
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePorphyrins as building blocks for single-molecule devices
Patrick
Zwick
 a,
Diana
Dulić
b,
Herre S. J.
van der Zant
*c and
Marcel
Mayor
*ade
a,
Diana
Dulić
b,
Herre S. J.
van der Zant
*c and
Marcel
Mayor
*ade
aDepartment of Chemistry, University of Basel, St Johanns-Ring 19, 4056 Basel, Switzerland. E-mail: marcel.mayor@unibas.ch
bDepartment of Physics and Department of Electrical Engineering, Faculty of Physical and Mathematical Sciences, University of Chile, Avenida Blanco Encalada 2008, Santiago 8330015, Chile
cKavli Institute of Nanoscience, Delft University of Technology, Lorentzweg 1, 2628 CJ Delft, The Netherlands
dInstitute for Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), P. O. Box 3640, 76021 Karlsruhe, Germany
eLehn Institute of Functional Materials (LIFM), School of Chemistry, Sun Yat-Sen University (SYSU), 510275 Guangzhou, China
First published on 19th August 2021
Abstract
Direct measurement of single-molecule electrical transparency by break junction experiments has become a major field of research over the two last decades. This review specifically and comprehensively highlights the use of porphyrins as molecular components and discusses their potential use for the construction of future devices. Throughout the review, the features provided by porphyrins, such as low level misalignments and very low attenuation factors, are shown with numerous examples, illustrating the potential and limitations of these molecular junctions, as well as differences emerging from applied integration/investigation techniques.
 Patrick Zwick | Patrick Zwick studied Chemistry at the University of Basel (Switzerland) and received his M.Sc. in 2017. He then completed his PhD in 2021 at the University of Basel under the supervision of Professor Marcel Mayor on the subject of tailor-made porphyrin cyclophanes for mechanosensitive single-molecule junction experiments. To expand his knowledge of supramolecular chemistry, Patrick will be a post-doctoral research associate in the group of Professor David A. Leigh at the University of Manchester (United Kingdom). |
 Diana Dulić | Diana Dulić received her PhD in 2001 from the University of Groningen (Netherlands). Since 2012, she has been a leader of the Molecular Nanoelectronics Group at the Department of Physics (FCFM) at the University of Chile (Chile). The main scope of her research has been studying electronic transport in molecular wires and bio-inspired molecules. She joined the field of the single molecule electronics at its very beginning and was one of the first to use the mechanically controllable break junction technique to study charge transport in conjugated functional molecular wires. |
 Herre S. J. van der Zant | Herre S. J. van der Zant received his PhD in 1991 from Delft University of Technology (TU Delft, the Netherlands). Afterwards, he was a post-doctoral researcher at the Massachusetts Institute of Technology (MIT, USA) and returned to the group of Hans Mooij at TU Delft to work on mesoscopic charge density waves. For this subject he received a five year fellowship from the Royal Academy for Sciences. In 2005, he cofounded the Molecular Electronics and Devices group at the Kavli Institute for Nanoscience at TU Delft. As a professor in this group, his recent research focuses on the nanomechanics of membranes and molecular scale electronics. |
 Marcel Mayor | Marcel Mayor received his PhD in 1995 from the University of Bern (Switzerland) under the supervision of Rolf Scheffold and Lorenz Walder. After working with Jean-Marie Lehn at the University Louis Pasteur in Strasbourg and at the Collège de France in Paris (France), he founded his own research group in the Institute of Nanotechnology (INT) at Karlsruhe Institute of Technology (KIT, Germany) in 1998. In 2004 he became Professor of Chemistry in the Department of Chemistry at the University of Basel (Switzerland), and in 2011 he became adjunct Professor of Chemistry at the School of Chemistry of Sun Yat-Sen University in Guangzhou (China). His research foci are beyond molecular electronics, molecular devices and machines, tailor-made model compounds for physical experiments, and molecular nano-architectures. |
1. Introduction and scope
The miniaturization of electronic components by top-down approaches proceeded so steadily that the development was described by Gordon Moore as a “law”.1 The ongoing size reduction will however hit physical, and probably sooner, economic limitations. Propelled by the quest for even smaller functional units, the visionary concept to integrate single molecules into electronic circuits was first proposed by Aviram and Ratner.2 While the merger between molecular structures and CMOS technology remains challenging, scientifically the investigation of the potential and limitations of single molecules in electronic circuits is intriguing. Molecules are not only the smallest building blocks that still provide the structural diversity required to implement particular electronic features and/or functions, they can also be designed and tailor-made for particular purposes by synthetic chemists. Since the hypothetical concept of molecular electronics2 was introduced until today, many efforts towards the realization of functional devices have been made and the blossoming of interdisciplinary research, covering chemistry, physics, and theory, allowed for investigations into molecular structures integrated in electric circuits: single-molecule junctions. Understanding the principles that determine electric transparency through single molecules is not only a priori interesting from a fundamental viewpoint, it impacts on the comprehension and development of entire research domains, ranging from molecular devices, over energy cascades in biological systems, to even bulk materials. The study of single-molecule junctions is at the heart of molecular electronics and has been extensively reviewed already, for example in ref. 3–6 and references therein. In most cases fundamental structure–property relationships are reported; these are extracted from a small set of synthesized molecules analyzed as single-molecule junctions in a particular experimental set-up.Here, we will review single-molecule junctions formed by derivatives of a particularly interesting class of aromatic organic compounds: porphyrins, which are key components in nature's machinery to store sunlight as chemical energy. Synthesis procedures for cyclic porphyrin arrays (mimicking natural light-harvesting antenna complexes),7 conjugated linear two- (2D) and three (3D) dimensional arrays,8,9 and asymmetric monomers by multi-component condensation10,11 or post-functionalization,12 or the strategic assembly of porphyrin architectures by transition-metal mediated cross-coupling13 have been the subject of many reviews already. The scope of this paper is charge transport studies of porphyrin based single-molecule junctions14 probed by direct electrical measurements with the structures being wired to two terminals. With a critical view to the differences in applied techniques and the molecular design of the investigated structures, this review is meant to serve as another puzzle-piece in the continuous quest to reveal the bigger picture of molecular electronics. Before we turn to the conductance experiments, we will have a brief look at the different measurement platforms, techniques, and conductance mechanisms.
Measurement platforms and techniques
In the past 20 years, a variety of techniques for measuring single-molecule electrical properties have been established. Break-junction techniques15,16 form the main direction and they are based either on mechanical control over the electrodes or are made by sending a large current through a narrow constriction (electromigrated junctions). Concerning mechanical break junctions, there are two types: mechanically controllable (MCBJ) and scanning tunneling microscopy (STM-BJ) break junctions, see panel (a) of Fig. 1 for an illustration of the three different set-ups. Both techniques (MCBJ and STM-BJ) can generate large data sets, and therefore statistical analysis enables the identification of transport properties at the single-molecule level. The main difference between the two techniques is the geometry: STM-BJs are formed by making and breaking a substrate–tip contact, whereas MCBJs are made by stretching a thin suspended metallic wire. Gold is in both cases the most commonly used metal. In both, the conductance (G) is recorded during breaking and making cycles of a contact at a fixed bias voltage. The breaking traces, during which G(s) is recorded, are plotted as one dimensional (1D) or two dimensional (2D) conduction histograms, where s is the distance between electrodes. Since 1D-histograms do not permit distance-dependent analysis, often, a more appropriate way to analyze data is the use of 2D-histograms, which show the evolution of conductance while moving the two electrodes further apart (see Fig. 1, panel (c) for an example of a 1D-histogram and (d) for a 2D-histogram). | ||
| Fig. 1 (a) Schematic representation of the set-ups of an MCBJ, an STM-BJ, and a junction formed by electromigration (from left to right). Mechanical control is depicted by red and blue arrows. (b) Illustration of the operating modus of the G(s), I(s) and I(t) method of an STM-BJ (upper) and an MCBJ (lower) set-up as a function of electrode spacing. (c) 1D-histogram and (d) 2D-histogram of the same data for molecular junctions formed by the MCBJ technique, adapted from ref. 17 published by The Royal Society of Chemistry. (e) I(V) characteristics of a porphyrin decorated electromigrated junction recorded at 5 K. From top to bottom, the applied gate biasing voltage Vg is −0.1, −0.08, −0.06, −0.04, −0.02 and 0.0 V, adapted from ref. 23 with permission from Elsevier. | ||
Related to this G(s) method of measuring is the I(s) technique18 (I is the current through the junction). This technique differs from the G(s) measurements in that the STM tip approaches the sample surface very closely, resulting in the stochastic formation of molecular bridges between the STM and surface. Since no metallic break junction is formed (in contrast to G(s) and MCBJ), the I(s) can be considered a non-contact mode of forming single-molecule junctions. Similar to the G(s) mode, the electrical properties of these junctions are statistically analyzed by repeated stretching, breaking and going back to the initial distance again. Another STM-based method for single-molecule conductance measurements is the so-called I(t) method: by placing the STM tip sufficiently close to the surface, current jumps, or “blinks”, may be observed in the current-versus-time profiles as molecules stochastically bridge between the tip and surface, forming junctions.19 The outlined methods of the junctions formed by mechanical control (STM-BJ and MCBJ) are shown in panel (b) of Fig. 1.
Another way to analyze molecular transparency is to measure the current–voltage (I(V)) characteristics at fixed electrode separation. Whereas low-bias conductance measurements can provide the conductance value of the metal–molecule–metal junction, I(V) characteristics contain richer information about the transport mechanism in the molecule, its density of states, and vibrational modes.20 However, molecular junctions stable enough to screen the voltage range of interest are required to record current–voltage (I(V)) characteristics. As stated above, single-molecule break junctions can also be made by an electromigration process; the main advantage of this technique is that a gate electrode can easily be implemented, giving control over the chemical potential of the molecule independent of that of the electrodes.21 By measuring I(V)s as a function of gate voltage, detailed spectroscopic information on the molecule can be obtained, especially at cryogenic temperatures. For example, reversible charging processes can be observed in such experiments. A disadvantage is the low molecular junction formation yield, which not only impedes the use of statistical analyses but also requires the selection of the junction of interest by the operator. An exemplary I(V) histogram is provided in panel (e) of Fig. 1. The electromigration method is not restricted to gold electrodes (or metal-based materials in general); graphene and carbon nanotubes have also been exploited to create atomically thin electrodes.22 As Joule heating plays a major role, the process is often called electroburning of carbon.
Transport mechanisms
It is well established that in two-terminal molecular junctions charge transport is influenced by intrinsic – often interdependent – properties of the embedded molecule,24–31 such as the positions of frontier orbitals, i.e., the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), the molecular conformation, and the molecular backbone composition (e.g., saturated versus unsaturated). In addition, the anchoring groups that connect a molecule to the metallic electrodes,32 the work function of the electrodes,33,34 and the contact geometry of the molecule–electrode interface35 also influence the charge transport properties of molecular junctions. Typically, charge transport can be described via a non-resonant tunneling process. The coherent transport of the charge-carrier (the LUMO for electrons, the HOMO for holes) can then be described as a function of level misalignment (i.e., the energetic distance of the transport involved orbital to the Fermi energy (EF) of metal based electrodes) and the contact resistance of the molecule–electrode interface (Γ, see Fig. 2). In the non-resonant tunneling regime, the conductance of molecular wires decays exponentially with increasing length and is expected to be basically temperature independent.36,37 However, recent experiments also suggest, for tunneling mechanisms, temperature-dependent transport regimes38e.g. due to improved π-coupling in the molecular backbone resulting from thermal excitation of interconnecting bonds and the concomitant variations in dihedral angles39,40 or thermal broadening of the Fermi energy of the contacts.41–43 In contrast, incoherent off-resonant transport, often described as “hopping”, typically exhibits a 1/d distance dependence and increases with increasing temperature, which can be depicted as a series of transitions between neighbouring sites consisting of localized states.44 | ||
| Fig. 2 Illustration of a molecule embedded in metal electrodes; EF, Γ, the HOMO and the LUMO are also depicted. | ||
2. Single-molecule porphyrin junctions
Over the past decade, there have been numerous charge-transport studies on single- or oligo-porphyrin based molecular junctions, using the measuring platforms described in the previous section. Only a few experiments were performed with carbon based electrodes,45–47 and thus, we will focus here on junctions made with metal electrodes, more specifically gold electrodes, unless noted otherwise. The experiments discussed in this review are organized by the nature of the molecular junction and the kind of measured data. The chronological order of experiments performed between 2003 and 2020 can be extracted from Table 1, which is complemented by the publication date of the experiment. Transport through the investigated molecules is usually envisioned in a “current-in-plane” (CIP) fashion, profiting from electrode-anchoring groups mounted at the periphery of the porphyrin scaffold. These exposed groups mechanically fix the molecule to the electrodes and ideally permit low-resistance charge carrier injection into the system (electrons or holes, for the LUMO or HOMO based transport, respectively). In three known studies published in the literature, electrode-coating with thiol functionalized pyridines allowed direct coordination to porphyrin-embedded metal ions, namely, Ni, Co, or Zn. With this approach, charge carriers are transmitted through the perpendicularly aligned porphyrin plane (with respect to the electrode's surface), the so-called “current-perpendicular-to-the-plane” (CPP) pathway. While this set-up of molecular junction seems rather unusual at first glance, it might even be instrumental for the comprehension of some CIP experiments. In many of the CIP junctions reported, a continuous evolution of different configurations is observed. It thus might be that in certain cases, the transition from a CPP junction at very small electrode spacing to the desired CIP configuration at larger electrode separation can be observed. Thus these CPP experiments complement this chapter.| Compound | Anchoring group | Platform | Conductance [G0] | Bias [mV] | Ref. | Year |
|---|---|---|---|---|---|---|
| 1 | Pyridine | STM-BJ/I(s) | 2.6 × 10−4 | 100 | 48 | 2012 |
| 2 | Pyridine | STM-BJ/I(s) | 2.5 × 10−4 | 100 | 48 | 2012 |
| 3 | Pyridine | STM-BJ/I(s) | 3.6 × 10−4 | 100 | 48 | 2012 |
| 4 | Sulfonate | STM-BJ | 6.7 × 10−5 | 100 | 49 | 2013 |
| 5 | Phenol | STM-BJ | — | 100 | 49 | 2013 |
| 6 | Nitrile | STM-BJ | 5.3 × 10−5 | 100 | 49 | 2013 |
| 7 | Amine | STM-BJ | 7.7 × 10−5 | 100 | 49 | 2013 |
| 8 | Benzoic acid | STM-BJ | 3.5 × 10−5 | 100 | 49 | 2013 |
| 9 | Benzene | STM-BJ | — | 100 | 49 | 2013 |
| 10 | Thiol | STM-BJ | 1.7 × 10−5 | 100 | 50 | 2017 |
| 11 | Thiol | STM-BJ | 4.2 × 10−4 | 100 | 50 | 2017 |
| 12 | Thiol | STM-BJ/I(s) | 1.8 × 10−5 | 500 | 53 | 2010 |
| 13 | Thiol | STM-BJ/I(s) | 2.7 × 10−5 | 500 | 53 | 2010 |
| 2.0 × 10−5 | ||||||
| 14 | Thioether | STM-BJ | 4.6 × 10−5 | 400 | 54 | 2014 |
| 15 | Thioether | STM-BJ | 1.9 × 10−5 | 400 | 54 | 2014 |
| 16 | Thioether | STM-BJ | 2.5 × 10−5 | 400 | 54 | 2014 |
| 17 | Thioether | STM-BJ | 3.6 × 10−5 | 400 | 54 | 2014 |
| 18 | Benzene | MCBJ/fast-breaking | HCR at ∼10−1 | 150 | 55 | 2011 |
| LCR at 10−3–10−5 | ||||||
| 19 | Thiol | MCBJ/fast-breaking | HCR at ∼10−1 | 150 | 55 | 2011 |
| LCR at 10−3–10−5 | ||||||
| 20 | Thiol | MCBJ/fast-breaking | 10−1 to 10−5 | 150 | 55 | 2011 |
| 21 | Thioacetate | MCBJ/fast-breaking | HC: 2.8 × 10−4 | 100 | 17 | 2019 |
| MC: 1.1 × 10−5 | ||||||
| LC: 1.0 × 10−6 | ||||||
| 22 | Thioacetate | MCBJ/fast-breaking | HC: 2.4 × 10−4 | 100–300 | 17 | 2019 |
| MC: 2.1 × 10−5 | ||||||
| LC: 4.1 × 10−6 | ||||||
| 23 | Thioacetate | MCBJ/fast-breaking | HC: 1.9 × 10−4 | 100 | 17 | 2019 |
| MC: 4.0 × 10−5 | ||||||
| LC: 1.7 × 10−6 | ||||||
| 24 | Thioacetate | MCBJ/fast-breaking | HC: 2.3 × 10−4 | 100–200 | 17 | 2019 |
| MC: 8.3 × 10−6 | ||||||
| LC: 1.0 × 10−6 | ||||||
| 25 | Thioacetate | MCBJ/fast-breaking | HC: — | 100–300 | 17 | 2019 |
| MC: — | ||||||
| LC: 1.22 × 10−6 | ||||||
| 26 | Phenyl | MCBJ/fast-breaking | HC: — | 100 | 17 | 2019 |
| MC: 5.1 × 10−5 | ||||||
| LC: 2.8 × 10−6 | ||||||
| 27 | Phenyl | MCBJ/fast-breaking | H: — | 100–200 | 17 | 2019 |
| MC: — | ||||||
| LC: — | ||||||
| 28 | Thioacetate | STM-BJ | HC: 3.2 × 10−3 | 230 | 56 | 2018 |
| LC: 7.9 × 10−5 | ||||||
| 29 | Thioether | STM-BJ | HC: 1.0 × 10−3 | 230 | 56 | 2018 |
| LC: 2.5 × 10−5 | ||||||
| 30 | Thioacetate | STM-BJ | HC: 2.5 × 10−3 | 230 | 56 | 2018 |
| LC: 7.9 × 10−5 | ||||||
| 31 | Thioether | STM-BJ | HC: 6.3 × 10−3 | 230 | 56 | 2018 |
| LC: 2.5 × 10−5 | ||||||
| 32 | Thioacetate | STM-BJ | HC: 3.2 × 10−3 | 230 | 56 | 2018 |
| LC: — | ||||||
| 33 | Thioacetate | MCBJ/self-breaking | 10−5–10−6 | 100 | 59 | 2020 |
| 34 | Thioacetate | MCBJ/fast-breaking | 4.1 × 10−6 | 100 | 60 | 2020 |
| 34 | Thioacetate | MCBJ/self-breaking | 4.1 × 10−6 | 100 | 60 | 2020 |
| 35 | PyrMT coated electrode | STM-BJ/l(s) | HC: — | 25 | 61 | 2014 |
| MC: 9.9 × 10−3 | ||||||
| LC: 2.7 × 10−3 | ||||||
| 36 | PyrMT coated electrode | STM-BJ/l(s) | HC: 2.9 × 10−2 | 25 | 61 | 2014 |
| 37 | PyrMT coated electrode | STM-BJ/l(s) | HC: 1.1 × 10−2 | 25 | 62 | 2016 |
| 38 | PyrMT coated electrode | STM-BJ/l(s) | HC: 4.6 × 10−2 | 25 | 62 | 2016 |
| 39 | PyrMT coated electrode | STM-BJ/l(s) | HC: 8.1 × 10−2 | 25 | 62 | 2016 |
| 40 | PyrMT coated electrode | STM-BJ/l(s) | HC: — | 7.5 | 63 | 2020 |
| MC: 2.24 × 10−4 | ||||||
| LC: — | ||||||
| 40 | PyrT coated electrode | STM-BJ/l(s) | HC: — | 7.5 | 63 | 2020 |
| MC: — | ||||||
| LC: — | ||||||
| 41 | PyrMT coated electrode | STM-BJ/l(s) | HC: — | 7.5 | 63 | 2020 |
| MC: 8.08 × 10−3 | ||||||
| LC: 2.31 × 10−3 | ||||||
| 41 | PyrT coated electrode | STM-BJ/l(s) | HC: — | 7.5 | 63 | 2020 |
| MC: — | ||||||
| LC: 1.60 × 10−5 | ||||||
| 42 | PyrMT coated electrode | STM-BJ/l(s) | HC: 9.31 × 10−3 | 15 | 63 | 2020 |
| MC: 3.01 × 10−4 | ||||||
| LC: — | ||||||
| 42 | PyrT coated electrode | STM-BJ/l(s) | HC: 1.51 × 10−4 | 7.5 | 63 | 2020 |
| MC: — | ||||||
| LC: — | ||||||
| 43 | PyrMT coated electrode | STM-BJ/l(s) | HC: 2.82 × 10−2 | 7.5 | 63 | 2020 |
| MC: 9.75 × 10−3 | ||||||
| LC: 3.02 × 10−3 | ||||||
| 43 | PyrT coated electrode | STM-BJ/l(s) | HC: 3.50 × 10−4 | 7.5 | 63 | 2020 |
| MC: — | ||||||
| LC: 1.70 × 10−5 | ||||||
| 44 | Phenyl | STM-spectroscopy | 6.0 × 10−5 | IV | 64 | 2003 |
| 45 | Phenyl | STM-spectroscopy | 2.6 × 10−4 | IV | 64 | 2003 |
| 46 | Thiol | STM-spectroscopy | 3.8 × 10−5 | IV | 67 | 2005 |
| 47 | Thiol | STM-spectroscopy | 1.3 × 10−5 | IV | 67 | 2005 |
| 48 | Thiol | STM-spectroscopy | 1.2 × 10−3 | IV | 67 | 2005 |
| 53 | Thiol | MCBJ | 10−3–10−5 | 100 | 71 | 2011 |
| 54 | Thioacetate | STM-BJ/l(s) | 1.5 × 10−5 | 600 | 83 | 2008 |
| 55 | Disulfide | conductive AFM/STM spectroscopy | 1.8 × 10−5 | IV | 75 | 2008 |
| 59 | Thioacetate | STM-BJ/l(s) | 2.7 × 10−5 | 600 | 83 | 2008 |
| 60 | Thioacetate | STM-BJ/l(s) | 7.7 × 10−6 | 600 | 83 | 2008 |
| 61 | Thiol | STM-BJ | HC: 1.3 × 10−4 | 100 | 84 | 2012 |
| LC: 2.9 × 10−5 | ||||||
| 62 | Thiol | STM-BJ | HC: 9.0 × 10−5 | 100 | 84 | 2012 |
| LC: 2.0 × 10−5 | ||||||
| 63 | Thiol | STM-BJ | HC: 6.0 × 10−5 | 100 | 84 | 2012 |
| LC: 1.4 × 10−5 | ||||||
| 64 | Pyridine | STM-BJ | 2.7 × 10−5 (25 °C) | 600 | 42 | 2011 |
| 5.2 × 10−5 (100 °C) | ||||||
| 64 | Pyridine | STM-BJ/l(s) | 2.2 × 10−5 | 600 | 85 | 2012 |
| 65 | Pyridine | STM-BJ | 1.6 × 10−5 (25 °C) | 600 | 42 | 2011 |
| 4.5 × 10−5 (100 °C) | ||||||
| 65 | Pyridine | STM-BJ/l(s) | 1.1 × 10−5 | 600 | 85 | 2012 |
| 66 | Pyridine | STM-BJ | 9.0 × 10−6 (25 °C) | 600 | 42 | 2011 |
| 3.9 × 10−5 (80 °C) | ||||||
| — (100 °C) | ||||||
| 66 | Pyridine | STM-BJ/l(s) | 0.7 × 10−5 | 600 | 85 | 2012 |
| 67 | Pyridine | STM-BJ/l(s) | 1.0 × 10−5 | 600 | 85 | 2012 |
| 68 | Pyridine | STM-BJ/l(s) | 3.5 × 10−6 | 600 | 85 | 2012 |
| 69 | Pyridine | STM-BJ/l(s) | 1.9 × 10−5 | 600 | 85 | 2012 |
| 70 | Pyridine | STM-BJ/l(s) | 1.6 × 10−5 | 600 | 85 | 2012 |
In this section, we will review the results obtained by break-junction techniques (STM- and MCBJ). We will start with single porphyrin junctions, envisioning CIP transport, followed by three CPP studies. In subsequent sections, we discuss the current–voltage (I–V) characteristics and thereafter β-values gathered from transport studies on single-to-oligo-porphyrin wires, all in a CIP transport configuration. The conductance values obtained for different compounds by the individual techniques are summarized in Table 1, which also provides the publication year of the study.
Borguet's group investigated conductance through highly symmetric tetra substituted porphyrin 1.48 This compound, bearing four 4-pyridyl substituents in the meso-positions for electrode contact, opens two possible junction configurations: the shorter, 90° angled “ortho-pathway” (where the electrodes are in contact with the pyridyl nitrogens of two neighbouring meso-positions); and the longer, linear “para-pathway” (where the electrodes are in contact with the pyridyl nitrogens of two opposite meso-positions). Interestingly, only one single molecular conductance peak was found for 1 in an STM-BJ experiment, located at 2.6 × 10−4G0 at a bias of 100 mV. Here, G0 is the quantum of conductance that is defined as e2/h with e being the elementary charge and h being Planck's constant. To identify the prominent pathway of conductance, two analogue structures to 1, where each of two pyridyls are substituted by benzenes in either a “para” (2) or “ortho” (3) fashion were analyzed (see Fig. 3). Assuming that contact with the electrodes is dominated by the interaction between pyridyl-nitrogens and the gold electrodes, electronic transmission preferably occurs through the “para-pathway” for 2, while 3 is designed to preferably form the junction configuration leading to the “ortho-pathway”. The measured conductance of 3.6 × 10−4G0 for 3 and 2.5 × 10−4G0 for 2 led to the conclusion that in 1 (where both pathways are possible) the longer and less conductive “para-pathway” is counterintuitively favored. This observation was attributed to the greater stability of the “para” connected junction geometry compared to the “ortho” connected one, when pulling up a molecule from the surface by the STM-tip during the breaking-process of the molecular junction. Consequently, in most studies on single-porphyrin BJ experiments, the compounds of interest are functionalized with anchoring groups in these opposite “para” meso-positions.
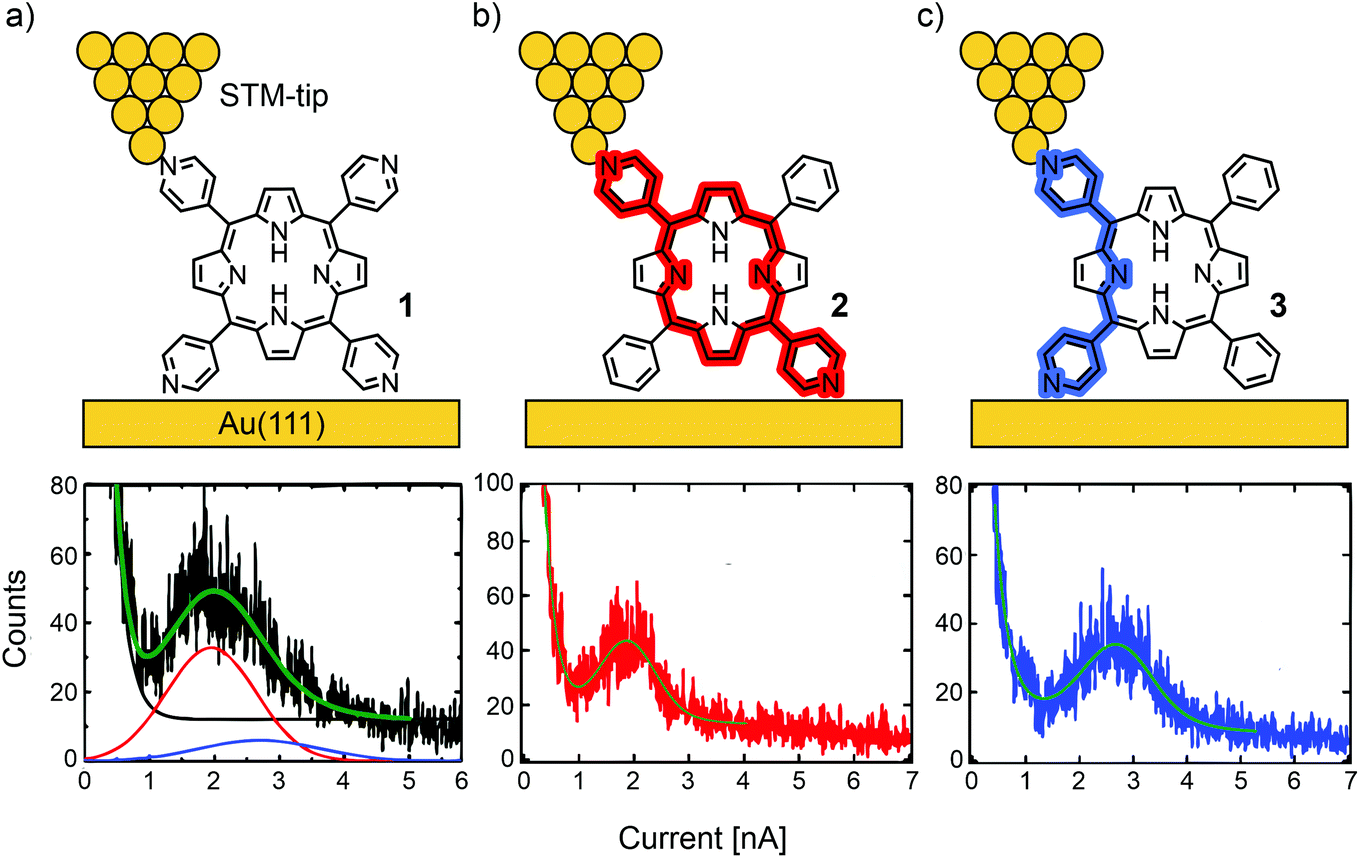 | ||
| Fig. 3 Upper: Chemical structures of 1 (a), 2 (b), and 3 (c) depicted in an STM-BJ. Lower: 1D-histograms of molecular junctions formed by 1 (a), 2 (b), and 3 (c), adapted with permission from ref. 48. Copyright 2012 American Chemical Society. The “para-pathway” is highlighted in red and the “ortho-pathway” in blue. | ||
In a subsequent study, Li et al. investigated the effect of different anchoring groups on single-porphyrin conductance in an STM-BJ set-up.49 Symmetrically tetra substituted porphyrins, bearing pyridyl (–C5H4N, 1), benzylsulfonate (–C6H4SO3−, 4), phenol (–C6H4OH, 5), benzonitrile (–C6H4CN, 6), aniline (–C6H4NH2, 7), benzoic acid (–C6H4CO2H, 8), and unfunctionalized phenyl (–C6H5, 9) in their meso-positions (where the functional groups are positioned para to the porphyrin), were analyzed (see Fig. 4). The measurements show that the conductance follows the sequence of 1 > 7 > 4 > 6 > 8 with values ranging from 2.6 × 10−4 to 3.5 × 10−5G0 (see Table 1). Interestingly, phenol bearing porphyrin 5 and phenyl substituted “unfunctionalized” porphyrin 9 showed no defined peaks in their conductance histograms. Electron-donating and electron-withdrawing groups can alter the orbital energies, but for the described series of porphyrins, the HOMO–LUMO gaps are almost the same. The authors stated that conductance differences should therefore be attributed to factors other than changes in the HOMO–LUMO gap, such as differences in the molecular junction geometries, level alignment, or overall molecular length. For example, the highest observed conductance of pyridyl-terminated porphyrin 1 can then be rationalized by the fact that the nitrogen atoms, being themselves part of the aromatic system, result in the most efficient coupling to the molecular structure combined with the shortest metal–molecule–metal distance.
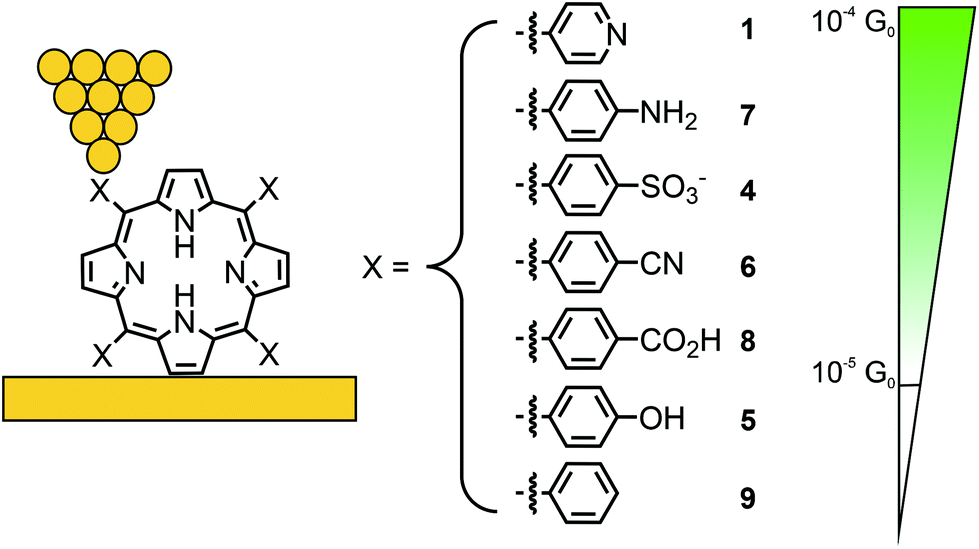 | ||
| Fig. 4 Chemical structures of symmetrically tetra-substituted porphyrins 1 and 4–9 depicted in an STM-BJ. The different anchoring groups used are listed in decreasing conductance from top to bottom; for 5 and 9 no molecular conductance features were observed. | ||
Fujii et al. compared the electrical properties of 4-thiobenzene functionalized Ni(II) porphyrin 10, bearing 3,5-dimethoxy benzene moieties in the lateral meso-positions, to norcorrole analogue 11 (see Fig. 5(a) and (b), respectively).50 The anti-aromaticity of the macrocyclic π-system of 11, comprising 16 π-electrons, leads to destabilization and thus, lowers the HOMO–LUMO gap compared to the aromatic 18 π-electron system of 10. Consequently, the frontier-orbital (the LUMO in this case) is pushed closer to the Fermi level, and an almost 25-fold increase in conductance is observed from 1.7 × 10−5G0 in 10 to 4.2 × 10−4G0 in the case of 11, as analyzed by the STM-BJ technique with an applied bias voltage of 100 mV (see Fig. 5(c)).
 | ||
| Fig. 5 Chemical structures of porphyrin 10 (a) and norcorrole 11 (b) depicted in an STM-BJ. The 18 π-electron aromatic macrocycle in 10 is highlighted in blue and the 16 π-electron antiaromatic macrocycle in 11 is highlighted in red. (c) 1D-histogram of molecular junctions of 10 and 11, adapted form ref. 50 with permission from Springer Nature. | ||
The thiol group is a promising gold anchoring group for single-porphyrin junctions, and molecular junctions in general. The formed covalent Au–S bond upon deposition of a thiol- (often acetate-masked thiol-groups are deposited and the thiol is liberated in situ) functionalized molecule onto a gold surface is stronger than the coordinative Au–pyridine bond, providing molecular junctions of considerable mechanical stability. Interestingly, the kinetics of in situ acetyl deprotection and Au–S bond formation was studied in a STM based break junction experiment with doubly acetyl masked porphyrin 12.51 The authors attribute a low conductance value of about 4–5 × 10−4G0 to junctions formed of porphyrins still bearing an acetyl protection group on one side. The drawbacks of thiol-anchored molecular junctions are the variety of possible binding configurations,52 the increased length of the bridging molecule and the less efficient integration of the anchoring group into the molecule's electronic system.
Acetate-masked arylthiol terminated porphyrin 12, bearing phenyls at the lateral meso-positions, and its Zn(II) metal complex 13 were analyzed by the STM-BJ technique at bias voltages of 500 and 1400 mV.53 While free-base porphyrin 12 showed only one clear conductance peak located at 1.78 × 10−5G0, two overlapping conductance peaks were observed for metal complex 13, located at 2.03 × 10−5 and 2.70 × 10−5G0 (see Fig. 6). The authors’ hypothesis to rationalize the two conductance states of 13 is based on variations of the coordinated Zn(II) ion: the expected conformation with Zn(II) in the porphyrin plane and a surprising one with Zn(II) pulled out of the porphyrin plane, forming a tetrahedral-like geometry, which seems rather unlikely, at least from the viewpoint of a coordination and/or porphyrin chemist.
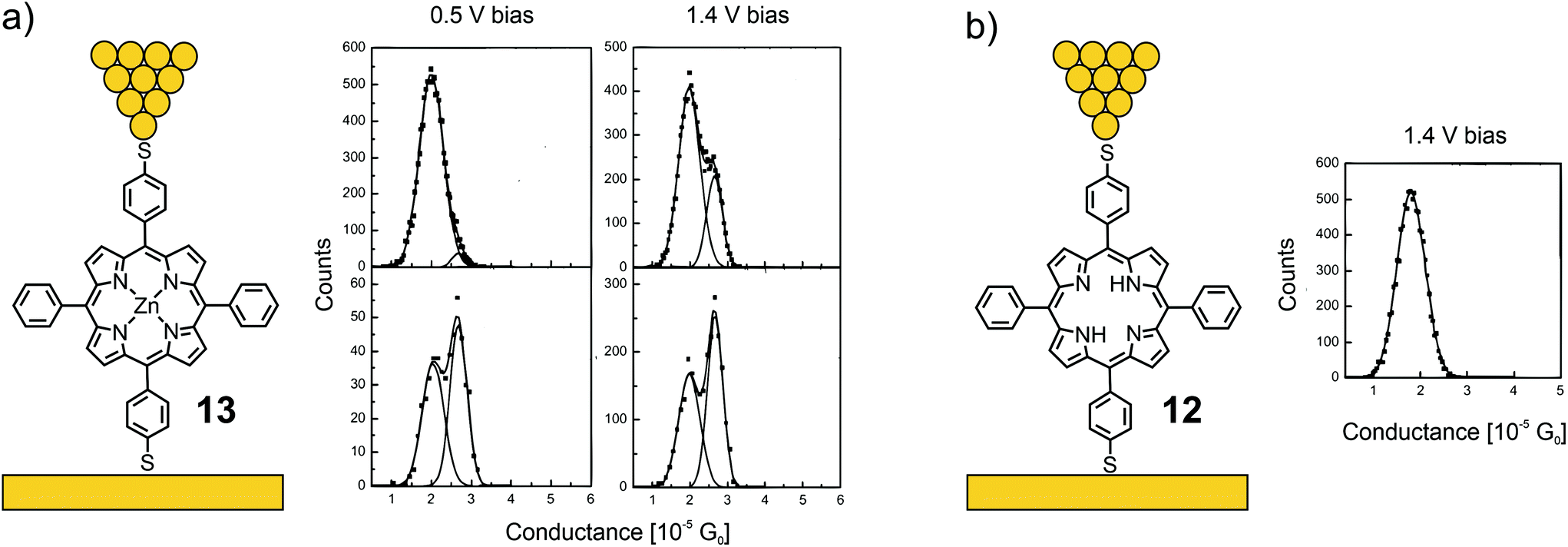 | ||
| Fig. 6 Left: Chemical structures of 13 (a) and 12 (b) depicted in an STM-BJ. Right: 1D-histograms of bistable 13 at 500 and 1400 mV bias (a) and 1D-histogram of 12 at 1400 mV bias (b), adapted from ref. 53 with permission from the American Institute of Physics. | ||
The influence of ligated metal ions on the conductance of single-porphyrin junctions was further studied by Liu et al.54 A dimethyl-thiochroman based anchoring group, installed the opposite meso-positions of the porphyrin (14), was used to form a contact with gold electrodes in a non-covalent fashion (in contrast to the covalent bond formed in the previously shown acetate-masked compounds 12 and 13). In the lateral meso-positions, 3,5-decanoxy substituted benzenes enrich the solubility and presumably hinder undesired molecular junction configurations (or through-space charge carrier injection into the π-electron cloud) as a result of bulkiness and increased van der Waals surface. Free base porphyrin 14 and its metalated Ni (15), Co (16), and Cu (17) analogues were analyzed by the STM-BJ technique at an applied bias voltage of 400 mV (see Fig. 7(a)). One single conductance peak was observed for free base porphyrin 14, the open-shell metal complexes 16 and 17, and the closed-shell metal complex 15, differing only by a factor of approximately 2, in the series of 14 (4.6 × 10−5G0) > 17 (3.6 × 10−5G0) > 16 (2.5 × 10−5G0) > 15 (1.9 × 10−5G0, see Fig. 7(b)). Small changes in transmission suggest a transport path being located on the aromatic porphyrin backbone, without much influence from the metal center.
 | ||
| Fig. 7 (a) Chemical structures of 14–17 depicted in an STM-BJ and (b) the obtained 1D-conductance histogram, adapted with permission from ref. 54. Copyright 2014 American Chemical Society. | ||
Perrin et al. used a series of porphyrin molecules to investigate the influence of the anchoring groups and molecular backbone-modification on single-molecule conductance using the MCBJ-technique.55 To study the former, the electrical properties of symmetrically tetra substituted phenyl porphyrin 18 (bearing no anchoring groups) and thiol functionalized analogue 19 (bearing two anchoring groups in opposite meso-positions) were compared (see Fig. 8). As a control experiment for the latter, octahedral Ru(II) complex 20 bearing two additional axially attached pyridine ligands was characterized. The intention was to sterically shield the π-clouds of the porphyrin plane by the coordinated protruding pyridine ligands.
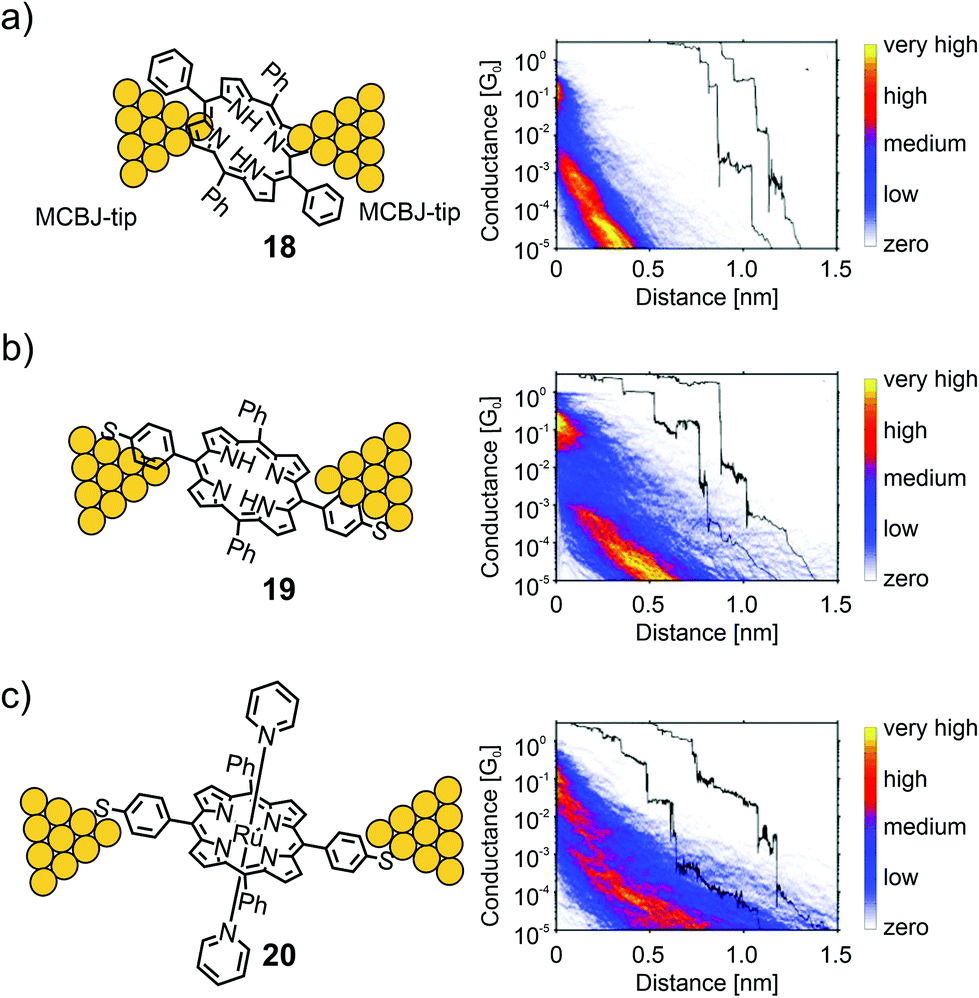 | ||
| Fig. 8 Left: Chemical structures of 18 (a), 19 (b), and 20 (c) depicted in an MCBJ. Right: Corresponding 2D-conductance vs. displacement histograms with off-set exemplary single-traces showing the high-conductance (HC) medium conductance (MC) and low conductance (LC) molecular features, adapted from ref. 55 with permission from John Wiley and Sons. | ||
For all three molecules, flat as well as sloped, not well defined, yet distinguishable plateaus were found at different conductance values in the sub-G0 regime. The sloped plateaus were interpreted as sliding of the molecule along the pointy electrodes, formed in the MCBJ, in combination with through space injection of charge carriers into the π-cloud of the molecule, while being pulled from the electrode's surfaces. In the trace histogram of 18 there are two distinct regions with high counts: a high-conductance region (HCR) around 10−1G0, and a sloped low-conductance region (LCR) below 10−3G0. For 19 both the HCR and LCR are longer than those for 18. Adding the thiol end groups upon changing from 18 to 19 not only increases the plateau length, but also reduces the slope of the LCR. In ruthenium porphyrin 20 the HCR and LCR can no longer be distinguished; only one long region sloping from 10−1G0 to 10−5G0 is present, which has a shallower slope compared to those of 18 and 19, and an increased length. The results showed that porphyrins could form, to some extent, stable bridging molecular junctions in MCBJ experiments. Even without thiol anchoring groups, molecular features different from through-space tunneling can be observed in the conductance versus displacement histograms. Adding thiol anchoring groups and increasing the bulkiness of the porphyrin backbone, respectively, increases the stability of the junctions and leads to an increased spread in conductance, indicating the formation of different junction configurations.
In a follow-up study, Abbassi et al. investigated the impact of the chemical structure of porphyrins on molecular junction configuration formations.17 A series of seven closely related porphyrin derivatives with distinct differences in chemical structure (21–27, see Fig. 9(a)) were analyzed using the same technique (MCBJ).
 | ||
| Fig. 9 (a) Chemical structures of 21–27. (b) Representative depiction of a molecular junction showing class A, B, and C paths. (b, c) Class A, B, and C conductances for the example of 21 in 2D- (b) and 1D- (c) histograms, adapted from ref. 17 published by The Royal Society of Chemistry. | ||
The occurrence of certain classes of conductance was analyzed by an unsupervised clustering algorithm. All four porphyrins exposing 4-thioacetatephenylacetylenes (22–24) in opposite meso-positions showed three different classes of molecular conductance, referred to as class A, B, and C. The difference in bulkiness of the lateral meso-positions and the presence of a central Zn(II) cation did not show much influence on the conductance and probability of the classes. Typically, conductance values in the order of 10−4G0, 10−5G0, and 10−6G0 were observed for class A, B, and C, respectively (see Fig. 9(d)). 25 as an analogue of 21, where one of the anchoring 4-thioacetatephenylacetylene moieties is exchanged for 4-tertbutylphenyl, showed only one class of molecular conductance at 1.2 × 10−6G0 and a sloped shape to the previously observed class C. 26, still exposing an opposite phenylacetylene backbone but missing the terminal thioacetates, showed class B (5.1 × 10−5G0) and class C (2.8 × 10−6G0). 27, exposing only bulky 4-tertbutylphenyls in all four meso-positions, yielded no molecular conductance plateau at all. With this information, class A with the highest conductance and a flat plateau in the 2D histogram (see Fig. 9(b)) was proposed to originate from the mechanically most stable sulfur-to-sulfur configuration with charge carrier injection directly through the anchoring groups (see Fig. 9(b)). Classes B and C were both proposed to originate from through-space charge carrier injection into the π-cloud. Since class B requires both acetylenes to be present (whereas class C does not), it was concluded that class B originated from the acetylene–acetylene and class C from the acetylene–aryl pathway.
Leary et al. studied two Zn(II) porphyrins having either an acetate-masked thiol (SAc) or a methyl thioether (SMe) anchoring group in the ortho-position of the benzenes, which are themselves attached in opposite meso-positions of the porphyrin scaffold.56 These structures exist as a pair of atropisomers, having the functional group either on the same side (28 for SAc, 29 for SMe) or on opposite sides (30 for SAc, 31 for SMe) with respect to the porphyrin plane, which are not interconvertible in solution at room temperature (rt, see panel a) of Fig. 10. Additionally, asymmetric Zn(II) porphyrin 32, where only one of the phenyls bears a gold-anchoring acetate-masked thiol group, was measured. This compound, being able to anchor to only one of the electrodes, served as a reference compound to identify certain configurations. For all five compounds (28–32) a sloped and short plateau at high conductance (1.0 to 6.3 × 10−3G0 with lengths of 0.76 to 1.02 nm, see Table 1) was observed, which was attributed to a tilted configuration (with respect to the substrate) and through-space charge-carrier injection at one electrode, as depicted for 32 in panel (b) of Fig. 10. Indeed, the four compounds each bearing two terminal anchoring groups (28–31) showed (less) sloped and longer plateaus at lower conductance around 10−5G0, which was attributed to the desired anchoring groups being connected via through-bond transport. While both atropisomers of methyl thioether functionalized porphyrins 29 and 31 showed the same conductance value of 2.5 × 10−5G0, a clearly distinguishable difference in the lengths of the two plateaus was observed (0.92 nm and 1.10 nm, respectively, see panel (c) of Fig. 10).
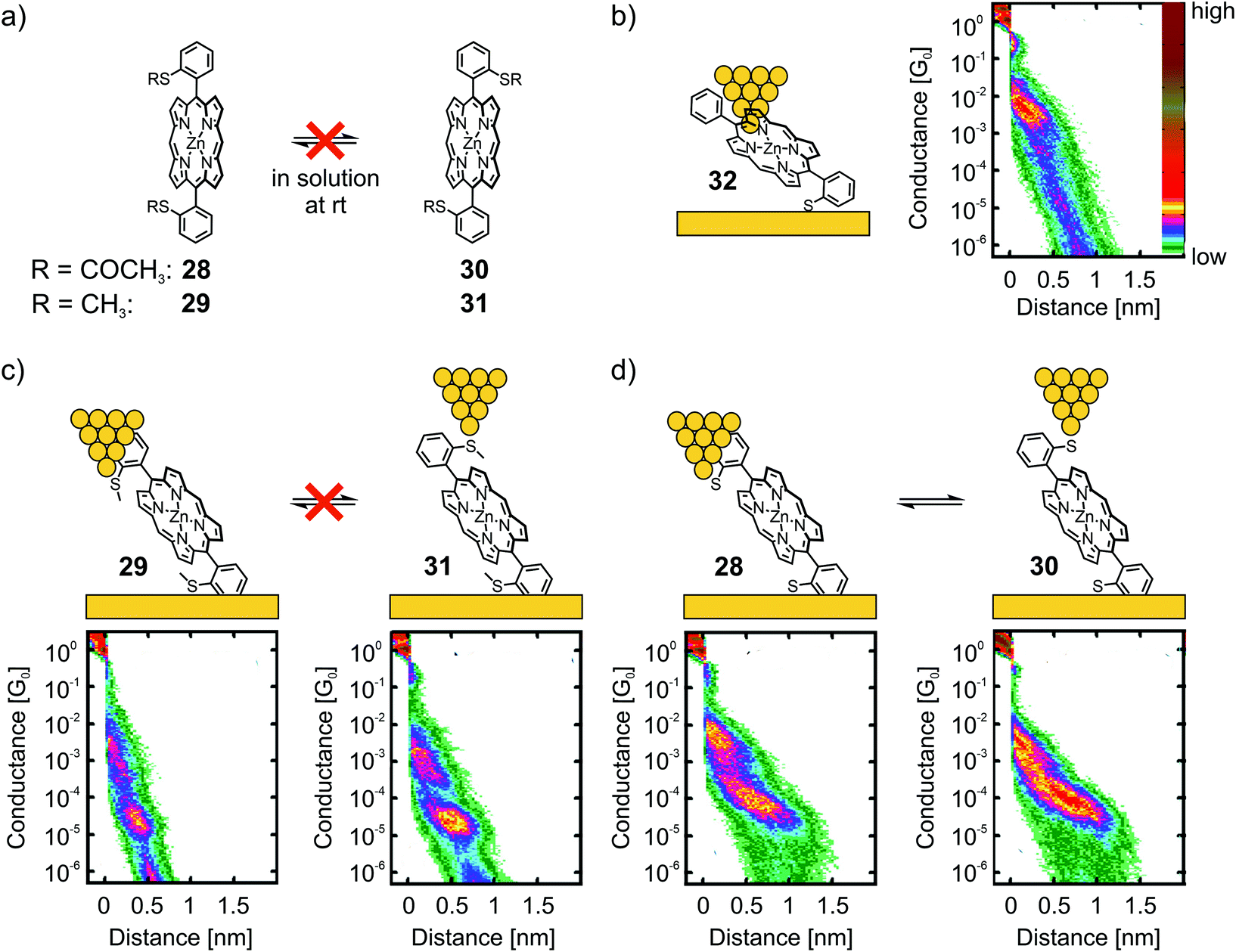 | ||
| Fig. 10 (a) Chemical structures of the atropisomeric pairs 28, 30 and 29, 31, highlighting their stability at room temperature in solution. (b) Chemical structure depicted in an STM-BJ (left) and 2D histogram (right) of reference compound 32. (c) Chemical structures depicted in a STM-BJ (upper) and corresponding 2D histograms (lower) of the non-interconvertible pair of atropisomers 29 and 31. (d) Chemical structures depicted in a STM-BJ (upper) and corresponding 2D histograms (lower) of the interconvertible pair of atropisomers 28 and 30. The 2D-histograms in (b)–(d) are adapted from ref. 56, https://pubs.acs.org/doi/10.1021/jacs.7b10542. Further queries related to the material excerpted should be directed to the ACS. | ||
Interesting differences were observed during the investigation of acetate-masked thiol functionalized porphyrins 28 and 30. Upon immobilization, the resulting molecular junction with thiol-connected porphyrins displayed similar conductance values of 7.9 × 10−5G0 (see panel (d) of Fig. 10). Interestingly, the plateaus are of comparable length (1.47 and 1.40 nm, respectively) and thus considerably longer than the plateaus observed for the previously mentioned pair of atropisomers (29 and 31). This observation was attributed to mechanochemical isomerization of 28 and 30 into each other, facilitated by deformation of the chemical structure when stress was applied to the molecular junction by increasing (or decreasing) the tip-to-surface distances. The plateaus of 28 and 30 were not only indistinguishable from each other, they were also significantly longer than the expected molecular length. This observation was interpreted to result from further deformations of the molecular junction prior to rupture of the molecular junction. This striking finding further corroborates the increased mechanical stability of the thiol–gold bond compared with the coordinative interaction of the methyl thioether anchoring group. Interestingly, the strong S–Au bond57 is, obviously, even strong enough to enable conformational- and configurational manipulation of molecular junctions by mechanical control.
Another approach to manipulate a single molecule junction mechanically was reported by Stefani et al.58 A porphyrin-inspired calix[4]pyrrole was decorated with a pair of para-quaterphenyl rods, mounted via benzylic linkers to the opposite nitrogen atoms of the calix[4]pyrrole macrocycle, while the exposed termini of the rods were functionalized with methyl thioethers as anchoring groups coordinating to the gold electrodes. An investigation of mechanically manipulated single molecule traces in a MCBJ experiment displayed long, slow decaying conductance traces, which were interpreted by the authors as a signature of the stretching deformation of the immobilized single molecule.
Zwick et al. developed the CIP concept further by considering model compounds that enabled them to analyze the interactions between two neighboring porphyrins, which were both functionalized to enable CIPs individually. Porphyrin cyclophanes with terminal thioacetates were designed and synthesized to analyze the mechanical sensitivity of the resulting molecular junction. A first design based on phenylethynyl-scaffolding yielded bicyclic structure 33 with a surprising C–C bond interlinking both porphyrin subunits after the key two-fold Sonogashira–Hagihara macrocyclization. The electronic transparency of the porphyrin dimer exposing thiol anchoring groups was analyzed using an MCBJ with a bias voltage of 100 mV under ambient conditions (see Fig. 11(a)).59 No clear conductance plateau was observed in the molecular traces (890 of 10000 consecutive traces), which was interpreted as an indication of numerous possible configurations of molecular junctions with comparable probability instead of the desired well-defined sulfur-to-sulfur molecular junction sketched in Fig. 11(b). From self-breaking experiments, the averaged conductance of the molecular junctions was estimated to be 10−5–10−6G0. Nevertheless, the experiment documented that in spite of the expanded structure with its remote anchoring groups, the multi-level porphyrin architecture still provided electronic transparency, enabling its analysis in MCBJ experiments.
 | ||
| Fig. 11 (a) Chemical structure of 33 depicted in an MCBJ. (b) 2D-histogram of the filtered conductance (all traces other than through-space tunneling between the two electrodes) vs. displacement of molecular junctions of 33, adapted with permission from ref. 59. Copyright 2020 American Chemical Society. | ||
In a subsequent study, the second generation of a porphyrin cyclophane was presented. In 34 (Fig. 12(a)) both porphyrins are interlinked by lateral xanthene acetylenes and expose a thiol anchoring group each.60 The revolving acetylene connections between porphyrin and xanthene are expected to enable the manipulation of the relative position of both porphyrins mechanically. In combination with the already mentioned mechanically stable thiol anchoring groups, this made the model compound particularly appealing for MCBJ experiments. The quite advanced mechanosensitive porphyrin cyclophane structure was assembled in a multi-step procedure with a four-fold Sonogashira–Hagihara macrocyclization as the key step. In the first investigation, the filtered molecular traces of this 3D structure showed a clear and flat plateau in the 2D conductance versus electrode displacement histogram with an average value of 4.1 × 10−6G0 reaching ∼2.5 nm with an applied bias voltage of 100 mV. This measured conductance value agrees with the one recorded in self-breaking experiments, indicating very comparable arrangements of the molecule in both experiments and most likely the desired sulfur-to-sulfur molecular junctions. According to the authors, mechano-sensitivity studies of these junctions are currently being performed and will be part of future work.
 | ||
| Fig. 12 (a) Chemical structure of 34 depicted in an MCBJ. (b) 2D-histogram of filtered molecular traces of 34 as adapted with permission from ref. 60. Copyright 2020 American Chemical Society. | ||
Díez-Pérez's group developed a method to build single-porphyrin STM based break junctions using electrodes coated with thiol functionalized pyridines. Upon deposition of a metal, embedded in the square-planar ligand field of a porphyrin, axial coordination to two opposite pyridyl nitrogens perpendicular to the porphyrin plane is executed, enabling direct connection to the embedded metal center (see Fig. 13).
 | ||
| Fig. 13 (a) Electrode coating demonstrated using 4-thiopyridine followed by deposition of a metal porphyrin. (b) Envisioned CPP transport pathway through the electrode-coordinated porphyrin plane, highlighted in red. | ||
The so-called “blinking method” was used, i.e., the I(t) method, to investigate charge transport perpendicular to the porphyrin plane in junctions connected this way. In their first study,61 they compared the conductance of a porphyrin bearing phenyls in opposite meso-positions (35), which has no metallic center and therefore no strong interaction with the pyridinyl groups on the electrode surfaces, and its Co(II) complex (36) with a divalent metal center allowing for coordination to the electrode's pyridinyl surface, affording a hexacoordinate system (see Fig. 14(a)). Both 35 and 36 show two common low (LC) and medium (MC) conductance peaks at around 10−3G0 (2.7 and 9.9 × 10−3G0 for 35). Cobalt complex 36 shows an extra high (HC) conductance located at 2.9 × 10−2G0 (see Fig. 14(b)). More insights into the origin of the three conductance levels were obtained by analyzing blinks (spontaneous formation of a molecular junction) for Co complex 36 at different, fixed tip-to-surface gap separations. When the tip-to-surface separation is large, the most frequently observed blinks are the lowest conductance ones, as the tip-to-surface distance diminishes, the medium conductance level dominates, whereas the high-conductance level dominates at the smallest separations. The high conductance level furthermore shows greater mechanical stability; the high conductance value is attributed to the generation of an extended electronic pathway between the two electrodes by molecular orbital delocalization along the junction's main axis as a consequence of pyridine–Co-pyridine coordination, i.e., yielding a pathway perpendicular to the plane of the porphyrin. The LC and MC conductance levels that are common for both 35 and 36 are attributed to hydrogen bonding interactions between the pyridine units and the porphyrin backbone, which could happen at multiple sites.
 | ||
| Fig. 14 (a) Pyridine–thiol used for electrode coating and the chemical structures of 35 and 36. (b) 1D-histograms of 35 and 36 in an STM-BJ, showing the low conductance (LC), medium conductance (MC), and high conductance (HC) peaks, adapted with permission from ref. 61. Copyright 2014 American Chemical Society. (c, d) Junction configuration yielding the HC peak depicted for the examples of 36–39 (c) and experimental and theoretical values of the HCs of 36–39 (d), adapted from ref. 62 with permission from Springer Nature. | ||
In a subsequent study,62 they investigated perpendicular transport in single-molecule junctions connected via different metal cores embedded in a porphyrin bearing phenyls in opposite meso-positions, namely, Co (36), Ni (37), Cu (38), and Zn (39, see Fig. 14(c)). The STM-based conductance measurements combined with quantum transport calculations demonstrate that CPP junctions have three-orders-of-magnitude higher electrical transparency than that of their CIP counterparts, ranging from 1.1 × 10−2G0 for Ni complex 37 up to 8.1 × 10−2G0 for Zn complex 39. The metal ion in the center of the diphenylpoprhyrin skeleton is strongly coordinated with the nitrogens of the pyridyl coated electrodes, with a binding energy that is sensitive to the choice of metal ion. They found that the binding energies of Zn complex 39 and Co complex 36 were significantly higher than those of Ni and Cu complexes 37 and 38, respectively. They identified 39 (Zn) as the favored candidate for high-conductance CPP single-molecule devices. Both theory and experiment reveal that the variation in conductance across this family of molecules increases in the order Ni < Co < Cu < Zn (see Fig. 14(d)).
More recently, they investigated the effect of different linkers (for electrode coating) on the configurations of supramolecular junctions using the same technique with an applied bias voltage of 7.5 mV.63 They used pyridine-4-thiol (PyrT) and the previously used pyridin-4-ylmethanethiol (PyrMT) as electrode coating materials to study junctions after exposition to porphyrin 40, 5,15-bisphenyl porphyrin 41, and their Co complexes 42 and 43, respectively (see Fig. 15(a)). Similar to previous experiments, HC, MC, and LC peaks were found for all porphyrins (40, 41, 42, 43), and MC and LC peaks for all free-base porphyrins (40 and 41), using PyrMT as the linker. Junctions formed by PyrT coated electrodes, showed HC peaks for all Co porphyrins (42 and 43). The previously observed MC peak was only found to be present with the PyrMT linker in all compounds. Using PyrT, however, the MC was found to be completely diminished for all compounds and LC peaks were only observed in phenyl bearing porphyrins (41 and 43). In general, conductance values were found to be two orders of magnitude higher for PyrMT coated electrodes compared to PyrT coated ones. Carefully comparing these findings, the authors proposed the formation of the responsible transport configurations, as supported by theory. While the HC peak still clearly seems to originate from the envisioned nitrogen–metal–nitrogen configured CPP transport, the MC peak most probably relies on the possibility of π–π interactions between the pyridine and the porphyrin. The absence of MC in PyrT coated junctions can then be explained by the rigidness of PyrT, standing up from the electrodes, unable to sufficiently approach the porphyrin spatially, in contrast to PyrMT, which has sufficient degrees of freedom. This π–π sandwich configuration between the pyridines and the metal (for HC), porphyrin backbone (for MC), or side-group phenyl (for LC) using PyrMT, in contrast to the laterally arranged architecture bonded to PyrT, is supposed to be responsible for the two orders of magnitude increase in conductance. A schematic diagram of the molecular junction configurations elucidated from the measurements, with the conductance values represented on the x-axis in G0 units, is given in Fig. 15(b). Conductance values of all configurations of all measured porphyrins (40, 41, 42, 43) using both electrode coating materials, PyrT and PyrMT, are listed in Table 1.
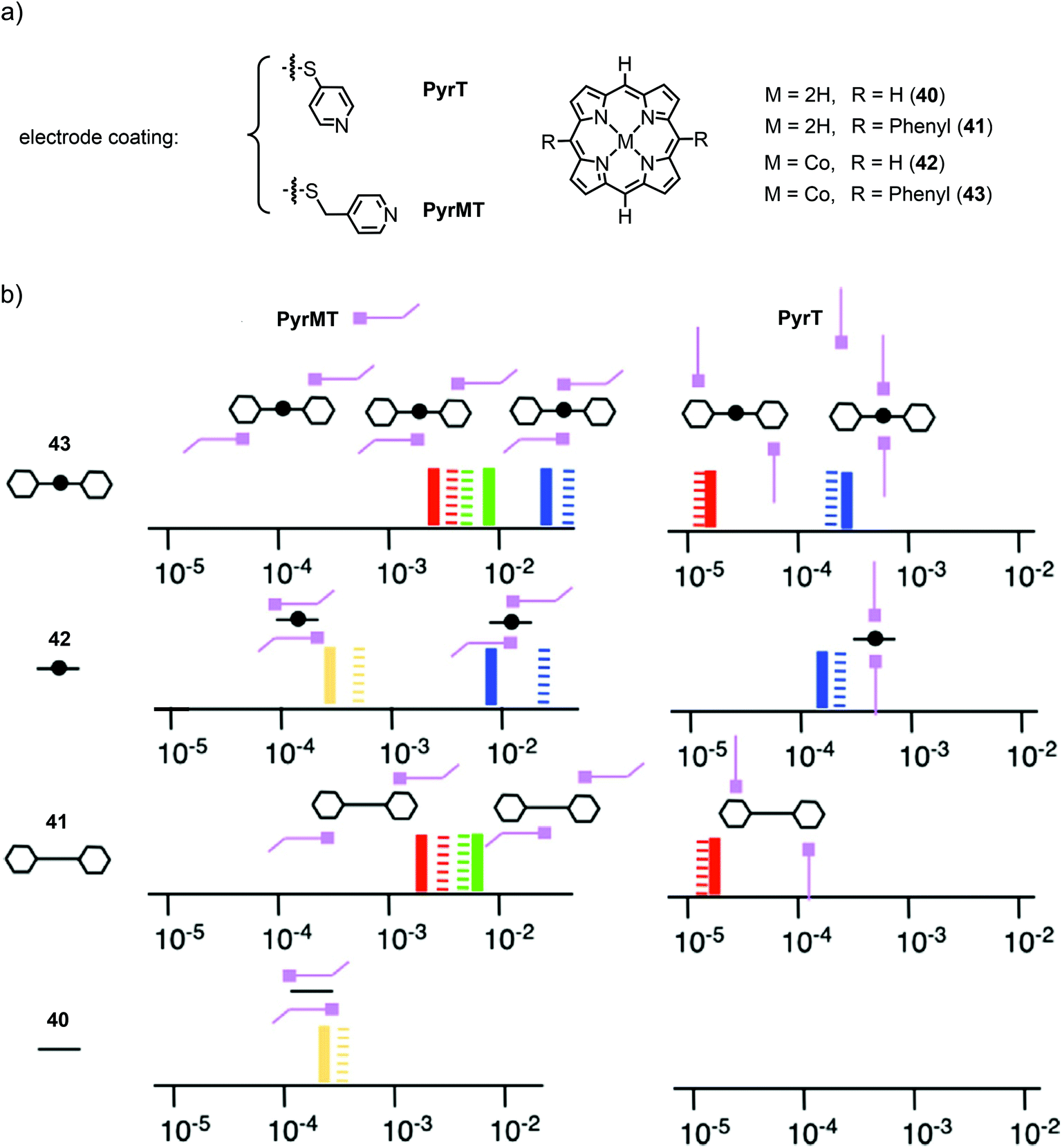 | ||
| Fig. 15 (a) Chemical structures of the electrode-coating materials PyrT and PyrMT, and the investigated free-base and Co(II) porphyrins 40–43. (b) Schematic diagram of the molecular junction configurations elucidated from measurements of the porphyrins. The conductance values are represented on the x-axis in G0 units (solid lines are the experimental values, striped lines the theoretical values) for both linkers (PyrT and PyrMT) and the four studied porphyrin systems 40, 41, 42, and 43. Schematic representations of the supramolecular structural motifs, confirmed by DFT, are depicted for each conductance signature. The color legend represents equivalent interactions across all junctions: blue corresponds to CPP transport (both pyridine linkers interacting with the metal center), green represents one pyridine interacting with the metal center and the other with a phenyl group, red indicates both pyridine linkers interacting with phenyl rings, and yellow corresponds to two pyridine linkers interacting with the aromatic ring of the porphyrin, as adapted with permission from ref. 63. | ||
In summary, the experiments show that several factors control charge transport in porphyrin molecules: the orientation of the molecule, the anchoring groups and the type of metal ion coordinated in its core. Specifically, charge transport in porphyrin molecular junctions is possible along two directions, either within the porphyrin plane with characteristic conducting values between 10−5 and 10−4G0 or perpendicular to the plane with stable junction formations achieved through the central metal incorporated into the porphyrin coordinated to pyridyl-coated electrodes. Conductance values reported in the latter case are higher, in the range of 10−2–10−1G0. The metal ion influences the conductance and the effect is more pronounced in CPP transport (a factor of four for the metals discussed above) compared to CIP, probably caused by a lack of communication between the charge carrier (being mainly localized on the aromatic π system of the porphyrin) and the incorporated metal cation in CIP transport. Interestingly, the variation in conductance upon changing the metal ion is consistent in both transport directions as it decreases in the order Zn > Cu > Co > Ni.
Anchoring groups provide a method to modify the molecular junction and charge transport through porphyrins, as reported in studies using the CIP transport pathway. The anchoring group triggered modulations were rather moderate with conductance variations of less than an order of magnitude for a number of different anchoring groups. However, in experiments investigating the mechanosensitivity of molecular junctions (like the ones with the porphyrin cyclophane),56 not only the electronic coupling of the molecule to the electrode has to be considered but also the mechanical stability of the molecular junction. Therefore, to optimize a model compound for a particular experiment, the choice of the anchoring groups and their position in the porphyrin structure have to be chosen by considering their role during the experiment as well as the electrode material.
In many of the shown experiments, multiple conductance values have been reported for the same set-up, indicating different molecular-junction configurations, even with the same anchoring groups. These include configurations with molecules lying flat on a surface (CPP transport), molecules in a tilted configuration with the substrate in between these two states, and molecules bridging two electrodes in a straight way (CIP transport), whereas these configurations continuously evolve into each other during the opening of the electrodes. Usually, high-conductive short plateaus are observed when the molecule is lifted up from the electrode's surface. During elevation of the tip, the molecular junction transforms into the anchoring-group connected configuration yielding the more stable, less conductive (and desired) in-plane transport. In addition to these possible flat-to-stretched conformations, the through-space injection of charge carriers via the π-clouds of the macrocycle or via side groups connected to the porphyrin ring create a plethora of possible junction configurations in both CIP and CPP transport. However, electron transmission through the porphyrin backbone is consistently efficient in all of the reported experiments and different probed transparencies arising by competitive pathways were generally readily distinguishable.
3. Current–voltage characteristics
In this section, we review current–voltage (I(V)) characteristic studies on porphyrins. The experiments are grouped into two subsections: one containing the results from planar device geometries and one with the results from the scanning probe techniques (STM and conductive-probe AFM (CP-AFM)), while largely omitting STM spectroscopy measurements on porphyrins lying flat on surfaces.Planar device geometries
The first I(V) spectroscopy experiments were performed by Yoon et al.64 on two types of porphyrin chains end-capped by non-functionalized phenyls: meso–meso-linked arrays consisting of 48 twisted Zn(II) porphyrins (44) and shorter and flat beta–beta, meso–meso, beta–beta linked arrays consisting of 8 fused Zn(II) porphyrins (45) in which the constituent porphyrin macrocycles possess a larger π-conjugation (see Fig. 16). Such meso–meso linked oligomers (like e.g.44) are typically synthesized by a repetitive dimerization strategy profiting from Ag(I) salt promoted oxidative coupling. However, 48-mer 44 was isolated as a side product during the dimerization of the 16-mer to the 32-mer.65 The triply linked tapes can then be obtained by oxidative double-ring closure (ODRC) from the respective meso–meso linked oligomer.66 For longer tapes, e-beam lithography was used to prepare gold electrodes with 20–30 nm spacing, whereas electrodes suitable for measuring the shorter molecules, with 7 nm spacing, were prepared using the electromigration-induced break-junction technique. Electrical contact between the porphyrin arrays and metal electrodes was achieved by using electrostatic trapping. | ||
| Fig. 16 Chemical structures of phenyl end-capped meso–meso linked porphyrin wire 44, terminally thiol-functionalized meso–meso linked wires 46 and 47, phenyl end-capped beta–beta, meso–meso, beta–beta linked tape 45, and terminally thiol-functionalized beta–beta, meso–meso, beta–beta linked tape 48. | ||
At room temperature, diode-like behavior and a hysteresis that depended on the voltage sweep direction was found in the I(V) curve of 44. This behavior was attributed to conformational heterogeneity arising from the dihedral angle distribution between neighboring porphyrin subunits in 44. On the other hand, the I(V) curves for 45 were nearly symmetrical, without hysteresis, with a higher conductivity and exhibiting a smaller gap in the current around zero voltage. The authors reported that these results indicated that the stronger π-electron conjugation resulted in better electrical conduction. In a follow-up study, using the same measuring platform,67 the length and temperature dependence in meso–meso linked arrays consisting of 24 and 64 Zn(II)porphyrins (46 and 47) end-capped by 4-thiophenyl gold-anchoring groups were studied and compared to the ones from a short porphyrin tape consisting of two beta–beta, meso–meso, beta–beta fused zinc(II)porphyrins (48, see Fig. 16). A higher conductivity and smaller voltage gap were found than in the fused porphyrin arrays (48). Concerning 46 and 47, the conductance decreased with increasing molecular length and thermally activated transport was found at high temperatures with an energy barrier of 0.35 eV for all samples. This behavior was attributed to thermally activated hopping, although one has to note that this conclusion is based on only two data points.
At low temperature (11 K), Noguchi et al. investigated the cobalt complex of a tetra phenyl porphyrin bearing bulky tert-butyl groups in opposite and acetate masked thiols in the lateral positions (49) using electromigrated break junctions.68I(V) characteristics of 158 junctions with 49 and 163 reference junctions without the molecules were measured. The conductance of the molecular junctions was significantly higher than that of empty junctions. They observed four types of I(V)s: (i) curves with currents below the detectable limit, (ii) curves with a staircase feature, (iii) asymmetric curves, and (iv) symmetric tunneling like curves (see Fig. 17(a)). This variation in I(V) characteristics was attributed to microscopic differences in the structure of the nanogaps, in the electrode/molecule interface, and in the molecular arrangement. Staircase I(V)s were observed in both (molecular and empty) junctions and can thus not serve as a hallmark for molecular behavior. A gate dependence was observed in some samples, but characteristic Coulomb diamonds expected for (molecular) quantum dots and resonant transport were found only in two of the junctions containing 49. From fitting the data, the tunneling barrier height or level-alignment (i.e., the distance to the closest orbital) was estimated to be smaller than 0.5 eV. Saha et al.69 also used the electromigration technique to investigate the zinc complex of a similar porphyrin structure, 50, with bare phenyls in the lateral positions. Room- and low (4.2 K) temperature experiments of molecular junctions were compared to those of empty control junctions (see Fig. 17(b)). Most of the samples (17 out of 25) showed an asymmetry at higher negative bias voltage, which was attributed to an asymmetric wave function distribution across the molecule. Half of the molecular junctions (two out of four that were tested for it) showed a gate dependence and the observed current suppression with increasing gate voltage was consistent with HOMO mediated charge transport.
 | ||
| Fig. 17 (a) Chemical structure of 49 (left) and four different observed I(V) curves at 11 K (right), as adapted from ref. 68 (permission from the Japanese Society of Applied Physics is currently under request). (b) Chemical structure of 50 (left) and molecular I(V) curves at room temperature and 4.2 K (right), adapted from ref. 69 with permission from the American Institute of Physics. | ||
The same group reported room temperature dependent bistability of an electromigrated molecular junction made with immobilized porphyrins 51 and 52, with triazole attached rigid rod-type subunits exposing a terminal anchor group. The synthesis strategy permits the anchoring group to be altered by 1 3-dipolar cycloaddition (also known as Huisgen or “click” reaction) from the same porphyrin precursor. While hysteretic bistability was observed at 300 K, a smooth and structureless I(V) was recorded at 4 K in the case of the oligo(phenylene-ethynylene) decorated 51 (Fig. 18). However, xylene decorated 52 displayed the opposite behavior: hysteretic bistability at 4 K and featureless I(V)s at 300 K. Molecular dynamics simulations suggest local conformation dynamics of dihedral angles of the molecular structure as a potential explanation for the temperature dependent phenomenon. In addition, the authors hypothesize that low frequency phonons may contribute to structural changes and bistability in the porphyrin systems.70
 | ||
| Fig. 18 (a) Chemical structures of 51 and 52. I(V) traces recorded with an electromigrated molecular junction with immobilized 51 at (b) 300 K and at (c) 4 K. The bistability in (b) at ±0.3 V was observed with a voltage sweep of 0→−0.5→0.5→0 (V). The I(V) curves of (b) and (c) are adapted with permission from ref. 70. Copyright 2015 American Chemical Society. | ||
The MCBJ technique at room- and low (6 K) temperature has also been used to study I(V) characteristics. The technique permits the combination of low-bias conductance statistical measurements with spectroscopy of the molecular levels to probe different junction configurations and monitor changes in the molecular-level alignment upon fusing and breaking of a molecular junction. With this method, Perrin et al.71 investigated charge transport through a Zn porphyrin with 4-thiophenyl anchors in opposite and p-tolyl groups in the lateral meso-positions (53, see Fig. 19(a)).
 | ||
| Fig. 19 (a) Chemical structure of 53 depicted in an MCBJ single-molecule junction. (b, c) I(V) characteristics (b) and differential conductance (c) for MCBJ devices that have been exposed to a solution of 53. (d, e) 2D-histograms of dI/dV for junctions containing 53 as a function of bias voltage and electrode displacement during fusing for the sample used in (d) (Sample A) and for three making/breaking cycles of a different sample, as plotted in (e) (Sample B). (f) I(V) characteristics of a control sample, which was not exposed to 53. (b)–(f) are adapted from ref. 72 with permission from Springer Nature. | ||
By repeatedly stretching and fusing the molecular junction, multiple stable single-molecule junction configurations were observed, which exhibited different spectroscopic features, while the low-bias conductance value remained approximately the same. In a subsequent study,7253 was studied using two- and three-terminal MCBJs at 6 K. Evidence of electrode-induced energy gap renormalization was demonstrated. When increasing the electrode separation, a substantial increase in the transport gap occurred with level shifts as large as several hundreds of meV for displacements of a few Å (see Fig. 19(b)–(f)). Analysis of this large and tunable gap renormalization with image-charge calculations73 confirmed the dominant role of image-charge effects in single-molecule junctions and the renormalization of their energy levels in the weak electrode-molecule coupling limit.
The measurements of symmetric junctions shown commonly demonstrate that the electrode configurations – even in the same measurement platform – permit different transparencies (arising from different geometries) to be probed. They typically indicate the presence of resonant transport with Coulomb blockade like features (weak electrode–molecule coupling with incoherent two-step charge carrier transport) or near-resonant transport with level misalignments in the order of 0.5 eV. The observed spectroscopic features are generally in line with HOMO–LUMO gaps that are much smaller than the ones expected from the gas phase. Gap renormalization by (dynamic) image charges has shown to be of crucial importance in explaining this reduction.
Scanning probe based spectroscopy measurements
I(V) characteristics of individual or a few porphyrin molecules isolated in an alkanethiol matrix have been studied using STM and conducting AFM methods. Bennett et al.74 employed STM spectroscopy to study thiol-terminated, butadiyne bridged porphyrin dimer 54 (see Fig. 20(a)) isolated in an alkanethiol matrix, and self-assembled monolayers of the same molecules. A large number (10000) of I(V) curves have been analyzed in terms of Fowler–Nordheim tunneling curves (ln(I/V2) versus 1/V). 70% of these curves display a minimum at a characteristic transition voltage, Vm. For isolated molecules, the transition voltage shows a multi-peaked distribution of values, indicating the presence of a small number of distinct molecular/contact configurations, each having a different transport characteristic. For self-assembled monolayers, in contrast, a single-peaked distribution is observed, which is consistent with parallel conduction through many molecules. Numerical calculations showed that bias asymmetry and variation in the Vm values could be caused by changes in the relative degree of coupling of the two ends of the molecule to the nanoscale contacts.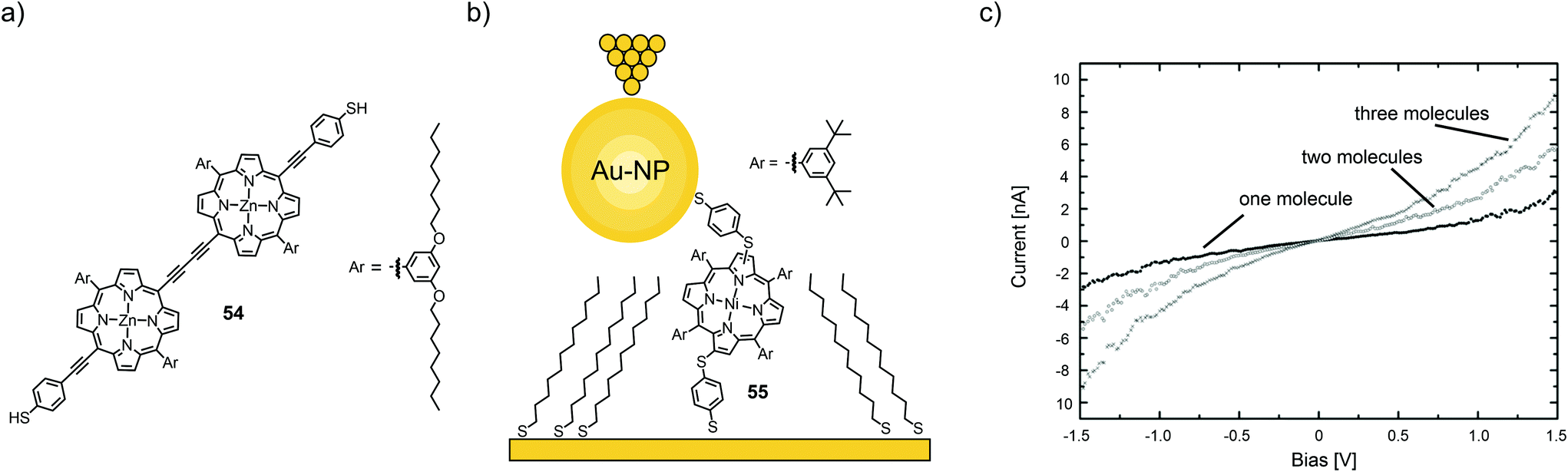 | ||
| Fig. 20 (a) Chemical structure of 54. (b) Chemical structure of 55 depicted in a thioalkane self-assembled monolayer (SAM) with opposite tip-contact via a chemisorbed gold nanoparticle (Au-NP) in an STM-BJ. (c) Three distinct I(V) characteristics observed for the junctions depicted in (a) corresponding to one, two, or three molecules bridging the circuit in parallel, adapted from ref. 75 with permission from The Royal Society of Chemistry. | ||
Using a combination of STM and CP-AFM, Xi et al.75 investigated a nickel-chelating porphyrin with antipodally arranged thiol anchors (55) and its self-assembly on gold electrodes as either films or isolated in a saturated alkyl thiol (1-tetradecanethiol) self-assembled monolayer and then capped with chemisorbed gold nanoparticles (see Fig. 20(b)). Statistical measurements (1000 junctions) revealed three distinct groups of I(V) curves, corresponding to the conductance of one, two, and three molecules, which exhibit integer multiples of a fundamental molecular conductance of 1.8 × 10−5G0 (see Fig. 20(c)). This value compares well with that obtained for isolated molecules and is significantly higher than the conductance of the host matrix. In all cases, I(V) trends are well described by a non-resonant tunneling model.
The Batteas group studied a free-base tripyridyl porphyrin attached to Au surfaces by a thiol tether through a tetrafluorophenyl linker (56, see Fig. 21(a)).76 When inserted into a background matrix of 1-dodecanethiol, the porphyrins tend to bind near the edges of defects within the thioalkane layer – either as single molecules or as small ensembles (ca. three to five molecules). I(V) curves from the single-molecule domains appeared roughly symmetrical, with an upturn in current flow around −1 V and +1.5 V. Larger (6 nm) domains showed a symmetrical upturn in current around 1 V, with two types of I(V) curves: one with a distinct current blockade and one without. The authors attributed the behavior to local intermolecular interactions shifting the HOMO–LUMO levels. From DFT calculations, the low observed tunneling efficiency can be attributed to the lack of effective orbital overlap between the frontier orbitals of the porphyrin macrocycle and the thiol tether, due to the orthogonal dihedral angle between the porphyrin ring and the perfluorophenyl linking group. In addition to the electronic effects on the phenyl ring, the fluorine atoms increase the rotation barrier and diminish the dynamics of the aryl–porphyrin bond (due to the increased dimensions of fluorine compared to a hydrogen atom). Thus, this group introduces an effective tunneling barrier within the molecule, despite the close proximity of the HOMO of the molecule to the Fermi level of Au.
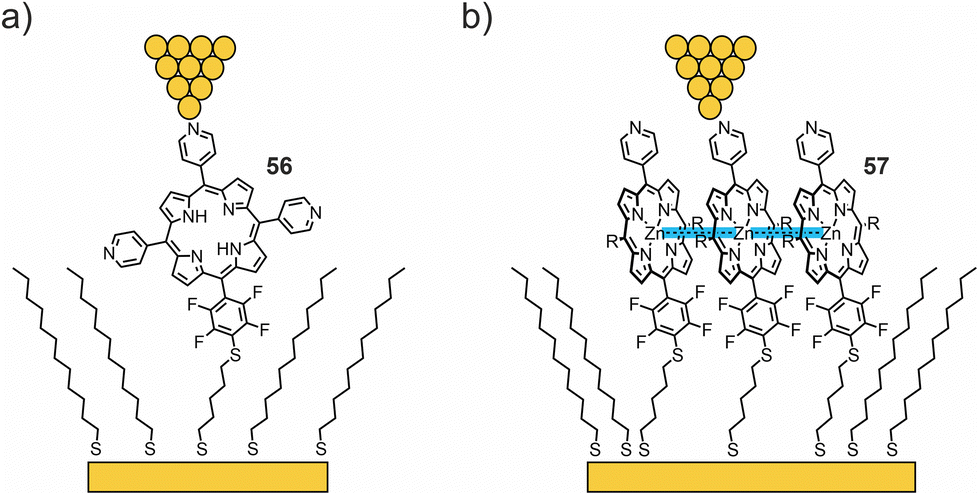 | ||
| Fig. 21 (a, b) Chemical structures of 56 (a) and 57 (b) depicted in a thioalkane SAM with opposite tip-contacts in an STM-BJ. Attractive interactions in zinc complex 57, facilitating the formation of large molecular islands, are highlighted in blue. | ||
In their more recent work,77 the same methodology was used to compare charge transport through free-base porphyrin 56 and its Zn analogue 57 (see Fig. 21(b)). Unlike the free-base porphyrin, where single-molecule through-bond tunneling was the dominant mechanism of charge transport, Zn complex 57 facilitated the formation of large molecular islands in an alkanethiol matrix on the gold surface. Bias-induced switching and increased tunneling efficiencies (compared to 56) observed by STM, as well as the Coulomb blockade observed in low-temperature crossed-wire tunnel junction measurements, indicate that charge hopping becomes the dominant mechanism of transport in molecular islands of 57. The enhancement of the nearest neighbor interactions is hypothesized to result in lateral delocalization of charge states in molecular islands and thereby favoring the more efficient, and more chemically tunable, charge-hopping regime.
Kuang et al.78 used STM spectroscopy at low temperature (4.9 K) to investigate single-molecule junctions of the Fe complex of tetraphenyl porphyrin 58 (see Fig. 22(a)). By stretching and fusing the junctions, they were able to reversibly tune the gap width and simultaneously conduct transport measurements. They observed a zero-bias resonance with a variable line shape as a function of electrode separation, which was attributed to a change in the spin density of the Fe center (see Fig. 22b). First-principles calculations revealed that the metal center underwent a spin transition from S = 2 to S = 1, when the junction gap was widened. This spin transition is associated with a conformational change of the metal porphyrin from saddle to planar. Even so, we initially excluded experiments studying porphyrins flat lying on a substrate by STM from this review; in this particular case, manipulation towards an upstanding porphyrin including spin flipping makes it an interesting supplement for this review.
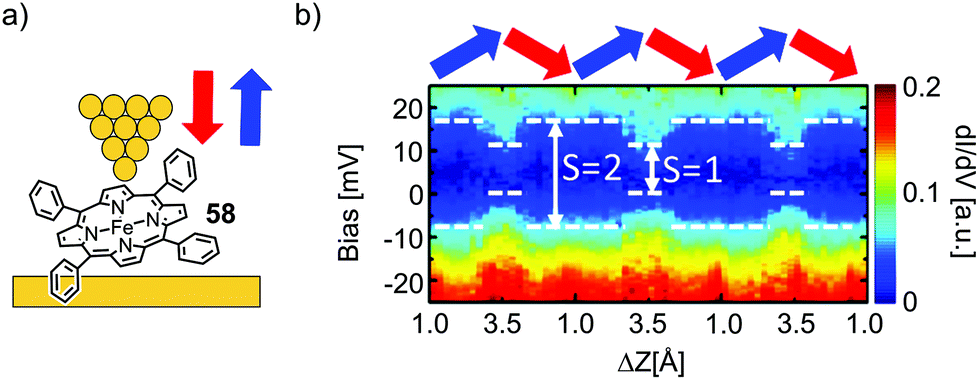 | ||
| Fig. 22 (a) Chemical structure of 58 depicted in an STM-BJ. (b) dI/dV spectra for the molecular junction depicted in (a) over the course of multiple tip up- and downward displacement cycles, as indicated by blue and red arrows, respectively, as adapted with permission from ref. 78. Copyright 2017 American Chemical Society. | ||
The results obtained by the scanning-probe techniques generally confirm the main conclusion of the previous sections, i.e., that single-molecule junctions can adapt to different conformations and that charge transport occurs by tunneling with small (less than 1 eV) level misalignments. Interestingly, the measurements on porphyrin clusters show that transport can proceed by hopping, indicating longer residence times of charge carriers on the molecules held together by π–π interactions. The delocalization of charge carriers increases the importance of charge–charge interactions and leads to the observed current reduction at low bias (Coulomb blockade).
4. Beta(β)-decay values of molecular porphyrin wires
Efficient charge transport through organic molecules over extended distances is an important aspect in molecular electronics, and a prerequisite for the design of robust molecular wires that could possibly be used for the fabrication and interconnection of devices. The decrease in transmission as a function of distance represents a key parameter in defining charge transport along (molecular) wires. In the coherent, off-resonant tunneling model, the molecular conductance (G) decreases exponentially with increasing molecular length, d, as:G = A0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e−βd, e−βd, |
There have been many studies that show an exponential decrease in conductance as a function of molecular length, which is consistent with charge transport being dominated by a coherent off-resonant tunneling mechanism. Corresponding experimental and theoretical β values are discussed for varying molecular structures, anchoring groups and electrode materials using various experimental platforms including both single-molecule and molecular assembly junctions.25–31,79,80 In most of the studies on short molecules (<3–5 nm), experimental β values are in the range of 2–12 nm−1 depending on the molecular backbone. Exceptions are oligothiophene with β values of around 1 nm−1,81 oligoyne with β values of around 0.6 nm−1,82 and porphyrins, which will be discussed further in this section. We further note that if the transport distance extends beyond the tunnelling regime (3–5 nm), electrons or holes may reside on the molecular wire for a long enough time to lose their phase information (e.g. when transport involves nuclear motion of the atoms that make up the molecule). In this case, incoherent “hopping” describes charge motion across the molecular wire by a series of transitions between neighbouring sites consisting of localized states. Unlike coherent tunnelling, hopping is thermally activated and typically exhibits a 1/d distance dependence instead of the exponential one characteristic of off-resonant tunneling. It is often observed in longer conjugated molecular wires where the direct tunneling process is strongly suppressed.
Next to efficient off-resonant transport along the backbone, a low β value can also be obtained experimentally when approaching the resonant transport regime, which occurs when the energy difference between one of the frontier molecular orbitals (HOMO or LUMO) and the Fermi level, EF, diminishes. In that case, the height of the tunnelling barrier is reduced and the charge can be rapidly transported through the molecular orbital. For the purpose of quantifying β decay values, porphyrin molecular wires of different length, with different connectivity in the backbone and different anchoring groups were studied. All structures involved in the studies reviewed in this chapter are given in Fig. 23(a)–(e); β values for the individual series of porphyrin wires and the respective measurement platforms are plotted in Fig. 23(f).
 | ||
| Fig. 23 Chemical structures of 54 and 59–70, building up Series 1–5 of porphyrin wires, depicted in an STM in (a)–(e). Thiol anchored Series 1 is constructed from acetylene bridged Zn porphyrins (a). Thiol anchored Series 2 (synthesized as thioacetate) is built up from butadiyne bridged Zn porphyrins (b). Similarly bridged Series 3 is anchored with terminal pyridines (c). Pyridine anchored twisted Series 4 and flat, tape-like Series 5 are constructed from meso–meso-connected (d) and beta–beta-, meso–meso-, beta–beta fused (e) Zn porphyrins, respectively. (f) Experimentally observed β values of Series 1–5 plotted from data collected from ref. 42 and 83–85 in decreasing order. | ||
In 2008, the first measurements of the length dependence of single-molecule conductance in thioacetate terminated butadiyne-bridged porphyrin molecular wires 59, 54, and 60 (Series 2 in Fig. 23(b)) using the STM break-junction technique were reported by the Nichols group83 using both the I(s) and I(t) modes. Both types of measurements were in good agreement with each other and revealed an exceptionally weak attenuation over distances up to 5 nm, with a β factor of 0.4 nm−1 at 600 mV applied bias (Fig. 23(f)). Although the distance dependent measurements were also found to be in agreement with a hopping mechanism (i.e., data could be fitted to a 1/d length dependence as well), the authors pointed out that the mechanisms were not mutually exclusive. However, the β factor was later re-evaluated to be 2.0 nm−1 at zero bias and 0.9 nm−1 at 900 mV.86
Borguet's group84 used the STM break junction technique (I(s)) to study thiol anchored acetylene-bridged Zn porphyrin wires (monomer 61, dimer 62, and trimer 63, Series 1 in Fig. 22(a)). They found two conductance peaks in their measurements, which were attributed to two different junction configurations: a fully stretched one leading to the low-conductance peak and a configuration in which the molecules remained under an angle with respect to the substrate (leading to the high-conductance peak). The ratio between the two observed configurations was about a factor of four (see Table 1). More importantly, both conductance peaks were found to decrease exponentially with molecular length with the same β values of 0.34 nm−1 at 100 mV applied bias (see Fig. 23(f)). Thus, in the low-bias regime, the acetylene bridged strands (Series 1) have about the same β attenuation factor as their butadiyne bridged analogues (Series 2). The conductance values of the two classes of porphyrin wires were further found to be in good agreement with each other (see Table 1).
With a low β value, the experimental distinction between hopping and coherent off-resonant tunneling transport mechanisms can be difficult, since linear and exponential fitting across the limited range of molecular lengths accessible for the experiment may be inconclusive. Next to analyzing the length dependence, the distinction between the two mechanisms can be achieved by measuring the temperature dependence of molecular conductance. Although off-resonant transport should be temperature independent, the temperature dependence of the Fermi distribution function can cause the overall process to be temperature dependent.30,32,41,42,87
For this purpose, the Nichols group studied the length dependence of conductance in pyridine-terminated butadiyne-bridged porphyrin wires 64, 65, and 66 (Series 3 in Fig. 23(c)) as a function of temperature.42 They obtained a similar β decay value of 0.4 nm−1 in the range of 25 to 100 °C (see Fig. 23(f)) at 600 mV bias voltage. Determination of the slopes of the Arrhenius plots yielded energy barriers of 0.08 eV for monomer 64, 0.14 eV for dimer 65, and 0.22 eV for trimer 66. The temperature dependence can be described within the coherent tunneling mechanism following Poot et al.:41 when the Fermi energy is positioned in the tails of one of the frontier orbitals (HOMO in this case), the exponential temperature dependence can be explained by considering the convolution of the smeared Fermi functions and the energy-dependent tail of the main transport channel. DFT calculations demonstrated the experimentally observed trend for the activation energies. For longer molecules the transmission of the HOMO is steeper. Thus, the experimental observations (exponential length and temperature dependence) are consistent with phase-coherent tunneling through the HOMO molecular orbital with EF as a single adjustable parameter. From comparison with experiments, the level misalignment was estimated to be 0.4 eV.
Subsequent measurements85 by the same group using the I(s) method were performed on three different types of porphyrin oligomers (monomer 64, dimer and trimer): butadiyne-linked chains (65 and 66, Series 3 in Fig. 23(c), similar to the previous study), meso–meso-linked twisted strands (67, 68, Series 4 in Fig. 23(d)) and beta–beta, meso–meso, beta–beta-fused planar tapes (69 and 70, Series 5 in Fig. 23(e)) with terminal pyridine anchoring groups. The β decay values of 0.4 nm−1 for Series 3; 1.1 nm−1 for Series 4; and 0.2 nm−1 for Series 5 were observed at an applied bias voltage of 600 mV. As expected, the twisted strands mediate (Series 4) transport least effectively. However, despite a considerable difference in the values and the length dependence of the HOMO–LUMO gap for different molecules, the conductances of all molecules were found to be within a factor of seven (see Table 1). It can therefore be speculated that the main obstacle to transport comes from the barrier at the contacts (Γ) and not from the intrinsic transmission through the backbone of the molecule. The coupling in this case was established through terminal pyridine anchoring groups attached to the porphyrin wires. Better coupling, i.e., highly transmissive metal–molecule contacts would be required to increase their conductance.
In general, the porphyrin backbones of the studied wires transport charges efficiently with ultra-low attenuation factors at low bias, especially if the porphyrin planes are not twisted with respect to each other (low dihedral angle distributions are favored). Experiments with high bias should be interpreted carefully, since approaching resonant transport increases the conductance and lowers the β values. However, the obtained β values reported herein gathered through experiments with applied biases from 100 to 600 mV are found to be in good agreement with each other.86,88
5. The future of porphyrins in molecular electronics
Due to their absorption and emission properties, appealing geometry, synthetic accessibility, suitable stability, and rich metal coordination chemistry, porphyrins have great potential for the construction of functional electronic devices. As summarized in this review, mainly basic issues concerning charge transport properties through single-porphyrin junctions have been studied during the past ten years, leading the way for the next level of molecular complexity, profiting to a larger extent from the features and functions of the porphyrin building blocks. The reported low β values at low biases recorded for oligo-porphyrins make them promising building blocks for molecular wires, as efficient charge transport over relatively long distances is an important issue in molecular electronics. With these promising building blocks, more complex molecular devices will become accessible, like e.g. porphyrin dimers varying the extent of communication between both porphyrins by an external trigger (an already realized example is mechanosensitive porphyrin cyclophane 34). Porphyrins can have a rich redox chemistry with a variety of charged states, which might be useful for information storage in molecular junctions,89 but might also serve as a trigger to manipulate intramolecular interactions electrostatically in more complex (super)structures, paving the way towards molecular muscles and machines.90,91 Even more visionary is the use of porphyrin based architectures as qubits integrated in a quantum computing set-up, and the first porphyrin complexes with f-orbital metals, namely, the lanthanides terbium and dysprosium, have already been reported.92 However, there are structural physicochemical challenges to be faced when extended molecular architectures, e.g. longer porphyrin wires, should be developed, such as the solubility required to guarantee wet chemical processability and chemical stability. Also, from the viewpoint of physical experiments, there are interesting challenges linked to long wire structures, like their tendency to interact with the electrode surface through their extended π-surface instead of forming molecular junctions of the fully stretched molecular wire. However, the steady contributions from neighboring disciplines will further push the limitations of accessible experimental data and explore the potential of these fascinating model compounds inspired by nature. The authors are convinced that the combination of unique physical properties and synthetic accessibility provides a bright future for molecular devices in single molecule junctions based on porphyrin subunits.Author contributions
All authors contributed to the manuscript. P. Z. provided the first draft, which was revised, complemented and corrected by D. D., H. S. J. van der Z., and M. M.Conflicts of interest
The authors declare that there are no competing interests.Acknowledgements
This study was supported by the EU through a RISE(DAFNEOX) project, SEP 210165479 and partially funded by the FET open project QuIET (no. 767187). D. D. acknowledges support by the Fondecyt Regular Project 1181080, H. v. d. Z. by the Dutch Science Foundation (NWO) and M. M. acknowledges generous financial support by the Swiss National Science Foundation (SNF grant number 200020-178808) and by the 111 project (90002-18011002).References
- R. R. Schaller, Moore's law: past, present and future, IEEE Spectr., 1997, 34, 52–59 Search PubMed.
- A. Aviram and M. A. Ratner, Molecular rectifiers, Chem. Phys. Lett., 1974, 29, 277–283 CrossRef CAS.
- N. Xin, J. Guan, C. Zhou, X. Chen, C. Gu, Y. Li, M. A. Ratner, A. Nitzan, J. F. Stoddart and X. Guo, Concepts in the design and engineering of single-molecule electronic devices, Nat. Rev. Phys., 2019, 1, 211–230 CrossRef.
- D. Vuillaume, Molecular Electronics: From Single-Molecule to Large-Area Devices, ChemPlusChem, 2019, 84, 1215–1221 CrossRef CAS PubMed.
- S. J. van der Molen, R. Naaman, E. Scheer, J. B. Neaton, A. Nitzan, D. Natelson, N. J. Tao, H. S. J. van der Zant, M. Mayor, M. Ruben, M. Reed and M. Calame, Visions for a molecular future, Nat. Nanotechnol., 2013, 8, 385–389 CrossRef PubMed.
- H. Chen and J. F. Stoddart, From molecular to supramolecular electronics, Nat. Rev. Mater., 2021, 1–25 Search PubMed.
- Y. Nakamura, N. Aratani and A. Osuka, Cyclic porphyrin arrays as artificial photosynthetic antenna: synthesis and excitation energy transfer, Chem. Soc. Rev., 2007, 36, 831–845 RSC.
- N. Aratani and A. Osuka, A New Strategy for Construction of Covalently Linked Giant Porphyrin Arrays with One, Two, and Three Dimensionally Arranged Architectures, Bull. Chem. Soc. Jpn., 2001, 74, 1361–1379 CrossRef CAS.
- T. Tanaka and A. Osuka, Conjugated porphyrin arrays: synthesis, properties and applications for functional materials, Chem. Soc. Rev., 2015, 44, 943–969 RSC.
- A. T. P. C. Gomes, Acid Catalysis in the Way to Porphyrins: Reaction of Pyrrole/Aldehydes in the Synthesis of meso-Substituted Porphyrins, Rev. Virtual Quim., 2013, 5, 312–317 CAS.
- T. D. Lash, What's in a name? The MacDonald condensation, J. Porphyrins Phthalocyanines, 2016, 20, 855–888 CrossRef CAS.
- S. Hiroto, Y. Miyake and H. Shinokubo, Synthesis and Functionalization of Porphyrins through Organometallic Methodologies, Chem. Rev., 2017, 117, 2910–3043 CrossRef CAS PubMed.
- K. Wang, A. Osuka and J. Song, Pd-Catalyzed Cross Coupling Strategy for Functional Porphyrin Arrays, ACS Cent. Sci., 2020, 6, 2159–2178 CrossRef CAS PubMed.
- M. Jurow, A. E. Schuckman, J. D. Batteas and C. M. Drain, Porphyrins as molecular electronic components of functional devices, Coord. Chem. Rev., 2010, 254, 2297–2310 CrossRef CAS PubMed.
- F. Schwarz and E. Lörtscher, Break-junctions for investigating transport at the molecular scale, J. Phys.: Condens. Matter, 2014, 26, 474201 CrossRef PubMed.
- P. Gehring, J. M. Thijssen and H. S. J. van der Zant, Single-molecule quantum-transport phenomena in break junctions, Nat. Rev. Phys., 2019, 1, 381–396 CrossRef.
- M. E. Abbassi, P. Zwick, A. Rates, D. Stefani, A. Prescimone, M. Mayor, H. S. J. van der Zant and D. Dulić, Unravelling the conductance path through single-porphyrin junctions, Chem. Sci., 2019, 10, 8299–8305 RSC.
- W. Haiss, H. van Zalinge, S. J. Higgins, D. Bethell, H. Höbenreich, D. J. Schiffrin and R. J. Nichols, Redox State Dependence of Single Molecule Conductivity, J. Am. Chem. Soc., 2003, 125, 15294–15295 CrossRef CAS PubMed.
- W. Haiss, S. Martín, E. Leary, H. van Zalinge, S. J. Higgins, L. Bouffier and R. J. Nichols, Impact of Junction Formation Method and Surface Roughness on Single Molecule Conductance, J. Phys. Chem. C, 2009, 113, 5823–5833 CrossRef CAS.
- Y. Isshiki, Y. Matsuzawa, S. Fujii and M. Kiguchi, Investigation on Single-Molecule Junctions Based on Current–Voltage Characteristics, Micromachines, 2018, 9, 67 CrossRef PubMed.
- M. L. Perrin, E. Burzurí and H. S. J. van der Zant, Single-molecule transistors, Chem. Soc. Rev., 2015, 44, 902–919 RSC.
- E. Lörtscher, Wiring molecules into circuits, Nat. Nanotechnol., 2013, 8, 381–384 CrossRef PubMed.
- Y. Noguchi, T. Nagase, T. Kubota, T. Kamikado and S. Mashiko, Fabrication of Au–molecule–Au junctions using electromigration method, Thin Solid Films, 2006, 499, 90–94 CrossRef CAS.
- M. Ratner, A brief history of molecular electronics, Nat. Nanotechnol., 2013, 8, 378–381 CrossRef CAS PubMed.
- R. L. McCreery, H. Yan and A. J. Bergren, A critical perspective on molecular electronic junctions: there is plenty of room in the middle, Phys. Chem. Chem. Phys., 2012, 15, 1065–1081 RSC.
- C. J. Lambert, Basic concepts of quantum interference and electron transport in single-molecule electronics, Chem. Soc. Rev., 2015, 44, 875–888 RSC.
- L. Sun, Y. A. Diaz-Fernandez, T. A. Gschneidtner, F. Westerlund, S. Lara-Avila and K. Moth-Poulsen, Single-molecule electronics: from chemical design to functional devices, Chem. Soc. Rev., 2014, 43, 7378–7411 RSC.
- R. M. Metzger, Unimolecular Electronics, Chem. Rev., 2015, 115, 5056–5115 CrossRef CAS PubMed.
- T. A. Su, M. Neupane, M. L. Steigerwald, L. Venkataraman and C. Nuckolls, Chemical principles of single-molecule electronics, Nat. Rev. Mater., 2016, 1, 1–15 Search PubMed.
- D. Xiang, X. Wang, C. Jia, T. Lee and X. Guo, Molecular-Scale Electronics: From Concept to Function, Chem. Rev., 2016, 116, 4318–4440 CrossRef CAS PubMed.
- E. Scheer and J. C. Cuevas, Molecular Electronics: An Introduction to Theory and Experiment, World Scientific, 2nd edn, 2017 Search PubMed.
- E. Leary, A. L. Rosa, M. T. González, G. Rubio-Bollinger, N. Agraït and N. Martín, Incorporating single molecules into electrical circuits. The role of the chemical anchoring group, Chem. Soc. Rev., 2015, 44, 920–942 RSC.
- T. Yelin, R. Korytár, N. Sukenik, R. Vardimon, B. Kumar, C. Nuckolls, F. Evers and O. Tal, Conductance saturation in a series of highly transmitting molecular junctions, Nat. Mater., 2016, 15, 444–449 CrossRef CAS PubMed.
- Z. Xie, I. Bâldea, C. E. Smith, Y. Wu and C. D. Frisbie, Experimental and Theoretical Analysis of Nanotransport in Oligophenylene Dithiol Junctions as a Function of Molecular Length and Contact Work Function, ACS Nano, 2015, 9, 8022–8036 CrossRef CAS PubMed.
- C. Jia and X. Guo, Molecule–electrode interfaces in molecular electronic devices, Chem. Soc. Rev., 2013, 42, 5642–5660 RSC.
- W. Wang, T. Lee and M. A. Reed, Mechanism of electron conduction in self-assembled alkanethiol monolayer devices, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 035416 CrossRef.
- M. Magoga and C. Joachim, Minimal attenuation for tunneling through a molecular wire, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1820–1823 CrossRef CAS.
- H. B. Li, Y.-F. Xi, Z.-W. Hong, J. Yu, X.-X. Li, W.-X. Liu, L. Domulevicz, S. Jin, X.-S. Zhou and J. Hihath, Temperature-Dependent Tunneling in Furan Oligomer Single-Molecule Junctions, ACS Sens., 2021, 6, 565–572 CrossRef CAS PubMed.
- S. H. Choi, C. Risko, M. C. R. Delgado, B. Kim, J.-L. Brédas and C. D. Frisbie, Transition from Tunneling to Hopping Transport in Long, Conjugated Oligo-imine Wires Connected to Metals, J. Am. Chem. Soc., 2010, 132, 4358–4368 CrossRef CAS PubMed.
- B. Capozzi, E. J. Dell, T. C. Berkelbach, D. R. Reichman, L. Venkataraman and L. M. Campos, Length-Dependent Conductance of Oligothiophenes, J. Am. Chem. Soc., 2014, 136, 10486–10492 CrossRef CAS PubMed.
- M. Poot, E. Osorio, K. O'Neill, J. M. Thijssen, D. Vanmaekelbergh, C. A. van Walree, L. W. Jenneskens and H. S. J. van der Zant, Temperature Dependence of Three-Terminal Molecular Junctions with Sulfur End-Functionalized Tercyclohexylidenes, Nano Lett., 2006, 6, 1031–1035 CrossRef CAS.
- G. Sedghi, V. M. García-Suárez, L. J. Esdaile, H. L. Anderson, C. J. Lambert, S. Martín, D. Bethell, S. J. Higgins, M. Elliott, N. Bennett, J. E. Macdonald and R. J. Nichols, Long-range electron tunnelling in oligo-porphyrin molecular wires, Nat. Nanotechnol., 2011, 6, 517–523 CrossRef CAS PubMed.
- I. Bâldea, Protocol for disentangling the thermally activated contribution to the tunneling-assisted charge transport. Analytical results and experimental relevance, Phys. Chem. Chem. Phys., 2017, 19, 11759–11770 RSC.
- D. Segal, A. Nitzan, M. Ratner and W. B. Davis, Activated Conduction in Microscopic Molecular Junctions, J. Phys. Chem. B, 2000, 104, 2790–2793 CrossRef CAS.
- J. A. Mol, C. S. Lau, W. J. M. Lewis, H. Sadeghi, C. Roche, A. Cnossen, J. H. Warner, C. J. Lambert, H. L. Anderson and G. A. D. Briggs, Graphene-porphyrin single-molecule transistors, Nanoscale, 2015, 7, 13181–13185 RSC.
- P. Gehring, J. K. Sowa, J. Cremers, Q. Wu, H. Sadeghi, Y. Sheng, J. H. Warner, C. J. Lambert, G. A. D. Briggs and J. A. Mol, Distinguishing Lead and Molecule States in Graphene-Based Single-Electron Transistors, ACS Nano, 2017, 11, 5325–5331 CrossRef CAS PubMed.
- B. Limburg, J. O. Thomas, J. K. Sowa, K. Willick, J. Baugh, E. M. Gauger, G. A. D. Briggs, J. A. Mol and H. L. Anderson, Charge-state assignment of nanoscale single-electron transistors from their current–voltage characteristics, Nanoscale, 2019, 11, 14820–14827 RSC.
- Z. Li and E. Borguet, Determining Charge Transport Pathways through Single Porphyrin Molecules Using Scanning Tunneling Microscopy Break Junctions, J. Am. Chem. Soc., 2012, 134, 63–66 CrossRef CAS PubMed.
- Z. Li, M. Smeu, M. A. Ratner and E. Borguet, Effect of Anchoring Groups on Single Molecule Charge Transport through Porphyrins, J. Phys. Chem. C, 2013, 117, 14890–14898 CrossRef CAS.
- S. Fujii, S. Marqués-González, J.-Y. Shin, H. Shinokubo, T. Masuda, T. Nishino, N. P. Arasu, H. Vázquez and M. Kiguchi, Highly-conducting molecular circuits based on antiaromaticity, Nat. Commun., 2017, 8, 15984 CrossRef CAS PubMed.
- Q. Zhou, A. Yamada, Q. Feng, A. Hoskins, B. D. Dunietz and K. M. Lewis, Modification of Molecular Conductance by in Situ Deprotection of Thiol-Based Porphyrin, ACS Appl. Mater. Interfaces, 2017, 9, 15901–15906 CrossRef CAS PubMed.
- H. Basch, R. Cohen and M. A. Ratner, Interface Geometry and Molecular Junction Conductance: Geometric Fluctuation and Stochastic Switching, Nano Lett., 2005, 5, 1668–1675 CrossRef CAS PubMed.
- G. Qian, S. Saha and K. M. Lewis, Two-state conductance in single Zn porphyrin molecular junctions, Appl. Phys. Lett., 2010, 96, 243107 CrossRef.
- Z.-F. Liu, S. Wei, H. Yoon, O. Adak, I. Ponce, Y. Jiang, W.-D. Jang, L. M. Campos, L. Venkataraman and J. B. Neaton, Control of Single-Molecule Junction Conductance of Porphyrins via a Transition-Metal Center, Nano Lett., 2014, 14, 5365–5370 CrossRef CAS PubMed.
- M. L. Perrin, F. Prins, C. A. Martin, A. J. Shaikh, R. Eelkema, J. H. van Esch, T. Briza, R. Kaplanek, V. Kral, J. M. van Ruitenbeek, H. S. J. van der Zant and D. Dulić, Influence of the Chemical Structure on the Stability and Conductance of Porphyrin Single-Molecule Junctions, Angew. Chem., Int. Ed., 2011, 50, 11223–11226 CrossRef CAS PubMed.
- E. Leary, C. Roche, H.-W. Jiang, I. Grace, M. T. González, G. Rubio-Bollinger, C. Romero-Muñiz, Y. Xiong, Q. Al-Galiby, M. Noori, M. A. Lebedeva, K. Porfyrakis, N. Agrait, A. Hodgson, S. J. Higgins, C. J. Lambert, H. L. Anderson and R. J. Nichols, Detecting Mechanochemical Atropisomerization within an STM Break Junction, J. Am. Chem. Soc., 2018, 140, 710–718 CrossRef CAS PubMed.
- Y. Xue, X. Li, H. Li and W. Zhang, Quantifying thiol–gold interactions towards the efficient strength control, Nat. Commun., 2014, 5, 4348 CrossRef CAS PubMed.
- D. Stefani, M. Perrin, C. Gutiérrez-Cerón, A. C. Aragonès, J. Labra-Muñoz, R. D. C. Carrasco, Y. Matsushita, Z. Futera, J. Labuta, T. H. Ngo, K. Ariga, I. Díez-Pérez, H. S. J. van der Zant, D. Dulić and J. P. Hill, Mechanical Tuning of Through-Molecule Conductance in a Conjugated Calix[4]pyrrole, ChemistrySelect, 2018, 3, 6473–6478 CrossRef CAS.
- P. Zwick, K. J. Weiland, J. Malinčík, D. Stefani, D. Häussinger, H. S. J. van der Zant, D. Dulić and M. Mayor, Mechanical Fixation by Porphyrin Connection: Synthesis and Transport Studies of a Bicyclic Dimer, J. Org. Chem., 2020, 85, 118–128 CrossRef CAS PubMed.
- P. Zwick, C. Hsu, M. El Abbassi, O. Fuhr, D. Fenske, D. Dulić, H. S. J. van der Zant and M. Mayor, Synthesis and Transport Studies of a Cofacial Porphyrin Cyclophane, J. Org. Chem., 2020, 85, 15072–15081 CrossRef CAS PubMed.
- A. C. Aragonès, N. Darwish, W. J. Saletra, L. Pérez-García, F. Sanz, J. Puigmartí-Luis, D. B. Amabilino and I. Díez-Pérez, Highly Conductive Single-Molecule Wires with Controlled Orientation by Coordination of Metalloporphyrins, Nano Lett., 2014, 14, 4751–4756 CrossRef PubMed.
- M. Noori, A. C. Aragonès, G. D. Palma, N. Darwish, S. W. D. Bailey, Q. Al-Galiby, I. Grace, D. B. Amabilino, A. González-Campo, I. Díez-Pérez and C. J. Lambert, Tuning the electrical conductance of metalloporphyrin supramolecular wires, Sci. Rep., 2016, 6, 37352 CrossRef CAS PubMed.
- A. C. Aragonès, A. Martín-Rodríguez, D. Aravena, J. Puigmartí-Luis, D. B. Amabilino, N. Aliaga-Alcalde, A. González-Campo, E. Ruiz and I. Díez-Pérez, Tuning Single-Molecule Conductance in Metalloporphyrin-Based Wires via Supramolecular Interactions, Angew. Chem., Int. Ed., 2020, 59, 19193–19201 CrossRef PubMed.
- D. H. Yoon, S. B. Lee, K.-H. Yoo, J. Kim, J. K. Lim, N. Aratani, A. Tsuda, A. Osuka and D. Kim, Electrical Conduction through Linear Porphyrin Arrays, J. Am. Chem. Soc., 2003, 125, 11062–11064 CrossRef CAS PubMed.
- N. Aratani, A. Osuka, Y. H. Kim, D. H. Jeong and D. Kim, Extremely Long, Discrete meso–meso-Coupled Porphyrin Arrays, Angew. Chem., Int. Ed., 2000, 39, 1458–1462 CrossRef CAS PubMed.
- A. Tsuda and A. Osuka, Fully Conjugated Porphyrin Tapes with Electronic Absorption Bands That Reach into Infrared, Science, 2001, 5527, 79–82 CrossRef PubMed.
- B. K. Kang, N. Aratani, J. K. Lim, D. Kim, A. Osuka and K.-H. Yoo, Length and temperature dependence of electrical conduction through dithiolated porphyrin arrays, Chem. Phys. Lett., 2005, 412, 303–306 CrossRef CAS.
- Y. Noguchi, T. Nagase, R. Ueda, T. Kamikado, T. Kubota and S. Mashiko, Fowler–Nordheim Tunneling in Electromigrated Break Junctions with Porphyrin Molecules, Jpn. J. Appl. Phys., 2007, 46, 2683 CrossRef CAS.
- S. Saha, J. R. Owens, V. Meunier and K. M. Lewis, Asymmetric electron transport and highest occupied molecular orbital assisted tunneling through Zn-porphyrin molecular junctions, Appl. Phys. Lett., 2013, 103, 173101 CrossRef.
- S. Saha, A. Nicolaï, J. R. Owens, A. Krawicz, P. H. Dinolfo, V. Meunier and K. M. Lewis, Temperature-Dependent and Bistable Current–Voltage Measurements in Zinc Porphyrin Molecular Junctions, ACS Appl. Mater. Interfaces, 2015, 7, 10085–10090 CrossRef CAS PubMed.
- M. L. Perrin, C. A. Martin, F. Prins, A. J. Shaikh, R. Eelkema, J. H. van Esch, J. M. van Ruitenbeek, H. S. J. van der Zant and D. Dulić, Charge transport in a zinc–porphyrin single-molecule junction, Beilstein J. Nanotechnol., 2011, 2, 714–719 CrossRef PubMed.
- M. L. Perrin, C. J. O. Verzijl, C. A. Martin, A. J. Shaikh, R. Eelkema, J. H. van Esch, J. M. van Ruitenbeek, J. M. Thijssen, H. S. J. van der Zant and D. Dulić, Large tunable image-charge effects in single-molecule junctions, Nat. Nanotechnol., 2013, 8, 282–287 CrossRef CAS PubMed.
- C. J. O. Verzijl, J. A. Celis Gil, M. L. Perrin, D. Dulić, H. S. J. van der Zant and J. M. Thijssen, Image effects in transport at metal-molecule interfaces, J. Chem. Phys., 2015, 143, 174106 CrossRef CAS PubMed.
- N. Bennett, G. Xu, L. J. Esdaile, H. L. Anderson, J. E. Macdonald and M. Elliott, Transition Voltage Spectroscopy of Porphyrin Molecular Wires, Small, 2010, 6, 2604–2611 CrossRef CAS PubMed.
- W. Xi, W. Zhang, B.-K. An, P. L. Burn and J. J. Davis, Tunnelling conductance of vectorial porphyrin monolayers, J. Mater. Chem., 2008, 18, 3109–3120 RSC.
- Y.-H. Chan, A. E. Schuckman, L. M. Pérez, M. Vinodu, C. M. Drain and J. D. Batteas, Synthesis and Characterization of a Thiol-Tethered Tripyridyl Porphyrin on Au(111), J. Phys. Chem. C, 2008, 112, 6110–6118 CrossRef CAS.
- A. E. Schuckman, B. W. Ewers, L. H. Yu, J. P. C. Tomé, L. M. Pérez, C. M. Drain, J. G. Kushmerick and J. D. Batteas, Utilizing Nearest-Neighbor Interactions To Alter Charge Transport Mechanisms in Molecular Assemblies of Porphyrins on Surfaces, J. Phys. Chem. C, 2015, 119, 13569–13579 CrossRef CAS.
- G. Kuang, Q. Zhang, T. Lin, R. Pang, X. Shi, H. Xu and N. Lin, Mechanically-Controlled Reversible Spin Crossover of Single Fe-Porphyrin Molecules, ACS Nano, 2017, 11, 6295–6300 CrossRef CAS PubMed.
- H. Liu, N. Wang, J. Zhao, Y. Guo, X. Yin, F. Y. C. Boey and H. Zhang, Length-Dependent Conductance of Molecular Wires and Contact Resistance in Metal–Molecule–Metal Junctions, ChemPhysChem, 2008, 9, 1416–1424 CrossRef CAS PubMed.
- G. Peng, M. Strange, K. S. Thygesen and M. Mavrikakis, Conductance of Conjugated Molecular Wires: Length Dependence, Anchoring Groups, and Band Alignment, J. Phys. Chem. C, 2009, 113, 20967–20973 CrossRef CAS.
- R. Yamada, H. Kumazawa, T. Noutoshi, S. Tanaka and H. Tada, Electrical Conductance of Oligothiophene Molecular Wires, Nano Lett., 2008, 8, 1237–1240 CrossRef CAS PubMed.
- C. Wang, A. S. Batsanov, M. R. Bryce, S. Martín, R. J. Nichols, S. J. Higgins, V. M. García-Suárez and C. J. Lambert, Oligoyne Single Molecule Wires, J. Am. Chem. Soc., 2009, 131, 15647–15654 CrossRef CAS PubMed.
- G. Sedghi, K. Sawada, L. J. Esdaile, M. Hoffmann, H. L. Anderson, D. Bethell, W. Haiss, S. J. Higgins and R. J. Nichols, Single Molecule Conductance of Porphyrin Wires with Ultralow Attenuation, J. Am. Chem. Soc., 2008, 130, 8582–8583 CrossRef CAS PubMed.
- Z. Li, T.-H. Park, J. Rawson, M. J. Therien and E. Borguet, ‘Quasi-Ohmic Single Molecule Charge Transport through Highly Conjugated meso-to-meso Ethyne-Bridged Porphyrin Wires, Nano Lett., 2012, 12, 2722–2727 CrossRef CAS PubMed.
- G. Sedghi, L. J. Esdaile, H. L. Anderson, S. Martin, D. Bethell, S. J. Higgins and R. J. Nichols, Comparison of the Conductance of Three Types of Porphyrin-Based Molecular Wires: β,meso,β-Fused Tapes, meso-Butadiyne-Linked and Twisted meso-meso Linked Oligomers, Adv. Mater., 2012, 24, 653–657 CrossRef CAS PubMed.
- E. Leary, B. Limburg, A. Alanazy, S. Sangtarash, I. Grace, K. Swada, L. J. Esdaile, M. Noori, M. T. González, G. Rubio-Bollinger, H. Sadeghi, A. Hodgson, N. Agraït, S. J. Higgins, C. J. Lambert, H. L. Anderson and R. J. Nichols, Bias-Driven Conductance Increase with Length in Porphyrin Tapes, J. Am. Chem. Soc., 2018, 140, 12877–12883 CrossRef CAS PubMed.
- L. Luo, S. H. Choi and C. D. Frisbie, Probing Hopping Conduction in Conjugated Molecular Wires Connected to Metal Electrodes, Chem. Mater., 2011, 23, 631–645 CrossRef CAS.
- E. Leary, G. Kastlunger, B. Limburg, L. Rincón-García, J. Hurtado-Gallego, M. T. González, G. R. Bollinger, N. Agrait, S. J. Higgins, H. L. Anderson, R. Stadler and R. J. Nichols, Long-lived charged states of single porphyrin-tape junctions under ambient conditions, Nanoscale Horiz., 2021, 6, 49–58 RSC.
- F. Anariba, H. Tiznado, J. R. Diers, I. Schmidt, A. Z. Muresan, J. S. Lindsey, F. Zaera and D. F. Bocian, Comprehensive Characterization of Hybrid Junctions Comprised of a Porphyrin Monolayer Sandwiched Between a Coinage Metal Overlayer and a Si(100) Substrate, J. Phys. Chem. C, 2008, 112, 9474–9485 CrossRef CAS.
- P. J. Gilissen, P. B. White, J. A. Berrocal, N. Vanthuyne, F. P. J. T. Rutjes, B. L. Feringa, J. A. A. W. Elemans and R. J. M. Nolte, Molecular motor-functionalized porphyrin macrocycles, Nat. Commun., 2020, 11, 1–10 Search PubMed.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Artificial Molecular Machines, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- J. J. Le Roy, J. Cremers, I. A. Thomlinson, M. Slota, W. K. Myers, P. H. Horton, S. J. Coles, H. L. Anderson and L. Bogani, Tailored homo- and hetero-lanthanide porphyrin dimers: a synthetic strategy for integrating multiple spintronic functionalities into a single molecule, Chem. Sci., 2018, 9, 8474–8481 RSC.
| This journal is © The Royal Society of Chemistry 2021 |