
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCucurbit[6]uril@MIL-101-Cl: loading polar porous cages in mesoporous stable host for enhanced SO2 adsorption at low pressures†
Yangyang
Sun
 a,
Jun
Liang
ab,
Philipp
Brandt
a,
Alex
Spieß
a,
Secil
Öztürk
a and
Christoph
Janiak
*ab
a,
Jun
Liang
ab,
Philipp
Brandt
a,
Alex
Spieß
a,
Secil
Öztürk
a and
Christoph
Janiak
*ab
aInstitut für Anorganische Chemie und Strukturchemie, Heinrich-Heine-Universität Düsseldorf, 40204 Düsseldorf, Germany. E-mail: janiak@hhu.de
bHoffmann Institute of Advanced Materials, Shenzhen Polytechnic, 7098 Liuxian Blvd, Nanshan District, Shenzhen 518055, China
First published on 7th September 2021
Abstract
The robust cucurbituril-MOF composite CB6@MIL-101-Cl was synthesized by a wet impregnation method and a concomitant OH-to-Cl ligand exchange {CB6 = cucurbit[6]uril, 31 wt% content in the composite, MIL-101-Cl = [Cr3(O)Cl(H2O)2(BDC)3], BDC = benzene-1,4-dicarboxylate}. MIL-101-Cl was formed postsynthetically from standard fluorine-free MIL-101 where Cr-OH ligands were substituted by Cl during treatment with HCl. CB6@MIL-101-Cl combines the strong SO2 affinity of the rigid CB6 macrocycles and the high SO2 uptake capacity of MIL-101, and shows a high SO2 uptake of 438 cm3 g−1 (19.5 mmol g−1) at 1 bar and 293 K (380 cm3 g−1, 17.0 mmol g−1 at 1 bar and 298 K). The captured SO2 amount is 2.2 mmol g−1 for CB6@MIL-101-Cl at 0.01 bar and 293 K (2.0 mmol g−1 at 298 K), which is three times higher than that of the parent MIL-101 (0.7 mmol g−1) under the same conditions. The near zero-coverage SO2 adsorption enthalpies of MIL-101 and CB6@MIL-101-Cl are −35 kJ mol−1 and −50 kJ mol−1, respectively, reflecting the impact of the incorporated CB6 macrocycles, having higher affinity towards SO2. FT-IR spectroscopy confirms the interactions of the SO2 with the cucurbit[6]uril moieties of the CB6@MIL-101-Cl composite and SO2 retention for a few minutes under ambient air. Comparative experiments demonstrated loss of crystallinity and porosity after dry SO2 adsorption for MIL-101, while CB6@MIL-101-Cl exhibits nearly complete retention of crystallinity and porosity under the exposure to both dry and wet SO2. Thus, CB6@MIL-101-Cl can be an attractive adsorbent for SO2 capture because of its excellent recycling stability, high capacity and strong affinity toward SO2 at low pressure.
Introduction
The large-scale emission of flue gases containing toxic SO2 arouses increasing worldwide concerns. As of 2010, global energy sector emissions of SO2 were estimated to be about 40 Tg.1 The majority of anthropogenic SO2 emissions originated from coal- and oil-burning for electricity generation.2 SO2 is responsible for human respiratory diseases including bronchitis, asthma as well as cardiovascular diseases,3,4 and SO2 along with NOX emissions lead to the formation of ‘acid rain’ that poses significant danger to the health of ecosystems, particularly by inhibiting plant growth and poisoning aquatic life.5 In addition, SO2 can react with other air pollutants to produce sulphate particles, which are a main component of fine particulate matter (PM2.5).6,7 Thus, developing new materials and technologies for SO2 removal is imperative.Limestone scrubbing, ammonia scrubbing and absorptive removal by organic solvents like monoethanol–amine (MEA) represent the majority of conventional technologies of SO2 removal from flue gases. A capture of more than 95% SO2 from the gas mixtures could be achieved via use of these traditional approaches.8–10 Improvements in energy efficiency and decrease of wastes suggest physisorption by solid adsorbents as the most promising alternatives, particularly due to the possibility to minimize energy requirements. It is worth noting that because of the corrosive nature of SO2, its complete as possible removal as the first step of flue gas processing allows broader possibilities for subsequent purification/sequestration steps.11,12 Therefore, the development of chemically resistant porous materials for adsorptive SO2 removal, especially with high uptake and selectivity at industrially relevant low partial pressures of SO2, is of great interest.
Metal–organic frameworks (MOFs) have drawn intensive attention in the context of gas storage and separation.13–15 Due to their high surface area, high pore volume and tunable surface functionality, MOFs can be designed for sorption of toxic chemicals including SO2,16–19 and the capture of CO2.20 The stability of MOFs against water vapors should not be overlooked during evaluation of potential MOF adsorbents for SO2 capture.21 Although some MOFs have been reported to be promising for SO2 adsorption and SO2/CO2 separation,22–30 the efficient MOF-based adsorbents with both high stability and high-capacity are still very limited.
MIL-101(Cr), which possesses excellent water stability, high surface area and micro/mesoporosity (29 and 34 Å inner diameter), is a particularly interesting adsorbent for practical applications (the guest-free form corresponds to [Cr3(μ3-O)(X)(H2O)2(BDC)3], X = F, OH, BDC = benzene-1,4-dicarboxylate). In 2020, Ibarra et al. reported that MIL-101(Cr)-4F(1%) (−4F = fully fluorinated BDC) shows a high SO2 uptake capacity of 18.4 mmol g−1 at 1 bar and 298 K and is chemically stable towards dry and humid SO2.31 The large pore volume, beneficial at higher pressures, is the primary reason for the superior performance of MIL-101(Cr) at 1 bar compared to other microporous MOFs. However, MIL-101(Cr) shows mediocre uptake at low pressure due to the lower density of strongly adsorbing sites, such as open-metal sites,32 and polar OH− and H2O ligands. MIL-101(Cr) has also no polar ligand substituents, such as –NH2 or –OH groups and lacks small micropores in the 4–8 Å region, both of which are advantageous for a low-pressure uptake of SO2.25,33–35 Importantly, it was also shown that the MIL-101(Cr) (formed by standard fluorine-free conditions), would gradually lose its crystallinity and, therefore the accessible surface area after SO2 sorption at 298 K even under nearly anhydrous conditions.31
One effective method to modulate the pore size of a host framework is to introduce functional molecules to obtain composite materials, which may exhibit synergetic performances towards certain adsorbates.36–40 However, MOF-based composites are not yet reported for SO2 sorption and separation until now to the best of our knowledge. Recently, our group studied the outstanding SO2 sorption and separation performance of a cucurbit[6]uril microporous hydrogen-bonded organic framework (HOF),41 nanoCB6-H (H stands for the solid-state honeycomb-like structure).42,43 It was found that the CB6 cages (“barrels”) have strong interactions with dry SO2 molecules by forming interactions both on the outer surfaces and in its intrinsic pores at a low pressure.41 However, the exposure of this CB6 HOF material to humid SO2 conditions led to a phase change and major loss of porosity (particularly its extrinsic pores) due to its weak hydrogen bonds. We propose that the shortcomings of CB6 HOF frameworks can be circumvented by dispersing CB6 molecules in a MOF matrix such as MIL-101 to obtain CB6@MIL-101 (Scheme 1).44 Herein, we report the preparation and characterizations of the targeted CB6@MIL-101-Cl material, which show enhanced SO2 sorption behavior due to the combined merits of high host surface area, optimized pore sizes, CB6 molecules with high SO2 affinity and increased stability toward SO2 molecules. Since CB6 molecules are confined in the pores of MIL-101 frameworks, the performance is less affected by moisture or humid SO2. Further, we discovered that hydrochloride could modify the MIL-101 framework and enhance the stability of the resultant CB6@MIL-101-Cl material toward corrosive SO2 molecules. This work demonstrates for the first time the potential of cage@MOF composite materials for enhanced SO2 removal and storage at low pressures.
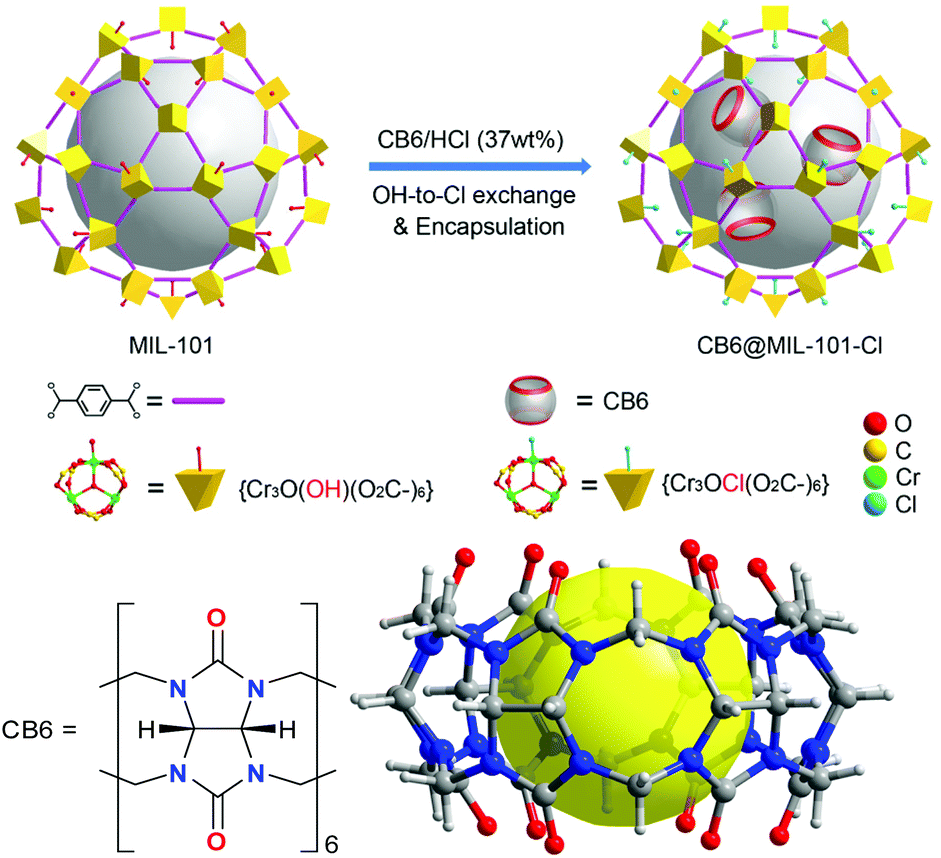 | ||
| Scheme 1 Top: Schematic representation of the encapsulation of CB6 molecules in MIL-101 with the formation of the CB6@MIL-101-Cl composite. The two coordinated aqua ligands on the trinuclear chromium SBU are not indicated for clarity. Bottom: Formula and structure of CB6 = cucurbit[6]uril. The yellow sphere represents the intrinsic pore. | ||
Experimental
Materials
All starting materials and solvents were obtained from commercial sources and used as delivered (Table S1†). CB6 was synthesized according to the literature.44Characterization methods
Powder X-ray diffraction (PXRD) data were collected at room temperature on Rigaku Miniflex 600 powder diffractometer using a low background silicon sample holder and Cu-Kα radiation (λ = 1.5418 Å). The measurements were performed over a 2θ = 2–50° range with a scan speed of 1.5 deg min−1 (600 W, 40 kV, 15 mA). The diffractograms were analysed using the software Match!.45Elemental analysis (CHNS) was carried out using an Elementar Analysensysteme vario MICRO cube instrument. The samples were dried at 150 °C under a vacuum for at least 20 h prior to the measurement.
Fourier transform infrared (FT-IR) spectra were measured on a Bruker FT-IR Tensor 37 spectrometer at room temperature in the range of 550–4000 cm−1 using an attenuated total reflection (ATR) technique (Platinum ATR-QL, diamond). The activated samples were exposed to dry SO2 for 15–30 minutes at room temperature (samples were activated prior to the procedure at 150 °C for 20 h). Then the sealed sample tubes with the MOF in SO2 atmosphere were stored at 77 K for the transfer to the FT-IR instrument room. The tube was allowed to heat up to ∼273 K and a sample was quickly transferred onto the crystal of the ATR unit, and the stamp of the ATR-unit was immediately closed afterwards. The change of the SO2 amount in the sample was monitored during up to 10 minutes (note that the goal of the experiment was to qualitatively monitor the spectral changes). Resolution: 2 cm−1; Sampling time: 16 scans; Background measuring time: 16 scans.
Scanning electron microscopy (SEM) was performed using a Jeol JSM-6510LV QSEM Advanced electron microscope with a LaB6 cathode operating at 20 keV. The microscope was equipped with a Bruker Xflash 410 silicon drift detector for energy-dispersive X-ray (EDX) spectroscopy.
The nitrogen adsorption isotherms were collected at 77 K on a Quantachrome Autosorb-6 automatic adsorption analyzer and evaluated with the AsiQwin V3 software. The samples were degassed in 10−2 mbar vacuum at 150 °C prior to the measurement. The Brunauer–Emmett–Teller (BET) surface areas were calculated using the data in the p/p0 range of 0.05–0.2. The total pore volumes were calculated from the adsorbed volume corresponding to p/p0 = 0.90. All gas uptake volume for N2 and SO2 is given at standard temperature and pressure, STP, i.e. the equivalent volume at 273.15 K and 1.013 bar.
The SO2 sorption isotherms were collected at 273 K and 293 K on a Quantachrome Autosorb iQ MP instrument in 1 × 10−3–1 bar pressure range. All samples were activated for at least 12 hours at 150 °C and <0.01 mbar vacuum prior to each experiment.
The cyclic SO2 sorption experiments were carried out on a Quantachrome Autosorb iQ MP at 293 K. For the three-cycle ad/desorption measurements, full isotherms (with 17 points for ads. and 12 points for des.) were collected for MIL-101, MIL-101-Cl and CB6@MIL-101-Cl. Between each individual isotherm sorption experiment the samples were activated at 150 °C under vacuum (1 × 10−3 mbar) for 12 hours. For the 10-cycle adsorption measurements for CB6@MIL-101-Cl, three points (at 0.01, 0.5 and 0.96 bar) were set for quick analysis of adsorption and desorption and also with activation at 150 °C under vacuum (1 × 10−3 mbar) for 30 minutes before the next cycle was started.
X-ray photoelectron spectroscopy (XPS) was conducted on a ULVAC-PHI VersaProbe II microfocus X-ray photoelectron spectrometer. The spectra were recorded using a polychromatic aluminium Kα X-ray source (1486.8 eV) and referenced to the carbon 1s orbital with a binding energy of 284.8 eV. Experimental XP spectra were fitted by the CasaXPS Software (version 2.3.19PR1.0).
Syntheses
Humid SO2 stability test (performed on MIL-101, nanoCB6-H, and CB6@MIL-101-Cl)
The method closely follows the description in the literature (Scheme S1, ESI†).25 A pre-dried air stream (2 L min−1) was bubbled through an aqueous sodium metabisulfite solution (0.4 g Na2S2O5 in 80 mL water) within a Schlenk flask (250 mL). The setup was equipped with a 3-way valve to regulate the outlet of the gas flow. The outlet of the flask was either connected to a desiccator, playing the role of a humidity chamber, or to the exhaust air when the concentration of SO2 in the chamber reached the target value. The diluted SO2 stream flowed through the desiccator and escaped through a vent. The desiccator contained a crystallizing dish filled with saturated sodium chloride solution (80 mL) ensuring a relative humidity (RH) near 75%. The RH value was monitored with a VWR TH300 hygrometer and the SO2 content with Dräger Pac 6000 electrochemical sensor.50 mg of each MIL-101, nanoCB6-H and CB6@MIL-101-Cl were pre-activated (≤0.01 mbar, 150 °C overnight) and exposed to humid SO2 (35 ± 5 ppm content in the gas stream) by placing them in the desiccator at 75% relative humidity in open broad-necked glass vials for 6 hours at room temperature. Afterwards, the samples were dried at 65 °C in the oven and activated (≤0.01 mbar, 150 °C overnight) for further measurements.
Results and discussion
MIL-101, chromium(III) terephthalate, is built of trinuclear {Cr3(μ3-O)X(H2O)2(O2C-)6}, X = F, OH secondary building units (SBUs), which are bridged by the terephthalate linkers (BDC2−), forming a porous zeotypic framework with two types of mesocages with diameters of 29 and 34 Å.47,48 The small cages have pentagonal windows with a diameter of 12 Å, and the large cages have hexagonal windows with diameters of 15 Å in addition to the pentagonal windows (Scheme S2, ESI†). The molecular size of CB6 is 14.4 Å with a height of 9.1 Å.44 Depending on the synthetic conditions the trinuclear unit has either a charge-compensating fluorido or a hydroxido ligand coordinated by one of the chromium atoms of the SBU.MIL-101, [Cr3(μ3-O)(OH)(H2O)2(BDC)3] was prepared according to the literature,44 and was used as a host to load CB6, which was performed via stirring of the MIL-101 in a CB6 solution in conc. HCl (37 wt%) (Scheme 1). We refer to this process as “wet impregnation”. It should be noted that cucurbit[6]uril was dissolved in conc. HCl (37 wt%) before being treated with MIL-101. A composite material designated as CB6@MIL-101-Cl was the result of the loading of CB6 into MIL-101. The treatment of the MIL-101 host with the HCl solution caused the simultaneous exchange of the charge-balancing hydroxido ligand by chlorido ligand, transforming the host framework to MIL-101-Cl, [Cr3(μ3-O)Cl(H2O)2(BDC)3] during the loading (Scheme 1). Thus, the formation of CB6@MIL-101-Cl, containing 31 wt% of CB6 according to the elemental analysis, and the conversion of the framework by exchange of the terminal anionic ligand, were performed as a concomitant “one-pot” process.
Powder X-ray diffraction (PXRD) confirmed the phase purity of MIL-101 and the retention of the MIL-101 framework structure in CB6@MIL-101-Cl (Fig. S1, ESI†). The nearly identical PXRD patterns confirmed that neither the hydroxido-to-chlorido exchange nor the encapsulation of CB6 into the pores influences the structure of the framework significantly. Compared with the Fourier transform infrared (FT-IR) spectroscopy of MIL-101, CB6@MIL-101-Cl shows additional peaks at 1740 cm−1 and at 1465 cm−1 due to the carbonyl and methylene groups of CB6 (Fig. S2, ESI†).44
The extent of the hydroxido-to-chlorido ligand exchange was quantified by energy dispersive X-Ray analysis (EDX) and X-ray photoelectron spectroscopy (XPS) measurements. For MIL-101 a negligible Cl content was confirmed by both EDX and XPS. EDX data (Fig. S3, Table S3, ESI†) suggests a [Cr3(μ3-O)Clx(OH)1−x(H2O)2(BDC)3] framework formula with x = 0.78 in the CB6@MIL-101-Cl composite. The Cl/Cr3 ratio was checked also after one-cycle of SO2 adsorption–desorption. As a result, the value x = 0.69 was found for CB6@MIL-101-Cl (Fig. S3, Table S3, ESI†), which are slightly lower compared to the initial materials, i.e., prior to the SO2 adsorption–desorption cycle, but still within experimental error.
XPS measurements were carried out after one-cycle of SO2 adsorption–desorption (Fig. S4–S5, ESI†). The Cl/Cr3 ratio or x in [Cr3(μ3-O)Clx(OH)1−x(H2O)2(BDC)3] was 1.08 in CB6@MIL-101-Cl (Table S4, ESI†). The XPS survey spectra show Cr and S for MIL-101, and Cr and Cl but no S for CB6@MIL-101-Cl (Fig. S4, ESI†). The S content in CB6@MIL-101-Cl was not detectable. The high-resolution Cl 2p spectrum for CB6@MIL-101-Cl features the Cl 2p1/2 and Cl 2p3/2 pair of peaks at 199.2 eV and 197.5 eV binding energies (Fig. S5, ESI†), which corresponds to a chloride ion,49–51 in line with the spin–orbit splitting value of 1.6 eV. The high-resolution Cr 2p XPS spectrum exhibits peaks at 587.3 eV and 577.8 eV for MIL-101, and 586.7 eV and 577.3 eV for CB6@MIL-101-Cl (Fig. S5, ESI†). These values correspond well to typical 587.4 eV and 577.8 eV binding energies for CrIII 2p (two Cr 2p peaks are observed due to spin–orbit splitting, which matches the expected value of 9.3 eV).52–54 The 0.5–0.6 eV shift to lower binding energy of Cr 2p for CB6@MIL-101-Cl compared with MIL-101 derives from the slight change of the average Cr environment due to the hydroxido-to-chlorido exchange.
The high-resolution S 2p spectrum for MIL-101 (Fig. S5, ESI†) exhibits the S 2p1/2 and S 2p3/2 pair of peaks at 170 eV and 168.8 eV binding energies with a peak separation of 1.2 eV, which verified the chemical oxidation state of S(IV).55 The S/Cr3 ratio is 4.23 in MIL-101 after one-cycle of SO2 adsorption–desorption (Table S4, ESI†). In contrast, sulfur could not be detected in CB6@MIL-101-Cl after SO2 sorption. Since XPS operates under high vacuum we suggest putatively an initial reaction of Cr-OH in MIL-101 with SO2 under formation of Cr-hydrogensulfite, Cr-OSO2H or -disulfite, Cr-OS2O4H leading to an opening up of Cr coordination sites for further SO2 chemisorption. The S 2p XPS spectrum of MIL-101 matches with metal(Ce, Ti)-sulfite/hydrogensulfite.21,56,57
Scanning electron microscopy (SEM) images further confirmed the retention of the morphology, thereby supporting the expected stability of the material also on the macroscopic level under mild adsorptive loading conditions. The synthesized MIL-101 and CB6@MIL-101-Cl phases have similar particle sizes (0.5–2.0 μm) and feature octahedral crystals typical for MIL-101.58,59 (Fig. S6, ESI†). The absence of other significant crystal forms or coverage of the octahedral surfaces indicates that CB6 has not crystallized separately on the outer surface of the MIL-101 crystallites.
The porosity of the samples was assessed by analysis of the N2 adsorption–desorption isotherms measured at 77 K. The isotherm types of both MIL-101 and CB6@MIL-101-Cl are similar and retain the characteristic two-step shape (corresponds to IUPAC type Ib isotherms)60 (Fig. 1). As expected, the uptake decreases after the encapsulation of CB6 in MIL-101. The Brunauer–Emmett–Teller (BET) surface areas for MIL-101 and CB6@MIL-101-Cl were found to be 3217 m2 g−1 and 2077 m2 g−1, respectively, their pore volumes 1.54 cm3 g−1 and 1.00 cm3 g−1 (assessed at p/p0 = 0.9; Table S2, ESI†). The BET surface area for nanoCB6-H was 435 m2 g−1.
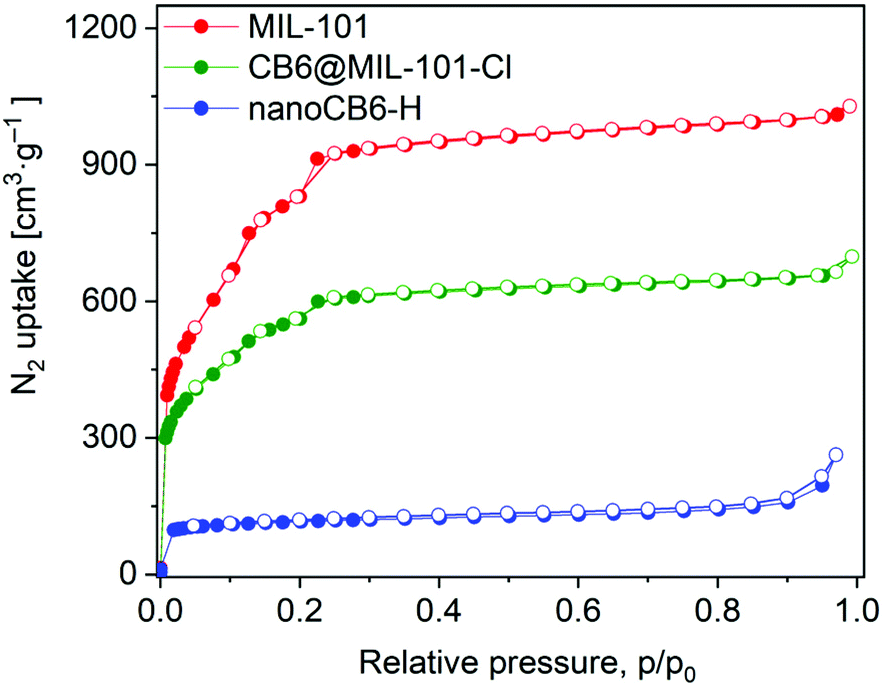 | ||
| Fig. 1 Nitrogen adsorption–desorption isotherms at 77 K of MIL-101, CB6@MIL-101-Cl and nanoCB6-H. Filled symbols, adsorption; empty symbols, desorption. | ||
The SO2 adsorption–desorption isotherms were measured for MIL-101 and CB6@MIL-101-Cl at 293 K and 298 K and nanoCB6-H at 293 K (Fig. 2a). All three materials feature a relatively narrow hysteresis for the whole measured range. MIL-101 has an almost linear SO2 uptake reaching 620 cm3 g−1 and 584 cm3 g−1 (27.7 mmol g−1 and 24.4 mmol g−1) at 293 K and 298 K, respectively, and 1 bar. Expectedly, as the temperature increases, the amount of gas adsorbed decreases. On the other hand, the uptake for the nanoCB6-H is 6.5 mmol g−1 (145 cm3 g−1) at 1 bar and 293 K, in line with its comparatively low total pore volume (Table S2, ESI†). However, about 43% of the total uptake of nanoCB6-H occurs in the low-pressure range of 0–0.01 bar, which reflects the strong affinity of CB6 toward SO2 (Fig. 2b, Fig. S7, Table S5, ESI†). “Nano” in nanoB6-H refers to a particle size of 100–500 nm. NanoCB6-H shows a higher SO2 uptake (63 cm3 g−1, 145 cm3 g−1) than micro-size crystallites of CB6 (43 cm3 g−1, 98 cm3 g−1) at both 0.01 bar and 0.97 bar and 293 K, due to the faster diffusion of SO2 molecules in nanoCB6-H with smaller CB6 particle size.41
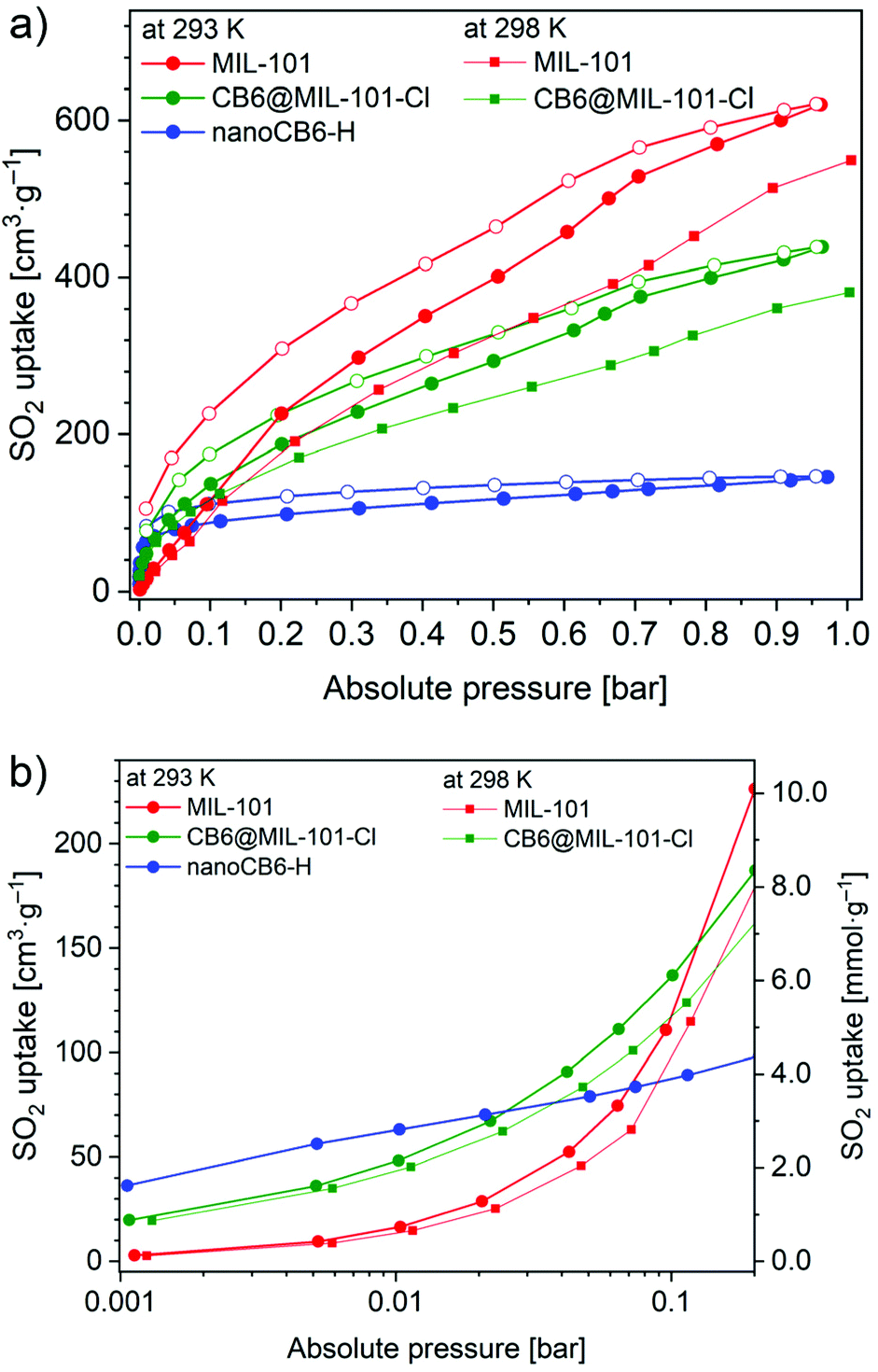 | ||
| Fig. 2 SO2 sorption isotherms for MIL-101 and CB6@MIL-101-Cl at 293 K (normal lines) and 298 K (thin lines, adsorption branch only), and for nanoCB6-H at 293 K: (a) 0–1 bar range. (b) 0.001–0.2 range, logarithmic scale (in b the desorption isotherms are omitted for clarity). | ||
The CB6@MIL-101-Cl composite combined the merits of the two materials for SO2 adsorption. It merges the strong affinity of the CB6 cages at low pressure and the high capacity of MIL-101. At 1 bar, a high SO2 uptake of 438 cm3 g−1 or 380 cm3 g−1 (19.5 mmol g−1, 17.0 mmol g−1) at 293 K or 298 K, respectively, is reached, which is better than for most other SO2-stable MOF materials (Table S6, ESI†), except of MIL-101 (27.7 mmol g−1 at 293 K), MOF-177 (25.7 mmol g−1 at 293 K)29 and close to MFM-170 (17.5 mmol g−1 at 298 K).24 However, MOF-177 and MIL-101 are unstable after SO2 exposure, while the composite CB6@MIL-101-Cl is stable after SO2 exposure. Understandably, the maximum uptake decreases somewhat in the CB6@MIL-101-Cl composite compared to the MIL-101 host, due to a lower total pore volume (Table S2, ESI†). The SO2 adsorption isotherms of MIL-101 and CB6@MIL-101-Cl at 1 bar did not reach saturation as they still have a high positive slope and are far from levelling off. Saturation will require a pressure above 1 bar.
At 0.01 bar and 293 K the captured SO2 amount was 2.2 mmol g−1 (2.0 mmol g−1 at 298 K) for CB6@MIL-101-Cl compared to 0.7 mmol g−1 (0.6 mmol g−1 at 298 K) for MIL-101, i.e., a remarkable improvement by a factor of three (Fig. 2b). For comparison, the SO2 uptake of nanoCB6-H was found to be 2.8 mmol g−1 at 0.01 bar. The important steep increase in the uptake of SO2 by CB6@MIL-101-Cl in the low-pressure zone demonstrates the desirable efficiency for SO2 removal at low partial pressure. The calculated mass-weighted SO2 uptake for a physical mixture of CB6 and MIL-101 would be only 1.3 mmol g−1 at 0.01 bar, indicating a synergistic effect in the composite CB6@MIL-101-Cl. At 0.01 bar, the SO2 uptake of CB6@MIL-101-Cl (2.2 mmol g−1 at 293 K and 2.0 mmol g−1 at 298 K) is significantly higher than that of most MOF sorbents in the literature (Fig. S8, Table S6, ESI†), and only lower than that of SIFSIX-2-Cu-i and MIL-160 (4.2 mmol g−1),28,29 SIFSIX-1-Cu (3.4 mmol g−1),28 DMOF-TM (3.8 mmol g−1),35 MFM-305 (3.3 mmol g−1),27 MFM-305-CH3, NH2-MIL-125(Ti) and mmen-MIL-101(Cr) (3.0 mmol g−1),27,29,61 as well as SIFSIX-3-Ni (2.4 mmol g−1).28 However, those materials show a lower uptake at 1 bar compared to CB6@MIL-101-Cl (Table S6, ESI†).
The isosteric enthalpy of adsorption (ΔHads) for SO2 adsorption on CB6@MIL-101-Cl and MIL-101 was calculated from the adsorption isotherms measured at 273 K and 293 K using the virial method (Fig. 3, Fig. S9 and S10, ESI†).62 The near zero-coverage adsorption enthalpy of SO2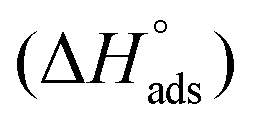 for CB6@MIL-101-Cl is ∼–50 kJ mol−1, while for MIL-101 it is ∼–35 kJ mol−1. The higher negative
for CB6@MIL-101-Cl is ∼–50 kJ mol−1, while for MIL-101 it is ∼–35 kJ mol−1. The higher negative  of SO2 for CB6@MIL-101-Cl proves that the incorporation of CB6 cages can indeed increase the affinity of materials towards SO2, probably by narrowing the pores towards multiple optimal gas-adsorbent interactions25,33,35,63 and providing intra/outer surfaces of the relatively polar CB6 for SO2 adsorption, even if the loading may block some of the external sites.
of SO2 for CB6@MIL-101-Cl proves that the incorporation of CB6 cages can indeed increase the affinity of materials towards SO2, probably by narrowing the pores towards multiple optimal gas-adsorbent interactions25,33,35,63 and providing intra/outer surfaces of the relatively polar CB6 for SO2 adsorption, even if the loading may block some of the external sites.
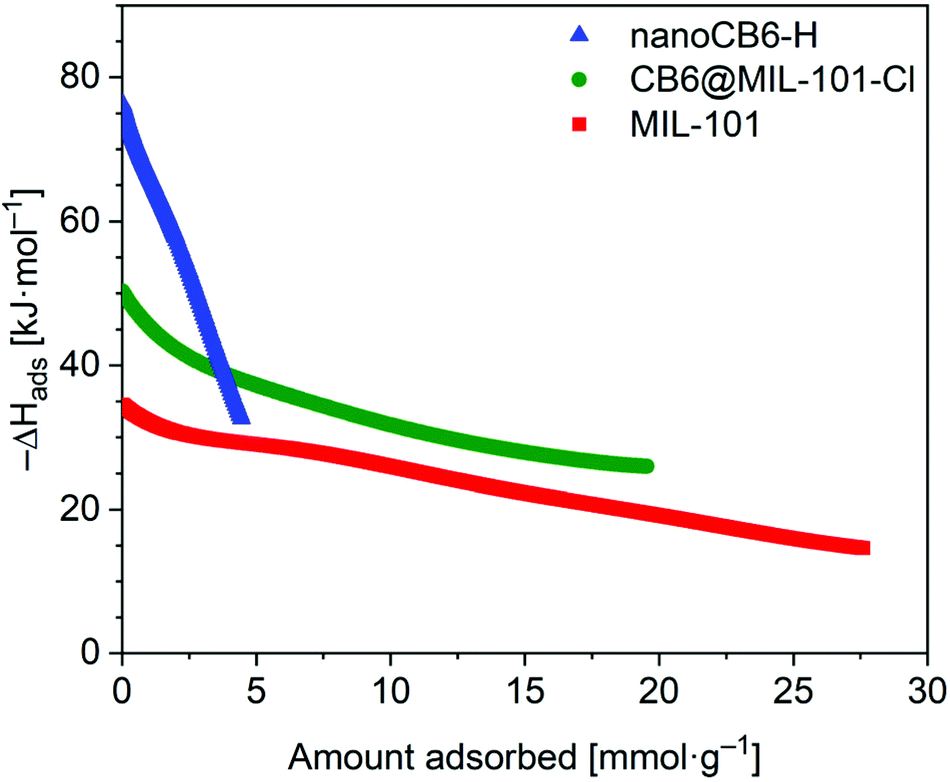 | ||
| Fig. 3 Isosteric enthalpy of adsorption dependence on the amount of SO2 adsorbed for MIL-101, CB6@MIL-101-Cl and nanoCB6-H. | ||
It should be noted that the  value of SO2 for nanoCB6-H was ∼–76 kJ mol−1 when the material was activated at 150 °C, at the same temperature as CB6@MIL-101-Cl and MIL-101 (in previous work,41 where nanoCB6-H was only activated at 100 °C,
value of SO2 for nanoCB6-H was ∼–76 kJ mol−1 when the material was activated at 150 °C, at the same temperature as CB6@MIL-101-Cl and MIL-101 (in previous work,41 where nanoCB6-H was only activated at 100 °C, 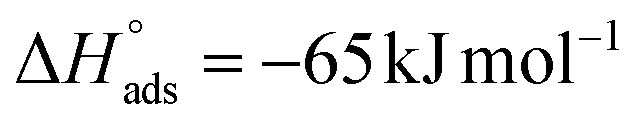 was given). From DFT-D3 (dispersion-corrected DFT) calculations on the possible binding sites for SO2 at CB6 macrocycles the cavity of the CB6 cage was the highest-energy binding site with a calculated binding energy of −82 kJ mol−1. There, SO2 molecules are primarily adsorbed through two electrostatic O2Sδ+⋯δ−O
was given). From DFT-D3 (dispersion-corrected DFT) calculations on the possible binding sites for SO2 at CB6 macrocycles the cavity of the CB6 cage was the highest-energy binding site with a calculated binding energy of −82 kJ mol−1. There, SO2 molecules are primarily adsorbed through two electrostatic O2Sδ+⋯δ−O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CB6) interactions at an S⋯O distance of 3.04 Å. The intrinsic pore of CB6 is large enough (5.8 Å center diameter, 3.9 Å window diameters, 9.1 Å height) to encapsulate two SO2 molecules, which can enter in these O2Sδ+⋯δ−OC(CB6) interactions in the confined space.41 The second highest-energy binding site with calculated −50 kJ mol−1 has SO2 located around the outer surface and involves electrostatic O2Sδ+⋯δ−OC(CB6), O2Sδ+⋯δ−N(CB6) and OSOδ−⋯δ+H–C(CB6) interactions.41
C(CB6) interactions at an S⋯O distance of 3.04 Å. The intrinsic pore of CB6 is large enough (5.8 Å center diameter, 3.9 Å window diameters, 9.1 Å height) to encapsulate two SO2 molecules, which can enter in these O2Sδ+⋯δ−OC(CB6) interactions in the confined space.41 The second highest-energy binding site with calculated −50 kJ mol−1 has SO2 located around the outer surface and involves electrostatic O2Sδ+⋯δ−OC(CB6), O2Sδ+⋯δ−N(CB6) and OSOδ−⋯δ+H–C(CB6) interactions.41
With regard to 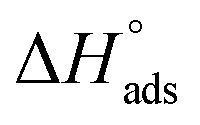 , the composite is closer to MIL-101 than to nanoCB6-H, while in the case of a non-interacting, physical mixture,
, the composite is closer to MIL-101 than to nanoCB6-H, while in the case of a non-interacting, physical mixture, 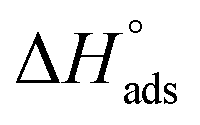 should be equal to the latter. The decrease could be interpreted as a blockage of active sites, e.g. via MIL-host⋯CB6-guest interactions. Also, the state of the CB6 guests in the MIL pores will be rather amorphous; thereby, the high values for the CB6-H crystalline state, could barely be expected. With increased loading of SO2, the isosteric enthalpy of adsorption (–ΔHads) of all three materials is decreased (Fig. 3), according to the occupation of binding sites in the order of decreasing binding energies, also indicating adsorbents with different sites.
should be equal to the latter. The decrease could be interpreted as a blockage of active sites, e.g. via MIL-host⋯CB6-guest interactions. Also, the state of the CB6 guests in the MIL pores will be rather amorphous; thereby, the high values for the CB6-H crystalline state, could barely be expected. With increased loading of SO2, the isosteric enthalpy of adsorption (–ΔHads) of all three materials is decreased (Fig. 3), according to the occupation of binding sites in the order of decreasing binding energies, also indicating adsorbents with different sites.
Comparative FT-IR spectroscopy was employed to assess the possible SO2 adsorption sites in CB6@MIL-101-Cl and MIL-101. A series of spectra of the SO2 loaded compounds with gradually decreasing SO2 content, as a result of contact with ambient atmosphere (0 to 10 min), were collected as described in the Experimental section, and compared with the spectrum of the activated compound (Fig. 4). The new bands, appearing as a result of adsorption, at 1328 and 1144 cm−1 in CB6@MIL-101-Cl and at 1332 and 1146 cm−1 in MIL-101 are assigned to the asymmetric and symmetric stretching vibrations of the physisorbed SO2 molecules.64,65 The new bands are slightly shifted compared to the bands at 1330 and 1149 cm−1 for free SO2 molecules, thereby indicating the interaction with the surface of the materials. The intensity of the bands assigned to the adsorbed SO2 gradually decreases with time. The bands are traceable for around 2 minutes for MIL-101 and around 5 minutes for CB6@MIL-101-Cl under the same experimental conditions (Fig. 4, Fig. S11, ESI†). This can be seen in line with the difference in isosteric adsorption enthalpy since the adsorption kinetics are judged as similar from the similar hysteresis widths of the desorption isotherms for MIL-101 and CB6@MIL-101-Cl.
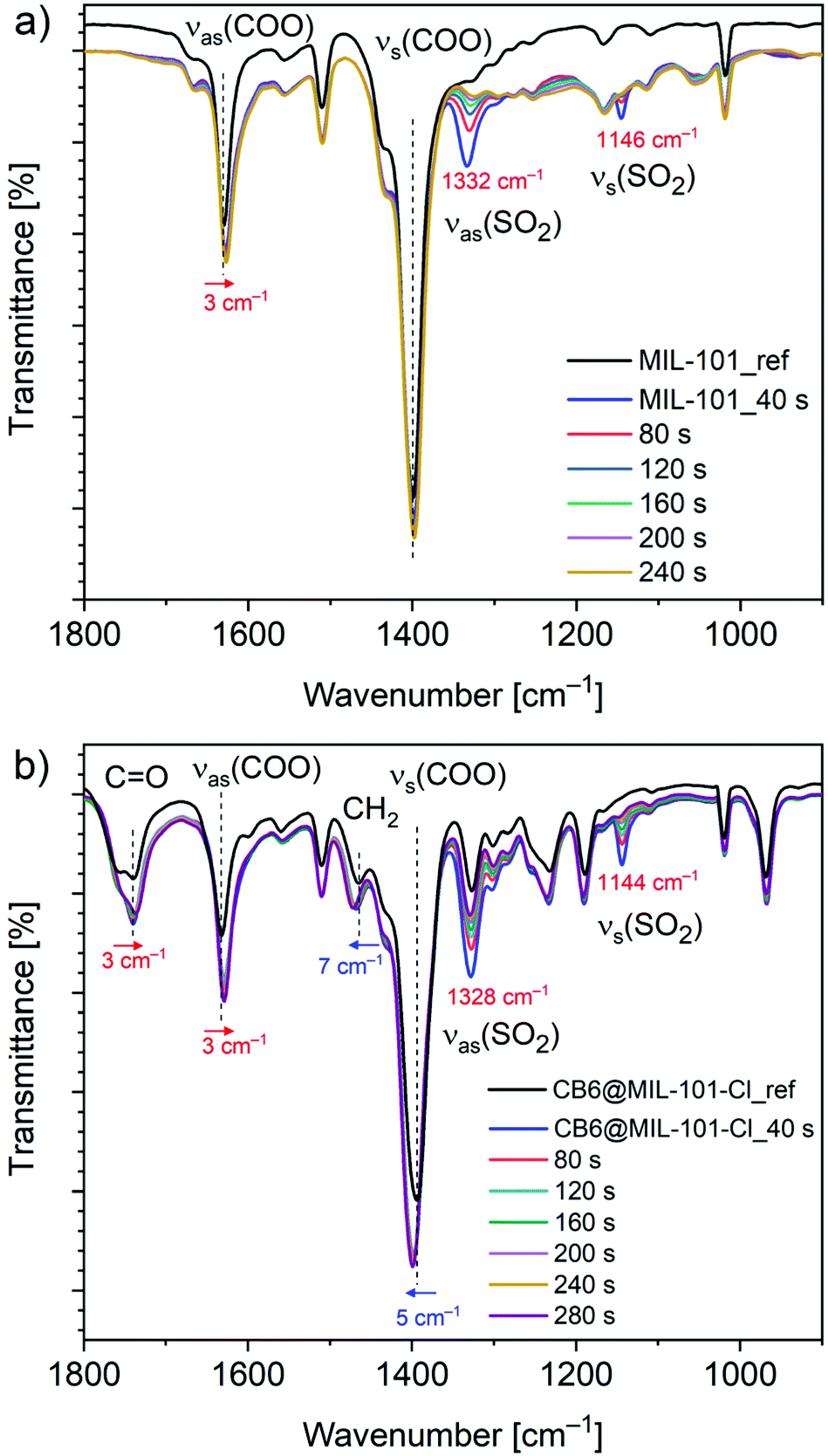 | ||
| Fig. 4 FT-IR spectra of (a) MIL-101 and (b) CB6@MIL-101-Cl before and after single exposure and loading with SO2 (see Exp. section for details). | ||
The comparison of the FT-IR spectra of the activated materials and the materials with variable SO2 contents also show vibrational changes of functional groups of the frameworks. The bands at 1630 cm−1 and 1398 cm−1 in MIL-101 and at 1632 cm−1 and 1394 cm−1 in CB6@MIL-101-Cl are attributed to the asymmetric νas and symmetric νs carboxylate (COO) stretching vibrations.61,66 In both materials, after exposed once to SO2, the carboxylate νas(COO) band is red-shifted (Δ = −3 cm−1), and the νs(COO) band is blue-shifted (Δ = 5 cm−1 for CB6@MIL-101-Cl and invariant within experimental error of ±2 cm−1 for MIL-101).
It is worth nothing that the carboxylate frequency shifts observed for CB6@MIL-101-Cl tend to be more pronounced compared to MIL-101. Additionally, the IR spectral response of CB6 in CB6@MIL-101-Cl upon SO2 exposure is also clearly traceable. Thus, the carbonyl stretching vibration ν(CO) = 1740 cm−1 of CB6 demonstrates a red shift (Δ = −3 cm−1) upon SO2 adsorption and the methylene bending vibration ν(CH2) = 1465 cm−1 a blue shift (Δ = 7 cm−1) indicating interactions between SO2 and CB6. In comparison, nanoCB6-H under the same conditions on the same instrument features a slightly stronger red shift of ν(CO) (Δ = −6 cm−1) and a slightly weaker blue shift of ν(CH2) (Δ = 4 cm−1).41
The FT-IR spectra (Fig. S12, ESI†) of MIL-101 and CB6@MIL-101-Cl after one full SO2 ad/desorption cycle followed by degassing at 150 °C and 1 × 10−3 mbar for 12 h were also measured. It was found that the spectrum of CB6@MIL-101-Cl was essentially unchanged within experimental error which supports the stability of CB6@MIL-101-Cl towards dry SO2 sorption. In MIL-101, the broad νas(COO) band in MIL-101, initially at ∼1630 cm−1, is remarkably red-shifted (Δ = −10 cm−1). The shift is comparable to the one observed in SO2 loaded MFM-170 carboxylate MOF (Δ = −10 cm−1), suggesting the interaction between SO2 and carboxylate groups in the framework.24 More importantly, new bands at 1100 cm−1 and 1049 cm−1 and a broad band at 900–1200 cm−1 appeared, which were attributed to the presence of sulfite and hydrogensulfite species.64,67,68 This is in line with the observation from XPS (Fig. S5, Table S4, ESI†). The broadening of the νas(COO) band indicates the partial collapse and decrease of the local uniformity in the MIL-101 framework.
To further investigate the stability of these materials against dry SO2, we performed cyclic adsorption–desorption at 293 K up to 0.96 bar for three runs for MIL-101, MIL-101-Cl and CB6@MIL-101-Cl and also for 10 runs for CB6@MIL-101-Cl (see the Experimental section). The uptake by MIL-101 gradually decreased to 40% from 620 to 245 cm3 g−1 (Fig. S13, ESI†), as expected from earlier reports,61,69 while CB6@MIL-101-Cl maintained 95% of the initial uptake amount also over 10 cycles (Fig. 5). It is worth noting here that statements about the limited stability of MIL-101 against dry SO2 can be found in a few publications.31,61 Ibarra et al. had noted that not only the chromium-hydroxide-containing MIL-101, i.e. [Cr3(O)(OH)(H2O)2(BDC)3], but also the more crystalline chromium-fluoride-containing MIL-101 compound, i.e. [Cr3(O)F(H2O)2(BDC)3], were unstable (cf. sample MIL-101(Cr)-HF, Fig. S22–S24 in the ESI of ref. 31†). A MIL-101 with stability towards dry and humid SO2 could only be obtained with partial incorporation of a fully fluorinated BDC ligand, giving the sample MIL-101(Cr)-4F(1%) which then showed a high SO2 uptake capacity of 18.4 mmol g−1 at 1 bar and 298 K.31 There is an ambiguity regarding the scattered literature data on the stability of Cr(III)-MOFs, but the instability against dry SO2 is the more surprising as Cr(III)-MOFs are in general considered to be highly kinetically inert.
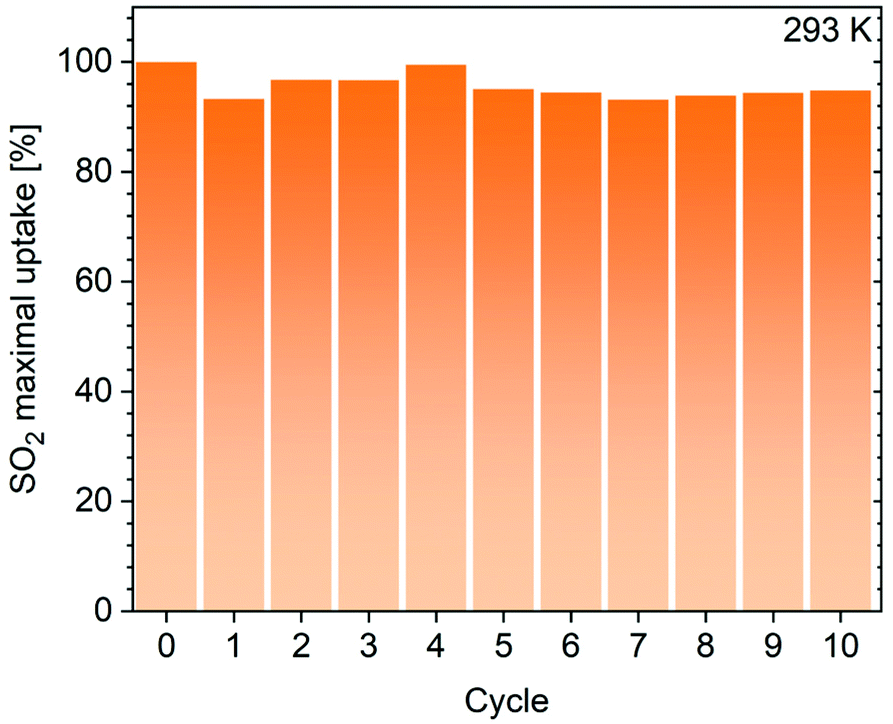 | ||
| Fig. 5 SO2 adsorption capacity of CB6@MIL-101-Cl at 293 K and 0.96 bar, measured over 10 repetitive adsorption–desorption cycles. | ||
The enhanced stability of the CB6@MIL-101-Cl composite towards dry SO2 could be explained by the hydroxido-to-chlorido ligand exchange, that is the substitution of the terminal Cr–OH ligand in the [Cr3(O)X(H2O)2(BDC)3] coordination framework of MIL-101 by Cr–Cl in CB6@MIL-101-Cl. To check the hypothesis a sample of MIL-101-Cl was prepared from MIL-101 in the absence of CB6 but otherwise under the same conditions as CB6@MIL-101-Cl, and submitted to SO2 adsorption–desorption cycling for three runs (Fig. S13, ESI†). The textural properties of MIL-101-Cl are listed in Table S7, ESI.† The BET surface area of MIL-101-Cl is somewhat higher than that of the parent MIL-101, which we reason with the decomposition of pore-filling residual coordination species by HCl and the subsequent removal through the washing processes.70 MIL-101-Cl have kept 97% adsorption capacity of the initial uptake after three adsorption–desorption runs at 293 K (Fig. S13, ESI†). The N2 sorption-based BET-surface area and total pore volume of MIL-101, CB6@MIL-101-Cl and MIL-101-Cl before and after multi-cycle SO2 sorption confirmed the above conclusions from the cyclic SO2 sorption experiments. The BET surface area decreased by 45% (1768 m2 g−1vs. 3217 m2 g−1) for MIL-101, but was retained within experimental error (2036 m2 g−1vs. 2077 m2 g−1) for CB6@MIL-101-Cl and even slightly increased (3541 m2 g−1vs. 3408 m2 g−1) for MIL-101-Cl after exposure to SO2 at 293 K (Table S7, ESI†). The comparison of the PXRD patterns before and after cyclic SO2 sorption for MIL-101, CB6@MIL-101-Cl and MIL-101-Cl verified the preservation of the crystallinity in the two later cases compared to the deterioration of the parent material MIL-101 (Fig. S14, ESI†). Thereby, the combined N2 adsorption and PXRD data confirm the enhanced stability of CB6@MIL-101-Cl and MIL-101-Cl against SO2 compared to the instability of the parent MIL-101. Further, the comparative experiments with MIL-101-Cl suggest that its increased stability is due to the facile one-step hydroxido-to-chlorido ligand exchange in the framework.
In this work we are focusing on the enhanced SO2 uptake and stability of CB6@MIL-101-Cl. The interesting and counterintuitive instability of the parent MIL-101 will be thoroughly addressed in a follow-up study.
Considering the inevitable moisture presence for any possible real-world applications, the stability of materials towards humid SO2 was also investigated by PXRD diffraction and N2 sorption analyses. The PXRD patterns of MIL-101 and CB6@MIL-101-Cl after humid SO2 treatment were retained, indicating their intact frameworks. In contrast, nanoCB6-H lost most of its crystallinity under acidic humid SO2 atmosphere condition. (Fig. S15, ESI†). N2 adsorption–desorption isotherms (Fig. 6) showed that MIL-101 and CB6@MIL-101-Cl kept the BET surface areas unchanged after exposure to humid SO2 (35 ppm) for 6 h.
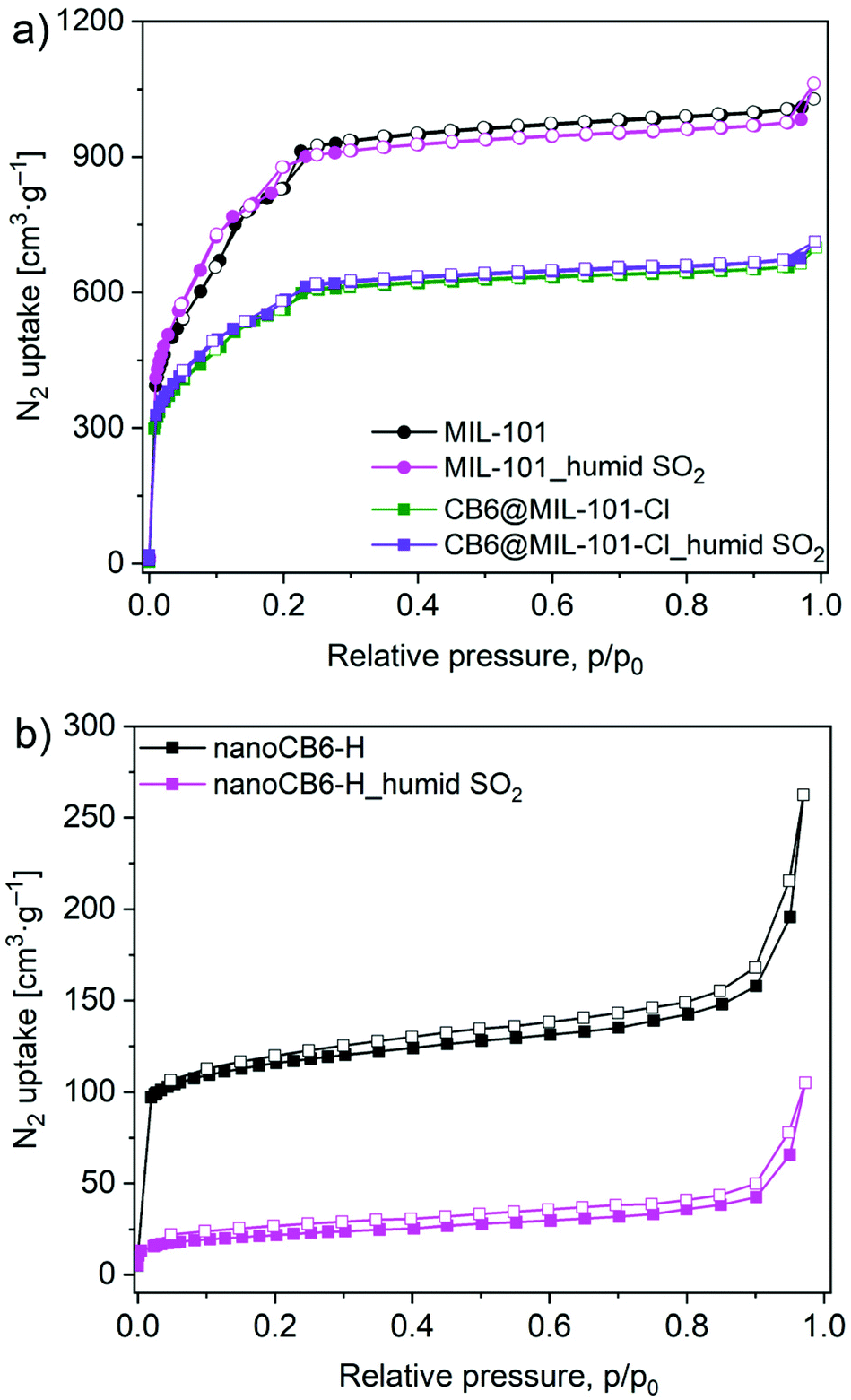 | ||
| Fig. 6 N2 adsorption isotherms (77 K) for (a) MIL-101 and CB6@MIL-101-Cl; (b) nanoCB6-H after exposure to humid SO2 for 6 h. | ||
The BET surface area of nanoCB6-H significantly decreased and only 18% of the initial BET surface area was retained (Table S7, ESI†). This result confirmed again that the hydrogen-bonded organic framework nanoCB6-H is unstable in humid conditions, which is in line with our previous study.41 The fact that MIL-101 was unstable in dry SO2 but stable in humid SO2 conditions could be related to the role of water vapor. We propose that under dry conditions, the Cr-OH site of MIL-101 is well accessible for the reaction with SO2 molecules, while under humid conditions this site (and possibly other reactive sites) is blocked and protected by water clusters. Note that MIL-101 has a medium hydrophilic surface and good stability in acidic solution.71–73 A similar counterintuitive observation was made in Zeolite Y, which exhibited a better stability performance under humid SO2 exposure conditions than toward dry SO2. Also for Zeolite Y the pre-adsorbed water in this highly hydrophilic framework may block the accessibility of SO2.25
In addition, the full SO2 sorption isotherms of these three samples (MIL-101, CB6@MIL-101-Cl and nanoCB6-H) after exposure to humid SO2 (35 ppm) for 6 h were measured (Fig. S16, ESI†). The SO2 uptake capacity of nanoCB6-H after exposure to humid SO2 decreased due to its decreased porosity (Fig. 7). Both MIL-101 and CB6@MIL-101-Cl showed unchanged or even slightly increased SO2 uptakes at 0.01 bar together with around 20% loss of total uptake capacity at 1 bar when compared to dry SO2 sorption (Fig. 7, Table S8, ESI†). Therefore, CB6@MIL-101-Cl exhibits enhanced stability and SO2 removal capability over MIL-101 under lower pressures in both dry SO2 and humid conditions. This stability under humid conditions is a necessity for realistic applications in view of the ubiquitous presence of water.
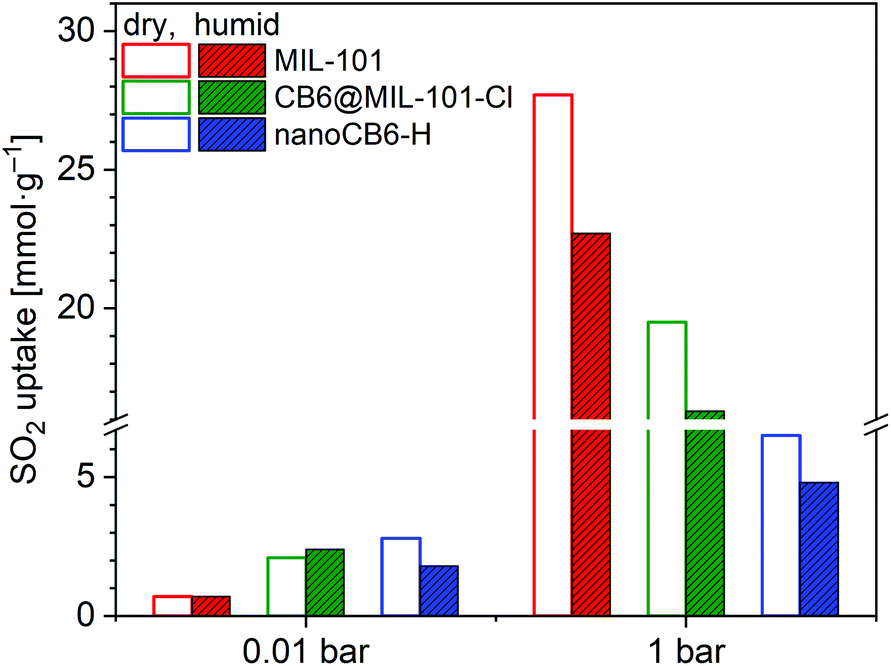 | ||
| Fig. 7 SO2 adsorption uptake (293 K) for MIL-101, CB6@MIL-101-Cl and nanoCB6-H after exposure to humid SO2 for 6 h compared to the behavior under dry conditions (Table S8, ESI†). | ||
Conclusions
We have systematically investigated a cage/MOF composite, named CB6@MIL-101-Cl, obtained by wet impregnation of cucurbituril, CB6 cages into the pores of MIL-101 for potential SO2 gas removal. It is found that CB6@MIL-101-Cl shows enhanced performance in the removal of SO2 gas under low pressure due to the combined merits of CB6 cages with high affinity towards SO2 and MIL-101 with hierarchical pores. The relatively strong interactions between SO2 molecules and CB6@MIL-101-Cl surfaces were supported by time-dependent FT-IR (ATR) spectra. MIL-101 was again confirmed to be unstable towards dry SO2. The stability of CB6@MIL-101-Cl was enhanced when compared with MIL-101 due to the unexpected hydroxido-to-chloride exchange in the {Cr3(O)X(BDC)3} metal node of MIL-101. This unexpected postsynthetic modification was supported by EDX and XPS analysis of related materials. The targeted CB6@MIL-101-Cl shows a high SO2 uptake of 19.5 mmol g−1 at 293 K and 1 bar and high chemical stability even under humid SO2 conditions. The counterintuitive instability of the parent MIL-101 and the observed enhanced stability of MIL-101-Cl is an unexpected finding, which will be thoroughly addressed in a follow-up study and for which the technological importance might be very high. The investigation of the comparable stabilities of different MIL-101-X materials, where X is a terminal counter-anion, is a task, which we are targeting in the near future.Conflicts of interest
There are no conflicts to declare.Author contributions
All the authors have significantly contributed to the paper and have given approval to the final version of the manuscript.Acknowledgements
We thank the China Scholarship Council (CSC) for granting a Ph.D. fellowship to Y. Y. S. J. L. acknowledges financial support of NSFC (22001178) and support from the Hoffmann Institute of Advanced Materials (HIAM), Shenzhen Polytechnic. The authors thank Dr István Boldog, Dr Shanghua Xing, Mr Dennis Woschko and Mrs. Stefanie Bügel for helpful assistance and discussion.References
- Z. Klimont, S. Smith and J. Cofala, Environ. Res. Lett., 2013, 8, 014003 CrossRef CAS.
- X. Xu, C. Chen, H. Qi, R. He, C. You and G. Xiang, Fuel Process. Technol., 2000, 62, 153–160 CrossRef CAS.
- P. Amoatey, H. Omidvarborna, M. S. Baawain and A. Al-Mamun, Process Saf. Environ. Prot., 2019, 123, 215–228 CrossRef CAS.
- M. Matooane and R. Diab, Arch. Environ. Health, 2003, 58, 763–770 CrossRef CAS PubMed.
- F. C. Menz and H. M. Seip, Environ. Sci. Policy, 2004, 7, 253–265 CrossRef CAS.
- J. Yang, L. Li, S. Wang, H. Li, J. S. Francisco, X. C. Zeng and Y. Gao, J. Am. Chem. Soc., 2019, 141, 19312–19320 CrossRef CAS PubMed.
- B. Wu, H. Tian, Y. Hao, S. Liu, X. Liu, W. Liu, X. Bai, W. Liang, S. Lin, Y. Wu, P. Shao, H. Liu and C. Zhu, Environ. Sci. Technol., 2018, 52, 14015–14026 CrossRef CAS PubMed.
- R. K. Srivastava and W. Jozewicz, J. Air Waste Manage. Assoc., 2001, 51, 1676–1688 CrossRef CAS PubMed.
- J.-Y. Lee, T. C. Keener and Y. J. Yang, J. Air Waste Manage. Assoc., 2009, 59, 725–732 CrossRef CAS PubMed.
- R. K. Srivastava, W. Jozewicz and C. Singer, Environ. Prog., 2001, 20, 219–228 CrossRef CAS.
- A. B. Rao and E. S. Rubin, Environ. Sci. Technol., 2002, 36, 4467–4475 CrossRef CAS PubMed.
- R. Pacciani, J. Torres, P. Solsona, C. Coe, R. Quinn, J. Hufton, T. Golden and L. F. Vega, Environ. Sci. Technol., 2011, 45, 7083–7088 CrossRef CAS PubMed.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- J. B. DeCoste and G. W. Peterson, Chem. Rev., 2014, 114, 5695–5727 CrossRef CAS PubMed.
- H. Wang, W. P. Lustig and J. Li, Chem. Soc. Rev., 2018, 47, 4729–4756 RSC.
- E. Martínez-Ahumada, M. L. Díaz-Ramírez, M. d. J. Velásquez-Hernández, V. Jancik and I. A. Ibarra, Chem. Sci., 2021, 12, 6772–6799 RSC.
- T. Islamoglu, Z. Chen, M. C. Wasson, C. T. Buru, K. O. Kirlikovali, U. Afrin, M. R. Mian and O. K. Farha, Chem. Rev., 2020, 120, 8130–8160 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- W. P. Mounfield, C. Han, S. H. Pang, U. Tumuluri, Y. Jiao, S. Bhattacharyya, M. R. Dutzer, S. Nair, Z. Wu, R. P. Lively, D. S. Sholl and K. S. Walton, J. Phys. Chem. C, 2016, 120, 27230–27240 CrossRef CAS.
- M. R. Tchalala, P. M. Bhatt, K. N. Chappanda, S. R. Tavares, K. Adil, Y. Belmabkhout, A. Shkurenko, A. Cadiau, N. Heymans, G. Weireld, G. Maurin, K. N. Salama and M. Eddaoudi, Nat. Commun., 2019, 10, 1328 CrossRef CAS PubMed.
- J. H. Carter, X. Han, F. Y. Moreau, I. da Silva, A. Nevin, H. G. W. Godfrey, C. C. Tang, S. Yang and M. Schröder, J. Am. Chem. Soc., 2018, 140, 15564–15567 CrossRef CAS PubMed.
- G. L. Smith, J. E. Eyley, X. Han, X. Zhang, J. Li, N. M. Jacques, H. G. W. Godfrey, S. P. Argent, L. J. McCormick McPherson, S. J. Teat, Y. Cheng, M. D. Frogley, G. Cinque, S. J. Day, C. C. Tang, T. L. Easun, S. Rudić, A. J. Ramirez-Cuesta, S. Yang and M. Schröder, Nat. Mater., 2019, 18, 1358–1365 CrossRef CAS PubMed.
- P. Brandt, A. Nuhnen, S. Öztürk, G. Kurt, J. Liang and C. Janiak, Adv. Sustainable Syst., 2021, 5, 2000285 CrossRef CAS.
- Y. Zhang, P. Zhang, W. Yu, J. Zhang, J. Huang, J. Wang, M. Xu, Q. Deng, Z. Zeng and S. Deng, ACS Appl. Mater. Interfaces, 2019, 11, 10680–10688 CrossRef CAS PubMed.
- L. Li, I. da Silva, D. I. Kolokolov, X. Han, J. Li, G. Smith, Y. Cheng, L. L. Daemen, C. G. Morris, H. G. W. Godfrey, N. M. Jacques, X. Zhang, P. Manuel, M. D. Frogley, C. A. Murray, A. J. Ramirez-Cuesta, G. Cinque, C. C. Tang, A. G. Stepanov, S. Yang and M. Schröder, Chem. Sci., 2019, 10, 1472–1482 RSC.
- X. Cui, Q. Yang, L. Yang, R. Krishna, Z. Zhang, Z. Bao, H. Wu, Q. Ren, W. Zhou, B. Chen and H. Xing, Adv. Mater., 2017, 29, 1606929 CrossRef PubMed.
- P. Brandt, A. Nuhnen, M. Lange, J. Möllmer, O. Weingart and C. Janiak, ACS Appl. Mater. Interfaces, 2019, 11, 17350–17358 CrossRef CAS PubMed.
- M. Savage, Y. Cheng, T. L. Easun, J. E. Eyley, S. P. Argent, M. R. Warren, W. Lewis, C. Murray, C. C. Tang, M. D. Frogley, G. Cinque, J. Sun, S. Rudić, R. T. Murden, M. J. Benham, A. N. Fitch, A. J. Blake, A. J. Ramirez-Cuesta, S. Yang and M. Schröder, Adv. Mater., 2016, 28, 8705–8711 CrossRef CAS PubMed.
- E. Martínez-Ahumada, M. L. Díaz-Ramírez, H. A. Lara-García, D. R. Williams, V. Martis, V. Jancik, E. Lima and I. A. Ibarra, J. Mater. Chem. A, 2020, 8, 11515–11520 RSC.
- Ü. Kökçam-Demir, A. Goldman, L. Esrafili, M. Gharib, A. Morsali, O. Weingart and C. Janiak, Chem. Soc. Rev., 2020, 49, 2751–2798 RSC.
- P. Brandt, S.-H. Xing, J. Liang, G. Kurt, A. Nuhnen, O. Weingart and C. Janiak, ACS Appl. Mater. Interfaces, 2021, 13, 29137–29149 CrossRef CAS PubMed.
- F. Chen, D. Lai, L. Guo, J. Wang, P. Zhang, K. Wu, Z. Zhang, Q. Yang, Y. Yang, B. Chen, Q. Ren and Z. Bao, J. Am. Chem. Soc., 2021, 143, 9040–9047 CrossRef CAS PubMed.
- S. Xing, J. Liang, P. Brandt, F. Schäfer, A. Nuhnen, T. Heinen, I. Boldog, J. Möllmer, M. Lange, O. Weingart and C. Janiak, Angew. Chem., Int. Ed., 2021, 60, 17998–18005 CrossRef CAS PubMed.
- Y. Lin, H. Lin, H. Wang, Y. Suo, B. Li, C. Kong and L. Chen, J. Mater. Chem. A, 2014, 2, 14658–14665 RSC.
- F. P. Kinik, A. Uzun and S. Keskin, ChemSusChem, 2017, 10, 2842–2863 CrossRef CAS PubMed.
- I. Cota and F. Fernandez Martinez, Coord. Chem. Rev., 2017, 351, 189–204 CrossRef CAS.
- D. K. Yoo, N. Abedin Khan and S. H. Jhung, J. CO2 Util., 2018, 28, 319–325 CrossRef CAS.
- Q.-X. Luo, B.-W. An, M. Ji and J. Zhang, Mater. Chem. Front., 2018, 2, 219–234 RSC.
- J. Liang, S. Xing, P. Brandt, A. Nuhnen, C. Schlüsener, Y. Sun and C. Janiak, J. Mater. Chem. A, 2020, 8, 19799–19804 RSC.
- S. Lim, H. Kim, N. Selvapalam, K.-J. Kim, S. J. Cho, G. Seo and K. Kim, Angew. Chem., Int. Ed., 2008, 47, 3352–3355 CrossRef CAS PubMed.
- J. Tian, J. Liu, J. Liu and P. K. Thallapally, CrystEngComm, 2013, 15, 1528–1531 RSC.
- J. Liang, A. Nuhnen, S. Millan, H. Breitzke, V. Gvilava, G. Buntkowsky and C. Janiak, Angew. Chem., Int. Ed., 2020, 59, 6068–6073 CrossRef CAS PubMed.
- Match! – phase analysis using powder diffraction, 2019 (v.3.5.3.109), H. Putz – Crystal Impact GbR, 53227 Bonn, Germany Search PubMed.
- T. Zhao, F. Jeremias, I. Boldog, B. Nguyen, S. K. Henninger and C. Janiak, Dalton Trans., 2015, 44, 16791–16801 RSC.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040 CrossRef PubMed.
- G. Férey, C. Mellot-Draznieks, C. Serre and F. Millange, Acc. Chem. Res., 2005, 38, 217–225 CrossRef PubMed.
- J. Liang, Y.-Q. Xie, X.-S. Wang, Q. Wang, T.-T. Liu, Y.-B. Huang and R. Cao, Chem. Commun., 2018, 54, 342–345 RSC.
- O. Buyukcakir, S. H. Je, S. N. Talapaneni, D. Kim and A. Coskun, ACS Appl. Mater. Interfaces, 2017, 9, 7209–7216 CrossRef CAS PubMed.
- S. M. Towsif Abtab, D. Alezi, P. M. Bhatt, A. Shkurenko, Y. Belmabkhout, H. Aggarwal, Ł. J. Weseliński, N. Alsadun, U. Samin, M. N. Hedhili and M. Eddaoudi, Chem, 2018, 4, 94–105 CAS.
- Y. Shen, T. Pan, P. Wu, J. Huang, H. Li, I. E. Khalil, S. Li, B. Zheng, J. Wu, Q. Wang, W. Zhang, D. Wei Wei and F. Huo, CCS Chem., 2020, 2, 1607–1614 Search PubMed.
- X. Huang, Q. Hu, L. Gao, Q. Hao, P. Wang and D. Qin, RSC Adv., 2018, 8, 27623–27630 RSC.
- M. Saikia and L. Saikia, RSC Adv., 2016, 6, 14937–14947 RSC.
- F. Zhang, Y. Jin, Y. Fu, Y. Zhong, W. Zhu, A. A. Ibrahim and M. S. El-Shall, J. Mater. Chem. A, 2015, 3, 17008–17015 RSC.
- S. H. Overbury, D. R. Mullins, D. R. Huntley and L. Kundakovic, J. Phys. Chem. B, 1999, 103, 11308–11317 CrossRef CAS.
- E. J. Romano and K. H. Schulz, Appl. Surf. Sci., 2005, 246, 262–270 CrossRef CAS.
- H. C. Yoon, P. B. S. Rallapalli, H. T. Beum, S. S. Han and J.-N. Kim, Energy Fuels, 2018, 32, 1365–1373 CrossRef CAS.
- H. B. Tanh Jeazet, C. Staudt and C. Janiak, Chem. Commun., 2012, 48, 2140–2142 RSC.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- Z. Zhang, B. Yang and H. Ma, Sep. Purif. Technol., 2021, 259, 118164 CrossRef CAS.
- A. Nuhnen and C. Janiak, Dalton Trans., 2020, 49, 10295–10307 RSC.
- A. M. Ebrahim, B. Levasseur and T. J. Bandosz, Langmuir, 2013, 29, 168–174 CrossRef CAS PubMed.
- A. L. Goodman, P. Li, C. R. Usher and V. H. Grassian, J. Phys. Chem. A, 2001, 105, 6109–6120 CrossRef CAS.
- K. Tan, P. Canepa, Q. Gong, J. Liu, D. H. Johnson, A. Dyevoich, P. K. Thallapally, T. Thonhauser, J. Li and Y. J. Chabal, Chem. Mater., 2013, 25, 4653–4662 CrossRef CAS.
- Z. Zhou, B. Cheng, C. Ma, F. Xu, J. Xiao, Q. Xia and Z. Li, RSC Adv., 2015, 5, 94276–94282 RSC.
- M. Waqif, A. M. Saad, M. Bensitel, J. Bachelier, O. Saur and J.-C. Lavalley, J. Chem. Soc., Faraday Trans., 1992, 88, 2931–2936 RSC.
- W. P. Mounfield, U. Tumuluri, Y. Jiao, M. Li, S. Dai, Z. Wu and K. S. Walton, Microporous Mesoporous Mater., 2016, 227, 65–75 CrossRef CAS.
- N. Tannert, Y. Sun, E. Hastürk, S. Nießing and C. Janiak, Z. Anorg. Allg. Chem., 2021, 647, 1124–1130 CrossRef CAS.
- K. Wang, D. Feng, T.-F. Liu, J. Su, S. Yuan, Y.-P. Chen, M. Bosch, X. Zou and H.-C. Zhou, J. Am. Chem. Soc., 2014, 136, 13983–13986 CrossRef CAS PubMed.
- K. Leus, T. Bogaerts, J. De Decker, H. Depauw, K. Hendrickx, H. Vrielinck, V. Van Speybroeck and P. Van Der Voort, Microporous Mesoporous Mater., 2016, 226, 110–116 CrossRef CAS.
- J. Canivet, A. Fateeva, Y. Guo, B. Coasne and D. Farrusseng, Chem. Soc. Rev., 2014, 43, 5594–5617 RSC.
- A. Khutia, H. U. Rammelberg, T. Schmidt, S. Henninger and C. Janiak, Chem. Mater., 2013, 25, 790–798 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Elemental analysis for CB6@MIL-101-Cl, setup for humid SO2 exposure experiments, PXRD, FT-IR, SEM, EDX and XPS analysis, details for SO2 sorption isotherm fitting, comparison of SO2 sorption for MOFs, isosteric enthalpy of adsorption, 3 cycles of full SO2 adsorption–desorption measurement, PXRD after cyclic measurement and exposure to humid SO2, SO2 adsorption isotherms after exposure to humid SO2. See DOI: 10.1039/d1nr04432j |
| This journal is © The Royal Society of Chemistry 2021 |