
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multi-component bioresponsive nanoparticles for synchronous delivery of docetaxel and TUBB3 siRNA to lung cancer cells†
Claudia
Conte
 *ab,
Patrícia F.
Monteiro
a,
Pratik
Gurnani
a,
Snow
Stolnik
a,
Francesca
Ungaro
b,
Fabiana
Quaglia
b,
Philip
Clarke
c,
Anna
Grabowska
c,
Maria
Kavallaris
def and
Cameron
Alexander
*a
*ab,
Patrícia F.
Monteiro
a,
Pratik
Gurnani
a,
Snow
Stolnik
a,
Francesca
Ungaro
b,
Fabiana
Quaglia
b,
Philip
Clarke
c,
Anna
Grabowska
c,
Maria
Kavallaris
def and
Cameron
Alexander
*a
aDivision of Molecular Therapeutics and Formulation, School of Pharmacy, University of Nottingham, NG7 2RD, UK. E-mail: claudia.conte@unina.it; cameron.alexander@nottingham.ac.uk
bDrug Delivery Laboratory, Department of Pharmacy, University of Napoli Federico II, 80131 Napoli, Italy
cThe BioDiscovery Institute and Faculty of Medicine & Health Sciences, University of Nottingham, NG7 2RD, UK
dChildren's Cancer Institute, Lowy Cancer Research Centre, UNSW Sydney, NSW 2052, Australia
eARC Centre of Excellence in Convergent Bio-Nano Science and Technology and Australian Centre for NanoMedicine, UNSW Sydney, NSW 2052, Australia
fSchool of Women's and Children's Health, Faculty of Medicine, UNSW Sydney, NSW, Australia 2052
First published on 23rd June 2021
Abstract
Bioresponsive nanoparticles (NPs) are of interest for anticancer nanomedicines, owing to the possibility to ‘design in’ selective modulation of drug release at target sites. Here we describe the double emulsion formulation of redox-responsive NPs based on modified polyethylene glycol (PEG)-co-poly(lactic-co-glycolic acid) (PLGA) block copolymers and oligo (β-aminoesters) (OBAE), both of which contained disulfide linkages, for the co-delivery of a cytotoxic small molecule drug and a nucleic acid. In particular, we focused our attention on docetaxel (DTX) and a siRNA against TUBB3, a gene that encodes for βIII-tubulin, in order to have a synergistic effect in the treatment of lung cancer. Spherical NPs of around 150 nm with negative zeta potential and high loading efficiencies of both drugs were obtained. Stability and release studies showed “on demand” drug release under reducing conditions. Unloaded NPs containing PEG-disulfide-PLGA and OBAE were well-tolerated by lung cancer cells, thus masking the intrinsic cytotoxicity of OBAE, while for intracellular siRNA delivery, redox responsive NPs demonstrated a higher cell internalization with a preferential cytoplasmic accumulation of siRNA, with a subsequent fast gene-silencing efficiency. The viability of cells treated with combined DTX/TUBB3-siRNA NPs significantly decreased as compared to NPs loaded only with DTX, thus showing an efficient combined anticancer effect, due to a substantial reduction of β-tubulin expression. Finally, in an in vivo feasibility study employing an orthotopic lung cancer model, NPs formulated with an anti-luciferase siRNA distributed throughout the lungs following oro-tracheal administration, and demonstrated effective gene knockdown and no apparent cytotoxicity. Taken together, these results show that the double emulsion formulated redox responsive PEG-PLGA and OBAE systems represent a promising new therapeutic approach for the local combined chemo- and gene-therapy of lung cancer.
Introduction
Lung cancer (LC) is one of the most common cancers worldwide and non-small cell lung cancer (NSCLC) accounts for approximately 85% of all LC cases.1 The simultaneous administration of two or more conventional anticancer drugs represents the first-line treatment for these patients, but the development of multidrug resistance (MDR) strongly limits the therapeutic effectiveness. Therefore, combinations of multiple therapeutic modalities (e.g. chemotherapy, gene therapy, immunotherapy, photodynamic therapy) with additive/synergistic mechanisms of cell killing are becoming standard in clinical settings for combating resistance mechanisms.2,3 In particular, regulation of gene expression through RNA-interference based therapeutics is currently of interest, due to the ability of these molecules to silence specific pathways involved in MDR.4,5 For instance, more than 2500 short-interfering RNA (siRNA) patents for LC have been developed so far.6 Nevertheless, the clinical application of siRNA in combination with other therapeutics is challenging due to the difference in chemical and biopharmaceutical profiles. Moreover, major obstacles to siRNA therapy arise from extracellular and intracellular barriers, including (i) in vivo degradation via nucleases (ii) inability to cross endothelial membranes and (iii) poor efficiency of endosomal escape to reach their cytoplasmic target. As a result, better delivery of siRNA is required in order to drive these delicate molecules into the body and to modulate their fate after administration.7,8 In this context, nanomedicine-based approaches for RNA delivery have been explored, aiming to improve pharmacological effectiveness at disease sites and reduce off-target effects.9–15 However, the features of nanoparticle (NP) delivery systems need to be refined carefully in terms of composition, structure, size, surface and stability, and to take into account (i) the drugs to be delivered, (ii) the characteristics of the tumor microenvironment, (iii) the administration route and (iv) the final target.16 Current research is focused on the design of carrier materials able to respond to intrinsic or extrinsic stimuli (pH decrease, redox gradients, enzyme action, light, magnetic fields, temperature, ultrasound or electric currents) for “on demand” delivery at a diseased site.13,17,18 In particular, by exploiting the differences in reduction potential between normal tissues and certain tumor microenvironments, redox-responsive NPs have been shown to release drugs selectively at specific subcellular locations.19 It is well-known that the cytosol, the mitochondria and the nucleus of tumor cells are characterized by high concentrations of reductive glutathione (GSH) tripeptides (approx. 10 mM, about 100 to 1000 times higher than those in the extracellular fluids and normal tissues). For this reason, GSH-responsive NPs have been recognized as promising intracellular delivery systems for cancer and gene therapy.20Finally, for the treatment of severe lung disorders, including LC, administration by the pulmonary route is commonly preferred, since it allows fast and “needle-free” direct access to the target sites, thus reducing systemic effects. However, it is vital to consider the anatomical and physiological features of the lung that can prevent drug and siRNA accumulation. These include mucociliary clearance and enzymatic barriers in the alveolar fluid that promote siRNA degradation.21 For these reasons, inhalable NPs for lung delivery must meet some demanding formulation criteria.22,23
In order to develop effective inhalable NPs for pulmonary delivery, we have focused on redox responsive NPs based on a biodegradable and amphiphilic diblock copolymer containing a reducible disulfide bridge between poly(lactic-co-glycolic acid) (PLGA) and polyethylene glycol (PEG) blocks intended for the treatment of LC. We have chosen as building blocks PEG and PLGA, for their well-established regulatory profile that renders our approach promising for future in vivo applications. PLGA and PEG are in fact both biodegradable and nontoxic polymers approved by the United States Food and Drug Administration (FDA) for medical uses.24 Moreover, this system has previously shown (i) good ability to entrap and trigger on-demand release of the conventional anticancer drug docetaxel (DTX) in conditions simulating intracellular reductive environments; (ii) high penetration properties through an artificial mucus layer due to the presence of the external PEG coating and (iii) enhanced uptake in 2D- and 3D- LC models due to a double (extra- and intra- cellular) redox responsive behavior.25 For this work, the desire to co-deliver siRNA for binary-drug therapy required modification of the carrier by the introduction of cationic oligo-β-aminoesters (OBAE). We chose OBAEs for their ability to condense nucleic acids at physiological pH, protect them in their passage across cell membranes, and enable payload release into cell interiors. The latter can occur within acidic endosomal or lysosomal environments, due to the presence of reversibly protonatable amine groups and acid or base-sensitive ester bonds, which are ultimately degraded into non-toxic small molecules.26 For this study, we have employed a disulfide-containing OBAE, which, as we recently reported, was able to condense siRNA into polyplexes through a fast inkjet printing method.27 The specific OBAE in this study was expected to protect siRNA against biological delivery barriers but also break down intracellularly to enable complete release of the nucleic acid cargo.28,29 Through this chemical strategy, we combined the mucus penetration properties of PEG-coated NPs,30 thus enhancing pulmonary delivery, with functionality for nucleic acid binding in the OBAE component, and bioresponsive chemistries in both components encoding for site-specific release.
We loaded NPs with DTX, clinically approved for LC treatment, and a therapeutic siRNA (TUBB3-siRNA) against βIII-tubulin, a protein involved in microtubule formation. A growing body of preclinical and clinical data suggests that increased expression of this protein can reduce the antimitotic activity of DTX and taxanes in general,31 thus conferring cancer resistance in different tumor types, including NSCLC.32–34 Therefore, we considered that combining knockdown of βIII-tubulin expression via siRNA with delivery of DTX would be a potential and promising strategy to enhance cancer cell kill with taxanes. After an in-depth evaluation of NP behavior in LC cells, proof-of-principle for the translational potential of these NPs was explored in vivo by assessing the tolerability, biodistribution and transfection of siRNA-loaded NPs after oro-tracheal administration in an orthotopic LC model. These studies represent the first steps in establishing the initial safety and efficacy profiles of the novel formulated redox-responsive NPs, in preparation for future full combination therapy studies, in view of a potential therapeutic alternative to the clinically used first-line lung cancer therapy.
Results and discussion
Bioresponsive NPs were effectively derived from polymers of PLGA and OBAE, each containing bioreducible disulfide bridges and with an outer PEG layer to allow good penetration through pulmonary barriers.35 OBAEs are able to condense siRNA and deliver it in the same cancer cell line due to its cationic charge, redox responsiveness and high buffering capacity to allow the endosomal escape of nucleic acids. As following step, herein we have decided to combine PLGA-block copolymers with OBAEs in the same nanoplatform to: (i) ensure the tolerability of the formulation due to the well-known safety of PLGA and PEG polymers; (ii) exploit the mucus penetrating properties of PEG, critical for a pulmonary administration; (iii) load with high efficiency a siRNA for the presence of the cationic OBAE in NP core, able to induce a proton sponge effect at cell level; (iv) load a second drug in the polymeric matrix of NPs, in order to have a combined effect with siRNA.Preparation and characterization of NPs
PLGA-PEG and PLGA-SS-PEG block copolymers were mixed with cationic disulfide containing OBAE and employed to prepare NPs which were either wholly (PLGA-SS-PEG, OBAE) or partially (PLGA-PEG, OBAE) redox-responsive, respectively. In particular, PLGA-based copolymers and OBAE were mixed in the same NP in order to have a carrier with a PEG outer layer to enhance pulmonary mucus penetration, and able to entrap simultaneously lipophilic drugs, such as DTX, and hydrophilic siRNA. The first drug is located in the PLGA core due its lipophilic profile, whereas the siRNA is entrapped through electrostatic interaction with the cationic charges of OBAE. For an appropriate set of comparative materials, we therefore prepared unloaded NPs, DTX-loaded NPs, siRNA-loaded NPs and combined siRNA/DTX-loaded NPs.NPs were prepared by a modified double emulsion-solvent evaporation technique25 and characterized in terms of size, PDI, ζ and morphology. A schematic representation of synthesis of the materials and NP preparation is shown in Fig. S1 and Scheme 1, respectively. After a preliminary formulation study by adjusting the processing parameters of the emulsification-solvent evaporation technique, we found that the PLGA/OBAE mass ratio 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was the best condition to obtain NPs with good colloidal properties and encapsulation of both DTX and siRNA. Initially, OBAE was dissolved in EtOH and then mixed with the DCM/PLGA solution at a volume ratio 1:9. DTX was co-dissolved in the organic phase with OBAE and PLGA-copolymers, and siRNA dissolved in the internal water phase before the emulsification process. Composition and properties of all the formulations are reported in Table 1. The yield for the formulations was high (∼70%) indicating that no extensive polymer/drug precipitation or NP aggregation occurred during preparation. The primary properties of the PLGA-PEG and PLGA-SS-PEG formulations were similar, thus highlighting a comparable assembling behavior of the copolymers. NPs displayed hydrodynamic diameters around 150 nm, with acceptable polydispersity indices and negative ζ potential values. The spherical morphology and the absence of NP aggregation was confirmed by TEM images (Fig. 1A). Entrapment of both drugs did not influence significantly surface properties and ζ potentials of NPs, whereas a slight increase of size as well as polydispersity index for both formulations was observed when DTX was loaded.
:1 was the best condition to obtain NPs with good colloidal properties and encapsulation of both DTX and siRNA. Initially, OBAE was dissolved in EtOH and then mixed with the DCM/PLGA solution at a volume ratio 1:9. DTX was co-dissolved in the organic phase with OBAE and PLGA-copolymers, and siRNA dissolved in the internal water phase before the emulsification process. Composition and properties of all the formulations are reported in Table 1. The yield for the formulations was high (∼70%) indicating that no extensive polymer/drug precipitation or NP aggregation occurred during preparation. The primary properties of the PLGA-PEG and PLGA-SS-PEG formulations were similar, thus highlighting a comparable assembling behavior of the copolymers. NPs displayed hydrodynamic diameters around 150 nm, with acceptable polydispersity indices and negative ζ potential values. The spherical morphology and the absence of NP aggregation was confirmed by TEM images (Fig. 1A). Entrapment of both drugs did not influence significantly surface properties and ζ potentials of NPs, whereas a slight increase of size as well as polydispersity index for both formulations was observed when DTX was loaded.
 | ||
| Scheme 1 Schematic illustration of materials and NP preparation technique. | ||
 | ||
| Fig. 1 NP characterization. (A) FEG-TEM images of TUBB3-DTX/NPs; (B) size distribution curves of NPs incubated in PBS 10 mM pH 7.4 with or without GSH (10 mM) at 37 °C for 30 min; (C) in vitro release of DTX and siRNA from TUBB3-DTX/NPs in PBS with or without GSH 10 mM. Results are expressed as release% over time ± SD of three experiments. | ||
| Code | OBAE (% w/w) | DTX (% w/w) | siRNA (% w/w) | Yield (%) | Mean DH (nm ± SD) | PDI | ζ (mV ± SD) | DTX actual loadinga (entr. eff%b) | siRNA actual loadinga (entr. eff%b) |
|---|---|---|---|---|---|---|---|---|---|
| a Actual loading is expressed as the amount (mg) of drug encapsulated per 100 mg of NPs. b Ratio between actual and theoretical loading × 100. | |||||||||
| PLGA-PEG | 10 | — | — | 75 | 153 ± 7 | 0.1 | −9.9 ± 2.3 | — | — |
| DTX/PLGA-PEG | 10 | 10 | — | 69 | 174 ± 5 | 0.2 | −9.2 ± 4.4 | 6.3 ± 0.4 (71.2 ± 4.8) | — |
| TUBB3-DTX/PLGA-PEG | 10 | 10 | 0.1 | 65 | 183 ± 9 | 0.2 | −9.7 ± 2.7 | 5.5 ± 1.1 (57.7 ± 8.3) | 0.1 ± 0.01 (98.1 ± 1.9) |
| PLGA-SS-PEG | 10 | — | — | 71 | 143 ± 2 | 0.1 | −11.2 ± 3.4 | — | — |
| DTX/PLGA-SS-PEG | 10 | 10 | — | 70 | 173.0 ± 7 | 0.2 | −12.2 ± 3.5 | 6.5 ± 2.4 (74.3 ± 7.1) | — |
| TUBB3-DTX/PLGA-SS-PEG | 10 | 10 | 0.1 | 73 | 178 ± 5 | 0.2 | −8.4 ± 3.3 | 6.2 ± 5.1 (69.7 ± 4.5) | 0.1 ± 0.03 (94.2 ± 3.3) |
However, the entrapment of siRNA in the systems did not influence the capability of PLGA-cores to load the lipophilic drug DTX. The complete entrapment of siRNA in all the formulations was confirmed by a quantitative assay through Nanodrop-UV measurements as well as by gel retardation assays (Fig. S2†). In order to investigate the redox responsiveness of NPs in a simulated intracellular reductive environment, different experimental techniques were performed, including stability and release studies. As clearly evidenced in Fig. 1B, PLGA-SS-PEG NPs showed low stability in GSH reducing media (10 mM), highlighted by the changing of the initial particle size and distribution curves, compared to PLGA-PEG NPs, in line with TEM images of NPs that we have already shown in our previous paper,36 where a strong deformation of PLGA-SS-PEG NP structure was clearly evidenced in GSH. At the same time, Fig. S2† evidenced a partial release of siRNA from PLGA-SS-PEG NPs after incubation in GSH. These results indicate the release of the surface-displayed PEG chains from the NPs in a simulated intracellular reductive environment through the cleavage of the disulfide bond between the PLGA and the PEG blocks, in line with prior data from similar polymers. To confirm these data, release studies of DTX from NPs retained in dialysis membranes were carried out in the presence of GSH 10 mM in the releasing medium. In Fig. 1C, it is possible to note that both PLGA-PEG NPs and PLGA-SS-PEG NPs exhibited a sustained release pattern of DTX in PBS. In particular, around 50% of the entrapped DTX was released in 14 days, compared with the free drug in the same conditions (inset). In contrast, with the addition of GSH in the releasing medium, complete drug release was observed from PLGA-SS-PEG NPs after 3 days of incubation. This trend was not significantly affected by the presence of OBAE in the formulations. The same trend was found for siRNA release.
Effects of NPs in lung cancer cells
Before moving to cell studies, we tried to predict the stability of NPs in simulated biological conditions. In fact, it is well known that on contact with physiological fluids, some NPs will interface with a wide range of biomacromolecules, including proteins that can strongly modify NP colloidal properties and identity. However, our system showed good stability in Fetal Bovin Serum (FBS), the major protein component of cell culture media as evident in Fig. S3,† which we attribute to the display of PEG-chains at the NP surfaces.A key obstacle to clinical translation of siRNA-based nanomedicine is the extensive cytotoxicity related to many cationic materials which have been tested, a problem which is partly due to the limited biodegradability of commonly used polycations.37 For this reason, the ‘bioreducible nanocarrier’ approach has been developed to resolve the efficacy–toxicity dilemma.38 However, the overall molar mass and molar mass distribution can significantly affect the transfection activity and cytotoxicity even of biodegradable polymers.39 We therefore compared the effects of free OBAE to unloaded NPs on cell metabolic activity, as a proxy for cell viability. These experiments were conducted in A549 and H1299 LC cells by the MTS assay as described in the Experimental section. Cells were incubated with a wide range of NP concentrations (0.05–5 mg mL−1) and tested after 24 h of treatment. At the same time, the effects on metabolic activity of free OBAE in the same concentration range entrapped into NPs (0.005 mg–0.5 mg) was evaluated. Results from MTS assays (Fig. 2A and S4†) show that 24 h after treatment, A549 and H1299 cells retained about 90% viability when treated with various concentrations of NPs as compared with free OBAE. In the latter case, a higher concentration dependent cytotoxicity was apparent, with a reduction in metabolic activity of ∼80%. The reduced toxicity of the PLGA-based NPs modified with cationic OBAE suggested that any cationic charges in these formulations were effectively shielded from the cells. To highlight the safety of these materials, NPs were tested on the same cell lines up to 50 mg mL−1 and no toxicity was found (Fig. S5†). Subsequently, the transfection efficiency of NPs loaded with a siRNA-based luciferase knock-down sequence was assessed on luciferase-expressing A549 cancer cells.
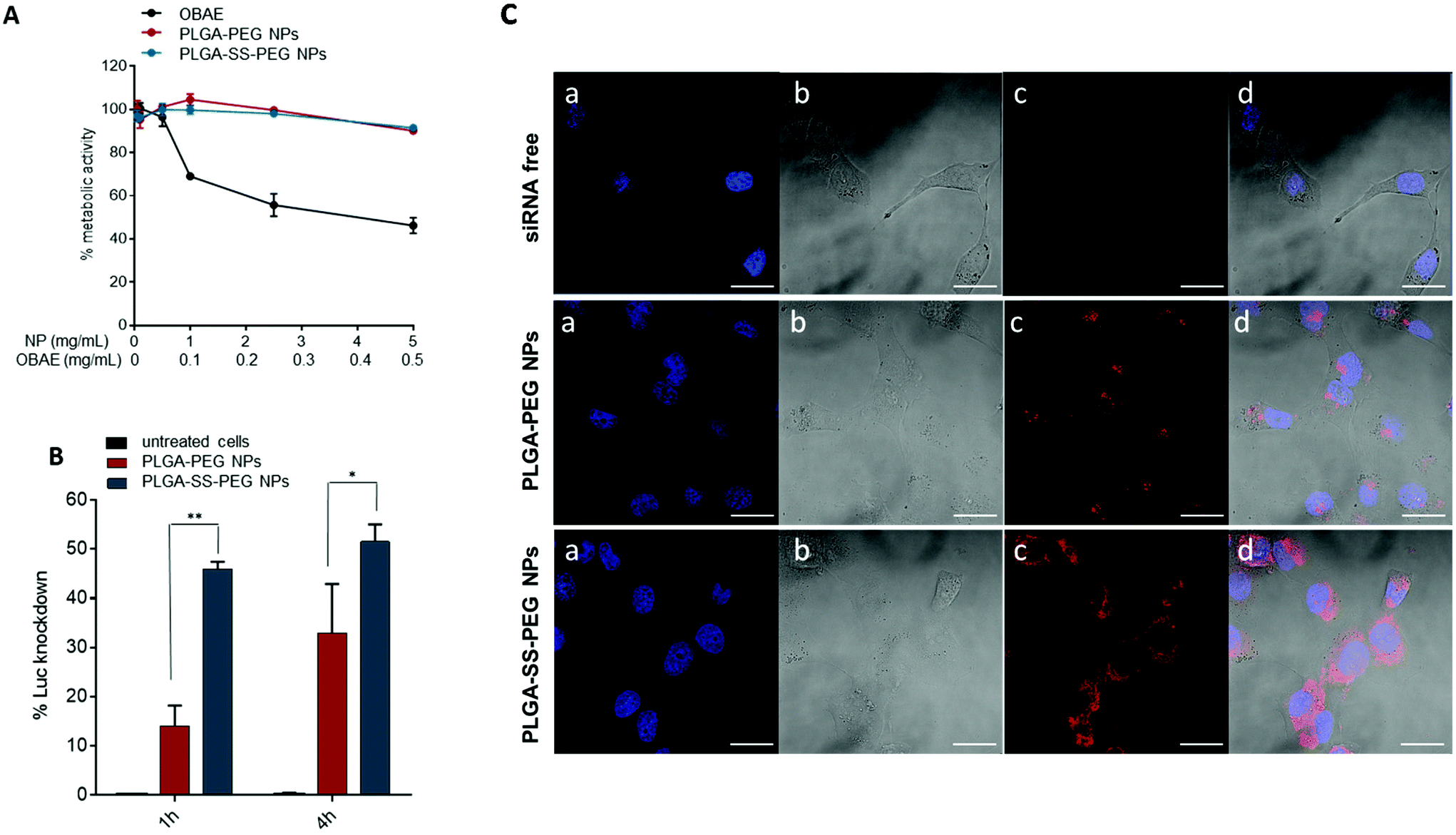 | ||
| Fig. 2 (A) Cytotoxicity of unloaded NPs against A549 lung cancer cells compared to free OBAE (24 h of treatment). OBAE concentration range: 0.005–0.5 mg mL−1. (B) In vitro luciferase siRNA transfection in A549-luciferase expressing cells after different NP/cell time contact. Results are expressed as mean ± SD of three experiments. **P < 0.01, *P < 0.05 two-way ANOVA test. (C) Confocal images of A549 cells after incubation with Cy™3-siRNA loaded NPs (125 μg mL−1) for 4 h: (a) nucleus stained with Hoechst 33258, (b) brightfield, (c) Cy™3-siRNA emission in red (λex: 555 nm; λem: LP 560 nm), (d) merge. Scale bar: 20 μm. Zen 2009 image Software was utilized for image processing. | ||
The primary properties of NPs loaded with Luc-siRNA are reported in Table S1† and are comparable to the other formulations. The silencing efficiency was evaluated after 1 h and 4 h of NP incubation into cancer cells and compared to untreated cells. As previously stated, we have already demonstrated the ability of free OBAEs to deliver siRNA in cancer cells,27 but due to the cytotoxicity of this cationic material, we decided to not include this control in the following experiments. For redox responsive PLGA-SS-PEG NPs, approximately 50% of silencing efficiency occurred after only 1 h of incubation, with a slight increase over time. In contrast, PLGA-PEG NPs showed a slower, but also time dependent, transfection efficiency, achieving 10% and 30% of Luciferase knockdown after 1 h and 4 h of contact time, respectively (Fig. 2B). In Fig. S6† these results are also reported as relative light units (RLU mg−1 protein) of Luciferase activity. This different behavior could be attributed to the reductive triggered degradation of disulfide linkages between the PLGA and PEGylated components, leading to subsequent NP instability and release of entrapped siRNA in the cellular environment in a shorter time.
In order to investigate the internalization properties of NPs into cells and to predict the fate of the drug cargoes, we carried out uptake experiments of labeled NPs against A549 cells with subsequent confocal microscopy analysis (Fig. 2C). For these experiments, we employed NPs loaded with a fluorescent Cy™3-siRNA (properties are reported in Table S2†). As evident in the images, the uptake of redox responsive NPs as well as the internalization of the siRNA (red) was higher as compared to non bioreducible NPs. This effect could be attributed to a partial extracellular responsiveness of the reducible linkage between PLGA and PEG blocks, as previously demonstrated25 and in line with some other studies.40,41 We subsequently probed the intracellular trafficking of NPs and the eventual endosomal escape facilitated by the cationic OBAE: A549 cells were incubated for 4 h with Cy™3-siRNA-NPs and then the cell nuclei and endosome were labeled with Hoechst 33258 (blue) and LysoTracker DND-26 (green), respectively. As evident in Fig. 3, we did not observe any overlapping of the red and green colors for both formulations, thus suggesting that the majority of NPs or of the released siRNA escaped from the lysosomes to the cytoplasm.
 | ||
| Fig. 3 Intracellular trafficking of NPs after 4 h of incubation. Plates (a–d) show PLGA-PEG NPs and plates (e–h) show PLGA-SS-PEG NPs. (a and e) Lysotracker emission in green (λex: 488 nm; λem: SP 555), (b and f) Cy3-siRNA emission in red (λex: 555 nm; λem: LP 560), (c and g) nucleus stained with Hoechst 33258. Images (d) and (h) show merged channels with enlarged sections contrast-enhanced for clarity. Scale bar: 20 μm. Zen 2009 image Software was utilized for image processing. | ||
Cell metabolic activity of TUBB3 siRNA/DTX-loaded NPs
To assess the effects of DTX and of βIII-tubulin knockdown on microtubule organization, immunofluorescence staining of βIII-tubulin was performed in LC cells after 72 h of NP treatment. As evident in Fig. 4 and Fig. S7,† untreated cells (the controls of the experiments) had an intact microtubule cytoskeleton. It is well established that microtubule bundle formation is a hallmark of taxane exposure. The dynamic reorganization of microtubules after treatment with docetaxel or paclitaxel causes the inhibition of major cellular events, and apoptosis has generally been accepted to be the predominant mechanism of cell death in response to taxane chemotherapy due to the activation of a ‘mitotic catastrophe’ by scoring of micronucleated cells and cells undergoing aberrant mitosis. In CLSM images, the treatment of cells with DTX, in free form or delivered from NPs, induced strong microtubule bundle formation in a manner not significantly different between the delivery system employed. Concerning the βIII-tubulin knockdown, TUBB3 siRNA in free form or delivered from PLGA-PEG NPs did not cause any significant modification of the microtubule networks, whereas a marked decrease of βIII-tubulin expression was evident from siRNA delivered from redox responsive NPs (Fig. 4). These results were in line with the metabolic activity of cells after NP treatment (Fig. 5) and the siRNA transfection properties previously discussed. | ||
| Fig. 4 Immunofluorescence microscopy of A549 cells treated with NPs for 72 h. βIII-tubulin was stained in green whereas nuclei were stained with Hoechst 33258 in blue. Scale bar: 10 μm. Zen 2009 image Software was utilized for image processing. | ||
 | ||
| Fig. 5 Cell metabolic activity of PLGA-PEG NPs and PLGA-SS-PEG NPs toward A549 cells after 72 h of incubation. Results are expressed as mean ± SD of three experiments. ***P < 0.001, **P < 0.01, *P < 0.05 free DTX, two-way ANOVA test. | ||
Concerning anticancer activity and effect of co-delivery, the in vitro cytotoxicity of NPs loaded with DTX, with or without TUBB3 siRNA, was evaluated. These MTS assays were carried out after different time exposures (24 h and 72 h), in comparison to free drugs alone or in combination.
In Fig. 5 and S8,† the cytotoxicity as interpreted from the loss in metabolic activity against A549 cells is reported (DTX concentration range: 0.1–100 μg mL−1; TUBB3-siRNA concentration range: 1–1000 ng mL−1). As expected, DTX, both in free form as well as entrapped into NPs, exhibited a time- and dose-dependent cytotoxicity due to its well-known antimitotic mechanism of action. Furthermore, neither PLGA-PEG and PLGA-SS-PEG NPs loaded with DTX induced statistically significant differences in cell growth inhibition when compared to free drug (P = 0.20).
The IC50 values of DTX against A549 cells at 72 h were calculated to be around 102, 132 and 79 μg mL−1, for free DTX, PLGA-PEG NPs and PLGA-SS-PEG NPs, respectively, with potentiating factor values (PF) lower than 1 (Table S3†). A similar trend was observed in H1299 cells (DTX concentration range: 0.0001–1 μg mL−1; TUBB3-siRNA concentration range: 0.001–10 ng mL−1) (Fig. S9†). The fact that DTX-NPs showed cytotoxicity similar to that of free DTX at the same dose is perhaps surprising, considering that the NPs were not expected to release all of the entrapped drug over the experimental timeframe. However, as previously reported, it can be speculated that this effect may be related to the differing mechanisms by which DTX entered the cancer cells when encapsulated in NPs. Thus endocytic transport of NPs would be expected to reduce the interaction of DTX with extracellular proteins in the cell culture media, and also bypass any membrane-associated DTX efflux pumps.42–44 In addition, the encapsulation and co-delivery of TUBB3-siRNA into bioresponsive NPs appeared to potentiate the anti-proliferative activity of DTX, especially in A549 cells which were otherwise less sensitive to DTX activity. Specifically, the IC50 values of PLGA-SS-PEG NPs notably decreased after 72 h (6.5 μg mL−1 in A549 cells), reaching PF values of 16 and 12 vs. free DTX and the corresponding bioreducible formulation loaded with only DTX, respectively. This different potentiation compared to non-responsive NPs indicated a clear DTX/TUBB3 synergistic activity, in line with the much higher efficacy of this formulation.
In vivo assays to assess potential of NPs for drug delivery after inhalation
On the basis of the above encouraging data for delivery and efficacy in cancer cell lines, we moved to preliminary in vivo studies. The aim of these experiments was to evaluate if the developed NPs were able to exert their silencing effect following pulmonary administration. To facilitate the study, we decided to look at an orthotopic model of lung cancer in mice established with luciferase-expressing cancer cells. siRNAs for the knock-down of luciferase activity are also readily available. These assays were expected to yield the key proof-of-principle that a biological target could be targeted by the NPs in an in vivo cancer setting, but without the complexities involved in evaluating the overall tumor response. In particular, since the combination of DTX and anti-TUBB3 therapies in NP formulations would require significant number of controls (single drugs NPs, free drugs, combinations) and thus of animals, it was essential for ethical reasons to carry out pilot experiments in as few mice as possible. Therefore, we applied anti-luc siRNA-containing NPs via intra-tracheal administration in an A549-luc bioluminescent orthotopic LC model, and assessed tolerability and biodistribution of the formulations, as well as transfection efficiency in the knock-down of luciferase.Tumors spontaneously grew in the lungs following intravenous injection of A549-luc tumor cells in the mice. Tumor growth and the effects of treatments were monitored by whole body in vivo bioluminescent imaging (IVIS) and confirmed by X-ray imaging (Fig. S10†). Data are presented in terms of mean tumor bioluminescent intensity (BLI) and typical IVIS images from one animal representative of each treatment group at each time point. Prior to the treatment with therapeutic NPs, acute tolerability of NPs loaded with a scrambled siRNA and without DTX was assessed in tumor bearing mice (three mice per treatment under dosing as per the therapy study for 14 days, followed by cessation of dosing and observation for a further 14 days). The properties of the NPs used in these experiments are reported in Table S4,† and as control a free scrambled siRNA at the same concentration was used. No adverse effects were noted in the mice (data not shown).
To assess NP biodistribution after intratracheal administration, tumor-bearing mice were randomized in 2 groups of 3 mice (n = 6). NPs loaded with the red fluorescent model drug DiR (DIR/PLGA-PEG NPs and DIR/PLGA-SS-PEG) were administered intratracheally and fluorescence images were collected live after 1 h and 24 h as well as after animal sacrifice by IVIS. The accumulation of DiR into the tumors after 1 h was higher in the case of PLGA-SS-PEG NPs with a more symmetrical distribution in the lungs (Fig. 6A and B). However, after 24 h, DiR fluorescence into the lungs strongly decreased compared to the control formulation, probably due to the redox responsiveness of the system that favours DiR release in the reductive tumor environment. These results are confirmed also by ex vivo analysis of the organs from sacrificed mice (Fig. 6C). After 24 h of treatment with NPs, DiR fluorescence was retained in the lungs (Fig. S11†), but fluorescence was found also in other organs (e.g. kidney and brain) only in the case of PLGA-SS-PEG formulation, which suggests again that DiR release from redox-responsive NPs was higher than that from PLGA-PEG NPs (Fig. S12†), as observed in the in vitro study.
 | ||
| Fig. 6 (A) Ex vivo fluorescence images and (B) quantification of the mean fluorescent intensity of DiR in excised lungs of the A549-luc tumor-bearing mice after 1 h and 24 h of treatment with DiR-loaded NPs; (C) quantitative presentation of organ biodistribution ex vivo at 24 h. | ||
Finally, to evaluate the ability of the NPs to deliver siRNA into the lung, gene silencing efficacy studies were carried out. Two weeks after tumor initiation, mice were treated with NPs loaded with a siRNA against Luciferase (antiLuc-siRNA) or with a scrambled siRNA (ctr-NPs). Mice were dosed again intratracheally and after 72 h, luminescence of the tumors was monitored in vivo as well as ex vivo. A reduction of lung bioluminescence after treatment with PLGA-SS-PEG and PLGA-PEG NPs (Fig. 7A). Moreover, the effect of anti-Luc-siRNA NPs was confirmed through the analysis of excised lungs after 72 h. The data showed that bioluminescence of the lungs strongly decreased compared to those treated with NPs loaded with a scrambled siRNA as control (Fig. 7B), thus demonstrating the efficacy of this system to reduce the lung tumor.
 | ||
| Fig. 7 (A) Ex vivo bioluminescence images and (B) quantitative luminescence of excised lungs of the A549-luc tumor-bearing mice after 72 h of treatment with NPs loaded with a siRNA against Luciferase. ***P < 0.001 vs. ctr-NPs, two-way ANOVA test. | ||
These preliminary data established the principle of delivering functional payloads to the lungs via combined PLGA-SS-PEG and OBAE NPs. The use of DiR as a fluorescent payload indirectly evaluated the efficiency of the NPs to release a small hydrophobic molecule such as DTX. Fluorescence from DiR was observed throughout the lungs within 1 h from both PLGA-PEG and the PLGA-SS-PEG formulations, indicating successful transport of the formulations, while the more rapid loss of the DiR signal in the lungs from the PLGA-SS-PEG NPs compared to the non-redox responsive variant implied a faster release of the payload across the lungs and into systemic circulation. The end-fate organ accumulation data showed that DiR was present mostly in the liver and kidneys at 24 h, as expected for small molecule transport, but were also indicative that for subsequent experiments relating to DTX delivery, some formulation tuning would be required to sustain release in the lungs and delay passage through to systemic circulation. For the siRNA delivery assays, the data showed effective knockdown of luciferase from the constitutively expressing A549 luc cells after 72 h for both formulations.
When considered together, cell culture and preliminary in vivo experiments demonstrated both the promise and complexity in using combined NPs. The cell data clearly demonstrated a combined effect of DTX and TUBB3 siRNA in LC cells, and indicated that the PLGA-SS-PEG redox-responsive formulation was the most effective. For the orthotopic lung cancers in mice, successful delivery of both a small molecule dye and a siRNA was demonstrated, with effective knockdown of luciferase showing that NPs were able to cross tissue and intracellular barriers to release an active biological cargo. However, the differences in the formulations were less apparent in the in vivo model, which may have related to the intricacies in transport of small and large molecules in lung tissue, and the impact of redox-responsiveness on dye/drug effective dose accumulating in cancer cells. There is an imperative to evaluate differential organ accumulation of these drugs when delivered in vivo as well as to establish routes to control release rates for each therapeutic from combination NPs. These questions are now under evaluation and will be reported subsequently.
Conclusions
Here we have reported novel redox-responsive PEGylated NPs, with the aim to deliver simultaneously into the lung the conventional anticancer drug docetaxel and a therapeutic siRNA against βIII-tubulin (TUBB3 siRNA). Both drugs were loaded inside the carrier and showed a complete and fast release at simulated intracellular levels of reductive agents. The redox-responsiveness of these systems led to higher silencing efficiency of delivered siRNA and cell uptake, in comparison to non-redox-responsive variant. Concerning anticancer activity, the co-delivery of TUBB3 siRNA through bioresponsive nanoparticles strongly increased DTX cytotoxicity in lung cancer cells, thus highlighting the effective combined anticancer activity of the compounds. In vivo, these systems were well tolerated after intratracheal administration in a bioluminescent A549-luc lung cancer model. The suitability of the system for inhalation delivery was also demonstrated, with data showing that NPs reached the lungs where an anti-Luc siRNA was released and exerted its silencing activity. These results indicate that combining DTX with TUBB3 siRNA in redox-responsive NPs as we have shown here could lead to promising candidates for the local and combined therapy of lung cancer with a specific final goal of reducing drug resistance related to the current clinically used therapeutics.Experimental
Materials
All solvents were of analytical or HPLC grade and purchased from Sigma or Fisher Scientific unless otherwise stated. Deuterated solvents, the two monomers glycolide (GL) and DL-lactide (LA) were from Sigma. Poly(ethylene glycol) methyl ether (mPEG) and α-methoxy-ω-amino poly(ethylene glycol) (mPEG-NH2) with Mn = 2.0 kDa were purchased from Iris Biotech GmbH (Germany). Dialysis membrane (MWCO 3500, regenerated cellulose) was used as received from Spectrapor. Tin(II)-2-ethylhexanoate (SnOct2), acryloyl chloride, ethylene-dioxy-bis-ethylamine, triethylamine (TEA), dithiodiethanol, Tris-Acetate-EDTA (TAE) buffer, poly(vinyl alcohol) (PVA, Mowiol®40–88, Mw ∼ 31000), sodium chloride, potassium phosphate dibasic and potassium phosphate monobasic, sodium azide, magnesium chloride, potassium chloride, sodium phosphate dibasic, sodium sulfate, calcium chloride dehydrated, sodium acetate, sodium bicarbonate, sodium citrate dehydrate, ethidium bromide, Hoechst 33258, BCA reagent, glutathione (GSH), were used as received from Sigma Aldrich. Docetaxel (DTX, MW = 807.88) was purchased from LC laboratories (USA). 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine Iodide (DiR) was acquired by Thermo Fisher Scientific (UK). siRNA against Luciferase (Luc-siRNA) (CCGCAAGAUCCGCGAGAUU), scrambled siRNA, Silencer®Cy™3 labeled Negative Control (λex max = 547, λem = 563 nm) and siRNA against βIII-tubulin (TUBB3-siRNA) (GUACGUGCCUCGAGCCAUUUU) were provided by Eurogentec (UK). Luciferase Assay System and CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) were provided by Promega (UK).
Synthesis of PLGA block copolymers and OBAE
:1.5) in DCM in the dark at 30 °C for 5 days under nitrogen. (Mn = 2.1 kDa at 25 °C in DMF; Đ = 1.2; yield = 72%).
1H- and 13C NMR, FTIR spectra, size exclusion chromatography (SEC) measurements and differential scanning calorimetry (DSC) of both PLGA-based copolymers as well as OBAE were performed as previously reported.25
Preparation and characterization of NPs
000 rpm, 30 min, 4 °C) after washing twice with distillated water to remove the excess of PVA and the unloaded drugs. Finally, NPs were re-dispersed in water (1 mL), freeze-dried for 24 h and kept at 4 °C. Unloaded and NPs loaded with a single drug were also prepared as controls. Recovery yield of production process was evaluated by weighing the solid residue after freeze-drying. Results are expressed as the ratio of the actual NPs mass to the theoretical polymer mass × 100.
:60 (v/v) mixture of 20 mM phosphate buffer at pH 4.5 and acetonitrile pumped at a flow rate of 1 mL min−1. The UV detector was set at 227 nm. A calibration curve for DTX in THF was constructed in the concentration range 1–200 μg mL−1.
The loading of siRNA was confirmed by agarose gel retardation assays. 1 mg of NPs containing 1 μg of siRNA were loaded on 2% agarose gel in Tris-Acetate-EDTA (TAE) buffer and subjected to electrophoresis for 45 min at 70 V. siRNA bands were stained with ethidium bromide and finally visualized with an UV illuminator. The entrapment efficiency of siRNA was assessed by dissolving 1 mg of freeze dried NPs in 500 μL of DCM under stirring for 1 h. Thereafter, 500 μL of water were added and the sample stirred for 1 h further. After centrifugation at 5000 rpm for 10 min, the aqueous phase was collected and analysed by UV spectrophotometry (Nanodrop 2000 spectrophotometer, Thermo Scientific, UK) at 290 nm. Potential interference of the polymers and DTX on siRNA absorbance was preliminary assessed spiking a siRNA solution in water with different amount of DTX-loaded NPs.
In vitro release of DTX in PBS with or without GSH 10 mM was investigated by a dialysis method. 2 mg of NPs were dispersed in 0.5 mL of PBS solution and placed in a dialysis bag (MWCO = 3500 Da, Spectra/Por®). The sample was plunged in 8 mL of PBS (sink condition) and kept at 37 °C. At selected time intervals, 1 mL of release medium was withdrawn and replaced with an equal volume of fresh medium. DTX quantitative analysis was performed by HPLC as described above. Release profile of the free drug is reported for comparison. Results are expressed as release% over time ± SD of three experiments.
For siRNA release studies, 500 μL of NP dispersions were diluted with PBS at pH 7.4 with or without GSH 20 mM to a total volume of 1 mL and incubated at 37 °C. At different time points aliquots of 500 μl of NP dispersions were collected and centrifuged at 16000g for 30 min at 4 °C. The supernatants were collected and analysed at 280 nm by UV-vis spectrometry (NanoDrop™ 2000). The values were calculated as mean ± SD of three experiments.
Cell studies
In vivo studies
Dosing was initiated in established tumors (mice six to seven weeks old), those whose volume was demonstrably larger than the original implantation volume. Mice were anesthetized with 10 ml g−1 ketamine/medatomidine (Ket 0.9 ml/Met 0.3 ml/NaCl 8.8 ml), restrained on a support ring and dosed intratracheally by a Microsprayer Aerosolizer (Model IA-IC, PennCentury, Philadelphia, PA, USA) (50 μL per dose). The administration was guided by a pediatric otoscope. All animals were sacrificed when tumor volume reached the maximum permissible size of 1.5 cm diameter in at least one mouse.
Data access statement
All relevant data are available from the University of Nottingham's Research Data Management Repository or via email from the corresponding authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
For this work, Claudia Conte was supported by a fellowship by Associazione Italiana per la Ricerca sul Cancro (AIRC) co-funded by the European Union (iCARE/Marie Curie 2014). Maria Kavallaris is supported by an NHMRC Principal Research Fellowship (APP1119152) and is funded by an Australian Research Council Centre of Excellence in Convergent Bio-Nano Science and Technology (CE140100036), and NHMRC Program Grant (APP1091261) and Cancer Australia Grant (1141485). This work was supported by the Engineering and Physical Sciences Research Council [grant numbers EP/H005625/1, EP/N03371X/1]; and the Royal Society [Wolfson Research Merit Award WM150086] to CA. We also thank Alison Ritchie and Giuseppe Inebria for assistance with in vivo experiments, Tom Booth, Paul Cooling, Esme Ireson and Douglas Crackett for technical support, the Nanoscale and Microscale Research Centre (NMRC) at UoN for TEM images and Carol Turrill for exceptional administrative and organizational contributions.References
- L. A. Torre, R. L. Siegel and A. Jemal, Adv. Exp. Med. Biol., 2016, 893, 1–19 CrossRef PubMed.
- R. Bayat Mokhtari, T. S. Homayouni, N. Baluch, E. Morgatskaya, S. Kumar, B. Das and H. Yeger, Oncotarget, 2017, 8, 38022–38043 CrossRef PubMed.
- M. J. Smyth, S. F. Ngiow, A. Ribas and M. W. Teng, Nat. Rev. Clin. Oncol., 2016, 13, 143–158 CrossRef CAS PubMed.
- B. Mansoori, S. Sandoghchian Shotorbani and B. Baradaran, Adv. Pharm. Bull., 2014, 4, 313–321 Search PubMed.
- N. S. Gandhi, R. K. Tekade and M. B. Chougule, J. Controlled Release, 2014, 194, 238–256 CrossRef CAS PubMed.
- C. Chakraborty, A. R. Sharma, G. Sharma, C. G. P. Doss and S. S. Lee, Mol. Ther.–Nucleic Acids, 2017, 8, 132–143 CrossRef CAS PubMed.
- C. Bouillon, Y. Bessin, F. Poncet, M. Gary-Bobo, P. Dumy, M. Barboiu, N. Bettache and S. Ulrich, J. Mater. Chem. B, 2018, 6, 7239–7246 RSC.
- F. Freitag and E. Wagner, Adv. Drug Delivery Rev., 2020, 168, 30–54 CrossRef PubMed.
- W. Viricel, S. Poirier, A. Mbarek, R. M. Derbali, G. Mayer and J. Leblond, Nanoscale, 2017, 9, 31–36 RSC.
- R. Kandil, Y. Xie, R. Heermann, L. Isert, K. Jung, A. Mehta and O. M. Merkel, Adv. Ther., 2019, 2, 1900047 CrossRef PubMed.
- L. J. Li, W. Sun, L. Li, Y. Y. Liu, L. Wu, F. L. Wang, Z. Zhou, Z. R. Zhang and Y. Huang, Nanoscale, 2017, 9, 314–325 RSC.
- J. Luo, E. Wagner and Y. Wang, J. Mater. Chem. B, 2020, 8, 2020–2031 RSC.
- S. Baek, R. K. Singh, D. Khanal, K. D. Patel, E. J. Lee, K. W. Leong, W. Chrzanowski and H. W. Kim, Nanoscale, 2015, 7, 14191–14216 RSC.
- F. Saviano, T. Lovato, A. Russo, G. Russo, C. R. Bouton, R. J. Shattock, C. Alexander, F. Quaglia, A. K. Blakney, P. Gurnani and C. Conte, J. Mater. Chem. B, 2020, 8, 4940–4949 RSC.
- W. J. Huang, X. X. Wang, C. R. Wang, L. L. Du, J. H. Zhang, L. D. Deng, H. Q. Cao and A. J. Dong, J. Mater. Chem. B, 2019, 7, 965–973 RSC.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- H. Sun, Y. Zhang and Z. Zhong, Adv. Drug Delivery Rev., 2018, 132, 16–32 CrossRef CAS.
- R. Cheng, F. Feng, F. H. Meng, C. Deng, J. Feijen and Z. Y. Zhong, J. Controlled Release, 2011, 152, 2–12 CrossRef CAS.
- R. Kandil and O. M. Merkel, Ther. Delivery, 2019, 10, 203–206 CrossRef CAS PubMed.
- I. d'Angelo, C. Conte, A. Miro, F. Quaglia and F. Ungaro, Curr. Top. Med. Chem., 2015, 15, 386–400 CrossRef PubMed.
- F. Ungaro, I. d'Angelo, A. Miro, M. I. La Rotonda and F. Quaglia, J. Pharm. Pharmacol., 2012, 64, 1217–1235 CrossRef CAS PubMed.
- S. Rezvantalab, N. I. Drude, M. K. Moraveji, N. Guvener, E. K. Koons, Y. Shi, T. Lammers and F. Kiessling, Front. Pharmacol., 2018, 9, 1260 CrossRef CAS.
- C. Conte, F. Mastrotto, V. Taresco, A. Tchoryk, F. Quaglia, S. Stolnik and C. Alexander, J. Controlled Release, 2018, 277, 126–141 CrossRef CAS PubMed.
- J. Karlsson, Y. Rui, K. L. Kozielski, A. L. Placone, O. Choi, S. Y. Tzeng, J. Kim, J. J. Keyes, M. I. Bogorad, K. Gabrielson, H. Guerrero-Cazares, A. Quifiones-Hinojosa, P. C. Searson and J. J. Green, Nanoscale, 2019, 11, 20045–20057 RSC.
- T. Lovato, V. Taresco, A. Alazzo, C. Sansone, S. Stolnik, C. Alexander and C. Conte, J. Mater. Chem. B, 2018, 6, 6550–6558 RSC.
- K. L. Kozielski and J. J. Green, Methods Mol. Biol., 2016, 1364, 79–87 CrossRef CAS PubMed.
- K. L. Kozielski, S. Y. Tzeng, B. A. De Mendoza and J. J. Green, ACS Nano, 2014, 8, 3232–3241 CrossRef CAS PubMed.
- L. M. Ensign, C. Schneider, J. S. Suk, R. Cone and J. Hanes, Adv. Mater., 2012, 24, 3887–3894 CrossRef CAS PubMed.
- C. Dumontet and B. I. Sikic, J. Clin. Oncol., 1999, 17, 1061–1070 CrossRef CAS PubMed.
- P. P. Gan, E. Pasquier and M. Kavallaris, Cancer Res., 2007, 67, 9356–9363 CrossRef CAS PubMed.
- J. A. McCarroll, P. P. Gan, R. B. Erlich, M. Liu, T. Dwarte, S. S. Sagnella, M. C. Akerfeldt, L. Yang, A. L. Parker, M. H. Chang, M. S. Shum, F. L. Byrne and M. Kavallaris, Cancer Res., 2015, 75, 415–425 CrossRef CAS PubMed.
- J. A. McCarroll, P. P. Gan, M. Liu and M. Kavallaris, Cancer Res., 2010, 70, 4995–5003 CrossRef CAS PubMed.
- C. S. Schneider, Q. Xu, N. J. Boylan, J. Chisholm, B. C. Tang, B. S. Schuster, A. Henning, L. M. Ensign, E. Lee, P. Adstamongkonkul, B. W. Simons, S. S. Wang, X. Gong, T. Yu, M. P. Boyle, J. S. Suk and J. Hanes, Sci. Adv., 2017, 3, e1601556 CrossRef PubMed.
- C. Conte, F. Mastrotto, V. Taresco, A. Tchoryk, F. Quaglia, S. Stolnik and C. Alexander, J. Controlled Release, 2018, 277, 126–141 CrossRef CAS PubMed.
- H. Y. Xue, S. Liu and H. L. Wong, Nanomedicine, 2014, 9, 295–312 CrossRef CAS PubMed.
- M. Breunig, U. Lungwitz, R. Liebl and A. Goepferich, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14454–14459 CrossRef CAS PubMed.
- A. A. Eltoukhy, D. J. Siegwart, C. A. Alabi, J. S. Rajan, R. Langer and D. G. Anderson, Biomaterials, 2012, 33, 3594–3603 CrossRef CAS PubMed.
- W. Sun and P. B. Davis, J. Controlled Release, 2010, 146, 118–127 CrossRef CAS PubMed.
- S. Y. Lee, J. Y. Tyler, S. Kim, K. Park and J. X. Cheng, Mol. Pharm., 2013, 10, 3497–3506 CrossRef CAS PubMed.
- C. Conte, F. Ungaro, G. Maglio, P. Tirino, G. Siracusano, M. T. Sciortino, N. Leone, G. Palma, A. Barbieri, C. Arra, A. Mazzaglia and F. Quaglia, J. Controlled Release, 2013, 167, 40–52 CrossRef CAS PubMed.
- G. Palma, C. Conte, A. Barbieri, S. Bimonte, A. Luciano, D. Rea, F. Ungaro, P. Tirino, F. Quaglia and C. Arra, Int. J. Pharm., 2014, 473, 55–63 CrossRef CAS PubMed.
- F. Ungaro, C. Conte, L. Ostacolo, G. Maglio, A. Barbieri, C. Arra, G. Misso, A. Abbruzzese, M. Caraglia and F. Quaglia, Nanomedicine, 2012, 8, 637–646 CrossRef CAS PubMed.
- C. Y. Hong, Y. Z. You, D. C. Wu, Y. Liu and C. Y. Pan, J. Am. Chem. Soc., 2007, 129, 5354–5355 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr02179f |
| This journal is © The Royal Society of Chemistry 2021 |