
Uncovering the encapsulation effect of reduced graphene oxide sheets on the hydrogen storage properties of palladium nanocubes†
Jinseok
Koh
 a,
Eunho
Choi
b,
Kouji
Sakaki
c,
Daeho
Kim
d,
Seung Min
Han
d,
Sangtae
Kim
*b and
Eun Seon
Cho
*a
a,
Eunho
Choi
b,
Kouji
Sakaki
c,
Daeho
Kim
d,
Seung Min
Han
d,
Sangtae
Kim
*b and
Eun Seon
Cho
*a
aDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: escho@kaist.ac.kr
bDepartment of Nuclear Engineering, Hanyang University, Seoul 04763, Republic of Korea. E-mail: sangtae@hanyang.ac.kr
cEnergy Process Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), 16-1 Onogawa, Tsukuba, Ibaraki 305-8569, Japan
dDepartment of Materials Science and Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea
First published on 17th September 2021
Abstract
Decades of research on solute-induced phase transformation of metal hydride systems have shown the possibility to enhance hydrogen storage properties through novel material design such as nanoconfinement engineering. Nevertheless, the fundamentals of mechanical stress effect on confined Pd nanoparticles remain yet to be elucidated due to the difficulty in linking with hydrogen sorption thermodynamics. Here, a thermodynamic tuning of Pd nanocubes associated with hydrogen sorption as a result of encapsulation by reduced graphene oxide (rGO) layers is demonstrated. Pd nanocubes are constrained by rGO to such a degree that the chemical potential and the pressure hysteresis of the system during hydrogen sorption drastically change while showing a size dependence. A thorough thermodynamic analysis elucidates the role of constraints on hydrogen uptake and release; despite the nanoscale regime, the thermodynamic parameters (enthalpy and entropy) during phase transition considerably increase, a phenomenon not seen before in unconstrained Pd nanoparticle systems.
 Eun Seon Cho | Professor Eun Seon Cho is an Associate Professor of Chemical and Biomolecular Engineering at Korea Advanced Institute of Science and Technology (KAIST). She received her PhD in Materials Science and Engineering in 2013 from Massachusetts Institute of Technology (MIT) and worked as a postdoctoral researcher at Lawrence Berkeley National Laboratory (LBNL) from 2013 to 2017. Her major research interests lie in the design and synthesis of functional hybrid nanomaterials for energy and environmental applications. Her current research particularly focuses on the development of nanostructured solid-state hydrogen storage materials, ranging from metal hydrides to porous frameworks. |
Introduction
Phase transformations in solids have received significant research attention in various applications such as hydrogen storage, lithium-ion batteries, and electronic devices.1–6 In particular, solute-driven phase transformations are critical for a number of energy conversion and storage systems, in which a substantial volume change induced by the atomic intercalation may dictate the structural changes and reaction thermodynamics.7 Metal hydrides are one such example where H atoms dissolve in the metal lattice (interstitial hydrides) or combine with metal cations (complex metal hydrides) to form metal hydride phases, allowing safe and efficient solid-state hydrogen storage.2,8The critical performance requirements of successful solid-state hydrogen storage include storage capacity, long-term storage duration against self-discharging or oxidation, and facile insertion/extraction kinetics.9,10 To enhance the storage performance of metal hydrides, researchers proposed structural engineering based on the confinement of metal hydrides into a matrix.2 For example, Parambhath et al. demonstrated that the hydrogen uptake capacity for N-doped graphene decorated by Pd nanoparticles could be enhanced under ambient conditions through facilitated migration of H atoms induced by strengthened interactions between Pd and the graphene matrix.11 Zhou et al. revealed that Pd/graphene nanocomposites have outstanding gravimetric storage density (up to 8.67 wt%) because the graphene matrix enables the homogeneous distribution of Pd nanoparticles while offering a porous space for hydrogen trapping.12 Li et al. showed that metal–organic framework coating altered the reactivity of Pd nanoparticles with hydrogen, leading to enhanced hydrogen storage capacity as well as expedited hydrogen sorption response.13 These reports confirm that host materials in composites play a crucial role in engineering the hydrogen sorption behaviour of metal hydrides.
However, many successful metal hydrides face intricate stability issues that critically depend on fundamental thermodynamics. For instance, lithium aluminium hydrides (LiAlH4 or Li3AlH6) suffer from occasional hydrogen loss or irreversibility under practical conditions due to the highly metastable hydride phases (LiAlH4), leading to impractical rehydrogenation pressures.14,15 On the other hand, the strong thermodynamic stability of magnesium hydrides (MgH2) results in high desorption temperatures of over 280 °C.16 These two contrasting examples show that the capability to tune the thermodynamics of metal hydride formation is essential for careful optimization. Yet, tuning the stability of metal hydrides remains challenging to accomplish, while at the same time satisfying the other performance requirements. Constraint-induced stress has been one potential thermodynamic handle for the tuning of hydride stability;17–19 for instance, an elastic clamping effect by matrix materials can tailor the thermodynamics of hydrogen absorption.20 However, how the external stress affects the hydrogen sorption thermodynamics reversibly remains yet to be demonstrated, especially in nanoconfined metal hydride composites, lacking the fundamental understanding of confinement strategy and the associated structure engineering in the de/hydriding phase transformation.
In this study, we demonstrate the reversible tuning of hydrogen sorption thermodynamics in metal nanoparticles via nanoconfinement-induced stresses. Using rGO-encapsulated Pd nanocubes (31, 45, and 65 nm size) as model systems, we reveal that during hydrogen sorption, mechanical constraints applied by rGO restrain the volume expansion of Pd lattice and induce a wider pressure hysteresis under isothermal conditions, compared to bare Pd nanocubes of similar sizes. Careful thermodynamic analyses show that both the desorption enthalpy and entropy increase upon rGO-confinement, and ab initio calculations quantitatively rationalize the results based on elastic strains.
Results and discussion
Cube-shaped Pd nanoparticles encapsulated by rGO sheets with three different edge sizes (31, 45, and 65 nm) are synthesized by a facile one-pot reduction method. In this method, the oxygen functional groups and the defect sites of GO act as the activation sites for Pd nanocube growth,21 followed by a co-reduction of GO and Pd precursors to rGO and Pd particles, respectively. Fig. 1a illustrates the rGO-Pd system where few rGO layers fully cover Pd nanocubes. Transmission electron microscopy (TEM) images show highly transparent GO sheets, indicating that GO layers are well exfoliated and dispersed to form few layers through an ultra-sonication process (Fig. S1†). Fig. 1b–d shows that Pd nanoparticles are grown in cube shapes with edge sizes ranging from 31 nm to 65 nm, depending on the amounts of surfactants. The composites with 31, 45, and 65 nm Pd nanocubes are labelled as rGO-Pd1, rGO-Pd2, and rGO-Pd3, respectively (see Fig. S2† for a low magnified view of the rGO-Pd samples). A Pd nanocube in the composite oriented along the [001], [013], and [112] zone axis is imaged by high-resolution TEM (HRTEM) to determine whether the nanocube is fully covered by the rGO layers. In all directions, few rGO layers appear to wrap the edges of the Pd nanocube (Fig. S3†). Another Pd nanocube from a different batch of similarly sized nanocomposite also confirms the fully encapsulating layers of rGO and perfect wrapping of Pd nanocubes (Fig. S4†). The perfectly wrapped rGO sheets are expected to provide mechanical constraint and pseudo-pressure effects22 during the H insertion-induced volume expansion. Since the pseudo-pressure effect can be affected by the number of rGO layers, the same commercial GO powders which have even layer-thickness distribution are used in all measurements. The bare-Pd nanocubes with similar edge sizes (30, 42, and 62 nm) are prepared and named Pd1, Pd2, and Pd3, respectively, to probe the encapsulation effect of rGO sheets on the hydrogenation behaviour. The size distribution of the bare-Pd nanocubes in each batch closely corresponds with that of the corresponding rGO-Pd batch (Fig. 1b–d and Fig. S5†), providing a nanoscale platform to study the sole effect of rGO encapsulation. In addition, the magnified TEM images and the corresponding FFTs (right panels of Fig. 1b–d) along with the selected area electron diffraction (SAED) patterns (Fig. S6†) show that Pd nanocubes are single-crystalline. This simplifies the analyses involving hydrogen uptake and release as no microstructural defects such as grain boundaries need to be considered.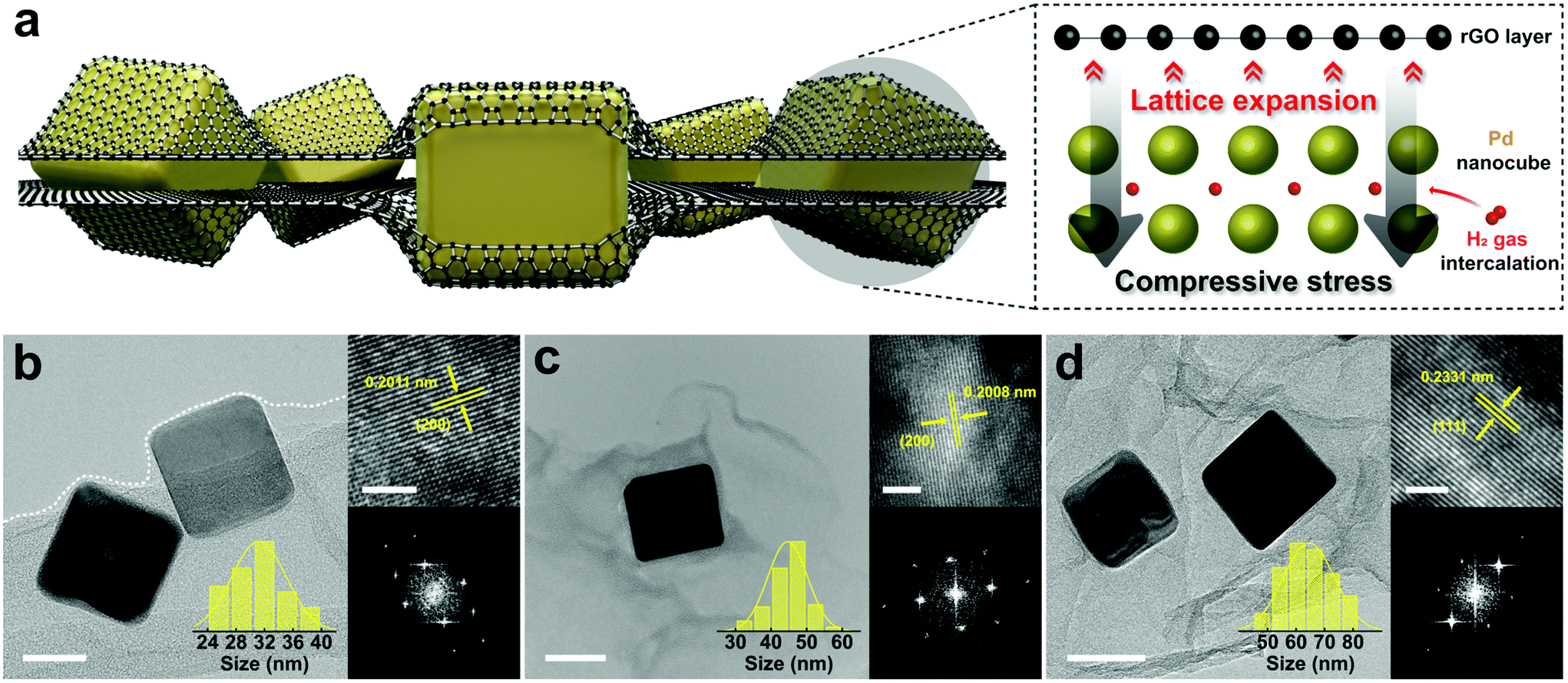 | ||
| Fig. 1 (a) A schematic illustration showing the rGO-Pd system in which compressive strain is applied to the inner Pd nanocubes by rGO layers. (b–d) TEM images of Pd nanocubes encapsulated by rGO sheets of side length (b) 31.2 ± 3.8 nm, (c) 44.7 ± 5.6 nm, and (d) 64.5 ± 9.0 nm (scale bars are 20 nm, 40 nm, and 50 nm, respectively). The right panels show the magnified views of nanocubes (a scale bar is 2 nm) and the corresponding FFT. | ||
While single-crystalline Pd nanocubes help us understand the effect of external stress on the hydriding thermodynamics without the complications of microstructural defects, surface stress becomes significant for Pd nanocubes smaller than 30 nm.23,24 This makes it difficult to solely compare the effect of extrinsic stress. Moreover, the enthalpy and entropy changes upon de/hydriding for particles below 10 nm size are reported to considerably decrease due to nanosizing effects,24,25 thus increasing the difficulty in performing independent thermodynamic analyses. The particle sizes of 31, 45, and 65 nm are thus selected for this study.
Furthermore, the interplanar spacing of Pd nanocubes is measured using HRTEM to clarify the mechanical constraints applied by the rGO sheets. As shown in Fig. S7,† the averaged spacing values (dhkl) are calculated from the intensity profiles of HRTEM images. The rGO-Pd samples of rGO-Pd1, rGO-Pd2 and rGO-Pd3 show characteristic (200) spacing of 2.011 Å, (200) spacing of 2.008 Å, and (111) spacing of 2.331 Å with the increase in Pd size, respectively, suggesting that the Pd lattices are tensile-strained in the in-plane direction and compressive-strained in the out-of-plane direction. The strains (ε) are determined using bulk Pd reference (d0, PDF # 00-046-1043), where ε = (dhkl − d0)/d0. Without any size-dependence, the strain values range from +3.24 to +3.78%, in line with the previous study on the rGO-confined spherical Pd nanoparticle system.26 This indicates that Pd nanocubes are under compressive stress from rGO layers, whose clamping effect induces a lateral expansion of Pd lattice in the in-plane orientation.
The X-ray diffraction (XRD) patterns show a clear crystallinity of the synthesized Pd nanocubes in both rGO-Pd and bare-Pd samples (Fig. S8†). The samples exhibit identical diffraction patterns in terms of the diffraction angles and relative intensities. No distinct palladium oxide (PdO) peak is observed in both patterns. The Pd loadings of rGO-Pd composites are measured to be approximately 82 wt% by inductively coupled plasma-atomic emission spectroscopy (ICP-AES).
X-ray photoelectron spectroscopy (XPS) measurements for rGO-Pd samples show decreased intensities of the oxygen functional groups and recovery of sp2 carbon intensities, confirming the successful reduction of GO (Fig. S9†). This change is critical for studying the hydriding phase transformation of Pd in our system because the removal of oxygen functional groups in GO decreases the graphene interlayer distances in the composite, leading to the generation of pseudo-pressure effects.22 The remaining oxygen functional groups result in a strong interaction between the Pd and GO layers, subsequently causing Pd to cover the surface of GO. This leads to an unintended worm-like, continuous Pd shape rather than the formation of individual particles.27 In Pd 3d XPS spectra of both rGO-Pd and bare-Pd (Fig. S10†), two strong peaks of elemental Pd without distinct PdO peaks are observed, in line with the XRD study. It is noteworthy that no obvious peak shift is observed for Pd binding energy, implying a weak chemical interaction between Pd and rGO sheets in the dehydrated states. This can be attributed to the relatively large Pd nanocubes in our system that reduce the proportion of surface Pd atoms in contact with graphene, in contrary to the sub-nanometer Pd particles that show strong interactions with C and/or O atoms from graphene.11,28,29 Such weak chemical interactions further help us focus on the stress-driven thermodynamic tuning as the hydrogen storage properties are significantly affected by chemical environments.11,13
Fig. 2 shows the hydrogen absorption and desorption behaviours of Pd nanocubes at three different temperatures. To determine the hydrogen sorption characteristics solely based on Pd, we normalize the data based on the Pd mass obtained from the ICP measurements. Considering that graphene derivatives rarely absorb hydrogen molecules,30 hydrogen sorption is only attributed to Pd nanocubes. Repeated re/dehydriding experiments on the rGO-Pd samples show nearly identical hydrogen sorption characteristics (Fig. S11†). Three distinctive regions, namely the hydrogen-poor α phase, two-phase equilibrium region, and the hydrogen-rich β phase, are observed in all the isotherms, in accordance with the first-order phase transition between Pd and PdHx.31 All the isotherms of bare-Pd show similar hydrogen absorption/desorption behaviours to those of bulk Pd (Fig. S12†), except for the narrower equilibrium pressure gaps. This is in good agreement with the previously reported study on unencapsulated Pd nanocubes.32
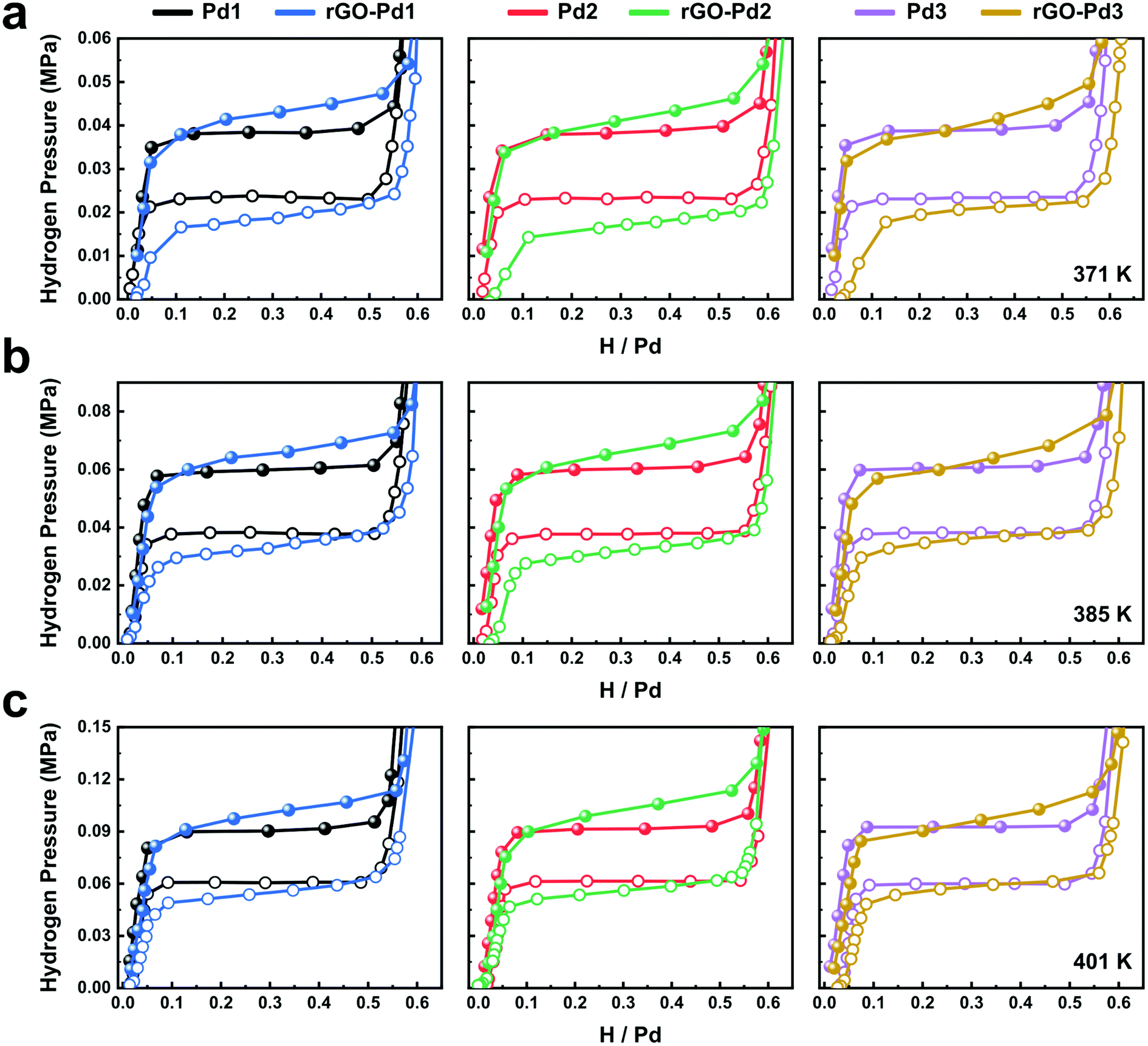 | ||
| Fig. 2 Pressure–composition–temperature (PCT) isotherms of rGO-Pd nanocubes and bare-Pd nanocubes at (a) 371 K, (b) 385 K, and (c) 401 K during hydrogen absorption (solid circles) and desorption (open circles). | ||
We note that the rGO-Pd isotherms show a sloped plateau between absorption (Pabs) and desorption (Pdes) compared to the bare-Pd isotherms. The sloped plateau during hydriding indicates that increasing H chemical potential (ΔμH(P) = RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln[P/P0]) is required as the hydrogen intake increases. Under isothermal conditions inside the rGO-Pd composites, the increasing chemical potential of H is explained by the hydrostatic stress-induced chemical potential change (ΔμH = σhydro[∂V/∂NH]T) due to the mechanical constraint. σhydro and [∂V/∂NH]T stand for the hydrostatic stress and partial molar volume of H, respectively. Since any stress state may be decomposed into hydrostatic stress and deviatoric stress, our uniaxial compressive stress with tensile Poisson stress also includes hydrostatic stress components. If we assume identical [∂V/∂NH]T at all H contents, as is typically observed in many intercalation systems, the increasing chemical potential according to H content indicates a non-linear increase of σhydro during hydriding.17 That is to say, mechanical constraints applied by rGO sheets suppress the volume expansion driven by interstitial H, leading to increasing pressure for the phase transformation,20,33 while inducing the sloped plateau due to the inhomogeneous stress.17 Similar sloping behaviour and non-linear stresses have been observed during the hydriding of Pd thin films constrained by the substrates.17
ln[P/P0]) is required as the hydrogen intake increases. Under isothermal conditions inside the rGO-Pd composites, the increasing chemical potential of H is explained by the hydrostatic stress-induced chemical potential change (ΔμH = σhydro[∂V/∂NH]T) due to the mechanical constraint. σhydro and [∂V/∂NH]T stand for the hydrostatic stress and partial molar volume of H, respectively. Since any stress state may be decomposed into hydrostatic stress and deviatoric stress, our uniaxial compressive stress with tensile Poisson stress also includes hydrostatic stress components. If we assume identical [∂V/∂NH]T at all H contents, as is typically observed in many intercalation systems, the increasing chemical potential according to H content indicates a non-linear increase of σhydro during hydriding.17 That is to say, mechanical constraints applied by rGO sheets suppress the volume expansion driven by interstitial H, leading to increasing pressure for the phase transformation,20,33 while inducing the sloped plateau due to the inhomogeneous stress.17 Similar sloping behaviour and non-linear stresses have been observed during the hydriding of Pd thin films constrained by the substrates.17
The rGO-Pd systems exhibit an increased pressure gap (hysteresis width) compared to those in bare Pd systems. The increased hysteresis gap is readily observed for rGO-Pd1 and rGO-Pd2, with 31 and 45 nm-sized Pd cubes (Fig. 2). It is generally regarded that a hysteresis gap opens up and widens due to a large nucleation energy barrier during the first-order phase transition (intrinsic hysteresis).32 It is expected that the compressive stress from rGO layers and Pd interaction increase the nucleation energy barrier for the hydride phase with increased volume per Pd atom, leading to the widened hysteresis gap (extrinsic hysteresis). The increased pressure hysteresis by the mechanical constraint can be found in other systems, such as clamped Pd–H films19 or nanoconfined Mg.18 Interestingly in rGO-Pd samples, these constraint-induced characteristics (the sloped plateau and the widened hysteresis width) are found to be reversible (Fig. S11†). This clearly contrasts with the previous research on stress-driven thermodynamic effects. For example, in clamped Pd–H films, stress drives irreversible change in the plateau and hysteresis widths as the H loading and unloading cycles continue.19 As shown in Fig. S11,† the rGO-Pd system exhibits nearly identical PC isotherms at 371 K even after repeated re/dehydrogenation cycles at elevated temperatures (385 K and 401 K), indicating that the nanocomposites can reversibly sustain the H-induced stresses.
Fig. 3a plots the hysteresis gap widths over the Pd nanocube size at three different temperatures. The hysteresis widths are defined in terms of the ratio between the two equilibrium pressures, ln(Pabs/Pdes). The equilibrium pressures for hydrogen absorption (Pabs) and desorption (Pdes) in rGO-Pd samples are obtained by averaging the pressure values at the beginning and end of the two-phase regions (Fig. S13a†). The hysteresis widths for both bare-Pd and rGO-Pd samples decrease with increasing temperature, reflecting that increased thermal energy makes it easier for the hydride phase to overcome the nucleation barrier.32 The hysteresis widths of bare-Pd decrease as the particle sizes become smaller, attributed to the small nucleation barriers for phase transition.32 The hysteresis width of rGO-Pd nanocubes, however, drops sharply as the nanocube size becomes larger than 60 nm. These size-dependent hysteresis widths of rGO Pd nanocubes can be rationalized by the size-dependent encapsulation regularity (Fig. 3b). For example, in the case of rGO-Pd3, Pd particles appear to be too large to be fully capped between the interlayer spacing of rGO sheets, as shown in Fig. S2;† thus the regularity of rGO layers may collapse, leading to an inefficient pseudo-pressure effect, consistent with the phenomenon observed in rGO-confined [Fe(Htrz)2(trz)](BF4) nanoparticle systems.22 It is reasonably expected that the pseudo-pressure effect increases with decreasing Pd nanocube size in rGO-Pd. These findings suggest that 2D layered-encapsulating structures can intensify the hysteresis behaviour during hydrogen sorption in a certain size range; thus, a proper size selection of the encapsulated nanoparticle should be taken into account for the optimization of the de/hydriding ability. Besides the layered-encapsulating structure, the size-dependent pressure effect has been reported in other material forms including thin films,19 alloys34 or patterned nanodots.18 However, the rGO-Pd systems in this work exhibit definite advantages compared to the above-mentioned structures, with high reversibility of hydrogen sorption offering the possibility to tune the thermodynamic properties according to the nanocube sizes.
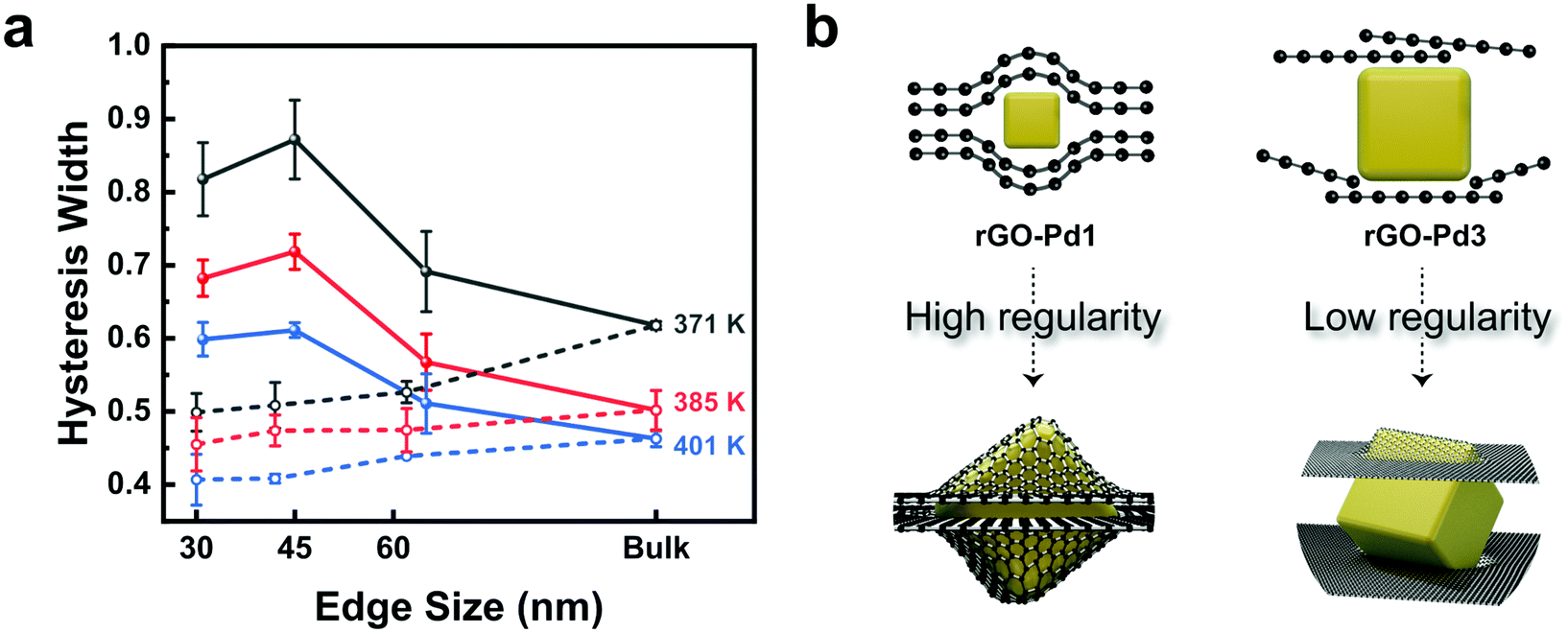 | ||
| Fig. 3 (a) Hysteresis widths, ln(Pabs/Pdes) over the nanocube size (solid line: rGO-Pd, dashed line: bare-Pd) at three different temperatures. (b) Schematic representations of the size-dependent layer regularity, playing an important role in affecting the hysteresis behaviour. | ||
To further investigate how the encapsulation affects rGO-Pd samples’ hydrogen sorption characteristics, we examine the enthalpy (ΔH) and entropy (ΔS) changes of H absorption and desorption using the van't Hoff equation. Table 1 shows the thermodynamic parameters obtained from fitting the isotherm data at three different temperatures. Repeated experiments exhibit a similar trend, demonstrating the reversibility of the phenomena. The fitting procedure is described in detail in Fig. S13b.† While the absorption enthalpy (ΔHabs) exhibits little difference between rGO-Pd and bare-Pd, the desorption enthalpy (ΔHdes) differs by 3.9–8.0 kJ (mol H2)−1 for rGO-Pd samples compared to the respective bare-Pd samples. It is well known that during the hydriding process, mechanical constraints can hinder the expansion of metal lattice, making it hard to insert H atoms into the metal lattice (destabilization of the hydride phase) as observed in several thin-film systems.20,33 Considering that ΔH reflects the Pd–H bond strength,25 the unaffected absorption enthalpy implies that rGO-confined PdHx is also stabilized at the same time. It is known that H atoms tend to be concentrated at the sub/surface for Pd nanocubes with the size of tens of nanometers.23,35 In our study, however, Pd nanocubes are fully covered by rGO layers as opposed to the previously reported ones (Fig. S3 and S4†). Accordingly, it is reasonable to suppose that this layered-encapsulating structure may minimize the surface energy and cause H atoms to occupy the core, thus strengthening the Pd–H bond (stabilization of the hydride phase). The increased desorption enthalpy compared to the unaffected absorption enthalpy suggests that the compressive interaction between Pd nanocubes and rGO decreases towards the end of hydrogenation, resulting in less strained Pd lattice as observed in the previous study.26 Owing to the weakened destabilization effect, the Pd–H bonds become strong and thus the desorption enthalpy increases upon hydrogen release. The unique change in the reaction enthalpy upon encapsulation suggests the potential engineering or tuning of hydride stability for various metastable metal hydrides such as AlH3 and LiAlH4.33
| Sample | ΔH [kJ (mol H2)−1] | ΔS [J K−1 (mol H2)−1] | ||
|---|---|---|---|---|
| −ΔHabs | ΔHdes | −ΔSabs | ΔSdes | |
| Pd1 | 35.6 ± 1.0 | 39.4 ± 1.0 | 87.9 ± 2.6 | 94.0 ± 2.5 |
| rGO-Pd1 | 35.1 ± 0.8 | 44.1 ± 2.2 | 87.4 ± 2.0 | 105.0 ± 5.6 |
| Pd2 | 35.6 ± 1.4 | 39.7 ± 0.8 | 87.9 ± 3.8 | 94.8 ± 2.0 |
| rGO-Pd2 | 37.0 ± 0.8 | 47.7 ± 2.0 | 92.3 ± 2.0 | 114.0 ± 5.3 |
| Pd3 | 35.6 ± 1.3 | 39.2 ± 1.8 | 88.1 ± 3.4 | 93.5 ± 4.6 |
| rGO-Pd3 | 35.7 ± 0.8 | 43.1 ± 2.5 | 88.7 ± 2.0 | 103.0 ± 6.5 |
| Bulk | 34.4 ± 0.7 | 40.5 ± 2.5 | 85.0 ± 1.9 | 96.7 ± 6.4 |
The rGO-Pd nanocubes also present increased desorption entropy (ΔSdes) in the range of 9.5–19.2 J K−1 (mol H2)−1 for all sizes compared to bare-Pd, showing a distinct change in de/hydriding thermodynamics. The rGO-Pd samples exhibit notably increased ΔSdes compared to the bulk in every particle size. This sharply contrasts the previous reports that nanoscale PdHx particles exhibit decreased reaction entropy as hydrogen absorbed in nanoscale Pd has higher entropy than that in the bulk lattice.25,36,37 The increases in ΔSdes suggest that rGO encapsulation reduces the entropy of hydrogen atoms in nanoscale Pd beyond the effect of intrinsic nanoscaling. In parallel with increased ΔHdes, the opposite effects on ΔSdes compared to bare-Pd imply that rGO encapsulation is the dominant factor governing the hydrogen sorption dynamics.
The kinetics of hydrogen absorption are measured at 303 K under 1.3 bar of H2 pressure for both bare-Pd and rGO-Pd nanocubes to clarify the rGO-encapsulation effect on the hydriding kinetics as shown in Fig. S14.† For all size samples, bare-Pd nanocubes uptake 0.3 H per Pd atom only after 10 min, whereas rGO-Pd nanocubes absorb more than 0.3 H per Pd atom within 10 min, demonstrating enhanced absorption kinetics. This can be attributed to the tensile-strained regions in the rGO-Pd system, observed by HRTEM measurements (Fig. S7†). During phase transformation from the α to the β phase, the β phase initially nucleates at the corners of Pd nanoparticles before forming a phase boundary. This β phase nucleation is known to be the rate-limiting step.38 Recent studies suggest that the β phase preferably nucleates at the region under tensile strain.38,39 Since Pd nanocubes are locally tensile-strained in the in-plane orientation by rGO-encapsulation, β phase nucleation may be accelerated, resulting in enhanced absorption kinetics. Since the nanosizing effect on hydrogen sorption behaviour is remarkable for particles below 10 nm size, no difference in kinetic profiles is observed in our samples. In addition, slightly increased capacities (less than 0.1 H per Pd atom) are observed in the rGO-Pd system. Earlier reports suggest that the charge transfer from Pd to rGO increases the number of holes in the 4d orbital which are closely related to the storage capability, enhancing the hydrogen capacity.25,40 Given that the rGO layers can act as an electron acceptor,26 the charge transfer between Pd and rGO may result in slightly increased hydrogen capacity due to a weak electron transfer observed in XPS measurements (Fig. S10†).
Ab initio computations reaffirm the experimental results on desorption entropy increase upon mechanical constraints. Fig. 4a shows the computed vibrational entropy changes of Pd, PdH0.625, and the desorption entropy of PdH0.625 at varied hydrostatic compressions. As the amount of compressive strain increases from 0% to 5%, the vibrational entropy of Pd and PdH0.625 both decrease steadily, yet at different paces. The vibrational entropy of PdH0.625 drops sharply after 1% compressive strain, reflecting the decreased vibrational entropy of H under compressed Pd lattice. This contrasts with the steady decrease of Pd and rationalizes the increased desorption entropy upon compressive strain. Fig. 4b plots the change in desorption entropy at varied temperatures between 270 K and 570 K. The desorption entropy curve monotonically shifts downward as temperature increases, yet the effect of compressive strain persists throughout all the temperatures we consider. Interestingly, the desorption entropy increases sharply only when the compressive strain exceeds 1% at all temperatures, indicating that over 1% compressive strain is required to experimentally observe the change in desorption entropy upon strain.
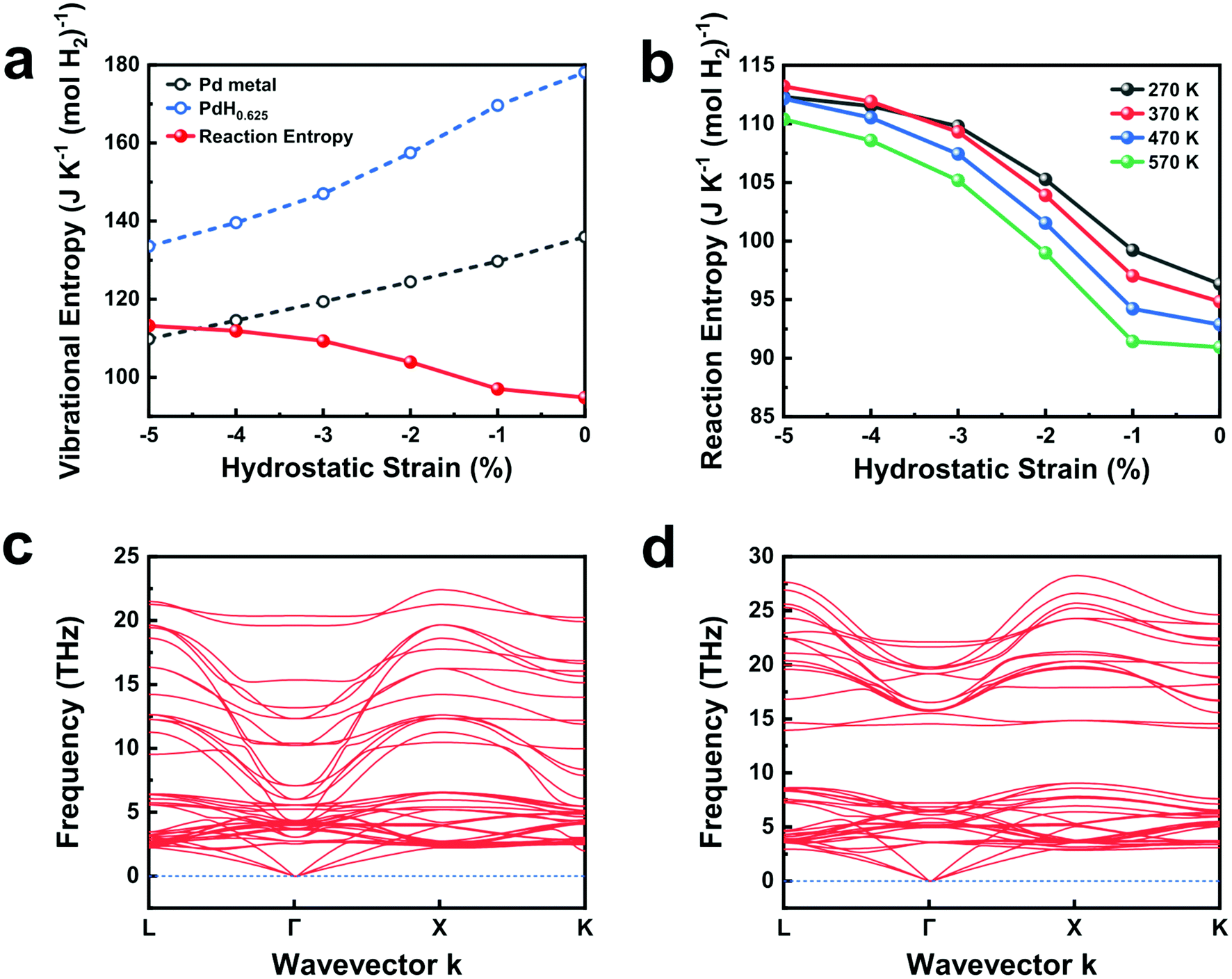 | ||
| Fig. 4 (a) Computed desorption entropy and vibrational entropy of PdH0.625 and Pd at varied hydrostatic compressions from 0 to 5%. (b) Desorption entropy computed at varied temperatures between 270 K and 570 K. (c and d) The computed phonon band structures of PdH0.625 at (c) 1% and (d) 5% compressive hydrostatic strain. | ||
Under uniaxial compressive strain with Poisson expansions in the other two axes, we observe similar trends in vibrational entropy and reaction entropy (Fig. S15†). Under this strain, however, the vibrational entropy of Pd remains almost identical, while that of PdH0.625 decreases steadily, causing the overall reaction entropy to increase upon uniaxial strain. The computed desorption entropies match well with the experimental results (Table 1) in terms of both trends and values, noting that a tensile Poisson strain of 3% roughly translates to the uniaxial compressive strain of 6%. The large estimated strain applied on Pd nanocubes far exceeds the elastic limit observed in typical metals. This implies that while the increase in desorption entropy upon strain is a bulk phenomenon, such strain under constraint may only be observed among metals in the nanoscale. A large strain greater than 1% matches well with the experimental observations that elastoplastic strains are present in β-PdHx.19
The phonon band structures of PdH0.625 computed at 1% and 5% compressive hydrostatic strain provide the rationale for the change in desorption entropy upon increased compression. At 1% hydrostatic compression, we observe the optical phonon modes at the Γ-point of the Brillouin zone reaching down to ∼5 THz frequency (Fig. 4c). Similar phonon dispersion curves have been reported for stress-free phonon dispersion curves of PdH in rocksalt structures.41 On the contrary, at 5% hydrostatic compression, PdH0.625 opens up a phonon bandgap by separating the optical and acoustic phonon modes (Fig. 4d). The sharp contrast in the optical phonon modes suggests that H atoms in octahedral sites decrease in vibrational degrees of freedom at compressive strains above 1%. This rationalizes the sharp reduction in vibrational entropy at above 1% hydrostatic compression. At tensile stresses of 1% or more, the computed phonon dispersion curves of PdH0.625 exhibit phonon modes at imaginary frequencies, indicating dynamic instability caused by unstable H atoms in enlarged octahedral sites. Previous literature reports that the computed phonon dispersion curves with H atoms in tetrahedral interstitial sites exhibit optical phonon modes above 25 THz frequency, further supporting this notion.41
In this respect, it is interesting to note that one should carefully choose the size of nanoparticles for encapsulation by graphene derivatives. Although decreasing the particle size towards nanoscale is effective in reducing hysteresis in general, nanoparticles in encapsulated composites exhibit increased hysteresis upon the addition of guest atoms such as H. In other words, being small does not necessarily provide advantages over bigger particles. In addition, the thermodynamics of hydrogenation may be tuned in rGO-Pd nanocomposites such that the hydride phase may be further stabilized in terms of desorption enthalpy and entropy. Considering that engineering the stability of metal hydride phases is technologically essential yet challenging to achieve, the nanocomposite route demonstrated in this work provides a unique strategy towards versatile engineering of hydride-based hydrogen storage.
Conclusions
In this study, we demonstrate that metal nanocomposites may be engineered to reversibly tune the thermodynamics of hydrogen storage. As opposed to several reported studies which show that the nanoscale effect is responsible for lowering the change of enthalpy and entropy, our elaborately designed rGO-Pd system exhibits significantly increased enthalpy and entropy of desorption, a direct result from the constraints by rGO. Ab initio computations demonstrate that compressive strain above 1% reduces the vibrational entropy of PdHx faster than that of Pd, subsequently verifying that the encapsulation effect is a predominant factor in hydrogen sorption dynamics. The concept of design for metal nanocrystals capped by host matrix provides unrivalled opportunities for a variety of energy storage/conversion systems in which a synergistic effect from both nanocrystals and the capping 2D materials such as graphene derivatives enhances the thermodynamic and/or electrochemical functionality of the system.Experimental section
Chemicals and reagents
Potassium chloride (KCl, ≥99.0%), palladium chloride (PdCl2, 99%), ascorbic acid (≥95%), potassium bromide (KBr, ≥99.0%), hexadecyltrimethylammonium bromide (CTAB, ≥99%), and hydrochloric acid (HCl, 36.5–38.0%) were purchased from Sigma-Aldrich. Graphene oxide powder (GO) was purchased from Standard Graphene. Deionized water was used in all experiments.rGO-Pd synthesis
Pd nanocubes encapsulated by rGO sheets were prepared via a modified one-pot co-reduction method reported earlier.42 38.4 mg of GO powders were dispersed in 32 mL of DI water and ultra-sonicated for 2 h to form a GO aqueous suspension (1.2 mg mL−1). To synthesize rGO-Pd of size 31 nm, 240 mg of ascorbic acid and 500 mg of KBr were dissolved in 32 mL of the GO aqueous suspension and stirred for 10 min. The mixture was injected into a 100 mL 3-neck flask and pre-heated at 80 °C for 10 min. 12.5 mL of the 0.0804 M K2PdCl4 solution was then quickly added (the K2PdCl4 solution was prepared by dissolving 182 mg of PdCl2 and 153 mg of KCl in 12.638 mL of DI water, followed by ultra-sonication for 2 h). The reaction mixture was stirred at 80 °C for 3 h and then cooled in a water bath. Preparing rGO-Pd of size 45 and 65 nm is similar to that of 31 nm except that 1000 mg of KBr was added in the former and 1500 mg in the latter. The resulting product was centrifuged at 10000 rpm for 20 min and washed with DI water once and ethanol once to remove excess reagents. The obtained product was dried at 60 °C under vacuum overnight.
Bare-Pd synthesis
Pd nanocubes without rGO encapsulation (bare-Pd) were prepared by modifying a previously reported procedure.43,44 Bare-Pd of size 30 nm was synthesized by mixing 215 mL of DI water, 5 mL of 0.1 M CTAB solution, and 25 mL of 0.01 M K2PdCl4 solution (the K2PdCl4 solution was prepared by dissolving 47 mg of PdCl2 and 39.5 mg of KCl in 26.239 mL of DI water, followed by ultra-sonication for 2 h). Subsequently, 5 mL of 0.1 M ascorbic acid was added, and the mixture was heated to 80 °C under magnetic stirring and allowed to proceed at 80 °C for 30 min. Bare-Pd of size 62 nm was synthesized using a seed-mediated growth method. The 11 nm-size cube seeds were prepared as indicated previously. 6.8 mL of the 0.403 M CTAB solution was mixed with 3.2 mL of 0.1 M H2PdCl4 solution (the H2PdCl4 solution was prepared by dissolving 60 mg of PdCl2 and 55 μL of HCl in 3.35 mL of DI water, followed by ultra-sonication for 2 h) in a water bath at 45 °C. While stirring vigorously, 10 mL of Pd seeds was added, followed by the addition of 5 mL of 1 M ascorbic acid. The reaction proceeded at 45 °C for 6 h under vigorous stirring. Bare-Pd of size 42 nm was synthesized similarly except that 40 mL of Pd seeds was added. The resulting product was centrifuged at 10000 rpm for 10 min, washed with water two times, and dried at 60 °C under vacuum overnight.
Characterization
The facet-dependent structures of both rGO-Pd and bare-Pd nanocubes were characterized by using a transmission electron microscope (TEM, Tecnai G2 F30 S-Twin) operated at 300 kV. The structures of bare-Pd nanocubes were also characterized by using a scanning electron microscope (SEM, Hitachi SU5000). For measuring nanocube size, at least 100 nanoparticles were measured based on TEM images using the ImageJ software. The X-ray diffraction (XRD) analyses were carried out by using a Rigaku SmartLab High Resolution Powder X-ray diffractometer equipped with Cu Kα-1 radiation (λ = 0.15406 nm). The Pd loading of rGO-Pd composites was analysed by using an inductively coupled plasma-atomic emission spectroscope (ICP-AES), Perkin-Elmer Avio500. X-ray photoelectron spectroscopy (XPS) measurements were carried out by using a Thermo VG Scientific spectrometer with an Al Kα source. Pressure–composition–temperature (PCT) measurements were performed at three different temperatures. For activation, the samples were evacuated at 373 K for more than 1 hour and then were hydrogenated at around 1 MPa. Before measuring each isotherm, the samples were evacuated at the target temperature for more than 1 hour. The TEM, SEM, XRD, and XPS analyses were conducted using equipment from the KAIST Analysis Center for Research Advancement (KARA).Ab initio computations
The reaction entropies and phonon dispersion curves were computed using density functional perturbation theory (DFPT) calculations as implemented in the Vienna ab initio simulation package (VASP).45 The Perdew–Burke–Erzenhof (PBE) functional46 with generalized-gradient approximation (GGA) was used by following the projector-augmented wave (PAW) methods.47 The β-PdH0.625 unit cell was obtained by partially filling the octahedral sites in a face-centred cubic Pd lattice. All symmetrically distinct H configurations up to 1 × 1 × 2 supercells at the fixed composition of PdH0.625 were obtained by using the enumeration library48 and relaxed in terms of all atomic coordinates to obtain the ground-state configuration. This process was repeated at each hydrostatic compressive strain from 0 to −5%. The K-point grid densities of 1000/atom for lattice relaxation and 4800/atom for phonon calculations were used. The vibrational entropies and phonon dispersion curves were obtained from the vibrational frequencies using statistical thermodynamic expressions as implemented in phonopy.49Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the International Energy Joint R&D Program of the Korea Institute of Energy Technology Evaluation and Planning (KETEP), granted financial resource from the Ministry of Trade, Industry & Energy, Republic of Korea (no. 20188520000570). E. Choi and S. Kim acknowledge the support from the Human Resources Development program (no. 20204010600090) of the Korea Institute of Energy Technology Evaluation and Planning (KETEP), funded by the Ministry of Trade, Industry, and Energy of the Korean Government.Notes and references
- J. Wan, S. D. Lacey, J. Dai, W. Bao, M. S. Fuhrer and L. Hu, Chem. Soc. Rev., 2016, 45, 6742–6765 RSC.
- A. Schneemann, J. L. White, S. Kang, S. Jeong, L. F. Wan, E. S. Cho, T. W. Heo, D. Prendergast, J. J. Urban, B. C. Wood, M. D. Allendorf and V. Stavila, Chem. Rev., 2018, 118, 10775–10839 CrossRef CAS PubMed.
- Y. Liu, W. Li, H. Wu and S. Lu, Acta Phys. – Chim. Sin., 2020, 2009082–2009080 Search PubMed.
- X. Zhou, L. J. Wan and Y. G. Guo, Adv. Mater., 2013, 25, 2152–2157 CrossRef CAS PubMed.
- Z. Zhang, Y. Du, Q. C. Wang, J. Xu, Y. N. Zhou, J. Bao, J. Shen and X. Zhou, Angew. Chem., 2020, 59, 17504–17510 CrossRef CAS PubMed.
- L. Li, Y. Huang, C. An and Y. Wang, Sci. China Mater., 2019, 62, 1597–1625 CrossRef.
- T. C. Narayan, A. Baldi, A. L. Koh, R. Sinclair and J. A. Dionne, Nat. Mater., 2016, 15, 768–774 CrossRef CAS PubMed.
- A. W. van den Berg and C. O. Arean, Chem. Commun., 2008, 668–681, 10.1039/b712576n.
- B. Sakintuna, F. Lamaridarkrim and M. Hirscher, Int. J. Hydrogen Energy, 2007, 32, 1121–1140 CrossRef CAS.
- E. Boateng and A. Chen, Mater. Today Adv., 2020, 6, 100022 CrossRef.
- V. B. Parambhath, R. Nagar and S. Ramaprabhu, Langmuir, 2012, 28, 7826–7833 CrossRef CAS PubMed.
- C. Zhou and J. A. Szpunar, ACS Appl. Mater. Interfaces, 2016, 8, 25933–25940 CrossRef CAS PubMed.
- G. Li, H. Kobayashi, J. M. Taylor, R. Ikeda, Y. Kubota, K. Kato, M. Takata, T. Yamamoto, S. Toh, S. Matsumura and H. Kitagawa, Nat. Mater., 2014, 13, 802–806 CrossRef CAS PubMed.
- X. Liu, G. S. McGrady, H. W. Langmi and C. M. Jensen, J. Am. Chem. Soc., 2009, 131, 5032–5033 CrossRef CAS PubMed.
- J. Graetz, ISRN Mater. Sci., 2012, 2012, 1–18 CrossRef.
- J. Lu, Y. J. Choi, Z. Z. Fang, H. Y. Sohn and E. Ronnebro, J. Am. Chem. Soc., 2009, 131, 15843–15852 CrossRef CAS PubMed.
- S. Wagner and A. Pundt, Int. J. Hydrogen Energy, 2016, 41, 2727–2738 CrossRef CAS.
- A. Molinari, F. D'Amico, M. Calizzi, Y. Zheng, C. Boelsma, L. Mooij, Y. Lei, H. Hahn, B. Dam and L. Pasquini, Int. J. Hydrogen Energy, 2016, 41, 9841–9851 CrossRef CAS.
- Y. Pivak, H. Schreuders, M. Slaman, R. Griessen and B. Dam, Int. J. Hydrogen Energy, 2011, 36, 4056–4067 CrossRef CAS.
- A. Baldi, M. Gonzalez-Silveira, V. Palmisano, B. Dam and R. Griessen, Phys. Rev. Lett., 2009, 102, 226102 CrossRef CAS PubMed.
- X. Chen, G. Wu, J. Chen, X. Chen, Z. Xie and X. Wang, J. Am. Chem. Soc., 2011, 133, 3693–3695 CrossRef CAS PubMed.
- Y. Sekimoto, R. Ohtani, M. Nakamura, M. Koinuma, L. F. Lindoy and S. Hayami, Sci. Rep., 2017, 7, 12159 CrossRef PubMed.
- A. Baldi, T. C. Narayan, A. L. Koh and J. A. Dionne, Nat. Mater., 2014, 13, 1143–1148 CrossRef CAS PubMed.
- S. Syrenova, C. Wadell, F. A. Nugroho, T. A. Gschneidtner, Y. A. Diaz Fernandez, G. Nalin, D. Switlik, F. Westerlund, T. J. Antosiewicz, V. P. Zhdanov, K. Moth-Poulsen and C. Langhammer, Nat. Mater., 2015, 14, 1236–1244 CrossRef CAS PubMed.
- M. Yamauchi, R. Ikeda, H. Kitagawa and M. Takata, J. Phys. Chem. C, 2008, 112, 3294–3299 CrossRef CAS.
- D. Kim, J. Koh, S. Kang, T. W. Heo, B. C. Wood, E. S. Cho and S. M. Han, J. Mater. Chem. A, 2021, 9, 11641–11650 RSC.
- J. M. Jeon, T. L. Kim, Y. S. Shim, Y. R. Choi, S. Choi, S. Lee, K. C. Kwon, S. H. Hong, Y. W. Kim, S. Y. Kim, M. Kim and H. W. Jang, Adv. Mater., 2017, 29, 1605929 CrossRef PubMed.
- X. Liu, L. Li, C. Meng and Y. Han, J. Phys. Chem. C, 2012, 116, 2710–2719 CrossRef CAS.
- C. M. Ramos-Castillo, J. U. Reveles, R. R. Zope and R. de Coss, J. Phys. Chem. C, 2015, 119, 8402–8409 CrossRef CAS.
- E. S. Cho, A. M. Ruminski, S. Aloni, Y. S. Liu, J. Guo and J. J. Urban, Nat. Commun., 2016, 7, 10804 CrossRef CAS PubMed.
- F. Hayee, T. C. Narayan, N. Nadkarni, A. Baldi, A. L. Koh, M. Z. Bazant, R. Sinclair and J. A. Dionne, Nat. Commun., 2018, 9, 1775 CrossRef PubMed.
- R. Bardhan, L. O. Hedges, C. L. Pint, A. Javey, S. Whitelam and J. J. Urban, Nat. Mater., 2013, 12, 905–912 CrossRef CAS PubMed.
- P. Ngene, A. Longo, L. Mooij, W. Bras and B. Dam, Nat. Commun., 2017, 8, 1846 CrossRef PubMed.
- Y. Oumellal, C. M. Ghimbeu, A. Martínez de Yuso and C. Zlotea, Int. J. Hydrogen Energy, 2017, 42, 1004–1011 CrossRef CAS.
- A. Ulvestad, M. J. Welland, S. S. E. Collins, R. Harder, E. Maxey, J. Wingert, A. Singer, S. Hy, P. Mulvaney, P. Zapol and O. G. Shpyrko, Nat. Commun., 2015, 6, 10092 CrossRef CAS PubMed.
- V. A. Vons, H. Leegwater, W. J. Legerstee, S. W. H. Eijt and A. Schmidt-Ott, Int. J. Hydrogen Energy, 2010, 35, 5479–5489 CrossRef CAS.
- M. Paskevicius, D. A. Sheppard and C. E. Buckley, J. Am. Chem. Soc., 2010, 132, 5077–5083 CrossRef CAS PubMed.
- N. J. J. Johnson, B. Lam, B. P. MacLeod, R. S. Sherbo, M. Moreno-Gonzalez, D. K. Fork and C. P. Berlinguette, Nat. Mater., 2019, 2, 454–458 CrossRef PubMed.
- K. Sytwu, F. Hayee, T. C. Narayan, A. L. Koh, R. Sinclair and J. A. Dionne, Nano Lett., 2018, 18, 5357–5363 CrossRef CAS PubMed.
- H. Kobayashi, M. Yamauchi, R. Ikeda, T. Yamamoto, S. Matsumura and H. Kitagawa, Chem. Sci., 2018, 9, 5536–5540 RSC.
- A. Houari, S. F. Matar and V. Eyert, J. Appl. Phys., 2014, 116, 9 CrossRef.
- S. Bai, C. Wang, M. Deng, M. Gong, Y. Bai, J. Jiang and Y. Xiong, Angew. Chem., Int. Ed., 2014, 53, 12120–12124 CrossRef CAS PubMed.
- X. Xie, G. Gao, Z. Pan, T. Wang, X. Meng and L. Cai, Sci. Rep., 2015, 5, 8515 CrossRef CAS PubMed.
- A. Klinkova, E. M. Larin, E. Prince, E. H. Sargent and E. Kumacheva, Chem. Mater., 2016, 28, 3196–3202 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. L. W. Hart, L. J. Nelson and R. W. Forcade, Comput. Mater. Sci., 2012, 59, 101–107 CrossRef CAS.
- A. Togo and I. Tanaka, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr04335h |
| This journal is © The Royal Society of Chemistry 2021 |