
Surface overgrowth on gold nanoparticles modulating high-energy facets for efficient electrochemical CO2 reduction†
Woong
Choi‡,§
a,
Joon Woo
Park§
a,
Woonghyeon
Park
b,
Yousung
Jung
 *b and
Hyunjoon
Song
*a
*b and
Hyunjoon
Song
*a
aDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: hsong@kaist.ac.kr
bDepartment of Chemical and Biomolecular Engineering (BK21 four), Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: ysjn@kaist.ac.kr
First published on 9th August 2021
Abstract
Electrochemical CO2 reduction reaction (eCO2RR) has been considered one of the potential technologies to store electricity from renewable energy sources into chemical energy. For this aim, designing catalysts with high surface activities is critical for effective eCO2RR. In this study, we introduced a surface overgrowth method on stable Au icosahedrons to generate Au nanostars with large bumps. As a catalyst for eCO2RR, the Au nanostars exhibited a maximum faradaic efficiency (FE) of 98% and a mass activity of 138.9 A g−1 for CO production, where the latter was one of the highest activities among Au catalysts. Despite the deducted electrochemically active surface area per mass, the high-energy surfaces from overgrowth provided a 3.8-fold larger specific activity than the original Au icosahedral seeds, resulting in superior eCO2RR performances that outweigh the trade-off of size and shape in nanoparticles. The Au nanostars also represented prolonged stability due to the durability of high-energy facets. The characterization of surface morphology and density functional theory calculations revealed that predominant Au(321) facets on the Au nanostars effectively stabilized *COOH adsorbates, thus lowering the overpotential and improving the FE for CO production. This overgrowth method is simple and universal for various materials, which would be able to extend into a wide range of electrochemical catalysts.
Introduction
The increasing atmospheric CO2 concentration by continuous consumption of fossil fuels has aroused serious concerns about global warming. To prevent further increase of the atmospheric CO2 level, the chemical conversion of CO2 to other valuable compounds has emerged as an ultimate solution to address this issue.1,2 Compared to other methods for converting CO2 such as photochemical or biological approaches, the direct electrochemical CO2 reduction reaction (eCO2RR) on a metal surface shows a more promising prospect owing to its mild reaction conditions in a simple device structure.1,3,4 Another advantage of eCO2RR is that it enables the storage of surplus electricity in the form of chemical energy, which covers inconsistent energy production of renewable energy sources including solar and wind energies.2,5,6For efficient eCO2RR, group 11 metals such as Au, Ag, and Cu have been employed as solid-state catalysts.3,7 In particular, Au has been intensively studied to generate CO2 to CO due to the optimal binding energy on the surface.8 This makes Au a promising eCO2RR catalyst since CO, especially when mixed with H2 to form syngas, serves as a crucial ingredient for hydrocarbon fuels.9–11 As well, since CO production only requires two electrons per molecule, it has been spotlighted as the most potential product from eCO2RR toward commercialization.10 However, the reaction always competes with the hydrogen evolution reaction (HER) in water, which lowers CO selectivity. The chemical inertness of the Au surface also leads to low catalytic activities with large overpotentials.12–14 In this aspect, it is necessary to enhance CO production activity by tailoring surface structures. Intrinsic high mass and specific activities are also essential to achieve an affordable price of a noble metal-based catalyst towards the actual application of eCO2RR.
Au nanoparticles (NPs) have larger surface areas compared to their bulk counterparts, leading to exceptionally high mass activities and current densities than bulk catalysts. In this regard, the size effect of Au NPs was extensively investigated for eCO2RR. Smaller Au NPs provided higher current densities by their large surface area. On the other hand, low coordination sites, such as corners and edges, were known as suitable surfaces for stabilizing reaction intermediates for CO2RR and suppressing HER;15,16 however, the unbalance of these sites in small Au NPs reduced CO faradaic efficiency (FE). Strasser et al. reported that the eCO2RR activity increased in small Au NPs, but the selectivity towards CO decreased simultaneously because the competing HER also enhanced at the low coordinated sites.17 Sun et al. investigated the size dependency of eCO2RR using carbon-supported Au NPs in the range of 4–10 nm.18 Among the different NP sizes, 8 nm was proven to be the optimum to yield high mass activity and FE for CO production at a moderate overpotential (−0.52 V vs. reference hydrogen electrode (RHE)).
The existence of an optimal size range reveals that the size and surface structure have a trade-off relationship, which limits catalytic performances. Regardless of the particle size, generating intrinsically more active surfaces would be a promising approach to overcome such an upper bound of the activity. For this aim, surface overgrowth from Au seeds in the presence of surface regulating reagents has been developed as a useful technique to yield unique shapes with specific high-energy surfaces.19 Nam et al. synthesized Au concave rhombic dodecahedrons by a seed-mediated overgrowth method. The particles showed a high CO FE compared to Au cubes and films,20 mainly rooted from the ensemble of multiple high-index facets on the surface. However, this overgrowth method also had a trade-off of size and shape since it also enlarged the NP size and decreased the active surface area compared to the initial seeds. The strategy is only feasible when the surface activity of newly generated facets is enough to compensate for the reduced surface area. In this regard, careful consideration of the size-shape trade-off is necessary to synthesize Au catalysts that are efficient for eCO2RR.
Here, we induced surface overgrowth from Au icosahedral seeds (Ih) and successfully generated a uniform nanostar morphology. The growth time was controlled to form Au nanostars with small (s-NS) and large (l-NS) bumps. In eCO2RR, l-NS exhibited excellent catalytic performances, including current density, mass activity, and FE for CO production, superior to Ih, s-NS, and foils, for the entire potential range. From Ih to l-NS, the electrochemical active surface area (ECSA) decreased. However, l-NS showed a massive specific activity 3.8 times larger than Ih, which highly exceeded the trade-off to get high mass activity. s-NS exhibited lower activity and selectivity than Ih because high-energy surface facets could not be fully developed. Structural analysis of l-NS indicated that {321} facets predominantly covered the NP surface. Density functional theory (DFT) calculations supported that these {321} facets effectively stabilized *COOH intermediates, resulting in high FE and activity for CO production.
Experimental
Chemicals and materials
Gold(I) chloride (AuCl), gold(III) chloride hydrate (HAuCl4·xH2O), gold film (99.99%, thickness of 0.1 mm), poly(vinyl pyrrolidone) (PVP, average Mw ∼ 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), dimethylamine (DMA, 45 wt% in aqueous solution), hydrochloric acid (HCl, 37%), potassium bicarbonate (KHCO3), Nafion® perfluorinated resin solution (5 wt%), N,N-dimethylformamide (DMF), and 1,5-pentanediol (PD) were purchased from Sigma-Aldrich. Isopropyl alcohol (IPA) and sulfuric acid (H2SO4, 95.0%) were purchased from Junsei. Ketjen black carbon was purchased from Lion Corporation. All chemicals were used as received without further purification. Ultrapure water (18.2 MΩ cm−1) was used for electrochemical experiments.
000), dimethylamine (DMA, 45 wt% in aqueous solution), hydrochloric acid (HCl, 37%), potassium bicarbonate (KHCO3), Nafion® perfluorinated resin solution (5 wt%), N,N-dimethylformamide (DMF), and 1,5-pentanediol (PD) were purchased from Sigma-Aldrich. Isopropyl alcohol (IPA) and sulfuric acid (H2SO4, 95.0%) were purchased from Junsei. Ketjen black carbon was purchased from Lion Corporation. All chemicals were used as received without further purification. Ultrapure water (18.2 MΩ cm−1) was used for electrochemical experiments.
Synthesis of Au icosahedral seeds
In a typical synthesis, 0.25 g of PVP was dissolved in 5.5 mL of PD and heated until refluxing. To this solution, 1.5 mL of 0.050 M AuCl in PD was quickly injected. After refluxing for 5 min, the reaction mixture was cooled down to room temperature and washed with ethanol three times. The final product was dispersed in 10 mL of ethanol.Synthesis of Au nanostars
Au nanostars were synthesized according to the literature with modifications.21 In a typical synthesis, 3.6 g of PVP was dissolved in 45 mL of DMF. 300 μL of DMA and 240 μL of 2.5 M HCl were added to the solution. 1.5 mL of Au icosahedral seeds dispersed in ethanol were collected by centrifugation and re-dispersed to 6.0 mL of DMF and then added to the reaction solution. 60 μL of 0.050 M HAuCl4 in DMF was added to the mixture, and the reaction mixture was stirred for 4 h in a 65 °C oil bath. The product was centrifuged and washed with ethanol twice, and finally dispersed in 5 mL of ethanol. For the nanostars with shorter bumps, the procedure was the same except for the reaction period being 1 h at 65 °C.Characterization
Transmission electron microscope (TEM) images were acquired with a Tecnai F30 ST (FEI) TEM operating at 300 kV. For the sample preparation, the Au nanoparticle dispersion was dropped onto a copper grid, and the sample was dried.Preparation of working electrodes
The working electrode was prepared by loading the carbon-supported Au nanoparticles on a glassy carbon electrode. The carbon-supported Au nanoparticles were prepared by adding colloidal Au NP dispersions in ethanol to a Ketjen black dispersion in ethylene glycol, followed by filtration and overnight drying in a vacuum oven at 60 °C. The content of Au was kept constant to be 11% by weight, measured by inductively coupled plasma optical emission spectroscopy (ICP-OES). This carbon-supported sample was dispersed in isopropanol (1 mg mL−1) with a few seconds of ultrasonication. The glassy carbon surface was masked by a friction tape to ensure a surface area of 0.50 cm2. On this surface area, 0.10 mg of the sample was drop-casted. Nafion® perfluorinated resin solution (5 wt%) was diluted 20 times with isopropanol, and 30 μL of the solution was drop-casted on the sample.Electrochemical measurements coupled with gas chromatography
All electrochemical measurements were performed with a Bi-potentiostat (CH Instruments, CHI760E). A 0.5 M KHCO3 aqueous solution was prepared and saturated with CO2 gas by bubbling CO2 at 200 cc min−1 for 1 h to be used as the electrolyte.A gas-tight H-cell with cathodic and anodic chambers was used for the eCO2RR system, where the cathode was the working electrode, and the anode was a Pt plate. A Ag/AgCl (in 3 M NaCl) electrode was used as the reference electrode. The two chambers were separated with a Nafion® 117 ion exchange membrane. Only the cathode chamber was stirred at 260 rpm. The CO2 flow rate was fixed to 60 cc min−1 using a mass flow controller (MFC, Line Tech M3030 V), except for electrochemical impedance spectroscopy (EIS) where the CO2 flow and stirring were turned off for the exact measurements. The out-line from the cathode chamber was connected directly to the in-line of a pulsed discharge detector gas chromatograph (PDD GC, YL6500GC), enabling a direct product analysis of the catalysis. Ultrahigh pure helium gas (99.9999%) was used as a carrier gas.
Before a typical eCO2RR measurement, a voltage of −1.0 V (vs. Ag/AgCl) was applied for 60 s to ensure the contact of the electrolyte with the catalyst. A linear sweep voltammetry (LSV) from −1.0 V to −2.2 V (vs. Ag/AgCl) was carried out to check the onset voltage for eCO2RR. The actual eCO2RR was carried out by applying from −1.0 to −2.2 V (vs. Ag/AgCl) with 0.1 V intervals (excluding −2.1 V) for 720 s. The concentration of gaseous products was analyzed by gas chromatography for each voltage step. This cycle was repeated once more to check the catalytic stability of the Au nanoparticles. After two cycles of eCO2RR, the LSV from −1.0 to −2.2 V (vs. Ag/AgCl) was conducted 3 times. Open circuit potential measurement and EIS were performed to calculate the resistance of the reaction cell.
The potential values from the experiment were converted to the potential values versus a reversible hydrogen electrode (RHE) according to the following equation. The resistance value from EIS was subtracted from this potential to calculate the real potential.
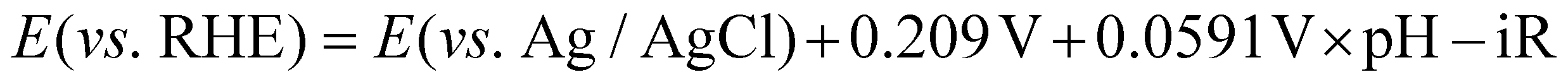
A separate eCO2RR experiment was conducted to check the stability of the Au nanoparticle catalysts. Instead of 12 steps of voltages, a constant voltage of −0.7 V (vs. RHE) was applied for 4 h to the working electrode loaded with Au nanoparticles.
Electrochemical active surface area (ECSA) measurement
A cyclic voltammogram (CV) was obtained using the working electrode in a 50 mM H2SO4 electrolyte. The converged CV data after repeated scans were acquired from 0 V to 1.5 V (vs. Ag/AgCl) at a scan rate of 50 mV s−1. The ECSAs of the Au catalyst electrodes were calculated from integrating the clear peak between 0.9 V and 1.0 V, which came from the reduction of mono-layered chemisorbed oxygen formed during the forward oxidation scan. The reduction charge per unit area was experimentally determined to be 448 μC cm−2, which was used for converting the integration value to ECSA.22Computational details
Spin-polarized density functional theory (DFT) calculations were performed using the Vienna Ab initio simulation package (VASP) code23,24 with the revised Perdew–Burke–Ernzerhof (RPBE) functional.25,26 The projector augmented wave (PAW) method was used for describing the potentials of the atoms.27 Geometries were optimized until the force on each atom was less than 0.05 eV Å−1 with cut-off energy of 400 eV.The lattice parameter of Au is 4.21 Å.28 The Au(111) and Au(211) were modeled by four atomic layers of a (3 × 3) surface unit cell, and Au(321) was represented by 12 atomic layers of a (4 × 4) surface unit cell. The Brillouin zone was sampled using (2 × 2 × 1) Monkhorst–Pack K-points for Au(321) and (3 × 3 × 1) for Au(111) and Au(211). Spacing more than 10 Å in the z-direction was applied and half of the layers from the bottom were fixed for all slab models. The computational hydrogen electrode (CHE) model29 was used to calculate free energies for the electrochemical reduction reactions. To convert electronic energies into the Gibbs free energies, thermal corrections were calculated only for gas molecules and adsorbates.30
Results and discussion
We considered multiple factors for designing efficient electrochemical catalysts based on Au NPs. First of all, Au seeds were designed as small as possible to minimize the loss of surface area from the large particle size. Secondly, the original Au seeds are better to have low-energy surfaces with uniform facets to induce a uniform surface structure with high stability. A large-scale synthesis should also be available for consistent electrochemical reactions. To meet these criteria, the Au icosahedron was selected as an ideal seed structure, where the surface facets were regularly composed of {111} and the morphology was close to a spherical form. The icosahedral NPs were prepared by a modified polyol process according to the literature.21 During the synthesis, AuCl was used instead of HAuCl4 to boost the reduction rate of Au ions and diminish the eventual size of NPs. Fig. 1a shows a transmission electron microscopy (TEM) image of the resulting Au icosahedrons (Ih) with an average diameter of 23 ± 2.5 nm. The shape was highly uniform with a fraction of over 90%. | ||
| Fig. 1 TEM images of Au (a) Ih, (b) s-NS, and (c) l-NS. The bars represent 20 nm. | ||
Surface overgrowth on Ih was induced by the addition of HAuCl4 in the presence of poly(vinyl pyrrolidone) (PVP) and dimethylamine (DMA) as surface regulating reagents in dimethylformamide (DMF). The reaction temperature was set relatively low at 65 °C, which slowed down the reduction rate of Au ions in the presence of HCl, ensuring the formation of the high-energy surface by the action of DMA.21 During this step, distinctive bumps started to grow on each surface. When the reaction was quenched at 1 h, Au nanostars with small bumps (s-NS) were generated (Fig. 1b) where the average diameter was measured to be 36 ± 2.3 nm. The bumps were fully grown at the reaction time of 4 h, yielding Au nanostars with large bumps (l-NS) with an average diameter of 52 ± 7.2 nm (Fig. 1c). The arrangement of the bumps in the Au nanostars was highly symmetric in their two-dimensional projections of the TEM images.
To investigate the effects of the surface morphology from overgrowth on catalytic performances, three representative Au morphologies, Ih, s-NS, and l-NS, were employed as catalysts for eCO2RR. The eCO2RR was performed in an H-cell with a typical three-electrode system using a Ag/AgCl reference electrode and a Pt plate counter electrode. The electrolyte was a 0.5 M KHCO3 aqueous solution at pH 7.3. Results with a Au foil cathode were also measured for comparison. Gas products from the reaction were analyzed by gas chromatography (GC).
Fig. 2a shows the iR-corrected potential-dependent CO partial current densities using a series of Ih, s-NS, l-NS, and Au foil as electrochemical catalysts. In general, the CO partial current density increased by increasing the negative applied potential. It was clear that the Au NPs showed CO partial current densities more considerable than those of the Au foil. Among the Au NPs, l-NS gave the highest CO partial current densities over the entire potential range. s-NS showed lower CO partial current densities than Ih despite having multiple bumps overgrown on the original Ih surface. The mass activity is a significant criterion for evaluating electrochemical performances, especially for expensive noble metal electrocatalysts. The iR-corrected potential-dependent CO mass activities were measured from the exact Au amount in the working electrode using ICP-OES (Fig. 2b). l-NS exhibited the highest mass activity for the entire potential range. In particular, the mass activity of l-NS was estimated to be 138.9 A g−1 at −0.7 V (vs. RHE) with a high FE of 97% for CO production, which was exceptionally high compared to those reported in the literature (Table S1, ESI†). Among the Au catalysts, l-NS showed one of the largest mass activities. Besides, l-NS exceeded either bare or modified Ag NPs, even though Ag NPs usually have higher mass activities due to the atomic mass of Ag being less than Au.
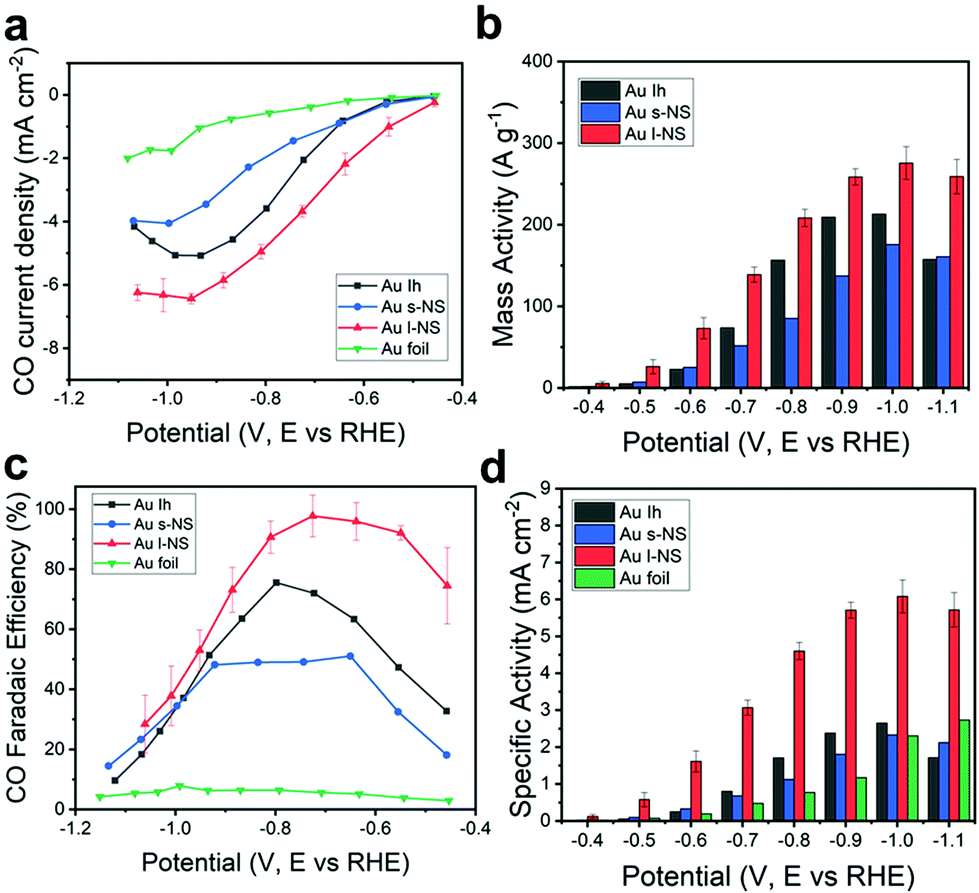 | ||
| Fig. 2 (a) Partial current densities, (b) mass activities, (c) FEs, and (d) specific activities of Ih (black), s-NS (blue), l-NS (red), and Au foil (green) for CO production versus applied potential. | ||
For FEs, CO and H2 were major products with a slight amount of CH4 from eCO2RR. The total sum of the FEs for detected gas products was nearly 100%, indicating that the catalysts generated only C1 products from CO2 (Fig. S1, ESI†). l-NS showed the highest CO FE up to 97.7% at −0.73 V (vs. RHE) (Fig. 2c). The other catalysts showed the optimal FEs of 75.5% for Ih, 51.1% for s-NS, and 7.86% for Au foil at the applied potentials of −0.80 V, −0.65 V, and −1.00 V (vs. RHE), respectively. In particular, l-NS still exhibited the CO FE of 92.1% at −0.55 V (vs. RHE), notably higher than <50% of the other catalysts. These trends were valid even when iR correction was not considered (Fig. S2, ESI†), meaning that the polarization effects were not significant in our experimental conditions.
The optimal loading ratio of Au on Ketjen black was also examined. In addition to the 11 wt% Au loading with l-NS, the 7 and 40 wt% samples were also employed under the identical eCO2RR conditions (Fig. S3, ESI†). When the Au content increased from 7 to 11 wt%, both FEs and CO partial current densities were enhanced. A further increase of Au loading to 40 wt% resulted in similar CO partial currents but lower FEs. It proved that an optimization of the catalyst loading was necessary for both activity and selectivity for CO.
To figure out the origin of high mass activity and selectivity of l-NS, a more precise surface characterization was carried out. The electrochemically active surface area (ECSA) of each Au catalyst was estimated using a cyclic voltammogram of the working electrode loaded with the catalyst in a 50 mM H2SO4 aqueous electrolyte.22,31 The clear peaks between 0.9 V and 1.0 V (vs. Ag/AgCl) represented the reduction of monolayered oxides on the Au surface (Fig. S4, ESI†). The ECSA was measured from the integral of the reduction peak area, which gave values of 1.06 cm2 for Ih, 0.88 cm2 for s-NS, 0.52 cm2 for l-NS, and 0.38 cm2 for Au foil (Table S2 and Supplementary Methods, ESI†). These ECSAs were in inverse order of the average particle size. The specific activity for CO production, which is the CO partial current density divided by the ECSA, was also estimated (Fig. 2d). In the experiment, the specific activity of l-NS (3.07 mA cm−2 at −0.7 V vs. RHE) was still distinctively larger than any other Au catalysts for the entire potential range. Considering that specific activities are strongly dependent on the surface structure, it is noteworthy that the unique high-energy surfaces of l-NS provided a high activity that vastly overcomes the reduced ECSA. The specific activities of Ih and s-NS (0.80 and 0.68 mA cm−2) were similar to that of Au foil (0.48 mA cm−2), indicating that their surface structures were not much different.
To understand how the overgrowth method affected catalytic performances, we simulated the relation of the NP diameter with the mass and specific activity values of Ih at −0.7 V (vs. RHE) (Fig. 3a). The mass activity was inversely proportional to the NP diameter, assuming that the total mass of Au was fixed and the specific activity value was the same as Ih. Therefore, a larger specific activity was necessary to compensate for the loss of mass activity of the original Ih seeds. With increased particle size, the specific activity should also increase to meet the mass activity of the dashed line of Fig. 3a. In our experiment for l-NS, although the diameter was enlarged by the overgrowth method, the specific activity was further enhanced to exceed this trade-off mass activity of the large NPs (red dot). Whereas, s-NS showed that the mass activity was sacrificed by the enlarged particle diameter without enhancing the specific activity. As a result, the mass activity was below Ih (blue dot). The relation between the particle size and specific activity was also depicted in Fig. 3b. The dashed line represents the specific activity needed to maintain the mass activity of Ih. For l-NS, a specific activity two times larger than that of Ih (1.8 mA cm−2) was required to reach the mass activity as large as Ih due to the large diameter. l-NS exhibited a remarkable 3.8-fold larger activity than Ih, leading to superior mass activity. In this graph, s-NS having a similar specific activity with Ih could not compensate for the trade-off trend with a larger diameter, resulting in low mass and specific activities. Overall, l-NS represented high CO current density and FE since the active surfaces could compensate for the enlarged NP diameter. In contrast, s-NS having overgrown surfaces without increasing specific activity sacrificed active surface area and showed lower catalytic performances. These results revealed that careful consideration for the trade-off of NP size and surface was necessary to evaluate catalytic performances of the overgrowth method.
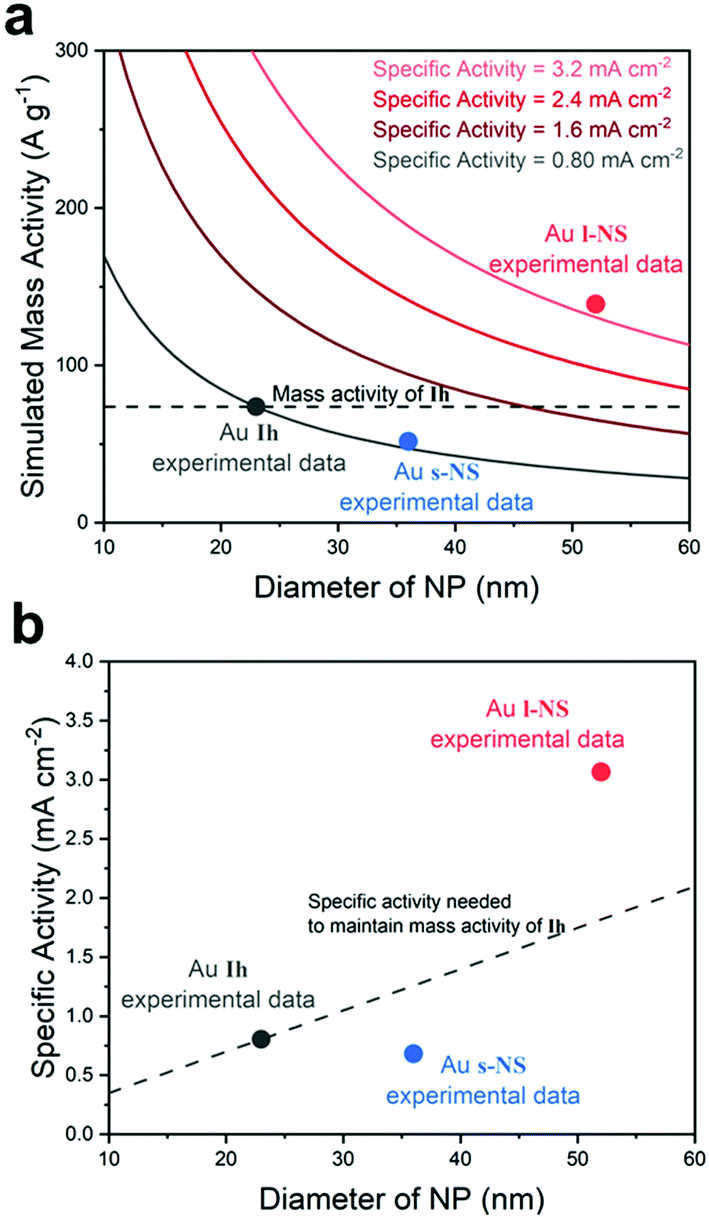 | ||
| Fig. 3 (a) The relation between the NP diameter and simulated mass activity. Solid lines indicate specific activities one, two, three, and four times larger than that of Ih (0.80 mA cm−2) at −0.7 V (vs. RHE). The dashed line is a trade-off line for retaining the mass activity of Ih. (b) The relation between the NP diameter and specific activity. The dashed line indicates the specific activity needed to maintain the mass activity of Ih. | ||
Catalyst stability during a prolonged electrochemical process is one of the most significant properties for actual applications.12 Durability tests were performed on Ih and l-NS at −0.65 V (vs. RHE) for 4 h (Fig. S5, ESI†). During the test, the CO FE of l-NS dropped from 90.7% to 81.3%, demonstrating a better consistency than Ih which showed a drastic decrease from 82.8% to 50.0%. After the test, the morphology of both catalysts did not change except that the edges and vertices were slightly rounded. The catalyst morphology did not show any severe agglomeration nor degradation.32–36 At this point, the rounding of edges and corner sites of Ih which significantly contribute to CO production was critical for the decreasing FE during the reaction. On the other hand, the active, high-energy facets of l-NS were maintained over 4 h giving significant CO selectivity despite the partial loss of surface homogeneity. It was attributed to the advantage of overgrowth, which transformed the entire particle surface into numerous low coordination sites highly active for eCO2RR. The edge or corner sites were limited in their population and were readily deactivated during the reaction. Instead, the large number of active facets generated on the faces by the overgrowth could replenish the catalytic loss from a certain number of deactivated sites. Thus, the overgrowth method could also enhance catalytic durability.
To investigate the origin of the high specific activity of l-NS, the surface morphology of l-NS was characterized by both high-resolution TEM (HRTEM) image analyses20,37,38 and simulated three-dimensional models of nanoparticles.21,39 The high-energy Au{321} facets on the surface of l-NS were identified from the HRTEM images, and the structural models covered with {321} facets also verified the uniform surface structure of l-NS. The HRTEM image in Fig. 4a is viewed from a [011] zone axis, which is determined based on the diffraction pattern of a bump from l-NS.40 If we view the Au{321} surface from the [011] axis, the two-dimensional projection of the most outer edges would have an alternating pattern of two and three steps as shown in Fig. 4d and e.41,42 Such patterns were marked on Fig. 4b and c based on the visible atomic arrangements of the HRTEM image and atomic distances. The auxiliary yellow lines representing the {321} facets were parallel to the outmost surface lines, indicating that the surface of each bump was majorly dominated by {321} facets except for the round tip region.
 | ||
| Fig. 4 (a) TEM image of a bump from l-NS and its corresponding FFT pattern indicating a [011] zone axis. The (111) (white) and (200) (red) planes are indicated. The areas in yellow boxes are magnified in (b) and (c). The yellow planes and auxiliary lines in (b) and (c) correspond to the atomic models in (d) and (e), respectively, depicting the top view of {321} surface planes. The bars represent 2 nm for (a) and 1 nm for (b) and (c). | ||
We also built an ideal three-dimensional model of the Au nanostars, where the surface only was bound by {321} facets (Fig. S6, ESI†). The two-dimensional projections of this model matched very well with the TEM images of individual particles, where the deviations of tip angles were in the range of 2 to 4 degrees.21 This comparison also confirmed that the {321} facets were the predominant surfaces on l-NS.
Tafel slopes of the Au catalysts were checked to verify that all catalysts shared the same reaction mechanism and intermediate (Fig. S7, ESI†). All Au catalysts showed similar slope values: 158, 201, 219, and 171 mV dec−1 for Ih, s-NS, l-NS, and Au foil, respectively. These slopes were higher than that of the polycrystalline Au surface, indicating that the rate-determining step was the initial electron transfer from CO2 to form an adsorbed *CO2−.43 Nyquist plots from EIS (Fig. S8, ESI†) also showed that our catalysts followed a common reaction pathway.44
It is reported that the relative energy level of the *COOH adsorbates on the surface can determine the electrocatalytic activity and selectivity of CO2RR for CO production.15,45,46 The low binding energy of *CO on the Au surface has an advantage in yielding gaseous CO due to the facile dissociation of CO produced on the Au surface. Besides, hydrogen evolution reaction (HER) primarily competes with eCO2RR in aqueous conditions, which should also be considered to understand the high selectivity toward CO production. Although the active sites on the Au catalyst surface effectively lower the free energy of the *COOH adsorbates, they may also be active towards HER by the occupation of *H which is an intermediate for HER. Hence, the reaction sites should stabilize the *COOH adsorbates and depress the formation of *H for an effective eCO2RR. All of these intermediates should be considered simultaneously.
A computational approach using density functional theory (DFT) could provide critical information on the energetics of reaction intermediates. The Au(321) facet was considered to represent l-NS, while the Au (111) was a surface model for Ih (Fig. 5). The conventional reaction mechanisms for CO2RR and HER are suggested as follows:47
For CO2 reduction to CO (CO2RR),
| CO2 (g) + (H+ + e−) → *COOH | (1) |
| *COOH + (H+ + e−) → *CO + H2O (l) | (2) |
| *CO → CO (g) | (3) |
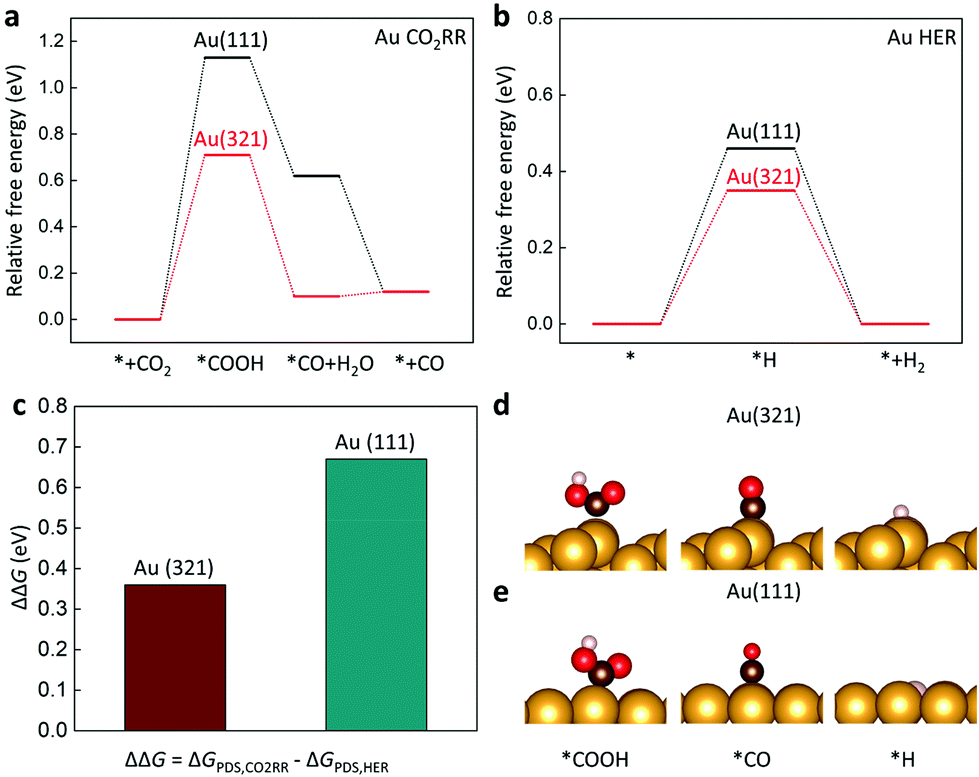 | ||
| Fig. 5 Free energy diagrams for (a) CO2RR producing CO and (b) HER on Au(111) (black) and Au(321) (red). (c) Relative reaction free energy (ΔΔG) for CO2RR compared to HER for Au(111) and Au(321). Optimized adsorption structures on (d) Au(321) and (e) Au(111) are presented. Asterisk (*) indicates adsorbed species on the surface. The yellow, brown, red, and pink balls indicate Au, C, O, and H atoms, respectively. | ||
For H2 evolution reaction (HER),
| *+(H+ + e−) → *H | (4) |
| *H + (H+ + e−) → * + H2 | (5) |
The free energy diagrams for CO2RR and HER are presented in Fig. 5a and b. To estimate the catalytic activity, we mainly focused on the potential-determining step (PDS), which requires the largest free energy since the free energy change at the PDS (ΔGPDS) determines the overall overpotential. We found that the *COOH formation (reaction (1)) was the PDS on both Au(111) and Au(321). Noticeably, the ΔGPDS of CO2RR on Au(321) (0.71 eV) was significantly lower than that on Au(111) (1.13 eV), indicating that CO2 activation was more facilitated on Au(321) (Fig. 5a). For HER, Au(321) showed a slightly reduced ΔGPDS (0.35 eV) than that of Au(111) (0.46 eV) (Fig. 5b). To estimate the relative faradaic efficiencies on the two catalyst facets, ΔΔG, which is defined as the difference of the free energy changes at the PDS for CO2RR and HER (ΔGPDS,CO2RR − ΔGPDS,HER), is plotted in Fig. 5c. The Au(321) facet (0.36 eV) showed a clear improvement compared to Au(111) (0.67 eV). The ΔGPDS and ΔΔG values revealed that the superior CO current density and FE of l-NS originated from the selective stabilization of *COOH binding energy over *H. In other words, the scaling relation between *COOH and *H bindings was broken on Au(321) facets of l-NS.48–51
The improved CO FE of l-NS resulted from the tuned binding energies of the reaction intermediates. The binding strengths of all adsorbates (*COOH and *H) became stronger on Au(321) relative to those on Au(111). However, *COOH showed a larger binding strength change over *H on Au(321). To clarify this trend, we considered the effects of local coordinate environments using the concept of generalized coordination number (GCN).52 (see Supplementary Method in ESI†). The previous study showed that the GCN had a linear correlation with the binding energies of adsorbates, including *COOH, *H, and *CO,28 with the smaller GCN of the adsorption site yielding the stronger binding energy. In particular, the GCN-dependence of the *COOH binding energy was four times greater than that of the *H binding energy,28 suggesting that the effect of low coordination sites was more critical for CO2RR than for HER. In the present cases, the GCNs of the Au(321) and Au(111) facets were calculated to be 4.9 and 7.5, respectively. Therefore, the lower GCN of the Au(321) led to improved activity and FE for CO production, consistent with our calculations and experimental results. It indicates that the low coordination sites of Au(321) in l-NS were the main reason for facile eCO2RR.
Conclusions
In summary, we designed and synthesized a unique morphology of Au nanostars through a simple overgrowth method on an icosahedron surface. For eCO2RR producing CO, l-NS showed a maximum FE of 98% at −0.73 V (vs. RHE), superior to the other catalysts: Ih, s-NS, and Au foil. Noticeably, the mass activity of l-NS (138.9 A g−1) was remarkably high compared to Au catalysts reported thus far. Analyzing both size and surface morphology effects indicated that the high activity of l-NS originated from the large specific activity of l-NS surfaces, which was more than enough to effectively compensate for the smaller surface area compared to the Ih seeds. Besides, l-NS also presented high durability for eCO2RR.The high activity and FE were attributed to the features of the high-energy {321} facets majorly covering the l-NS surface. According to DFT calculations, Au(321) selectively stabilized *COOH, the major intermediate for CO2RR, due to the low coordination number of the local surface. The difference between the free energy changes for CO2RR and HER on Au(321) was smaller than on Au(111), supporting the high FE towards CO production.
This work successfully demonstrated that inducing high-energy facets on an existing Au surface by a simple overgrowth step is feasible for providing high activity and selectivity on CO production. The facets generated by overgrowth had the specific activity large enough to outweigh the losses from the reduced surface area. This synthetic strategy is simple and universal for various materials; therefore, it would be readily extended to other catalytic surfaces such as wires, grids, and thin films to promote catalytic performances. Moreover, it would help to reduce the catalyst cost to an affordable range for various applications.
Author contributions
W. Choi and J.W. Park contributed equally to this work. W. Choi, J.W. Park, and H. Song designed the experiments. W. Choi and J.W. Park performed synthesis, characterization, and reaction studies. W. Park and Y. Jung carried out theoretical calculations and analysis. All authors joined to write the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Saudi-Aramco and KAIST CO2 management center. This work was also supported by the National Research Foundation (NRF) funded by the Ministry of Science and ICT, Republic of Korea (NRF-2018R1A2B3004096 and 2018R1A5A1025208). Y. Jung acknowledges the financial support from NRF Korea (2017R1A2B3010176) and generous supercomputing time from Korea Institute of Science and Technology Information (KISTI). J.W. Park is grateful for financial support from Hyundai Motor Chung Mong-Koo Foundation. The authors acknowledge KARA (KAIST Analysis center for Research Advancement) for characterization.Notes and references
- M. Li, S. Garg, X. Chang, L. Ge, L. Li, M. Konarova, T. E. Rufford, V. Rudolph and G. Wang, Small Methods, 2020, 4, 2000033 CrossRef CAS.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C.-T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef PubMed.
- A. Liu, M. Gao, X. Ren, F. Meng, Y. Yang, L. Gao, Q. Yang and T. Ma, J. Mater. Chem. A, 2020, 8, 3541–3562 RSC.
- R. Daiyan, W. H. Saputera, H. Masood, J. Leverett, X. Lu and R. Amal, Adv. Energy Mater., 2020, 10, 1902106 CrossRef CAS.
- F. Pan and Y. Yang, Energy Environ. Sci., 2020, 13, 2275–2309 RSC.
- K. R. Gustavsen and K. Wang, Phys. Chem. Chem. Phys., 2021, 23, 12514–12532 RSC.
- J. He, N. J. J. Johnson, A. Huang and C. P. Berlinguette, ChemSusChem, 2018, 11, 48–57 CrossRef CAS PubMed.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 14107–14113 CrossRef CAS PubMed.
- F.-Y. Gao, R.-C. Bao, M.-R. Gao and S.-H. Yu, J. Mater. Chem. A, 2020, 8, 15458–15478 RSC.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- S. Zhao, R. Jin and R. Jin, ACS Energy Lett., 2018, 3, 452–462 CrossRef CAS.
- U. O. Nwabara, E. R. Cofell, S. Verma, E. Negro and P. J. A. Kenis, ChemSusChem, 2020, 13, 855–875 CrossRef CAS PubMed.
- M. Cho, J. M. Kim, B. Kim, S. Yim, Y. J. Kim, Y. S. Jung and J. Oh, J. Mater. Chem. A, 2019, 7, 6045–6052 RSC.
- D.-R. Yang, L. Liu, Q. Zhang, Y. Shi, Y. Zhou, C. Liu, F.-B. Wang and X.-H. Xia, Sci. Bull., 2020, 65, 796–802 CrossRef CAS.
- S. Back, M. S. Yeom and Y. Jung, ACS Catal., 2015, 5, 5089–5096 CrossRef CAS.
- W. Zhu, Y.-J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS PubMed.
- H. Mistry, R. Reske, Z. Zeng, Z.-J. Zhao, J. Greeley, P. Strasser and B. Roldan Cuenya, J. Am. Chem. Soc., 2014, 136, 16473–16476 CrossRef CAS PubMed.
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- Y. Ma, J. Wang, J. Yu, J. Zhou, X. Zhou, H. Li, Z. He, H. Long, Y. Wang, P. Lu, J. Yin, H. Sun, Z. Zhang and Z. Fan, Matter, 2021, 4, 888–926 CrossRef CAS.
- H.-E. Lee, K. D. Yang, S. M. Yoon, H.-Y. Ahn, Y. Y. Lee, H. Chang, D. H. Jeong, Y.-S. Lee, M. Y. Kim and K. T. Nam, ACS Nano, 2015, 9, 8384–8393 CrossRef CAS PubMed.
- W. Niu, Y. A. A. Chua, W. Zhang, H. Huang and X. Lu, J. Am. Chem. Soc., 2015, 137, 10460–10463 CrossRef CAS PubMed.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- S. Back, M. S. Yeom and Y. Jung, J. Phys. Chem. C, 2018, 122, 4274–4280 CrossRef CAS.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- F. Illas, Angew. Chem., Int. Ed., 2015, 54, 10404–10405 CrossRef CAS.
- X. Bin, E. H. Sargent and S. O. Kelley, Anal. Chem., 2010, 82, 5928–5931 CrossRef CAS PubMed.
- H. Yun, J. Kim, W. Choi, M. H. Han, J. H. Park, H.-s. Oh, D. H. Won, K. Kwak and Y. J. Hwang, Electrochim. Acta, 2021, 371, 137795 CrossRef CAS.
- K. Manthiram, Y. Surendranath and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 7237–7240 CrossRef CAS PubMed.
- J. Huang, N. Hörmann, E. Oveisi, A. Loiudice, G. L. De Gregorio, O. Andreussi, N. Marzari and R. Buonsanti, Nat. Commun., 2018, 9, 3117 CrossRef PubMed.
- L. Zhang, Z. Wei, S. Thanneeru, M. Meng, M. Kruzyk, G. Ung, B. Liu and J. He, Angew. Chem., Int. Ed., 2019, 58, 15834–15840 CrossRef CAS PubMed.
- W. Choi, D. H. Won and Y. J. Hwang, J. Mater. Chem. A, 2020, 8, 15341–15357 RSC.
- Y. Yu, Q. Zhang, J. Xie, X. Lu and J. Y. Lee, Nanoscale, 2011, 3, 1497–1500 RSC.
- J. Y. Kim, W. Park, C. Choi, G. Kim, K. M. Cho, J. Lim, S. J. Kim, A. Al-Saggaf, I. Gereige, H. Lee, W.-B. Jung, Y. Jung and H.-T. Jung, ACS Catal., 2021, 11, 5658–5665 CrossRef CAS.
- J. W. Hong, S.-U. Lee, Y. W. Lee and S. W. Han, J. Am. Chem. Soc., 2012, 134, 4565–4568 CrossRef CAS PubMed.
- B. Glam, D. Moreno, S. Eliezer and D. Eliezer, J. Nucl. Mater., 2009, 393, 230–234 CrossRef CAS.
- S. W. Im, H.-Y. Ahn, R. M. Kim, N. H. Cho, H. Kim, Y.-C. Lim, H.-E. Lee and K. T. Nam, Adv. Mater., 2020, 32, 1905758 CrossRef CAS PubMed.
- W. Zhang, Y. Shen, F. Pang, D. Quek, W. Niu, W. Wang and P. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 41613–41619 CrossRef PubMed.
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS PubMed.
- M. N. Hossain, J. Wen and A. Chen, Sci. Rep., 2017, 7, 3184 CrossRef PubMed.
- Z. Cao, D. Kim, D. Hong, Y. Yu, J. Xu, S. Lin, X. Wen, E. M. Nichols, K. Jeong, J. A. Reimer, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 8120–8125 CrossRef CAS PubMed.
- C. Kim, H. S. Jeon, T. Eom, M. S. Jee, H. Kim, C. M. Friend, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2015, 137, 13844–13850 CrossRef CAS PubMed.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- M. R. Singh, J. D. Goodpaster, A. Z. Weber, M. Head-Gordon and A. T. Bell, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E8812 CrossRef CAS PubMed.
- Y. Li and Q. Sun, Adv. Energy Mater., 2016, 6, 1600463 CrossRef.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, Catal. Today, 2017, 288, 74–78 CrossRef CAS.
- D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. Wang, J. Wang and X. Bao, J. Am. Chem. Soc., 2015, 137, 4288–4291 CrossRef CAS PubMed.
- F. Calle–Vallejo, J. I. Martínez, J. M. García–Lastra, P. Sautet and D. Loffreda, Angew. Chem., Int. Ed., 2014, 53, 8316–8319 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr03928h |
| ‡ Present Address: Clean Energy Research Center, Korea Institute of Science and Technology (KIST), Seoul 02792, Republic of Korea. |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |