heerklotz
cdeg and
Maximilian H.
Ulbrich
*eh
aFaculty of Biology, University of Freiburg, Germany
bInstitute of Physical Chemistry, University of Freiburg, Germany
cCIBSS Centre for Integrative Biological Signalling Studies, University of Freiburg, Germany
dInstitute of Pharmaceutical Sciences, University of Freiburg, Germany
eBIOSS Centre for Biological Signalling Studies, University of Freiburg, Germany. E-mail: max.ulbrich@bioss.uni-freiburg.de
fCore Facility Signalling Factory & Robotics, University of Freiburg, Germany
gLeslie Dan Faculty of Pharmacy, University of Toronto, Germany
hInternal Medicine IV, University of Freiburg Medical Center and Faculty of Medicine, University of Freiburg, Germany
First published on 8th December 2021
Abstract
Many membrane proteins utilize dimerization to transmit signals across the cell membrane via regulation of the lateral binding affinity. The complexity of natural membrane proteins hampers the understanding of this regulation on a biophysical level. We designed simplified membrane proteins from well-defined soluble dimerization domains with tunable affinities, flexible linkers, and an inert membrane anchor. Live-cell single-molecule imaging demonstrates that their dimerization affinity indeed depends on the strength of their binding domains. We confirm that as predicted, the 2-dimensional affinity increases with the 3-dimensional binding affinity of the binding domains and decreases with linker lengths. Models of extended and coiled linkers delineate an expected range of 2-dimensional affinities, and our observations for proteins with medium binding strength agree well with the models. Our work helps in understanding the function of membrane proteins and has important implications for the design of synthetic receptors.
Introduction
Most membrane proteins laterally interact with other membrane proteins. In contrast to ion channels that form rigid multi-subunit structures spanning the extracellular, transmembrane, and intracellular regions, proteins that use a shift in the monomer/dimer equilibrium as a tool to transduce signals across the membrane, e.g. receptor tyrosine kinases, often have interaction domains that are well separated by flexible connections. In this way, the propensity to form dimers can be regulated by soluble extra- or intracellular ligands, or a change in the membrane composition.1,2 Effectively, the regulation is a change of the molecules’ 2-dimensional affinity for each other. For several proteins, this 2-dimensional affinity on the cell surface, and the influence of regulators, ligands and mutations are measured in cells or artificial bilayers.3–7 Although quantifying a protein's properties is of essential relevance for understanding its physiology, it often remains elusive how its structure shapes the biophysics of the lateral interactions because the interplay of the subdomains and conformational changes inside the protein are difficult to assess.Therefore, we set out to design a simple model system for the heterodimerization of two membrane proteins that mimic the lateral interaction of naturally occurring membrane proteins. We chose a modular design with exchangeable components that carry the functionalities of dimerization domains, membrane anchors, and fluorescent markers for microscopy-based readout. A leucine zipper pair, which is a well-defined, α-helical protein interaction motif, mediates the dimerization, and the membrane anchor is a single-pass transmembrane domain. The markers were green and red fluorescent fusion tags that we used to visualize the interacting proteins in the plasma membrane of living cells by single-molecule imaging using total internal reflection fluorescence microscopy. With a low density of the proteins in the membrane, heterodimers appeared as yellow spots, whereas monomers were green and red spots.
We found that with a 47 amino acid (aa) long leucine zipper, the affinity of the interaction was high, resulting in a fraction of yellow spots comparable to a construct with green and red tags in the same protein. When shortening the leucine-zipper domains, the equilibrium shifted towards an increasing fraction of monomers, resulting in less yellow spots and more green and red spots. In analogy to a titration assay, we measured the fraction of dimers from cells with different densities of protein molecules in the membrane, and obtained 2-dimensional dissociation constants (a smaller 2-dimensional dissociation constant is equivalent to stronger binding). Extending the linker regions between the membrane anchor (the transmembrane domain) and the dimerization domain reduced the binding affinity.
In a biophysical model, the length of the linker between the membrane and binding domain determines the binding domain's local concentration in the vicinity of the membrane. The 2-dimensional affinity depends linearly on the affinity of the soluble binding domain and the linker length. For experimentally testing this relationship, we first constructed soluble versions of the binding domains and measured their affinities by isothermal titration calorimetry (ITC). Since we cannot determine the linker length directly, we established two different models that suggest upper and lower limits for the possible range. In the model that gives the upper limit for the linker length, the binding domain can freely move in volume given by the fully extended linker. The lower limit for the linker length is modeled by a chain with randomly oriented links; then the linker mostly resides in a contracted state, and its effective length scales with the square root of the number of its residues. For the proteins with shortened leucine-zipper domains, the measured 2-dimensional affinities agree well with the range predicted by the models. For the proteins with full-length binding domains, the binding is 20- to 100-fold weaker than that predicted by the models.
Results
Modular design of a transmembrane heterodimer
Leucine zippers are well-defined motifs for multimerization and are easy to manipulate due to their simple alpha-helical structure. In a previous study, 22 synthetic leucine-zipper domains, designed to form heterodimers, were characterized extensively.8,9 From this study, we selected a 47 aa long leucine zipper pair called “SYNZIP1′′ and “SYNZIP2”, which showed the best combination of affinity and specificity, and also had no propensity to homodimerize when expressed alone (ESI Note 1†). As a transmembrane anchor, we chose the transmembrane domain of the platelet-derived growth factor receptor alpha (PDGFR), which forms a helix from mainly hydrophobic residues, does not contain identified dimerization motifs, and has previously been used in a scaffold for sensitive readout and characterization of membrane protein dimerization.10,11 As we will see later, it remains monomeric also in our hands.Using single-molecule imaging as a readout imposes the requirements of high efficiency and low background on the labeling approach. In the fluorescent proteins we had tested so far, only intracellularly fused GFP satisfied these conditions.12,13 Since we needed a second fluorescent marker in a different wavelength range, we resorted to the extracellular SNAP-tag, which we labeled with a fluorescent substrate that consists of the SNAP-tag binding moiety benzylguanine covalently bound to the orange-red organic dye DY-549P1 (BG-DY549) (ESI Note 2†). The GFP version we used was monomeric EGFP, which has virtually no propensity to dimerize.14 Likewise, the SNAP-tag has also not been reported to form dimers or clusters. In a test alongside with many other fluorescent SNAP-tag substrates, BG-DY549 had been demonstrated to display the lowest non-specific background labeling and virtually no membrane permeation.15 In the following, to simplify the names of the constructs, we will use the abbreviation S549 for the SNAP-tag after labeling with the BG-DY549 substrate.
For the final design of our two proteins, we added short flexible linker sequences between the modules, and to maintain symmetry, an extracellular (unlabeled) HALO-tag to the GFP-containing construct, and a non-fluorescent GFP (Y66L mutant) to the intracellular side of the SNAP-tag construct (Fig. 1A). The placement of the SNAP-tag, HALO-tag and leucine zipper domains in the extracellular space was achieved by signal peptides; their effectiveness was predicted computationally by the prediction tool SignalP-5.0.16 The first protein we will use for the heterodimerization experiment will therefore consist (from N- to C-terminus) of the signal peptide, an unlabeled HALO-tag, SYNZIP1, the transmembrane domain of PDGFR, and GFP (HALO-SZ1-TM-GFP); the second protein will contain the signal peptide, the labeled SNAP-tag, SYNZIP2, the transmembrane domain of PDGFR, and a non-fluorescent GFP (S549-SZ2-TM-xGFP).
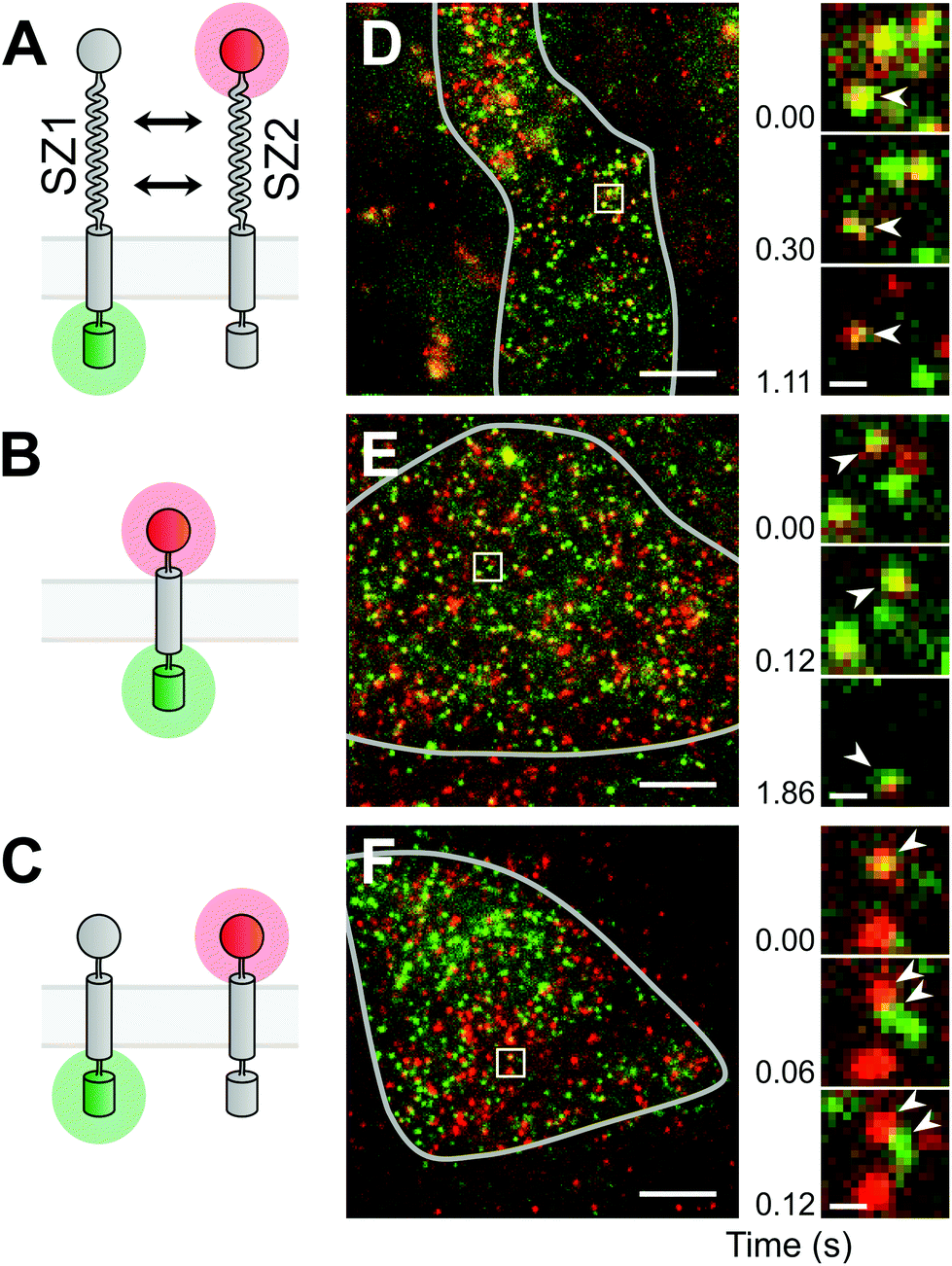 | ||
| Fig. 1 Single molecule imaging of a designed membrane protein interaction. (A) The designed proteins consist of the extracellular SZ1 and SZ2 binding domains, a membrane anchor and green (GFP) or red (S549) reporter tags. (B) A constitutive heterodimer was mimicked by directly fused green and red tags. (C) Omitting the binding domains yields a mix of monomers. (D) Representative image from a cell expressing the designed heterodimer pair. The grey line marks the area of the attached cell. Bright spots are single molecules of the green and red labeled proteins. The boxed region is magnified at different time points of the movie, as indicated by the time index. Arrowheads mark a long lasting yellow spot. (E) Image from the constitutive heterodimer mimic. Arrowheads mark a long lasting yellow spot. (F) Image from the monomer mix. Arrowheads mark a yellow spot that quickly separated into green and red spots. Scale bars: 5 μm (full view), 500 nm (magnified view). | ||
Single-molecule imaging of synthetic membrane proteins in living cells
Before measuring the dimer formation of the newly designed SYNZIP-based heterodimer constructs, we should give thought to the results expected from a single-molecule experiment in a living cell. The formation of heterodimers between GFP- and S549-labeled constructs should ideally result in the appearance of diffusing fluorescent spots at the plasma membrane that emit light in both the green and the red channel (referred to as yellow spots hereafter). However, we know from previous experiments and reports from other groups that a fraction of GFP tags is non-fluorescent, and labeling the SNAP-tag with a substrate usually does not proceed to completion; therefore, some heterodimers would appear as green spots without red fluorescence, or as red spots without green fluorescence. On the other hand, the coincidental encounter of an only-green fluorescent and an only-red fluorescent monomer would result in a yellow spot, although they do not interact. Therefore, as control experiments, we should first determine the maximal and minimal fractions of yellow spots we obtain when observing a pure heterodimer or a mix of pure monomers. To mimic the first condition of a pure heteromer, we used a fusion protein containing the SNAP-tag, the transmembrane domain of PDGFR, and GFP, and labeled it with the SNAP-tag substrate (S549-TM-GFP) (Fig. 1B). The second condition, the monomer mix, was achieved by co-expressing the two proteins designed above, but without the leucine-zipper dimerization domains, i.e. HALO-TM-GFP and S549-TM-xGFP (Fig. 1C).Single-molecule imaging was done using a custom built objective-type total internal reflection fluorescence (TIRF) microscopy setup.12 We simultaneously imaged GFP and S549 during illumination with a 488 nm and a 561 nm laser, and detected emission through a device that splits light into the green and the red channel. In CHO-K1 cells, transient expression of the constructs resulted in membrane densities ranging from below 0.1 μm−2 up to 100 μm−2 or more. We selected cells with a membrane density below 5 μm−2, where the labeled proteins were visible as diffusing fluorescent spots that could be well separated from each other (Fig. 1D–F). In the recorded movies of the heterodimer-mimicking S549-TM-GFP construct, we observed a majority of yellow spots, but also some green and red spots (Fig. 1E, ESI Movie 1†). In contrast, when co-expressing HALO-TM-GFP and S549-TM-xGFP, which should stay monomeric, we observed a large excess of green and red spots. We also observed a smaller fraction of yellow spots that quickly separated into green and red, suggesting that the molecules interacted only transiently or not at all (Fig. 1F, ESI Movie 2†).
After establishing the controls mimicking the pure heterodimer or a mix of two monomers, we imaged cells where we co-expressed the designed SYNZIP constructs HALO-SZ1-TM-GFP and S549-SZ2-TM-xGFP. We again observed a majority of yellow spots and smaller fractions of green and red spots (Fig. 1D, ESI Movie 3†), similar to the case of the heterodimer mimic. The large fraction of yellow spots and the sustained co-localization of green and red spots during diffusion confirmed that we successfully had designed a pair of heterodimerizing membrane proteins based on leucine zippers. Finally, as a control to exclude the possibility that SYNZIP1 or SYNZIP2 form dimers on their own, we also co-expressed HALO-SZ1-TM-GFP with S549-SZ1-TM-xGFP, or HALO-SZ2-TM-GFP with S549-SZ2-TM-xGFP, i.e. a SYNZIP1 pair in green and red, or a SYNZIP2 pair in green and red. In both cases, we observed a large excess of green-only and red-only spots, suggesting that there was no significant homodimerization of SYNZIP1 or SYNZIP2, consistent with results from previous studies.9
To quantify the degree of interaction, we counted the number of green, red and yellow spots (NG, NR, and NY) in a rectangular area of the cell surface that displayed an even distribution and similar number of red and green spots. As an initial measure for the degree of interaction, we used the term fD = 2NY/(2NY + NG + NR), which would describe the fraction of the fluorescent molecules bound in dimers, if the assumptions are true that no homo-dimers or higher order oligomers are present, and that there is no co-localization due to random encounters (i.e. without interaction) of multiple protein molecules. Obviously, random encounters can occur, and the latter assumption is usually not met, which should lead to an offset fD > 0 also in the case of non-interacting proteins. In addition, we manually tracked individual yellow spots and determined the time tD the green and red spots stayed co-localized.
For the heterodimer mimic control, we obtained fD = 55.6 ± 5.4% (s.e.m., n = 3 regions of interest) and tD = 0.97 ± 0.11 s (n = 66 yellow spots), for the monomer mix fD = 25.5 ± 3.4% (n = 3) and tD = 0.24 ± 0.05 s (n = 68), and for the SYNZIP1/SYNZIP2 pair fD = 56.8 ± 6.8% (n = 3) and tD = 1.09 ± 0.12 s (n = 61). For the SYNZIP1 green/red pair, we obtained fD = 23.1 ± 2.8% (n = 3) and tD = 0.29 ± 0.05 s (n = 74), and for the SYNZIP2 green/red pair fD = 24.1 ± 2.6% (n = 3) and tD = 0.32 ± 0.05 s (n = 59) (Fig. 2A and B). Both the similar fraction of co-localization and the longer time of co-localization for the SYNZIP1/SYNZIP2 pair as for the pure heterodimer mimic, and the large differences to the monomer mix, support the notion that our designed protein pair yields predominantly heteromers. Also, to exclude the possibility that GFP, the SNAP-tag or the transmembrane domains themselves cause the formation of dimers or higher order multimers, we inspected the intensities along the trajectories of individual green or red spots in the heterodimer mimic experiment. The lack of multiple bleaching steps from both green and red, and the virtually identical shapes of the intensity histograms from the beginning and the end of all trajectories confirm that the proteins are monomeric (ESI Fig. 1†).
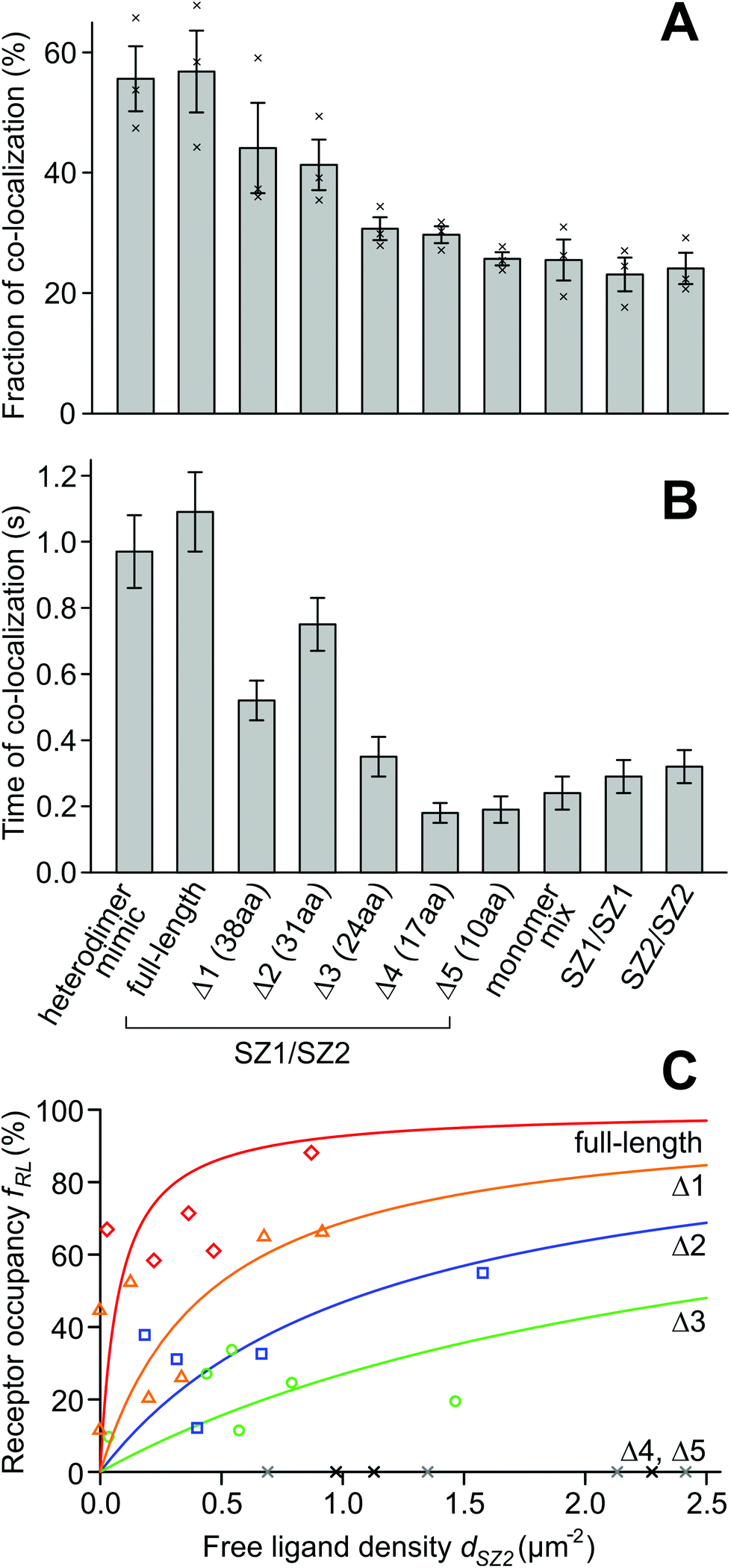 | ||
| Fig. 2 Fraction, time of co-localization, and dissociation constants for the designed membrane proteins. (A) The fraction of binding for the protein pair carrying full-length SZ1/SZ2 binding domains is equal to the constitutive heterodimer mimic, while for the shortest binding domain pair Δ5, the proteins resemble the monomer mix. SZ1 and SZ2 alone have no propensity to form homodimers. (B) The time of co-localization behaves similar to the fraction of binding. (C) Dissociation constants of full-length and truncated heterodimers. The receptor occupancy fRL is the fraction of SZ1 ‘receptor’ molecules that carries a bound SZ2 ‘ligand’. dSZ2 is the density of the unbound SZ2 ligand. For the full-length construct (red diamonds), the receptor is almost fully occupied by the ligand even at low densities. For Δ1 (orange triangles), Δ2 (blue squares) and Δ3 (green circles), the receptor is partially occupied at the accessible ligand densities, whereas for Δ4 and Δ5 (grey and black crosses), the ligand did not bind to the receptor at the accessible densities. Lines are fits of binding curves with the single free parameter K2D. | ||
In the heterodimer mimic, where each protein carries a GFP and a SNAP-tag, one would, in principle, expect a co-localization fraction of 100%. However, due to the limited labeling efficiency of the SNAP-tag and a dark fraction of the GFP molecules, some molecules appeared as only green and others as only red, leading to the reduced co-localization. Likewise, in the monomer mix, we would only expect green and red, but we also observed yellow spots. These can be explained by a coincidental overlap of green and red spots. The fraction of 25% results from the spot density of 2.5 μm−2, and an approximate threshold distance of 250 nm below which red and green spots appear yellow. This fraction was also confirmed in a Monte Carlo simulation of non-interacting green and red spots (ESI Fig. 2†).
Shortening the binding domains reduces the binding strength of the designed heterodimer
The designed heteromer pair based on SYNZIP1 and SYNZIP2 displayed the same fraction and time of co-localization as the heterodimer mimic where GFP and SNAP-tag were on the same protein; therefore, we assume it indeed forms a constitutive heterodimer. Next, we want to test if weakening the strength of the binding domains by shortening the length of the leucine zippers reduces the affinity of the proteins in the heteromer; an intermediate length of the leucine zippers should result in a mix of monomers and dimers. To experimentally observe the transition from strong to weak binding, we progressively shortened the SYNZIP domains, which have a length of 47 aa, from the N-terminal end, and obtained 5 truncations for each binding domain, with lengths of 38 aa (Δ1), 31 aa (Δ2), 24 aa (Δ3), 17 aa (Δ4), and 10 aa (Δ5) (ESI Note 1†).The resulting designed protein pairs with shortened leucine zippers were HALO-SZ1Δ1-TM-GFP/S549-SZ2Δ1-TM-xGFP (Δ1 pair), HALO-SZ1Δ2-TM-GFP/S549-SZ2Δ2-TM-xGFP (Δ2 pair), HALO-SZ1Δ3-TM-GFP/S549-SZ2Δ3-TM-xGFP (Δ3 pair), HALO-SZ1Δ4-TM-GFP/S549-SZ2Δ4-TM-xGFP (Δ4 pair), and HALO-SZ1Δ5-TM-GFP/S549-SZ2Δ5-TM-xGFP (Δ5 pair). After transfection of the respective pair of constructs into CHO cells, we measured the fraction and time of co-localization. For the Δ1 pair, we obtained fD = 44.1 ± 7.5% (s.e.m., n = 3 cells) and tD = 0.52 ± 0.06 s (n = 54 yellow spots), for Δ2 fD = 41.3 ± 4.2% (n = 3) and tD = 0.75 ± 0.08 s (n = 58), for Δ3 fD = 30.7 ± 1.9% (n = 3) and tD = 0.35 ± 0.06 s (n = 63), for Δ4 fD = 29.7 ± 1.4% (n = 3) and tD = 0.18 ± 0.03 s (n = 67), and for Δ5 fD = 25.7 ± 1.1% (n = 3) and tD = 0.19 ± 0.04 s (n = 62) (Fig. 2A and B). It seems unexpected that from Δ1 to Δ2, the time of co-localization increases, although we would expect a decrease. However, the significance of the difference is not given (p < 0.14, Mann–Whitney U test).
As predicted, both the yellow fraction and co-localization time decreased with shortening the binding domains. The Δ1 pair already shows a decreased fraction and time of co-localization compared to the full-length construct. For the Δ2 pair, the fraction of co-localization lies even lower, roughly in the middle between the full-length construct and the monomer mix, whereas the Δ3, Δ4 and Δ5 constructs behave similar to the monomer mix. Therefore, while the pair with the full-length SYNZIP domains is virtually dimeric, the constructs with the shortest binding domains are almost exclusively monomeric, and the transition point from dimers to monomers is around the length of the Δ2 domains.
2D binding affinity of the designed heteromer pairs
The previous analysis is insufficient to accurately characterize the dimerization for several reasons: first, the fraction of dimerizing receptors depends on the ratio of green and red receptors; if one species is less frequent, it sets a limit to the number of possible dimers. Second, the monomer/dimer equilibrium depends on the membrane density. And finally, the assessment of yellow spot fraction is only meaningful when comparing other proteins (e.g. the positive and negative controls), but does not result in a value that can be used to characterize the binding strength, like a dissociation constant. Therefore, we intended to find a more informative way to quantify their properties than determining the fraction of co-localization.In analogy to an affinity measurement for soluble substances, we can define one species as the receptor and the other one as the ligand, and obtain a dissociation constant from measuring the fraction of receptors occupied by ligands in dependence of the free ligand concentration. This is not directly possible from the co-localization we observed, since non-fluorescent GFP and unlabeled SNAP-tag, and overlap of green with red spots lead to a deviation of the counted green, red, and yellow spot numbers from the number of GFP- and SNAP-labeled receptor monomers and dimers.
We can account for these deviations in a model. It accounts for the coincidental overlap of green and red spots, and the fractions pG and pR of non-fluorescent GFP and unlabeled SNAP-tags. The model accepts the densities dSZ1 of free receptor (SYNZIP1), dSZ2 of free ligand (SYNZIP2), dSZ12 of SYNZIP1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SYNZIP2 receptor–ligand complexes, and pG and pR as parameters, to calculate the densities dG, dR and dY of green, red, and yellow spots (ESI Note 3†). If pG and pR are known and dG, dR and dY are measured in an experiment, it is possible to solve the model equations for dSZ1, dSZ2, and dSZ12. To determine pG and pR, we used the heteromer mimic S549-TM-GFP, where no monomers are present and dSZ1 = dSZ2 = 0. From cells with spot densities in the range of 1–3 μm−2, we obtained estimates of pG = 0.61 ± 0.08 (s.e.m., n = 3) and pR = 0.71 ± 0.03 (n = 3) (ESI Note 4†). With these values, we can calculate the ligand-occupied receptor fraction fRL = dSZ12/(dSZ12 + dSZ1) and the free ligand density dSZ2 for the full-length and the five truncated SYNZIP constructs (Fig. 3). For the full-length constructs HALO-SZ1-TM-GFP and S549-SZ2-TM-xGFP, the receptor occupancy was already high at low free ligand concentrations and increased up to fRL = 88% at dSZ2 = 0.87 μm−2. For the Δ1, Δ2 and Δ3 constructs, fRL assumed intermediate values, and for Δ4 and Δ5, it remained zero at all free ligand densities.
:SYNZIP2 receptor–ligand complexes, and pG and pR as parameters, to calculate the densities dG, dR and dY of green, red, and yellow spots (ESI Note 3†). If pG and pR are known and dG, dR and dY are measured in an experiment, it is possible to solve the model equations for dSZ1, dSZ2, and dSZ12. To determine pG and pR, we used the heteromer mimic S549-TM-GFP, where no monomers are present and dSZ1 = dSZ2 = 0. From cells with spot densities in the range of 1–3 μm−2, we obtained estimates of pG = 0.61 ± 0.08 (s.e.m., n = 3) and pR = 0.71 ± 0.03 (n = 3) (ESI Note 4†). With these values, we can calculate the ligand-occupied receptor fraction fRL = dSZ12/(dSZ12 + dSZ1) and the free ligand density dSZ2 for the full-length and the five truncated SYNZIP constructs (Fig. 3). For the full-length constructs HALO-SZ1-TM-GFP and S549-SZ2-TM-xGFP, the receptor occupancy was already high at low free ligand concentrations and increased up to fRL = 88% at dSZ2 = 0.87 μm−2. For the Δ1, Δ2 and Δ3 constructs, fRL assumed intermediate values, and for Δ4 and Δ5, it remained zero at all free ligand densities.
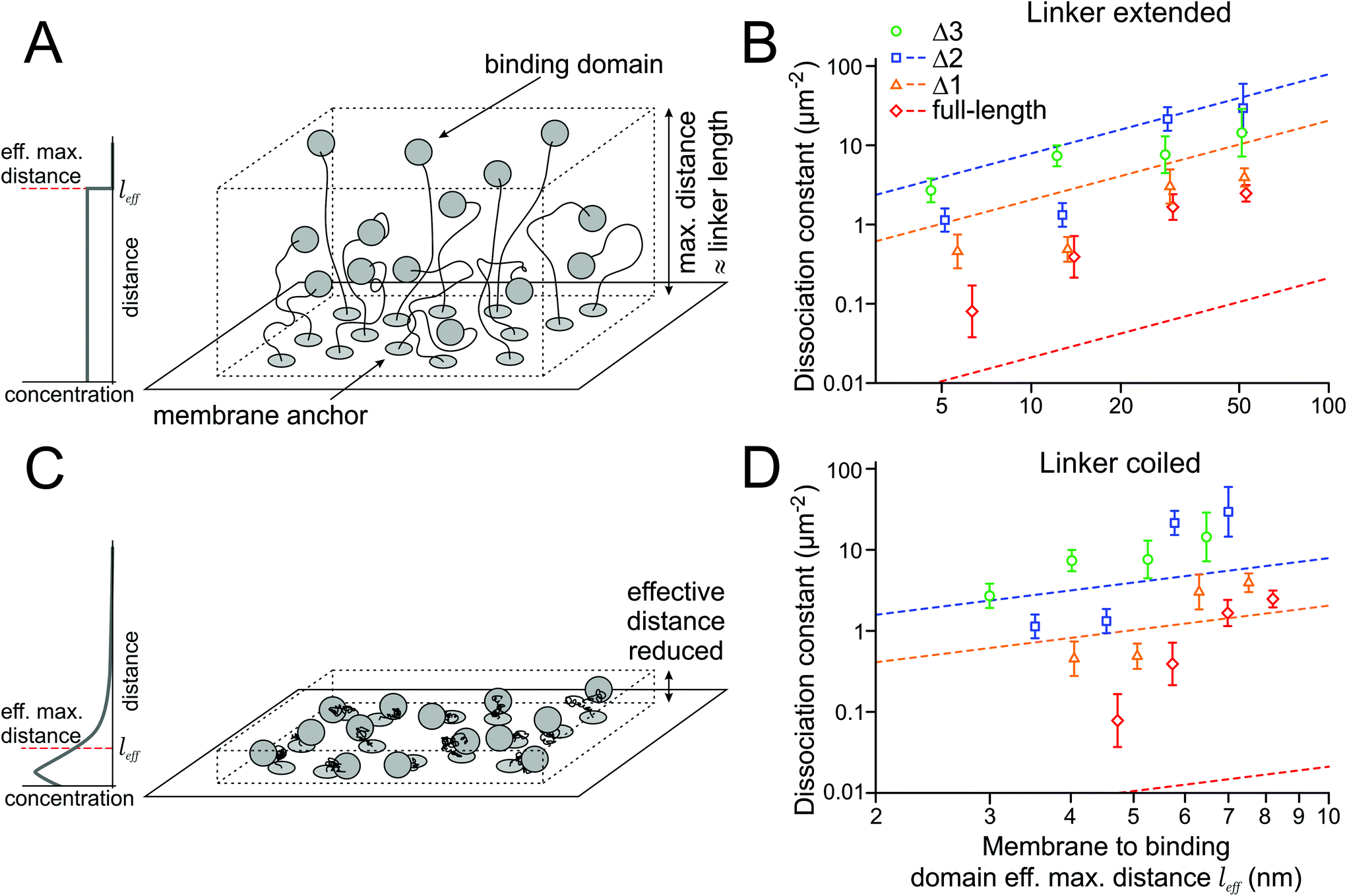 | ||
| Fig. 3 Relation of linker length and the 2D binding affinity. (A) In a first model, the binding domain is restricted to a certain volume due to the membrane linker, with a constant concentration throughout the accessible space. (B) 2D dissociation constants increase with the linker length. Dashed lines indicate the relation predicted from the measured 3D affinities of the soluble binding domains (no measurement was done for Δ3). (C) In a second model, the linkers are coiled due to entropic reasons. Therefore, the effective distance between the membrane and binding domain is reduced. (D) For shorter linkers, the predicted affinities are higher and dissociation constants (dashed lines) lower. | ||
In principle, the dependence of receptor occupancy fRL on the free ligand concentration dSZ2 should follow a binding curve. This curve is described by the Hill-Langmuir equation fRL (dSZ2) = dSZ2/(K2Dd + dSZ2), which contains the dissociation constant K2Dd as its only free parameter. A fit of the binding curve to the data yields an estimate for the 2D dissociation constant of K2Dd = 0.078 ± 0.059 μm−2 (68% CI, n = 5 regions of interest) for the full-length constructs. For the truncations Δ1 (38 aa), Δ2 (31 aa) and Δ3 (24 aa), we obtained K2Dd = 0.45 ± 0.22 μm−2 (n = 7), K2Dd = 1.1 ± 0.4 μm−2 (n = 5), and K2Dd = 2.7 ± 0.9 μm−2 (n = 6). For the truncations Δ4 (17 aa) and Δ 5 (10 aa), the dissociation constant was infinity since all values for receptor occupancies were zero (Fig. 2C).
Our model that estimates binding affinities for the designed membrane proteins yielded increasing K2Dd values (meaning reduced lateral binding affinities) for decreasing length of the binding domains. In contrast to the calculation of the fraction of co-localization (Fig. 2A and B), the model also accounts for unequal expression levels of green and red labeled molecules.
Extending the linker length reduces binding affinities
The association rate of the binding domains depends on their local concentration, while the dissociation rate is constant. Since the local concentration is inversely proportional to the accessible volume, the affinity should decrease when the linker length between the transmembrane domain and the binding domain increases. To experimentally assess the impact of changing the linker length on the dissociation constant, we designed proteins with extended linkers (Fig. 3A).The HALO-SZ1-TM-GFP/S549-SZ2-TM-xGFP pair has a short linker of 7 aa between the transmembrane domain and the binding domain. The full-length SYNZIP binding domains are 47 aa long, and, as an approximation, we assume that the points where they bind each other are at their centers. With a length of 0.4 nm per aa for the flexible short linker and 0.15 nm per aa for the translation in an alpha helix, we obtain an estimate of 6.3 nm for the maximal distance of the binding domain to the membrane. We chose three different linker lengths of 26 aa, 66 aa, and 123 aa, which extend the maximal distance for the full-length binding domain to about 14 nm, 30 nm, and 53 nm (factors 2.2, 4.7, and 8.4). Since we expected that extending the linkers would decrease the affinities and the binding affinity of Δ4 and Δ5 were already too low to be measured by our single molecule approach, we only did experiments for the full-length construct, Δ1, Δ2, and Δ3; due to the shorter binding domains of the latter three, the maximal distances between the membrane and the binding domains are shorter than those for the full-length construct by 0.7 nm, 1.2 nm and 1.7 nm, respectively.
We measured the numbers of green, red, and yellow spots for several combinations of linkers (7 aa, 26 aa, 66 aa, 123 aa) and the full-length and Δ1–Δ3 binding domains, and calculated the dissociation constants with the model established in the previous section (Table 1). As predicted, the affinities decreased with increasing linker length (Fig. 3B).
| linker length (fully extended) | |||||
|---|---|---|---|---|---|
| 7 aa/3 nm | 26 aa/10 nm | 66 aa/26 nm | 123 aa/49 nm | ||
| Binding domain | Full-length (47 aa) | 0.078 ± 0.059 (5) | 0.39 ± 0.24 (7) | 1.7 ± 0.6 (4) | 2.5 ± 0.6 (5) |
| Δ1 (38 aa) | 0.45 ± 0.22 (7) | 0.49 ± 0.17 (6) | 3.0 ± 1.5 (5) | 3.9 ± 1.0 (4) | |
| Δ2 (31 aa) | 1.1 ± 0.4 (5) | 1.3 ± 0.5 (6) | 21 ± 7 (3) | 29 ± 21 (4) | |
| Δ3 (24 aa) | 2.7 ± 0.9 (6) | 7.4 ± 2.2 (5) | 7.6 ± 4.1 (6) | 14 ± 10 (5) | |
| Δ4 (17 aa) | ∞ | — | — | — | |
| Δ5 (10 aa) | ∞ | — | — | — | |
Relation of 2D to 3D affinities
The membrane protein can also be seen as a binding domain in solution, with its intrinsic 3D dissociation constant K3Dd, that is tethered to the membrane. We want to understand how the 2D dissociation constants K2Dd of the membrane proteins depend on these 3D dissociation constants K3Dd of the binding domains in solution. In a first model, we assume the simplest thinkable relationship, where the binding domains are homogeneously and isotropically distributed within the layer determined by the linker length (Fig. 3A). Then, the local concentration c3D of the binding domains in the vicinity of the membrane is constant up to the maximal distance from the membrane that the linker can extend to. In this case, c3D is related to the 2D concentration, i.e. the density d2D, of the protein in the membrane, and the maximal distance l from the membrane through the equation c3D = d2D/l.For understanding the relation between 2D and 3D dissociation constants, it is critical to remember that when the ligand concentration equals the dissociation constant, half of the receptors are bound to ligands. This is true for the 2D perspective, where the receptor and ligand concentrations relate to their densities in the membrane, as much as for the 3D perspective, where we refer to the 3D concentrations of the soluble, but membrane-tethered, binding domains. Therefore
| K2Dd = K3Dd·l | (1) |
To test relation (1), we set out to measure the 3D dissociation constants K3Dd of the binding domains in their soluble form. To this end, we fused the binding domains via a 10 aa flexible linker to the maltose binding protein (MBP) and a SUMO protein, which are protein tags frequently used for increasing the solubility and stability of proteins. A TEV cleavage site and a 10xHis-Tag were fused for purification via Ni-NTA affinity columns and cleaved afterwards. The resulting constructs were SZ1-MBP-TEV-10xHis, SZ2-SUMO-TEV-10xHis and the corresponding truncated Δ1 and Δ2 versions. After expression in E. coli and purification, we determined their dissociation constants by isothermal titration calorimetry (ESI Note 5†). For the binding domains of the full-length SYNZIP pair, we obtained K3Dd = 3.5 ± 1.4 nM (for calculation of CI, see ESI Note 5†), which is in the range of <10 nM previously measured by the group that designed the SYNZIPs.8,9 The truncations yielded K3Dd = 340 ± 100 nM (Δ1) and K3Dd = 1310 ± 220 nM (Δ2).
The measured K3Dd values define the linear relation (1) between the 2D dissociation constant K2Dd and the length l of the linker. In a double logarithmic graph, this relation is a line with slope 1, and K3Dd determines the shift of the line (Fig. 3B). The values of the dissociation constants measured for different linker lengths (Table 1) should, in principle, lie on these lines. However, we find that the measured K2Dds for the constructs with the full-length binding domain are (in the average) a factor of 20 higher than those predicted (Table 2). Based on the measured K3Dd of 3.5 nM for the soluble full-length SYNZIP domains, their membrane-tethered versions should always be dimerized at the densities we used, independent of the linker length; but we observe only little dimerization for long linkers. In contrast, for the Δ1 and Δ2 binding domains, the K2Dds are smaller than predicted by a factor of about 3 (Table 2).
| 7 aa | 26 aa | 66 aa | 123 aa | Δfac | |
|---|---|---|---|---|---|
| Full-length (47 aa) | 0.013 ± 0.005 | 0.029 ± 0.012 | 0.063 ± 0.025 | 0.11 ± 0.04 | 20.4 |
| Δ1 (38 aa) | 1.2 ± 0.3 | 2.7 ± 0.8 | 6.0 ± 1.8 | 11 ± 3 | 0.32 |
| Δ2 (31 aa) | 4.0 ± 0.7 | 10 ± 2 | 23 ± 4 | 41 ± 7 | 0.35 |
Contracted state of the linker
In the previous section, we assumed a constant concentration of the binding domains from the membrane up to the maximal distance given by the linker length. However, this assumption is inadequate for several reasons: (i) within the volume of a half-sphere around the anchor, there are more points close to the membrane than farther away that the binding domain can access, (ii) due to entropic reasons, the linker will spend (much) more time in a contracted than in an extended shape (Fig. 3C). This behavior can be modeled by a flexible chain with links of constant length, which has been found to be close to 0.4 nm per aa for unfolded proteins.17 In addition, (iii) the extracellular matrix might have an impact on the extension of the chain, e.g. due to crowding effects.We estimated the influence of (i) and (ii) by modeling the linker by a flexible chain with the number of chain links given by the number of amino acids, and the angles of the chain links randomly oriented (ESI Note 6†). We find that for 7, 26, 66, and 123 aa long linkers, their effective lengths are l7 = 1.2 nm, l26 = 2.2 nm, l66 = 3.5 nm, and l123 = 4.7 nm, i.e. they are shortened by factors of 3.4, 4.7, 7.7 and 10.5 compared to the first model, where the binding domain's concentration is constant between the membrane and the maximal possible distance allowed by the linker, as explained in the previous section.
With the shorter effective linker length, the measured K3Dds of the soluble binding domains would predict lower dissociation constants K2Dd of the membrane-tethered constructs, and therefore stronger binding (Table 3 and Fig. 3D). In this model, the average K2Dd values of the Δ1 and Δ2 binding domains match the predictions from the ITC measurements well, and the binding of the tethered full-length binding domain seems weakened by a factor of ∼100 compared to the binding of its soluble counterpart.
| 7 aa | 26 aa | 66 aa | 123 aa | Δfac | |
|---|---|---|---|---|---|
| Full-length (47 aa) | 0.010 ± 0.004 | 0.012 ± 0.005 | 0.015 ± 0.006 | 0.017 ± 0.007 | 98 |
| Δ1 (38 aa) | 0.83 ± 0.24 | 1.0 ± 0.3 | 1.3 ± 0.4 | 1.5 ± 0.5 | 1.3 |
| Δ2 (31 aa) | 2.8 ± 0.5 | 3.6 ± 0.6 | 4.6 ± 0.8 | 5.5 ± 0.9 | 1.0 |
Discussion
Because a single transmembrane helix is unable to transmit a signal across the membrane, single-pass transmembrane proteins rely on lateral interactions; therefore, dimerization and clustering of membrane proteins play an important role in physiology. In particular, receptor tyrosine kinases and many cytokine receptors form ligand-induced dimers that are signaling competent.5,7,18 When we try to understand the regulation of the receptor's dimerization, we quickly realize that its structural complexity impedes analysis on a biophysical level. First, several interaction domains usually control the receptor's dimerization; e.g. for the EGF receptor, the extracellular, intracellular, transmembrane and juxtamembrane domains all have a tendency to dimerize under certain conditions.18,19 Second, these binding domains often couple allosterically to each other, e.g. the juxtamembrane and intracellular domain of the EGF receptor. Third, regulatory proteins – which might even be unknown – can modulate the dimerization. Although dimerization has been investigated for several membrane proteins previously, usually the studies were limited to comparing the proteins of interest to known monomers and dimers and measuring the fraction of dimers under varying conditions.20,21 In a few cases, values for lateral affinities have been determined, but the molecular basis for the absolute value of the affinity remained elusive.In this work, we designed a simple model system to obtain a biophysical understanding of membrane protein dimerization. In particular, we wanted to relate the molecular features of the model protein to the lateral dimerization affinity in the membrane. The model system was designed to have an inert transmembrane domain and intracellular domain, such that the dimerization is exclusively mediated by the properties of the extracellular domain. The extracellular domain had two characteristics that affected the dimerization propensity of the full protein: the heterodimerization affinity of the binding domain and the length of the linker to the membrane. As predicted, an increase of the affinity of the binding domain increased the dimerization of the full protein, and an extension of the linker reduced dimerization. We also modeled how the 2D binding affinity of the membrane protein depends on the soluble 3D affinity of the free binding domain and the linker length.
The dimerization was directly observed on a single-molecule level in living cells and therefore did not rely on downstream signaling. Although single-molecule imaging yields a direct readout, several caveats need to be considered for the interpretation of the results. First, the small number of molecules that contributes to an individual data point bears an intrinsic error from the Poisson statistics associated with counting randomly occurring events. A particularly high impact of this error appears in conditions where one of the counted fractions is small, i.e., for very strong dimerization (low number of free molecules) or very weak dimerization (low number of dimerized molecules). Second, errors from contamination with green or red dyes, which might originate from non-specific binding of the SNAP-tag substrate or GFP from dead cells, increase the number of spots appearing only green or red. Similarly, pre-bleaching of tags during search for a new cell increases the fraction of non-fluorescent tags. Nevertheless, single-molecule imaging allows a direct measurement of the membrane density of the different molecule species, which is crucial to determining affinities. Improvements to the approach we chose for this work can be expected from more photostable dyes that allow collection of more data from each cell, reduce pre-bleaching, and could even facilitate imaging of interactions via FRET, thereby obviating the need to account for the random co-localization of red and green, since FRET occurs only at distances of a few nanometers.
For the membrane proteins with truncated binding domains Δ1 and Δ2, the dissociation constants that we observed were close to the predictions based on the ITC measurement of the soluble domains. However, for the strongest, the full-length binding domain, we observed a different behavior: the 2D dissociation constants that we predicted from the measured 3D values of the soluble domains and the linker length were higher by a factor of 20 or more than the ones measured in the single-molecule experiment. This effect could not originate from a measurement error, since for the longest linker and the full-length binding domain, we would predict near complete dimerization, but observed primarily monomers. One explanation would be a cleavage or rupture of the linker due to its length, leaving the transmembrane domain with the GFP but without the binding domain and the SNAP-tag. We exclude this possibility because control experiments with constructs carrying both GFP and SNAP-tag yielded a high degree of green/red co-localization, also for the longest linker, suggesting that the linker stayed intact. An alternative explanation for the reduced dimer fraction is that one or both of the binding domains have a certain affinity for the membrane, since they are leucine zippers that contain a hydrophobic side. We used a prediction tool for amphipathic in-plane membrane anchor prediction and indeed found that full-length SYNZIP1 has a predicted affinity to the membrane.22,23
We devised two models for the linker topology that suggest a possible range of effective linker lengths. In the first model, the linker can fully extend and allows the binding domain to visit every accessible point with equal probability. In the second model, the distance of the binding domain to the membrane is strongly reduced due to the much higher state density in a curled-up state of the linker. This second model also prompted us to establish a definition of the effective linker length that also allows for a non-uniform membrane-to-binding domain distance distribution, such that eqn (1), originally setup for the first model, still applies. With increasing linker length, the factor by which the linker is contracted increases. Although, after all, our data did not allow to clearly decide which model is more accurate, we found that the model used for the topology of long, flexible domains has a strong impact on the binding of the domains at the linker ends.
The tuning of transmembrane protein affinities has important implications for signal transduction. Proteins that use lateral interactions (like dimerization) to initiate a signaling process upon appearance of a cue should have their density and affinity tuned close to the point of activation. In this way, only a small affinity increase is needed to dimerize them and activate the signal. At receptor densities of 1–100 μm−2 and a distance of 5 nm to the membrane, one can calculate an optimal affinity of 0.33–33 μM (depending on receptor density) to facilitate activation. Indeed, affinity values for binding domains of natural receptors in solution are in this range, e.g. 0.2 μM for the kinase domain of the EGFR and 5 μM for the interferon receptor subunits IFNAR1 and IFNAR2 upon ligand addition.5,24 Also, for the new design of artificial receptors, the affinity of the binding domain must be chosen carefully and with respect to the expected membrane density of the receptor to achieve optimal switching properties. Therefore, our work will help to understand the function of naturally occurring membrane proteins and demonstrate practical and theoretical progress towards the design of synthetic, switchable membrane receptors.
Materials and methods
Plasmids
Membrane bound constructs were assembled from SYNZIP1 and SYNZIP2 from ref. 8, the transmembrane domain of PDGFRα, the SNAP- (enhanced SNAPf version) and HALO-tags, and GFP containing the A206 K mutation, and cloned into the pWHE636 vector (gift by Christian Berens) that contains a tetracycline inducible promoter25 (ESI Note 1†). An N-terminal signal peptide was fused to the SNAP-tag and HALO-tag for extracellular targeting. GFP was rendered non-fluorescent with the Y66L mutation. For soluble proteins, SYNZIP1 and SYNZIP2 were fused to MBP or a SUMO domain and His-tagged by cloning into the pET303/CT-His vector.Cell culture and transient transfection
CHO-K1 cells were grown in DMEM added with 10% FBS, 100 units per mL penicillin, 100 μg mL−1 streptomycin, 2 mM L-glutamate and 50 nM β-mercaptoethanol at 37 °C and 5% CO2. 2–4 × 104 cells were seeded on coverslips with a refractive index of 1.78 in 12-well culture plates to grow for 24 hours before transfection. Coverslips were cleaned in 1 M KOH in an ultrasonic bath for 15 min and washed in DPBS before seeding cells. Constructs were co-transfected at 60–80% confluency. Per well, 0.5–1 μg of DNA was mixed with 174 μL of DMEM and 8 μL of polyethylene imine (PEI) solution (100 μg ml−1) for transfection. Cells were incubated with PEI-DNA for 3 hours, washed with ice-cold DPBS three times, and incubated in growth media containing 1 μg mL−1 doxycycline for 2 hours to induce protein expression. Finally, cells were washed again in ice-cold DPBS and grown for 12 hours before labeling and imaging.SNAP-tag labeling and single-molecule imaging
Cells were labeled with 1–2 μM (final concentration) commercial (NEB) or home-made SNAP-Surface 549 for 15 min at 37 °C (ESI Note 2†). Then, cells were washed in DPBS at least five times and one time in cell-culture media. Cells were imaged within 1 hour after labeling in DPBS at room-temperature on an Olympus IX71 microscope in total internal reflection mode (objective: Olympus NA 1.70 APON100xHO TIRF) with an EMCCD camera (Andor iXon 897). The recorded area was 25.6 × 25.6 μm2 at 160× magnification. GFP and SNAP-Surface 549 were excited simultaneously or in alternating excitation mode by 488 nm and 561 nm lasers and at power densities of 100–500 W cm−2 and were imaged with an image splitter (OptoSplit II, CAIRN Research) to view green and red emission on the same camera.Data evaluation
In the first frame of a movie, a rectangular area with appropriate densities of green and red labeled molecules was evaluated. Visible yellow, green and red spots were manually counted. The fraction of co-localization was calculated as 2NY/(2NY + NG + NR), where NY, NG and NR are the numbers of yellow, green and red spots, respectively. Tracking of spots was manually done with help of the ImageJ plugin MTrackJ.26 To check for bias in manual counting, we also used an automated spot recognition routine that incorporates the particle tracking algorithm from;27 spots were considered co-localized (yellow) when their distance was smaller than 110 nm (for two-color simultaneous illumination) or 174 nm (for two-color alternating excitation) (ESI Note 7†). Due to the high spot density we required, the automated routine failed to recognize many spots. However, since the pG and pR values were also lower for automated counting (0.42 and 0.48), the affinities obtained from automated evaluation were close to the results from manual counting (ESI Note 7†).Isothermal titration calorimetry
SZ1-SUMO-His and MBP-SZ2-His were purified after expression in E. coli, and the His tag was removed from MBP-SZ2-His with TEV protease. ITC experiments were performed with a MicroCal VP-ITC instrument (Malvern Instruments, UK). The sample cell had a volume of 1.4 mL. ITC experiments were conducted by repeating injections of 7 μL aliquots of 6.5 μM MBP-SZ2 into the sample cell that contained 0.6 μM SZ1-SUMO-His. The heterodimer affinities of the full length, Δ1 and Δ2 constructs were determined taking into account also the homomerization of each construct in the cell and syringe volumes before mixing (ESI Note 5†).Data availability
All data generated or analyzed during this study are included in this published article or are available from the corresponding author upon request.Author contributions
C. L. performed all single-molecule experiments. C. L. and M. H. U. conceived the study, designed experiments, analyzed the single-molecule imaging data and wrote the manuscript. A. S., H. H., and M. H. U. designed the ITC measurement and analyzed the data. P. S. expressed and purified all proteins. S. L. performed pilot experiments for the determination of affinities. N. P. F. B. and M. J. established the synthesis and synthesized the fluorescent SNAP-tag substrate.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by Deutsche Forschungsgemeinschaft (DFG) grants UL 312/6-1 (Project ID 278267951), RTG 2202 (Project ID 278002225), and CRC 992 (Project ID 192904750), the Excellence Initiative of the German Federal and State Governments (EXC 294), and Germanýs Excellence Strategy (EXC-2189 – Project ID 390939984).References
- S. Löchte, S. Waichman, O. Beutel, C. You and J. Piehler, Live cell micropatterning reveals the dynamics of signaling complexes at the plasma membrane, J. Cell Biol., 2014, 207, 407–418 CrossRef PubMed.
- R. Maeda, T. Sato, K. Okamoto, M. Yanagawa and Y. Sako, Lipid-protein interplay in dimerization of juxtamembrane domains of epidermal growth factor receptor, Biophys. J., 2018, 114, 893–903 CrossRef CAS PubMed.
- R. S. Kasai, et al., Full characterization of GPCR monomer-dimer dynamic equilibrium by single molecule imaging, J. Cell Biol., 2011, 192, 463–480 CrossRef CAS PubMed.
- S. Sarabipour and K. Hristova, Mechanism of FGF receptor dimerization and activation, Nat. Commun., 2016, 7, 10262 CrossRef CAS PubMed.
- O. Beutel, et al., Two-dimensional trap for ultrasensitive quantification of transient protein interactions, ACS Nano, 2015, 9, 9783–9791 CrossRef CAS PubMed.
- R. Chadda, et al., The dimerization equilibrium of a ClC Cl−/H+ antiporter in lipid bilayers, eLife, 2016, 5, e17438 CrossRef PubMed.
- S. Wilmes, et al., Mechanism of homodimeric cytokine receptor activation and dysregulation by oncogenic mutations, Science, 2020, 367, 643–652 CrossRef CAS PubMed.
- A. W. Reinke, R. A. Grant and A. E. Keating, A synthetic coiled-coil interactome provides heterospecific modules for molecular engineering, J. Am. Chem. Soc., 2010, 132, 6025–6031 CrossRef CAS PubMed.
- K. E. Thompson, C. J. Bashor, W. A. Lim and A. E. Keating, SYNZIP protein interaction toolbox: in vitro and in vivo specifications of heterospecific coiled-coil interaction domains, ACS Synth. Biol., 2012, 1, 118–129 CrossRef CAS PubMed.
- E. Li, W. C. Wimley and K. Hristova, Transmembrane helix dimerization: Beyond the search for sequence motifs, Biochim. Biophys. Acta, 2012, 1818, 183–193 CrossRef CAS PubMed.
- M. C. Wehr, et al., Monitoring regulated protein-protein interactions using split TEV, Nat. Methods, 2006, 3, 985–993 CrossRef CAS PubMed.
- M. H. Ulbrich and E. Y. Isacoff, Subunit counting in membrane-bound proteins, Nat. Methods, 2007, 4, 319–321 CrossRef CAS PubMed.
- T. Bartoi, K. Augustinowski, G. Polleichtner, S. Gründer and M. H. Ulbrich, Acid-sensing ion channel (ASIC) 1a/2a heteromers have a flexible 2:1/1:2 stoichiometry, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8281–8286 CrossRef CAS PubMed.
- D. A. Zacharias, et al., Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells, Science, 2002, 296, 913–916 CrossRef CAS PubMed.
- P. J. Bosch, et al., Evaluation of fluorophores to label SNAP-tag fused proteins for multicolor single-molecule tracking microscopy in live cells, Biophys. J., 2014, 107, 803–814 CrossRef CAS PubMed.
- J. J. A. Armenteros, et al., SignalP 5.0 improves signal peptide predictions using deep neural networks, Nat. Biotechnol., 2019, 37, 420–423 CrossRef PubMed.
- A. Hoffmann, et al., Mapping protein collapse with single-molecule fluorescence and kinetic synchrotron radiation circular dichroism spectroscopy, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 105–110 CrossRef CAS PubMed.
- N. F. Endres, et al., Conformational coupling across the plasma membrane in activation of the EGF receptor, Cell, 2013, 152, 543–556 CrossRef CAS PubMed.
- A. Arkhipov, et al., Architecture and membrane interactions of the EGF receptor, Cell, 2013, 152, 557–569 CrossRef CAS PubMed.
- D. Calebiro, et al., Single-molecule analysis of fluorescently labeled G-protein-coupled receptors reveals complexes with distinct dynamics and organization, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 743–748 CrossRef CAS PubMed.
- A. Tabor, et al., Visualization and ligand-induced modulation of dopamine receptor dimerization at the single molecule level, Sci. Rep., 2016, 6, 33233 CrossRef CAS PubMed.
- C. Combet, C. Blanchet, C. Geourjon and G. Deléage, NPS@: Network protein sequence analysis, Trends Biochem. Sci., 2000, 25, 147–150 CrossRef CAS PubMed.
- N. Sapay, Y. Guermeur and G. Deléage, Prediction of amphipathic in-plane membrane anchors in monotopic proteins using a SVM classifier, BMC Bioinform., 2006, 7, 255 CrossRef PubMed.
- N. Jura, et al., Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment, Cell, 2009, 137, 1293–1307 CrossRef PubMed.
- C. Danke, et al., Adjusting transgene expression levels in lymphocytes with a set of inducible promoters, J. Gene Med., 2010, 2, 501–515 CrossRef PubMed.
- E. Meijering, O. Dzyubachyk and I. Smal, Methods for cell and particle tracking, Meth. Enzymol, 2012, 504, 183–200 CrossRef PubMed.
- J. C. Crocker and D. G. Grier, Methods of digital video microscopy for colloidal studies, J. Colloid Interface Sci., 1996, 179, 298–310 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr06574b |
| This journal is © The Royal Society of Chemistry 2021 |